
KYNTHEUM injektioneste, liuos, esitäytetty ruisku 210 mg
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty ruisku sisältää 210 mg brodalumabia 1,5 ml:ssa liuosta.
1 ml liuosta sisältää 140 mg brodalumabia (brodalumab.).
Brodalumabi on kiinanhamsterin munasarjasoluissa (CHO) rekombinantti-DNA-tekniikalla tuotettu ihmisen monoklonaalinen vasta‑aine.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Kyntheum on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon aikuisille, joille harkitaan systeemistä hoitoa.
Ehto
Valmiste on tarkoitettu käytettäväksi psoriaasin diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Kyntheum on tarkoitettu käytettäväksi psoriaasin diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus
Suositeltu annos on 210 mg injektiona ihon alle viikkoina 0, 1 ja 2, ja tämän jälkeen 210 mg annettuna kahden viikon välein.
Jos potilaalla ei ole vastetta 12–16 viikon hoidon jälkeen, hoidon lopettamista on harkittava. Osa potilaista, joilla on aluksi osittainen vaste, voi parantua myöhemmin, kun hoitoa jatketaan yli 16 viikkoa.
Iäkkäät potilaat (vähintään 65 vuoden ikäiset)
Iäkkäiden potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten ja maksan vajaatoiminta
Kyntheum‑valmistetta ei ole tutkittu näissä potilasryhmissä. Annossuosituksia ei voida antaa.
Pediatriset potilaat
Kyntheum‑valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Kyntheum annetaan injektiona ihon alle. Jokainen esitäytetty ruisku on tarkoitettu vain kertakäyttöön. Kyntheum-valmistetta ei saa pistää ihoalueille, joilla on kipua, mustelmia, punoitusta, kovettumia, paksuuntumaa, hilseilyä tai psoriaasimuutoksia. Esitäytettyä ruiskua ei saa ravistaa.
Kun potilaalle on opetettu ihon alle pistämisessä käytettävä asianmukainen tekniikka, hän voi pistää Kyntheum‑valmisteen itse, jos lääkäri katsoo sen olevan tarkoituksenmukaista. Potilasta on neuvottava pistämään koko Kyntheum‑valmistemäärä pakkausselosteen ohjeiden mukaisesti. Pakkausselosteen lopussa on valmisteen tarkat käyttöohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiivinen Crohnin tauti.
Kliinisesti merkittävät aktiiviset infektiot (esim. aktiivinen tuberkuloosi, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Tulehduksellinen suolistosairaus (mukaan lukien Crohnin tauti ja haavainen koliitti)
IL‑17:n estäjien käytön yhteydessä on ilmoitettu uusista tulehduksellisen suolistosairauden tapauksista tai sairauden pahenemisesta. Siksi brodalumabia ei suositella potilaille, joilla on tulehduksellinen suolistosairaus (ks. kohta Haittavaikutukset). Jos potilaalle kehittyy tulehduksellisen suolistosairauden oireita ja merkkejä tai hänen tulehduksellinen suolistosairautensa pahentuu, hoito tulisi lopettaa ja suolistosairautta tulisi alkaa hoitamaan asianmukaisesti.
Itsemurha‑ajatukset ja ‑käyttäytyminen
Brodalumabilla hoidetuilla potilailla on raportoitu olleen itsemurha‑ajatuksia ja ‑käyttäytymistä, mukaan lukien toteutettu itsemurha. Suurimmalla osalla potilaista, joilla oli itsemurhakäyttäytymistä, oli ollut aiemminkin masennusta ja/tai itsemurha‑ajatuksia tai ‑käyttäytymistä. Brodalumabihoidon ja lisääntyneen itsemurha‑ajatus‑ ja ‑käyttäytymisriskin välistä syy‑yhteyttä ei ole todettu.
Brodalumabihoidon riskit ja hyödyt on punnittava huolellisesti niiden potilaiden kohdalla, joilla on ollut masennusta ja/tai itsemurha‑ajatuksia tai ‑käyttäytymistä tai joille kehittyy kyseisiä oireita. Potilaita, hoitajia ja perheitä on neuvottava seuraamaan mahdollista masennuksen, itsemurha‑ajatusten, ahdistuneisuuden tai muiden mielialamuutosten ilmenemistä tai pahenemista, ja heidän on otettava yhteyttä hoitopaikkaansa, jos tällaisia tapahtumia ilmenee. Jos potilaalla on uusia tai pahenevia masennuksen oireita, ja/tai itsemurha-ajatuksia tai -käyttäytymistä havaitaan, hoidon lopettaminen on suositeltavaa.
Yliherkkyysreaktiot
Markkinoille tulon jälkeen on raportoitu esiintyvyydeltään harvinaisia yliherkkyysreaktioita. Anafylaktisen reaktion tai muun allergisen reaktion esiintyessä brodalumabin annostelu tulee lopettaa ja aloittaa oireenmukainen hoito.
Infektiot
Brodalumabi saattaa lisätä infektioiden riskiä.
12 viikon lumelääkekontrolloidun kliinisen tutkimuksen jaksolla, johon osallistui psoriaasipotilaita, vakavia infektioita havaittiin 0,5 %:lla brodalumabia saaneista potilaista (ks. kohta Haittavaikutukset).
Varovaisuutta on noudatettava, kun harkitaan brodalumabin käyttöä potilaille, joilla on krooninen infektio tai joilla on ollut toistuvia infektioita. Potilaita on neuvottava hakeutumaan lääkärin hoitoon, jos infektion oireita tai merkkejä ilmenee. Jos potilaalle kehittyy vakava infektio, häntä on seurattava huolellisesti eikä brodalumabia saa antaa, ennen kuin infektio on parantunut.
Brodalumabia ei saa antaa potilaille, joilla on aktiivinen tuberkuloosi. Tuberkuloosilääkitystä on harkittava ennen hoidon aloittamista potilaille, joilla on latentti tuberkuloosi.
Rokotukset
Potilaiden rokotukset on suositeltavaa saattaa ajan tasalle paikallisten rokotussuositusten mukaisesti ennen hoidon aloittamista. Eläviä rokotteita ei saa antaa brodalumabihoidon aikana (ks. kohta Yhteisvaikutukset). Tietoja brodalumabia saavien potilaiden vasteesta eläviä viruksia sisältäviin rokotteisiin, infektioriskistä tai infektion tarttumisesta eläviä viruksia sisältävien rokotteiden antamisen jälkeen ei ole saatavilla.
Imeväisten rokotukset
Kolmannen raskauskolmanneksen aikana brodalumabille altistuneiden imeväisten rokottamisesta elävillä rokotteilla on keskusteltava lääkärin kanssa (ks. myös kohta Raskaus ja imetys).
Samainaikainen immunosuppressiivinen hoito
Brodalumabin turvallisuutta ja tehoa yhdessä immunosuppressanttien, mukaan lukien biologiset lääkkeet, tai valohoidon kanssa, ei ole tutkittu.
Yhteisvaikutukset
Eläviä rokotteita ei saa antaa brodalumabihoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
CYP450‑entsyymien muodostumisessa voi tapahtua muutoksia, kun tiettyjen sytokiinien (esim. IL‑1, IL‑6, IL‑10, TNFα, IFN) pitoisuus kasvaa kroonisen tulehduksen yhteydessä. Vaikka interleukiinien (IL)‑17A ja IL‑17RA roolia ei ole raportoitu CYP450‑entsyymien säätelyssä, brodalumabin vaikutusta CYP3A4/3A5:n aktiivisuuteen arvioitiin sairaus‑lääke‑lääke‑yhteisvaikutustutkimuksessa.
Yksi ihon alle annettu 210 mg:n annos brodalumabia lisäsi keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavien potilaiden altistumista midatsolaamille, CYP3A4/3A5‑substraatille, 24 %:lla. Midatsolaamille altistumisen muutoksen laajuuden perusteella CYP3A4/3A5‑substraattien annoksen muuttaminen ei ole tarpeen, käytettäessä samanaikaisesti brodalumabin kanssa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 12 viikkoa hoidon päättymisen jälkeen.
Raskaus
Ei ole tietoja tai on vain vähän tietoja brodalumabin käytöstä raskaana oleville naisille.
Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kappale Prekliiniset tiedot turvallisuudesta).
Ihmisen IgG2:n tiedetään läpäisevän veri‑istukkaesteen, ja brodalumabi on ihmisen IgG2:ta. Tämän vuoksi brodalumabi saattaa siirtyä äidiltä kehittyvälle sikiölle. Varmuuden vuoksi Kyntheum‑valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Koska brodalumabin metaboloitumista imeväisillä ei tunneta, hyödyistä ja riskeistä, jotka liittyvät imeväisen altistumiseen eläviä viruksia sisältäville rokotteille kolmannen raskauskolmanneksen aikana tapahtuneen Kyntheum‑altistuksen jälkeen, on keskusteltava lääkärin kanssa.
Imetys
Ei tiedetä, erittyykö brodalumabi ihmisen rintamaitoon. Brodalumabi on monoklonaalinen vasta‑aine, ja sitä odotetaan olevan ensimaidossa ja pieninä pitoisuuksina myöhemmin.
Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea.
On päätettävä lopetetaanko rintaruokinta vai lopetetaanko/pidättäydytäänkö Kyntheum‑hoidosta ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Brodalumabin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Eläinkokeissa ei ole havaittu vaikutuksia urosten tai naarasten sukupuolielimiin tai siittiöiden määrään, liikkuvuuteen tai morfologiaan (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kyntheum‑valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitetut haittavaikutukset olivat nivelkipu (4,6 %), päänsärky (4,3 %), väsymys (2,6 %), ripuli (2,2 %) ja suunielun kipu (2,1 %).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa ja markkinoille tulon jälkeen ilmoitetut haittavaikutukset (Taulukko 1) esitetään MedDRA‑elinjärjestelmäluokituksen mukaisesti. Haittavaikutukset on luokiteltu kussakin elinjärjestelmäluokassa yleisyyden mukaan yleisimmistä alkaen. Kunkin haittavaikutuksen kohdalla mainittu yleisyysluokka perustuu seuraavaan käytäntöön: hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1 000, <1/100), harvinainen (≥1/10 000, <1/1 000), hyvin harvinainen (<1/10 000) ja ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kunkin yleisyysluokan haittavaikutukset on esitetty vakavuusjärjestyksessä vakavimmasta alkaen.
Taulukko 1: Kliinisissä tutkimuksissa ja markkinoille tulon jälkeen todetut haittavaikutukset
| Elinjärjestelmä | Esiintymistiheys | Haittavaikutus |
| Infektiot | Yleinen | Influenssa Silsainfektiot (mukaan lukien jalkasilsa, savipuoli, nivussilsa) |
| Melko harvinainen | Candida‑infektiot (mukaan lukien suun, sukupuolielinten ja ruokatorven infektiot) | |
| Veri ja imukudos | Melko harvinainen | Neutropenia |
| Immuunijärjestelmä | Harvinainen | Anafylaktinen reaktio* |
| Hermosto | Yleinen | Päänsärky |
| Silmät | Melko harvinainen | Sidekalvotulehdus |
| Hengityselimet, rintakehä ja välikarsina | Yleinen | Suunielun kipu |
| Ruoansulatuselimistö | Yleinen | Ripuli Pahoinvointi |
| Luusto, lihakset ja sidekudos | Yleinen | Nivelsärky Lihassärky |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Väsymys Pistoskohdan reaktiot (mukaan lukien pistoskohdan punoitus, kipu, kutina, mustelmat, verenvuoto) |
| Ihon ja ihonalaisen kudoksen häiriöt | Tuntematon | Pyoderma gangraenosum |
Tiettyjen haittavaikutusten kuvaus
Tulehduksellinen suolistosairaus
IL‑17:n estäjien käytön yhteydessä on ilmoitettu uusista tulehduksellisen suolistosairauden (mukaan lukien Crohnin tauti ja haavainen koliitti) tapauksista tai sairauden pahenemisesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infektiot
12 viikon lumelääkekontrolloidulla kliinisen läiskäpsoriaasitutkimuksen jaksolla infektioita ilmoitettiin esiintyneen 28,2 %:lla brodalumabilla hoidetuista potilaista ja 23,4 %:lla lumelääkkeellä hoidetuista potilaista. Valtaosa infektiotapauksista oli nenänielun tulehduksia, ylähengitystieinfektioita, nielutulehduksia, virtsatietulehduksia, keuhkoputkentulehduksia, influenssaa ja nenän sivuonteloiden tulehduksia, jotka eivät edellyttäneet hoidon lopettamista. Vakavia infektioita esiintyi 0,5 %:lla brodalumabihoitoa saaneista potilaista ja 0,1 %:lla lumehoitoa saaneista potilaista. Brodalumabipotilailla havaittiin kliinisissä tutkimuksissa suurempia määriä sieni‑infektioita (2,5 %), ensisijaisesti ei‑vakavia ihon ja limakalvojen Candida‑infektioita, verrattuna lumelääkepotilaisiin (1,0 %).
Viikkoon 52 mennessä infektioille vakioidut määrät 100:aa potilasvuotta kohden olivat brodalumabilla hoidetuilla potilailla 134,7 ja ustekinumabilla hoidetuilla potilailla 124,1. Vakaville infektioille vakioidut määrät 100 potilasvuotta kohden olivat brodalumabilla hoidetuilla potilailla 2,4 ja ustekinumabilla hoidetuilla potilailla 1,2. Kliinisissä tutkimuksissa havaittiin yksi vakava kryptokokkimeningiittitapaus ja yksi vakava coccidioides-infektiotapaus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Neutropenia
Kliinisten tutkimusten 12 viikon lumelääkekontrolloidulla jaksolla neutropeniaa raportoitiin esiintyneen 0,9 %:lla brodalumabilla hoidetuista potilaista verrattuna 0,5 %:iin lumelääkkeellä hoidetuista potilaista. Useimmat brodalumabiin liittyvät neutropeniat olivat lieviä, tilapäisiä ja paranevia.
Luokan 3 (<1,0 × 109/l - 0,5 × 109/l) neutropeniaa raportoitiin esiintyneen 0,5 %:lla brodalumabia saavista potilaista verrattuna 0 %:iin ustekinumabia tai lumelääkettä saaneista potilaista. Luokan 4 (<0,5 × 109/l) neutropeniaa ei raportoitu brodalumabia tai lumelääkettä saavilla potilailla verrattuna 0,2 %:iin ustekinumabia saaneista potilaista. Neutropeniaan ei liittynyt vakavia infektioita.
Immunogeenisuus
Brodalumabille kehittyi vasta‑aineita 2,2 %:lla (88/3935) brodalumabilla enintään 52 viikkoa kliinisissä psoriaasitutkimuksissa hoidetuista potilaista (0,3 %:lla näistä potilaista oli brodalumabin vasta‑aineita lähtötilanteessa). Näistä potilaista yhdelläkään ei ollut neutraloivia vasta‑aineita.
Brodalumabin vasta‑aineiden muodostumiseen ei liittynyt todisteita muuttuneesta farmakokineettisestä profiilista, kliinisestä vasteesta tai turvallisuusprofiilista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa laskimoon on annettu enintään 700 mg:n annoksia ilman annosta rajoittavaa toksisuutta. Yliannostustapauksissa on suositeltavaa seurata potilaan vointia haittavaikutusten merkkien ja oireiden varalta ja aloittaa asianmukainen oireenmukainen hoito viipymättä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunosuppressantit, interleukiinin estäjät, ATC‑koodi: L04AC12
Vaikutusmekanismi
Brodalumabi on rekombinantti ihmisen monoklonaalinen IgG2‑immunoglobuliinivasta‑aine, joka sitoutuu suurella affiniteetilla ihmisen interleukiini-17 reseptori A (IL‑17RA):han estäen proinflammatoristen sytokiinien IL‑17A, IL‑17F, IL‑17A/F‑heterodimeeri, IL-17C ja IL-17E (kutsutaan myös IL‑25:ksi) biologisia toimintoja, jolloin psoriaasiin liittyvä tulehdus ja kliiniset oireet estyvät. IL‑17RA on proteiini, joka esiintyy solun pinnalla. Se on useiden IL‑17‑perheen sytokiinien käyttämien reseptorikompleksien välttämätön osa. IL‑17‑perheen sytokiinipitoisuuksien on raportoitu kasvaneen psoriaasin yhteydessä. IL‑17A:lla, IL‑17F:llä ja IL‑17A/F‑heterodimeerillä on pleiotrooppisia toimintoja, mukaan lukien proinflammatoristen välittäjien, kuten IL‑6:n, GROα:n ja G‑CSF:n, induktio epiteelisoluista, endoteelisoluista ja fibroblasteista, mikä edistää kudostulehdusta. IL-17C:n on osoitettu saavan aikaan keratinosyyteissä samanlaisen vasteen kuin IL-17A ja IL-17F. IL‑17RA:n estäminen estää IL‑17‑sytokiinin aiheuttamia vasteita, mistä seuraa ihon tulehdustilan normalisoituminen.
Farmakodynaamiset vaikutukset
IL‑17A‑, IL‑17C‑ ja IL‑17F‑geeniekspression on todettu lisääntyneen psoriaasiläiskissä. Psoriaasiläiskissä esiintyy myös IL‑23:n (IL‑17A:n ja IL-17F:n ylävirran aktivaattori) kahden alayksikön geenien IL‑12B:n ja IL‑23A:n suurentuneita ilmentymäpitoisuuksia. Psoriaasipotilaiden hoitamisen brodalumabilla on osoitettu pienentävän IL‑17A:n sekä solujen proliferaation ja epidermaalisen paksuuntumisen merkkiaineiden pitoisuuksia psorileesio-ihobiopsioissa terveen ihon biopsioiden pitoisuuksien tasolle enintään 12 viikkoa hoidon jälkeen.
Kliininen teho ja turvallisuus
Brodalumabin tehoa ja turvallisuutta arvioitiin kolmessa monikansallisessa, satunnaistetussa, kaksoissokkoutetussa, vaiheen 3 lumelääkekontrolloidussa kliinisessä tutkimuksessa (AMAGINE‑1, AMAGINE‑2, and AMAGINE‑3), joihin osallistui 4 373 aikuista läiskäpsoriaasipotilasta. AMAGINE‑2‑ ja AMAGINE‑3‑tutkimukset olivat myös aktiivisella vertailuvalmisteella (ustekinumabi) kontrolloituja. Kaikki kolme tutkimusta sisälsivät 12 viikon lumelääkekontrolloidun induktiovaiheen, 52 viikkoa kestäneen kaksoissokkoutuksen ja avoimen pitkäaikaisen jatkovaiheen.
Mukaan otetuille potilaille oli harkittu systeemistä hoitoa, mukaan lukien valohoitoa, biologisia ja ei-biologisia systeemisiä hoitoja. Noin 21 %:lla potilaista oli ollut psoriaasiartriitti. Noin 30 % potilaista oli saanut aiemmin biologista hoitoa ja 13 %:lla potilaista biologinen hoito oli epäonnistunut.
Potilaat olivat pääasiassa miehiä (70 %) ja valkoisia (91 %). Keski-ikä oli 45 vuotta (ikähaarukka 18–86 vuotta) ja näistä 6,4 % oli ≥65-vuotiaita ja 0,3 % oli >75-vuotiaita. Kaikissa hoitoryhmissä lähtötilanteen psoriaasin vaikeusindeksi (PASI) -pisteet vaihtelivat välillä 9,4–72 (mediaani: 17,4), ja lähtötilanteessa kehon pinta-ala (BSA) vaihteli välillä 10–97 (mediaani: 21). Lähtötilanteen lääkärin staattisen kokonaisarvion (sPGA) pisteet vaihtelivat välillä ”3 (keskivaikea)” (58 %) ja ”5 (erittäin vaikea)” (5 %).
AMAGINE‑1‑tutkimukseen osallistui 661 potilasta. Tutkimus sisälsi 12 viikon kaksoissokkoutetun, lumelääkekontrolloidun induktiovaiheen, jota seurasi enintään 52 viikon kaksoissokkoutettu poistumis‑ ja uudelleenhoitovaihe. Potilaat, jotka satunnaistettiin saamaan brodalumabia, saivat 210 mg tai 140 mg viikolla 0 (päivänä 1), viikolla 1 ja viikolla 2, minkä jälkeen he saivat saman annoksen 2 viikon välein. Viikolla 12 potilaat, jotka satunnaistettiin alun perin saamaan brodalumabia ja jotka saavuttivat onnistuneen hoitotuloksen sPGA-vasteen (0 tai 1), satunnaistettiin uudelleen saamaan joko lumelääkettä tai brodalumabia aloitusannoksellaan. Potilaat, jotka satunnaistettiin alunperin saamaan lumelääkettä ja potilaat, jotka eivät täyttäneet uudelleensatunnaistamisen kriteereitä, saivat brodalumabia 210 mg kahden viikon välein alkaen viikosta 12. Uudelleenhoitoa oli saatavilla viikolla 16 tai sen jälkeen potilaille, joiden sairaus palasi, ja pelastushoitoa oli saatavilla 12 viikon jälkeen uudelleenhoidosta.
AMAGINE‑2 ja AMAGINE‑3 olivat identtisiä lumelääke‑ ja ustekinumabikontrolloituja tutkimuksia, joihin osallistui 1 831 ja 1 881 potilasta. Molempiin tutkimuksiin kuului 12 viikon kaksoissokkoutettu, lumelääke‑ ja ustekinumabikontrolloitu induktiovaihe, jota seurasi kaksoissokkoutettu ylläpitovaihe, joka kesti enintään 52 viikkoa. Potilaat, jotka satunnaistettiin induktiovaiheessa saamaan brodalumabia, saivat 210 mg tai 140 mg viikolla 0 (päivänä 1), viikolla 1 ja viikolla 2, minkä jälkeen he saivat saman annoksen 2 viikon välein. Potilaat, jotka satunnaistettiin saamaan ustekinumabia, saivat 45 mg, jos painoivat enintään 100 kg, ja 90 mg, jos painoivat yli 100 kg, viikolla 0, 4 ja 16, minkä jälkeen he saivat saman annoksen aina 12 viikon välein. Viikolla 12 potilaat, jotka satunnaistettiin alunperin saamaan brodalumabia, satunnaistettiin uudelleen saamaan joko 210 g kahden viikon välein tai 140 mg kahden viikon välein tai 140 mg neljän viikon välein tai 140 mg kahdeksan viikon välein ylläpitovaiheen aikana. Potilaat, jotka satunnaistettiin alunperin saamaan lumelääkettä, saivat brodalumabia 210 mg kahden viikon välein alkaen viikosta 12. Viikolla 12 ustekinumabiryhmän potilaat saivat edelleen ustekinumabia, minkä jälkeen ustekinumabi vaihdettiin 210 mg:aan brodalumabia kahden viikon välein viikolla 52. Pelastushoitoa oli saatavilla viikolla 16 tai sen jälkeen potilaille, joilla oli riittämätön yksittäinen sPGA‑vaste ≥3 tai jatkuva sPGA‑vaste 2 vähintään 4 viikon jakson ajan.
Taulukko 2: Tärkeimpien tehotulosten yleiskatsaus
| AMAGINE-1 | AMAGINE-2 ja AMAGINE-3 | ||||
| Lumelääke | Brodalumabi 210 mg Q2W | Lumelääke | Brodalumabi 210 mg Q2W | Ustekinumabi | |
| n-satunnaistettu | 220 | 222 | 624 | 1 236 | 613 |
| n-viikolla12 | 209 | 212 | 601 | 1 205 | 594 |
| n-ylläpitovaihe | 84 | 83 | – | 339 | 590 |
| n-viikolla52 | 2 | 74 | – | 236 | 300 |
| PASI | |||||
| PASI lähtötilanteen pisteet (keskiarvo ± SD) | 19,7±7,7 | 19,4±6,6 | 20,2±8,4 | 20,3±8,3 | 20,0±8,4 |
| PASI 75, viikko 12 (%) | 3 | 83* | 7 | 86* | 70* |
| PASI 75, viikko 52 (%) | 0 | 87* | – | 65 | 48 |
| sPGA (%) | |||||
| sPGA 0 tai 1, viikko 12 | 1 | 76* | 4 | 79* | 59* |
| sPGA 0 tai 1, viikko 52 | 0 | 83* | – | 65 | 45 |
| PSI | |||||
| PSIlähtötilanteen pisteet (keskiarvo ± SD) | 19,0±6,7 | 18,9±6,7 | 18,8±6,9 | 18,7±7,0 | 18,8±6,9 |
| PSIvast. saan. viikko 12 (%) | 4 | 61* | 7 | 64* | 54* |
Q2W = kahden viikon välein. PSI = Psoriasis Symptom Inventory. PSI-vasteen saaneet: kokonaispisteet ≤8 ilman >1 pisteitä; SD: keskihajonta. Puuttuvat tiedot tulkittiin vastetta saamattomaksi tulokseksi. Muihin annostuksiin uudelleensatunnaistuksen vuoksi n-ylläpitovaihe on merkittävästi pienempi kuin n-satunnaistettu useissa ryhmissä. AMAGINE-2- ja -3-tutkimusten ylläpitovaihe ei sisältänyt lumelääkettä. *p-arvo vs. vastaava lumelääke, mukautettu ositustekijöille <0,001 | |||||
Vaiheen 3 tutkimuksissa PASI 75 -vasteen saavutti 2 viikon kohdalla 20–25 % potilaista lumelääkkeeseen (0–0,6 %) ja ustekinumabiin (3–3,5 %) verrattuna.
Kuva 1: PASI 100 brodalumabin ja ustekinumabin induktio‑ ja ylläpitovaiheen aikana (AMAGINE‑2 ja AMAGINE‑3, yhdistetyt tulokset)
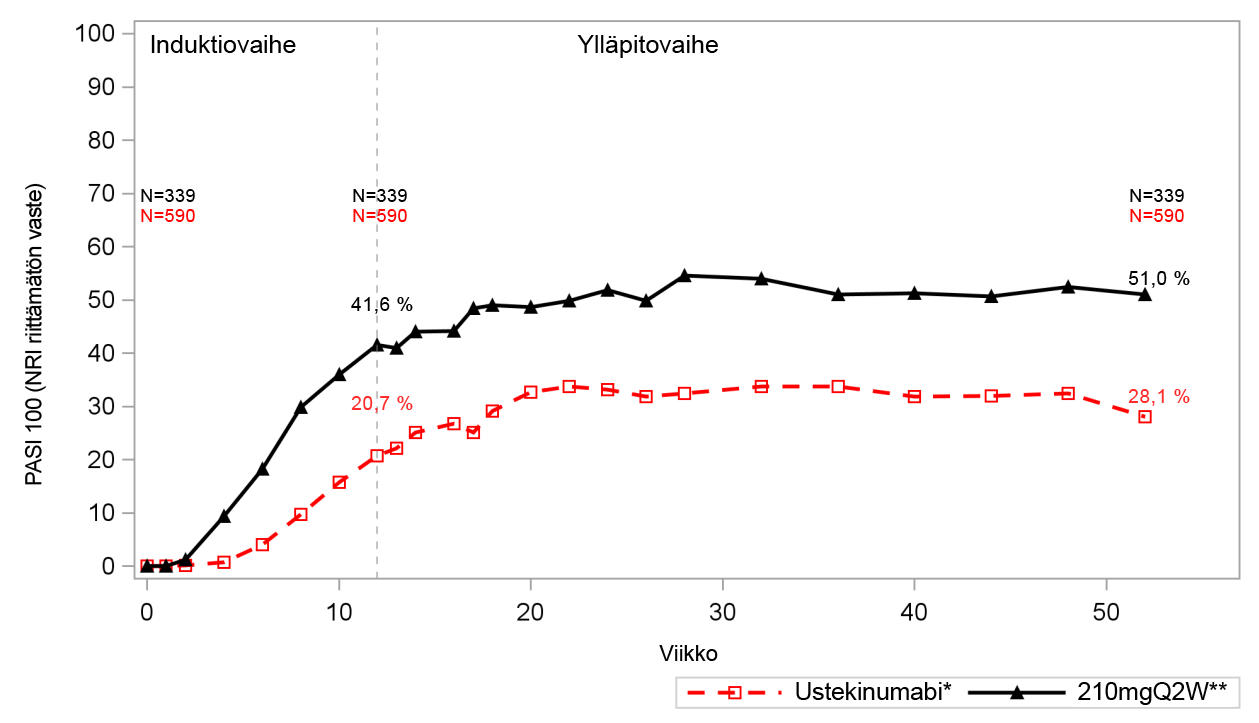
N = potilaiden määrä, joka ilmaistaan lähtötilanteessa, viikolla 12 ja viikolla 52.
Q2W = kahden viikon välein.
*Potilaille annettiin ustekinumabia induktiovaiheessa, ja he jatkoivat ustekinumabihoitoa ylläpitovaiheessa.
**Potilaille annettiin 210 mg brodalumabia kahden viikon välein induktiovaiheessa ja heidät uudelleensatunnaistettiin saamaan 210 mg brodalumabia kahden viikon välein ylläpitovaiheessa.
NRI = non-responder imputation (puuttuvat tiedot on tulkittu vastetta saamattomaksi tulokseksi)
Alaryhmä-analyyseissä iän, sukupuolen, rodun, systeemisen hoidon, valohoidon tai biologisen hoidon aiemman käytön ja biologisen hoidon epäonnistumisen suhteen ei havaittu eroja brodalumabin vasteessa kaikissa tärkeimmissä päätetapahtumissa (PASI 75, PASI 100, sPGA-vaste onnistunut hoitotulos [0 tai 1] ja sPGA puhdas [0]) kaikissa kolmessa kliinisessä tutkimuksessa.
Tehon ensisijaisten päätetapahtumien lisäksi kliinisesti tärkeitä parannuksia havaittiin päänahan psoriaasin indeksissä (Psoriasis Scalp Severity Index, PSSI) viikolla 12 (AMAGINE-1) ja kynsipsoriaasin indeksissä (Nail Psoriasis Severity Index, NAPSI) viikoilla 12 ja 52 (AMAGINE-1, -2 ja -3).
Elämänlaatu / potilaiden ilmoittamat vaikutukset
PSI-pisteet 0 (ei ollenkaan) tai 1 (lievä) saavuttaneiden potilaiden osuus kunkin oireen osalta (kutina, polttelu, kirvely, kipu, punoitus, suomuilu, halkeilu ja hilseily) viikolla 12 on esitetty taulukossa 2.
DLQI-pisteet (Dermatology Life Quality Index) 0 tai 1 viikolla 12 saavuttaneiden potilaiden osuus oli 56 %, 61 %, 59 % brodalumabi 210 mg:n ryhmässä ja 5 %, 5 %, 7 % lumelääkeryhmässä AMAGINE-1-, -2- ja -3-tutkimuksissa (säädetty p-arvo <0,001) ja 44 % ustekinumabiryhmissä (AMAGINE-2 ja -3).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset brodalumabin käytöstä läiskäpsoriaasin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Populaatiofarmakokineettisen mallinnuksen perusteella arvioitu akkumulaatiosuhde on 20 viikon annostuksen jälkeen 2,5‑kertainen. Keskivaikeassa tai vaikeassa läiskäpsoriaasissa yhden, ihon alle annetun 210 mg:n brodalumabiannoksen jälkeen keskimääräinen enimmäisseerumipitoisuus (Cmax) oli 13,4 mikrog/ml (keskihajonta [SD] = 7,29 mikrog/ml). Mediaaniaika enimmäispitoisuuteen (Tmax) oli 3,0 päivää (vaihtelualue: 2,0–4,0 päivää) ja keskimääräinen pitoisuus‑aika‑käyrän alle jäävä pinta‑ala viimeiseen mitattavissa olevaan pitoisuuteen (AUClast) oli 111 mikrog*vrk/ml (SD = 64,4 mikrog*vrk/ml). Brodalumabin ihonalainen biologinen hyötyosuus populaatiofarmakokineettisen mallinnuksen mukaan arvioituna oli 55 %.
Havaitut farmakokineettiset parametrit vakaan tilan aikana (viikoilla 10–12) olivat: keskimääräinen vakaan tilan pitoisuus‑aika‑käyrän alle jäävä pinta‑ala yli annostusintervallin (AUCtau) oli 227,4 mikrog*vrk/ml (SD = 191,7 mikrog*vrk/ml), mikä vastaa keskimääräistä pitoisuutta (Cav,ss) 16,2 mikrog/ml; keskimääräinen Cmax oli 20,9 mikrog/ml (SD = 17,0 mikrog/ml) ja viikon 12 keskimääräinen vähimmäisseerumipitoisuus (Ctrough) oli 9,8 mikrog/ml (SD = 11,2 mikrog/ml).
Jakautuminen
Populaatiofarmakokineettisen mallinnuksen perusteella brodalumabin arvioitu keskimääräinen vakaan tilan jakautumistilavuus oli noin 7,24 l.
Biotransformaatio
Ihmisen monoklonaalisena IgG2‑vasta‑aineena brodalumabin odotetaan hajoavan pieniksi peptideiksi ja aminohapoiksi katabolisten reittien kautta samalla tavalla kuin endogeenisen IgG:n.
Eliminaatio
Kun brodalumabia on annettu 210 mg ihon alle, se noudattaa epälineaarista farmakokinetiikkaa, joka on tyypillinen kohdevälitteisen jakautumisen läpikäyvälle monoklonaaliselle vasta‑aineelle.
Brodalumabin puhdistuma vähenee, kun annos suurenee, ja altistus suurenee annosta suuremmassa suhteessa. Kun brodalumabin ihon alle annettava annos nousi 3‑kertaiseksi 70 mg:sta 210 mg:aan, vakaan tilan seerumin brodalumabin Cmax nousi 18‑kertaiseksi ja AUC0‑t 25‑kertaiseksi.
Kun brodalumabia annetaan yksi 210 mg:n annos ihon alle läiskäpsoriaasipotilaille, ilmeinen puhdistuma (CL/F) on 2,95 l/vrk.
Populaatiofarmakokineettinen mallinnus ennusti, että seerumin brodalumabipitoisuudet laskivat 95%:lla potilaista alle mittausrajan (0,05 mikrog/ml) 63 päivää sen jälkeen, kun vakaan tilan 210 mg:n annos brodalumabia annettuna joka toinen viikko lopetettiin. LLOQ:n (mittauksen alaraja) alapuolella olevat brodalumabipitoisuudet liittyivät kuitenkin IL‑17‑reseptorin enintään 81 %:een sitoutumiseen.
Populaatiofarmakokineettisen mallinnuksen perusteella brodalumabin arvioitu puoliintumisaika oli 10,9 päivää vakaassa tilassa sen jälkeen kun sitä oli annettu 210 mg:n annos ihon alle joka toinen viikko.
Painon vaikutus farmakokinetiikkaan
Populaatiofarmakokineettinen mallinnus osoitti, että altistus väheni ruumiinpainon kasvaessa. Annoksen muuttamista ei suositella.
Iäkkäät potilaat
Populaatiofarmakokineettinen mallinnus osoitti, ettei iällä ollut vaikutusta brodalumabin farmakokinetiikkaan, mikä perustui siihen, että 259 (6 %) potilaista oli 65–74-vuotiaita ja 14 (0,3 %) potilaista oli ≥75-vuotiaita yhteensä 4 271 läiskäpsoriaasipotilaan farmakokineettisessä populaatiossa.
Munuaisten tai maksan vajaatoiminta
Farmakokineettisiä tietoja ei ole saatavilla munuaisten tai maksan vajaatoimintaa sairastavista potilaista. Pilkkoutumattoman brodalumabin, monoklonaalisen IgG‑vasta‑aineen, munuaispuhdistuman odotetaan olevan vähäistä ja vähämerkityksistä. Brodalumabin odotetaan eliminoituvan pääasiassa katabolisesti, eikä maksan vajaatoiminnan odoteta vaikuttavan puhdistumaan.
Muut ryhmät
Brodalumabin farmakokinetiikka oli samankaltainen psoriaasia sairastavien japanilaisten ja ei‑japanilaisten potilaiden välillä.
Populaatiofarmakokineettinen analyysi osoitti, että sukupuolella ei ollut vaikutusta brodalumabin farmakokinetiikkaan.
Farmakokineettiset/farmakodynaamiset suhteet
Populaatiofarmakokineettinen/‑farmakodynaaminen malli, joka kehitettiin käyttämällä kaikkia saatavilla olevia tietoja, osoitti, että kun potilaille annettiin 210 mg:n annos kahden viikon välein, 90 %:n ennustettiin ylläpitävän suurempaa vähimmäisseerumipitoisuutta kuin arvioitu IC90‑arvo 1,51 mikrog/ml. Eksploratiivisten kuvailevien analyysien perusteella altistuksen ja vakavien infektioiden, Candida‑infektioiden, virusinfektioiden ja itsemurha‑ajatusten ja ‑käyttäytymistapahtumien välillä ei havaittu suhdetta. Altistus‑vaste‑analyysi osoittaa, että suuret brodalumabipitoisuudet liittyvät parempaan PASI‑ ja sPGA‑vasteeseen.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta (mukaan lukien farmakologista turvallisuutta koskevien ja hedelmällisyyteen liittyvien päätetapahtumien arvioinnit) ja lisääntymis‑ ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Brodalumabilla ei ole tehty karsinogeenisuustutkimuksia. Jaavanmakakeissa ei kuitenkaan tapahtunut proliferatiivisia muutoksia, kun niille annettiin viikoittain 90 mg/kg brodalumabia ihon alle 6 kuukauden ajan (AUC‑altistus oli 47‑kertaisesti suurempi kuin ihmispotilailla, jotka saivat brodalumabia 210 mg kahden viikon välein). Brodalumabin mutageenistä potentiaalia ei arvioitu, mutta monoklonaalisten vasta‑aineiden ei odoteta muuntavan DNA:ta tai kromosomeja.
Naaras‑ ja urosjaavanmakakien sukupuolielimissä tai siittiöiden määrässä, liikkuvuudessa ja morfologiassa ei esiintynyt muutoksia sen jälkeen, kun brodalumabia annettiin enintään 90 mg/kg kerran viikossa 6 kuukauden ajan (AUC‑altistus enintään 47‑kertaa suurempi kuin brodalumabia 210 mg kahden viikon välein saavilla ihmispotilailla).
Jaavanmakakeilla alkion/sikiön kehitykseen tai synnytyksen jälkeiseen (6 kuukauden ikään asti) kehitykseen kohdistuvia vaikutuksia ei havaittu, kun brodalumabia annettiin ihon alle koko raskauden ajan enintään 27 kertaa suuremmilla altistustasoilla kuin mitä saavutettiin ihmispotilaissa, joille annettiin brodalumabia 210 mg kahden viikon välein, pitoisuuskäyrän alle jäävän pinta‑alan (AUC) perusteella. Apinaimeväisten ja kaniinin sikiöiden seerumipitoisuudet osoittivat brodalumabin merkittävää siirtymistä emolta sikiölle raskauden loppuvaiheessa.
Kun jaavanmakakeille oli annettu brodalumabia ihon alle viikoittain enintään 90 mg/kg 6 kuukauden ajan, brodalumabiin liittyvät vaikutukset rajoittuivat pistoskohdan reaktioihin ja mukokutaaniseen tulehdukseen, joka sopi isäntäorganismin seurannan farmakologiseen modulaatioon kommensaalisen mikroflooran suhteen. Vaikutuksia perifeeriseen veren immunofenotyypitykseen ja T‑soluriippuvaiseen vasta‑ainevasteen määritykseen ei ollut. Kaniineilla tehdyssä paikallisessa siedettävyystestissä havaittiin keskivaikeaa tai vaikeaa edeemaa, kun niille oli annettu ihon alle injektio, joka sisälsi kliinisen pitoisuuden, 140 mg/ml, verran brodalumabia.
Farmaseuttiset tiedot
Apuaineet
Proliini
Glutamaatti
Polysorbaatti 20
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
4 vuotta
Säilytys
Säilytä jääkaapissa (2°C ‑ 8°C).
Ei saa jäätyä.
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Kyntheum‑valmistetta voi säilyttää huoneenlämmössä (enintään 25°C) ulkopakkauksessaan yhden enintään 14 vuorokauden pituisen jakson ajan. Kun Kyntheum on otettu pois jääkaapista ja se on saavuttanut huoneenlämmön (enintään 25°C), se on käytettävä 14 vuorokauden kuluessa tai hävitettävä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KYNTHEUM injektioneste, liuos, esitäytetty ruisku
210 mg (L:ei) 2 x 1,5 ml (140 mg/ml) (975,00 €)
PF-selosteen tieto
1,5 ml liuosta tyypin I lasisessa esitäytetyssä ruiskussa, jossa on ruostumattomasta teräksestä valmistettu 27 G x ½ tuuman neula ja joka on suojattu elastomeerineulakorkilla.
Kyntheum-valmistetta on saatavilla yksikköpakkauksissa, joissa on 2 esitäytettyä ruiskua ja monipakkauksissa, joissa on 6 (3 kahden ruiskun pakkausta) esitäytettyä ruiskua.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Liuos on kirkasta tai hieman opaalinhohtoinen, väritöntä tai hieman kellertävää, eikä siinä ole hiukkasia.
Käyttö- ja käsittelyohjeet
Epämukavaa tunnetta pistoskohdassa vältetään odottamalla vähintään 30 minuuttia ennen pistämistä, jotta esitäytetty ruisku saavuttaisi huoneenlämmön. Esitäytettyä ruiskua ei saa lämmittää muulla tavoin. Esitäytettyä ruiskua ei saa ravistaa. Esitäytetyn ruiskun neulakorkkia ei saa poistaa, kun ruiskun odotetaan saavuttavan huoneenlämmön.
Kyntheum on tarkistettava silmämääräisesti hiukkasten ja värjäytymien varalta ennen käyttöä. Tätä lääkevalmistetta ei saa käyttää, jos liuos on sameaa tai värjäytynyttä tai jos se sisältää kappaleita, hiutaleita tai hiukkasia.
Esitäytettyä ruiskua ei saa käyttää, jos se on pudonnut kovalle pinnalle.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
KYNTHEUM injektioneste, liuos, esitäytetty ruisku
210 mg 2 x 1,5 ml
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Adalimumabi, bimekitsumabi, brodalumabi, etanersepti, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sekukinumabi, sertolitsumabipegoli, tildrakitsumabi ja ustekinumabi (ihopsoriaasi): Vaikean kroonisen ihopsoriaasin hoito erityisin edellytyksin (319).
ATC-koodi
L04AC12
Valmisteyhteenvedon muuttamispäivämäärä
12.08.2025
Yhteystiedot
Karhumäentie 3
01530 Vantaa
020 721 8440
www.leo-pharma.fi
info.fi@leo-pharma.com