
LUCENTIS injektioneste, liuos, esitäytetty ruisku 10 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Lucentis® (ranibitsumabi) – Proliferatiivisen diabeettisen retinopatian (PDR) ja/tai diabeettisesta makulaturvotuksesta johtuvan näkökyvyn heikentymisen (DME) hoitoon / För proliferativ diabetesretinopati (PDR) och/eller synnedsättning till följd av diabetiskt makulaödem (DME)
Lucentis® (ranibitsumabi) – Silmän suonikalvon uudissuonittumisen (CNV) aiheuttamaan näkökyvyn heikkenemiseen / För nedsatt syn på grund av koroidal neovaskularisering (CNV)
Lucentis® (ranibitsumabi) – Silmänpohjan kosteaan ikärappeumaan (AMD) / För våt makuladegeneration (AMD)
Lucentis® (ranibitsumabi) – Verkkokalvon laskimotukoksesta (RVO) (laskimohaara- tai keskuslaskimotukoksesta) johtuvan makulaturvotuksen aiheuttamaan näkökyvyn heikentymiseen / För nedsatt syn på grund av makulaödem till följd av retinal venocklusion (RVO)
Vaikuttavat aineet ja niiden määrät
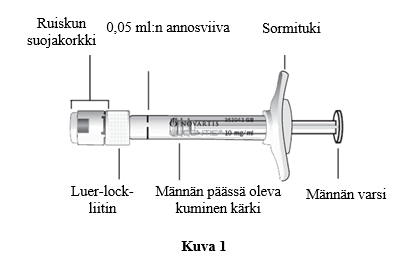
Yksi millilitra liuosta sisältää 10 mg ranibitsumabia*. Yksi esitäytetty ruisku sisältää 0,165 ml liuosta, mikä vastaa 1,65 mg ranibitsumabia. Esitäytetystä ruiskusta saatava kokonaismäärä on 0,1 ml. Tästä saadaan tarvittava määrä 0,05 ml eli 0,5 mg ranibitsumabia sisältävän kerta-annoksen annosteluun.
*Ranibitsumabi on humanisoitu monoklonaalinen vasta-ainefragmentti, joka on valmistettu Escherichia coli ‑soluissa rekombinaatio-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos
Kliiniset tiedot
Käyttöaiheet
Lucentis on tarkoitettu aikuisten:
- ikään liittyvän neovaskulaarisen (kostean) verkkokalvon makulan rappeuman (AMD) hoitoon
- diabeettisen makulaturvotuksen (DME) aiheuttaman näkökyvyn heikkenemisen hoitoon
- proliferatiivisen diabeettisen retinopatian (PDR) hoitoon
- verkkokalvon laskimotukoksesta (verkkokalvon laskimohaara- tai keskuslaskimotukoksesta) johtuvan makulaturvotuksen aiheuttaman näkökyvyn heikkenemisen hoitoon
- silmän suonikalvon uudissuonittumisen (CNV) aiheuttaman näkökyvyn heikkenemisen hoitoon.
Ehto
Injektion antavalla silmälääkärillä on oltava kokemusta lasiaisen sisään annettavista injektioista.
Annostus ja antotapa
Lucentis-valmisteen antavalla silmälääkärillä on oltava kokemusta lasiaiseen annettavista injektioista.
Annostus
Suositeltu Lucentis-annos on 0,5 mg silmän lasiaiseen annettuna kertainjektiona. Tämä vastaa injektiotilavuutta 0,05 ml. Kahden samaan silmään annettavan pistoksen antovälin tulee olla vähintään neljä viikkoa.
Hoito aloitetaan antamalla yksi pistos kerran kuukaudessa kunnes maksimaalinen näöntarkkuus on saavutettu ja/tai silmässä ei ole havaittavissa tautiaktiivisuuden merkkejä, eli näöntarkkuudessa ei todeta muutoksia eikä potilaalla havaita muutoksia muissa sairauden merkeissä tai oireissa jatkuvan hoidon aikana. Potilaille, joilla on ikään liittyvä kostea verkkokalvon makulan rappeuma, diabeettinen makulaturvotus, proliferatiivinen diabeettinen retinopatia tai verkkokalvon laskimotukos, voi aluksi olla tarpeen antaa peräkkäisiä kuukausittaisia injektioita kolme tai useampia.
Tämän jälkeen lääkäri määrittää sopivat seuranta- ja hoitovälit sairauden aktiivisuuden mukaan, mikä arvioidaan näöntarkkuuden ja/tai anatomisten parametrien perusteella.
Jos visuaaliset ja anatomiset parametrit lääkärin arvion mukaan osoittavat, että potilas ei hyödy hoidon jatkamisesta, Lucentis-hoito tulee lopettaa.
Sairauden aktiivisuuden seurantaan voivat kuulua kliininen tutkiminen, toiminnalliset kokeet tai kuvaustekniikat (esim. valokerroskuvaus tai fluoreseiiniangiografia).
Kun maksimaalinen näöntarkkuus on saavutettu ja/tai tautiaktiivisuuden merkkejä tai oireita ei ole havaittavissa, voidaan pistosten antoväliä asteittain pidentää, jos potilaan hoito noudattaa annosteluohjelmaa, jossa hoitovälejä voidaan pidentää (TE, treat-and extend). Hoitovälien pidentämistä voidaan jatkaa, kunnes merkkejä tautiaktiivisuudesta tai näkökyvyn heikkenemistä jälleen ilmenee. Ikään liittyvän kostean verkkokalvon makulan rappeuman (AMD) hoidossa ei hoitoväliä saa pidentää kerralla kahta viikkoa enempää. Diabeettisen makulaturvotuksen (DME) hoidossa hoitoväliä saa pidentää enintään kuukauden verran kerrallaan. Proliferatiivisen diabeettisen retinopatian ja verkkokalvon laskimotukoksen hoidossa hoitoväliä voidaan myös asteittain pidentää, mutta hoitovälien pituuden määrittämiseksi ei ole saatavilla riittäviä tietoja. Jos tauti jälleen muuttuu aktiiviseksi, on hoitovälejä lyhennettävä sen mukaisesti.
Silmän suonikalvon uudissuonittumisen (CNV) aiheuttaman näkökyvyn heikkenemisen hoito on määriteltävä yksilöllisesti, taudin aktiivisuusasteen perusteella. Joillekin potilaille yksi pistos ensimmäisten 12 kuukauden aikana voi riittää, kun toiset puolestaan saattavat tarvita tiheämmin toistuvaa hoitoa, mukaan lukien kerran kuukaudessa annettavat pistokset. Patologisesta likitaittoisuudesta (PM) johtuvan silmän suonikalvon uudissuonittumisen (CNV) aiheuttaman näkökyvyn heikkenemisen hoidossa moni potilas voi tarvita vain yhden tai kaksi pistosta ensimmäisen hoitovuotensa aikana (ks. kohta Farmakodynamiikka).
Lucentis ja laserkoagulaatio diabeettisen makulaturvotuksen sekä verkkokalvon laskimohaaratukoksen (BRVO) aiheuttaman makulaturvotuksen hoidossa
Lucentis-valmisteen samanaikaisesta käytöstä laserkoagulaation kanssa on jonkin verran kokemusta (ks. kohta Farmakodynamiikka). Jos nämä hoidot annetaan samana päivänä, on Lucentis-hoito annettava aikaisintaan 30 minuuttia laserkoagulaation jälkeen. Lucentis-valmistetta voidaan antaa potilaille, jotka aiemmin ovat saaneet laserkoagulaatiohoitoa.
Lucentis-valmisteen ja verteporfiinilla toteutetun fotodynaamisen hoidon käyttö patologisesta likitaittoisuudesta (PM) johtuvan silmän suonikalvon uudissuonittumisen (CNV) aiheuttaman näkökyvyn heikkenemisen hoitoon
Kokemusta Lucentisin ja verteporfiinin samanaikaisesta käytöstä ei ole.
Erityispotilasryhmät
Maksan vajaatoiminta
Lucentis-valmisteen käyttöä ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla, mutta erityistoimenpiteet eivät ole tarpeen.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Iäkkäät henkilöt
Iäkkäiden henkilöiden annoksia ei tarvitse muuttaa. Yli 75-vuotiaista diabeettista makulaturvotusta sairastavista potilaista on rajallisesti tietoa.
Pediatriset potilaat
Lucentis-valmisteen turvallisuutta ja tehoa lasten ja alle 18-vuotiaiden nuorten hoidossa ei ole varmistettu. Nuorten, 12 - 17-vuotiaiden potilaiden silmän suonikalvon uudissuonittumisen (CNV) aiheuttaman näkökyvyn heikkenemisen hoidosta käytettävissä olevat tiedot on kuvattu kohdassa Farmakodynamiikka.
Antotapa
Kertakäyttöinen esitäytetty ruisku. Tarkoitettu annettavaksi vain silmän lasiaiseen. Esitäytetty ruisku sisältää enemmän lääkettä kuin 0,5 mg:n suositusannoksen. Esitäytetystä ruiskusta saatavaa kokonaismäärää (0,1 ml) ei tule käyttää kokonaan. Liuosylimäärä on poistettava ennen annostelua. Esitäytetyn ruiskun kokonaismäärän injisointi silmään voi johtaa yliannostukseen. Poista ilmakuplat ja ylimääräinen lääkevalmiste painamalla mäntää hitaasti kunnes männän kuperan kärjen reuna on ruiskussa olevan mustan annosviivan kohdalla (vastaa 0,05 ml eli 0,5 mg ranibitsumabia).
Lucentis-valmiste on tarkistettava silmämääräisesti ennen käyttöä mahdollisten hiukkasten tai värimuutoksien havaitsemiseksi.
Injektio on annettava aseptisissa olosuhteissa, mikä tarkoittaa kirurgista käsien desinfiointia, steriilien käsineiden, steriilien suojavaatteiden ja steriilin luomilevittimen (tai vastaavan) käyttöä ja tarvittaessa mahdollisuutta steriiliin parasenteesiin. Potilaan aiemmat yliherkkyysreaktiot on selvitettävä tarkoin ennen intravitreaalista toimenpidettä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Ennen injektiota silmää ympäröivä iho, silmäluomi ja silmän pinta desinfioidaan laajakirjoisella paikallisella mikrobisidilla sekä annetaan riittävä puudutus paikallisen hoitokäytännön mukaisesti.
Lisätietoja Lucentis-injektionesteen käyttövalmiiksi saattamiseksi, ks. kohta Käyttö- ja käsittelyohjeet.
Injektioneula pistetään 3,5‑4,0 mm limbuksesta posteriorisesti lasiaiseen, vältetään horisontaalista meridiaania ja tähdätään silmämunan keskikohtaan. Annettava 0,05 ml:n määrä injisoidaan. Kovakalvon pistoskohtaa vaihdetaan seuraavissa injektioissa. Yhtä esitäytettyä ruiskua tulee käyttää vain yhden silmän hoitoon.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Okulaarinen tai periokulaarinen infektio tai sen epäily.
Vakava silmän sisäinen tulehdus.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Intravitreaalisen injektion antoon liittyvät reaktiot
Intravitreaali-injektioiden, myös Lucentis-injektioiden, yhteydessä on esiintynyt endoftalmiittia, silmänsisäistä tulehdusta, regmatogeenistä verkkokalvon irtaumaa ja repeytymää sekä hoidosta johtuvaa traumaattista kaihia (ks. kohta Haittavaikutukset). Asianmukaista aseptista injektiotekniikkaa on käytettävä aina, kun Lucentis-valmistetta annetaan. Lisäksi tilannetta tulee seurata viikon ajan injektion jälkeen, jotta mahdollisen infektion hoito voidaan aloittaa ajoissa. Potilaita neuvotaan ilmoittamaan viipymättä endoftalmiittiin tai muihin edellä mainittuihin viittaavista oireista.
Silmänsisäisen paineen nousu
Ohimenevää silmänpaineen (IOP) nousua on havaittu 60 minuutin kuluessa Lucentis-injektion annosta. Silmänpaineen jatkuvaa koholla pysymistä on myös havaittu (ks. kohta Haittavaikutukset). Sekä silmänpainetta että näköhermon perfuusiota on tarkkailtava ja tarvittaessa hoidettava asianmukaisesti.
Potilaille on kerrottava näiden mahdollisten haittavaikutusten oireista, ja heidät on ohjeistettava kertomaan lääkärilleen, jos heillä ilmenee oireita, kuten silmäkipua tai lisääntynyttä epämukavuuden tunnetta silmässä, pahenevaa silmän punoitusta, näön hämärtymistä tai heikkenemistä, pienten hiukkasten määrän lisääntymistä näkökentässä tai lisääntynyttä silmän valoherkkyyttä (ks. kohta Haittavaikutukset).
Molempien silmien samanaikainen hoito
Rajoitettu tieto molempien silmien samanaikaisesta Lucentis-hoidosta (sisältäen annostelun saman päivän aikana) ei viittaa systeemisten haittavaikutusten riskin suurenemiseen verrattuna yhden silmän hoitoon.
Immunogeenisuus
Lucentis-valmisteeseen voi liittyä immunogeenisuutta. Systeeminen altistus saattaa olla korkeampi diabeettista makulaturvotusta sairastavilla potilailla, joten tavallista suurempi riski yliherkkyysreaktioiden kehittymiselle tässä potilaspopulaatiossa ei ole poissuljettu. Potilaita on myös kehotettava ilmoittamaan, jos silmänsisäinen tulehdus pahenee, koska se voi olla kliininen merkki silmänsisäisten vasta-aineiden muodostumisesta.
Muiden anti-VEGF-lääkkeiden (endoteelikasvutekijän estäjien) samanaikainen käyttö
Lucentis-valmistetta ei saa antaa samanaikaisesti muiden anti-VEGF-lääkevalmisteiden (systeemisten tai silmälääkevalmisteiden) kanssa.
Tilanteet, jolloin Lucentis-annosta ei tule antaa
Lääkeannosta ei saa antaa eikä hoitoa jatkaa ennen seuraavaa lääkkeenottoajankohtaa, jos jokin seuraavista ilmenee:
- parhaan lasikorjatun näöntarkkuuden (best-corrected visual acuity, BCVA) heikkeneminen 30 kirjainta tai enemmän verrattuna edelliseen näöntarkkuuden tarkastukseen;
- silmänpaine ≥ 30 mmHg;
- verkkokalvoreikä;
- verkkokalvonalainen verenvuoto, joka ulottuu verkkokalvon keskikuoppaan (fovea) tai jos verenvuodon laajuus on ≥ 50 % leesion kokonaisalasta;
- suunniteltu tai tehty silmäleikkaus edeltävien tai seuraavien 28 vuorokauden aikana.
Verkkokalvon pigmenttiepiteelin repeytymä
Hoidettaessa kosteaa ikärappeumaa ja mahdollisesti myös muita silmän suonikalvon uudissuonittumisen (CNV) muotoja, VEGF-hoidon jälkeiseen verkkokalvon pigmenttiepiteelin repeytymän kehittymiseen liittyviä riskitekijöitä ovat laaja-alainen ja/tai korkea verkkokalvon pigmenttiepiteelin irtauma. Aloitettaessa ranibitsumabihoitoa, varovaisuutta on noudatettava niiden potilaiden kohdalla, joilla on näitä verkkokalvon pigmenttiepiteelin repeytymän riskitekijöitä.
Regmatogeeninen verkkokalvon repeytymä tai makulan reikä
Hoito on lopetettava, jos potilaalla on regmatogeeninen verkkokalvon repeytymä tai makulan 3. tai 4. asteen reikä.
Potilasryhmät, joiden hoidosta on vain rajallisesti kokemusta
Tyypin I diabeteksen aiheuttaman makulaturvotuksen hoidosta on vain rajallisesti kokemusta. Lucentis-hoitoa ei ole tutkittu potilailla, jotka ovat aiemmin saaneet intravitreaali-injektioita; potilailla, joilla on jokin aktiivisessa vaiheessa oleva yleisinfektio; tai potilailla, joilla on muita samanaikaisia silmäsairauksia, kuten verkkokalvon irtauma tai reikä makulassa. Lucentisin käytöstä on rajallisesti kokemusta sellaisten diabeetikkojen hoidossa, joiden HbA1c on yli 108 mmol/mol (12 %), eikä kokemusta ole sellaisten potilaiden hoidosta, joilla on kontrolloimaton hypertensio. Tällaisia potilaita hoitavien lääkäreiden on harkittava puuttuvien tietojen merkitystä.
Tietoja ei ole riittävästi johtopäätösten tekemiseen Lucentis-hoidon tehosta potilailla, joilla on verkkokalvon laskimotukos ja kliinisiä merkkejä palautumattomasta iskeemisestä näön menetyksestä.
Tiedot Lucentis-valmisteen tehosta ovat rajalliset sellaisten patologisesta likitaittoisuudesta (PM) kärsivien potilaiden osalta, joiden aikaisempi verteporfiinilla suoritettu fotodynaaminen (vPDT) hoito on epäonnistunut. Tehon todettiin olevan yhtä hyvä potilailla, joilla oli fovean alla tai sen vieressä sijaitsevia leesioita. Tiedot sellaisten patologisesta likitaittoisuudesta (PM) kärsivien potilaiden osalta, joilla on fovean ulkopuolella sijaitsevia leesioita, ovat liian puutteelliset päätelmien tekemiseen Lucentis-hoidon tehosta näiden potilaiden hoidossa.
Systeemisiä haittavaikutuksia silmän lasiaiseen tapahtuneen annon jälkeen
Systeemisiä haittavaikutuksia, mukaan lukien verenvuototapahtumia muualla kuin silmässä ja valtimotromboembolisia tapahtumia, on raportoitu VEGF-estäjien silmän lasiaiseen tapahtuneen annon jälkeen.
Tietoja tämän hoidon turvallisuudesta diabeettista makulaturvotusta, verkkokalvon laskimotukoksesta (RVO) johtuvaa makulaturvotusta ja patologisesta likitaittoisuudesta (PM) johtuvaa silmän suonikalvon uudissuonittumista (CNV) sairastavilla potilailla, joilla on aiemmin ollut aivohalvaus tai ohimeneviä aivoverenkiertohäiriöitä, on vain rajallisesti. Varovaisuutta noudatettava tällaisia potilaita hoidettaessa (ks. kohta Haittavaikutukset).
Yhteisvaikutukset
Varsinaisia yhteisvaikutustutkimuksia ei ole tehty.
Verteporfiinilla tehostetun fotodynaamisen hoidon (PDT) ja Lucentis-valmisteen käyttö liitännäislääkityksenä ikään liittyvän verkkokalvon kostean makulan rappeuman ja patologisen likitaittoisuuden (PM) hoitoon, ks. kohta Farmakodynamiikka.
Laserkoagulaation ja Lucentis-valmisteen käyttö liitännäishoitona diabeettisessä makulaturvotuksessa ja verkkokalvon laskimohaaratukoksessa, ks. kohdat Annostus ja antotapa ja Farmakodynamiikka.
Diabeettisen makulaturvotuksen (DME) aiheuttaman näkökyvyn heikkenemisen hoidossa suoritetuissa kliinisissä tutkimuksissa samanaikainen tiatsolidinedionihoito ei vaikuttanut Lucentis-hoitoa saaneiden potilaiden näöntarkkuutta tai keskeisen makulan paksuutta (CSFT) koskeviin tuloksiin.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset/ehkäisy naisille
Hedelmällisessä iässä olevien naisten on käytettävä tehokasta ehkäisyä hoidon aikana.
Raskaus
Ranibitsumabille altistuneista raskauksista ei ole kliinisiä tietoja. Eläinkokeissa cynomolgus-apinoilla ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia raskauteen tai alkion/sikiön kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Systeeminen altistuminen ranibitsumabille on vähäistä intraokulaarisen annon jälkeen, mutta ranibitsumabin vaikutusmekanismin perusteella se voi olla teratogeeninen ja alkio-/sikiötoksinen. Sen vuoksi ranibitsumabia ei saa käyttää raskauden aikana, ellei odotettavissa oleva hyöty ole suurempi kuin sikiölle mahdollisesti aiheutuva haitta. Ranibitsumabihoitoa saaneiden, raskautta toivovien naisten suositellaan odottavan ainakin 3 kuukautta viimeisen ranibitsumabiannoksen jälkeen ennen kuin he yrittävät tulla raskaaksi.
Imetys
Saatavilla olevien hyvin vähäisten tietojen perusteella pieniä määriä ranibitsumabia saattaa erittyä ihmisen rintamaitoon. Ei ole tietoja ranibitsumabin vaikutuksista vastasyntyneeseen/imeväiseen. Varotoimena Lucentis-hoidon aikana ei ole suositeltavaa imettää.
Hedelmällisyys
Tiedot mahdollisista vaikutuksista hedelmällisyyteen puuttuvat.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Hoitotoimenpide voi aiheuttaa tilapäisiä näköhäiriöitä, jotka voivat vaikuttaa ajokykyyn tai koneidenkäyttökykyyn (ks. kohta Haittavaikutukset). Jos potilaalla on tilapäisten näköhäiriöiden merkkejä, hän ei saa ajaa eikä käyttää koneita ennen kuin ne häviävät.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Suurin osa Lucentis-valmisteen annon jälkeen raportoiduista haittavaikutuksista liittyvät intravitreaalisen injektion antoon.
Kaikista yleisimmin Lucentis-injektion jälkeen raportoidut silmään liittyvät haittavaikutukset ovat: silmäkipu, silmän verekkyys, kohonnut silmänsisäinen paine, lasiaisen tulehdus, lasiaisirtauma, verkkokalvoverenvuoto, näköhäiriöt, lasiaiskellujat, sidekalvon verenvuoto, silmä-ärsytys, vierasesineen tunne silmässä, lisääntynyt kyynelnesteen eritys, luomitulehdus, kuivasilmäisyys ja silmän kutina.
Kaikista yleisimmät raportoidut silmään liittymättömät haittavaikutukset ovat päänsärky, nenänielutulehdus ja nivelkivut.
Harvemmin raportoituja, mutta vakavampia haittavaikutuksia ovat endoftalmiitti, sokeutuminen, verkkokalvon irtauma ja repeytymä sekä hoidosta johtuva traumaattinen kaihi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yhteenveto kliinisissä tutkimuksissa Lucentis-valmisteen annon jälkeen esiintyneistä haittavaikutuksista on esitetty alla olevassa taulukossa.
Taulukkomuotoinen yhteenveto haittavaikutuksista#
Haittavaikutukset on lueteltu elinluokan ja esiintyvyyden mukaan seuraavan luokituksen mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Infektiot | |
Hyvin yleiset | Nasofaryngiitti |
Yleiset | Virtsatieinfektio* |
Veri ja imukudos | |
Yleiset | Anemia |
Immuunijärjestelmä | |
Yleiset | Yliherkkyys |
Psyykkiset häiriöt | |
Yleiset | Ahdistuneisuus |
Hermosto | |
Hyvin yleiset | Päänsärky |
Silmät | |
Hyvin yleiset | Vitreiitti, lasiaisirtauma, verkkokalvon verenvuoto, näköhäiriöt, silmäkipu, lasiaissamentumat, sidekalvon verenvuoto, silmän ärsytys, roskan tunne silmässä, kyynelnesteen erityksen lisääntyminen, luomitulehdus, silmien kuivuminen, silmän verekkyys, silmien kutina. |
Yleiset | Verkkokalvon rappeuma, verkkokalvon sairaudet, verkkokalvon irtauma, verkkokalvon repeytymä, verkkokalvon pigmenttiepiteelin irtoaminen, verkkokalvon pigmenttiepiteelin repeytymä, näöntarkkuuden heikkeneminen, lasiaisen verenvuoto, lasiaissairaudet, uveiitti, iriitti, iridosykliitti, harmaakaihi, kapselinalainen kaihi, takakapselin samentuma, pilkukas sarveiskalvontulehdus, sarveiskalvon naarmut, etukammion valotie, näön sumeneminen, injektiokohdan verenvuoto, silmän verenvuoto, sidekalvotulehdus, allerginen sidekalvotulehdus, silmien rähmiminen, fotopsia, valonarkuus, silmävaivat, silmäluomen turvotus, silmäluomen kipu, sidekalvon verekkyys. |
Melko harvinaiset | Sokeutuminen, endoftalmiitti, etukammion märkäsakka, etukammioverenvuoto, keratopatia, värikalvon kiinnitakertuminen, sarveiskalvon saostumat, sarveiskalvon turvotus, sarveiskalvon arpijuovat, injektiokohdan kipu, injektiokohdan ärsytys, poikkeavat tuntemukset silmässä, silmäluomen ärsytys. |
Hengityselimet, rintakehä ja välikarsina | |
Yleiset | Yskä |
Ruoansulatuselimistö | |
Yleiset | Pahoinvointi |
Iho ja ihonalainen kudos | |
Yleiset | Allergiset reaktiot (ihottuma, nokkosihottuma, kutina, punoitus) |
Luusto, lihakset ja sidekudos | |
Hyvin yleiset | Nivelkipu |
Tutkimukset | |
Hyvin yleiset | Kohonnut silmänpaine |
# Haittavaikutuksiksi määriteltiin sellaiset haittatapahtumat (vähintään 0,5 prosenttiyksiköllä potilaista), joita ilmeni useammin (vähintään 2 prosenttiyksikköä) potilailla, jotka saivat 0,5 mg:aa Lucentista kuin potilailla, jotka saivat vertailuhoitoa (PDT-hoito lumelääkkeellä tai verteporfiinilla). * havaittu vain diabeettista makulaturvotusta sairastavilla potilailla | |
Lääkeaineryhmään liittyvät haittavaikutukset
Ikään liittyvää verkkokalvon kosteaa makulan rappeumaa koskevissa vaiheen III tutkimuksissa silmään liittymättömän verenvuodon (haittavaikutuksen, joka mahdollisesti liittyy systeemiseen VEGF:n estoon) kokonaisesiintyvyys oli hieman kohonnut ranibitsumabia saaneilla potilailla. Kuitenkaan johdonmukaisuutta erityyppisten verenvuotojen välillä ei löytynyt. VEGF-estäjien intravitreaaliseen antoon liittyy valtimotromboembolian, myös aivohalvauksen ja sydäninfarktin, teoreettinen vaara. Valtimoiden tromboemboliatapahtumia havaittiin Lucentis-valmisteella tehdyissä, ikään liittyvää makulan rappeumaa, diabeettista makulaturvotusta, proliferatiivista diabeettista retinopatiaa, verkkokalvon laskimotukosta ja silmän suonikalvon uudissuonittumista koskevissa kliinisissä lääketutkimuksissa vähän, eikä ranibitsumabia ja kontrollivalmistetta saaneiden ryhmien välillä havaittu merkittäviä eroja.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ikään liittyvää kosteaa verkkokalvon makulan rappeumaa koskevista kliinisistä tutkimuksista ja myyntiluvan saamisen jälkeisessä haittavaikutusseurannassa on raportoitu vahingossa tapahtuneita yliannostustapauksia. Näihin raportoituihin tapauksiin liittyneitä haittavaikutuksia olivat silmänpaineen kohoaminen, ohimenevä sokeutuminen, näöntarkkuuden heikkeneminen, sarveiskalvon turvotus, sarveiskalvon kipu ja silmäkipu. Jos yliannostapauksia ilmenee, silmänpainetta on tarkkailtava ja hoidettava, mikäli hoitava lääkäri katsoo sen tarpeelliseksi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Silmätautien lääkkeet, uudissuonittumisen estoon käytettävät lääkkeet, ATC-koodi: S01LA04.
Vaikutusmekanismi
Ranibitsumabi on humanisoitu rekombinantti monoklonaalinen vasta-ainefragmentti, jonka kohteena on ihmisen vaskulaarinen endoteelikasvutekijä A (VEGF-A). Sillä on suuri taipumus sitoutua VEGF-A-isoformeihin (esim. VEGF110, VEGF121 ja VEGF165), jolloin se estää VEGF-A:n sitoutumisen sen omiin reseptoreihin VEGFR-1 ja VEGFR-2. VEGF-A:n sitoutuminen sen omiin reseptoreihin johtaa endoteelisolujen lisääntymiseen ja uudissuonittumiseen samoin kuin uudissuonivuotoihin. Näiden kaikkien uskotaan edistävän ikään liittyvän neovaskulaarisen (kostean) verkkokalvon makulan rappeuman, patologisen likitaittoisuuden sekä silmän suonikalvon uudissuonittumisen etenemistä tai edistävän näön heikentymistä, joka johtuu diabeettisesta makulaturvotuksesta tai verkkokalvon laskimotukoksen aiheuttamasta makulaturvoksesta.
Kliininen teho ja turvallisuus
Ikään liittyvän kostean verkkokalvon makulan rappeuman hoito
Lucentis-valmisteen kliininen teho ja turvallisuus ikään liittyvän kostean verkkokalvon makulan rappeuman hoitoon on arvioitu kolmessa satunnaistetussa lumelääke- tai vaikuttavaan aineeseen vertailevassa, 24 kuukautta kestäneessä kaksoissokkotutkimuksessa, jotka tehtiin neovaskulaarista AMD:tä sairastavilla potilailla. Kaikkiaan 1 323 potilasta (879 vaikuttavaa lääkeainetta ja 444 lumelääkettä saanutta potilasta) oli mukana tutkimuksissa.
Tutkimuksessa FVF2598g (MARINA) 716 potilasta, joilla oli minimaalisesti klassinen tai piilevä ei-klassinen leesio satunnaistettiin suhteessa 1:1:1 saamaan kerran kuukaudessa annettavat 0,3 mg:n tai 0,5 mg:n Lucentis-injektiot tai lumelääkeinjektiot.
Tutkimuksessa FVF2587g (ANCHOR) 423 potilasta, joilla oli pääasiassa klassinen CNV-leesio, satunnaistettiin suhteessa 1:1:1 saamaan kuukausittaiset 0,3 mg:n tai 0,5 mg:n Lucentis-injektiot tai PDT-hoidot verteporfiinilla (lähtötasossa ja sen jälkeen joka kolmas kuukausi, jos fluoreseiiniangiografiassa näkyi pysyvä tai uusiutuva uudissuonivuoto).
Tutkimusten olennaisimmat tulokset on esitetty taulukossa 1 ja kuvassa 1.
Taulukko 1 Tutkimusten FVF2598g (MARINA) ja FVF2587g (ANCHOR) 12kk:n ja 24kk:n tulokset
FVF2598g (MARINA) | FVF2587g (ANCHOR) | ||||
Tuloksen mittaus | Kuukausi | Lumelääke (n = 238) | Lucentis 0,5 mg (n = 240) | Verteporfiini-PDT (n = 143) | Lucentis 0,5 mg (n = 140) |
< 15 kirjaimen menetys näöntarkkuudesta (%)a (näön säilyminen, ensisijainen päätetapahtuma) | 12. kuukausi | 62 % | 95 % | 64 % | 96 % |
24. kuukausi | 53 % | 90 % | 66 % | 90 % | |
≥ 15 kirjaimen lisäys näöntarkkuuteen (%)a | 12. kuukausi | 5 % | 34 % | 6 % | 40 % |
24. kuukausi | 4 % | 33 % | 6 % | 41 % | |
Näöntarkkuuden keskimääräinen muutos (kirjaimet) (SD)a | 12. kuukausi | ‑10,5 (16,6) | +7,2 (14,4) | ‑9,5 (16,4) | +11,3 (14,6) |
24. kuukausi | ‑14,9 (18,7) | +6,6 (16,5) | ‑9,8 (17,6) | +10,7 (16,5) | |
a p < 0,01 | |||||
Kuva 1 Näöntarkkuuden keskimääräinen muutos lähtötilanteesta 24. kk:n loppuun tutkimuksessa FVF2598g (MARINA) ja tutkimuksessa FVF2587g (ANCHOR)
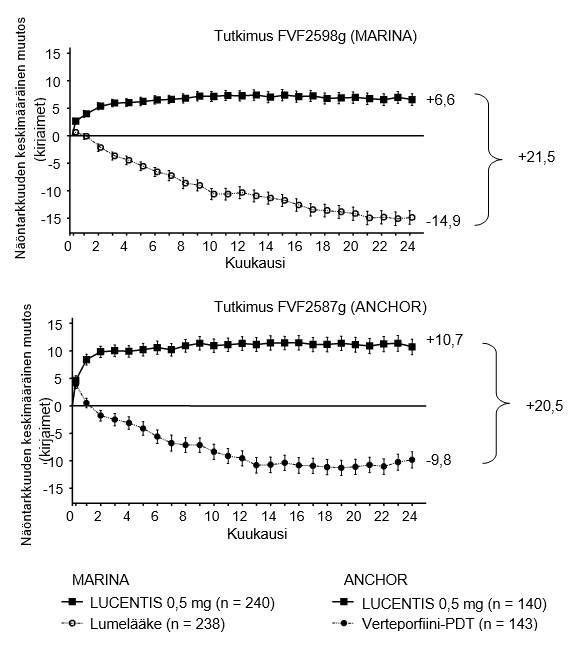
Kummastakin tutkimuksesta saadut tulokset viittaavat siihen, että myös ne potilaat, jotka menettivät parhaasta lasikorjatusta näöntarkkuudestaan (BCVA) vähintään 15 kirjainta ensimmäisen hoitovuoden aikana, saattavat hyötyä ranibitsumabihoidon jatkamisesta.
Potilaiden itsensä raportoimaa, tilastollisesti merkitsevää näkökyvyn toimintaan kohdistuvaa etua todettiin ranibitsumabihoitoa saaneiden ryhmissä verrattuna vertailuryhmiin sekä MARINA- että ANCHOR-tutkimuksissa, kun tulokset mitattiin NEI VFQ-25 -kyselyn avulla.
Tutkimuksessa FVF3192g (PIER) 184 potilasta, joilla oli kaikkia eri neovaskulaarisen AMD:n muotoja, satunnaistettiin suhteessa 1:1:1 saamaan 0,3 mg:n, tai 0,5 mg:n Lucentis-injektioita tai lumelääkeinjektioita kerran kuukaudessa kolmen peräkkäisen kuukauden ajan, minkä jälkeen he saivat annoksen kerran kolmessa kuukaudessa. Lumelääkehoitoa saaneilla potilailla oli 14. tutkimuskuukauden jälkeen mahdollisuus saada ranibitsumabihoitoa, ja 19. tutkimuskuukaudesta lähtien tiheämmin tapahtuvat hoidot olivat mahdollisia. PIER-tutkimuksessa Lucentis-potilaat saivat keskimäärin kymmenen hoitoa.
Alun näöntarkkuuden paranemisen jälkeen (kerran kuukaudessa annostelun jälkeen) potilaiden näöntarkkuus keskimäärin huononi, kun lääkettä annettiin neljännesvuosittain, ja näöntarkkuus palasi lähtötasolle kuukaudella 12, ja vaikutus säilyi useimmilla ranibitsumabihoitoa saaneilla potilailla (82 %:lla) 24 kuukauden ajan. Rajalliset tiedot lumelääkettä saaneesta tutkimuspotilasjoukosta, joka myöhemmin sai ranibitsumabia, viittaavat siihen, että hoidon aloittaminen varhaisessa vaiheessa saattaa olla yhteydessä näöntarkkuuden säilymiseen parempana.
Kahden myyntiluvan jälkeen suoritetun tutkimuksen (MONT BLANC, BPD952A2308 ja DENALI, BPD952A2309) tulokset vahvistivat Lucentis-valmisteen tehon, mutta niissä ei nähty lisätehoa potilailla, jotka saivat verteporfiinia (Visudyne PDT) yhdessä Lucentis-valmisteen kanssa verrattuna potilaisiin, jotka saivat Lucentis-valmistetta monoterapiana.
Patologisesta likitaittoisuudesta (PM) johtuvan silmän suonikalvon uudissuonittumisen (CNV) aiheuttaman näkökyvyn heikkenemisen hoito
Lucentis-valmisteen kliinistä tehoa ja turvallisuutta patologisesta likitaittoisuudesta johtuvan silmän suonikalvon uudissuonittumisen aiheuttaman näkökyvyn heikkenemisen hoidossa on arvioitu 12 kuukautta kestäneessä, kaksoissokkoutetussa, kontrolloidussa tutkimuksessa F2301 (RADIANCE). Tässä tutkimuksessa 227 potilasta satunnaistettiin seuraaviin tutkimushaaroihin suhteessa 2:2:1:
- Ryhmä I (0,5 mg ranibitsumabia; antotiheys määriteltiin ”vakaa”-kriteerin perusteella, jonka mukaan parhaassa lasikorjatussa näöntarkkuudessa (BCVA) ei saanut esiintyä muutoksia kahteen edelliseen kuukausittaiseen arvioon verrattuna.).
- Ryhmä II (0,5 mg ranibitsumabia; antotiheys määriteltiin ”tautiaktiivisuus”-kriteerin perusteella, jonka määritelmänä oli näön heikkeneminen johtuen uudissuonittumisen (CNV) aiheuttamasta ja OCT:llä ja/tai FA:lla havaitusta verkkokalvon alaisesta tai verkkokalvon sisäisestä vuodosta tai uudissuonileesion aktiivisesta vuodosta).
- Ryhmä III (vPDT - potilaille sallittiin ranibitsumabihoito kuukaudesta 3 lähtien).
Ryhmän II potilaista, joiden annostusohjelma vastasi suositeltua annostusta (ks. kohta Annostus ja antotapa), 50,9 % tarvitsivat 1 ‑ 2 pistosta; 34,5 % tarvitsi 3 ‑ 5 pistosta ja 14,7 % tarvitsi 6 ‑ 12 pistosta 12 kuukautta kestäneen tutkimusjakson aikana. Ryhmään II kuuluneista potilaista 62,9 % ei tarvinnut lainkaan pistoksia tutkimusjakson jälkimmäisten 6 kuukauden aikana.
RADIANCE-tutkimuksen keskeisimmät tulokset on esitetty taulukossa 2 ja kuvassa 2.
Taulukko 2 Tutkimustulokset kuukausina 3 ja 12 (RADIANCE)
Ryhmä I Ranibitsumabi 0,5 mg ”näkökyky vakaa” (n = 105) | Ryhmä II Ranibitsumabi 0,5 mg ”tautiaktiivisuus” (n = 116) | Ryhmä III vPDTb (n = 55) | |
Kuukausi 3 | |||
Parhaan lasikorjatun näöntarkkuuden (BCVA) keskimääräinen muutos kuukaudesta 1 kuukauteen 3 verrattuna lähtötasoona (kirjaimet) | +10,5 | +10,6 | +2,2 |
Osuus potilaista, jotka saavuttivat näöntarkkuuden paranemisen: ≥ 15 kirjainta, tai saavuttivat ≥ 84 kirjainta BCVA:ssa | 38,1 % | 43,1 % | 14,5 % |
Kuukausi 12 | |||
Pistosten lukumäärä kuukauteen 12 mennessä: Keskiarvo Mediaani | 4,6 4,0 | 3,5 2,5 | N/A N/A |
Parhaan lasikorjatun näöntarkkuuden (BCVA) keskimääräinen muutos kuukaudesta 1 kuukauteen 12 verrattuna lähtötasoon (kirjaimet) | +12,8 | +12,5 | N/A |
Osuus potilaista, jotka saavuttivat näöntarkkuuden paranemisen: ≥ 15 kirjainta, tai saavuttivat ≥ 84 kirjainta BCVA:ssa | 53,3 % | 51,7 % | N/A |
a p < 0,00001 verrattuna vPDT-ryhmään
b Vertaileva kontrolli oli käytössä kuukauteen 3 saakka. Visudynellä suoritettavaan fotodynaamiseen hoitoon (vPDT) satunnaistetuille potilaille sallittiin ranibitsumabihoito kuukaudesta 3 alkaen (38 ryhmän III potilaista saivat ranibitsumabia kuukaudesta 3 lähtien).
Kuva 2 Parhaan lasikorjatun näöntarkkuuden (BCVA) keskimuutos lähtötilanteesta 12. tutkimuskuukauteen mennessä (RADIANCE)
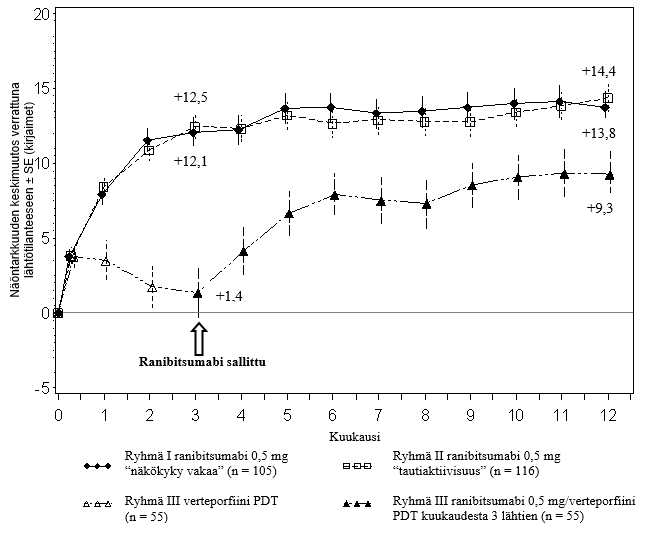
Näkökyvyn paranemiseen liittyi verkkokalvon keskiosan paksuuden vähenemistä.
Potilaiden raportoimia, ranibitsumabilla saavutettuja hyötyjä suhteessa vPDT-hoitoon (p-arvo < 0,05) todettiin yhteenlaskettujen pisteiden ja useampien osa-alueiden tulosten (yleinen näkökyky, lähinäkö, mielenterveys ja riippuvuus muiden ihmisten avusta) osalta, kun näitä mitattiin the National Eye Institute Visual Function Questionnaire (NEI VFQ-25) -kyselyn pisteiden avulla.
Silmän suonikalvon uudissuonittumisen (CNV) aiheuttaman näkökyvyn heikkenemisen hoito (kun tämä ei ole sekundaarista patologiselle likitaittoisuudelle eikä ikään liittyvälle kostealle verkkokalvon makulan rappeumalle)
Lucentis-valmisteen kliinistä turvallisuutta ja tehoa hoidettaessa potilaita, joilla on silmän suonikalvon uudissuonittumisesta aiheutuvaa näkökyvyn heikkenemistä, on arvioitu 12 kuukautta kestäneen, kaksoissokkoutetun, lumelääkekontrolloidun päätutkimuksen G2301 (MINERVA) tulosten perusteella. Kyseistä tutkimusta varten 178 aikuista potilasta satunnaistettiin suhteessa 2:1 saamaan jompaakumpaa seuraavista hoidoista:
- 0,5 mg ranibitsumabia lähtötasossa, jonka jälkeen yksilöllinen, näöntarkkuuden ja/tai anatomisten parametrien perusteella määritellyn taudin aktiivisuuden (esim. näöntarkkuuden heikkeneminen, verkkokalvon sisäinen/alainen neste, verenvuoto tai tihkuminen) mukaan säädetty hoito-ohjelma
- lumelääkepistos tutkimuksen lähtötasossa, jonka jälkeen yksilöllinen, taudin aktiivisuuden mukaan määritelty hoito-ohjelma.
Kuukaudesta 2 alkaen kaikki potilaat saivat avointa ranibitsumabihoitoa tarpeensa mukaan.
MINERVA-tutkimuksen keskeisimmät tulokset on esitetty taulukossa 3 ja kuvassa 3. Näkökyvyn paranemista, johon liittyi keskeisen makulan paksuuden vähenemistä, todettiin koko 12 kk:n jakson ajan.
Keskimääräinen pistosten lukumäärä 12 kuukauden aikana oli 5,8 ranibitsumabiryhmässä ja 5,4 niillä potilailla, jotka kuuluivat lumelääkeryhmään ja jotka soveltuivat saamaan ranibitsumabihoitoa kuukaudesta 2 eteenpäin. Lumelääkeryhmän yhteensä 59 potilaasta 7 potilasta ei saanut lainkaan ranibitsumabihoitoa tutkittavaan silmään koko 12 kuukauden jakson aikana.
Taulukko 3 Tutkimustulokset kuukautena 2 (MINERVA)
Ranibitsumabi 0,5 mg (n = 119) | Lumelääke (n = 59) | |
Parhaan lasikorjatun näöntarkkuuden (BCVA) keskimääräinen muutos lähtötasosta kuukauteen 2 mennessäa | 9,5 kirjainta | -0,4 kirjainta |
Osuus potilaista, jotka olivat saavuttaneet näöntarkkuuden paranemisen: ≥ 15 kirjainta, tai jotka olivat saavuttaneet ≥ 84 kirjainta BCVA:ssa kuukauteen 2 mennessä | 31,4 % | 12,3 % |
Osuus potilaista, jotka eivät olleet menettäneet > 15 kirjainta lähtötasosta kuukauteen 2 mennessäa | 99,2 % | 94,7 % |
CSFT:nb väheneminen lähtötasosta kuukauteen 2 mennessäa | 77 µm | -9,8 µm |
a Yhdensuuntainen p < 0,001 verrattuna lumelääkekontrolliin
b CSFT - keskeisen makulan paksuus
Kuva 3 Parhaan lasikorjatun näöntarkkuuden (BCVA) keskimuutos lähtötilanteesta 12. tutkimuskuukauteen mennessä (MINERVA)
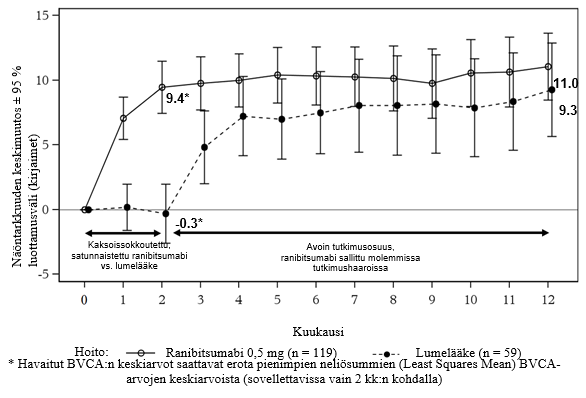
Kun ranibitsumabia verrattiin lumelääkekontrolliin tutkimuskuukauden 2 kohdalla, havaittiin yhdenmukainen teho sekä kaiken kaikkiaan että kaikkien lähtötason etiologisten alaryhmien osalta.
Taulukko 4 Hoidon kokonaisteho sekä teho eri lähtötason etiologisten alaryhmien osalta
Kokonaistulos ja tulokset eri etiologisten ryhmien osalta | Hoitoteho lumelääkkeeseen nähden (kirjaimet) | Potilaiden lukumäärä (n) (hoito + lumelääke) |
Kaikki osallistujat | 9,9 | 178 |
Verisuonia muistuttavat juosteet silmänpohjassa (Angioid Streaks) | 14,6 | 27 |
Tulehduksen jälkeinen retinokoroidopatia | 6,5 | 28 |
Keskeinen, seroosi korioretinopatia | 5,0 | 23 |
Idiopaattinen korioretinopatia | 11,4 | 63 |
Sekalaiset etiologiata | 10,6 | 37 |
a Käsittää erilaisia etiologioita, joiden esiintyvyys oli alhainen ja jotka eivät kuuluneet muihin alaryhmiin
Päätutkimuksessa G2301 (MINERVA) viisi nuorta, 12 - 17-vuotiasta potilasta, joilla ilmeni silmän suonikalvon uudissuonittumisesta johtuvaa näkökyvyn heikkenemistä, saivat tutkimuksen lähtötasossa avointa ranibitsumabihoitoa (0,5 mg) ja sen jälkeen samalla tavalla yksilöllisesti säädettyä hoitoa kuin aikuisten tutkimuksessa. Paras lasikorjattu näöntarkkuus (BCVA) parani kaikilla viidellä potilaalla lähtötasosta kuukauteen 12 mennessä yhteensä 5 - 38 kirjainta (keskiarvo: 16,6 kirjainta). Näkökyvyn paranemiseen liittyi keskeisen makulan paksuuden vakautuminen tai väheneminen 12 kuukauden jakson aikana. Keskimääräinen ranibitsumabipistosten lukumäärä kaikkien osallistujien tutkittavaan silmään 12 kuukauden aikana oli 3 (vaihteluväli: 2 - 5). Kaiken kaikkiaan ranibitsumabihoito oli hyvin siedetty.
Diabeettisesta makulaturvotuksesta aiheutuvan näöntarkkuuden heikkenemisen hoito
Lucentis-valmisteen tehoa ja turvallisuutta arvioitiin kolmessa satunnaistetussa, kontrolloidussa, vähintään 12 kuukautta kestäneessä tutkimuksessa. Näissä tutkimuksissa oli mukana yhteensä 868 potilasta (708 potilasta saivat vaikuttavaa ainetta ja 160 potilasta olivat kontrolliryhmissä).
Vaiheen II tutkimuksessa D2201 (RESOLVE), 151 potilaalle annettiin joko ranibitsumabia (6 mg/ml, n = 51; 10 mg/ml, n = 51) tai lumelääkettä (n = 49) kuukauden välein annettavina injektioina silmän lasiaiseen. Ranibitsumabihoitoa saaneiden potilaiden (n = 102) yhdistettyjen tulosten perusteella parhaan lasikorjatun näöntarkkuuden (BCVA) keskimääräinen muutos kuukaudesta 1 kuukauteen 12 verrattuna lähtötasoon oli +7,8 (± 7,72) kirjainta verrattuna -0,1 (± 9,77) kirjaimen muutokseen lumelääkettä saaneisiin; ja vastaavat keskimääräiset parhaan lasikorjatun näöntarkkuuden muutokset lähtötasosta kuukauteen 12 mennessä olivat 10,3 (± 9,1) verrattuna -1,4 (± 14,2) kirjaimeen (p < 0,0001 hoitojen erolle).
Vaiheen III tutkimuksessa D2301 (RESTORE) 345 potilasta satunnaistettiin suhteessa 1:1:1 saamaan 0,5 mg ranibitsumabia monoterapiana yhdistettynä lumelaserkoagulaatioon, 0,5 mg ranibitsumabia yhdistettynä laserkoagulaatioon, tai lumelääkeinjektiot ja laserkoagulaatiohoidon. 240 potilasta, jotka aiemmin olivat osallistuneet koko 12 kuukautta kestäneeseen RESTORE-tutkimukseen, otettiin mukaan avoimeen, 24 kuukautta kestäneeseen monikeskusjatkotutkimukseen (RESTORE Extension). Potilaat saivat 0,5 mg:n ranibitsumabiannokset pro re nata (PRN) -hoitona samaan silmään, johon pistokset oli annettu päätutkimuksen yhteydessä (D2301 RESTORE).
Yhteenveto tärkeimmistä tuloksista esitetään taulukossa 5 (RESTORE ja sen jatkotutkimus RESTORE Extension) sekä kuvassa 4 (RESTORE).
Kuva 4 Näöntarkkuuden keskimuutos ajan mittaan lähtötilanteesta tutkimuksessa D2301 (RESTORE)
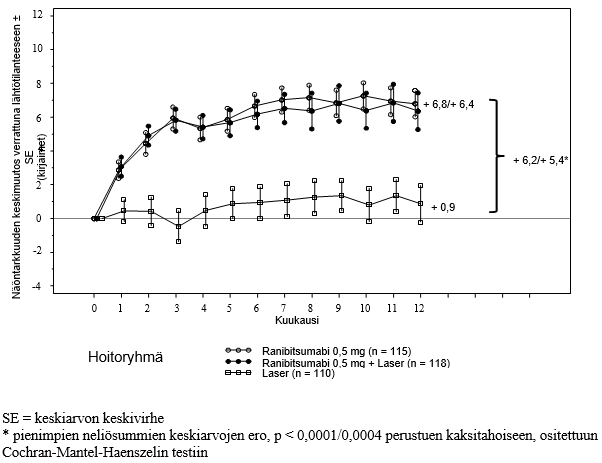
12. kuukauden kohdalla hoidon teho oli yhdenmukainen useimmissa alaryhmissä. Potilaat, joilla lähtötason paras lasikorjattu näöntarkkuus oli > 73 kirjainta ja makulaturvotus, jossa verkkokalvon keskiosan paksuus oli < 300 μm, eivät kuitenkaan näyttäneet saavan suurempaa hyötyä ranibitsumabihoidosta verrattuna laserkoagulaatiohoitoon.
Taulukko 5 Tulokset 12 kuukauden kohdalla tutkimuksessa D2301 (RESTORE) ja 36 kuukauden kohdalla tutkimuksessa D2301-E1 (RESTORE Extension -jatkotutkimus)
Hoitotulokset 12 kuukauden kohdalla verrattuna lähtötasoon tutkimuksessa D2301 (RESTORE) | Ranibitsumabi 0,5 mg n = 115 | Ranibitsumabi 0,5 mg + laserkoagulaatiohoito n = 118 | Laserkoagulaatiohoito n = 110 |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos kuukaudesta 1 kuukauteen 12a (±SD) | 6,1 (6,4)a | 5,9 (7,9)a | 0,8 (8,6) |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos kuukautena 12 (±SD) | 6,8 (8,3)a | 6,4 (11,8)a | 0,9 (11,4) |
Näöntarkkuuden paraneminen ≥ 15 kirjainta tai paras lasikorjattu näöntarkkuus ≥ 84 kirjainta kuukautena 12(%) | 22,6 | 22,9 | 8,2 |
Keskimääräinen pistosten lukumäärä (kuukaudet 0 - 11) | 7,0 | 6,8 | 7,3 (lumelääke) |
Tutkimuksen D2301-E1 (RESTORE Extension) hoitotulokset kuukautena 36 verrattuna D2301 (RESTORE) -tutkimuksen lähtötasoon | Aikaisempi 0,5 mg:n ranibitsumabihoito n = 83 | Aikaisempi 0,5 mg:n ranibitsumabihoito + laserkoagulaatiohoito n = 83 | Aikaisempi laserkoagulaatiohoito n = 74* |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos kuukautena 24 (SD) | 7,9 (9,0) | 6,7 (7,9) | 5,4 (9,0) |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos kuukautena 36 (SD) | 8,0 (10,1) | 6,7 (9,6) | 6,0 (9,4) |
Näöntarkkuuden paraneminen ≥ 15 kirjainta tai paras lasikorjattu näöntarkkuus ≥ 84 kirjainta kuukautena 36 (%) | 27,7 | 30,1 | 21,6 |
Keskimääräinen pistosten lukumäärä (kuukaudet 12 - 35) | 6,8 | 6,0 | 6,5 |
ap < 0,0001 ranibitsumabia ja laserkoagulaatiohoitoa saaneiden ryhmien vertailussa
Tutkimuksessa D2301-E1 (RESTORE Extension) n on se lukumäärä potilaita, joilta oli tiedossa olevat arvot sekä tutkimuksen D2301 (RESTORE) lähtötasossa (kuukausi 0) että kuukauden 36 käynnin yhteydessä.
* Osuudet potilaista, jotka eivät saaneet yhtään ranibitsumabihoitoa jatkovaiheen aikana olivat 19 % aiemmin ranibitsumabiryhmään kuuluneista, 25 % aiemmin ranibitsumabia + laserkoagulaatiohoitoa saaneista sekä 20 % aiemmin laserkoagulaatiohoitoa saaneista potilaista.
Tilastollisesti merkitsevää, potilaiden itsensä raportoimaa etua, joka kohdistui useimpiin näköön liittyviin toimintoihin todettiin ranibitsumabilla (laserkoagulaatiohoidon kanssa tai ilman sitä) suhteessa vertailuryhmien tuloksiin, kun NEI VFQ-25 kyselyä käytettiin tulosten mittarina. Muiden tämän kyselytutkimuksen ala-asteikkojen osalta ei nähty eroja hoitojen välillä.
24 kuukautta kestäneen jatkotutkimuksen aikana todettu ranibitsumabin pitkäaikaishoidon turvallisuusprofiili on yhdenmukainen Lucentis-valmisteen aikaisemmin tunnetun turvallisuusprofiilin kanssa.
Vaiheen IIIb tutkimuksessa D2304 (RETAIN) 372 potilasta satunnaistettiin suhteessa 1:1:1 saamaan jotakin alla mainituista hoidoista:
- 0,5 mg ranibitsumabia yhdistettynä laserkoagulaatiohoitoon annostusohjelmassa, jossa hoitovälejä voidaan pidentää (TE, treat-and-extend)
- 0,5 mg ranibitsumabia monoterapiana annostusohjelmassa, jossa hoitovälejä voidaan pidentää (TE, treat-and-extend)
- 0,5 mg ranibitsumabia monoterapiana PRN-annostelun (pro re nata) mukaan.
Kaikissa ryhmissä ranibitsumabihoito annettiin kuukauden välein, kunnes paras lasikorjattu näöntarkkuus pysyi vakaana ainakin kolmen peräkkäisen kuukausittaisen arvioinnin yhteydessä. TE-hoidossa ranibitsumabia annettiin 2 - 3 kuukauden välein. Kaikissa hoitoryhmissä kuukausittaiset hoidot aloitettiin uudestaan parhaan lasikorjatun näöntarkkuuden alkaessa heikentyä diabeettisen makulaturvotuksen etenemisen vuoksi. Tällöin hoitoa jatkettiin kunnes vakaa tilanne parhaan lasikorjatun näöntarkkuuden osalta jälleen saavutettiin.
Kolmen ensimmäisen pistoksen jälkeen suunniteltujen hoitokäyntien määrä oli TE-ryhmässä 13 ja PRN-ryhmässä 20. Molemmissa TE-ryhmissä yli 70 % potilaista säilytti parhaan lasikorjatun näöntarkkuutensa keskimäärin ≥ 2 kuukauden välein toteutetuin tutkimuskäynnein.
Yhteenveto tärkeimmistä tuloksista esitetään taulukossa 6.
Taulukko 6 Tutkimuksen D2304 (RETAIN) tulokset
Tulokset verrattuna lähtötasoon | TE-ranibitsumabihoito 0,5 mg + laserkoagulaatio n = 117 | TE-ranibitsumabihoito 0,5 mg yksinään n = 125 | PRN-ranibitsumabihoito 0,5 mg n = 117 |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos kuukaudesta 1 kuukauteen 12 (SD) | 5,9 (5,5) a | 6,1 (5,7) a | 6,2 (6,0) |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos kuukaudesta 1 kuukauteen 24 (SD) | 6,8 (6,0) | 6,6 (7,1) | 7,0 (6,4) |
Lasikorjatun näöntarkkuuden keskimääräinen muutos kuukautena 24 (SD) | 8,3 (8,1) | 6,5 (10,9) | 8,1 (8,5) |
Näöntarkkuuden paraneminen ≥ 15 kirjainta tai paras lasikorjattu näöntarkkuus ≥ 84 kirjainta kuukautena 24(%) | 25,6 | 28,0 | 30,8 |
Keskimääräinen pistosten lukumäärä (kuukaudet 0 - 23) | 12,4 | 12,8 | 10,7 |
ap < 0,0001 arvioitaessa vertailukelposuutta (non-inferiority) PRN-hoitoa vastaan.
Diabeettista makulaturvotusta koskevissa tutkimuksissa parhaan lasikorjatun näöntarkkuuden paranemiseen liittyi kaikissa hoitoryhmissä keskeisen makulan paksuuden (CSFT) väheneminen ajan kuluessa.
Proliferatiivisen diabeettisen retinopatian hoito
Lucentisin kliinistä turvallisuutta ja tehoa proliferatiivista diabeettista retinopatiaa sairastavien potilaiden hoidossa arvioitiin Protocol S –tutkimuksessa, jossa verrattiin lasiaiseen annettuja 0,5 mg ranibitsumabi-injektioita ja panretinaalista valopolttohoitoa (PRP, panretinal photocoagulation). Ensisijaisena päätetapahtumana oli näöntarkkuuden keskimuutos 2 vuoden kohdalla. Lisäksi arvioitiin diabeettisen retinopatian vaikeusasteen muutosta silmänpohjan valokuvien perusteella DRSS-asteikkoa (Diabetic retinopathy severity score; diabeettisen retinopatian vaikeusasteikko) käyttäen.
Protocol S oli vaiheen III monikeskuksinen satunnaistettu, aktiivikontrolloitu, rinnakkaisryhmä-, vertailukelpoisuus (non inferiority) –tutkimus. Tutkimukseen otettiin 305 potilasta (394 tutkittavaa silmää), joilla oli proliferatiivinen diabeettinen retinopatia, johon lähtötilanteessa liittyi tai ei liittynyt diabeettista makulaturvotusta (DME). Tutkimuksessa verrattiin lasiaiseen annettuja 0,5 mg ranibitsumabi-injektioita ja standardihoitoa eli panretinaalista valopolttohoitoa (PRP). Yhteensä 191 silmää (48,5 %) satunnaistettiin saamaan 0,5 mg ranibitsumabihoitoa ja 203 silmää (51,5 %) saamaan panretinaalista valopolttohoitoa. Yhteensä 88 silmässä (22,3 %) oli lähtötilanteessa diabeettista makulaturvotusta: 42:ssa (22,0 %) ranibitsumabiryhmän ja 46:ssa (22,7 %) panretinaalisen valopolttohoitoryhmän silmässä.
Tässä tutkimuksessa näöntarkkuuden keskimuutos 2 vuoden kohdalla oli +2,7 kirjainta ranibitsumabiryhmässä ja ‑0,7 kirjainta panretinaalisen valopolttohoidon ryhmässä. Pienimmän neliösumman keskiarvojen ero oli 3,5 kirjainta (95 % lv: [0,2; 6,7]).
1 vuoden kohdalla DRSS oli parantunut ≥ 2 astetta 41,8 %:lla ranibitsumabilla hoidetuista silmistä (n = 189) ja 14,6 %:lla panretinaalisella valohoidolla hoidetuista silmistä (n = 199). Ranibitsumabin ja laserhoidon arvioitu ero oli 27,4 % (95 % lv: [18,9; 35,9]).
Taulukko 7 DRSS:n paraneminen tai huononeminen ≥ 2 tai ≥ 3 asteella 1 vuoden kohdalla Protocol S –tutkimuksessa (LOCF-menetelmä)
Muutoksen luokittelu lähtötilanteeseen verrattuna | Protocol S | ||
Ranibitsumabi 0,5 mg (N = 189) | Panretinaalinen valopolttohoito (N = 199) | Osuuksien ero (%), lv | |
≥ 2 asteen paraneminen | |||
n (%) | 79 (41,8 %) | 29 (14,6 %) | 27,4 (18,9; 35,9) |
≥ 3 asteen paraneminen | |||
n (%) | 54 (28,6 %) | 6 (3,0 %) | 25,7 (18,9; 32,6) |
≥ 2 asteen huononeminen | |||
n (%) | 3 (1,6 %) | 23 (11,6 %) | ‑9.9 (‑14,7; ‑5,2) |
≥ 3 asteen huononeminen | |||
n (%) | 1 (0,5 %) | 8 (4,0 %) | ‑3.4 (‑6.3; ‑0.5) |
DRSS = diabetic retinopathy severity score; diabeettisen retinopatian vaikeusasteikko, n = käynnillä määritelmän täyttäneiden potilaiden lukumäärä, N = tutkittavien silmien kokonaislukumäärä. | |||
1 vuoden kohdalla ranibitsumabihoitoa Protocol S –tutkimuksessa saaneessa ryhmässä DRSS-aste oli parantunut ≥ 2 asteella johdonmukaisesti sekä silmissä, joissa ei ollut diabeettista makulaturvotusta (39,9 %) että silmissä, joissa lähtötilanteessa oli diabeettista makulaturvotusta (48,8 %).
Protocol S –tutkimuksen 2 vuoden tietojen analyysi osoitti, että DRSS-aste parani ≥ 2 asteella lähtötilanteeseen verrattuna 42,3 %:ssa (n = 80) silmistä ranibitsumabihoitoa saaneessa ryhmässä ja 23,1 %:ssa (n = 46) silmistä panretinaaliseta valohoitoa saaneista silmistä. Ranibitsumabihoitoa saaneessa ryhmässä ≥ 2 asteen paraneminen DRSS-asteessa lähtötilanteeseen verrattuna havaittiin 58,5 %:ssa (n = 24) niistä silmistä, joissa lähtötilanteessa oli diabeettista makulaturvotusta, ja 37,8 %:ssa (n = 56) niistä silmistä, joissa diabeettista makulaturvotusta ei ollut.
Diabeettisen retinopatian vaikeusasteen muutosta arvioitiin DRSS-asteikon avulla myös kolmessa erillisessä diabeettista makulaturvotusta koskevassa aktiivikontrolloidussa vaiheen III tutkimuksessa (ranibitsumabi 0,5 mg tarpeen mukaan verrattuna laserhoitoon). Tutkimuksiin osallistui yhteensä 875 potilasta, joista noin 75 % oli aasialaista alkuperää. Lähtötasoltaan keskivaikean tai sitä vaikeamman ei-proliferatiivisen DR:n (NPDR) alaryhmässä arvioitiin 315 potilaan DRSS-aste. Tutkimustulosten meta-analyysi osoitti, että 48,4 %:lla ranibitsumabihoitoa saaneista potilaista (n = 192) ja 14,6 %:lla laserhoitoa saaneista potilaista (n = 123) DRSS parantui ≥ 2 astetta kuukauden 12 kohdalla. Ranibitsumabihoidon ja laserhoidon arvioitu ero oli 29,9 % (95 % lv: [20,0; 39,7]). Vaikeusasteeltaan kohtalaisen tai sitä lievemmän NPDR:n alaryhmässä arvioitiin 405 potilaan DRSS-aste. DRSS parantui ≥ 2 astetta 1,4 %:lla ranibitsumabihoitoa saaneista potilaista ja 0,9 %:lla laserhoitoa saaneista potilaista.
Verkkokalvon laskimotukoksesta johtuvan makulaturvotuksen aiheuttaman näkökyvyn heikkenemisen hoito
Lucentis-valmisteen tehoa ja turvallisuutta verkkokalvon laskimotukoksesta aiheutuneen makulaturvotuksen aiheuttaman näkökyvyn heikkenemisen hoidossa on arvioitu satunnaistetuissa, kaksoissokkoutetuissa, kontrolloiduissa BRAVO- ja CRUISE-tutkimuksissa, joista ensimmäiseen osallistui verkkokalvon laskimohaaratukosta sairastavia (n = 397) ja toiseen verkkokalvon keskuslaskimotukosta sairastavia (n = 392) potilaita. Molemmissa tutkimuksissa potilaat saivat joko 0,3 mg:n tai 0,5 mg:n ranibitsumabi–injektiot tai lumelääkeinjektiot. Kuuden kuukauden jälkeen lumelääkettä saaneiden potilaiden lääkitys vaihdettiin 0,5 mg:aan ranibitsumabia.
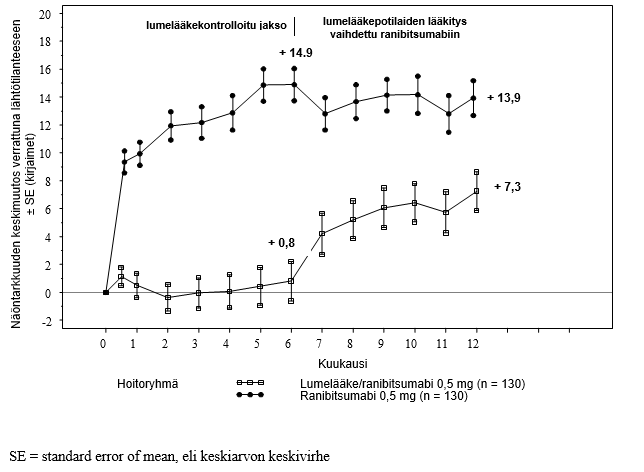
BRAVO- ja CRUISE-tutkimusten keskeisimmät tulokset on esitetty taulukossa 8 sekä kuvissa 5 ja 6.
Taulukko 8 Tutkimustulokset kuukausina 6 ja 12 (BRAVO ja CRUISE)
BRAVO | CRUISE | |||
Lumelääke/Lucentis 0,5 mg (n = 132) | Lucentis 0,5 mg (n = 131) | Lumelääke/Lucentis 0,5 mg (n = 130) | Lucentis 0,5 mg (n = 130) | |
Näöntarkkuuden keskimääräinen muutos kuukautena 6a (kirjaimet) (SD) (ensisijainen päätetapahtuma) | 7,3 (13,0) | 18,3 (13,2) | 0,8 (16,2) | 14,9 (13,2) |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos tutkimuskuukautena 12 (kirjaimet) (SD) | 12,1 (14,4) | 18,3 (14,6) | 7,3 (15,9) | 13,9 (14,2) |
Näöntarkkuuden paraneminen ≥ 15 kirjainta kuukautena 6a (%) | 28,8 | 61,1 | 16,9 | 47,7 |
Näöntarkkuuden paraneminen ≥ 15 kirjainta kuukautena 12 (%) | 43,9 | 60,3 | 33,1 | 50,8 |
Laserkoagulaatiota hätätoimenpiteenä 12 kuukauden aikana saaneiden potilaiden osuus (%) | 61,4 | 34,4 | N/A | N/A |
a p < 0,0001 molemmissa tutkimuksissa
N/A = ei sovellettavissa.
Kuva 5 Parhaimman lasikorjatun näöntarkkuuden (BCVA) keskimuutos ajan mittaan lähtötilanteesta 6. sekä 12. tutkimuskuukauteen mennessä (BRAVO)
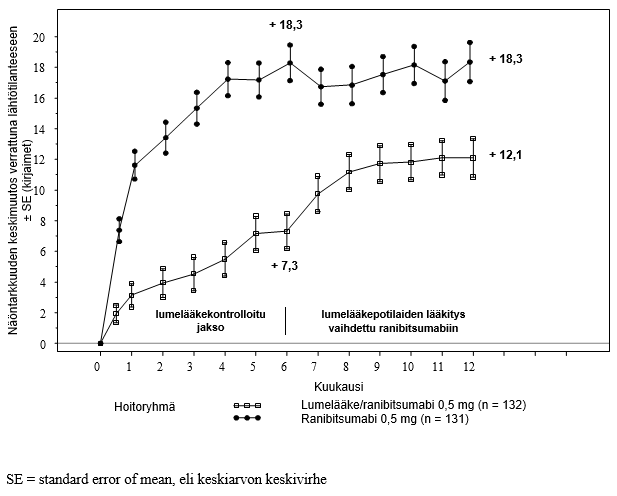
Kuva 6 Parhaimman lasikorjatun näöntarkkuuden (BCVA) keskimuutos ajan mittaan lähtötilanteesta 6. sekä 12. tutkimuskuukauteen mennessä (CRUISE)
Molemmissa tutkimuksissa näkökyvyn paranemiseen liittyi jatkuva ja merkitsevä, verkkokalvon keskiosan paksuuden mittausten avulla todennettu makulaturvotuksen väheneminen.
Verkkokalvon keskuslaskimotukosta (CRVO) sairastavat potilaat (CRUISE-tutkimus ja jatkotutkimus HORIZON): Näöntarkkuuden paraneminen (noin 6 kirjainta) kuukautena 24 sellaisilla potilailla, jotka ensimmäisen kuuden kuukauden aikana saivat lumelääkettä ja sen jälkeen ranibitsumabia, ei ollut yhtä hyvä kuin tutkimuksen alusta asti ranibitsumabihoitoa saaneilla potilailla (noin 12 kirjainta).
Ranibitsumabilla todettiin tilastollisesti merkitsevää, potilaiden itsensä raportoimaa etua lähi- ja kaukonäköön liittyvissä osa-alueissa suhteessa kontrolliryhmään, kun tulosten mittarina käytettiin NEI VFQ-25 -kyselyä.
BRIGHTER (BRVO) ja CRYSTAL (CRVO) –tutkimuksissa arvioitiin Lucentis-hoidon pitkäaikaista (24 kuukautta) kliinistä turvallisuutta ja tehoa niiden potilaiden hoidossa, joiden näkökyky on heikentynyt verkkokalvon laskimotukoksen aiheuttaman makulaturvotuksen vuoksi. Molemmissa tutkimuksissa potilaat saivat 0,5 mg ranibitsumabia yksilöllisten vakauttamiskriteerien perusteella määritellyn PRN-annosteluohjelman mukaisesti. BRIGHTER oli kolmen tutkimushaaran satunnaistettu aktiivikontrolloitu tutkimus, jossa 0,5 mg ranibitsumabia monoterapiana tai yhdistettynä laserkoagulaatiohoitoon verrattiin pelkkään laserkoagulaatiohoitoon. Kuuden kuukauden hoidon jälkeen laser-tutkimushaaran potilaille sallittiin 0,5 mg:n ranibitsumabiannos. CRYSTAL-tutkimuksessa potilaat saivat 0,5 mg ranibitsumabia monoterapiana.
BRIGHTER ja CRYSTAL –tutkimusten keskeisimmät tulokset on esitetty taulukossa 9.
Taulukko 9 Tutkimustulokset kuukausina 6 ja 24 (BRIGHTER and CRYSTAL)
BRIGHTER | CRYSTAL | |||
Lucentis 0,5 mg n = 180 | Lucentis 0,5 mg + Laser n = 178 | Laser* n = 90 | Lucentis 0,5 mg n = 356 | |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos kuukautena 6a (kirjaimet) (SD) | +14,8 (10,7) | +14,8 (11,13) | +6,0 (14,27) | +12,0 (13,95) |
Parhaan lasikorjatun näöntarkkuuden keskimääräinen muutos kuukautena 24b (kirjaimet) (SD) | +15,5 (13,91) | +17,3 (12,61) | +11,6 (16,09) | +12,1 (18,60) |
Parhaan lasikorjatun näöntarkkuuden paraneminen ≥15 kirjainta kuukautena 24 (%) | 52,8 | 59,6 | 43,3 | 49,2 |
Keskimääräinen injektioiden lukumäärä (SD) (kuukausina 0‑23) | 11,4 (5,81) | 11,3 (6.02) | N/A | 13,1 (6,39) |
a p < 0,0001 molemmille vertailuille BRIGHTER-tutkimuksessa 6 kuukauden kohdalla: Lucentis 0,5 mg vs laser ja Lucentis 0,5 mg + laser vs pelkkä laser. b p < 0,0001 CRYSTAL-tutkimuksen nollahypoteesille, jossa keskimääräinen muutos 24 kuukauden kohdalla verrattuna lähtötilanteeseen olisi ollut 0. * Kuukaudesta 6 lähtien ranibitsumabi 0,5 mg –hoito sallittiin (24 potilasta hoidettiin pelkällä laserilla). | ||||
BRIGHTER –tutkimuksessa 0,5 mg ranibitsumabin teho laserhoitoon yhdistettynä oli vertailukelpoinen (non-inferior) ranibitsumabimonoterapian kanssa lähtötilanteesta 24 kuukauteen saakka (95 % lv: [-2,8; 1,4]).
Molemmissa tutkimuksissa havaittiin nopeaa ja tilastollisesti merkitsevää pienenemistä keskeisen makulan paksuudessa hoitokuukautena 1. Tämä vaste säilyi kuukauteen 24 saakka.
Ranibitsumabi-hoidon vaste oli riippumaton verkkokalvon iskemiasta. BRIGHTER –tutkimuksessa ranibitsumabimonoterapialla hoidetuilla potilailla 24 kuukauden kohdalla keskimääräinen muutos lähtötilanteeseen verrattuna oli +15,3 kirjainta potilailla, joilla oli iskemiaa (n = 46) ja +15,6 kirjainta potilailla, joilla ei ollut iskemiaa (n = 133). CRYSTAL –tutkimuksessa ranibitsumabimonoterapialla hoidetuilla potilailla keskimääräinen muutos lähtötilanteeseen verrattuna oli +15,0 kirjainta potilailla, joilla oli iskemiaa (n = 53) ja +11,5 kirjainta potilailla, joilla ei ollut iskemiaa (n = 300).
BRIGHTER ja CRYSTAL –tutkimuksissa näkökykyä parantava vaikutus saavutettiin kaikilla 0,5 mg ranibitsumabimonoterapialla hoidetuilla potilailla riippumatta sairauden kestosta. Potilailla, joiden sairaus oli kestänyt < 3 kuukautta havaittiin näöntarkkuuden parantuneen kuukautena 1 BRIGHTER –tutkimuksessa 13,3 ja CRYSTAL –tutkimuksessa 10,0 kirjainta, ja vastaavasti kuukautena 24 BRIGHTER –tutkimuksessa 17,7 ja CRYSTAL –tutkimuksessa 13,2 kirjainta. Vastaava näöntarkkuuden paraneminen potilailla, joiden sairaus oli kestänyt ≥12 kuukautta, oli BRIGHTER –tutkimuksessa 8,6 ja CRYSTAL –tutkimuksessa 8,4 kirjainta. Hoidon aloittamista pitäisi harkita diagnoosihetkellä.
Ranibitsumabin 24 kuukautta kestäneissä tutkimuksissa osoitettu pitkäaikaisturvallisuusprofiili on yhdenmukainen Lucentis-valmisteen tunnetun turvallisuusprofiilin kanssa.
Pediatriset potilaat
Ranibitsumabin (0,5 mg esitäytetyssä ruiskussa) turvallisuutta ja tehoa ei ole tutkittu lapsipotilailla.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Lucentis-valmisteen käytöstä neovaskulaarisen verkkokalvon makulan rappeuman (AMD), diabeettisen makulaturvotuksen (DME), verkkokalvon laskimotukoksesta (RVO) johtuvan makulaturvotuksen, tai silmän suonikalvon uudissuonittumisen (CNV) aiheuttaman näkökyvyn heikkenemisen ja diabeettisen retinopatian hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Kuukausittain annetun Lucentis-intravitreaalihoidon jälkeen neovaskulaarista AMD:tä sairastavien potilaiden seerumin ranibitsumabipitoisuudet olivat yleensä alhaiset ja huippupitoisuus (Cmax) yleensä pienempi kuin ranibitsumabipitoisuus, joka tarvitaan estämään VEGF:n biologista aktiivisuutta 50 prosentilla (11‑27 ng/ml, joka on määritetty soluproliferaatiokokeessa in vitro). Huippupitoisuus (Cmax) oli annosriippuvainen annosvälillä 0,05‑1,0 mg/silmä. Seerumin lääkepitoisuudet rajallisella määrällä diabeettista makulaturvotusta sairastavilla potilailla viittasivat siihen, että hieman suurempaa systeemistä altistusta ei voida sulkea pois verrattaessa neovaskulaarista AMD:tä sairastaviin potilaisiin. RVO-potilailla ranibitsumabipitoisuudet seerumissa olivat samankaltaiset tai hieman korkeammat kuin kosteaa AMD:tä sairastavilla potilailla.
Ranibitsumabin keskimääräinen puoliintumisaika oli lasiaisessa noin 9 vuorokautta tutkimuksen mukaan, joka perustui populaatiofarmakokinetiikkaan ja ranibitsumabin poistumiseen seerumista hoidettaessa neovaskulaarista AMD:tä sairastavia potilaita 0,5 mg annoksella. Kuukausittaisella intravitreaaliannoksella 0,5 mg/silmä ranibitsumabin huippupitoisuus (Cmax) saavutettiin seerumissa noin vuorokauden kuluttua annosta. Huippupitoisuuden ennustetaan olevan yleensä 0,79‑2,90 ng/ml ja pienimmän pitoisuuden (Cmin) yleensä 0,07‑0,49 ng/ml. Seerumin ranibitsumabipitoisuuden arvioidaan olevan noin 90 000 kertaa pienempi kuin lasiaisen ranibitsumabipitoisuus.
Munuaisten vajaatoiminta: Lucentis-valmisteen farmakokinetiikasta ei ole tehty varsinaisia tutkimuksia munuaisten vajaatoimintaa sairastavilla potilailla. Kosteaa AMD:tä sairastavilla potilailla suoritetussa populaatiofarmakokineettisessa analyysissä 68 % (136 potilasta 200:sta) potilaista sairasti munuaisten vajaatoimintaa (46,5 % lievää [50‑80 ml/min], 20 % kohtalaisen vaikeaa [30‑50 ml/min] ja 1,5 % vakavaa [< 30 ml/min]). RVO-potilaista 48,2 %:lla (253 potilaalla 525:stä) potilaista oli munuaisten vajaatoiminta (joista 36,4 %:lla lievä; 9,5 %:lla keskivaikea ja 2,3 %:lla vaikea). Systeeminen puhdistuma oli hieman pienempi, mutta ei kliinisesti merkittävästi.
Maksan vajaatoiminta: Lucentis-valmisteen farmakokinetiikasta ei ole tehty varsinaisia tutkimuksia maksan vajaatoimintaa sairastavilla potilailla.
Prekliiniset tiedot turvallisuudesta
Kun ranibitsumabia annettiin makaki-apinoille molempiin silmiin intravitreaalisesti annoksina 0,25‑2,0 mg/silmä joka toinen viikko enintään 26 viikon ajan, se aiheutti silmiin annosriippuvaisia vaikutuksia.
Intraokulaarisessa annossa etukammion valotie ja solut lisääntyivät annosriippuvaisesti, ja tämän lisääntymisen huippu oli kaksi päivää injektiosta. Tulehdusvasteen voimakkuus lievenee yleensä seuraavien injektioiden myötä tai toipumisvaiheessa. Silmän takaosassa havaittiin lasiaissolujen infiltraatiota ja lasiaissamentumia, jotka näyttivät olevan annoksen suuruudesta riippuvaisia, ja joita yleensä esiintyi hoitojakson loppuun asti. Lasiaistulehdukset pahenivat 26 viikkoa kestäneessä tutkimuksessa injektiomäärien myötä. Toipumisen jälkeen niiden havaittiin kuitenkin hävinneen. Silmän takaosan tulehduksen ajoitus ja luonne näyttäisi viittaavan immuunivälitteiseen vasta-ainevasteeseen, joka voi olla kliinisesti merkityksetöntä. Joillakin eläimillä havaittiin kaihin kehittymistä suhteellisen pitkään kestäneen vakavan tulehduksen jälkeen, mikä näyttäisi viittaavan siihen, että linssin muutokset olivat seurausta vakavasta tulehduksesta. Intravitreaalisen injektion jälkeen havaittiin annoksesta riippumatta ohimenevää silmänpaineen nousua.
Mikroskooppiset silmämuutokset liittyivät tulehdukseen, eivätkä viitanneet rappeuttaviin muutoksiin. Granulomatoottisia tulehdusmuutoksia havaittiin joidenkin silmien näköhermon nystyissä. Nämä silmän takaosan muutokset vähenivät ja joissain tapauksissa hävisivät kokonaan toipumisvaiheen aikana.
Intravitreaalisen annon jälkeen ei havaittu merkkejä systeemisestä toksisuudesta. Seerumissa ja lasiaisessa havaittiin ranibitsumabin vasta-aineita valmisteella hoidettujen eläinten alaryhmästä.
Karsinogeenisuuteen tai mutageenisuuteen liittyviä tietoja ei ole saatavilla.
Tiineille apinoille silmän lasiaiseen annettu ranibitsumabihoito, joka aiheutti 0,9‑7 kertaisen maksimaalisen systeemisen altistuksen verrattuna suurimpaan kliiniseen altistukseen, ei aiheuttanut kehitystoksisuutta eikä teratogeenisuutta eikä se vaikuttanut istukan painoon tai rakenteeseen, vaikka ranibitsumabin tulisi farmakologisten vaikutustensa perusteella katsoa olevan mahdollisesti teratogeeninen tai alkio-/sikiötoksinen.
Ranibitsumabivälitteisiä vaikutuksia alkion ja sikiön kehitykseen ei esiintynyt, ja se johtuu todennäköisesti lähinnä siitä, ettei Fab-fragmentti kyennyt läpäisemään istukkaa. Yksi tapaus kuitenkin kuvattiin, jossa emon seerumissa esiintyi suuria ranibitsumabipitoisuuksia ja ranibitsumabia oli havaittavissa sikiön seerumissa, mikä viittaa siihen, että ranibitsumabivasta-aine (Fc-osan sisältävä) toimi ranibitsumabin kantajaproteiinina ja pienensi siten emon seerumipuhdistumaa ja mahdollisti ranibitsumabin kulkeutumisen istukan läpi. Koska alkion ja sikiön kehitykseen liittyvät tutkimukset tehtiin terveillä tiineillä eläimillä, sairaus (esim. diabetes) saattaa muuttaa istukan läpäisevyyttä Fab-fragmentin osalta, joten tutkimus tulisi tulkita varauksella.
Farmaseuttiset tiedot
Apuaineet
α,α-trehaloosidihydraatti
Histidiinihydrokloridi, monohydraatti
Histidiini
Polysorbaatti 20
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Säilytä jääkaapissa (2°C − 8°C).
Ei saa jäätyä.
Pidä esitäytetty ruisku avaamattomassa repäisypakkauksessaan ja pahvikotelossa. Herkkä valolle. Avaamaton repäisypakkaus voi olla huoneenlämmössä (25 °C) enintään 24 tunnin ajan ennen käyttöä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LUCENTIS injektioneste, liuos, esitäytetty ruisku
10 mg/ml (L:ei) 0,165 ml (1206,80 €)
PF-selosteen tieto
0,165 ml steriiliä liuosta sisältävä esitäytetty ruisku (tyypin I lasia), jossa kumitulppa (bromobutyyliä) männän päässä ja valkoinen, jäykkä, mahdolliset avaamisyritykset paljastava sinettirengas sekä harmaa, kuminen suojakorkki (bromobutyyliä) ruiskun päässä, jossa on myös Luer-lock-liitin. Esitäytetyssä ruiskussa on lisäksi männän varsi sekä sormituki ja se on pakattu kiinnisaumattuun repäisypakkaukseen.
Pakkaus sisältää yhden esitäytetyn ruiskun.
Valmisteen kuvaus:
Kirkas, väritön tai vaalea ruskeankeltainen vesiliuos.
Käyttö- ja käsittelyohjeet
Esitäytetty ruisku on tarkoitettu vain kertakäyttöön. Esitäytetty ruisku on steriili. Älä käytä valmistetta, jos repäisypakkaus on vaurioitunut. Esitäytetyn ruiskun steriiliyttä ei voida taata, jos repäisypakkaus ei ole ehjä. Älä käytä esitäytettyä ruiskua, jos liuos on värjäytynyt, samea tai sisältää hiukkasia.
Esitäytetty ruisku sisältää enemmän lääkettä kuin 0,5 mg:n suositusannoksen. Esitäytetystä ruiskusta saatavaa kokonaismäärää (0,1 ml) ei tule käyttää kokonaan. Liuosylimäärä on poistettava ennen annostelua. Esitäytetyn ruiskun kokonaismäärän injisointi silmään voi johtaa yliannostukseen. Poista ilmakuplat ja ylimääräinen lääkevalmiste painamalla mäntää hitaasti kunnes männän kuperan kärjen reuna on ruiskussa olevan mustan annosviivan kohdalla (vastaa 0,05 ml eli 0,5 mg ranibitsumabia).
Lasiaiseen annettavaan injektioon tulee käyttää steriiliä, 30G x ½″-kokoista injektioneulaa.
Noudata käyttöohjeita valmistaessasi Lucentis-valmistetta annettavaksi silmän lasiaiseen:
Johdanto | Lue kaikki ohjeet huolellisesti ennen esitäytetyn ruiskun käyttöä. Esitäytetty ruisku on tarkoitettu vain kertakäyttöön. Esitäytetty ruisku on steriili. Älä käytä valmistetta, jos repäisypakkaus on vaurioitunut. Repäisypakkauksen avaaminen ja kaikki sen jälkeen tehtävät toimenpiteet on suoritettava aseptisissa olosuhteissa. Huom! Annos on asetettava 0,05 millilitraan. | |
Esitäytetyn ruiskun kuvaus |
| |
Esivalmistelut | 1. Varmista, että pakkaus sisältää:
2. Revi kansi pois repäisypakkauksen päältä ja poista ruisku pakkauksestaan aseptista tekniikkaa noudattaen. | |
Tarkista ruisku | 3. Tarkista, että:
4. Jos jokin edellä mainituista kohdista ei täyty, kyseinen esitäytetty ruisku on hävitettävä ja annostelu suoritettava toisesta ruiskusta. | |
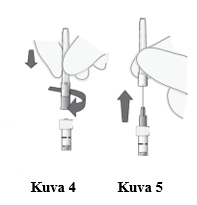
Ruiskun suojakorkin poistaminen | 5. Napsauta (älä käännä tai kierrä) ruiskun suojakorkki irti (ks. kuva 2). 6. Heitä ruiskun suojakorkki roskiin (ks. kuva 3). |
|
Kiinnitä neula | 7. Kiinnitä steriili 30G x ½″-kokoinen injektioneula tukevasti ruiskuun kiertämällä se tiiviisti Luer-lock-liittimeen (ks. kuva 4). 8. Poista varovasti neulan suojus pystysuoraan vetämällä (ks. kuva 5). Huom! Älä pyyhi neulaa missään vaiheessa. |
|
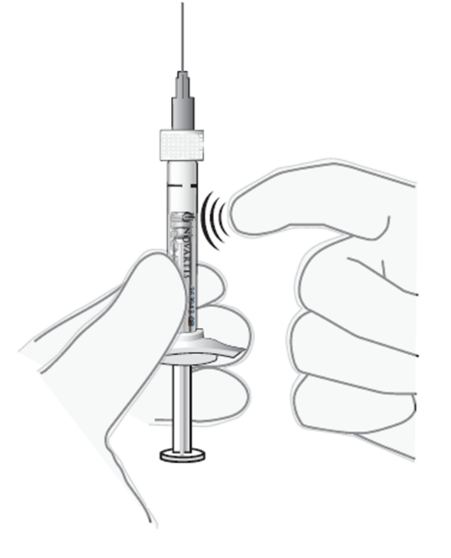
Poista ilmakuplat | 9. Pidä ruisku pystyasennossa. 10. Jos liuoksessa on ilmakuplia, naputtele varovasti ruiskun kylkeä kunnes kuplat nousevat liuoksen pintaan (ks. kuva 6). |
Kuva 6 |
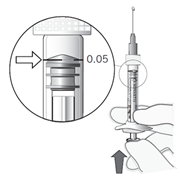
Säädä annos | 11. Nosta ruisku silmiesi korkeudelle ja paina varovasti mäntää, kunnes männän kumisen kärjen reuna on ruiskun mustan annosviivan kohdalla (ks. kuva 7). Näin ruiskussa oleva ilma ja ylimääräinen liuos poistuvat ja annos asettuu määrään 0,05 ml. Huom! Männän varsi ei ole kiinni männän kumisessa päässä. Tämä estää ilman vetämisen ruiskuun. |
Kuva 7 |
Injektion anto | Injektiotoimenpide on annettava aseptisissa olosuhteissa. 12. Injektioneula pistetään 3,5 - 4,0 mm limbuksesta posteriorisesti lasiaiseen, vältetään horisontaalista meridiaania ja tähdätään silmämunan keskikohtaan. 13. Liuos injisoidaan hitaasti, kunnes männän kuminen kärki saavuttaa ruiskun pohjan ja silmään on siirtynyt 0,05 ml:n annos. 14. Kovakalvon pistoskohtaa vaihdetaan seuraavissa injektioissa. 15. Kun injektio on annettu, neulansuojusta ei tule asettaa takaisin eikä neulaa irrottaa ruiskusta. Ruisku ja siinä oleva neula hävitetään heittämällä ne viiltävälle jätteelle tarkoitettuun astiaan tai paikallisten vaatimusten mukaisesti. | |
Korvattavuus
LUCENTIS injektioneste, liuos, esitäytetty ruisku
10 mg/ml 0,165 ml
- Ei korvausta.
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
S01LA04
Valmisteyhteenvedon muuttamispäivämäärä
13.05.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com