
IVEMEND infuusiokuiva-aine, liuosta varten 150 mg
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää fosaprepitanttidimeglumiinia, joka vastaa 150 mg fosaprepitanttia, joka vastaa 130,5 mg aprepitanttia. Liuottamisen ja laimentamisen jälkeen 1 ml liuosta sisältää 1 mg fosaprepitanttia (1 mg/ml) (ks. kohta Käyttö- ja käsittelyohjeet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokuiva-aine, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Pahoinvoinnin ja oksentelun ehkäisyyn voimakkaasti ja kohtalaisesti pahoinvointia aiheuttavan syöpäsairauksien solunsalpaajalääkityksen yhteydessä aikuisille ja vähintään 6 kuukauden ikäisille pediatrisille potilaille.
IVEMEND 150 mg annetaan yhdistelmähoidon osana (ks. kohta Annostus ja antotapa).
Annostus ja antotapa
Annostus
Aikuiset
Suositeltu 150 mg:n annos annetaan 20–30 minuuttia kestävänä infuusiona ensimmäisenä päivänä, ja infuusio aloitetaan noin 30 minuuttia ennen solunsalpaajahoitoa (ks. kohta Käyttö- ja käsittelyohjeet). IVEMEND on annettava yhdessä kortikosteroidin ja 5-HT3-antagonistin kanssa alla olevissa taulukoissa kuvatulla tavalla.
Seuraavia hoito-ohjelmia suositellaan pahoinvoinnin ja oksentelun ehkäisyyn pahoinvointia aiheuttavan syöpäsairauksien solunsalpaajalääkityksen yhteydessä:
Taulukko 1: Suositeltu annostus aikuisille pahoinvoinnin ja oksentelun ehkäisyyn voimakkaasti pahoinvointia aiheuttavan solunsalpaajahoidon yhteydessä
1. päivä | 2. päivä | 3. päivä | 4. päivä | |
| IVEMEND | 150 mg laskimoon | ei lainkaan | ei lainkaan | ei lainkaan |
Deksametasoni | 12 mg suun kautta | 8 mg suun kautta | 8 mg suun kautta kahdesti vuorokaudessa | 8 mg suun kautta kahdesti vuorokaudessa |
5-HT3-antagonistit | Vakioannos 5-HT3-antagonisteja. Ks. sopiva annos valitun 5-HT3-antagonistin tuotetiedoista | ei lainkaan | ei lainkaan | ei lainkaan |
Deksametasoni annetaan 30 minuuttia ennen solunsalpaajahoitoa ensimmäisenä päivänä sekä aamuisin 2.–4. päivänä. Kolmantena ja neljäntenä päivänä deksametasonia annetaan myös iltaisin. Deksametasoniannoksessa on huomioitu lääkeaineiden yhteisvaikutukset.
Taulukko 2: Suositeltu annostus aikuisille pahoinvoinnin ja oksentelun ehkäisyyn kohtalaisesti pahoinvointia aiheuttavan solunsalpaajahoidon yhteydessä
1. päivä | |
| IVEMEND | 150 mg laskimoon |
Deksametasoni | 12 mg suun kautta |
5-HT3-antagonistit | Vakioannos 5-HT3-antagonisteja. Ks. sopiva annos valitun 5-HT3-antagonistin tuotetiedoista |
Deksametasoni annetaan 30 minuuttia ennen solunsalpaajahoitoa ensimmäisenä päivänä. Deksametasoniannoksessa on huomioitu lääkeaineiden yhteisvaikutukset.
Pediatriset potilaat
Vähintään 6 kuukauden ikäiset ja vähintään 6 kg painavat pediatriset potilaat
Taulukossa 3 on IVEMENDin suositeltu hoito-ohjelma, joka yhdistetään 5‑HT3‑antagonistiin ja mahdollisesti myös kortikosteroidiin pahoinvoinnin ja oksentelun ehkäisemiseksi, kun potilas saa voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä (Highly Emetogenic Chemotherapy (HEC)) tai kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajalääkitystä (Moderately Emetogenic Chemotherapy (MEC)) yhden tai useamman päivän hoito-ohjelmana. Yhden päivän solunsalpaajahoito-ohjelmia ovat hoito-ohjelmat, joissa voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajahoitoa annetaan vain yhden vuorokauden ajan. Useamman päivän solunsalpaajahoito-ohjelmia ovat hoito-ohjelmat, joissa voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajahoitoa annetaan kahden tai useamman vuorokauden ajan.
Taulukossa 4 on vaihtoehtoinen hoito-ohjelma, jota voidaan käyttää yhden päivän solunsalpaajahoito-ohjelmien yhteydessä.
Annostus yhden tai useamman päivän solunsalpaajahoito-ohjelman yhteydessä
Pediatrisille potilaille, jotka saavat voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajahoitoa yhden tai useamman päivän hoito-ohjelmana, IVEMEND annetaan infuusiona laskimoon keskuslaskimokatetrin kautta 1., 2. ja 3. päivänä. EMEND-kapseleita tai EMEND-oraalisuspensiota voidaan antaa IVEMENDin sijasta 2. tai 3. päivänä taulukossa 3 annettujen ohjeiden mukaisesti. EMEND-kapseleiden ja EMEND-oraalisuspension annosteluohjeet on tarkistettava näiden valmisteiden valmisteyhteenvedoista.
Taulukko 3: Suositeltu annostus lapsille pahoinvoinnin ja oksentelun ehkäisyyn voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavien yhden tai useamman päivän solunsalpaajahoito-ohjelmien yhteydessä
| Potilaat | 1. päivä | 2. päivä | 3. päivä |
IVEMEND* | 12 vuotta täyttäneet pediatriset potilaat | 115 mg laskimoon | 80 mg laskimoon TAI 80 mg suun kautta (EMEND-kapselit) | 80 mg laskimoon TAI 80 mg suun kautta (EMEND-kapselit) |
6 kuukauden – alle 12 vuoden ikäiset ja vähintään 6 kg painavat pediatriset potilaat
| 3 mg/kg laskimoon
Enimmäisannos 115 mg
| 2 mg/kg laskimoon TAI 2 mg/kg suun kautta (EMEND-oraalisuspensio)
Enimmäisannos 80 mg | 2 mg/kg laskimoon TAI 2 mg/kg suun kautta (EMEND-oraalisuspensio)
Enimmäisannos 80 mg | |
Deksametasoni** | Kaikki pediatriset potilaat | Jos annetaan samanaikaisesti kortikosteroidia, kuten deksametasonia, 50 % suositellusta kortikosteroidiannoksesta annetaan 1.–4. päivänä | ||
5-HT3-antagonisti | Kaikki pediatriset potilaat | Suositeltu annostus on tarkistettava valitun 5-HT3-antagonistin tuotetiedoista | ||
| * 12 vuotta täyttäneille pediatrisille potilaille IVEMEND annetaan 30 minuutin infuusiona laskimoon siten, että infuusio päättyy noin 30 minuuttia ennen solunsalpaajahoitoa. Alle 12-vuotiaille pediatrisille potilaille IVEMEND annetaan 60 minuutin infuusiona laskimoon siten, että infuusio päättyy noin 30 minuuttia ennen solunsalpaajahoitoa. ** Deksametasoni annetaan 30 minuuttia ennen solunsalpaajahoitoa ensimmäisenä päivänä. | ||||
Vaihtoehtoinen annostus yhden päivän solunsalpaajahoito-ohjelmien yhteydessä
Pediatrisille potilaille, jotka saavat voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajahoitoa yhden päivän hoito-ohjelmana, IVEMEND voidaan antaa infuusiona laskimoon keskuslaskimokatetrin kautta ensimmäisenä päivänä.
Taulukko 4: Vaihtoehtoinen annostus lapsille pahoinvoinnin ja oksentelun ehkäisyyn voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavien yhden päivän solunsalpaajahoito-ohjelmien yhteydessä
| Potilaat | 1. päivä |
IVEMEND* | 12 vuotta täyttäneet pediatriset potilaat | 150 mg laskimoon |
2-vuotiaat – alle 12-vuotiaat pediatriset potilaat | 4 mg/kg laskimoon
Enimmäisannos 150 mg | |
6 kuukauden – alle 2 vuoden ikäiset ja vähintään 6 kg painavat pediatriset potilaat | 5 mg/kg laskimoon
Enimmäisannos 150 mg | |
Deksametasoni** | Kaikki pediatriset potilaat | Jos annetaan samanaikaisesti kortikosteroidia, kuten deksametasonia, 50 % suositellusta kortikosteroidiannoksesta annetaan 1. ja 2. päivänä. |
5-HT3-antagonisti | Kaikki pediatriset potilaat | Suositeltu annostus on tarkistettava valitun 5-HT3-antagonistin tuotetiedoista |
| * 12 vuotta täyttäneille pediatrisille potilaille IVEMEND annetaan 30 minuutin infuusiona laskimoon siten, että infuusio päättyy noin 30 minuuttia ennen solunsalpaajahoitoa. Alle 12-vuotiaille pediatrisille potilaille IVEMEND annetaan 60 minuutin infuusiona laskimoon siten, että infuusio päättyy noin 30 minuuttia ennen solunsalpaajahoitoa. ** Deksametasoni annetaan 30 minuuttia ennen solunsalpaajahoitoa ensimmäisenä päivänä. | ||
IVEMENDin turvallisuutta ja tehoa alle 6 kuukauden ikäisten imeväisten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Yleistä
Yhteiskäytön tehosta muiden kortikosteroidien ja 5-HT3-antagonistien kanssa on vain vähän tietoja. Lisätietoja yhteiskäytöstä kortikosteroidien kanssa, ks. kohta Yhteisvaikutukset.
Tutustu samanaikaisesti käytettävien 5-HT3-antagonistien valmisteyhteenvetoihin.
Erityisryhmät
Iäkkäät (≥ 65-vuotiaat)
Annoksen muuttaminen ei ole tarpeen iäkkäitä potilaita hoidettaessa (ks. kohta Farmakokinetiikka).
Sukupuoli
Annostuksen muuttaminen ei ole tarpeen sukupuolen perusteella (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen hoidettaessa potilaita, joilla on munuaisten vajaatoiminta tai hemodialyysihoitoa vaativa munuaissairaus (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen lievässä maksan vajaatoiminnassa. Kohtalaista maksan vajaatoimintaa sairastavien potilaiden hoidosta on vain vähän tietoja ja vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidosta ei lainkaan. IVEMENDin käytössä on noudatettava varovaisuutta näissä potilasryhmissä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Antotapa
IVEMEND 150 mg on annettava laskimoon. Sitä ei saa antaa lihakseen eikä ihon alle. Se tulisi antaa aikuisille 20–30 minuuttia kestävänä jatkuvana infuusiona laskimoon. Vähintään 6 kuukauden ikäisille pediatrisille potilaille se tulisi antaa keskuslaskimokatetrin kautta: 30 minuutin infuusiona 12 vuotta täyttäneille potilaille ja 60 minuutin infuusiona alle 12-vuotiaille potilaille (ks. kohta Käyttö- ja käsittelyohjeet). Älä anna IVEMENDiä bolusinjektiona äläkä laimentamattomana liuoksena.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle, polysorbaatti 80:lle tai kohdassa Apuaineet mainituille muille apuaineille.
IVEMENDiä ei saa antaa samanaikaisesti pimotsidin, terfenadiinin, astemitsolin eikä sisapridin kanssa (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavat potilaat
Kohtalaista maksan vajaatoimintaa sairastavien potilaiden hoidosta on vain vähän tietoja ja vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidosta ei lainkaan. IVEMENDin käytössä on noudatettava varovaisuutta näissä potilasryhmissä (ks. kohta Farmakokinetiikka).
CYP3A4-yhteisvaikutukset
IVEMENDin käytössä on noudatettava varovaisuutta, jos potilas saa samanaikaisesti muita lääkkeitä, jotka metaboloituvat ensisijaisesti CYP3A4-entsyymin välityksellä ja joilla on kapea terapeuttinen alue, kuten siklosporiinia, takrolimuusia, sirolimuusia, everolimuusia, alfentaniilia, ergotamiinijohdoksia, fentanyyliä ja kinidiiniä (ks. kohta Yhteisvaikutukset). On myös syytä olla erityisen varovainen annettaessa samanaikaisesti irinotekaania, koska yhteiskäyttö voi lisätä sen toksisuutta.
Yhteiskäyttö varfariinin (CYP2C9-substraatin) kanssa
Pitkäaikaista varfariinihoitoa saavien potilaiden INR-arvoa (International Normalised Ratio) on seurattava tarkoin 14 vuorokauden ajan fosaprepitantin antamisen jälkeen (ks. kohta Yhteisvaikutukset).
Yhteiskäyttö hormonaalisten ehkäisyvalmisteiden kanssa
Hormonaalisten ehkäisyvalmisteiden teho saattaa heikentyä fosaprepitantin käytön aikana ja 28 päivän ajan hoidon jälkeen. Vaihtoehtoisia ei-hormonaalisia täydentäviä ehkäisymenetelmiä on käytettävä fosaprepitantin käytön aikana ja kahden kuukauden ajan sen jälkeen (ks. kohta Yhteisvaikutukset).
Yliherkkyysreaktiot
Fosaprepitantti-infuusion aikana tai pian sen jälkeen on esiintynyt välittömiä yliherkkyysreaktioita mukaan lukien punoitusta, eryteemaa, hengenahdistusta ja anafylaksiaa/anafylaktista sokkia. Nämä yliherkkyysreaktiot ovat yleensä hävinneet, kun infuusio on keskeytetty ja asianmukainen hoito aloitettu. Infuusion aloittamista uudelleen yliherkkyysreaktioita saaville potilaille ei suositella.
Lääkkeen anto ja infuusiokohdan reaktiot
Infuusiokohdan reaktioita on raportoitu käytettäessä IVEMENDiä (ks. kohta Haittavaikutukset). Vaikeita reaktioita, mukaan lukien tromboflebiittia ja vaskuliittia, raportoitiin useimmiten samanaikaisen vesikkelimuodostusta aiheuttavan (esim. antrasykliinipohjaisen) kemoterapian käytössä, erityisesti laskimonviereisen annostelun yhteydessä. Nekroosia on myös raportoitu joillakin samanaikaista rakkulamuodostusta aiheuttavaa kemoterapiaa saavilla potilailla. Käytettäessä suurempia annoksia ilman samanaikaista rakkuloita aiheuttavaa kemoterapiaa on todettu lievä tromboosi injektiokohdassa.
IVEMENDiä ei saa antaa bolusinjektiona, vaan se on aina laimennettava ja annettava hitaana infuusiona laskimoon (ks. kohta Annostus ja antotapa). IVEMENDiä ei saa antaa lihakseen eikä ihon alle (ks. kohta Prekliiniset tiedot turvallisuudesta). Jos paikallisen ärsytyksen merkkejä tai oireita ilmenee, injektio tai infuusio on keskeytettävä ja annettava toiseen laskimoon.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Laskimoon annettu fosaprepitantti muuttuu nopeasti aprepitantiksi.
Kerta-annoksena annettu fosaprepitantti 150 mg on CYP3A4:n heikko estäjä. Fosaprepitantilla ei näytä olevan yhteisvaikutuksia kuljetusproteiini P-glykoproteiinin kanssa, mistä on osoituksena se, ettei suun kautta annettavalla aprepitantilla ole yhteisvaikutuksia digoksiinin kanssa. Fosaprepitantin oletetaan aiheuttavan lievempää tai enintään yhtä voimakasta CYP2C9:n, CYP3A4:n ja glukuronidaation induktiota kuin suun kautta annettavan aprepitantin. Vaikutuksesta CYP2C8- ja CYP2C19-entsyymeihin ei ole tietoa.
Laskimoon annettavan fosaprepitantin mahdolliset yhteisvaikutukset muiden lääkkeiden kanssa liittyvät todennäköisesti samoihin lääkeaineisiin, joilla on yhteisvaikutuksia suun kautta annettavan aprepitantin kanssa. Yhteisvaikutusten todennäköisyyden ei odoteta olevan suurempi useamman päivän fosaprepitanttihoito-ohjelmien kuin suun kautta annettavien aprepitanttihoito-ohjelmien yhteydessä. Siksi IVEMENDin ja muiden lääkevalmisteiden yhteiskäyttöä koskevat suositukset pediatristen potilaiden hoidossa perustuvat aikuispotilailla tehtyjen fosaprepitantti- ja aprepitanttitutkimusten tietoihin. IVEMENDin ja EMENDin yhdistelmähoito-ohjelmia käytettäessä on perehdyttävä EMEND-kapseleiden tai EMEND-oraalisuspension valmisteyhteenvedon kohtaan Yhteisvaikutukset.
Seuraavat tiedot perustuvat suun kautta annettavalla aprepitantilla tehtyihin tutkimuksiin ja tutkimuksiin, joissa laskimoon kerta-annoksena annettua fosaprepitanttia annettiin yhdessä deksametasonin, midatsolaamin tai diltiatseemin kanssa.
Fosaprepitantin vaikutus muiden lääkeaineiden farmakokinetiikkaan
CYP3A4:n estyminen
Heikkona CYP3A4:n estäjänä fosaprepitantin 150 mg:n kerta-annos voi suurentaa ohimenevästi muiden samanaikaisesti annettujen CYP3A4-entsyymin välityksellä metaboloituvien lääkeaineiden pitoisuuksia plasmassa. CYP3A4:n substraattien aikaansaama kokonaisaltistus voi nousta jopa kaksinkertaiseksi ensimmäisenä ja toisena päivänä sen jälkeen, kun niitä on annettu yhdessä fosaprepitantin 150 mg:n kerta-annoksen kanssa. Fosaprepitanttia ei saa käyttää samanaikaisesti pimotsidin, terfenadiinin, astemitsolin eikä sisapridin kanssa. Fosaprepitantin aiheuttama CYP3A4:n toiminnan estyminen voi suurentaa näiden lääkeaineiden pitoisuuksia plasmassa, mikä saattaa johtaa vakaviin tai hengenvaarallisiin reaktioihin (ks. kohta Vasta-aiheet). Varovaisuutta on noudatettava annettaessa samanaikaisesti fosaprepitanttia ja ensisijaisesti CYP3A4-entsyymin välityksellä metaboloituvia lääkkeitä, joilla on kapea terapeuttinen alue, kuten siklosporiinia, takrolimuusia, sirolimuusia, everolimuusia, alfentaniilia, dihydroergotamiinia, ergotamiinia, fentanyyliä ja kinidiiniä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kortikosteroidit
Deksametasoni: Suun kautta annettavan deksametasonin annosta tulisi pienentää noin 50 %, kun sitä annetaan yhdessä fosaprepitantin kanssa (ks. kohta Annostus ja antotapa). Ensimmäisenä päivänä kerta-annoksena laskimoon annettu fosaprepitantti 150 mg suurensi deksametasonin, CYP3A4:n substraatin, AUC0–24h-arvoa 100 % ensimmäisenä päivänä, 86 % toisena päivänä ja 18 % kolmantena päivänä, kun deksametasonia annettiin 8 mg:n kerta-annoksena suun kautta 1.–3. päivänä.
Solunsalpaajat
Fosaprepitantin 150 mg:n vahvuudella ei ole tehty yhteisvaikutustutkimuksia solunsalpaajien kanssa. Suun kautta annetulla aprepitantilla tehdyt tutkimukset doketakselin ja vinorelbiinin kanssa viittaavat kuitenkin siihen, ettei IVEMEND 150 mg -valmisteella ole todennäköisesti kliinisesti merkittäviä yhteisvaikutuksia laskimoon annetun doketakselin eikä vinorelbiinin kanssa. Yhteisvaikutusta suun kautta annettujen, pääasiassa tai osittain CYP3A4-entsyymin välityksellä metaboloituvien solunsalpaajien (esim. etoposidin, vinorelbiinin) kanssa ei voida sulkea pois. Pääasiassa tai osittain CYP3A4:n välityksellä metaboloituvia lääkevalmisteita saavien potilaiden hoidossa on syytä noudattaa varovaisuutta ja heidän tilaansa tulisi seurata tavanomaista tarkemmin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Myyntiluvan myöntämisen jälkeen on ilmoitettu neurotoksisia tapahtumia, jotka ovat mahdollisia ifosfamidin haittavaikutuksia, kun aprepitanttia ja ifosfamidia on annettu samanaikaisesti.
Immunosuppressiiviset aineet
Altistuminen CYP3A4-entsyymin välityksellä metaboloituville immunosuppressiivisille lääkkeille (esim. siklosporiinille, takrolimuusille, everolimuusille ja sirolimuusille) saattaa ensin suurentua kohtalaisesti ja ohimenevästi kahden päivän ajan fosaprepitantin 150 mg:n kerta-annoksen jälkeen ja mahdollisesti pienentyä sitten jonkin verran. Koska altistus suurenee vain lyhytaikaisesti, hoitoannosten seurannan (Therapeutic Dose Monitoring) perusteella ei suositella immunosuppressiivisten lääkkeiden annostuksen pienentämistä IVEMENDin antopäivänä eikä sitä seuraavana päivänä.
Midatsolaami
Ensimmäisenä päivänä kerta-annoksena laskimoon annettu fosaprepitantti 150 mg suurensi midatsolaamin AUC0-∞-arvoa 77 % ensimmäisenä päivänä, ja neljäntenä päivänä sillä ei ollut enää vaikutusta, kun midatsolaamia annettiin 2 mg kerta-annoksena suun kautta ensimmäisenä ja neljäntenä päivänä. Kerta-annoksena ensimmäisenä päivänä annettu fosaprepitantti 150 mg on heikko CYP3A4:n estäjä, ja neljäntenä päivänä ei ole enää havaittavissa CYP3A4:n estymistä eikä induktiota.
Kun midatsolaamia tai muita CYP3A4-entsyymin välityksellä metaboloituvia bentsodiatsepiineja (alpratsolaamia, triatsolaamia) annetaan yhdessä IVEMENDin kanssa, niiden pitoisuus plasmassa saattaa suurentua, minkä mahdolliset vaikutukset on syytä ottaa huomioon.
Diltiatseemi
Fosaprepitantin 150 mg:n vahvuudella ei ole tehty yhteisvaikutustutkimuksia diltiatseemin kanssa. Seuraava tutkimus, jossa käytettiin 100 mg:n fosaprepitanttiannoksia, on kuitenkin otettava huomioon, kun IVEMEND 150 mg -valmistetta käytetään yhdessä diltiatseemin kanssa. Kun potilaille, joilla oli lievä tai kohtalainen hypertensio, annettiin 100 mg fosaprepitanttia 15 minuutin infuusiona laskimoon ja diltiatseemia 120 mg kolmesti päivässä, diltiatseemin AUC-arvo suureni 1,4-kertaiseksi ja verenpaine laski vähän mutta kliinisesti merkitsevästi; sydämen syke tai PR-väli ei kuitenkaan muuttunut kliinisesti merkitsevästi.
Induktio
Yhteisvaikutustutkimuksessa midatsolaamin kanssa 150 mg:n kerta-annoksena annettu fosaprepitantti ei indusoinut CYP3A4-entsyymin toimintaa ensimmäisenä eikä neljäntenä päivänä. IVEMENDin aiheuttaman CYP2C9:n, CYP3A4:n ja glukuronidaation induktion odotetaan olevan lievempää tai enintään yhtä voimakasta kuin kolme vuorokautta kestävän suun kautta annettavan aprepitanttihoidon aiheuttama ohimenevä induktio, jonka vaikutuksen on todettu olevan voimakkaimmillaan 6–8 vuorokauden kuluttua ensimmäisestä aprepitanttiannoksesta. Kolmen vuorokauden hoito suun kautta annettavalla aprepitantilla pienensi CYP2C9:n substraattien AUC-arvoa noin 30–35 % ja etinyyliestradiolin minimipitoisuuksia enintään 64 %. Vaikutuksesta CYP2C8- ja CYP2C19-entsyymeihin ei ole tietoa. Varovaisuutta on syytä noudattaa, mikäli varfariinia, asenokumarolia, tolbutamidia, fenytoiinia tai muita lääkeaineita, joiden tiedetään metaboloituvan CYP2C9-entsyymin välityksellä, annetaan IVEMENDin kanssa.
Varfariini
Pitkäaikaista varfariinihoitoa saavien potilaiden tromboplastiiniaikaa (INR) on seurattava tarkoin solunsalpaajahoidon aiheuttaman pahoinvoinnin ja oksentelun ehkäisyyn annetun IVEMEND-hoidon aikana ja 14 vuorokauden ajan sen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hormonaaliset ehkäisyvalmisteet
Hormonaalisten ehkäisyvalmisteiden teho saattaa heikentyä fosaprepitantin käytön aikana ja 28 päivän ajan hoidon jälkeen. Vaihtoehtoisia ei-hormonaalisia täydentäviä ehkäisymenetelmiä on käytettävä fosaprepitantin käytön aikana ja kahden kuukauden ajan sen jälkeen.
5-HT3-antagonistit
Fosaprepitantin 150 mg:n vahvuudella ei ole tehty yhteisvaikutustutkimuksia 5-HT3-antagonistien kanssa. Kliinisissä yhteisvaikutustutkimuksissa suun kautta annetulla aprepitanttihoidolla ei ollut kuitenkaan kliinisesti merkitseviä vaikutuksia ondansetronin, granisetronin eikä hydrodolasetronin (dolasetronin aktiivisen metaboliitin) farmakokinetiikkaan. Ei siis ole havaittu viitteitä IVEMEND 150 mg -valmisteen ja 5-HT3-antagonistien yhteisvaikutuksesta.
Muiden lääkevalmisteiden vaikutus 150 mg:n fosaprepitanttiannoksesta peräisin olevan aprepitantin farmakokinetiikkaan
Jos fosaprepitanttia annetaan yhdessä CYP3A4:n toimintaa estävien lääkeaineiden (esim. ketokonatsolin, itrakonatsolin, vorikonatsolin, posakonatsolin, klaritromysiinin, telitromysiinin, nefatsodonin ja proteaasinestäjien) kanssa, on noudatettava varovaisuutta, koska yhteiskäyttö saattaa suurentaa aprepitantin pitoisuuden plasmassa moninkertaiseksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Ketokonatsoli pidensi suun kautta annetun aprepitantin terminaalisen puoliintumisajan noin kolminkertaiseksi.
Fosaprepitantin samanaikaista antoa yhdessä CYP3A4:n toimintaa voimakkaasti indusoivien lääkkeiden (esim. rifampisiinin, fenytoiinin, karbamatsepiinin, fenobarbitaalin) kanssa on vältettävä, koska yhteiskäyttö saattaa pienentää aprepitantin pitoisuutta plasmassa ja heikentää siten hoidon tehoa. Fosaprepitantin ja mäkikuismaa (Hypericum perforatum) sisältävien rohdosvalmisteiden yhteiskäyttöä ei suositella. Rifampisiini pienensi suun kautta annetun aprepitantin terminaalisen puoliintumisajan keskiarvoa 68 %.
Diltiatseemi
Fosaprepitantin 150 mg:n vahvuudella ei ole tehty yhteisvaikutustutkimuksia diltiatseemin kanssa. Seuraava tutkimus, jossa käytettiin 100 mg:n fosaprepitanttiannoksia, on kuitenkin otettava huomioon, kun IVEMEND 150 mg -valmistetta käytetään yhdessä diltiatseemin kanssa. Kun 100 mg fosaprepitanttia annettiin 15 minuutin infuusiona laskimoon ja diltiatseemia 120 mg kolmesti päivässä, aprepitantin AUC-arvo suureni 1,5-kertaiseksi. Tällä vaikutuksella ei katsottu olevan kliinistä merkitystä.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuispotilailla.
Raskaus ja imetys
Ehkäisy miehille ja naisille
Hormonaalisten ehkäisyvalmisteiden teho saattaa heikentyä fosaprepitanttihoidon aikana ja 28 päivän ajan hoidon jälkeen. Vaihtoehtoisia ei-hormonaalisia täydentäviä ehkäisymenetelmiä on käytettävä fosaprepitanttihoidon aikana ja kahden kuukauden ajan viimeisen fosaprepitanttiannoksen jälkeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Raskaus
Fosaprepitantin ja aprepitantin käytöstä raskauden aikana ei ole kliinistä tietoa. Fosaprepitantin ja aprepitantin mahdollisia toksisia vaikutuksia lisääntymiseen ei ole täysin selvitetty, koska eläinkokeissa ei onnistuttu saavuttamaan suurempaa altistusta kuin saadaan aikaan ihmisessä käytettäessä terapeuttista annosta. Näissä tutkimuksissa ei saatu viitteitä suorista tai epäsuorista haitallisista vaikutuksista raskauteen, alkion/sikiön kehitykseen, synnytykseen tai postnataaliseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Neurokiniinisäätelyn muutosten mahdollisia vaikutuksia lisääntymiseen ei tunneta. IVEMENDiä ei pidä käyttää raskauden aikana, ellei se ole aivan välttämätöntä.
Imetys
Aprepitantti erittyy imettävien rottien maitoon sekä laskimoon annetun fosaprepitantin että suun kautta annetun aprepitantin jälkeen. Ei tiedetä, erittyykö aprepitantti äidinmaitoon. Siksi imettämistä ei suositella IVEMEND-hoidon aikana.
Hedelmällisyys
Fosaprepitantin ja aprepitantin mahdollisia vaikutuksia hedelmällisyyteen ei ole täysin selvitetty, koska eläinkokeissa ei onnistuttu saavuttamaan suurempaa altistusta kuin saadaan aikaan ihmisessä käytettäessä terapeuttista annosta. Näissä hedelmällisyystutkimuksissa ei havaittu viitteitä välittömistä eikä välillisistä haitallisista vaikutuksista paritteluun, hedelmällisyyteen, alkion-/sikiönkehitykseen eikä siittiöiden lukumäärään ja liikkuvuuteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
IVEMENDillä voi olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. IVEMENDin käytön jälkeen voi esiintyä heitehuimausta ja uupumusta (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kliinisissä tutkimuksissa fosaprepitantin erilaisia lääkemuotoja on annettu kaikkiaan 2687 aikuiselle, mukaan lukien 371 terveelle tutkittavalle ja 2084 potilaalle, sekä 299 lapselle ja nuorelle, joilla oli solunsalpaajahoidon aiheuttamaa pahoinvointia ja oksentelua. Koska fosaprepitantti muuttuu aprepitantiksi, aprepitanttiin liittyviä haittavaikutuksia esiintyy todennäköisesti myös fosaprepitantin käytön yhteydessä. Aprepitantin turvallisuutta on arvioitu noin 6500 aikuisesta ja 184 lapsesta ja nuoresta saatujen tietojen perusteella.
Aprepitantti suun kautta
Kun potilaat olivat saaneet voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä (HEC), yleisimmät haittavaikutukset, joita raportoitiin esiintyneen aprepitanttihoito-ohjelmaa saaneilla aikuisilla useammin kuin tavanomaista hoitoa saaneilla, olivat: nikottelu (4,6 %:lla aprepitanttia saaneista, 2,9 %:lla tavanomaista hoitoa saaneista), alaniiniaminotransferaasiarvon (ALAT) kohoaminen (2,8 %, 1,1 %), ruoansulatushäiriöt (2,6 %, 2,0 %), ummetus (2,4 %, 2,0 %), päänsärky (2,0 %, 1,8 %) ja vähentynyt ruokahalu (2,0 %, 0,5 %). Kun potilaat olivat saaneet kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajalääkitystä (MEC), uupumus oli yleisin haittavaikutus, jota raportoitiin olleen aprepitanttihoito-ohjelmaa saaneilla potilailla useammin (1,4 %) kuin tavanomaista hoitoa saaneilla (0,9 %).
Kun potilaat olivat saaneet pahoinvointia aiheuttavaa solunsalpaajalääkitystä, yleisimmät haittavaikutukset, joita raportoitiin aprepitanttihoito-ohjelmaa saaneilla pediatrisilla potilailla useammin kuin vertailuhoitoa saaneilla, olivat nikottelu (3,3 %:lla aprepitanttia saaneista, 0,0 %:lla vertailuhoitoa saaneista) ja kasvojen ja kaulan punoitus (1,1 %, 0,0 %).
Haittavaikutustaulukko – aprepitantti
Seuraavia haittavaikutuksia todettiin HEC- ja MEC-tutkimusten yhdistetyssä analyysissä useammin aprepitanttia suun kautta saaneilla kuin tavanomaista hoitoa saaneilla aikuisilla tai pediatrisilla potilailla, tai lääkkeen markkinoille tulon jälkeen.
Taulukossa esitetyt yleisyysluokat perustuvat aikuisilla tehtyihin tutkimuksiin. Pediatrisissa tutkimuksissa havaitut esiintymistiheydet olivat samalla tasolla tai pienempiä, ellei niitä mainita taulukossa. Joitakin aikuisilla harvemmin esiintyneitä haittavaikutuksia ei havaittu lainkaan pediatrisissa tutkimuksissa.
Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1000, < 1/100), harvinainen (≥ 1/10 000, < 1/1000) ja hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Taulukko 5: Haittavaikutustaulukko – aprepitantti
Elinjärjestelmä | Haittavaikutus | Esiintymistiheys |
Infektiot | kandidiaasi, stafylokokki-infektiot | harvinainen |
Veri ja imukudos | kuumeinen neutropenia, anemia | melko harvinainen |
Immuunijärjestelmä | yliherkkyysreaktiot, mukaan lukien anafylaktiset reaktiot | tuntematon |
Aineenvaihdunta ja ravitsemus | vähentynyt ruokahalu | yleinen |
jatkuva jano | harvinainen | |
Psyykkiset häiriöt | ahdistuneisuus | melko harvinainen |
ajan ja paikan tajun hämärtyminen, euforinen mieliala | harvinainen | |
Hermosto | päänsärky | yleinen |
heitehuimaus, uneliaisuus | melko harvinainen | |
kognitiiviset häiriöt, horros, makuaistin häiriöt | harvinainen | |
Silmät | sidekalvotulehdus | harvinainen |
Kuulo ja tasapainoelin | korvien soiminen | harvinainen |
Sydän | sydämentykytys | melko harvinainen |
bradykardia, sydän- ja verisuonihäiriöt | harvinainen | |
Verisuonisto | kuumat aallot / kasvojen ja kaulan punoitus | melko harvinainen |
Hengityselimet, rintakehä ja välikarsina | nikottelu | yleinen |
suunielun kipu, aivastelu, yskä, lima nielussa, kurkun ärsytys | harvinainen | |
Ruoansulatuselimistö | ummetus, ruoansulatushäiriöt | yleinen |
röyhtäily, pahoinvointi*, oksentelu*, gastroesofageaalinen refluksitauti, vatsakivut, suun kuivuminen, ilmavaivat | melko harvinainen | |
puhjennut pohjukaissuolihaava, suutulehdus, vatsan pingotus, kova uloste, neutropeeninen koliitti | harvinainen | |
Iho ja ihonalainen kudos | ihottuma, akne | melko harvinainen |
valoherkkyysreaktio, voimakas hikoilu, talivuoto, ihon haavaumat, kutiava ihottuma, Stevens-Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysi | harvinainen | |
kutina, nokkosihottuma | tuntematon | |
Luusto, lihakset ja sidekudos | lihasheikkous, lihasspasmit | harvinainen |
Munuaiset ja virtsatiet | dysuria | melko harvinainen |
tiheä virtsaamistarve | harvinainen | |
Yleisoireet ja antopaikassa todettavat haitat | uupumus | yleinen |
voimattomuus, yleinen huonovointisuus | melko harvinainen | |
edeema, epämukava tunne rinnassa, kävelyhäiriöt | harvinainen | |
Tutkimukset | ALAT-arvon nousu | yleinen |
ASAT-arvon nousu, alkalisen fosfataasin nousu veressä | melko harvinainen | |
punasoluja virtsassa, vähentynyt veren natriumpitoisuus, painon lasku, neutrofiilimäärän pieneneminen, glukoosia virtsassa, lisääntynyt virtsamäärä | harvinainen | |
| * Pahoinvointi ja oksentelu olivat tehon muuttujia ensimmäisten viiden päivän ajan solunsalpaajahoidon jälkeen ja ne ilmoitettiin haittavaikutuksina vasta sen jälkeen. | ||
Valittujen haittavaikutusten kuvaus
HEC- ja MEC-tutkimusten aikuisten jatkohoitotutkimuksessa, jossa annettiin vielä kuusi jaksoa solunsalpaajahoitoa, haittavaikutukset olivat yleisesti samanlaisia kuin ensimmäisen hoitojakson aikana.
Aktiivikontrolloidussa kliinisessä lisätutkimuksessa, johon osallistui 1169 aprepitanttia ja voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saavaa aikuispotilasta, haittavaikutusprofiili oli yleisesti samanlainen kuin muissa aprepitantilla tehdyissä HEC-tutkimuksissa.
Muut kuin solunsalpaajahoidon aiheuttamaa pahoinvointia ja oksentelua koskevat tutkimukset
Aikuispotilailla, jotka saivat aprepitanttia leikkauksen jälkeisen pahoinvoinnin ja oksentelun ehkäisyyn, todettiin lisäksi seuraavia haittavaikutuksia, joita esiintyi yleisemmin kuin ondansetronia saaneilla potilailla: ylävatsakipu, epänormaalit äänet suolistosta, ummetus*, dysartria, hengenahdistus, hypestesia, unettomuus, mioosi, pahoinvointi, tuntohäiriöt, vatsavaivat, subileus*, heikentynyt näontarkkuus, hengityksen vinkuminen.
*Raportoitu potilailla, jotka käyttivät suurempia annoksia aprepitanttia.
Fosaprepitantti
Aktiivikontrolloidussa kliinisessä tutkimuksessa, johon osallistui voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saavia aikuispotilaita, turvallisuutta arvioitiin vertaamalla IVEMEND 150 mg -valmistetta yhden päivän hoito-ohjelmassa saavia potilaita (1143) aprepitanttia kolmen päivän hoito-ohjelmassa saaviin potilaisiin (1169). Turvallisuutta arvioitiin myös MEC-lääkitystä saaneilla aikuispotilailla tehdyssä plasebokontrolloidussa kliinisessä tutkimuksessa, jossa verrattiin kerta-annoksen IVEMEND 150 mg ‑valmistetta saaneita 504 potilasta 497 potilaaseen, jotka saivat vertailuhoitoa.
Laskimoon annettavan hoidon turvallisuutta yhden päivän hoito-ohjelmassa tuki yhdistetty analyysi kolmesta vaikuttavalla vertailuaineella kontrolloidusta kliinisestä tutkimuksesta, joihin osallistuneet 139 pediatrista potilasta (ikäjakauma 6 kuukautta – 17 vuotta) saivat voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajahoitoa ja kerta-annoksen IVEMENDiä yhden päivän hoito-ohjelman suositeltuna tai sitä suurempana annoksena.
Laskimoon annettavan hoidon turvallisuutta kolmen päivän hoito-ohjelmassa tukee yhden hoitohaaran kliininen tutkimus, johon osallistuneet 100 pediatrista potilasta (ikäjakauma 6 kuukautta – 17 vuotta) saivat voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajahoitoa ja IVEMENDiä kolmen päivän hoito-ohjelmassa suositellulla annoksella (ks. kohta Annostus ja antotapa). Laskimoon annettavan fosaprepitanttihoidon kolmen päivän hoito-ohjelman turvallisuusprofiili on pediatrisilla potilailla samanlainen kuin yhden päivän hoito-ohjelman.
Fosaprepitantin turvallisuusprofiili oli aikuisilla ja pediatrisilla potilailla yleisesti samanlainen kuin aprepitantin turvallisuusprofiili.
Haittavaikutustaulukko – fosaprepitantti
Seuraavassa on esitetty haittavaikutuksia, joita on raportoitu fosaprepitanttia saaneilla aikuispotilailla kliinisissä tutkimuksissa tai lääkkeen markkinoille tulon jälkeen ja joita ei ole raportoitu aprepitanttia saaneilla potilailla (ks. yllä olevat tiedot). Taulukossa esitetyt yleisyysluokat perustuvat aikuisilla tehtyihin tutkimuksiin. Pediatrisissa tutkimuksissa havaitut esiintymistiheydet olivat samalla tasolla tai pienempiä. Joitakin aikuisilla yleisesti havaittuja haittavaikutuksia ei havaittu lainkaan pediatrisissa tutkimuksissa. Infuusiokohdan reaktioita on raportoitu käytettäessä IVEMENDiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1000, < 1/100), harvinainen (≥ 1/10 000, < 1/1000) ja hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Taulukko 6: Haittavaikutustaulukko – fosaprepitantti
Elinjärjestelmä | Haittavaikutus | Esiintymistiheys |
Verisuonisto | punoitus, tromboflebiitti (lähinnä infuusiokohdan tromboflebiitti) | melko harvinainen |
Iho ja ihonalainen kudos | eryteema | melko harvinainen |
Yleisoireet ja antopaikassa todettavat haitat | infuusiokohdan eryteema, infuusiokohdan kipu, infuusiokohdan kutina | melko harvinainen |
infuusiokohdan kovettuminen | harvinainen | |
välittömät yliherkkyysreaktiot mukaan lukien punoitus, eryteema, hengenahdistus, anafylaktiset reaktiot/anafylaktinen sokki | tuntematon | |
Tutkimukset | kohonnut verenpaine | melko harvinainen |
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksissa fosaprepitanttihoito on keskeytettävä ja potilaalle on annettava yleistä elintoimintoja tukevaa hoitoa. Potilaan tilaa on tarkkailtava. Aprepitantin antiemeettisen vaikutuksen vuoksi oksennuttaminen lääkkeiden avulla ei ehkä tehoa.
Aprepitanttia ei voida poistaa hemodialyysin avulla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Pahoinvointilääkkeet, ATC-koodi: A04AD12.
Fosaprepitantti on aprepitantin aihiolääke, joka laskimoon annettaessa muuttuu nopeasti aprepitantiksi (ks. kohta Farmakokinetiikka). Fosaprepitantin osuutta antiemeettisessä vaikutuksessa ei ole vielä täysin selvitetty, mutta on mahdollista että se vaikuttaa osaltaan heti lääkkeenannon jälkeen. Aprepitantti on selektiivinen antagonisti, jolla on voimakas affiniteetti ihmisen substanssi P:n neurokiniini 1 (NK1) -reseptoreihin. Fosaprepitantin farmakologisen vaikutuksen katsotaan johtuvan aprepitantista.
Yhden päivän fosaprepitanttihoito-ohjelma aikuisilla
Voimakkaasti pahoinvointia aiheuttava solunsalpaajalääkitys
Satunnaistetussa, rinnakkaisryhmissä tehdyssä, kaksoissokkoutetussa, vaikuttavalla vertailuaineella kontrolloidussa tutkimuksessa IVEMEND 150 mg -valmistetta (n = 1147) verrattiin kolmen päivän aprepitanttihoito-ohjelmaan (n = 1175). Tutkimukseen osallistuneet aikuispotilaat saivat voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä, joka sisälsi sisplatiinia (≥ 70 mg/m2). Fosaprepitanttihoito-ohjelmassa annettiin fosaprepitanttia 150 mg ensimmäisenä päivänä ja ondansetronia 32 mg laskimoon ensimmäisenä päivänä sekä deksametasonia 12 mg ensimmäisenä päivänä, 8 mg toisena päivänä ja 8 mg kaksi kertaa päivässä kolmantena ja neljäntenä päivänä. Aprepitanttihoito-ohjelmassa annettiin aprepitanttia 125 mg ensimmäisenä päivänä ja 80 mg/vrk toisena ja kolmantena päivänä ja ondansetronia 32 mg laskimoon ensimmäisenä päivänä sekä deksametasonia 12 mg ensimmäisenä päivänä ja 8 mg/vrk 2.–4. päivänä. Fosaprepitantin kaltaista plaseboa, aprepitantin kaltaista plaseboa ja deksametasonin kaltaista plaseboa (kolmannen ja neljännen päivän iltana) käytettiin sokkoutuksen säilyttämiseksi (ks. kohta Annostus ja antotapa). Vaikka ondansetronin 32 mg:n annosta laskimoon käytettiin kliinisissä tutkimuksissa, ei tätä annosta enää suositella käytettäväksi. Katso valitun 5-HT3-antagonistin tuotetiedoista sopiva annostus.
Tehon arviointi perustui seuraaviin yhdistettyihin kriteereihin: täydellinen hoitovaste sekä koko arviointijakson että viivästyneen vaiheen aikana ja ei oksentelua koko arviointijakson aikana. IVEMEND 150 mg osoitettiin yhdenvertaiseksi (non-inferior) kolmen päivän aprepitanttihoito-ohjelman kanssa. Taulukossa 7 on yhteenveto ensisijaisista ja toissijaisista päätetapahtumista.
Taulukko 7: Voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saaneet aikuispotilaat. Vasteprosentti hoitoryhmittäin eri vaiheissa — Ensimmäinen solunsalpaajahoitojakso
PÄÄTETAPAHTUMAT* | Fosaprepitanttihoito-ohjelma | Aprepitanttihoito-ohjelma | Ero† |
Täydellinen hoitovaste‡ | |||
Yhteensä§ | 71,9 | 72,3 | -0,4 (-4,1; 3,3) |
Viivästynyt vaihe§§ | 74,3 | 74,2 | 0,1 (-3,5; 3,7) |
Ei oksentelua | |||
Yhteensä§ | 72,9 | 74,6 | -1,7 (-5,3; 2,0) |
| *Ensisijainen päätetapahtuma esitetään lihavoituna. **n:Täydellisen hoitovasteen primaarisessa analyysissä mukana olleiden aikuispotilaiden lukumäärä. †Ero ja luottamusväli (CI) laskettiin Miettisen ja Nurmisen ehdottamaa menetelmää käyttäen ja korjattiin sukupuolen suhteen. ‡Täydellinen hoitovaste = ei oksentelua eikä varalääkkeiden käyttöä. §Yhteensä = 0–120 tuntia sisplatiinia sisältävän solunsalpaajahoidon alkamisen jälkeen. §§Viivästynyt vaihe = 25–120 tuntia sisplatiinia sisältävän solunsalpaajahoidon alkamisen jälkeen. | |||
Kohtalaisesti pahoinvointia aiheuttava solunsalpaajalääkitys
Satunnaistetussa, rinnakkaisryhmillä tehdyssä, kaksoissokkoutetussa, plasebokontrolloidussa tutkimuksessa verrattiin ondansetroniin ja deksametasoniin yhdistettyä IVEMEND 150 mg ‑valmistetta (n = 502) yksinään annettuun ondansetroniin ja deksametasoniin (vertailuhoito) (n = 498) kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saaneilla aikuispotilailla. Fosaprepitanttihoito-ohjelmassa annettiin ensimmäisenä päivänä fosaprepitanttia 150 mg ja ondansetronia 8 mg suun kautta kahden annoksen ajan ja deksametasonia 12 mg suun kautta. Toisena ja kolmantena päivänä fosaprepitanttiryhmän potilaat saivat ondansetronin kaltaista plaseboa 12 tunnin välein. Vertailuhoito-ohjelmassa annettiin fosaprepitantin kaltaista plaseboa 150 mg laskimoon ensimmäisenä päivänä ja ondansetronia 8 mg suun kautta kahden annoksen ajan ja deksametasonia 20 mg suun kautta. Toisena ja kolmantena päivänä vertailuryhmän potilaat saivat 8 mg ondansetronia suun kautta 12 tunnin välein. Fosaprepitantin kaltaista plaseboa ja deksametasonin kaltaista plaseboa (ensimmäisenä päivänä) käytettiin sokkoutuksen säilyttämiseksi.
Fosaprepitantin tehoa arvioitiin taulukossa 8 lueteltujen ensisijaisten ja toissijaisten päätetapahtumien perusteella ja sen osoitettiin olevan vertailuhoidon tehoa parempi, kun tarkasteltiin täydellistä hoitovastetta viivästyneessä vaiheessa ja koko arviointijakson aikana.
Taulukko 8: Kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saaneet aikuispotilaat. Vasteprosentti hoitoryhmittäin eri vaiheissa
PÄÄTETAPAHTUMAT* | Fosaprepitanttihoito-ohjelma | Vertailuhoito-ohjelma | p-arvo |
Täydellinen hoitovaste† | |||
Viivästynyt vaihe‡ | 78,9 | 68,5 | < 0,001 |
Täydellinen hoitovaste† | |||
Yhteensä§ | 77,1 | 66,9 | < 0,001 |
Akuutti vaihe§§ | 93,2
| 91
| 0,184
|
| *Ensisijainen päätetapahtuma esitetään lihavoituna. **n: Hoitoaiepopulaatiossa mukana olleiden aikuispotilaiden lukumäärä. †Täydellinen hoitovaste = ei oksentelua eikä varalääkkeiden käyttöä. ‡Viivästynyt vaihe = 25–120 tuntia solunsalpaajahoidon alkamisen jälkeen. §Yhteensä = 0–120 tuntia solunsalpaajahoidon alkamisen jälkeen. §§Akuutti = 0–24 tuntia solunsalpaajahoidon alkamisen jälkeen. | |||
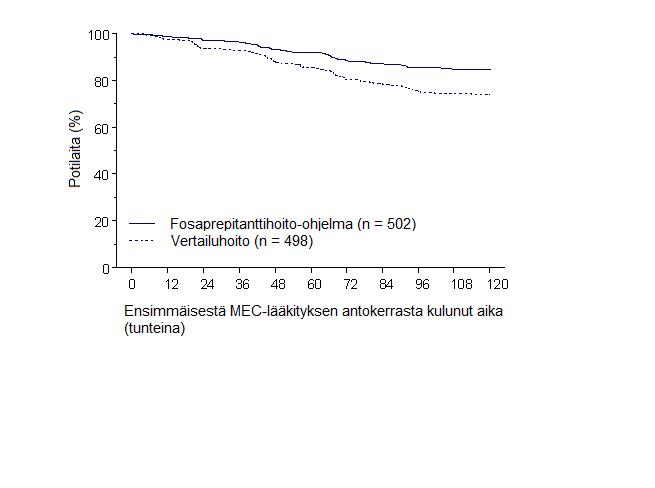
Ensimmäisen pahoinvointikohtauksen arvioitu ilmaantumisajankohta on kuvattuna Kaplan-Meierin käyrällä kuvassa 1.
Kuva 1: Niiden kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saaneiden aikuispotilaiden prosentuaalinen osuus ajan funktiona, joilla ei ollut pahoinvointia
Pediatriset potilaat
Kolmessa vaikuttavalla vertailuaineella kontrolloidussa avoimessa kliinisessä tutkimuksessa 6 kuukauden – 17 vuoden ikäiset pediatriset potilaat saivat joko voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajahoitoa ja kerta-annoksen fosaprepitanttia yhden päivän hoito-ohjelman suositeltuna tai sitä suurempana annoksena (139 potilasta) tai kolmen päivän hoito-ohjelmana (199 potilasta) yhdessä ondansetronin ja mahdollisesti myös deksametasonin kanssa.
Yhden päivän fosaprepitanttihoito-ohjelma pediatrisilla potilailla
Yhden päivän fosaprepitanttihoito-ohjelman teho pediatristen potilaiden hoidossa perustuu aikuisilla potilailla todettuun yhden päivän fosaprepitanttihoito-ohjelman tehoon, jota on kuvattu kohdassa Yhden päivän fosaprepitanttihoito-ohjelma aikuisilla.
Yhden päivän fosaprepitanttihoito-ohjelman tehon odotetaan olevan pediatrisilla potilailla samanlainen kuin aikuisilla potilailla.
Kolmen päivän fosaprepitanttihoito-ohjelma pediatrisilla potilailla
Kolmen päivän fosaprepitanttihoito-ohjelman teho pediatristen potilaiden hoidossa perustuu suun kautta annetun kolmen päivän aprepitanttihoito-ohjelman todettuun tehoon pediatrisilla potilailla.
Kolmen päivän fosaprepitanttihoito-ohjelman tehon odotetaan olevan pediatrisilla potilailla samanlainen kuin suun kautta annetun kolmen päivän aprepitanttihoito-ohjelman teho. EMEND kapseleiden ja EMEND jauheen oraalisuspensiota varten valmisteyhteenvedoissa on täydelliset kliiniset tiedot suun kautta annettavalla aprepitantilla tehdyistä tutkimuksista.
Farmakokinetiikka
Fosaprepitantti on aprepitantin aihiolääke, ja laskimoon annettuna se muuttuu nopeasti aprepitantiksi. Fosaprepitantin pitoisuus plasmassa laskee määritysrajan alapuolelle 30 minuutin kuluessa infuusion päättymisestä.
Aprepitantti fosaprepitantti-infuusion jälkeen
Kun 150 mg:n kerta-annos fosaprepitanttia annettiin terveille vapaaehtoisille aikuisille 20 minuutin infuusiona laskimoon, aprepitantin AUC0-∞-arvon keskiarvo oli 35,0 µg•h/ml ja aprepitantin maksimipitoisuuden keskiarvo oli 4,01 µg/ml.
Jakautuminen
Aprepitantti sitoutuu voimakkaasti proteiineihin. Sitoutumisaste on keskimäärin 97 %. Fosaprepitantin 150 mg:n laskimoon annetun kerta-annoksen perusteella arvioitu aprepitantin jakautumistilavuuden geometrinen keskiarvo vakaan tilan aikana (Vdss) on ihmisessä noin 82 l.
Biotransformaatio
Fosaprepitantti muuttui nopeasti aprepitantiksi, kun sitä inkuboitiin ihmisen maksakudospreparaateissa in vitro. Lisäksi fosaprepitantti muuttui aprepitantiksi nopeasti ja lähes täydellisesti muissa ihmiskudosten, kuten munuaisten, keuhkojen ja ileumin, S9-preparaateissa. Näyttää siis siltä, että fosaprepitantti voi muuttua aprepitantiksi monissa kudoksissa. Ihmisen laskimoon annettu fosaprepitantti muuttui aprepitantiksi nopeasti, 30 minuutin kuluessa infuusion päättymisen jälkeen.
Aprepitantti metaboloituu tehokkaasti. Kun terveille nuorille aikuisille annettiin [14C]-merkittyä fosaprepitanttia, aprepitantin aihiolääkettä, 100 mg:n kerta-annoksena laskimoon, aprepitantin osuus oli noin 19 % plasmassa tavatusta radioaktiivisuudesta 72 tunnin kuluessa lääkkeenannosta. Tämä osoittaa, että plasmassa oli huomattava määrä metaboliitteja. Ihmisen plasmasta on tunnistettu 12 aprepitantin metaboliittia. Aprepitantti metaboloituu suurelta osin morfoliinirenkaan ja sen sivuketjujen oksidaation kautta. Nämä metaboliitit ovat vain heikosti aktiivisia. Ihmisen maksan mikrosomeilla tehdyt in vitro -tutkimukset ovat osoittaneet, että aprepitantti metaboloituu pääasiassa CYP3A4-entsyymin välityksellä ja mahdollisesti vähäisessä määrin CYP1A2:n ja CYP2C19:n välityksellä.
Kaikkia metaboliitteja, joita tavattiin laskimoon annetun 100 mg:n [14C]-merkityn fosaprepitanttiannoksen jälkeen virtsassa, ulosteessa ja plasmassa, esiintyi myös suun kautta annetun [14C]-merkityn aprepitanttiannoksen jälkeen. Kun 245,3 mg fosaprepitanttidimeglumiinia (vastaa 150 mg fosaprepitanttia) muuttuu aprepitantiksi, vapautuu 23,9 mg fosforihappoa ja 95,3 mg meglumiinia.
Eliminaatio
Aprepitantti ei erity muuttumattomana virtsaan. Metaboliitit erittyvät virtsaan ja sapen kautta ulosteeseen. Kun terveille koehenkilöille annettiin [14C]-merkittyä fosaprepitanttia 100 mg:n kerta-annoksena laskimoon, 57 % radioaktiivisuudesta todettiin virtsassa ja 45 % ulosteessa.
Aprepitantin farmakokinetiikka on epälineaarinen koko kliinisellä annosalueella. Aprepitantin terminaalinen puoliintumisaika oli laskimoon annetun fosaprepitantin 150 mg:n kerta-annoksen jälkeen noin 11 tuntia. Aprepitantin plasmapuhdistuman geometrinen keskiarvo oli laskimoon annetun 150 mg:n fosaprepitanttiannoksen jälkeen noin 73 ml/min.
Farmakokinetiikka erityisryhmissä
Maksan vajaatoiminta: Fosaprepitantti metaboloituu useissa maksan ulkopuolisissa kudoksissa, joten maksan vajaatoiminnan ei odoteta vaikuttavan fosaprepitantin muuttumiseen aprepitantiksi. Lievä maksan vajaatoiminta (Child-Pugh-luokka A) ei vaikuta aprepitantin farmakokinetiikkaan kliinisesti merkittävästi. Annoksen muuttaminen ei ole tarpeen lievässä maksan vajaatoiminnassa. Saatavilla olevien tietojen perusteella ei voi tehdä johtopäätöksiä keskivaikean maksan vajaatoiminnan (Child-Pugh-luokka B) vaikutuksista aprepitantin farmakokinetiikkaan. Käytettävissä ei ole kliinisiä eikä farmakokineettisiä tietoja vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavien potilaiden hoidosta.
Munuaisten vajaatoiminta: Aprepitanttia annettiin suun kautta 240 mg kerta-annoksena potilaille, joilla oli vaikea munuaisten vajaatoiminta (CrCl < 30 ml/min), ja potilaille, joilla oli hemodialyysihoitoa vaativa munuaissairaus.
Vaikeassa munuaisten vajaatoiminnassa aprepitantin kokonaispitoisuuden (sitoutumattoman ja proteiiniin sitoutuneen) AUC0-∞ oli 21 % pienempi ja Cmax oli 32 % pienempi kuin terveissä tutkittavissa. Hemodialyysihoitoa saavissa munuaistautia sairastavissa potilaissa aprepitantin kokonaispitoisuuden AUC0-∞ oli 42 % pienempi ja Cmax oli 32 % pienempi. Koska aprepitantin sitoutuminen proteiiniin heikkenee vain vähän munuaisten vajaatoiminnan aikana, munuaisten vajaatoimintaa sairastaneista potilaista mitatun farmakologisesti aktiivisen sitoutumattoman lääkeaineen AUC ei poikennut merkitsevästi terveistä tutkittavista mitatuista vastaavista arvoista. Hemodialyysihoito, jota annettiin 4 tai 48 tuntia annoksen jälkeen, ei vaikuttanut merkitsevästi aprepitantin farmakokinetiikkaan – dialysaatissa tavattiin alle 0,2 % annoksesta.
Annoksen muuttaminen ei ole tarpeen hoidettaessa potilaita, joilla on munuaisten vajaatoiminta tai hemodialyysihoitoa vaativa munuaissairaus.
Pediatriset potilaat: Taulukossa 9 on kuvattu laskimoon annetun kolmen päivän hoito-ohjelman yhteydessä simuloitu aprepitantin AUC0-24h-arvojen mediaani, huippupitoisuuksien (Cmax) mediaani plasmassa 1. päivänä ja mediaanipitoisuudet 1. päivän, 2. päivän ja 3. päivän päättyessä pediatrisilla potilailla (ikäjakauma 6 kuukautta – 17 vuotta).
Taulukko 9: Aprepitantin farmakokineettiset parametrit laskimoon annetun 3 päivän fosaprepitanttihoito-ohjelman yhteydessä pediatrisilla potilailla
Potilaat | Kolmena päivänä laskimoon annetut annokset | AUC0-24 h. (ng*h/ml) | Cmax (ng/ml) | C24 (ng/ml) | C48 (ng/ml) | C72 (ng/ml) |
12–17-vuotiaat | 115 mg, 80 mg, 80 mg | 21 172 | 2 475 | 454 | 424 | 417 |
6 – < 12-vuotiaat |
3 mg/kg, 2 mg/kg, 2 mg/kg
| 25 901 | 2 719 | 518 | 438 | 418 |
2 – < 6-vuotiaat | 20 568 | 2 335 | 336 | 248 | 232 | |
6 kuukauden – | 16 979 | 1 916 | 256 | 179 | 167 |
Taulukossa 10 on kuvattu laskimoon annettuun yhden päivän fosaprepitanttihoito-ohjelmaan liittyvä simuloitu aprepitantin AUC0-24h-arvon mediaani, huippupitoisuuksien (Cmax) mediaani plasmassa 1. päivänä ja mediaanipitoisuudet 1. päivän, 2. päivän ja 3. päivän päättyessä pediatrisilla potilailla (ikäjakauma 6 kuukautta – < 12 vuotta) sekä havaittu AUC0-24h-keskiarvo, huippupitoisuuksien (Cmax) mediaani plasmassa 1. päivänä ja pitoisuuksien keskiarvo 1. päivän, 2. päivän ja 3. päivän päättyessä pediatrisilla potilailla (ikäjakauma 12–17 vuotta).
Taulukko 10: Aprepitantin farmakokineettiset parametrit laskimoon annetun 1 päivän fosaprepitanttihoito-ohjelman yhteydessä pediatrisilla potilailla
Potilaat | Yhtenä päivänä laskimoon annettu annos | AUC0-24 h. (ng*h/ml) | Cmax (ng/ml) | C24 (ng/ml) | C48 (ng/ml) | C72 (ng/ml) |
12–17-vuotiaat | 150 mg | 30 400 | 3 500 | 735 | NR | NR |
6 – < 12-vuotiaat |
4 mg/kg
| 35 766 | 3 637 | 746 | 227 | 69,2 |
2 – < 6-vuotiaat | 28 655 | 3 150 | 494 | 108 | 23,5 | |
6 kuukauden – | 5 mg/kg | 30 484 | 3 191 | 522 | 112 | 24,4 |
| NR = Ei raportoitu | ||||||
Aprepitantin populaatiofarmakokineettinen analyysi pediatrisista potilaista (ikäjakauma 6 kuukautta – 17 vuotta) viittaa siihen, ettei sukupuolella tai etnisellä taustalla ole kliinisesti merkittävää vaikutusta aprepitantin farmakokinetiikkaan.
Pitoisuuden suhde tehoon
Positroniemissiotomografiatutkimuksissa (PET), joissa käytettiin erittäin spesifistä NK1-reseptorin merkkiainetta, annettiin terveille nuorille miehille laskimoon 150 mg:n kerta-annos fosaprepitanttia (N=8). Tutkimuksessa osoitettiin, että aivojen NK1-reseptorien sitoutumisaste oli ≥ 100 % Tmax-ajankohtana ja 24 tunnin kuluttua, ≥ 97 % 48 tunnin kuluttua ja 41–75 % 120 tunnin kuluttua annoksen antamisesta. Aivojen NK1-reseptorien sitoutumisaste tässä tutkimuksessa korreloi hyvin plasman aprepitanttipitoisuuksien kanssa.
Prekliiniset tiedot turvallisuudesta
Laskimoon annettavaa fosaprepitanttia ja suun kautta annettavaa aprepitanttia koskevat prekliiniset tiedot, jotka perustuvat kerta-annoksen ja toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta (myös in vitro -testit) sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tuloksiin, eivät viittaa erityiseen vaaraan ihmisille.
Karsinogeenisuutta jyrsijöillä tutkittiin vain suun kautta annetulla aprepitantilla. On kuitenkin syytä ottaa huomioon, että jyrsijöillä, kaniineilla ja apinoilla tehtyjen toksisuustutkimusten, myös lisääntymistoksisuutta koskevien tutkimusten, arvo on vähäinen, koska näissä tutkimuksissa systeeminen fosaprepitantti- ja aprepitanttialtistus oli sama tai jopa pienempi kuin hoitoannosten aikaansaama altistus aikuisilla ihmisillä. Turvallisuusfarmakologiaa ja toistetun altistuksen aiheuttamaa toksisuutta selvittävissä tutkimuksissa koirilla fosaprepitantin Cmax-arvo oli enintään kolme kertaa ja aprepitantin AUC-arvo 40 kertaa suurempi kuin kliiniset arvot.
Nuorilla koirilla tehdyssä toksisuustutkimuksessa, jossa annettiin fosaprepitanttia syntymän jälkeen päivästä 14 päivään 42 saakka, havaittiin uroksilla kivesten painon ja Leydigin solujen pienenemistä annoksella 6 mg/kg/vrk ja naarailla kohdun painon suurenemista, kohdun ja kohdunkaulan hypertrofiaa ja emätinkudosten turvotusta annoksesta 4 mg/kg/vrk lähtien. Nuorilla rotilla tehdyssä toksisuustutkimuksessa aprepitantin anto syntymän jälkeen päivästä 10 päivään 63 saakka aiheutti varhaisempaa emättimen avautumista naarailla annoksesta 250 mg/kg kahdesti vuorokaudessa alkaen ja viivästynyttä esinahan separaatiota uroksilla annoksesta 10 mg/kg kahdesti vuorokaudessa alkaen. Hoitoon liittyviä vaikutuksia paritteluun, hedelmällisyyteen tai alkioiden/sikiöiden selviytymiseen ei ollut eikä lisääntymiselimissä todettu patologisia muutoksia. Kliinisesti merkitykselliselle aprepitanttialtistukselle ei ollut asetettu marginaaleja. Lyhytkestoisen hoidon yhteydessä katsotaan, että nämä löydökset eivät todennäköisesti ole kliinisesti merkityksellisiä.
Ei-kaupallisina formulaatioina annettu fosaprepitantti aiheutti koe-eläimille verisuoniin kohdistuvia toksisia vaikutuksia ja hemolyysiä pitoisuuden ollessa alle 1 mg/ml tai suurempi, formulaatiosta riippuen. Ei-kaupalliset formulaatiot aiheuttivat myös ihmisen pestyissä verisoluissa hemolyysiin viittaavia merkkejä, kun fosaprepitanttipitoisuus oli 2,3 mg/ml tai suurempi, vaikka ihmisen kokoverellä tehdyissä testeissä tulos oli negatiivinen. Kaupallista formulaatiota käytettäessä hemolyysiä ei havaittu ihmisen kokoveressä eikä pestyissä ihmisen punasoluissa, kun fosaprepitanttipitoisuus oli enintään 1 mg/ml.
Fosaprepitantti aiheutti alkuvaiheessa ohimenevän paikallisen akuutin tulehduksen, kun sitä annettiin kaniineille laskimon viereen, ihon alle ja lihakseen. Seurantajakson päättyessä (kahdeksantena päivänä annoksen jälkeen) laskimon viereen ja lihakseen annetun annoksen jälkeen havaittiin enintään vähäistä paikallista subakuuttia tulehdusta ja lihakseen annetun annoksen jälkeen lisäksi enintään kohtalaista fokaalista lihasten degeneraatiota/nekroosia ja lihasten regeneraatiota.
Farmaseuttiset tiedot
Apuaineet
Dinatriumedetaatti (E386)
Polysorbaatti 80 (E433)
Vedetön laktoosi
Natriumhydroksidi (E524) (pH:n säätöön) ja/tai
Laimea kloorivetyhappo (E507) (pH:n säätöön)
Yhteensopimattomuudet
IVEMEND ei ole yhteensopiva kaksiarvoisia kationeja (esim. Ca2+, Mg2+) sisältävien liuosten kanssa, jollaisia ovat esimerkiksi Hartmanin liuos ja laktaattia sisältävä Ringerin liuos. Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
2 vuotta.
Kun infuusiokuiva-aine on liuotettu ja laimennettu, kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan 24 tuntia 25 °C:ssa.
Mikrobiologisista syistä lääkevalmiste tulee käyttää heti. Ellei sitä käytetä heti, käytönaikaiset säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla, ja normaalisti ne saavat olla enintään 24 tuntia 2–8 °C:ssa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Liuotetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
IVEMEND infuusiokuiva-aine, liuosta varten
150 mg (L:kyllä) 1 kpl (10 ml) (94,62 €)
PF-selosteen tieto
10 ml:n lasinen injektiopullo (tyypin I kirkasta lasia), jossa klooributyyli- tai bromobutyylikumitulppa ja alumiinisuljin, jossa harmaa muovinen repäisykorkki.
Pakkauskoot: 1 tai 10 injektiopulloa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai vaalea amorfinen jauhe.
Käyttö- ja käsittelyohjeet
IVEMEND on liuotettava ja laimennettava ennen annostelua.
IVEMEND 150 mg -vahvuuden valmistaminen laskimoon antoa varten:
1. Lisää 5 ml 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionesteliuosta injektiopulloon. Suuntaa natriumkloridi-injektionesteliuos injektiopullon seinämään, jotta pulloon ei muodostu vaahtoa. Pyörittele pulloa varovasti. Älä ravista äläkä suihkuta natriumkloridi-injektionesteliuosta voimakkaasti injektiopulloon.
2. Valmista infuusiopussi, jossa on 145 ml 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionesteliuosta (esim. poistamalla 105 ml 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionesteliuosta infuusiopussista, jossa on 250 ml 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionesteliuosta).
3. Vedä injektiopullon sisältö kokonaan ruiskuun ja siirrä se infuusiopussiin, jossa on 145 ml 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionesteliuosta, minkä jälkeen liuoksen kokonaistilavuus on 150 ml ja lopullinen pitoisuus 1 mg/ml. Käännä pussi varovasti ylösalaisin 2–3 kertaa.
4. Määritä tästä käyttövalmiiksi saatetusta infuusiopussista potilaalle annettava määrä suositellun annoksen perusteella (ks. kohta Annostus ja antotapa).
Aikuiset
Käyttövalmiiksi saatetun infuusiopussin koko sisältö (150 ml) annetaan potilaalle.
Pediatriset potilaat
12 vuotta täyttäneille potilaille annettava tilavuus lasketaan seuraavasti:
- Annettava tilavuus (ml) on sama kuin suositeltu annos (mg)
6 kuukauden – alle 12 vuoden ikäisille potilaille annettava tilavuus lasketaan seuraavasti:
-
Annettava tilavuus (ml) = suositeltu annos (mg/kg) x paino (kg)
- Huom: Enimmäisannoksia ei saa ylittää (ks. kohta Annostus ja antotapa).
5. Jos laskettu tilavuus on alle 150 ml, se voidaan tarvittaessa siirtää sopivan kokoiseen pussiin tai ruiskuun ennen kuin se annetaan infuusiona.
Käyttövalmiiksi sekoitettu liuos on ulkonäöltään samanlaista kuin liuotin.
Liuotettu ja laimennettu lääkevalmiste on tarkastettava silmämääräisesti ennen annostelua hiukkasten ja värimuutosten havaitsemiseksi.
Jäljelle jäänyt liuos ja jätemateriaali on hävitettävä. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Lääkevalmistetta ei saa liuottaa eikä sekoittaa sellaisiin liuoksiin, joiden fysikaalista ja kemiallista yhteensopivuutta ei ole varmistettu (ks. kohta Yhteensopimattomuudet).
Korvattavuus
IVEMEND infuusiokuiva-aine, liuosta varten
150 mg 1 kpl
- Ei korvausta.
ATC-koodi
A04AD12
Valmisteyhteenvedon muuttamispäivämäärä
25.08.2022
Yhteystiedot
 MSD FINLAND OY
MSD FINLAND OY Keilaniementie 1, PL 46
02151 Espoo
09 804 650
www.msd.fi
info@msd.fi