
CYRAMZA infuusiokonsentraatti, liuosta varten 10 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi millilitra infuusiokonsentraattia sisältää 10 mg ramusirumabia.
Yksi 10 ml injektiopullo sisältää 100 mg ramusirumabia.
Yksi 50 ml injektiopullo sisältää 500 mg ramusirumabia.
Ramusirumabi on ihmisen monoklonaalinen IgG-1-vasta-aine, joka on tuotettu hiiren (NS0) soluissa yhdistelmä-DNA-tekniikalla.
Apuaine, jonka vaikutus tunnetaan
Yksi 10 ml injektiopullo sisältää noin 17 mg natriumia ja 1 mg polysorbaatti 80:tä.
Yksi 50 ml injektiopullo sisältää noin 85 mg natriumia ja 5 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Mahasyöpä
Cyramza on tarkoitettu yhdessä paklitakselin kanssa pitkälle edenneen mahasyövän tai ruokatorvi-mahalaukkurajan adenokarsinooman hoitoon aikuisille potilaille, joiden tauti on edennyt aiemman platina- ja fluoropyrimidiinipohjaisen solunsalpaajahoidon jälkeen (ks. kohta Farmakodynamiikka).
Cyramza monoterapia on tarkoitettu pitkälle edenneen mahasyövän tai ruokatorvi-mahalaukkurajan adenokarsinooman hoitoon aikuisille potilaille, joiden tauti on edennyt aiemman platina- tai fluoropyrimidiinipohjaisen solunsalpaajahoidon jälkeen ja joilla hoito yhdessä paklitakselin kanssa ei ole tarkoituksenmukaista (ks. kohta Farmakodynamiikka).
Kolorektaalisyöpä
Cyramza on tarkoitettu yhdessä FOLFIRI-yhdistelmän (irinotekaani, foliinihappo ja 5-fluorourasiili) kanssa metastasoitunutta kolorektaalisyöpää (mCRC) sairastavien aikuispotilaiden hoitoon, joiden tauti on edennyt aiemman bevasitsumabi-, oksaliplatiini- ja fluoropyrimidiinipohjaisen hoidon aikana tai sen jälkeen.
Ei-pienisoluinen keuhkosyöpä
Cyramza on tarkoitettu yhdessä erlotinibin kanssa metastasoituneen ei-pienisoluisen keuhkosyövän ensilinjan hoitoon aikuisille potilaille, joilla on aktivoiva epidermaalisen kasvutekijäreseptorin (EGFR) mutaatio (ks. kohta Farmakodynamiikka).
Cyramza on tarkoitettu yhdessä dosetakselin kanssa paikallisesti levinneen tai metastasoituneen ei-pienisoluisen keuhkosyövän hoitoon aikuisille potilaille, joiden tauti on edennyt aiemman platinapohjaisen solunsalpaajahoidon jälkeen.
Maksasolukarsinooma
Cyramza on tarkoitettu käytettäväksi ainoana lääkkeenä pitkälle edennyttä tai leikkaukseen soveltumatonta maksasolukarsinoomaa sairastavien aikuispotilaiden hoitoon, kun alfafetoproteiinin (AFP) pitoisuus seerumissa on ≥ 400 ng/ml ja potilas on saanut aiemmin sorafenibihoitoa.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Ramusirumabihoidon aloittaa ja sitä valvoo lääkäri, jolla on kokemusta syöpähoitojen toteuttamisesta.
Annostus
Mahasyöpäja ruokatorvi-mahalaukkurajan adenokarsinooma
Cyramza yhdessä paklitakselin kanssa
Suositeltu ramusirumabiannos on 8 mg/kg 28 päivän syklin päivinä 1 ja 15 ennen paklitakseli-infuusiota. Suositeltu paklitakseliannos on 80 mg/m2 infuusiona laskimoon noin 60 minuutin aikana 28 päivän syklin päivinä 1, 8 ja 15. Potilaan täydellinen verenkuva ja veren kemiallinen koostumus on tutkittava maksan toiminnan selvittämiseksi ennen jokaista paklitakseli-infuusiota. Taulukossa 1 on ilmoitettu kriteerit, joiden on täytyttävä ennen jokaista paklitakseli-infuusiota.
Taulukko 1: Kriteerit, joiden on täytyttävä ennen jokaista paklitakseli-infuusiota
| Kriteerit | |
| Neutrofiilit | Päivä 1: ≥ 1,5 x 109/l Päivät 8 ja 15: ≥ 1,0 x 109/l |
| Trombosyytit | Päivä 1: ≥ 100 x 109/l Päivät 8 ja 15: ≥ 75 x 109/l |
| Bilirubiini | ≤ 1,5 x viitealueen yläraja (ULN) |
| Aspartaattiaminotransferaasi (ASAT)/Alaniiniaminotransferaasi (ALAT) | Ei maksametastaaseja: ALAT/ASAT ≤ 3 x ULN Maksametastaaseja: ALAT/ASAT ≤ 5 x ULN |
Cyramza ainoana lääkkeenä
Suositeltu ramusirumabiannos monoterapiana on 8 mg/kg 2 viikon välein.
Kolorektaalisyöpä
Suositeltu ramusirumabiannos on 8 mg/kg 2 viikon välein laskimoinfuusiona annosteltuna, ennen FOLFIRI-yhdistelmän antoa. Ennen solunsalpaajahoidon aloittamista potilaalta on otettava täydellinen verenkuva. Taulukossa 2 on ilmoitettu kriteerit, joiden on täytyttävä ennen FOLFIRI-yhdistelmän antoa.
Taulukko 2: Kriteerit, joiden on täytyttävä ennen FOLFIRI-yhdistelmän antoa
| Kriteerit | |
| Neutrofiilit | ≥ 1,5 x 109/l |
| Trombosyytit | ≥ 100 x 109/l |
| Solunsalpaajahoitoon liittyvä ruoansulatuskanavan toksisuus | ≤ Aste 1 (National Cancer Institute -instituutin Common Terminology Criteria for Adverse Events -kriteerit [NCI CTCAE]) |
Ei-pienisoluinen keuhkosyöpä
Cyramza yhdessä erlotinibin kanssa ei-pienisoluisen keuhkosyövän hoidossa, kun kasvaimessa on aktivoiva EGFR-mutaatio
Suositeltu ramusirumabiannos yhdessä erlotinibin kanssa on 10 mg/kg 2 viikon välein.
EGFR-mutaatiostatus on määritettävä validoidulla testimenetelmällä ennen ramusirumabi- ja erlotinibihoidon aloittamista. Erlotinibin annostus ja antotapa, ks. erlotinibin valmisteyhteenveto.
Cyramza yhdessä dosetakselin kanssa ei-pienisoluisen keuhkosyövän hoidossa, kun potilas on saanut aiemmin platinapohjaista solunsalpaajahoitoa
Suositeltu ramusirumabiannos on 10 mg/kg 21 päivän syklin päivänä 1 ennen dosetakseli-infuusiota. Suositeltu dosetakseliannos on 75 mg/m2 infuusiona laskimoon noin 60 minuutin aikana 21 päivän syklin päivänä 1. Itä-Aasialaisille potilaille tulee harkita pienempää dosetakselin aloitusannosta 60 mg/m2 21 päivän syklin päivänä 1. Tarkat annosteluohjeet ks. dosetakselin valmisteyhteenveto.
Maksasolukarsinooma
Suositeltu ramusirumabiannos monoterapiana on 8 mg/kg 2 viikon välein.
Alfafetoproteiinin (AFP) määritys maksasolukarsinoomaa sairastavilla
Ennen ramusirumabihoitoa maksasolukarsinoomaa sairastavat potilaat valitaan hoitoon, kun validoidulla AFP-testillä mitattu seerumin AFP-pitoisuus on ≥ 400 ng/ml (ks. kohta Farmakodynamiikka).
Hoidon kesto
Hoidon jatkaminen on suositeltavaa, kunnes tauti etenee tai kehittyy sietämätöntä toksisuutta.
Esilääkitys
Esilääkitystä H1-antihistamiinilla (esim. difenhydramiinilla) suositellaan ennen ramusirumabi-infuusiota. Jos potilaalle kehittyy asteen 1 tai 2 infuusioreaktio, esilääkitys on annettava kaikkien tulevien infuusioiden yhteydessä. Jos potilaalle kehittyy toinen asteen 1 tai 2 infuusioreaktio, hänelle on annettava deksametasonia (tai vastaavaa). Seuraavien infuusioiden yhteydessä esilääkityksenä annetaan seuraavia tai vastaavia lääkevalmisteita: H1-antihistamiini (esim. difenhydramiinihydrokloridi) laskimoon, parasetamoli ja deksametasoni.
Esilääkitystä koskevat vaatimukset ja lisätiedot, ks. paklitakselin, FOLFIRI-yhdistelmän komponenttien ja dosetakselin valmisteyhteenvedot.
Ramusirumabiannostuksen muuttaminen
Infuusioreaktiot
Ramusirumabin infuusionopeus on puolitettava infuusion ajaksi ja kaikkien tulevien infuusioiden ajaksi, jos potilaalle kehittyy asteen 1 tai 2 infuusioreaktio. Jos potilaalle kehittyy asteen 3 tai 4 infuusioreaktio, ramusirumabin anto on lopetettava välittömästi ja pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hypertensio
Potilaan verenpainetta on seurattava ennen jokaista ramusirumabin antokertaa ja hoidettava kliinisen tilanteen mukaan. Jos potilaalle kehittyy vaikea hypertensio, ramusirumabihoito on keskeytettävä tilapäisesti, kunnes verenpaine saadaan lääkityksellä hallintaan. Jos potilaalle kehittyy lääketieteellisesti merkittävä hypertensio, jota ei saada turvallisesti hallintaan verenpainelääkityksellä, ramusirumabihoito on lopetettava pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Proteinuria
Potilaita on seurattava ramusirumabihoidon aikana proteinurian kehittymisen tai pahenemisen varalta. Jos virtsan proteiinipitoisuus on liuskatestissä ≥ 2+, potilaalta on kerättävä vuorokausivirtsa. Ramusirumabihoito on keskeytettävä väliaikaisesti, jos virtsan proteiinipitoisuus on ≥ 2 g/24 h. Kun virtsan proteiinipitoisuus on palautunut tasolle < 2 g/24 h, hoito aloitetaan uudestaan pienemmällä annoksella (ks. Taulukko 3). Annoksen pienentäminen uudestaan (ks. Taulukko 3) on suositeltavaa, jos virtsan proteiinipitoisuus palautuu tasolle ≥ 2 g/24 h.
Ramusirumabihoito on lopetettava pysyvästi, jos virtsan proteiinipitoisuus on > 3 g/24 h tai jos potilaalle kehittyy nefroottinen oireyhtymä.
Taulukko 3: Ramusirumabiannostuksen pienentäminen proteinurian takia
| Alkuperäinen ramusirumabiannos | Annoksen pienentäminen ensimmäisen kerran | Annoksen pienentäminen toisen kerran |
| 8 mg/kg | 6 mg/kg | 5 mg/kg |
| 10 mg/kg | 8 mg/kg | 6 mg/kg |
Elektiivinen leikkaus tai haavojen huono paraneminen
Ramusirumabihoito on keskeytettävä väliaikaisesti vähintään 4 viikon ajaksi ennen elektiivistä leikkausta. Jos potilaalla on haavan paranemiskomplikaatioita, ramusirumabihoito on keskeytettävä väliaikaisesti, kunnes haava on täysin parantunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lopettaminen pysyvästi
Ramusirumabihoito on lopetettava pysyvästi, jos potilaalle ilmaantuu:
Vaikea tromboembolinen valtimotapahtuma (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maha-suolikanavan perforaatio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vaikea verenvuoto: NCI CTCAE -asteen 3 tai 4 verenvuoto (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Spontaanisti kehittynyt fisteli (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hepaattinen enkefalopatia tai hepatorenaalinen oireyhtymä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Paklitakseliannoksen muuttaminen
Paklitakseliannosta voidaan pienentää potilaan kokemasta toksisuuden asteesta riippuen. Jos potilaalla on NCI CTCAE -asteen 4 hematologinen toksisuus tai paklitakseliin liittyvä asteen 3 ei-hematologinen toksisuus, on suositeltavaa pienentää paklitakseliannosta 10 mg/m2 kaikissa seuraavissa sykleissä. Jos toksisuus pitkittyy tai uusiutuu, on suositeltavaa pienentää annosta toistamiseen 10 mg/m2.
FOLFIRI-annoksen muuttaminen
FOLFIRI-yhdistelmän yksittäisten komponenttien annoksia voidaan pienentää spesifisten toksisuuksien vuoksi. Kunkin FOLFIRI-komponentin annosmuutokset tulisi tehdä toisistaan riippumatta taulukon 4 mukaisesti. Taulukossa 5 on esitetty yksityiskohtia FOLFIRI-yhdistelmän komponenttien annosten siirtämisestä tai pienentämisestä seuraavassa syklissä spesifisten haittavaikutusten korkeimman asteen perusteella.
Taulukko 4: FOLFIRI-annoksen pienentäminen
| FOLFIRI komponenttia | Annostaso | |||
| Aloitusannos | -1 | -2 | -3 | |
| Irinotekaani | 180 mg/m2 | 150 mg/m2 | 120 mg/m2 | 100 mg/m2 |
| 5‑FU bolus | 400 mg/m2 | 200 mg/m2 | 0 mg/m2 | 0 mg/m2 |
| 5‑FU infuusio | 2400 mg/m2 yli 46‑48 tuntia | 2000 mg/m2 yli 46‑48 tuntia | 1600 mg/m2 yli 46‑48 tuntia | 1200 mg/m2 yli 46‑48 tuntia |
a 5-FU = 5-fluorourasiili
Taulukko 5: FOLFIRI-komponenttien annosmuutokset spesifisten haittavaikutusten perusteella
| Haittavaikutus | NCI CTCAE -aste | Annoksen muuttaminen haittavaikutusta seuraavan syklin ensimmäisenä päivänä | |
| Ripuli | 2 | Jos ripuli on korjaantunut asteeseen ≤ 1, pienennä 5-FU:n annostasoa yhdellä. Toistuvan asteen 2 ripulin tapauksessa pienennä 5-FU:n ja irinotekaanin annostasoa yhdellä. | |
| 3 | Jos ripuli on korjaantunut asteeseen ≤ 1, pienennä 5-FU:n ja irinotekaanin annostasoa yhdellä. | ||
| 4 | Jos ripuli on korjaantunut asteeseen ≤ 1, pienennä 5-FU:n ja irinotekaanin annostasoa kahdella. Jos asteen 4 ripuli ei korjaannu asteeseen ≤ 1, siirrä 5-FU:n ja irinotekaanin antamista korkeintaan 28* päivän ajan, kunnes ripuli on lievittynyt asteeseen ≤ 1. | ||
| Neutropenia tai Trombosytopenia | Taulukon 2 hematologiset kriteerit täyttyvät | Taulukon 2 hematologiset kriteerit eivät täyty | |
| 2 | Ei annosmuutoksia. | Pienennä 5-FU:n ja irinotekaanin annostasoa yhdellä. | |
| 3 | Pienennä 5-FU:n ja irinotekaanin annostasoa yhdellä. | Siirrä 5-FU:n ja irinotekaanin antamista korkeintaan 28* päivän ajan, kunnes lievittynyt asteeseen ≤ 1, sitten pienennä 5-FU:n ja irinotekaanin annosta yhdellä tasolla. | |
| 4 | Pienennä 5-FU:n ja irinotekaanin annostasoa kahdella. | Siirrä 5-FU:n ja irinotekaanin antamista korkeintaan 28* päivän ajan, kunnes lievittynyt asteeseen ≤ 1, sitten pienennä 5-FU:n ja irinotekaanin annosta kahdella tasolla. | |
| Stomatiitti/Mukosiitti | 2 | Jos stomatiitti/mukosiitti on korjaantunut asteeseen ≤ 1, pienennä 5-FU:n annostasoa yhdellä. Toistuvan asteen 2 stomatiitin tapauksessa pienennä 5-FU:n annostasoa kahdella. | |
| 3 | Jos stomatiitti/mukosiitti on korjaantunut asteeseen ≤ 1, pienennä 5-FU:n annostasoa yhdellä. Jos asteen 3 mukosiitti/stomatiitti ei lievity asteeseen ≤ 1, siirrä 5-FU:n antamista korkeintaan 28* päivän ajan, kunnes lievittynyt asteeseen ≤ 1, sitten pienennä 5-FU:n annosta kahdella tasolla. | ||
| 4 | Siirrä 5-FU:n antamista korkeintaan 28* päivän ajan, kunnes lievittynyt asteeseen ≤ 1, sitten pienennä 5-FU:n annosta kahdella tasolla. | ||
| Kuumeinen neutropenia | Taulukon 2 hematologiset kriteerit täyttyvät ja kuume on laskenut | Taulukon 2 hematologiset kriteerit eivät täyty ja kuume on laskenut | |
| Pienennä 5-FU:n ja irinotekaanin annostasoa kahdella. | Siirrä 5-FU:n ja irinotekaanin antamista korkeintaan 28* päivän ajan, kunnes lievittynyt asteeseen ≤ 1, sitten pienennä 5-FU:n ja irinotekaanin annosta kahdella tasolla. Harkitse valkosolujen kasvutekijän käyttöä ennen seuraavaa sykliä. | ||
* 28 päivän aikajakso alkaa haittavaikutusta seuraavan syklin ensimmäisenä päivänä.
Dosetakseliannoksen muuttaminen
Dosetakseliannosta voidaan pienentää potilaan kokemasta toksisuuden asteesta riippuen. Jos potilaalla on joko kuumeinen neutropenia, neutrofiileja < 500 solua/mm3 yli 1 viikon ajan, vaikeita tai kumulatiivisia ihoreaktioita tai muu asteen 3 tai 4 ei-hematologinen toksisuus dosetakselihoidon aikana, dosetakselihoito tulee keskeyttää, kunnes toksisuus on lieventynyt. On suositeltavaa pienentää dosetakseliannosta 10 mg/m2 kaikissa seuraavissa sykleissä. Jos toksisuus pitkittyy tai uusiutuu, on suositeltavaa pienentää annosta toistamiseen 15 mg/m2. Tässä tapauksessa Itä-Aasialaisten potilaiden, joiden aloitusannos oli 60 mg/m2, tulee keskeyttää dosetakselihoito (katso Annostus).
Erityisryhmät
Iäkkäät
Avaintutkimuksissa on rajallisesti näyttöä siitä, että 65-vuotiailla tai vanhemmilla potilailla on suurempi haittavaikutusten riski kuin alle 65-vuotiailla potilailla. Annoksen pienentämistä iäkkäillä potilailla ei suositella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Munuaisten vajaatoiminta
Cyramza-hoitoa munuaisten vajaatoimintapotilailla ei ole tutkittu. Kliiniset tiedot viittaavat siihen, että annosta ei tarvitse muuttaa, jos potilaalla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Annoksen pienentämistä ei suositella.
Maksan vajaatoiminta
Cyramza-hoitoa maksan vajaatoimintapotilailla ei ole tutkittu. Kliiniset tiedot viittaavat siihen, että annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta. Ramusirumabin käytöstä vaikeaa maksan vajaatoimintaa sairastavilla potilailla ei ole tietoja (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Annoksen pienentämistä ei suositella.
Pediatriset potilaat
Cyramzan turvallisuutta ja tehoa lasten ja alle 18-vuotiaiden nuorten hoidossa ei ole varmistettu. Tällä hetkellä saatavilla olevat tiedot on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka. Rajallisten tietojen takia annossuosituksia ei voida antaa.
Ei ole asianmukaista käyttää ramusirumabia pediatristen potilaiden pitkälle edenneen mahasyövän, ruokatorvi-mahalaukkurajan adenokarsinooman, paksu- ja peräsuolen adenokarsinooman, keuhkosyövän eikä maksasolukarsinooman hoidossa.
Antotapa
Cyramza annetaan laskimoon. Cyramza annetaan laimentamisen jälkeen noin 60 minuuttia kestävänä infuusiona laskimoon. Valmistetta ei saa antaa laskimoboluksena. Tarvittavan infuusion keston (noin 60 min) saavuttamiseksi infuusion enimmäisnopeutta (25 mg/min) ei saa ylittää, vaan infuusion kestoa on pidennettävä. Potilasta on seurattava infuusion aikana infuusioreaktioiden merkkien varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) ja asianmukaisten elvytysvälineiden saatavuus on varmistettava.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Ramusirumabi on vasta-aiheinen potilaille, jotka sairastavat ei-pienisoluista keuhkosyöpää ja joiden kasvaimessa on onteloita tai kasvain kiinnittyy pääsuoniin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Tromboemboliset valtimotapahtumat
Kliinisissä tutkimuksissa on ilmoitettu vakavia, joskus kuolemaan johtaneita tromboembolisia valtimotapahtumia, mm. sydäninfarkti, sydämenpysähdys, aivoverisuonitapahtuma ja aivoiskemia. Ramusirumabihoito on lopetettava pysyvästi, jos potilaalle kehittyy vaikea tromboembolinen valtimotapahtuma (ks. kohta Annostus ja antotapa).
Maha-suolikanavan perforaatiot
Ramusirumabi on angiogeneesiä estävä hoito, joka saattaa suurentaa maha-suolikanavan perforaatioiden riskiä. Maha-suolikanavan perforaatioita on ilmoitettu ramusirumabilla hoidetuilla potilailla. Ramusirumabihoito on lopetettava pysyvästi, jos potilaalle kehittyy maha-suolikanavan perforaatio (ks. kohta Annostus ja antotapa).
Vaikea verenvuoto
Ramusirumabi on angiogeneesiä estävä hoito, joka saattaa suurentaa vaikean verenvuodon riskiä. Ramusirumabihoito on lopetettava pysyvästi, jos potilaalle kehittyy asteen 3 tai 4 verenvuoto (ks. kohta Annostus ja antotapa). Verenkuvaa ja hyytymisarvoja on seurattava, jos potilaalla on verenvuodoille altistava sairaus tai jos potilas saa antikoagulaatiohoitoa tai samanaikaisesti muita verenvuotoriskiä suurentavia lääkevalmisteita. Maksasolukarsinoomaa sairastavat potilaat, joilla on näyttöä porttilaskimohypertensiosta tai anamneesissa ruokatorven laskimolaajentumien verenvuotoa, on seulottava ruokatorven laskimolaajentumien varalta, ja mahdolliset ruokatorven laskimolaajentumat on hoidettava tavanomaisen käytännön mukaisesti ennen ramusirumabihoidon aloittamista.
Ramusirumabin ja paklitakselin yhdistelmää saaneilla mahasyöpäpotilailla sekä ramusirumabin ja FOLFIRIN-yhdistelmää saaneilla metastasoitunutta kolorektaalisyöpää sairastavilla potilailla ilmoitettiin vaikeita maha-suolikanavan verenvuotoja, mukaan lukien kuolemaan johtaneita tapahtumia.
Keuhkoverenvuoto ei-pienisoluisessa keuhkosyövässä
Potilailla, joilla on levyepiteelimäinen histologia, on suurempi riski saada vakava keuhkoverenvuoto. REVEL-tutkimuksessa ei kuitenkaan havaittu lisääntynyttä asteen 5 keuhkoverenvuotoa ramusirumabilla hoidetuilla potilailla, joilla oli levyepiteelimäinen histologia. Kliinisiin tutkimuksiin ei otettu mukaan ei-pienisoluista keuhkosyöpää sairastavia potilaita, joilla oli ollut äskettäin keuhkoverenvuotoa (> 2,5 ml tai kirkkaan punaista verta) tai potilaita, joilla oli histologiasta riippumatta merkkejä kasvaimen onteloista lähtötilanteessa tai joilla oli merkkejä kasvaimen invaasiosta tai koteloitumisesta pääverisuoniin (ks. kohta Vasta-aiheet). Ei-pienisoluista keuhkosyöpää arvioineesta kliinisestä REVEL-tutkimuksesta suljettiin pois potilaat, jotka saivat mitä tahansa terapeuttista antikoagulanttia, ja ei-pienisoluista keuhkosyöpää arvioineista kliinisistä REVEL- ja RELAY-tutkimuksista suljettiin pois potilaat, jotka saivat pitkäaikaisesti tulehduskipulääkkeitä tai muita verihiutaleiden estäjiä. Aspiriinin käyttö sallittiin annokseen 325 mg/päivässä saakka (ks. kohta Farmakodynamiikka).
Infuusioreaktiot
Kliinisissä ramusirumabitutkimuksissa ilmoitettiin infuusioreaktioita. Valtaosa tapahtumista ilmaantui ensimmäisen tai toisen ramusirumabi-infuusion aikana tai sen jälkeen. Potilaita on seurattava infuusion aikana yliherkkyyden merkkien varalta. Oireita olivat mm. vapina/väristys, selkäkipu/spasmit, rintakipu ja/tai rinnan kireys, vilunväristykset, kuumat aallot, hengenahdistus, hengityksen vinkuminen, hypoksia ja parestesiat. Vaikeissa tapauksissa oireita olivat mm. bronkospasmi, supraventrikulaarinen takykardia ja hypotensio. Ramusirumabihoito on lopetettava välittömästi ja pysyvästi, jos potilaalle kehittyy asteen 3 tai 4 infuusioreaktio (ks. kohta Annostus ja antotapa).
Hypertensio
Vaikean hypertension esiintyvyyden ilmoitettiin kasvaneen ramusirumabia saaneilla potilailla verrattuna lumelääkettä saaneisiin potilaisiin. Useimmissa tapauksissa hypertensiota hoidettiin tavanomaisella verenpainelääkityksellä. Tutkimuksiin ei otettu mukaan potilaita, joiden hypertensio ei ollut hallinnassa. Ramusirumabihoitoa ei saa aloittaa potilailla, joiden aiemmin todettu hypertensio ei ole hallinnassa. Ramusirumabia saavien potilaiden verenpainetta on seurattava. Jos potilaalle kehittyy vaikea hypertensio, ramusirumabihoito on keskeytettävä väliaikaisesti, kunnes verenpaine saadaan lääkityksellä hallintaan. Ramusirumabihoito on lopetettava pysyvästi, jos lääketieteellisesti merkittävää hypertensiota ei saada hallintaan verenpainelääkityksellä (ks. kohta Annostus ja antotapa).
Posteriorinen reversiibeli enkefalopatiaoireyhtymä
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES) -tapauksia, myös kuolemaan johtaneita, on ilmoitettu harvoin ramusirumabia saaneilla potilailla. PRES-oireisiin saattaa kuulua kouristuskohtaus, päänsärky, pahoinvointi/oksentelu, näön menetys tai tajunnantason muutos, johon saattaa liittyä kohonnut verenpaine. PRES-diagnoosi voidaan vahvistaa aivojen kuvantamisella (esim. magneettikuvaus). Ramusirumabihoito tulee keskeyttää potilailla, joilla todetaan PRES.
Ramusirumabin uudelleenaloituksen turvallisuudesta potilailla, joille kehittyy PRES ja jotka toipuvat siitä, ei ole tietoa.
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen Cyramza-hoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Haavojen huono paraneminen
Ramusirumabin vaikutusta ei ole arvioitu potilailla, joilla on vakavia haavoja tai haavoja, jotka eivät parane. Eläimillä tehdyssä tutkimuksessa ramusirumabi ei heikentänyt haavojen paranemista. Ramusirumabihoito on kuitenkin keskeytettävä vähintään 4 viikoksi ennen elektiivistä leikkausta, sillä angiogeneesiä estävänä hoitona ramusirumabihoito saattaa vaikuttaa haavojen paranemiseen haitallisesti. Leikkauksen jälkeistä ramusirumabihoidon jatkamista koskevan päätöksen tulee perustua kliiniseen arvioon riittävästä haavan paranemisesta.
Jos potilaalla on haavan paranemiskomplikaatioita hoidon aikana, ramusirumabihoito on keskeytettävä, kunnes haava on täysin parantunut (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Ramusirumabia on käytettävä varoen potilailla, joilla on vaikea maksakirroosi (Child–Pugh-luokka B tai C), kirroosi, johon liittyy hepaattinen enkefalopatia, kliinisesti merkittävä kirroosista johtuva askites tai hepatorenaalinen oireyhtymä. Näiden potilaiden hoidosta on saatavilla hyvin niukasti teho- ja turvallisuustietoa. Ramusirumabia tulee käyttää näillä potilailla vain, jos hoidon mahdollisten hyötyjen arvioidaan olevan mahdollisen progressiivisen maksan vajaatoiminnan riskiä suurempia.
Maksasolukarsinoomapotilaiden ryhmässä hepaattista enkefalopatiaa ilmoitettiin useammin ramusirumabihoitoa saaneilla kuin lumehoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Potilaita on seurattava hepaattisen enkefalopatian kliinisten oireiden ja löydösten varalta. Ramusirumabihoito on lopetettava pysyvästi, jos potilaalle kehittyy hepaattinen enkefalopatia tai hepatorenaalinen oireyhtymä (ks. kohta Annostus ja antotapa).
Sydämen vajaatoiminta
Ramusirumabilla tehdyistä kliinisistä tutkimuksista saatujen yhdistettyjen tietojen perusteella sydämen vajaatoimintaa raportoitiin määrällisesti useammin potilailla, jotka saivat ramusirumabia yhdistelmänä erilaisten solunsalpaajahoitojen tai erlotinibin kanssa verrattuna kemoterapiaan tai erlotinibiin yksinään. Tätä lisääntynyttä ilmaantuvuutta ei havaittu ramusirumabia saaneilla potilailla verrattuna lumelääkkeeseen kliinisissä monoterapiatutkimuksissa. Markkinoille tulon jälkeen sydämen vajaatoimintaa todettiin ramusirumabilla hoidetuilla potilailla, useimmiten yhdessä paklitakselin kanssa. Potilaita tulee seurata sydämen vajaatoiminnan kliinisten merkkien ja oireiden varalta hoidon aikana, ja hoidon keskeyttämistä tulee harkita, jos sydämen vajaatoiminnan kliinisiä merkkejä ja oireita ilmaantuu. Katso kohta Haittavaikutukset.
Fisteli
Potilailla saattaa olla suurentunut fisteliriski Cyramza-hoidon aikana. Ramusirumabihoito on lopetettava, jos potilaalle kehittyy fisteli (ks. kohta Annostus ja antotapa).
Proteinuria
Proteinurian esiintyvyyden ilmoitettiin kasvaneen ramusirumabia saaneilla potilailla lumelääkettä saaneisiin potilaisiin verrattuna. Potilaita on seurattava ramusirumabihoidon aikana proteinurian kehittymisen tai pahenemisen varalta. Jos virtsan proteiinipitoisuus on liuskatestissä > 2+, potilaalta on kerättävä vuorokausivirtsa. Ramusirumabihoito on keskeytettävä väliaikaisesti, jos virtsan proteiinipitoisuus on ≥ 2 g/24 h. Kun virtsan proteiinipitoisuus on palautunut tasolle < 2 g/24 h, hoito aloitetaan uudestaan pienemmällä annoksella. Annoksen pienentäminen toistamiseen on suositeltavaa, jos virtsan proteiinipitoisuus palautuu tasolle ≥ 2 g/24 h. Ramusirumabihoito on lopetettava pysyvästi, jos virtsan proteiinipitoisuus on > 3 g/24 h tai jos potilaalle kehittyy nefroottinen oireyhtymä (ks. kohta Annostus ja antotapa).
Stomatiitti
Stomatiitin esiintyvyyden ilmoitettiin kasvaneen ramusirumabia ja kemoterapiaa saaneilla potilailla verrattuna lumelääkettä ja kemoterapiaa saaneisiin potilaisiin. Oireenmukainen hoito tulee aloittaa viipymättä, jos potilaalla ilmenee stomatiittia.
Munuaisten vajaatoiminta
Ramusirumabihoitoa koskevia turvallisuustietoja on saatavilla vaikeaa munuaisten vajaatoimintaa sairastavista potilaista rajallisesti (kreatiniinipuhdistuma 15-29 ml/min) (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Iäkkäät ei-pienisoluista keuhkosyöpää sairastavat potilaat
Potilaiden, jotka saivat ramusirumabia ja dosetakselia edenneen ei-pienisoluisen keuhkosyövän hoitoon ja joiden tauti oli edennyt aiemman platinapohjaisen solunsalpaajahoidon jälkeen, korkeamman iän havaittiin viittaavan heikompaan tehoon (ks. kohta Farmakodynamiikka). Korkeampaan ikään, toimintakykyluokkaan ja kemoterapian todennäköiseen siedettävyyteen liittyvät komorbiditeetit tulee siksi arvioida huolellisesti ennen hoidon aloittamista iäkkäillä potilailla (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Kun ramusirumabia käytettiin yhdessä erlotinibin kanssa ensilinjan hoitona ei-pienisoluisessa keuhkosyövässä, jossa kasvaimessa oli aktivoiva EGFR-mutaatio, vähintään 70-vuotiailla potilailla oli suurempi asteen ≥ 3 haittavaikutusten sekä kaikkien asteiden vakavien haittavaikutusten ilmaantuvuus verrattuna alle 70-vuotiaisiin potilaisiin.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 10 ml injektiopullo, eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää noin 85 mg natriumia per 50 ml injektiopullo. Tämä vastaa noin 4 %:a WHO:n suosittelemasta natriumin 2 g:n enimmäisvuorokausiannoksesta aikuiselle.
Polysorbaatti
Tämä lääkevalmiste sisältää noin 1 mg polysorbaatti 80:tä per 10 ml injektiopullo ja 5 mg polysorbaatti 80:tä per 50 ml injektiopullo.
Yhteisvaikutukset
Ramusirumabin ja paklitakselin välillä ei havaittu yhteisvaikutuksia. Ramusirumabin samanaikainen anto ei vaikuttanut paklitakselin farmakokinetiikkaan eikä paklitakselin samanaikainen anto ramusirumabin farmakokinetiikkaan. Ramusirumabin samanaikainen anto ei vaikuttanut irinotekaanin ja sen aktiivisen metabolitiin, SN-38:n, farmakokinetiikkaan. Ramusirumabin samanaikainen anto ei vaikuttanut dosetakselin eikä erlotinibin farmakokinetiikkaan.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Raskauden ehkäisy
Naisia, jotka voivat tulla raskaaksi, on kehotettava välttämään raskautta Cyramza-hoidon aikana, ja heille on kerrottava raskauteen ja sikiöön mahdollisesti kohdistuvista riskeistä. Hedelmällisessä iässä olevien naisten on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 3 kuukauden ajan viimeisen ramusirumabiannoksen jälkeen.
Raskaus
Ramusirumabin käytöstä raskaana olevilla naisilla ei ole olemassa tietoja. Riittäviä eläinkokeita lisääntymistoksisuuden selvittämiseksi ei ole tehty (ks. kohta Prekliiniset tiedot turvallisuudesta). Angiogeneesi on välttämätöntä raskauden jatkumisen ja sikiön kehityksen kannalta, joten angiogeneesin estyminen ramusirumabin annon jälkeen saattaa vaikuttaa haitallisesti raskauteen ja sikiöön. Cyramzaa saa käyttää raskauden aikana vain, jos hoidon mahdolliset hyödyt äidille ylittävät siihen mahdollisesti liittyvät riskit. Jos potilas tulee raskaaksi ramusirumabihoidon aikana, hänelle on kerrottava raskauden jatkumiseen ja sikiöön mahdollisesti kohdistuvasta riskistä. Cyramzan käyttö ei ole suositeltavaa raskauden aikana eikä naisilla, jotka voivat saada lapsia mutta eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyykö ramusirumabi ihmisen rintamaitoon. Erittyminen maitoon ja imeytyminen suun kautta on todennäköisesti vähäistä. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea, joten imetys on lopetettava Cyramza-hoidon ajaksi ja vähintään 3 kuukaudeksi viimeisen annoksen jälkeen.
Hedelmällisyys
Ramusirumabin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Eläintutkimusten perusteella on todennäköistä, että naisten hedelmällisyys heikkenee ramusirumabihoidon aikana (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Cyramzalla ei ole haitallista vaikutusta ajokykyyn eikä koneiden käyttökykyyn. Jos potilaalla on oireita, jotka vaikuttavat keskittymis- ja reaktiokykyyn, on suositeltavaa, että potilas ei aja eikä käytä koneita ennen kuin vaikutus häviää.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Vakavimpia ramusirumabihoitoon (monoterapia tai yhdistelmähoito solunsalpaajan kanssa) liittyviä haittavaikutuksia olivat:
- Maha-suolikanavan perforaatio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
- Vaikea maha-suolikanavan verenvuoto (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
- Tromboembolinen valtimotapahtuma (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
- Posteriorinen reversiibeli enkefalopatiaoireyhtymä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Yleisimmät haittavaikutukset ramusirumabihoitoa monoterapiana saaneilla potilailla olivat ääreisosien turvotus, hypertensio, ripuli, vatsakipu, päänsärky, proteinuria ja trombosytopenia.
Yleisimmät haittavaikutukset ramusirumabihoitoa yhdessä solunsalpaajahoidon kanssa saaneilla potilailla olivat uupumus/voimattomuus, neutropenia, ripuli, nenäverenvuoto ja stomatiitti.
Yleisimmät haittavaikutukset ramusirumabihoitoa yhdessä erlotinibin kanssa saaneilla potilailla olivat infektiot, ripuli, hypertensio, stomatiitti, proteinuria, hiustenlähtö ja nenäverenvuoto.
Haittavaikutustaulukko
Taulukoissa 6 ja 7 luetellaan ramusirumabin käyttöön liittyneet haittavaikutukset vaiheen III lumekontrolloiduissa kliinisissä tutkimuksissa, joissa ramusirumabia käytettiin joko monoterapiana mahasyövän ja maksasolukarsinooman hoitoon tai yhdessä eri solunsalpaajahoitojen tai erlotinibin kanssa mahasyövän, metastasoituneen kolorektaalisyövän ja ei-pienisoluisen keuhkosyövän hoitoon. Haittavaikutukset on lueteltu alla MedDRA-elinjärjestelmäluokan mukaan. Haittavaikutukset on luokiteltu seuraavan yleisyysluokituksen mukaan:
Hyvin yleiset (≥ 1/10)
Yleiset (≥ 1/100, < 1/10)
Melko harvinaiset (≥ 1/1000, < 1/100)
Harvinaiset (≥ 1/10000, < 1/1000)
Hyvin harvinaiset (< 1/10000)
Tuntematon (saatavissa oleva tieto ei riitä arvointiin)
Kussakin yleisyysluokassa haittavaikutukset on esitetty vakavuusasteen mukaan alenevassa järjestyksessä.
Taulukko 6: Ilmoitetut haittavaikutukset potilailla, jotka saivat ramusirumabia monoterapiana vaiheen 3 kliinisissä tutkimuksissa (REGARD-, REACH-2- ja REACH-tutkimusten potilaat, joilla alfafetoproteiinipitoisuus oli ≥400ng/ml)
| Elinjärjestelmä (MedDRA) | Hyvin yleinen | Yleinen | Melko harvinainen |
| Veri ja imukudos | Trombosytopeniaa | Neutropeniaa | |
| Aineenvaihdunta ja ravitsemus | Hypokalemiaa,b Hyponatremiaa Hypoalbuminemiaa | ||
| Hermosto | Päänsärky | Hepaattinen enkefalopatiac | |
| Verisuonisto | Hypertensioa,d | Tromboemboliset valtimotapahtumata | |
| Hengityselimet, rintakehä ja välikarsina | Nenäverenvuoto | ||
| Ruoansulatuselimistö | Vatsakipua,e Ripuli | Suolitukosa | Ruoansulatuskanavan perforaatioa |
| Iho ja ihonalainen kudos | Ihottumaa | ||
| Munuaiset ja virtsatiet | Proteinuriaa,f | ||
| Yleisoireet ja antopaikassa todettavat haitat | Ääreisosien turvotus | Infuusioreaktiota |
a Termit kuvaavat tapahtumaryhmiä ja pikemminkin lääketieteellistä käsitettä kuin yksittäistä tapahtumaa tai haittavaikutustermiä.
b Mukana hypokalemia ja pienentynyt veren kaliumpitoisuus.
c Perustuu REACH-2- ja REACH-tutkimuksiin (ramusirumabi monoterapiana maksasolukarsinooman
hoidossa). Mukana hepaattinen enkefalopatia ja maksakooma.
d Mukana verenpaineen nousu ja hypertensio.
e Mukana vatsakipu, alavatsakipu, ylävatsakipu ja maksan kipu.
f Mukana yksi nefroottinen syndrooma -tapaus
Taulukko 7: Ilmoitetut haittavaikutukset potilailla, jotka saivat ramusirumabia yhdessä solunsalpaajahoidon tai erlotinibin kanssa vaiheen 3 kliinisissä tutkimuksissa (RAINBOW, REVEL, RAISE ja RELAY)
| Elinjärjestelmä (MedDRA) | Hyvin yleinen | Yleinen | Melko harvinainen |
| Infektiot | Infektiotj,k | Sepsisa,b | |
| Veri ja imukudos | Neutropeniaa Leukopeniaa,c Trombosytopeniaa Anemiaj | Kuumeinen neutropeniad | |
| Aineenvaihdunta ja ravitsemus | Hypoalbuminemiaa Hyponatremiaa | ||
| Hermosto | Päänsärkyj | ||
| Sydän | Sydämen vajaatoiminta | ||
| Verisuonisto | Hypertensioa,e | Valtimoiden tromboemboliset tapahtumata | |
| Hengityselimet, rintakehä ja välikarsina | Nenäverenvuoto | Keuhkoverenvuotoj,l | |
| Ruoansulatuselimistö | Suutulehdus Ripuli | Ruoansulatuskanavan verenvuototapahtumata,f Ruoansulatuskanavan perforaatioa Ienten verenvuotoj | |
| Iho ja ihonalainen kudos | Hiustenlähtöj | Palmoplantaarinen erytrodysestesia-oireyhtymäg | |
| Munuaiset ja virtsatiet | Proteinuriaa,h | ||
| Yleisoireet ja antopaikassa todettavat haitat | Uupumusa,i Mukosiittid Ääreisosien turvotus |
a Termit kuvaavat tapahtumaryhmiä ja pikemminkin lääketieteellistä käsitettä kuin yksittäistä tapahtumaa tai haittavaikutustermiä.
b Perustuu RAINBOW-tutkimukseen (ramusirumabi ja paklitakseli).
c Perustuu RAINBOW-tutkimukseen (ramusirumabi ja paklitakseli). Mukana leukopenia ja valkosolumäärän pieneneminen.
d Perustuu REVEL-tutkimukseen (ramusirumabi ja dosetakseli).
e Mukana verenpaineen nousu, hypertensio ja hypertensiivinen kardiomyopatia.
f Perustuu RAINBOW-tutkimukseen (ramusirumabi ja paklitakseli), REVEL-tutkimukseen (ramusirumabi ja dosetakseli) ja RAISE-tutkimukseen (ramusirumabi ja FOLFIRI).Mukana peräaukon verenvuoto, veriripuli, mahalaukun verenvuoto, ruoansulatuskanavan verenvuoto, verioksennus, veriuloste, peräpukamaverenvuoto, Mallory–Weissin oireyhtymä, meleena, ruokatorven verenvuoto, peräsuolen verenvuoto ja ruoansulatuskanavan yläosan verenvuoto.
g Perustuu RAISE-tutkimukseen (ramusirumabi ja FOLFIRI).
h Mukana nefroottisen oireyhtymän tapauksia.
i Perustuu RAINBOW-tutkimukseen (ramusirumabi ja paklitakseli) ja REVEL-tutkimukseen (ramusirumabi ja dosetakseli). Mukana uupumus ja voimattomuus.
j Perustuu RELAY-tutkimukseen (ramusirumabi ja erlotinibi).
k Infektiot-termi kattaa kaikki Infektiot-elinjärjestelmäluokkaan kuuluvat haittavaikutustermit. Yleisimpiä (≥ 1 %) asteen ≥ 3 infektioita ovat keuhkokuume, selluliitti, kynnenvierustulehdus, ihon infektio ja virtsatieinfektio.
l Mukana veriyskökset, verenvuoto kurkunpäästä, hemothorax (kuolemaan johtava tapahtuma on todettu) ja keuhkoverenvuoto.
Kliinisissä tutkimuksissa angiogeneesiä estävään hoitoon liittyneitä kliinisesti merkittäviä vaikutuksia (mukaan lukien asteen ≥ 3 vaikutukset) ramusirumabia saaneilla potilailla olivat maha-suolikanavan perforaatiot, infuusioreaktiot ja proteinuria (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kolorektaalisyöpä
Ramusirumabi yhdessä FOLFIRI-yhdistelmän kanssa
RAISE-tutkimuksessa ramusirumabin ja FOLFIRIn yhdistelmällä hoidetuilla metastasoitunutta kolorektaalisyöpää sairastavilla potilailla yleisin (≥ 1 %) haittavaikutus, joka johti ramusirumabihoidon keskeyttämiseen, oli proteinuria (1,5 %). Yleisimmät haittavaikutukset (≥ 1 %), jotka johtivat yhden tai useamman FOLFIRI-komponentin keskeyttämiseen, olivat neutropenia (12,5 %), trombosytopenia (4,2 %), ripuli (2,3 %) ja stomatiitti (2,3 %). FOLFIRI-komponenteista yleisimmin keskeytettiin 5-FU bolus.
Haittavaikutukset muista lähteistä
Taulukko 8: Ramusirumabiin yhdistetyt haittavaikutukset kliinisissä tutkimuksissa ja markkinoille tulon jälkeen tehdyissä ilmoituksissa
| Elinjärjestelmä (MedDRA) | Yleiset | Melko harvinaiset | Harvinaiset | Tuntematon |
| Kasvaimet, hyvänlaatuinen, pahanlaatuinen ja täsmentämätön (mukaan lukien kystat ja polyypit) | Hemangiooma | |||
| Veri ja imukudos | Tromboottinen mikroangiopatia | |||
| Umpieritys | Kilpirauhasen vajaatoiminta | |||
| Hermosto | Posteriorinen reversiibeli enkefalopatia-oireyhtymä | |||
| Sydän | Sydämen vajaatoimintaa | |||
| Verisuonisto | Aneurysmat ja valtimon dissekaatiot | |||
| Hengityselimet, rintakehä ja välikarsina | Dysfonia |
a Markkinoille tulon jälkeen sydämen vajaatoimintaa on havaittu ramusirumabilla enimmäkseen yhdessä paklitakselin kanssa. Katso kohta Varoitukset ja käyttöön liittyvät varotoimet.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 Fimea
Pediatriset potilaat
Uusia turvallisuuteen liittyviä ongelmia ei havaittu tutkimuksessa I4T-MC-JVDA rajallisella määrällä pediatrisia potilaita, jotka saivat ramusirumabi-valmistetta monoterapiana (ks. kohta Farmakodynamiikka). Yhdellä potilaalla kyseisessä tutkimuksessa esiintyi distaalisen reisiluun kasvulevyn progressiivista levenemistä. Tämän löydöksen vaikutusta kasvuun ei tunneta. Uusia turvallisuuteen liittyviä ongelmia ei ilmoitettu tutkimuksissa J1S-MC-JV01 ja J1S-MC-JV02 rajallisella määrällä pediatrisia potilaita, jotka saivat ramusirumabi-valmistetta yhdistelmähoitona (ks. kohta Farmakodynamiikka).
Yliannostus
Yliannostuksesta ihmisillä ei ole tietoa. Cyramzaa on annettu vaiheen 1 tutkimuksessa enintään 10 mg/kg kahden viikon välein. Siedettyä enimmäisannosta ei saavutettu. Yliannostustapauksessa on käytettävä elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: solunsalpaajat, VEGF/VEGFR (Vaskulaarisen endoteelikasvutekijän/endoteelikasvutekijäreseptorin) -estäjät, ATC-koodi: L01FG02.
Vaikutusmekanismi
Verisuonten endoteelikasvutekijän (VEGF) reseptori 2 on VEGF:n indusoiman angiogeneesin keskeinen välittäjä. Ramusirumabi on kohdereseptorin humaani vasta-aine, joka sitoo spesifisesti VEGF-reseptori 2:n ja estää VEGF-A:n, VEGF-C:n ja VEGF-D:n sitoutumisen. Tämän seurauksena ramusirumabi estää VEGF-reseptori 2:n ja siitä alavirtaan olevien signalointikomponenttien ligandin stimuloimaa aktivaatiota, mukaan lukien p44/p42-mitogeeniaktivoidut proteiinikinaasit, neutralisoiva ligandin indusoima proliferaatio ja ihmisen endoteelisolujen migraatio.
Kliininen teho ja turvallisuus
Mahasyöpä
RAINBOW
Maailmanlaajuisessa, satunnaistetussa, kaksoissokkoutetussa RAINBOW-tutkimuksessa Cyramzan ja paklitakselin yhdistelmää verrattiin lumelääkkeen ja paklitakselin yhdistelmään. Tutkimus tehtiin 665 potilaalla, joilla oli aiemman platinaa ja fluoropyrimidiiniä ja mahdollisesti myös antrasykliiniä sisältävän solunsalpaajahoidon jälkeen paikallisesti uusiutunut ja leikkauskelvoton tai metastasoitunut mahasyöpä (mukaan lukien ruokatorvi-mahalaukkurajan adenokarsinooma). Ensisijainen päätetapahtuma oli kokonaiselossaoloaika ja toissijaisia päätetapahtumia olivat etenemisvapaa elossaoloaika ja kokonaisvasteprosentti. Vaatimuksena oli, että potilaan tauti oli edennyt ensilinjan hoidon aikana tai 4 kk kuluessa viimeisestä ensilinjan hoidon annoksesta ja ECOG-toimintakykyluokka oli 0–1. Potilaat satunnaistettiin suhteessa 1:1 saamaan Cyramzaa ja paklitakselia (n = 330) tai lumelääkettä ja paklitakselia (n = 335). Satunnaistaminen stratifioitiin maantieteellisen alueen, ensilinjan hoidon aloittamisesta taudin etenemiseen kuluneen ajan (< 6 kk vs. ≥ 6 kk) ja taudin mitattavuuden mukaan. Cyramzaa (8 mg/kg) tai lumelääkettä annettiin infuusiona laskimoon 2 viikon välein 28 päivän syklin aikana (päivinä 1 ja 15). Paklitakselia (80 mg/m2) annettiin infuusiona laskimoon kunkin 28 päivän syklin päivinä 1, 8 ja 15.
Valtaosa (75 %) tutkimuksessa satunnaistetuista potilaista oli saanut aiempaa platinan ja fluoropyrimidiinin yhdistelmähoitoa ilman antrasykliiniä. Loput (25 %) olivat saaneet aiemmin platinan ja fluoropyrimidiinin yhdistelmähoitoa yhdessä antrasykliinin kanssa. Tauti eteni ensilinjan hoidon aikana kahdella potilaalla kolmesta (66,8 %). Potilaiden lähtötilanteen demografiset tiedot ja taudin ominaisuudet olivat hoitoryhmissä yleisesti ottaen samankaltaiset: iän mediaani oli 61 vuotta, 71 % potilaista oli miehiä, 61 % oli valkoihoisia ja 35 % aasialaisia, 39 %:lla potilaista ECOG-toimintakykyluokka oli 0 ja 61 %:lla 1, 81 %:lla potilaista tauti oli mitattavissa, 79 %:lla potilaista oli mahasyöpä ja 21 %:lla ruokatorvi-mahalaukkurajan adenokarsinooma. Valtaosalla potilaista (76 %) tauti oli edennyt 6 kk kuluessa ensilinjan hoidon aloittamisesta. Cyramzan ja paklitakselin yhdistelmää saaneilla potilailla hoidon keston mediaani oli 19 viikkoa ja lumelääkettä ja paklitakselia saaneilla potilailla 12 viikkoa. Cyramzan suhteellisen annosintensiteetin mediaani oli 98,6 % ja lumeen 99,6 %. Paklitakselin suhteellisen annosintensiteetin mediaani oli 87,7 % Cyramza + paklitakseli -ryhmässä ja 93,2 % lumelääke + paklitakseli -ryhmässä. Haittatapahtumien takia hoidon lopettaneiden potilaiden prosenttiosuus oli samaa luokkaa: 12 % Cyramzaa ja paklitakselia saaneista potilaista ja 11 % lumelääkettä ja paklitakselia saaneista. Systeemistä syöpähoitoa annettiin lopettamisen jälkeen 47,9 %:lle Cyramzaa ja paklitakselia saaneista potilaista ja 46,0 %:lle lumelääkettä ja paklitakselia saaneista.
Kokonaiselossaoloaika parani tilastollisesti merkitsevästi Cyramzan ja paklitakselin yhdistelmää saaneilla potilailla lumelääkettä ja paklitakselia saaneisiin potilaisiin verrattuna (HR 0,807; 95 % lv: 0,678–0,962; p = 0,0169). Elossaolon mediaani piteni 2,3 kk enemmän Cyramza + paklitakseli -ryhmässä: 9,63 kk Cyramza + paklitakseli -ryhmässä ja 7,36 kk lumelääke + paklitakseli -ryhmässä. Etenemisvapaa elossaolo parani tilastollisesti merkitsevästi Cyramzan ja paklitakselin yhdistelmää saaneilla potilailla lumelääkettä ja paklitakselia saaneisiin potilaisiin verrattuna (HR 0,635; 95 % lv: 0,536–0,752; p < 0,0001). Etenemisvapaan elossaolon mediaani piteni 1,5 kk enemmän Cyramza + paklitakseli -ryhmässä: 4,4 kk Cyramza + paklitakseli -ryhmässä ja 2,9 kk lume + paklitakseli -ryhmässä. Objektiivinen vasteprosentti (täydellinen vaste + osittainen vaste) parani tilastollisesti merkitsevästi Cyramzan ja paklitakselin yhdistelmää saaneilla potilailla lumetta ja paklitakselia saaneisiin potilaisiin verrattuna (ristitulosuhde 2,140; 95 % lv: 1,499–3,160; p = 0,0001). Objektiivinen vasteprosentti oli Cyramza + paklitakseli -ryhmässä 27,9 % ja lumelääke + paklitakseli -ryhmässä 16,1 %. Kokonaiselossaoloaika ja etenemisvapaa elossaoloaika paranivat johdonmukaisesti ennalta määritellyissä ikään, sukupuoleen ja etniseen taustaan perustuvissa alaryhmissä ja useimmissa muissa ennalta määritellyissä alaryhmissä. Tehotulokset esitetään taulukossa 9.
Taulukko 9: Tehotietojen yhteenveto – hoitoaikomuspopulaatio (ITT-populaatio)
| Cyramza + paklitakseli N = 330 | Lumelääke + paklitakseli N = 335 | ||
| Kokonaiselossaoloaika (kk) | |||
| Mediaani (95 % lv) | 9,6 (8,5; 10,8) | 7,4 (6,3; 8,4) | |
| Riskisuhde (95 % lv) | 0,807 (0,678; 0,962) | ||
| Stratifioitu log-rank-testin p-arvo | 0,0169 | ||
| Etenemisvapaa elossaoloaika (kk) | |||
| Mediaani (95 % lv) | 4,4 (4,2; 5,3) | 2,9 (2,8; 3,0) | |
| Riskisuhde (95 % lv) | 0,635 (0,536; 0,752) | ||
| Stratifioitu log-rank-testin p-arvo | < 0,0001 | ||
| Objektiivinen vasteprosentti (täydellinen vaste + osittainen vaste) | |||
| Prosenttiosuus (95 % lv) | 27,9 (23,3; 33,0) | 16,1 (12,6; 20,4) | |
| Ristitulosuhde | 2,140 (1,449; 3,160) | ||
| CMH-testin stratifioitu p-arvo | 0,0001 | ||
Lyhenteet: lv = luottamusväli, CMH = Cochran–Mantel–Haenszel
Kuva 1: Kokonaiselossaoloajan Kaplan–Meier -käyrät, Cyramza + paklitakseli vs. lumelääke + paklitakseli, RAINBOW-tutkimus
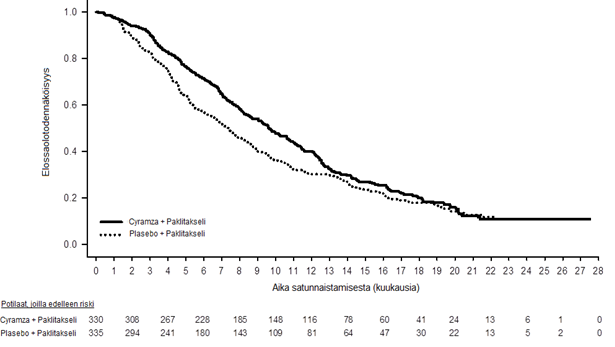
Kuva 2: Etenemisvapaan elossaoloajan Kaplan–Meier -käyrät, Cyramza + paklitakseli vs. lumelääke + paklitakseli, RAINBOW-tutkimus
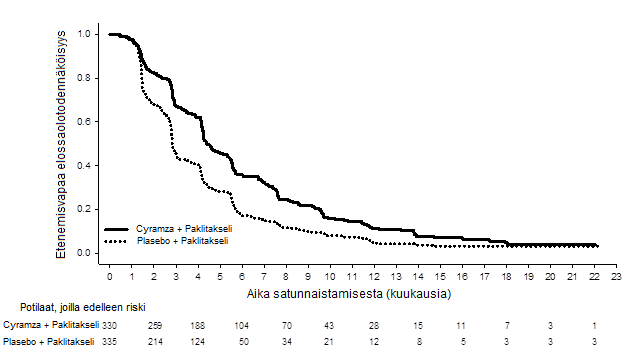
REGARD
Monikansallisessa, satunnaistetussa, kaksoissokkoutetussa REGARD-tutkimuksessa Cyramzan ja parhaan mahdollisen tukihoidon yhdistelmää verrattiin lumelääkkeen ja parhaan mahdollisen tukihoidon yhdistelmään. Tutkimus tehtiin 355 potilaalla, joilla oli aiemman platina- tai fluoropyrimidiinipohjaisen solunsalpaajahoidon jälkeen paikallisesti uusiutunut ja leikkauskelvoton tai metastasoitunut mahasyöpä (mukaan lukien ruokatorvi-mahalaukkurajan adenokarsinooma). Ensisijainen päätetapahtuma oli kokonaiselossaoloaika ja toissijaisia päätetapahtumia oli etenemisvapaa elossaoloaika. Vaatimuksena oli, että potilaan tauti oli edennyt 4 kk kuluessa metastasoituneen taudin viimeisestä ensilinjan hoidon annoksesta, liitännäishoidon aikana tai 6 kk kuluessa viimeisestä liitännäishoitoannoksesta ja potilaan ECOG-toimintakykyluokka oli 0–1. Potilaiden tutkimukseenottokriteerinä oli, että kokonaisbilirubiini oli ≤ 1,5 mg/dl ja ASAT ja ALAT olivat ≤ 3 x ULN, tai jos potilaalla oli maksametastaaseja, ≤ 5 x ULN.
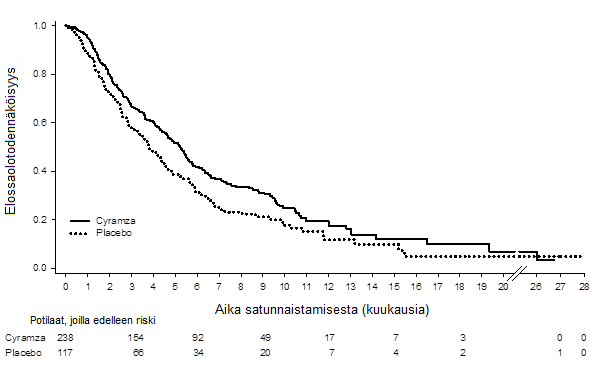
Potilaat satunnaistettiin suhteessa 2:1 saamaan infuusiona laskimoon 8 mg/kg Cyramzaa (n = 238) tai lumelääkettä (n = 117) 2 viikon välein. Satunnaistaminen stratifioitiin edeltävien 3 kuukauden aikaisen painonlaskun (≥ 10 % vs. < 10 %), maantieteellisen alueen ja primaarikasvaimen sijainnin (mahalaukku vs. ruokatorvi-mahalaukkuraja) mukaan. Lähtötilanteen demografiset tiedot ja taudin ominaisuudet olivat tasapainotettu. ECOG-toimintakykyluokka oli 72 %:lla potilaista 1. REGARD-tutkimukseen ei otettu potilaita, joilla oli Child–Pugh -luokan B tai C maksakirroosi. Cyramza-hoitoa saaneista potilaista 11 % ja lumelääkettä saaneista 6 % lopetti hoidon haittatapahtumien takia. Kokonaiselossaoloaika parani tilastollisesti merkitsevästi Cyramzaa saaneilla potilailla lumelääkettä saaneisiin potilaisiin verrattuna (riskisuhde [HR] 0,776; 95 % lv: 0,603–0,998; p = 0,0473), eli kuoleman riski pieneni 22 % ja elossaoloajan mediaani piteni lumehoidon 3,8 kuukaudesta Cyramza-hoidon 5,2 kuukauteen. Etenemisvapaa elossaoloaika parani tilastollisesti merkitsevästi Cyramzaa saaneilla potilailla lumelääkettä saaneisiin potilaisiin verrattuna (HR 0,483; 95 % lv: 0,376–0,620; p < 0,0001), eli taudin etenemisen tai kuoleman riski pieneni 52 % ja etenemisvapaan elossaoloajan mediaani piteni lumehoidon 1,3 kuukaudesta Cyramza-hoidon 2,1 kuukauteen. Tehotulokset esitetään taulukossa 10.
Taulukko 10: Tehotietojen yhteenveto – ITT-hoitoaikomuspopulaatio
| Cyramza N = 238 | Lumelääke N = 117 | ||
| Kokonaiselossaoloaika (kk) | |||
| Mediaani (95 % lv) | 5,2 (4,4; 5,7) | 3,8 (2,8; 4,7) | |
| Riskisuhde (95 % lv) | 0,776 (0,603; 0,998) | ||
| Stratifioitu log-rank-testin p-arvo | 0,0473 | ||
| Etenemisvapaa elossaoloaika (kk) | |||
| Mediaani (95 % lv) | 2,1 (1,5; 2,7) | 1,3 (1,3; 1,4) | |
| Riskisuhde (95 % lv) | 0,483 (0,376; 0,620) | ||
| Stratifioitu log-rank-testin p-arvo | < 0,0001 | ||
| 12 viikon etenemisvapaa elossaoloaika (%) (95 % lv) | 40,1 (33,6; 46,4) | 15,8 (9,7; 23,3) | |
Lyhenteet: lv = luottamusväli
Kuva 3: Kokonaiselossaoloajan Kaplan–Meier -käyrät, Cyramza vs. lumelääke, REGARD-tutkimus
Perustuen REGARD-tutkimuksen rajalliseen määrään HER2-positiivisia, mahasyöpää tai ruokatorvi-mahalaukkurajan adenokarsinoomaa sairastavia potilaita ja aiemmin trastutsumabilla hoidettuja potilaita (RAINBOW-tutkimuksessa), voidaan pitää epätodennäköisenä, että Cyramza vaikuttaisi haitallisesti tai ei tehoaisi potilaisiin, joilla on HER2-positiivinen mahasyöpä. Post hoc, stratifioimaton alaryhmä-analyysi RAINBOW-tutkimuksen potilaista, joita oli aiemmin hoidettu trastutsumabilla (n = 39), osoitti potilaille mahdollista etua elossaoloajassa (HR 0,679, 95 % CI 0,327, 1,419) ja etenemisvapaassa elossaoloajassa (PFS)(HR 0,399, 95 % CI 0,194, 0,822).
Kolorektaalisyöpä
RAISE
RAISE oli maailmanlaajuinen, satunnaistettu, kaksoissokkoutettu tutkimus, jossa Cyramzan ja FOLFIRI-hoidon yhdistelmää verrattiin lumelääkkeen ja FOLFIRI-hoidon yhdistelmään etäpesäkkeistä kolorektaalisyöpää sairastaneilla potilailla, joilla tauti oli edennyt ensilinjan bevasitsumabi-, oksaliplatiini- ja fluoropyrimidiinihoidon aikana tai sen jälkeen. Vaatimuksena oli, että potilaiden ECOG-toimintakykyluokka oli 0 tai 1 ja että tauti oli edennyt 6 kuukauden kuluessa ensilinjan hoidon viimeisestä annoksesta. Potilaiden maksa-, munuais- ja koagulaatiotoiminnan oli oltava riittävä. Tutkimuksesta suljettiin pois potilaat, joilla oli anamneesissa hallitsematon perinnöllinen tai hankinnainen verenvuoto tai tromboottinen häiriö tai joilla oli ollut äskettäin vaikea (asteen ≥ 3) verenvuoto tai valtimotromboositapahtuma satunnaistamista edeltävien 12 kuukauden aikana. Tutkimuksesta suljettiin pois myös potilaat, joilla oli ollut valtimotromboositapahtuma, asteen 4 hypertensio, asteen 3 proteinuria, asteen 3–4 verenvuototapahtuma tai suolen perforaatio ensilinjan bevasitsumabihoidon aikana.
Yhteensä 1072 potilasta satunnaistettiin (1:1) saamaan joko Cyramzaa (n = 536) annoksena 8 mg/kg tai lumelääkettä (n = 536) yhdessä FOLFIRI-hoidon kanssa. Kaikki lääkevalmisteet annettiin laskimoon.
FOLFIRI-hoidossa annettiin 180 mg/m2 irinotekaania 90 minuutin aikana ja samanaikaisesti 400 mg/m2 foliinihappoa 120 minuutin aikana, minkä jälkeen 5-fluorourasiilia annettiin ensin boluksena 400 mg/m2 2–4 minuutin aikana ja sitten 2400 mg/m2 jatkuvana infuusiona 46–48 tunnin aikana. Hoitojaksot toistettiin molemmissa ryhmissä 2 viikon välein. Jos potilas lopetti yhden tai useamman hoitokomponentin haittatapahtuman takia, hän sai jatkaa hoitoa yhdellä tai useammalla muulla hoitokomponentilla, kunnes tauti eteni tai ilmeni sietämätöntä toksisuutta. Ensisijainen päätetapahtuma oli kokonaiselossaoloaika ja toissijaisia päätetapahtumia olivat mm. etenemisvapaa elossaoloaika, objektiivinen vasteprosentti ja elämänlaatu EORTC:n (European Organisation for Research and Treatment of Cancer) QLQ‑C30-kyselylomakkeella mitattuna. Satunnaistaminen stratifioitiin maantieteellisen alueen, kasvaimen KRAS-statuksen (mutantti tai villityyppi) ja ensilinjan hoidon aloittamisen jälkeen taudin etenemiseen kuluneen ajan mukaan (< 6 kk vs. ≥ 6 kk).
Hoitoryhmien hoitoaikomuspopulaatioiden demografiset ja lähtötilanteen tiedot olivat samanlaiset. Iän mediaani oli 62 vuotta, ja 40 % potilaista oli ≥ 65-vuotiaita, 57 % potilaista oli miehiä, 76 % oli valkoihoisia ja 20 % aasialaisia, 49 %:lla ECOG-toimintakykyluokka oli 0, 49 %:lla kasvaimessa oli KRAS-mutaatio ja 24 %:lla potilaista taudin etenemiseen kulunut aika ensilinjan hoidon aloittamisen jälkeen oli < 6 kuukautta. Systeemistä syöpähoitoa annettiin lopettamisen jälkeen 54 %:lle Cyramzaa ja FOLFIRI-hoitoa saaneista potilaista ja 56 %:lle lumetta ja FOLFIRI-hoitoa saaneista.
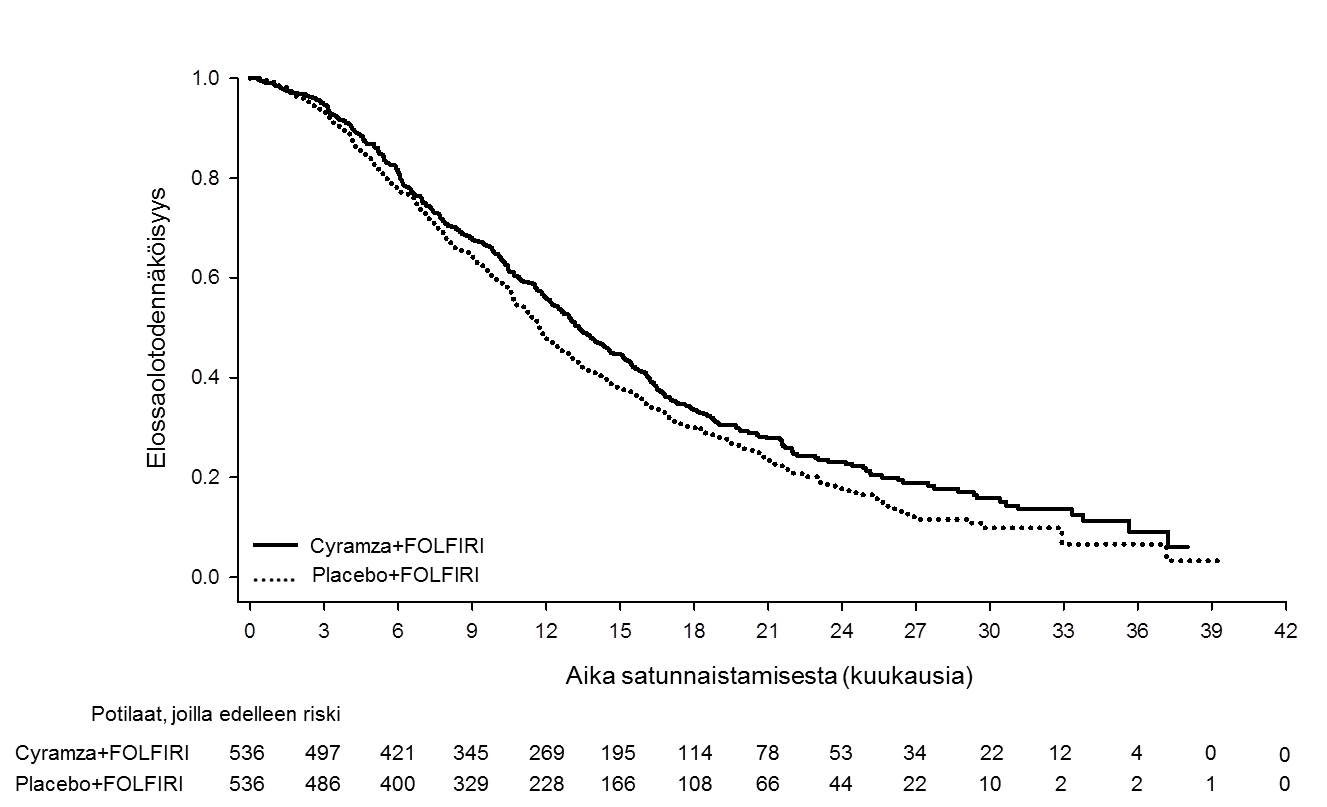
Kokonaiselossaoloaika parani tilastollisesti merkitsevästi Cyramzan ja FOLFIRI-hoidon yhdistelmää saaneilla potilailla verrattuna lumetta ja FOLFIRI-hoitoa saaneisiin (HR 0,844; 95 % lv: 0,730–0,976; p = 0,0219). Elossaoloajan mediaani piteni 1,6 kk enemmän Cyramza + FOLFIRI ‑ryhmässä: 13,3 kk Cyramza + FOLFIRI ‑ryhmässä ja 11,7 kk lume + FOLFIRI ‑ryhmässä. Etenemisvapaa elossaoloaika parani tilastollisesti merkitsevästi Cyramzan ja FOLFIRI-hoidon yhdistelmää saaneilla potilailla verrattuna lumetta ja FOLFIRI-hoitoa saaneisiin (HR 0,793; 95 % lv: 0,697–0,903; p = 0,0005). Etenemisvapaan elossaoloajan mediaani piteni 1,2 kk enemmän Cyramza + FOLFIRI ‑ryhmässä: 5,7 kk Cyramza + FOLFIRI ‑ryhmässä ja 4,5 kk lume + FOLFIRI ‑ryhmässä. Tehotulokset esitetään taulukossa 11 ja kuvissa 4 ja 5.
Kokonaiselossaoloajasta ja etenemisvapaasta elossaoloajasta tehtiin ennalta määritellyt analyysit stratifiointitekijöiden mukaan. Kokonaiselossaoloajan HR oli 0,82 (95 % lv: 0,67–1,0) potilailla, joilla kasvaimessa oli KRAS-villityyppi, ja 0,89 (95 % lv: 0,73–1,09) potilailla, joilla kasvaimessa oli KRAS-mutaatio. Kokonaiselossaoloajan HR oli 0,86 (95 % lv: 0,73–1,01) potilailla, joilla taudin etenemiseen kulunut aika ensilinjan hoidon aloittamisen jälkeen oli ≥ 6 kk, ja 0,86 (95 % lv: 0,64–1,13) potilailla, joilla taudin etenemiseen kulunut aika ensilinjan hoidon aloittamisen jälkeen oli < 6 kk. Molemmat ennalta määritellyt etenemisvapaan elossaoloajan ja kokonaiselossaoloajan alaryhmäanalyysit osoittivat, että Cyramza + FOLFIRI ‑hoidolla saavutettiin parempi hoitovaikutus kuin lume + FOLFIRI -hoidolla. Alaryhmien muodostusperusteet olivat ikä (< 65 ja ≥ 65 vuotta), sukupuoli, etninen tausta, ECOG-toimintakykyluokka (0 tai ≥ 1), etäpesäkkeitä sisältävien elinten lukumäärä, etäpesäkkeiden sijainti vain maksassa, primaarikasvaimen sijainti (paksusuoli tai peräsuoli) ja karsinoembryonaalisen antigeenin pitoisuus (< 200 mikrog/l, ≥ 200 mikrog/l). 32:ssa ennalta määritellyssä kokonaiselossaoloajan alaryhmäanalyysissä 33:sta HR oli < 1,0. Ainoa alaryhmä, jossa HR oli > 1, koski potilaita, joilla tauti oli edennyt < 3 kuukauden kuluessa ensilinjan bevasitsumabihoidon aloittamisesta (HR 1,02 [95 % lv: 0,68–1,55]). Tällä ainoalla alaryhmällä voidaan katsoa olevan aggressiivinen tauti, joka reagoi suhteellisen huonosti ensilinjan hoitoon. Molemmissa hoitoryhmissä potilailla, joilla oli neutropeniaa, oli pidempi kokonaiselossaoloajan mediaani kuin niillä, joilla ei ollut neutropeniaa. Kokonaiselossaoloajan mediaani potilailla, joilla oli minkä tahansa asteista neutropeniaa, oli suurempi ramusirumabiryhmässä (16,1 kk) kuin lumelääkeryhmässä (12,6 kk). Kokonaiselossaoloajan mediaani potilailla, joilla ei ollut neutropeniaa, oli 10,7 kk molemmissa ryhmissä.
Taulukko 11: Tehotietojen yhteenveto – ITT-populaatio
| Cyramza + FOLFIRI N = 536 | Lumelääke + FOLFIRI N = 536 | |
| Kokonaiselossaoloaika (kk) | ||
| Mediaani (95 % lv) | 13,3 (12,4; 14,5) | 11,7 (10,8; 12,7) |
| Riskisuhde (95 % lv) | 0,84 (0,73; 0,98) | |
| Stratifioitu log-rank-testin p-arvo | 0,022 | |
| Etenemisvapaa elossaoloaika (kk) | ||
| Mediaani (95 % lv) | 5,7 (5,5; 6,2) | 4,5 (4,2; 5,4) |
| Riskisuhde (95 % lv) | 0,79 (0,70; 0,90) | |
| Stratifioitu log-rank-testin p-arvo | < 0,001 | |
Lyhenteet: lv = luottamusväli
Kuva 4: Kokonaiselossaoloajan Kaplan–Meier -käyrät, Cyramza + FOLFIRI vs. lumelääke + FOLFIRI, RAISE-tutkimus
Kuva 5: Etenemisvapaan elossaoloajan Kaplan–Meier -käyrät, Cyramza + FOLFIRI vs. lumelääke + FOLFIRI, RAISE-tutkimus
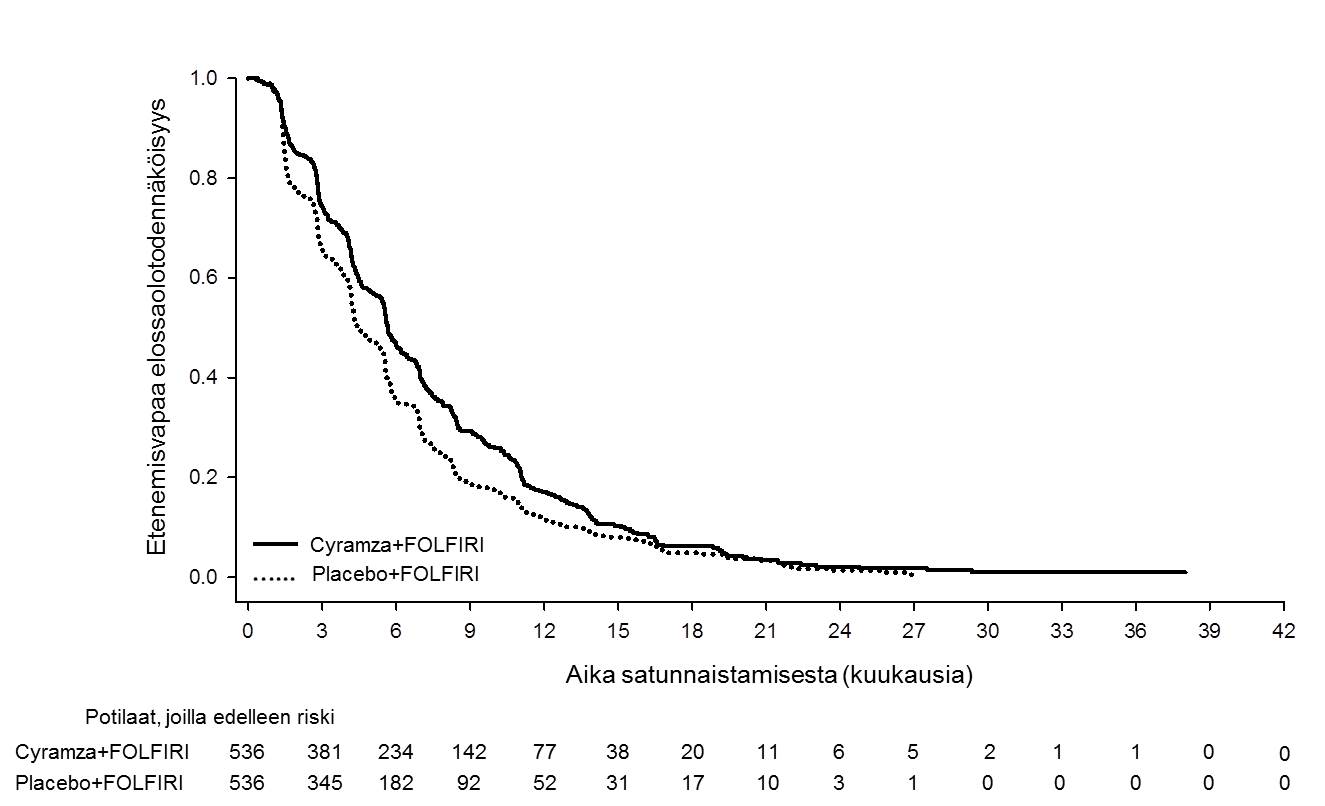
Objektiivinen vasteprosentti oli samaa luokkaa molemmissa hoitoryhmissä (13,4 % ramusirumabi + FOLFIRI ‑ryhmässä vs. 12,5 % lume + FOLFIRI ‑ryhmässä). Taudin hallintaprosentti (täydellinen vaste + osittainen vaste + stabiili tauti) oli numeerisesti suurempi ramusirumabi + FOLFIRI ‑ryhmän potilailla (74,1 %) kuin lume + FOLFIRI ‑ryhmässä (68,8 %). Ramusirumabi + FOLFIRI ‑hoitoryhmän potilaat ilmoittivat elämänlaadun heikentyneen ohimenevästi useimmilla EORTC QLQ‑C30 ‑kyselylomakkeen asteikoilla verrattuna lume + FOLFIRI ‑hoitoryhmän potilaisiin. Ryhmien välisiä eroja ilmoitettiin vähän ensimmäisen hoitokuukauden jälkeen.
Ei-pienisoluinen keuhkosyöpä
RELAY
RELAY oli maailmanlaajuinen, satunnaistettu, kaksoissokkoutettu vaiheen 3 tutkimus, jossa Cyramzan ja erlotinibin yhdistelmää verrattiin lumelääkkeen ja erlotinibin yhdistelmään. Tutkimukseen satunnaistettiin (suhteessa 1:1) 449 aiemmin hoitamatonta potilasta, joilla oli metastasoitunut ei-pienisoluinen keuhkosyöpä ja joilla oli tutkimukseenottohetkellä epidermaalisen kasvutekijäreseptorin (EGFR) eksonin 19 deleetio tai eksonin 21 (L858R) aktivoivia mutaatioita. Tutkimukseen soveltuvien potilaiden ECOG-toimintakykyluokka oli 0 tai 1. Tutkimuksesta suljettiin pois potilaat, joilla oli keskushermostometastaaseja tai tiedossa oleva T790M-EGFR-mutaatio lähtötilanteessa. Tutkimuksesta suljettiin pois myös potilaat, joilla oli suuri verenvuotoriski tai suuri kardiovaskulaaritapahtumien riski, mm. potilaat, joilla oli ollut jokin valtimotromboositapahtuma tutkimukseenottoa edeltävien 6 kuukauden aikana.
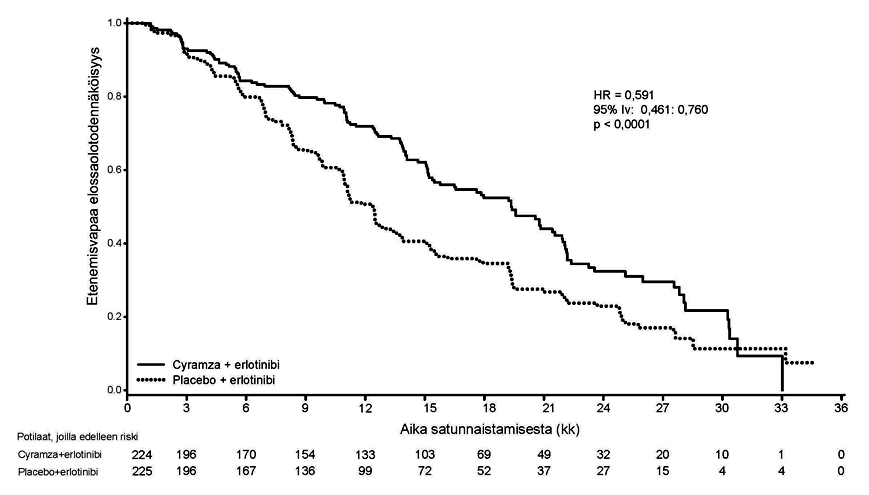
Demografiset tiedot ja lähtötilanteen tiedot olivat molemmissa ryhmissä samankaltaiset. Potilaista 77 % oli aasialaisia ja 22 % valkoihoisia. Cyramzaa ja erlotinibia saaneilla potilailla etenemisvapaa elossaoloaika (PFS) oli tilastollisesti merkitsevästi parempi kuin lumelääkettä ja erlotinibia saaneilla (taulukko 12). Tutkimuksessa havaittiin yhdenmukaiset tulokset eri alaryhmissä, joiden muodostamisperusteisiin kuuluivat eksonin 19 deleetiot ja eksonin 21 (L858R) substituutiot, ikä ja etninen tausta (HR valkoihoisilla 0,618; HR aasialaisilla 0,638), sekä tupakoivien ja koko elämänsä ajan tupakoimattomien tutkittavien alaryhmissä. Kokonaiselossaolotiedot eivät olleet valmiita ensisijaisen PFS-analyysin ajankohtana (maturiteetti 17,6 %). RELAY-tutkimuksen tehotulokset esitetään taulukossa 12 ja kuvassa 6.
Taulukko12: RELAY-tutkimuksen tehotietojen yhteenveto – ITT-populaatio
| Cyramza + erlotinibi N = 224 | Lumelääke + erlotinibi N = 225 | |
| Etenemisvapaa elossaoloaika (PFS) | ||
| Tapahtumia (%) | 122 (54,5) | 158 (70,2) |
| Mediaani, kk (95 % lv) | 19,4 (15,38; 21,55) | 12,4 (10,97; 13,50) |
| Riskisuhde (95 % lv) | 0,591 (0,461; 0,760) | |
| Stratifioitu log-rank-testin p-arvo | < 0,0001 | |
| Objektiivinen vasteprosentti (täydellinen vaste + osittainen vaste) | ||
| Prosenttiosuus (95 % lv) | 76 (70,8; 81,9) | 75 (69,0; 80,3) |
| Täydellinenvaste, n (%) | 3 (1,3) | 2 (0,9) |
| Osittainen vaste, n (%) | 168 (75,0) | 166 (73,8) |
| Vasteen kesto | N = 171 | N = 168 |
| Tapahtumia (%) | 101 (59,1) | 128 (76,2) |
| Mediaani, kk (95 % lv) | 18,0 (13,86; 19,78) | 11,1 (9,69: 12,29) |
| Riskisuhde (95 % lv) | 0,619 (0,477; 0,805) | |
| Stratifioimaton log-rank-testin p-arvo | 0,0003 | |
Lyhenteet: ES = ei saavutettu, lv = luottamusväli.
Päätetapahtumien kohdalla käytettiin alfan suojausta.
Kuva 6: Etenemisvapaan elossaoloajan Kaplan–Meier -käyrät, Cyramza + erlotinibi vs. lumelääke + erlotinibi, RELAY-tutkimus
Kokonaiselossaoloajan lopullinen analyysi, johon tutkimuksen tilastollinen voima ei riittänyt, toteutettiin 297 tapahtumalla (kypsyysaste 66,1 %). Stratifioitu kokonaiselossaoloajan riskisuhde oli 0,98 [95 %:n luottamusväli 0,78–1,24, p = 0,864], ja kokonaiselossaoloajan mediaani 51,1 kuukautta [95 %:n luottamusväli 44,85–57,26] Cyramza- ja erlotinibi-valmisteilla vs. 46,0 kuukautta [95 %:n luottamusväli 43,56–53,03] lumelääkkeellä ja erlotinibi-valmisteella.
REVEL
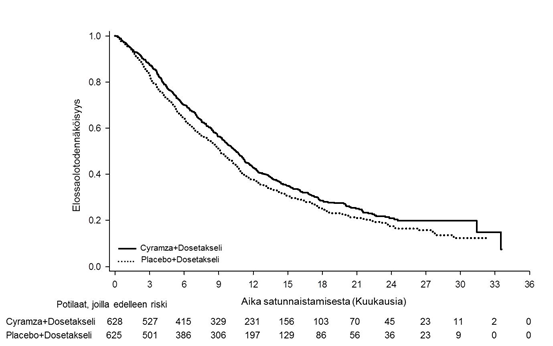
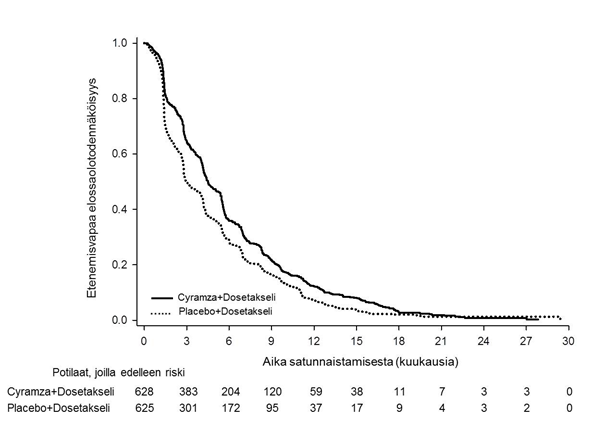
REVEL oli satunnaistettu, kaksoissokkoutettu tutkimus, jossa Cyramzan ja dosetakselin yhdistelmää verrattiin lumelääkkeen ja dosetakselin yhdistelmään 1253 potilaalla, joilla oli paikallisesti edennyt tai metastasoitunut levyepiteelimäinen tai ei-levyepiteelimäinen keuhkosyöpä, joka oli edennyt yhden platinapohjaisen hoidon aikana tai sen jälkeen. Ensisijainen päätetapahtuma oli kokonaiselossaoloaika. Potilaat satunnaistettiin suhteessa 1:1 saamaan Cyramzaa ja dosetakselia (n = 628) tai lumetta ja dosetakselia (n = 625). Satunnaistaminen stratifioitiin maantieteellisen alueen, sukupuolen, aiemman ylläpitohoidon ja ECOG-toimintakykyluokan mukaan. Cyramzaa (10 mg/kg) tai lumelääkettä ja dosetakselia (75 mg/m2) annettiin infuusiona laskimoon 21-päiväisen hoitojakson ensimmäisenä päivänä. Itä-Aasiassa sijaitsevissa tutkimuskeskuksissa annettiin pienempi dosetakseliannos (60 mg/m2) 21 päivän välein. Tutkimuksesta suljettiin pois potilaat, joilla oli ollut äskettäin vakava keuhkoverenvuoto, maha-suolikanavan verenvuoto tai postoperatiivinen verenvuoto, tai joilla oli merkkejä keskushermoston verenvuodosta, suuren ilmatien tai verisuonen kasvain, ontelo kasvaimessa tai anamneesissa merkittävä vuoto tai hallitsematon tromboottinen häiriö. Tutkimuksesta suljettiin pois myös potilaat, jotka saivat mitään terapeuttista antikoagulanttia ja/tai pitkäaikaisesti tulehduskipulääkkeitä tai muita verihiutaleiden estäjiä tai joilla oli hoitamaton, kliinisesti epävakaa aivo/keskushermoston metastaasi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Aspiriinin käyttö sallittiin annokseen 325 mg/päivä saakka. Tutkimuksessa oli mukana rajallinen määrä muita kuin valkoihoisia potilaita, erityisesti tummaihoisia potilaita (2,6 %). Siksi tässä potilasryhmässä ramusirumabin ja dosetakselin yhdistelmän käytöstä on rajallisesti kokemusta edenneen ei-pienisoluisen keuhkosyövän hoidossa, kuten myös potilailla, joilla on munuaisten vajaatoiminta, kardiovaskulaarinen sairaus tai lihavuus.
Potilaiden lähtötilanteen demografiset tiedot ja taudin ominaisuudet olivat hoitoryhmissä yleisesti ottaen samankaltaiset: iän mediaani oli 62 vuotta, 67 % potilaista oli miehiä, 82 % oli valkoihoisia ja 13 % aasialaisia, 32 %:lla potilaista ECOG-toimintakykyluokka oli 0 ja 67 %:lla 1, 73 %:lla potilaista tauti oli histologialtaan muunlainen kuin levyepiteelimäinen ja 26 %:lla levyepiteelimäinen. Yleisimpiä aiempia hoitoja olivat pemetreksedi (38 %), gemsitabiini (25 %), taksaani (24 %) ja bevasitsumabi (14 %). 22 % potilaista sai aiempaa ylläpitohoitoa. Dosetakselihoidon keston mediaani oli 14,1 viikkoa ramusirumabi + dosetakseli ‑ryhmässä (saatujen infuusioiden mediaani 4,0) ja 12,0 viikkoa lume + dosetakseli ‑ryhmässä (saatujen infuusioiden mediaani 4,0).
Kokonaiselossaoloaika parani tilastollisesti merkitsevästi Cyramzan ja dosetakselin yhdistelmää saaneilla potilailla verrattuna lumetta ja dosetakselia saaneisiin (HR 0,857; 95 % lv: 0,751–0,979; p = 0,024). Elossaoloajan mediaani piteni 1,4 kk enemmän Cyramza + dosetakseli ‑ryhmässä: 10,5 kk Cyramza + dosetakseli ‑ryhmässä ja 9,1 kk lume + dosetakseli ‑ryhmässä. Etenemisvapaa elossaoloaika parani tilastollisesti merkitsevästi Cyramzan ja dosetakselin yhdistelmää saaneilla potilailla verrattuna lumetta ja dosetakselia saaneisiin (HR 0,762; 95 % lv: 0,677–0,859; p < 0,001). Etenemisvapaan elossaoloajan mediaani piteni 1,5 kk enemmän Cyramza + dosetakseli ‑ryhmässä: 4,5 kk Cyramza + dosetakseli ‑ryhmässä ja 3 kk lume + dosetakseli ‑ryhmässä. Objektiivinen vasteprosentti parani merkitsevästi Cyramzan ja dosetakselin yhdistelmää saaneilla potilailla verrattuna lumetta ja dosetakselia saaneisiin (22,9 % vs. 13,6 %; p < 0,001). Ensisijainen elämänlaatuanalyysi osoitti, että elämänlaadun heikentymiseen kulunut aika oli hoitoryhmissä samaa luokkaa kaikilla keuhkosyövän oireasteikoilla (Lung Cancer Symptom Scale, LCSS) mitattuna.
Johdonmukaista edistymistä (ramusirumabi + dosetakseli vs. lume + dosetakseli) havaittiin tärkeissä etenemisvapaan elossaoloajan ja kokonaiselossaoloajan alaryhmissä. Kokonaiselossaoloaikaa koskevan alaryhmän tulokset olivat seuraavanlaiset: histologialtaan muunlainen kuin levyepiteelimäinen syöpä (HR 0,83; 95 % lv: 0,71–0,97; kokonaiselossaoloajan mediaani: 11,1 vs. 9,7 kk) ja levyepiteelimäinen syöpä (HR 0,88; 95 % lv: 0,69–1,13; kokonaiselossaoloajan mediaani: 9,5 vs. 8,2 kk); potilaat, jotka saivat aiempaa ylläpitohoitoa (HR 0,69; 95 % lv: 0,51–0,93; kokonaiselossaoloajan mediaani: 14,4 vs. 10,4 kk); aiemman hoidon aloittamisesta kulunut aika < 9 kk (HR 0,75; 95 % lv: 0,64–0,88; kokonaiselossaoloajan mediaani: 9,3 vs. 7,0 kk); < 65-vuotiaat potilaat (HR 0,74, 95 % lv: 0,62–0,87; kokonaiselossaoloajan mediaani 11,3 vs. 8,9 kk). Potilaiden, jotka saivat ramusirumabia ja dosetakselia edenneen ei-pienisoluisen keuhkosyövän hoitoon ja joiden tauti oli edennyt aiemman platinapohjaisen solunsalpaajahoidon jälkeen, korkeamman iän havaittiin viittaavan heikompaan tehoon (ks. kohta Farmakodynamiikka). Tehossa ei havaittu eroja hoitoryhmien välillä alaryhmissä ≥ 65-vuotiaat potilaat (kokonaiselossaoloajan HR 1,10, 95 % lv: 0,89-1,36; kokonaiselossaoloajan mediaani: 9,2 vs. 9,3 kk, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), ennen hoitoa taksaania saaneet potilaat (HR 0,81; 95 % lv: 0,62-1,07; kokonaiselossaoloajan mediaani: 10,8 vs. 10,4 kk) ja potilaat, joiden aiemman hoidon aloittamisesta kulunut aika oli ≥ 9 kk (HR 0,95; 95 % lv: 0,75-1,2; kokonaiselossaoloajan mediaani: 13,7 vs. 13,3 kk). Tehotulokset esitetään taulukossa 13.
Taulukko 13: Tehotietojen yhteenveto – ITT-populaatio
| Cyramza + dosetakseli N = 628 | Lumelääke + dosetakseli N = 625 | |
| Kokonaiselossaoloaika (kk) | ||
| Mediaani (95 % lv) | 10,5 (9,5; 11,2) | 9,1 (8,4; 10,0) |
| Riskisuhde (95 % lv) | 0,857 (0,751; 0,979) | |
| Stratifioitu log-rank-testin p-arvo | 0,024 | |
| Etenemisvapaa elossaoloaika (kk) | ||
| Mediaani (95 % lv) | 4,5 (4,2; 5,4) | 3,0 (2,8; 3,9) |
| Riskisuhde (95 % lv) | 0,762 (0,677; 0,859) | |
| Stratifioitu log-rank-testin p-arvo | < 0,001 | |
| Objektiivinen vasteprosentti (täydellinen vaste + osittainen vaste) | ||
| Prosenttiosuus (95 % lv) | 22,9 (19,7; 26,4) | 13,6 (11,0; 16,5) |
| CMH-testin stratifioitu p-arvo | < 0,001 | |
Lyhenteet: lv = luottamusväli, CMH = Cochran-Mantel-Haenszel
Kuva 7: Kokonaiselossaoloajan Kaplan–Meier -käyrät, Cyramza + dosetakseli vs. lumelääke + dosetakseli, REVEL-tutkimus
Kuva 8: Etenemisvapaan elossaoloajan Kaplan–Meier -käyrät, Cyramza + dosetakseli vs. lumelääke + dosetakseli, REVEL-tutkimus
Maksasolukarsinooma
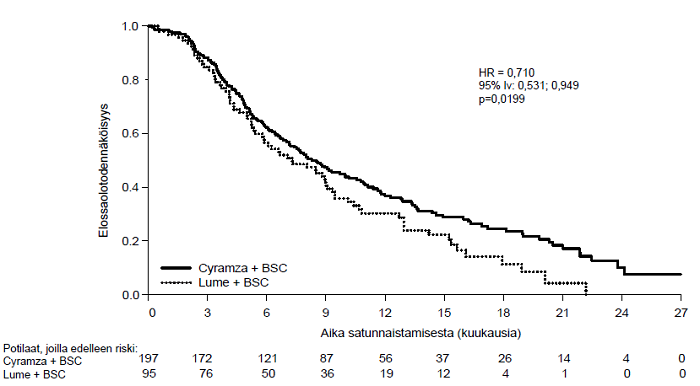
REACH-2
REACH-2 oli maailmanlaajuinen, satunnaistettu, kaksoissokkoutettu tutkimus, jossa Cyramzan ja parhaan mahdollisen tukihoidon (Best Supportive Care, BSC) yhdistelmää verrattiin lumelääkkeen ja parhaan mahdollisen tukihoidon yhdistelmään. Tutkimukseen satunnaistettiin (2:1) 292 potilasta, joilla oli maksasolukarsinooma ja joiden seerumin AFP-pitoisuus oli tutkimukseenottohetkellä ≥ 400 ng/ml. Tutkimukseen otettiin potilaita, joiden tauti oli edennyt aiemman sorafenibihoidon aikana tai sen jälkeen, tai jotka eivät sietäneet sorafenibia. Soveltuvien potilaiden Child–Pugh-luokka oli A (pisteet < 7), kreatiniinipuhdistuma ≥ 60 ml/min ja ECOG-toimintakykyluokka 0 tai 1. Lisäksi potilaat joko kuuluivat Barcelona Clinic Liver Cancer -luokituksen (BCLC) luokkaan B eikä lokoregionaalinen hoito ollut enää mahdollinen, tai he kuuluivat BCLC-luokkaan C. Tutkimuksesta suljettiin pois potilaat, joilla oli aivometastaaseja, leptomeningeaalinen tauti, hallitsematonta selkäydinkompressiota, aiempaa tai ajankohtaista hepaattista enkefalopatiaa tai kliinisesti merkittävää askitesta, vaikeaa laskimolaajentumien verenvuotoa hoitoa edeltävien 3 kuukauden aikana, tai mahan tai ruokatorven laskimolaajentumia, joiden verenvuotoriski oli suuri. Ensisijainen päätetapahtuma oli kokonaiselossaoloaika. REACH-2-tutkimuksen mukaanottokriteerinä toiminut suurentuneen AFP-pitoisuuden kynnysarvo määritettiin REACH-tutkimuksen ennalta määritellyn eksploratiivisen alaryhmäanalyysin elossaolotulosten perusteella. REACH oli aiemmin päättynyt, supportiivinen vaiheen 3 kliininen tutkimus, jossa 565 maksasolukarsinoomaa sairastavaa potilasta satunnaistettiin (1:1) saamaan joko Cyramzan ja parhaan mahdollisen tukihoidon yhdistelmää tai lumelääkkeen ja parhaan mahdollisen tukihoidon yhdistelmää. Potilaiden tauti oli edennyt aiemman sorafenibihoidon aikana tai sen jälkeen.
REACH-2-tutkimuksessa potilaiden lähtötilanteen demografiset tiedot ja taudin piirteet olivat yleisesti ottaen samanlaiset molemmissa ryhmissä; poikkeuksena oli AFP-pitoisuus, joka oli lumeryhmässä pienempi. Cyramza-hoitoa saaneiden potilaiden kokonaiselossaoloaika oli tilastollisesti merkitsevästi pidempi lumehoitoon verrattuna (taulukko 14). REACH-2-tutkimuksen tärkeää tehon tulosmuuttujaa tuki se, että etenemisvapaa elossaoloaika oli Cyramza-hoitoa saaneilla potilailla tilastollisesti merkitsevästi pidempi kuin lumehoitoa saaneilla. Cyramzan suhteellinen hoitovaikutus (riskisuhteen perusteella arvioituna) verrattuna lumelääkkeeseen oli yleisesti ottaen johdonmukainen eri alaryhmissä, joiden määrittelyperusteisiin kuuluivat ikä, etninen tausta, taudin etiologia ja sorafenibihoidon lopettamisen syy (taudin eteneminen vs. huono siedettävyys). REACH-2-tutkimuksessa ramusirumabialtistus korreloi kokonaiselossaoloajan paranemiseen (ks. kohta Farmakokinetiikka). REACH-2-tutkimuksen tehotulokset esitetään taulukossa 14 ja kuvassa 9.
Taulukko 14: REACH-2-tutkimuksen tehotietojen yhteenveto – ITT-populaatio
| Cyramza N = 197 | Lumelääke N = 95 | |
| Kokonaiselossaoloaika, kk | ||
| Mediaani (95 % lv) | 8,51 (7,00; 10,58) | 7,29 (5,42; 9,07) |
| Riskisuhde (95 % lv) | 0,710 (0,531; 0,949) | |
| Stratifioitu log-rank-testin p-arvo | 0,0199 | |
| Etenemisvapaa elossaoloaika (kk) | ||
| Mediaani (95 % lv) | 2,83 (2,76; 4,11) | 1,61 (1,45; 2,69) |
| Riskisuhde (95 % lv) | 0,452 (0,339; 0,603) | |
| Stratifioitu log-rank-testin p-arvo | < 0,0001 | |
| Objektiivinen vasteprosentti (täydellinen vaste + osittainen vaste) | ||
| Objektiivinen vasteprosentti, % (95 % lv) | 4,6 (1,7; 7,5) | 1,1 (0,0; 3,1) |
| p-arvo | 0,1697 | |
Lyhenteet: lv = luottamusväli
Kuva 9: Kokonaiselossaoloajan Kaplan–Meier-käyrät, Cyramza vs. lumelääke, REACH-2-tutkimus
Potilaat, joiden ECOG-toimintakykyluokka (Eastern Cooperative Oncology Group) on ≥ 2
Avaintutkimuksiin ei otettu millään indikaatiolla mukaan potilaita, joiden ECOG-toimintakykyluokka oli ≥ 2. Tästä syystä Cyramzan turvallisuutta ja tehoa ei tunneta tässä potilaspopulaatiossa.
Immunogeenisyys
Lääkevasta-aineita tutkittiin potilailta useina ajankohtina kahdessa vaiheen 3 tutkimuksessa (RAINBOW ja REGARD). Näytteet tutkittiin 956 potilaalta: 527:ltä ramusirumabia saaneelta potilaalta ja 429:ltä vertailuhoitoa saaneelta potilaalta. Lääkevasta-aineita kehittyi 11:llä (2,2 %) ramusirumabia saaneista potilaista ja kahdella (0,5 %) vertailuhoitoa saaneista potilaista. Infuusioreaktioita ei esiintynyt kenelläkään potilaista, joille kehittyi lääkevasta-aineita. Yhdellekään potilaalle ei kehittynyt ramusirumabia neutraloivia vasta-aineita. Lääkevasta-aineiden vaikutusta ramusirumabin tehoon ja turvallisuuteen ei voida arvioida, sillä tietoja ei ole riittävästi.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Cyramza-valmisteen käytöstä kaikkien pediatristen potilasryhmien hoidossa mahalaukun adenokarsinoomassa, paksu- ja peräsuolen adenokarsinoomassa, keuhkosyövässä ja maksasyövässä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Monoterapiana käytetyn ramusirumabin turvallisuutta ja farmakokinetiikkaa arvioitiin avoimessa vaiheen 1 monikeskustutkimuksessa I4T-MC-JVDA pediatrisilla ja nuorilla aikuispotilailla (ikä 1–21 vuotta) vaiheen 2 annossuosituksen määrittämiseksi. Tutkimus koostui kahdesta osasta. Osassa A ramusirumabia annosteltiin 8 mg/kg tai 12 mg/kg laskimonsisäisesti 60 minuutin aikana 2 viikon välein 23 potilaalle, joilla oli uusiutuneita tai vaikeahoitoisia kasvaimia keskushermoston ulkopuolella. Maksimaalista siedettyä annosta ei saavutettu. Vaiheeseen 2 suositelluksi annokseksi määritettiin 12 mg/kg kahden viikon välein annosteltuna. Osassa B ramusirumabia annosteltiin vaiheen 2 annossuosituksen mukaisesti 6 potilaalle, joilla oli uusiutuneita tai vaikeahoitoisia kasvaimia keskushermostossa, lääkkeen siedettävyyden arvioimiseksi tässä populaatiossa. Kasvainvasteita ei havaittu kummassakaan osassa A tai B.
Ramusirumabin turvallisuutta ja tehoa yhdessä syklofosfamidin ja vinorelbiinin kanssa verrattuna syklofosfamidiin ja vinorelbiiniin yksinään arvioitiin satunnaistetussa, maailmanlaajuisessa vaiheen 2 monikeskustutkimuksessa J1S-MCJV01 (JV01). Tutkimukseen otettiin 30 pediatrista tai nuorta aikuispotilasta (ikä 36 kk–29 vuotta), joilla oli relapsoitunut, uusiutunut tai refraktorinen desmoplastinen pienipyörösolukasvain. Satunnaistaminen (2:1) stratifioitiin levinneisyyden perusteella (metastaattinen sairaus vs. paikallisesti edennyt sairaus) uusiutumishetkellä. JV01 ei saavuuttanut tutkimukselle ennalta määriteltyä onnistumiskriteeriä, joka vaati 99 %:n posteriorisen todennäköisyyden paremmuudesta (HR<1) hoidon onnistumisen toteamiseksi. Lopullisessa analyysissä (frequentist-analyysin mukaan), etenemisvapaan elossaoloajan mediaani oli kokeellisessa ryhmässä 6,75 kuukautta ja vertailuryhmässä 1,71 kuukautta (HR 0,465; 80 %:n lv: 0,261–0,827). Kokeellisen hoidon ryhmässä havaittiin yksi osittainen vaste ja yksi täydellinen vaste. Vertailuryhmässä havaittiin yksi osittainen vaste, eikä yhtään täydellistä vastetta. Tämän tutkimuksen rajallisen koon vuoksi ei ole mahdollista päätellä, että käytön hyödyt olisivat riskejä suuremmat.
Ramusirumabin turvallisuutta ja tehoa yhdessä gemsitabiinin ja dosetakselin kanssa verrattuna gemsitabiiniin ja dosetakseliin yksinään arvioitiin satunnaistetussa, maailmanlaajuisessa vaiheen 2 monikeskustutkimuksessa J1S-MC-JV02 (JV02). Tutkimukseen otettiin 23 pediatrista tai nuorta aikuispotilasta (ikä 36 kk–29 vuotta), jotka sairastivat relapsoitunutta, uusiutunutta tai progressiivista synoviaalisarkoomaa. Satunnaistaminen (2:1) stratifioitiin levinneisyyden perusteella (metastaattinen sairaus vs. paikallisesti edennyt sairaus) uusiutumishetkellä. JV02-tutkimus lopetettiin ilman ensisijaisen päätetapahtuman (etenemisvapaa elossaoloaika) muodollista arviointia, koska välivaiheen hyödyllisyysanalyysissa tutkimus ei saavuttanut ennaltamääritettyä 60 %:n luotettavuutta kokeellisen hoidon paremmuudessa (etenemisvapaan elossaoloajan riskisuhde vähemmän kuin 1 synoviaalisarkoomapotilailla). Tutkimuksessa havaittiin yksi osittainen vaste, mutta ei yhtään täydellistä vastetta kokeelliselle hoidolle. Vertailuryhmässä ei havaittu vastetta, ei täydellistä eikä osittaista.
Farmakokinetiikka
Kun annostus oli 8 mg/kg 2 viikon välein, monoterapiana käytetyn ramusirumabin geometrinen Cmin-keskiarvo pitkälle edennyttä mahasyöpää sairastavien potilaiden seerumissa ennen neljättä ramusirumabiannosta oli 49,5 μg/ml (vaihteluväli 6,3–228 μg/ml) ja ennen seitsemättä ramusirumabiannosta 74,4 μg/ml (vaihteluväli 13,8–234 μg/ml). Ramusirumabin geometrinen Cmin-keskiarvo maksasolukarsinoomaa sairastavien potilaiden seerumissa ennen toista ramusirumabiannosta oli 23,5 μg/ml (vaihteluväli 2,9–76,5 μg/ml), ennen neljättä annosta 44,1 μg/ml (vaihteluväli 4,2–137 μg/ml) ja ennen seitsemättä annosta 60,2 μg/ml (vaihteluväli 18,3–123 μg/ml)
Kun ramusirumabin annostus oli 8 mg/kg 2 viikon välein yhdessä FOLFIRI-yhdistelmän kanssa, ramusirumabin geometriset Cmin-keskiarvot seerumissa olivat 46,3 µg/ml (vaihteluväli 7,7–119 µg/ml) ennen kolmatta annosta ja 65,1 µg/ml (vaihteluväli 14,5–205 µg/ml) ennen viidettä annosta, metastasoitunutta kolorektaalisyöpää sairastaneilla potilailla.
Kun annostus oli 10 mg/kg 3 viikon välein, ramusirumabin geometriset Cmin-keskiarvot seerumissa olivat 28,3 μg/ml (vaihteluväli 2,5–108 μg/ml) ennen kolmatta annosta ja 38,4 μg/ml (vaihteluväli 3,1–128 μg/ml) ennen viidettä annosta, kun ramusirumabia annettiin yhdessä dosetakselin kanssa potilaille, joilla oli ei-pienisoluinen keuhkosyöpä.
Kun annostus oli 10 mg/kg ramusirumabia 2 viikon välein, ramusirumabin geometriset Cmin-keskiarvot seerumissa olivat 68,5 μg/ml (vaihteluväli 20,3–142 μg/ml) ennen neljättä annosta ja 85,7 μg/ml (vaihteluväli 36,0–197 μg/ml) ennen seitsemättä annosta, kun ramusirumabia annettiin yhdessä erlotinibin kanssa potilaille, joilla oli ei-pienisoluinen keuhkosyöpä.
Imeytyminen
Cyramza annetaan infuusiona laskimoon. Muita antoreittejä ei ole tutkittu.
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella ramusirumabin vakaan tilan jakautumistilavuuden keskiarvo (% variaatiokerroin [CV %]) oli 5,4 l (15 %).
Biotransformaatio
Ramusirumabin metaboliaa ei ole tutkittu. Vasta-aineet häviävät pääosin katabolian vaikutuksesta.
Eliminaatio
Populaatiofarmakokineettisen analyysin perusteella ramusirumabin keskipuhdistuma (CV %) oli 0,015 l/h (30 %) ja puoliintumisajan keskiarvo 14 päivää (20 %).
Riippuvaisuus ajasta ja annoksesta
Ramusirumabin (6–20 mg/kg) farmakokinetiikassa ei ollut selviä annossuhteen poikkeamia. Kumuloitumissuhde oli 1,5, kun ramusirumabia annettiin 2 viikon välein. Populaatiofarmakokineettisen mallin avulla tehtyjen simulaatioiden perusteella vakaa tila saavutetaan kuudenteen annokseen mennessä.
Iäkkäät
Populaatiofarmakokineettisen analyysin perusteella ramusirumabialtistus ≥ 65-vuotiailla potilailla ei eronnut altistuksesta < 65-vuotiailla.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminnan vaikutusta ramusirumabin farmakokinetiikkaan ei ole tutkittu. Populaatiofarmakokineettisen analyysin perusteella ramusirumabialtistus oli samaa luokkaa lievässä munuaisten vajaatoiminnassa (kreatiniinipuhdistuma ≥ 60 – < 90 ml/min), keskivaikeassa munuaisten vajaatoiminnassa (kreatiniinipuhdistuma ≥ 30 – < 60 ml/min) tai vaikeassa munuaisten vajaatoiminnassa (kreatiniinipuhdistuma 15-29 ml/min) verrattuna normaaliin munuaistoimintaan (kreatiniinipuhdistuma ≥ 90 ml/min).
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutusta ramusirumabin farmakokinetiikkaan ei ole tutkittu. Populaatiofarmakokineettisen analyysin perusteella ramusirumabialtistus lievässä maksan vajaatoiminnassa (kokonaisbilirubiini > 1,0–1,5 x viitealueen yläraja [ULN] ja mikä tahansa ASAT tai kokonaisbilirubiini ≤ 1,0 ULN ja ASAT > ULN) tai keskivaikeassa maksan vajaatoiminnassa (kokonaisbilirubiini > 1,5–3,0 x ULN ja mikä tahansa ASAT) oli samaa luokkaa kuin normaalissa maksatoiminnassa (kokonaisbilirubiini ja ASAT ≤ ULN). Ramusirumabia ei ole tutkittu vaikeassa maksan vajaatoiminnassa (kokonaisbilirubiini > 3,0 x ULN ja mikä tahansa ASAT).
Pediatriset potilaat
Keskimääräinen huippupitoisuus oli 165 μg/ml 8 mg/kg annoksen jälkeen (tutkimus JVDA), 231 μg/ml 9 mg/kg annoksen jälkeen (tutkimus JV02), 238 µg/ml (CV % = 35) 12 mg/kg annoksen jälkeen (tutkimus JV01) ja 285 μg/ml (CV % = 26) 12 mg/kg annoksen jälkeen (tutkimus JVDA). Viidentenätoista päivänä 1. syklissä (n = 19), jäännöspitoisuus (Ctrough) oli 41,6 µg/ml (CV % = 57) tutkimuksessa JV01 ja 48,3 µg/ml (CV % = 41) tutkimuksessa JVDA. Ramusirumabialtistus oli pediatrisilla ja nuorilla aikuispotilailla (yli 12 kuukautta vanhat lapset, alle 21-vuotiaat), joilla oli vaikeahoitoisia kiinteitä kasvaimia, mukaan lukien keskushermostokasvaimet, yhden tai usean 8 mg/kg tai 12 mg/kg annoksen jälkeen samankaltainen kuin aikuispotilailla. Ramusirumabialtistus 12 mg/kg annoksen jälkeen oli samankaltainen koko ikäryhmässä yli 12 kuukauden ikäisistä alle 21-vuotiaisiin potilaisiin.
Muut erityisryhmät
Populaatiofarmakokineettisen analyysin perusteella seuraavien muuttujien ei todettu vaikuttavan ramusirumabin jakautumiseen: ikä, sukupuoli, etninen tausta ja albumiiniarvo. Näiden ja muiden tutkittujen tekijöiden vaikutus ramusirumabin jakautumiseen oli < 20 %. Painon katsotaan olevan ramusirumabin farmakokinetiikan merkitsevä kovariaatti, mikä tukee painoon perustuvan annostelun käyttöä.
Altistus-vastesuhde
Teho
Altistus-vasteanalyysit osoittavat, että ramusirumabin teho korreloi ramusirumabialtistuksen kanssa avaintutkimuksissa. Teho (kokonaiselossaoloajan pidentymisenä mitattuna) liittyi ramusirumabin altistuksen suurenemiseen, kun ramusirumabia annettiin 8 mg/kg 2 viikon välein ja 10 mg/kg 3 viikon välein. Etenemisvapaan elossaoloajan pidentyminen liittyi myös ramusirumabialtistuksen suurenemiseen edenneessä mahasyövässä, ei-pienisoluisessa keuhkosyövässä taudin edettyä platinapohjaisen solunsalpaajahoidon jälkeen ja metastasoituneessa kolorektaalisyövässä.
Maksasolukarsinooman hoitoa koskeneessa REACH-2-tutkimuksessa ramusirumabin altistuksen ja tehon välillä havaittiin oleellinen yhteys. Kokonaiselossaoloaika piteni lumehoitoon verrattuna vain potilailla, joiden ramusirumabialtistus oli mediaania suurempi, ja altistuksen ja tehon välinen suhde säilyi, kun muut ennustavat tekijät pyrittiin huomioimaan. Etenemisvapaan ajan kohdalla todettiin hoitovaikutus kaikilla altistustasoilla, jotka saavutettiin annettaessa 8 mg/kg ramusirumabia 2 viikon välein. Tällaista yhteyttä ei havaittu ei-pienisoluista keuhkosyöpää arvioineessa RELAY-tutkimuksessa, jossa annettiin 10 mg/kg ramusirumabia ja erlotinibia 2 viikon välein.
Turvallisuus
RAINBOW–tutkimuksessa asteen ≥ 3 hypertension, neutropenian ja leukopenian ilmaantuvuus lisääntyi ramusirumabialtistuksen suurentuessa.
RAISE-tutkimuksessa asteen ≥ 3 neutropenian ilmaantuvuus lisääntyi ramusirumabialtistuksen suurentuessa.
RELAY-tutkimuksessa ei todettu altistus-turvallisuussuhdetta valikoitujen turvallisuuspäätetapahtumien kohdalla. Näitä olivat mm. asteen ≥ 3 hypertensio, ripuli, proteinuria ja aknen kaltainen dermatiitti.
REVEL-tutkimuksessa asteen ≥ 3 kuumeisen neutropenian ja hypertension ilmaantuvuus lisääntyi ramusirumabialtistuksen suurentuessa.
REACH-2- ja REACH-tutkimusten (potilaat, joilla alfafetoproteiinipitoisuus ≥ 400 ng/ml) yhdistettyjen tietojen mukaan asteen ≥ 3 hypertension ilmaantuvuus suureni ramusirumabialtistuksen suurentuessa.
Prekliiniset tiedot turvallisuudesta
Ramusirumabin karsinogeenisuutta tai geenitoksisuutta ei ole tutkittu eläinkokeissa.
Toistuvilla annoksilla toteutetuissa toksisuustutkimuksissa makakiapinoilla tunnistetut kohde-elimet olivat munuainen (glomerulonefriitti), luusto (epifyysilevyjen paksuuntuminen ja poikkeava endokondraalinen luutuminen) ja naaraiden sukuelimet (munasarjojen ja kohdun painon pienentyminen). Useissa elimissä havaittiin erittäin lieväasteista tulehdusta ja/tai mononukleaaristen solujen infiltraatiota.
Ramusirumabin lisääntymistoksisuutta ei ole tutkittu, mutta eläinmalleissa angiogeneesiin, VEGF:ään ja VEGF-reseptori-2:een on havaittu liittyvän naaraiden lisääntymistä, alkion ja sikiön kehitystä ja postnataalista kehitystä koskevia kriittisiä seikkoja. Ramusirumabin vaikutusmekanismin perusteella on todennäköistä, että ramusirumabi estää eläimillä angiogeneesiä ja vaikuttaa haitallisesti hedelmällisyyteen (ovulaatioon), istukan kehitykseen, sikiön kehitykseen ja postnataaliseen kehitykseen.
Kerta-annos ramusirumabia ei heikentänyt haavojen paranemista apinoilla koko paksuutta koskevassa viiltomallissa.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Histidiinimonohydrokloridi
Natriumkloridi
Glysiini (E640)
Polysorbaatti 80 (E433)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Cyramzaa ei saa antaa eikä sekoittaa glukoosiliuoksen kanssa.
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Laimennuksen jälkeen
Kun Cyramza valmistetaan ohjeiden mukaan, infuusioliuos ei sisällä antimikrobisia säilöntäaineita.
Cyramzan on osoitettu säilyvän 9 mg/ml (0,9 %) NaCl-injektionesteessä kemiallisesti ja fysikaalisesti stabiilina 24 tunnin ajan 2–8 °C lämpötilassa tai 4 tunnin ajan 25 ºC lämpötilassa. Mikrobiologiselta kannalta valmiste tulee käyttää välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikainen säilytysaika ja käyttöä edeltävät säilytysolosuhteet ovat käyttäjän vastuulla, kuitenkin yleensä enintään 24 tuntia 2–8 °C:n lämpötilassa, mikäli laimennus ei ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
CYRAMZA infuusiokonsentraatti, liuosta varten
10 mg/ml (L:ei) 10 ml (674,25 €), 50 ml (3096,55 €)
PF-selosteen tieto
10 ml liuosta injektiopullossa (tyypin I lasia), jossa on klorobutyylikumitulppa, alumiinisinetti ja polypropeenikorkki.
50 ml liuosta injektiopullossa (tyypin I lasia), jossa on klorobutyylikumitulppa, alumiinisinetti ja polypropeenikorkki.
Pakkaus, jossa yksi 10 ml:n injektiopullo.
Pakkaus, jossa kaksi 10 ml:n injektiopulloa.
Pakkaus, jossa yksi 50 ml:n injektiopullo.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Konsentraatti on kirkasta tai hiukan opalisoivaa, väritöntä tai hiukan kellertävää liuosta, jonka pH on 6,0.
Käyttö- ja käsittelyohjeet
Älä ravista injektiopulloa.
Valmista infuusioliuos aseptista tekniikkaa käyttäen, jotta valmistettu liuos pysyy steriilinä.
Injektiopullot on tarkoitettu vain yhtä käyttökertaa varten. Tarkasta injektiopullon sisältö hiukkasten ja värimuutosten varalta (infuusiokonsentraatin pitäisi olla kirkasta tai hiukan opalisoivaa, väritöntä tai hiukan kellertävää, eikä siinä saa olla näkyviä hiukkasia) ennen laimentamista. Jos havaitset hiukkasia tai värimuutoksia, hävitä injektiopullo.
Laske infuusioliuoksen valmistamista varten tarvittava ramusirumabin annos ja määrä. Injektiopullo sisältää joko 100 mg tai 500 mg 10 mg/ml ramusirumabiliuosta. Laimennukseen saa käyttää vain 9 mg/ml (0,9 %) NaCl-injektionestettä.
Jos käytössä on esitäytetty laskimoinfuusiopakkaus
Poista ramusirumabin lasketun määrän perusteella vastaava määrä 9 mg/ml (0,9 %) NaCl-injektionestettä esitäytetystä 250 ml laskimoinfuusiopakkauksesta. Siirrä laskettu määrä ramusirumabia aseptisesti laskimoinfuusiopakkaukseen. Pakkauksen sisältämän kokonaismäärän pitäisi olla lopuksi 250 ml. Kääntele pakkausta varovasti, jotta liuos sekoittuu riittävästi. Infuusioliuos ei saa jäätyä. Infuusioliuosta ei saa ravistaa. Valmistetta ei saa laimentaa muilla liuoksilla eikä antaa samassa infuusiossa muiden elektrolyyttien tai lääkevalmisteiden kanssa.
Jos käytössä on tyhjä laskimoinfuusiopakkaus
Siirrä laskettu määrä ramusirumabia aseptisesti tyhjään laskimoinfuusiopakkaukseen. Lisää riittävä määrä 9 mg/ml (0,9 %) NaCl-injektionestettä pakkaukseen, jotta kokonaistilavuudeksi tulee 250 ml. Kääntele pakkausta varovasti, jotta liuos sekoittuu riittävästi. Infuusioliuos ei saa jäätyä. Infuusioliuosta ei saa ravistaa. Valmistetta ei saa laimentaa muilla liuoksilla eikä antaa samassa infuusiossa muiden elektrolyyttien tai lääkevalmisteiden kanssa.
Parenteraaliset lääkevalmisteet tulee tarkastaa silmämääräisesti hiukkasten varalta ennen niiden antamista potilaalle. Jos valmisteessa näkyy hiukkasia, infuusioliuos on hävitettävä.
Hävitä injektiopulloon käyttämättä jäänyt ramusirumabi, sillä valmiste ei sisällä antimikrobisia säilöntäaineita.
Anna lääke infuusiopumpulla. Infuusio on annettava erillisen infuusioletkun kautta. Letkussa on oltava 0,22 mikronin proteiinisuodatin ja letku on huuhdeltava 9 mg/ml (0,9 %) NaCl-injektionesteellä infuusion lopussa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
CYRAMZA infuusiokonsentraatti, liuosta varten
10 mg/ml 10 ml, 50 ml
- Ei korvausta.
ATC-koodi
L01FG02
Valmisteyhteenvedon muuttamispäivämäärä
08.01.2026
Yhteystiedot
 OY ELI LILLY FINLAND AB
OY ELI LILLY FINLAND AB Mannerheimintie 117
00280 Helsinki
09 854 5250
www.lilly.com/fi
medinfo_finland@lilly.com
Lääketietopalvelu puh. 0800-140 240