
LEMTRADA infuusiokonsentraatti, liuosta varten 12 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Opas sisältää tärkeää turvallisuutta koskevaa tietoa LEMTRADA-hoitoa (alemtutsumabi) aloittaville potilaille.
Potilaan varoituskortti sisältää tärkeitä tietoja LEMTRADA-valmisteesta (alemtutsumabi).
Terveydenhuollon ammattilainen
Muistilista terveydenhuollon ammattilaiselle.
Opas sisältää tärkeitä turvallisuutta koskevia tietoja LEMTRADA-valmistetta määrääville terveydenhuollon ammattilaisille.
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 12 mg alemtutsumabia 1,2 ml:ssa (10 mg/ml).
Alemtutsumabi on yhdistelmä-DNA-tekniikalla nisäkässolususpensioviljelmässä (kiinanhamsterinmunasarjasolut) - ravintoaineliuoksessa tuotettu monoklonaalinen vasta-aine.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol kaliumia (39 mg) yhdessä infuusiossa, eli sen voidaan sanoa olevan ”kaliumiton”.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) yhdessä infuusiossa, eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää 0,12 mg polysorbaatti 80:tä (E433) yhdessä injektiopullossa, jonka nimellinen täyttötilavuus on 1,2 ml, mikä vastaa 0,1 mg/1,0 ml.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Lemtrada on tarkoitettu käytettäväksi yksinään taudinkulkuun vaikuttavana hoitona aikuisille, joilla on erittäin aktiivinen aaltomainen MS-tauti (relapsoiva-remittoiva multippeliskleroosi, RRMS), seuraavissa potilasryhmissä:
- potilaat, joilla tauti on erittäin aktiivinen huolimatta asianmukaisesti toteutetusta hoitojaksosta vähintään yhdellä taudin kulkuun vaikuttavalla hoidolla, tai
- potilaat, joilla on vaikea ja nopeasti etenevä relapsoiva-remittoiva multippeliskleroosi, joka määritellään niin, että yhden vuoden sisällä on ilmennyt vähintään kaksi toimintakykyä heikentävää relapsia ja aivojen magneettikuvauksessa on todettu vähintään yksi gadoliniumilla tehostuva leesio tai T2-leesiokuorman huomattava suureneminen aiempaan tuoreeseen magneettikuvaukseen verrattuna.
Ehto
Hoito on aloitettava MS-potilaiden hoitoon perehtyneen neurologin valvonnassa.
Annostus ja antotapa
Lemtrada-hoidon saa aloittaa vain multippeliskleroosipotilaiden (MS-potilaiden) hoitoon perehtyneen neurologin valvonnassa ja sairaalassa, jossa on valmius tehohoitoon. Haittavaikutusten, erityisesti sydänlihasiskemian ja sydäninfarktin, aivoverisuoniin liittyvien haittavaikutusten, autoimmuunitilojen ja infektioiden, oikea-aikaiseen diagnosointiin ja hoitoon erikoistuneita lääkäreitä ja tarvittavia laitteita on oltava saatavilla.
Sytokiinioireyhtymän, yliherkkyys- ja/tai anafylaksiareaktioiden hoitoon tarvittavien valmiuksien on oltava saatavilla.
Lemtrada-valmisteella hoidetuille potilaille on annettava potilasvaroituskortti ja potilaan opas, ja heille on kerrottava Lemtrada-hoidon riskeistä (ks. myös pakkausseloste).
Annostus
Suositeltu alemtutsumabiannos on 12 mg/vrk laskimoinfuusiona kahdessa aloitushoitojaksossa ja tarvittaessa enintään kahdessa lisähoitojaksossa.
Aloitushoito kahdessa jaksossa:
- Ensimmäinen hoitojakso: 12 mg vuorokaudessa viitenä peräkkäisenä päivänä (60 mg:n kokonaisannos)
- Toinen hoitojakso: 12 mg vuorokaudessa kolmena peräkkäisenä päivänä (36 mg:n kokonaisannos) annettuna 12 kuukautta ensimmäisen hoitojakson jälkeen.
Voidaan harkita enintään kahta lisähoitojaksoa tarpeen mukaan (ks. kohta Farmakodynamiikka):
- Kolmas tai neljäs hoitojakso: 12 mg vuorokaudessa kolmena peräkkäisenä päivänä (36 mg:n kokonaisannos) annettuna vähintään 12 kuukautta edellisen hoitojakson jälkeen (ks. kohdat Käyttöaiheet ja Farmakodynamiikka).
Väliin jääneitä annoksia ei saa antaa samana päivänä kuin aikataulun mukainen annos annetaan.
Potilaiden seuranta
Aloitushoito suositellaan annettavaksi kahtena hoitojaksona ja tarvittaessa enintään kaksi lisähoitojaksoa (ks. annostus) ja potilaita on turvallisuuden vuoksi seurattava ensimmäisen hoitojakson aloittamisesta lähtien koko hoidon ajan ja vähintään 48 kuukautta toisen hoitojakson viimeisen infuusion jälkeen. Jos annetaan lisäksi kolmas tai neljäs hoitojakso, turvallisuusseurantaa on jatkettava vähintään 48 kuukautta viimeisen infuusion jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Esihoito
Potilaille on annettava esihoitona kortikosteroidia juuri ennen Lemtrada-valmisteen antamista jokaisen hoitojakson kolmena ensimmäisenä päivänä. Kliinisissä tutkimuksissa potilaita esihoidettiin 1 000 mg:n metyyliprednisoloniannoksella jokaisen Lemtrada-hoitojakson kolmena ensimmäisenä päivänä.
Esihoitoa antihistamiineilla ja/tai kuumelääkkeillä ennen Lemtrada-valmisteen antamista voidaan harkita.
Suun kautta otettavaa herpesinfektiota ennaltaehkäisevää lääkitystä on annettava kaikille potilaille hoitojakson ensimmäisestä päivästä lähtien ja sitä on jatkettava vähintään kuukauden ajan Lemtrada-hoitojakson jälkeen (ks. myös kohtaa "Infektiot" kohdassa Varoitukset ja käyttöön liittyvät varotoimet). Kliinisissä tutkimuksissa potilaille annettiin asikloviiriä 200 mg kaksi kertaa päivässä tai vastaavaa.
Erityispotilasryhmät
Iäkkäät potilaat
Kliinisissä tutkimuksissa ei ollut mukana yli 61-vuotiaita potilaita. Ei ole tutkittu, esiintyykö näillä potilailla erilainen hoitovaste kuin nuoremmilla potilailla.
Munuaisten tai maksan toimintahäiriö
Lemtrada-valmisteen käyttöä munuaisten tai maksan toimintahäiriötä sairastavilla potilailla ei ole tutkittu.
Pediatriset potilaat
Lemtrada-valmisteen turvallisuutta ja tehoa 0–18-vuotiaiden MS-tautia sairastavien lasten hoidossa ei ole vielä varmistettu. Alemtutsumabivalmisteella ei ole relevanttia käyttötarkoitusta alle 10-vuotiailla lapsipotilailla multippeliskleroosin hoidossa. Tietoja ei ole saatavilla.
Antotapa
Lemtrada on laimennettava ennen infuusiota. Laimennettu liuos on annettava laskimoinfuusiona noin neljän tunnin kuluessa.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
HIV-infektio.
Potilaat, joilla on vaikea infektio, sen täydelliseen parantumiseen asti.
Potilaat, joilla on huonossa hoitotasapainossa oleva hypertensio.
Potilaat, joilla on aiemmin todettu pään ja kaulan alueen valtimon dissekoituminen.
Potilaat, joilla on aiemmin ollut aivohalvaus.
Potilaat, joilla on aiemmin ollut angina pectoris tai sydäninfarkti.
Potilaat, joilla tiedetään olevan koagulopatia tai jotka saavat verihiutaleiden estäjiä tai antikoagulantteja.
Potilaat, joilla on muita samanaikaisia autoimmuunisairauksia (MS-taudin lisäksi).
Varoitukset ja käyttöön liittyvät varotoimet
Lemtrada-valmistetta ei suositella potilaille, joilla tauti ei ole aktiivinen, ja joiden tila on vakaa nykyisellä hoidolla.
Lemtrada-valmisteella hoidettaville potilaille on annettava pakkausseloste, potilasvaroituskortti ja potilaan opas. Ennen hoitoa potilaille on kerrottava riskeistä ja hyödyistä sekä tarpeesta sitoutua seurantaan, joka alkaa hoidon aloittamisesta ja kestää vähintään 48 kuukautta toisen Lemtrada-hoitojakson viimeisen infuusion jälkeen. Jos lisähoitojakso annetaan, turvallisuusseurantaa on jatkettava vähintään 48 kuukautta viimeisen infuusion jälkeen.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Autoimmuniteetti
Hoito voi aiheuttaa autovasta-aineiden muodostumista ja lisätä mahdollisesti vakavien ja hengenvaarallisten autoimmuunivälitteisten tilojen riskiä. Autoimmuunisairauksia, kuten kilpirauhassairauksia, immunologista trombosytopeenista purppuraa (ITP), munuaissairauksia (esim. tyvikalvovasta-aineglomerulonefriittia), autoimmuunihepatiittia (AIH), hankinnaista A-hemofiliaa, tromboottista trombosytopeenista purppuraa, sarkoidoosia ja autoimmuunienkefaliittia on ilmoitettu. Myyntiluvan myöntämisen jälkeen on todettu tapauksia, joissa potilaalle on kehittynyt useita autoimmmuunisairauksia Lemtrada-hoidon jälkeen. Jos potilaalle kehittyy autoimmuniteetti, hänet on tutkittava autoimmuunivälitteisten sairauksien varalta (ks. kohta Vasta-aiheet). Potilaiden ja lääkärien on oltava tietoisia autoimmuunisairauksien mahdollisesta kehittymisestä myöhäisemmässä vaiheessa, 48 kuukauden seurantajakson jälkeen.
Hankinnainen A-hemofilia
Hankinnaista A-hemofiliaa (hyytymistekijä VIII:n vasta-aineita) on ilmoitettu sekä kliinisissä tutkimuksissa että myyntiluvan myöntämisen jälkeen. Potilailla on tyypillisesti spontaaneja ihonalaisia hematoomia ja laajoja mustelmia, mutta myös hematuriaa, nenäverenvuotoa, maha-suolikanavan verenvuotoa tai muuntyyppistä verenvuotoa saattaa esiintyä. Veren hyytymistutkimus, johon sisältyy APTT-aika, on tehtävä kaikille potilaille, joilla on tällaisia oireita. Jos potilaalla todetaan pidentynyt APTT-aika, hänet on lähetettävä hematologin hoitoon. Potilaille on kerrottava hankinnaisen A-hemofilian oireista ja löydöksistä ja heitä on kehotettava hakeutumaan välittömästi hoitoon, jos tällaisia oireita ilmenee.
Tromboottinen trombosytopeeninen purppura (TTP)
Lemtrada-hoitoa saaneilla potilailla on ilmoitettu TTP:n kehittymistä lääkkeen myyntiluvan myöntämisen jälkeen, ja yksi tapauksista johti kuolemaan. TTP on vakava tila, joka vaatii välitöntä arviointia ja pikaista hoitoa ja voi kehittyä useiden kuukausien kuluttua viimeisen Lemtrada-infuusion annon jälkeen. TTP:n tunnusomaisia piirteitä saattavat olla trombosytopenia, mikroangiopaattinen hemolyyttinen anemia, neurologiset oireet, kuume ja munuaisten vajaatoiminta.
Autoimmuunienkefaliitti
Lemtrada-valmistetta saaneilla potilailla on ilmoitettu autoimmuunienkefaliittia. Autoimmuunienkefaliitille on tyypillistä subakuutisti alkava (kuukausien kuluessa nopeasti etenevä) muistin heikkeneminen, psyykkisen tilan muuttuminen tai psyykkinen oireilu, johon yleensä liittyy uutena ilmenneitä fokaalisia neurologisia löydöksiä ja kouristuskohtauksia. Jos potilaalla epäillään autoimmuunienkefaliittia, hänelle on järjestettävä neurokuvantaminen (magneettikuvaus), EEG-tutkimus, lannepisto ja asianmukaisten biomarkkerien (esim. neuronaalisten autovasta-aineiden) serologisia tutkimuksia diagnoosin varmistamiseksi ja muiden vaihtoehtoisten syiden poissulkemiseksi.
Immunologinen trombosytopeeninen purppura (ITP)
Vakavia immunologisen trombosytopeenisen purppuran tapauksia esiintyi 12 hoidetulla potilaalla (1 %) MS-taudin kontrolloiduissa kliinisissä tutkimuksissa (vastaa vuositasolla 4,7 tapahtumaa 1 000 potilasvuotta kohti). Lisäksi 12 vakavaa ITP-tapausta on todettu seurannassa, jonka keston mediaani oli 6,1 vuotta (enimmäiskesto 12 vuotta) (kumulatiivinen vuosittainen määrä oli 2,8 tapahtumaa 1 000 potilasvuotta kohti). Yhdelle potilaalle kehittyi ITP, joka jäi tunnistamatta, koska kuukausittaista veriarvojen seurantaa ei oltu vielä toteutettu, ja potilas kuoli aivoverenvuotoon. 79,5 %:ssa tapauksista ITP alkoi 4 vuoden sisällä ensimmäisen Lemtrada-hoidon jälkeen. Joissain tapauksissa ITP kehittyi kuitenkin vuosia myöhemmin. ITP:n oireita voivat olla mm. helposti ilmaantuvat mustelmat, petekiat, spontaani mukokutaaninen verenvuoto (esim. nenäverenvuoto, veriyskä) sekä normaalia runsaampi tai epäsäännöllinen kuukautisvuoto. Veriyskä voi myös viitata tyvikalvovasta-aineglomerulonefriittiin (ks. jäljempänä) ja asianmukainen erotusdiagnoosi on tehtävä. Potilasta on muistutettava pysymään valppaana näiden oireiden varalta ja hakeutumaan välittömästi lääkäriin, jos jokin oire huolestuttaa häntä.
Täydellinen verenkuva ja valkosolujen erittelylaskenta on otettava ennen hoidon aloittamista ja kuukausittain hoidon aloittamisen jälkeen ja aina vähintään 48 kuukauden ajan viimeisen infuusion antamisesta. Tämän ajanjakson jälkeen tarvittavia laboratoriokokeita on otettava ITP:hen viittaavien kliinisten löydösten perusteella. Jos epäillään ITP:tä, täydellinen verenkuva on otettava välittömästi.
Jos ITP varmistuu, asianmukaisiin hoitotoimenpiteisiin on ryhdyttävä viipymättä ja erikoislääkäriä tulee konsultoida välittömästi. Kliinisten MS-taudin tutkimusten tiedot ovat osoittaneet, että veriarvojen seurantavaatimusten noudattaminen ja koulutus ITP:hen liittyvistä oireista ja löydöksistä on johtanut ITP:n varhaiseen havaitsemiseen ja hoitamiseen, ja useimmissa tapauksissa ensilinjan lääkehoidolla on saatu hoitovaste.
Munuaissairaudet
Munuaissairauksia, kuten tyvikalvovasta-aineglomerulonefriittiä, on havaittu 6 potilaalla (0,4 %) kliinisten MS-taudin tutkimusten seurannassa, jonka keston mediaani oli 6,1 vuotta (enimmäiskesto 12 vuotta), ja ne ovat yleensä esiintyneet 39 kuukauden sisällä viimeisen Lemtrada-annoksen jälkeen. Kliinisissä tutkimuksissa esiintyi kaksi tyvikalvovasta-aineglomerulonefriittitapausta. Molemmat tapaukset olivat vakavia ja ne tunnistettiin varhaisessa vaiheessa kliinisen ja laboratorioseurannan avulla. Molemmissa tapauksissa hoitotulos oli hyvä.
Munuaissairauden kliinisiä merkkejä voivat olla seerumin kreatiniiniarvon nousu, hematuria ja/tai proteinuria. Veriysköksinä ilmenevää keuhkorakkuloiden verenvuotoa voi esiintyä tyvikalvovasta-aineglomerulonefriitin yhteydessä, vaikka sitä ei havaittukaan kliinisissä tutkimuksissa. Veriyskä voi myös viitata ITP:hen (ks. edellä) tai hankinnaiseen A-hemofiliaan, ja asianmukainen erotusdiagnoosi on tehtävä. Potilasta on muistutettava pysymään valppaana ilmenevien oireiden varalta ja hakeutumaan välittömästi lääkärinhoitoon, jos hänellä on huolta aiheuttavia oireita. Tyvikalvovasta-aineglomerulonefriitti voi aiheuttaa dialyysiä ja/tai elinsiirtoa vaativan munuaisten vajaatoiminnan, jos sitä ei hoideta nopeasti, ja se voi olla hengenvaarallinen, jos se jätetään hoitamatta.
Seerumin kreatiniinipitoisuudet on tarkistettava ennen hoidon aloittamista ja kuukausittain hoidon aloittamisen jälkeen ja vähintään 48 kuukauden ajan viimeisen infuusion antamisesta. Virtsakoe, mukaan lukien virtsan mikroskopointi, on tehtävä ennen hoidon aloittamista ja sen jälkeen kuukauden välein aina vähintään 48 kuukauden ajan viimeisen infuusion jälkeen. Mikäli havaitaan kliinisesti merkittäviä muutoksia seerumin kreatiniiniarvoissa, selittämätöntä hematuriaa ja/tai proteinuriaa, munuaissairauden lisäarviointi on aloitettava ja potilas lähetettävä viipymättä erikoislääkärille. Munuaissairauksien varhainen tunnistus ja hoito voi pienentää huonojen hoitotulosten vaaraa. Tämän ajanjakson jälkeen kokeita on tehtävä munuaissairauksiin viittaavien kliinisten löydösten perusteella.
Kilpirauhashäiriöt
Endokriinisiä kilpirauhashäiriöitä mukaan lukien kilpirauhasen autoimmuunihäiriöt on havaittu 36,8 %:lla potilaista, joita hoidettiin 12 mg:n Lemtrada-annoksella MS-taudin kliinisissä tutkimuksissa, joissa seurannan keston mediaani oli 6,1 vuotta (enimmäiskesto 12 vuotta) ensimmäisen Lemtrada-annoksen jälkeen. Kilpirauhastapahtumia esiintyi enemmän sekä Lemtrada- että interferonibeeta 1a (IFNB-1a) ‑hoitoryhmissä silloin, kun potilailla oli ollut aiemmin kilpirauhassairaus. Havaittuja kilpirauhasen autoimmuunihäiriöitä ovat kilpirauhasen liika- tai vajaatoiminta. Useimmat tapahtumat olivat lieviä tai keskivaikeita. Vakavia endokriinisiä tapahtumia esiintyi 4,4 %:lla potilaista ja Basedowin tautia (tunnettu myös Gravesin tautina), liikatoimintaa, vajaatoimintaa, autoimmuunia kilpirauhastulehdusta ja struumaa esiintyi enemmän kuin yhdellä potilaalla. Useimmat kilpirauhastapahtumat saatiin hallintaan tavallisella lääkehoidolla, mutta jotkut potilaat tarvitsivat leikkaushoitoa. Myyntiluvan myöntämisen jälkeen useille potilaille, joille kehittyi koepalalla varmistettu autoimmuunihepatiitti, oli aiemmin kehittynyt kilpirauhasen autoimmuunihäiriöitä.
Kilpirauhasen toimintakokeet, kuten S-TSH-arvo, on tarkistettava ennen hoidon aloittamista, hoidon aikana ja sen jälkeen kolmen kuukauden välein 48 kuukauden ajan viimeisen infuusion antamisesta. Tämän ajanjakson jälkeen lisätutkimuksia tehdään kilpirauhasen toimintahäiriöön viittaavien kliinisten löydösten perusteella tai jos potilas on raskaana.
Kilpirauhassairaus aiheuttaa erityisen riskin raskaana oleville naisille (ks. kohta Raskaus ja imetys).
Kliinisissä tutkimuksissa kilpirauhaseen liittyvä haittatapahtuma kehittyi 74 %:lle potilaista, joilla anti-tyreoideaperoksidaasi- (anti-TPO‑) vasta-ainestatus oli lähtötilanteessa positiivinen, ja 38 %:lle potilaista, joilla vasta-ainestatus oli lähtötilanteessa negatiivinen. Suurin osa (noin 80%) potilaista, joilla esiintyi kilpirauhastapahtuma hoidon jälkeen, oli anti-TPO-vasta-ainenegatiivisia lähtötilanteessa. Täten riippumatta potilaan hoitoa edeltävästä anti-TPO-vasta-ainestatuksesta, heille voi kehittyä kilpirauhaseen liittyvä haittavaikutus ja potilaille on tehtävä kaikki kilpirauhaskokeet säännöllisesti edellä kuvatun mukaisesti.
Sytopeniat
Epäiltyjä autoimmuunisytopenioita, kuten neutropeniaa, hemolyyttistä anemiaa ja pansytopeniaa, on ilmoitettu harvoin kliinisissä MS-taudin tutkimuksissa. Täydellisen verenkuvan tuloksia (ks. yllä kohta ITP) käytetään seurattaessa potilaita sytopenioiden, kuten neutropenian, varalta. Jos varmistuu, että potilaalla on sytopenia, asianmukaisiin hoitotoimenpiteisiin on ryhdyttävä viipymättä ja potilas on lähetettävä erikoislääkärille.
Autoimmuunihepatiitti ja maksavauriot
Lemtrada-valmistetta saaneilla potilailla on ilmoitettu autoimmuunihepatiittia (myös kuolemaan johtaneita tapauksia ja maksansiirtoa vaatineita tapauksia) ja infektioihin liittyviä maksavaurioita (ks. kohta Vasta-aiheet). Maksan toimintakokeet on tehtävä ennen hoidon aloittamista ja kuukauden välein vähintään 48 kuukauden ajan viimeisen infuusion antamisesta. Potilaille on kerrottava autoimmuunihepatiitin, maksavaurion ja maksavaurioon liittyvien oireiden riskistä.
Hemofagosyyttinen lymfohistiosytoosi (HLH)
Lemtrada-valmistetta saaneilla potilailla on ilmoitettu hemofagosyyttistä lymfohistiosytoosia (myös kuolemaan johtaneita tapauksia) myyntiluvan myöntämisen jälkeen. Hemofagosyyttinen lymfohistiosytoosi on patologiseen immuuniaktivaatioon liittyvä hengenvaarallinen oireyhtymä, jolle ovat tunnusomaisia erittäin voimakkaan systeemisen tulehduksen kliiniset oireet ja löydökset. Hemofagosyyttinen lymfohistiosytoosi aiheuttaa tyypillisesti kuumetta, hepatomegaliaa ja sytopenioita. Hemofagosyyttiseen lymfohistiosytoosiin liittyy korkea kuolleisuus, ellei tilaa todeta varhaisessa vaiheessa ja hoideta. Oireiden on ilmoitettu ilmenevän muutaman kuukauden – neljän vuoden kuluessa hoidon aloittamisesta. Jos potilaalle ilmaantuu patologisen immuuniaktivaation varhaisia oireita, potilas on tutkittava välittömästi ja hemofagosyyttisen lymfohistiosytoosin diagnoosi on otettava huomioon.
Infuusioon liittyvät reaktiot
Kliinisissä tutkimuksissa infuusioon liittyviksi reaktioiksi katsottiin kuuluviksi kaikki haittavaikutukset, joita esiintyi Lemtrada-infuusion aikana tai 24 tunnin sisällä sen antamisesta. Suurin osa näistä voi johtua sytokiinien vapautumisesta infuusion aikana. Kliinisissä MS-tautitutkimuksissa useimmat Lemtrada-valmisteella hoidetut potilaat kokivat lieviä tai keskivaikeita infuusioon liittyviä reaktioita 12 mg:n Lemtrada-annoksen antamisen aikana ja/tai 24 tuntia sen jälkeen. Infuusioon liittyviä reaktioita esiintyi enemmän ensimmäisellä hoitojaksolla kuin seuraavilla hoitojaksoilla. Kaiken saatavilla olevan seurantatiedon perusteella, mukaan lukien lisähoitojaksoja saaneet potilaat, yleisimpiä infuusioon liittyviä reaktioita olivat päänsärky, ihottuma, kuume, pahoinvointi, urtikaria, kutina, unettomuus, vilunväristykset, punoitus, uupumus, hengenahdistus, makuaistin häiriöt, epämukava tunne rinnassa, yleistynyt ihottuma, takykardia, bradykardia, dyspepsia, huimaus ja kipu. Vakavia reaktioita, kuten päänsärkyä, kuumetta, urtikariaa, takykardiaa, eteisvärinää, pahoinvointia, rintakipua ja hypotensiota, esiintyi 3 %:lla potilaista. Anafylaksian kliiniset löydökset voivat olla infuusioreaktioiden löydösten kaltaisilta, mutta ne ovat yleensä vaikeampia tai voivat olla hengenvaarallisia. Anafylaksiaan liittyviä reaktioita on ilmoitettu harvoin, toisin kuin infuusioon liittyviä reaktioita.
On suositeltavaa antaa potilaille esilääkitys Lemtrada-valmisteen infuusioreaktioiden lieventämiseksi (ks. kohta Annostus ja antotapa).
Kontrolloiduissa kliinisissä tutkimuksissa useimmat potilaat saivat antihistamiineja ja/tai kuumelääkkeitä vähintään ennen yhtä Lemtrada-infuusiota. Potilailla voi esiintyä infuusioon liittyviä reaktioita esilääkityksestä huolimatta. Infuusioreaktioiden tarkkailua suositellaan Lemtrada-infuusion aikana ja vähintään kaksi tuntia sen jälkeen. Pidennettyä seuranta-aikaa (sairaalahoitoa) on harkittava tarpeen mukaan. Jos vaikeita infuusioreaktioita esiintyy, infuusio on välittömästi keskeytettävä. Anafylaktisten tai vakavien reaktioiden hoitoon on oltava tarvittavat valmiudet (ks. jäljempänä).
Aikuisten Stillin tauti (AOSD)
Myyntiluvan myöntämisen jälkeen Lemtrada-hoitoa saaneilla potilailla on ilmoitettu aikuisten Stillin tautia (AOSD). Aikuisten Stillin tauti on harvinainen tulehduksellinen sairaus, joka vaatii kiireellistä arviointia ja hoitoa. Aikuisten Stillin tautia sairastavilla potilailla saattaa ilmetä merkkeinä ja oireina kuumetta, niveltulehdusta, ihottumaa ja leukosytoosia, ilman että ilmenee infektioita, maligniteetteja tai muita reumasairauksia. Lemtrada-hoidon keskeyttämistä tai lopettamista on harkittava, jos merkeille tai oireille ei voida todeta muita syitä.
Muut vakavat reaktiot, joilla on ajallinen yhteys Lemtrada-infuusioon
Myyntiluvan myöntämisen jälkeen on ilmoitettu harvinaisia, vakavia, joskus kuolemaan johtaneita ja ennalta-arvaamattomia haittatapahtumia eri elinjärjestelmissä. Useimmissa tapauksissa reaktiot ilmaantuivat 1–3 vuorokauden kuluessa Lemtrada-infuusion antamisesta. Reaktioita on ilmaantunut minkä tahansa annostelupäivän sekä myös toisen hoitojakson jälkeen. Potilaita on kehotettava hakeutumaan välittömästi hoitoon, jos tällaisia oireita ilmenee, ja heille on kerrottava mahdollisista viiveellä ilmaantuvista reaktioista.
Aivoverenvuoto
Ilmoitetuissa tapauksissa useat potilaat olivat alle 50-vuotiaita eikä heillä ollut aiemmin ollut hypertensiota tai verenvuotohäiriöitä eivätkä he olleet aiemmin saaneet samanaikaisesti antikoagulantteja tai verihiutaleiden estäjiä. Joillakin potilailla verenpaine oli kohonnut lähtötilanteesta ennen verenvuotoa.
Sydänlihasiskemia ja sydäninfarkti
Ilmoitetuissa tapauksissa useat potilaat olivat alle 40-vuotiaita eikä heillä ollut iskeemisen sydänsairauden riskitekijöitä. Joillakin potilailla oli todettu tilapäisesti epänormaali verenpaine ja/tai sydämen syke infuusion aikana.
Pään ja kaulan alueen valtimon dissekoituminen
Pään ja kaulan alueen valtimon dissekoitumista, myös useampia dissekoitumisia, on ilmoitettu sekä ensimmäisten päivien aikana Lemtrada-infuusion jälkeen että myöhemmin ensimmäisen kuukauden sisällä infuusion jälkeen.
Keuhkorakkuloiden verenvuoto
Ilmoitetut tapaukset, joissa tapahtumilla oli ajallinen yhteys, eivät liittyneet tyvikalvovasta-aineglomerulonefriittiin (Goodpasturen oireyhtymään).
Trombosytopenia
Ilmoitetut trombosytopeniatapaukset ilmaantuivat ensimmäisten päivien aikana infuusion jälkeen (toisin kuin ITP). Ne paranivat usein itsestään ja olivat suhteellisen lieviä, vaikka monissa tapauksissa vaikeusaste ja lopputulos eivät ole tiedossa.
Sydänpussitulehdus
Sydänpussitulehdusta, sydänpussin nestekertymää ja muita sydänpussiin liittyviä tapahtumia on ilmoitettu harvinaisina tapauksina, sekä akuutin infuusioreaktion yhteydessä että myöhemmin ilmaantuneina.
Keuhkotulehdus
Keuhkotulehdusta on ilmoitettu Lemtrada-infuusioita saaneilla potilailla. Useimmat tapauksista ilmaantuivat yhden kuukauden sisällä Lemtrada-hoidosta. Potilaita on neuvottava ilmoittamaan keuhkotulehduksen oireet, joihin voi kuulua hengenahdistusta, yskää, vinkunaa, rintakipua tai puristuksen tunnetta rinnassa ja veriyskää.
Infuusioon liittyvät ohjeet Lemtrada-infuusioon ajallisesti liittyvien vakavien reaktioiden vähentämiseksi
-
Tutkimukset ennen infuusiota:
- EKG-tutkimus ja vitaalitoimintojen, kuten sykkeen ja verenpaineen, mittaaminen lähtötilanteessa
- laboratoriokokeet (täydellinen verenkuva ja erittelylaskenta, seerumin transaminaasit, seerumin kreatiniini, kilpirauhasen toimintakoe ja virtsakoe, mukaan lukien virtsan mikroskopointi)
-
Infuusion aikana:
- sykkeen, verenpaineen ja potilaan kliinisen tilan jatkuva/tiheä seuranta (vähintään tunnin välein)
- Infuusio on keskeytettävä
- jos ilmenee vakava haittatapahtuma
- jos potilaalla ilmenee kliinisiä oireita, jotka viittaavat infuusioon liittyvän vakavan haittavaikutuksen kehittymiseen (sydänlihasiskemia, aivoverenvuoto, pään ja kaulan alueen valtimon dissekoituminen tai keuhkorakkuloiden verenvuoto)
- Infuusio on keskeytettävä
- sykkeen, verenpaineen ja potilaan kliinisen tilan jatkuva/tiheä seuranta (vähintään tunnin välein)
-
Infuusion jälkeen:
- Seurantaa infuusioreaktioiden varalta on suositeltavaa jatkaa vähintään kahden tunnin ajan Lemtrada-infuusion jälkeen. Potilaita, joilla on kliinisiä oireita, jotka viittaavat infuusioon ajallisesti liittyvän vakavan haittatapahtuman (sydänlihasiskemian, aivoverenvuodon, pään ja kaulan alueen valtimon dissekoitumisen tai keuhkorakkuloiden verenvuodon) kehittymiseen, on tarkkailtava huolellisesti, kunnes oireet häviävät kokonaan. Seuranta-aikaa on tarpeen mukaan pidennettävä (sairaalahoito). Potilaille on kerrottava mahdollisista viiveellä ilmaantuvista infuusioon liittyvistä reaktioista, ja heitä on kehotettava ilmoittamaan oireista ja hakeutumaan lääkärin hoitoon.
- Verihiutalemäärä on tutkittava välittömästi infuusion jälkeen, ensimmäisen infuusiohoitojakson päivinä 3 ja 5 sekä välittömästi infuusion jälkeen kaikkien seuraavien hoitojaksojen päivänä 3. Kliinisesti merkittävää trombosytopeniaa on seurattava sen paranemiseen asti. Potilaan lähettämistä hematologin hoitoon on harkittava.
Infektiot
Kaksi vuotta kestäneissä kontrolloiduissa MS-taudin kliinisissä tutkimuksissa infektioita esiintyi 71 %:lla 12 mg:n Lemtrada-annoksella hoidetuista potilaista verrattuna 53 %:iin potilaista, jotka saivat ihonalaisesti interferoni beeta-1a:ta [IFNB 1a] (44 mikrog 3 kertaa viikossa). Infektiot olivat lieviä tai keskivaikeita. Infektioita, joita esiintyi useammin Lemtrada-valmisteella kuin IFNB-1a:lla hoidetuilla potilaita, olivat nasofaryngiitti, virtsatieinfektio, ylempien hengitysteiden infektio, sinuiitti, suuherpes, influenssa ja bronkiitti. Kontrolloiduissa kliinisissä MS-taudin tutkimuksissa esiintyi vakavia infektioita 2,7 %:lla Lemtrada-valmisteella hoidetuista verrattuna 1 %:iin IFNB-1a:lla hoidetuista potilaista. Lemtrada-ryhmän vakavia infektioita olivat: umpisuolentulehdus, gastroenteriitti, keuhkokuume, vyöruusu ja hammasinfektio. Infektiot olivat yleensä kestoltaan tavallisia ja hoidettavissa vakiintuneella lääkehoidolla.
Infektioiden kumulatiivinen vuosittainen määrä oli 0,99, kun seurannan keston mediaani oli 6,1 vuotta (enimmäiskesto 12 vuotta) ensimmäisen Lemtrada-annoksen jälkeen. Kontrolloiduissa kliinisissä tutkimuksissa vastaava arvo oli 1,27.
Kliinisissä tutkimuksissa vakavia varicella zoster ‑virusinfektioita, kuten primääriä vesirokkoa ja varicella zoster ‑viruksen uudelleenaktivoitumista, on esiintynyt useammin 12 mg:n Lemtrada-annoksella hoidetuilla potilailla (0,4 %) verrattuna INFB-1a:lla hoidettuihin potilaisiin (0 %). Kohdunkaulan ihmisen papilloomavirusinfektiota (HPV-infektiota), kuten kohdunkaulan dysplasiaa ja anogenitaalisia syyliä, on myös ilmoitettu 12 mg:n Lemtrada-annoksilla hoidetuilla potilailla (2 %). On suositeltavaa, että naispotilaille tehdään HPV-seulonta vuosittain.
Lemtrada-valmisteella hoidetuilla potilailla on ilmoitettu sytomegalovirusinfektioita ja niiden uudelleenaktivoitumista. Useimmat tapaukset ilmenivät kahden kuukauden kuluessa alemtutsumabihoidon aloittamisesta. Ennen hoidon aloittamista voidaan harkita immunologisen serostatuksen arviointia paikallisten ohjeiden mukaan.
Epstein-Barrin virus (EBV) -infektioita, mukaan lukien uudelleenaktivoitumista sekä vaikeita ja joskus kuolemaan johtaneita EBV-hepatiittitapauksia, on ilmoitettu Lemtrada-valmistetta saaneilla potilailla.
Kliinisissä tutkimuksissa on ilmoitettu tuberkuloosia Lemtrada-valmisteella ja IFNB-1a:lla hoidetuilla potilailla. Aktiivista ja latenttia tuberkuloosia, mukaan lukien harvoja tapauksia yleistynyttä tuberkuloosia, on ilmoitettu 0,3 %:lla Lemtrada-valmisteella hoidetuista potilaista, useimmiten sellaisilla maantieteellisillä alueilla, joissa sitä esiintyy yleisesti. Ennen hoidon aloittamista kaikki potilaat on arvioitava sekä aktiivisen että inaktiivisen (latentin) tuberkuloosi-infektion varalta paikallisten ohjeiden mukaan.
Listerioosia/Listeriameningiittiä on ilmoitettu Lemtrada-hoitoa saaneilla potilailla yleensä yhden kuukauden sisällä Lemtrada-infuusiosta. Infektioriskin pienentämiseksi Lemtrada-hoitoa saaneiden potilaiden on vältettävä kypsentämättömän tai huonosti kypsennetyn lihan, pehmeiden juustojen ja pastöroimattomien maitotuotteiden nauttimista kaksi viikkoa ennen hoitoa, hoidon aikana ja vähintään yhden kuukauden ajan Lemtrada-infuusion jälkeen.
MS-taudin kontrolloiduissa kliinisissä tutkimuksissa pinnallisia sieni-infektioita, erityisesti suun ja emättimen hiivatulehdusta, esiintyi useammin Lemtrada-valmisteella hoidetuilla (12 %) kuin IFNB‑1a:lla hoidetuilla potilailla (3 %).
Lemtrada-hoidon aloitusta on siirrettävä potilailla, joilla on vaikea aktiivinen infektio, sen paranemiseen asti. Lemtrada-valmistetta saaneita potilaita on neuvottava ilmoittamaan infektioiden oireet lääkärille.
Suun kautta otettava herpestä ennaltaehkäisevä hoito on aloitettava Lemtrada-hoidon ensimmäisenä päivänä ja sitä on jatkettava vähintään kuukauden ajan jokaisen hoitojakson jälkeen. Kliinisissä tutkimuksissa potilaille annettiin asikloviiriä 200 mg kaksi kertaa päivässä tai vastaavaa.
Lemtrada-valmistetta ei ole käytetty MS-taudin hoitoon samanaikaisesti antineoplastisen tai immunosuppressiivisen hoidon kanssa tai niiden jälkeen. Kuten muidenkin immunomoduloivien hoitojen kohdalla, mahdolliset yhteisvaikutukset potilaan immuunijärjestelmään on otettava huomioon harkittaessa Lemtrada-valmisteen antamista. Lemtrada-valmisteen käyttö samanaikaisesti näiden hoitojen kanssa voi lisätä immunosuppression vaaraa.
Tietoja Lemtrada-valmisteen liittymisestä B-hepatiittiviruksen (HBV) tai C-hepatiittiviruksen (HCV) uudelleenaktivoitumiseen ei ole, koska potilaat, joilla oli merkkejä aktiivisista tai kroonisista infektioista, suljettiin pois kliinisistä tutkimuksista. HBV- ja/tai HCV-infektioriskipotilaiden seulomista ennen Lemtrada-hoidon aloittamista on harkittava ja varovaisuutta on noudatettava määrättäessä Lemtrada-valmistetta potilaille, jotka on tunnistettu HBV- ja/tai HCV-viruksen kantajiksi, koska näillä potilailla voi olla palautumattoman maksavaurion vaara viruksen mahdollisen uudelleenaktivoitumisen vuoksi.
Progressiivinen multifokaalinen leukoenkefalopatia (PML)
Progressiivista multifokaalista leukoenkefalopatiaa (myös kuolemaan johtanutta) on ilmoitettu harvinaisina tapauksina MS-potilailla alemtutsumabihoidon jälkeen. Alemtutsumabia saavia potilaita täytyy tarkkailla mahdollisesti progressiiviseen multifokaaliseen leukoenkefalopatiaan liittyvien merkkien varalta. Erityisen merkittäviä riskitekijöitä ovat aiemmat immunosuppressiiviset hoidot, erityisesti muut MS-hoidot, joihin tiedetään liittyvän progressiivisen multifokaalisen leukoenkefalopatian kehittymisen riski.
Magneettikuvauksessa saattaa ilmetä löydöksiä ennen kliinisiä merkkejä tai oireita. Ennen alemtutsumabihoidon aloittamista tai uudelleen antamista on tehtävä magneettikuvaus, jossa arvioidaan progressiiviseen multifokaaliseen leukoenkefalopatiaan viittaavat merkit. Tarvittaessa on tehtävä lisätutkimuksia, kuten selkäydinnesteen tutkiminen JC-viruksen DNA:n varalta ja toistuvia neurologisia arviointeja. Lääkärin on tarkkailtava erityisesti progressiiviseen multifokaaliseen leukoenkefalopatiaan viittaavia merkkejä, joita potilas ei välttämättä havaitse (esim. kognitiivisia, neurologisia ja psykiatrisia oireita). Potilasta on myös kehotettava kertomaan hoidostaan sukulaisilleen tai huoltajilleen, koska he saattavat havaita oireita, joita potilas ei tiedosta. Erotusdiagnoosissa on otettava huomioon progressiivisen multifokaalisen leukoenkefalopatian mahdollisuus alemtutsumabia saavilla MS-potilailla, joilla ilmenee neurologisia oireita ja/tai joilla todetaan magneettikuvauksessa uusia aivoleesioita.
Jos potilaalla todetaan progressiivinen multifokaalinen leukoenkefalopatia, alemtutsumabihoitoa ei pidä aloittaa tai antaa potilaalle uudelleen.
Akuutti kivetön sappirakkotulehdus
Lemtrada-hoito saattaa suurentaa akuutin kivettömän sappirakkotulehduksen riskiä. Kontrolloiduissa kliinisissä tutkimuksissa 0,2 %:lle Lemtrada-hoitoa saaneista MS-potilaista kehittyi akuutti kivetön sappirakkotulehdus, kun vastaava osuus oli IFNB-1a:lla hoidetuilla potilailla 0 %. Myyntiluvan myöntämisen jälkeen Lemtrada-valmistetta saaneilla potilailla on ilmoitettu lisää akuutin kivettömän sappirakkotulehduksen tapauksia. Aika oireiden puhkeamiseen vaihteli alle 24 tunnista 2 kuukauteen Lemtrada-infuusion jälkeen. Useimpia potilaita hoidettiin konservatiivisesti antibiooteilla ja he toipuivat ilman kirurgisia toimenpiteitä, kun taas osalta potilaista poistettiin sappirakko. Akuutin kivettömän sappirakkotulehduksen oireita ovat vatsakipu, vatsan arkuus, kuume, pahoinvointi ja oksentelu. Akuutti kivetön sappirakkotulehdus on tila, johon saattaa liittyä suuri sairastuvuus ja kuolleisuus, ellei sitä todeta varhaisessa vaiheessa ja hoideta. Jos epäillään akuuttia kivetöntä sappirakkotulehdusta, potilas on tutkittava ja hoidettava nopeasti.
Maligniteetti
Kuten muidenkin immunomodulaatiohoitojen kohdalla, varovaisuutta on noudatettava aloitettaessa Lemtrada-hoito potilaille, joilla on aiempi tai samanaikainen maligniteetti. Tällä hetkellä ei tiedetä, aiheuttaako Lemtrada suuremman kilpirauhasmaligniteettien kehittymisen vaaran, koska kilpirauhasen autoimmuniteetti voi sinänsä olla riskitekijä kilpirauhasen maligniteeteille.
Ehkäisyvalmisteet:
Istukan läpi kulkeutumista ja mahdollista Lemtrada-valmisteen farmakologista aktiviteettia havaittiin hiirillä tiineyden ja poikimisen jälkeen. Hedelmällisessä iässä olevien naisten on käytettävä tehokkaita ehkäisymenetelmiä hoidon aikana ja 4 kuukauden ajan Lemtrada-hoitojakson jälkeen (ks. kohta Raskaus ja imetys).
Rokotteet
On suositeltavaa, että potilaat saavat paikallisten vaatimusten mukaisen immunisaation vähintään 6 viikkoa ennen Lemtrada-hoitoa. Kykyä kehittää immuunivastetta rokotteisiin Lemtrada-hoidon jälkeen ei ole tutkittu.
Eläviä viruksia sisältävillä rokotteilla tehdyn immunisaation turvallisuutta Lemtrada-hoitojakson jälkeen ei ole tutkittu virallisesti kontrolloiduissa kliinisissä MS-taudin tutkimuksissa, eikä rokotteita saa antaa MS‑potilaille, jotka ovat äskettäin saaneet Lemtrada-hoitojakson.
Varicella zoster -viruksen vasta-ainetestaus/‑rokotus
Kuten minkä tahansa immunomoduloivan lääkevalmisteen kohdalla, ennen Lemtrada-hoitojakson aloittamista potilaat, joilla ei ole ollut vesirokkoa tai jotka eivät ole saaneet rokotusta varicella zoster ‑virusta (VZV) vastaan, on testattava VZV-vasta-aineiden varalta. Vasta-ainenegatiivisten potilaiden vesirokkorokotusta on harkittava ennen Lemtrada-hoidon aloittamista. Jotta vesirokkorokotuksen täysi teho voidaan saavuttaa, Lemtrada-hoitoa on siirrettävä 6 viikkoa rokotuksen jälkeen.
Suositellut laboratoriokokeet potilaiden seuraamiseen
Potilaalle on tehtävä kliininen tutkimus ja laboratoriokokeita on otettava säännöllisin väliajoin vähintään 48 kuukauden ajan viimeisen Lemtrada-hoitojakson jälkeen autoimmuunisairauksien varhaisten merkkien havaitsemiseksi:
- Täydellinen verenkuva ja erittelylaskenta, seerumin transaminaasi- ja kreatiniinipitoisuudet (ennen hoidon aloittamista ja kuukausittain sen jälkeen)
- Virtsakoe, mukaan lukien virtsan mikroskopointi (ennen hoidon aloittamista ja kuukausittain sen jälkeen)
- Kilpirauhasen toimintakoe, kuten TSH-pitoisuus (ennen hoidon aloittamista ja joka 3. kuukausi sen jälkeen).
Tietoa alemtutsumabin käytöstä ennen Lemtrada-valmisteen myyntiluvan saamista yrityksen sponsoroimien tutkimusten ulkopuolelta
Seuraavia haittavaikutuksia esiintyi ennen Lemtrada-valmisteen myyntiluvan saamista, kun alemtutsumabia käytettiin kroonisen lymfaattisen leukemian (B-CLL) sekä muiden sairauksien hoitoon, yleensä suuremmilla ja tiheämmin annetuilla annoksilla (esim. 30 mg) kuin mitä suositellaan MS-taudin hoitoon. Koska nämä haittavaikutukset on ilmoitettu vapaaehtoisesti tuntemattoman kokoisesta ryhmästä, ei ole aina mahdollista arvioida luotettavasti niiden esiintyvyyttä tai määrittää niiden syy-seuraussuhdetta alemtutsumabihoitoon.
Autoimmuunisairaus
Alemtutsumabihoitoa saaneilla potilailla ilmoitettuja autoimmuunitapahtumia ovat neutropenia, hemolyyttinen anemia (mukaan lukien kuolemaan johtanut tapaus), hankinnainen hemofilia, tyvikalvovasta-aineglomerulonefriitti ja kilpirauhassairaus. Vakavia ja joskus kuolemaan johtaneita autoimmuunitapahtumia, kuten autoimmuunista hemolyyttista anemiaa, autoimmuunia trombosytopeniaa, aplastista anemiaa, Guillain-Barrén oireyhtymää ja kroonista tulehduksellista demyelinoivaa polyradikuloneuropatiaa on ilmoitettu alemtutsumabilla hoidetuilla potilailla, jotka sairastivat muuta kuin MS-tautia. Yhdellä alemtutsumabilla hoidetulla syöpäpotilaalla on ilmoitettu positiivinen Coombsin testin tulos. Yhdellä alemtutsumabilla hoidetulla syöpäpotilaalla on ilmoitettu kuolemaan johtanut verensiirtoon liittyvä käänteishyljintäsairaus.
Infuusioon liittyvät reaktiot
Vakavia ja joskus kuolemaan johtaneita infuusioon liittyviä reaktioita, kuten bronkospasmeja, hypoksiaa, keuhkoinfiltraatteja, akuuttia hengitysvaikeusoireyhtymää, hengityksen pysähdystä, sydäninfarktia, rytmihäiriöitä, akuuttia sydämen vajaatoimintaa ja sydämenpysähdystä on esiintynyt potilailla, jotka sairastivat muuta kuin MS-tautia ja joita hoidettiin alemtutsumabilla suuremmilla ja useammin otetuilla annoksilla kuin mitä MS-taudissa käytetään. Vaikeaa anafylaksiaa ja muita yliherkkyysreaktioita, kuten anafylaktista sokkia ja angioedeemaa, on myös ilmoitettu.
Infektiot
Vakavia ja joskus kuolemaan johtaneita virus-, bakteeri-, alkueläin- ja sieni-infektioita, myös latenttien infektioiden uudelleenaktivoitumisesta johtuvia, on ilmoitettu MS-tautia sairastamattomilla potilailla, joita hoidettiin alemtutsumabilla suuremmilla ja useammin otetuilla annoksilla kuin mitä MS-taudissa käytetään.
Veri ja imukudos
Vaikeita verenvuotoreaktioita on ilmoitettu potilailla, jotka eivät sairasta MS-tautia.
Sydän
Sydämen vajaatoimintaa, kardiomyopatiaa ja ejektiofraktion pienenemistä on ilmoitettu alemtutsumabilla hoidetuilla MS-tautia sairastamattomilla potilailla, joita oli aiemmin hoidettu mahdollisesti sydäntoksisilla aineilla.
Epstein-Barrin virukseen liittyvät lymfoproliferatiiviset sairaudet
Epstein-Barrin virukseen liittyviä lymfoproliferatiivisia sairauksia on havaittu muussa kuin yrityksen sponsoroimien tutkimusten yhteydessä.
Lemtrada sisältää natriumia, kaliumia ja polysorbaattia
Tämä lääkevalmiste sisältää alle 1 mmol kaliumia (39 mg) yhdessä infuusiossa, eli sen voidaan sanoa olevan ”kaliumiton”.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) yhdessä infuusiossa, eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää 0,12 mg polysorbaatti 80:tä (E433) yhdessä injektiopullossa, jonka nimellinen täyttötilavuus on 1,2 ml, mikä vastaa 0,1 mg/1,0 ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Sellaisia Lemtrada-valmistetta koskevia virallisia yhteisvaikutustutkimuksia, joissa olisi käytetty MS-potilaille suositeltua annosta, ei ole tehty. Kontrolloidussa kliinisessä tutkimuksessa MS-potilaiden, joita oli äskettäin hoidettu beetainterferonilla ja glatirameeriasetaatilla, täytyi keskeyttää hoito 28 päivän ajaksi ennen Lemtrada-hoidon aloittamista.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset
Seerumin lääkepitoisuudet olivat pieniä tai havaitsemattomia noin 30 päivän sisällä kunkin hoitojakson jälkeen. Siksi hedelmällisessä iässä olevien naisten on käytettävä tehokasta ehkäisyä Lemtrada-hoitojakson aikana ja 4 kuukauden ajan jokaisen hoitojakson jälkeen.
Raskaus
On vain vähän tietoja alemtutsumabin käytöstä raskaana olevilla naisilla. Lemtrada-valmistetta saa antaa raskaana oleville naisille ainoastaan, kun odotettavissa olevat hyödyt ylittävät sikiölle mahdollisesti koituvan haitan.
Ihmisen IgG:n tiedetään läpäisevän istukan; myös alemtutsumabi voi läpäistä istukan ja siten mahdollisesti aiheuttaa riskin sikiölle. Eläinkokeet ovat osoittaneet lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Ei tiedetä, voiko alemtutsumabi aiheuttaa haittaa sikiölle, kun sitä annetaan raskaana olevalle naiselle, tai voiko se vaikuttaa lisääntymiskykyyn.
Kilpirauhassairaus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet Kilpirauhassairaudet) aiheuttaa erityisen riskin raskaana oleville naisille. Mikäli kilpirauhasen vajaatoimintaa ei hoideta raskauden aikana, on olemassa suurentunut keskenmenon ja sikiöön kohdistuvien vaikutusten, kehitysvammaisuuden ja lyhytkasvuisuuden, vaara. Gravesin tautia sairastavilla äideillä kilpirauhasta stimuloivan hormonin (TSH) reseptorin vasta-aineet voivat siirtyä kehittyvään sikiöön ja aiheuttaa vastasyntyneelle ohimenevän Gravesin taudin.
Imetys
Alemtutsumabia havaittiin imettävien naarashiirien maidossa ja poikasissa.
Ei tiedetä, erittyykö alemtutsumabi ihmisen rintamaitoon. Imetettävään vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida sulkea pois. Siksi imetys on keskeytettävä jokaisen Lemtrada-hoitojakson ajaksi ja 4 kuukauden ajaksi kunkin hoitojakson viimeisen infuusion jälkeen. Rintamaidon kautta saadun immuniteetin hyödyt voivat kuitenkin olla suurempia imetettävälle vastasyntyneelle/imeväiselle kuin potentiaalinen altistus alemtutsumabille.
Hedelmällisyys
Ei ole olemassa riittäviä kliinisiä turvallisuustietoja Lemtrada-valmisteen vaikutuksesta hedelmällisyyteen. Alatutkimuksessa, johon osallistui 13 Lemtrada-valmisteella hoidettua miespotilasta (joita hoidettiin joko 12 mg:n tai 24 mg:n annoksella), ei esiintynyt merkkejä siemennesteen puuttumisesta, siittiökadosta, jatkuvasti matalasta siittiömäärästä, liikkuvuushäiriöistä ja siittiöiden morfologisten poikkeavuuksien lisääntymisestä.
Ihmisen ja jyrsijöiden lisääntymiskudoksissa tiedetään olevan CD52:a. Eläinkokeet ovat osoittaneet vaikutuksia humanisoitujen hiirten hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Kuitenkaan saatavilla olevien tietojen perusteella mahdollista vaikutusta ihmisen hedelmällisyyteen altistusaikana ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lemtrada-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Useimmilla potilailla ilmenee infuusioon liittyviä reaktioita Lemtrada-hoidon aikana tai 24 tunnin kuluessa hoidosta. Jotkut infuusioreaktiot (esim. huimaus) voivat väliaikaisesti vaikuttaa potilaan ajokykyyn tai koneiden käyttökykyyn, ja varovaisuutta on noudatettava, kunnes reaktiot ovat hävinneet.
Haittavaikutukset
Kliinisten tutkimusten turvallisuusprofiilin yhteenveto
Yhteensä 1 486 Lemtrada-valmisteella (12 mg tai 24 mg) hoidettua potilasta muodosti turvallisuusryhmän yhdistetyssä MS-taudin kliinisten tutkimusten analyysissä, jossa seurannan keston mediaani oli 6,1 vuotta (enimmäiskesto 12 vuotta). Tämä tuotti 8 635 potilasvuotta turvallisuusseurantaa.
Tärkeimpiä haittavaikutuksia ovat autoimmuunisairaudet (ITP, kilpirauhassairaudet, munuaissairaudet, sytopeniat), infuusioreaktiot ja infektiot. Nämä on kuvattu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Lemtrada-valmisteeseen liittyviä yleisimpiä haittavaikutuksia (≥ 20 %:lla potilaista) olivat ihottuma, päänsärky, kuume ja hengitystieinfektiot.
Haittavaikutustaulukko
Seuraava taulukko perustuu kaikkien Lemtrada-hoitoa 12 mg:n annoksella saaneiden potilaiden yhdistettyihin turvallisuustietoihin kaikissa saatavilla olevissa kliinisten tutkimusten seurantatiedoissa. Haittavaikutukset on lueteltu MedDRAn elinjärjestelmäluokan ja suositellun termin mukaisesti. Esiintyvyydet on määritetty seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Tutkimuksissa 1, 2, 3 ja 4 Lemtrada-valmisteen 12 mg:n annoksella hoidetuilla potilailla ja kauppaantuonnin jälkeisessä seurannassa havaitut haittavaikutukset
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Tuntematon |
| Infektiot | Ylähengitystieinfektio, virtsatieinfektio, herpesvirusinfektio1 | Vyöruusuinfektiot2, alahengitystieinfektiot, gastroenteriitti, suun hiivatulehdus, vulvovaginaalinen hiivatulehdus, influenssa, korvainfektio, keuhkokuume, emätininfektio, hammasinfektio | Kynsisilsa, ientulehdus, ihon sieni-infektio, tonsilliitti, akuutti sinuiitti, selluliitti, tuberkuloosi, sytomegalovirusinfektio | Listerioosi/listeria-meningiitti Epstein-Barrin virus (EBV) ‑infektio (mukaan lukien uudelleenaktivoituminen) | |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Ihon papillooma | ||||
| Veri ja imukudos | Lymfopenia, leukopenia, mukaan lukien neutropenia | Lymfadenopatia, immuuni trombosytopeeninen purppura, trombosytopenia, anemia, hematokriittiarvon lasku, leukosytoosi | Pansytopenia, hemolyyttinen anemia, hankinnainen A-hemofilia | Hemofagosyyttinen lymfohistiosytoosi (HLH), tromboottinen trombosytopeeninen purppura (TTP) | |
| Immuunijärjestelmä | Sytokiinioireyhtymä*, yliherkkyys, mukaan lukien anafylaksia* | Sarkoidoosi | |||
| Umpieritys | Basedowin tauti, kilpirauhasen liikatoiminta, kilpirauhasen vajaatoiminta | Autoimmuunityroidiitti mukaan lukien subakuutti tyroidiitti, struuma, kilpirauhasen vasta-ainepositiivisuus | |||
| Aineenvaihdunta ja ravitsemus | Heikentynyt ruokahalu | ||||
| Psyykkiset häiriöt | Unettomuus*, ahdistus, masennus | ||||
| Hermosto | Päänsärky* | MS:n pahenemisvaihe, huimaus*, hypoestesia, parestesia, vapina, makuhäiriöt*, migreeni* | Aistihäiriöt, hyperestesia, jännityspäänsärky, autoimmuunienkefaliitti | Aivoverenvuoto**, pään ja kaulan alueen valtimon dissekoituminen** | |
| Silmät | Sidekalvotulehdus, endokriininen silmäoireyhtymä, näön sumeneminen | Diplopia | |||
| Kuulo ja tasapainoelin | Pyörrytys | Korvakipu | |||
| Sydän | Takykardia* | Bradykardia*, sydämentykytykset* | Eteisvärinä* | Sydänlihasiskemia**, sydäninfarkti** | |
| Verisuonisto | Punoitus* | Hypotensio*, hypertensio* | |||
| Hengityselimet, rintakehä ja välikarsina | Hengenahdistus*, yskä, nenäverenvuoto, hikka, suunielun kipu, astma | Kurkun kiristys*, kurkun ärsytys, keuhkotulehdus | Keuhkorakkuloiden verenvuoto** | ||
| Ruoansulatus-elimistö | Pahoinvointi* | Vatsakipu, oksentelu, ripuli, ruoansulatushäiriö*, suutulehdus | Ummetus, gastroesofageaalinen refluksitauti, ikenien verenvuoto, suun kuivuus, nielemishäiriö, gastrointestinaalinen häiriö, hematoketsia | ||
| Maksa ja sappi | ASAT-arvon nousu, ALAT-arvon nousu | Sappirakkotulehdus, mukaan lukien kivetön sappirakkotulehdus ja akuutti kivetön sappirakkotulehdus | Autoimmuunihepatiitti, hepatiitti (EBV-infektioon liittyvä) | ||
| Iho ja ihonalainen kudos | Urtikaria*, ihottuma*, kutina*, yleistynyt ihottuma* | Eryteema*, ekkymoosi, alopesia, liikahikoilu, akne, ihovaurio, ihotulehdus | Rakkulat, yöhikoilu, kasvojen turvotus, ekseema, vitiligo, pälvikalju | ||
| Luusto, lihakset ja sidekudos | Lihaskipu, lihasheikkous, nivelkipu, selkäkipu, raajakipu, lihasspasmit, niskakipu, muskuloskeletaalinen kipu | Muskuloskeletaalinen jäykkyys, epämiellyttävät tuntemukset raajoissa | Aikuisten Stillin tauti (AOSD) | ||
| Munuaiset ja virtsatiet | Proteinuria, hematuria | Munuaiskivitauti, ketonuria, munuaissairaudet mukaan lukien tyvikalvovasta-aineglomerulonefriitti | |||
| Sukupuolielimet ja rinnat | Menorragia, epäsäännöllinen kuukautiskierto | Kohdunkaulan dysplasia, amenorrea | |||
| Yleisoireet ja antopaikassa todettavat haitat | Kuume*, uupumus*, vilunväristykset* | Epämukava tunne rinnassa*, kipu*, ääreisturvotus, astenia, influenssaa muistuttava sairaus, pahoinvointi, antopaikan kipu | |||
| Tutkimukset | Veren kreatiniinipitoisuuden suureneminen | Painon lasku, painon nousu, punasolumäärän pieneneminen, positiivinen bakteerikokeen tulos, veren glukoosipitoisuuden suureneminen, solujen keskimääräisen tilavuuden suureneminen | |||
| Vammat ja myrkytykset | Ruhje, infuusioon liittyvät reaktiot |
1 Herpesvirusinfektioihin mukaan luettavat suositellut termit: suun herpes, yskänrokko, herpesvirusinfektio, genitaaliherpes, herpesihottuma, silmänseudun herpes, herpes simplex seropositiivisuus.
2 Vyöruusuinfektioihin mukaan luettavat suositellut termit: Vyöruusu, hajapesäkkeinen vyöruusu (disseminoitunut), silmänseudun vyöruusu, silmänseudun herpes, neurologinen vyöruusu, meningiitti vyöruusu.
Kuvaus valikoiduista haittavaikutuksista
Taulukossa 1 tähdellä (*) merkityt termit sisältävät haittavaikutukset, joita ilmoitettiin infuusioon liittyvinä reaktioina. 4.4).
Taulukossa 1 kahdella tähdellä (**) merkityt termit sisältävät haittavaikutukset, joita on havaittu myyntiluvan myöntämisen jälkeen ja jotka useimmissa tapauksissa ilmaantuivat 1–3 vuorokauden kuluessa Lemtrada-infuusion antamisesta minkä tahansa annoksen saamisen jälkeen hoidon aikana.
Neutropenia
Vaikea-asteisen (myös kuolemaan johtaneen) neutropenian tapauksia on ilmoitettu ilmenneen kahden kuukauden kuluessa Lemtrada-infuusion antamisesta.
Turvallisuusprofiili pitkäaikaisessa seurannassa
Lemtrada-hoitoryhmissä kaikissa saatavilla olevissa seurantatiedoissa havaittujen haittavaikutusten tyyppi, mukaan lukien vakavuus ja vaikeusaste, olivat samanlaiset verrattuna aktiivikontrolloituihin tutkimuksiin. Infuusioon liittyvät reaktiot olivat yleisempiä 1. hoitojakson kuin sitä seuraavien hoitojaksojen jälkeen.
Potilailla, jotka jatkoivat kontrolloiduista kliinisistä tutkimuksista ja jotka eivät saaneet yhtään lisäannosta Lemtrada-valmistetta kahden aloitushoitojakson jälkeen, useimpien haittavaikutusten määrä (tapahtumat henkilöä kohti vuodessa) oli vuosina 3–6 vastaavanlainen tai pienempi kuin vuosina 1–2. Kilpirauhaseen liittyvien haittavaikutusten määrä oli suurimmillaan kolmantena vuotena ja pieneni sen jälkeen.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kontrolloiduissa kliinisissä tutkimuksissa kaksi MS-potilasta sai vahingossa jopa 60 mg Lemtrada-valmistetta (ts. aloitushoitojakson koko annoksen) yhtenä infuusiona ja heillä ilmeni vakavia reaktioita (päänsärkyä, ihottumaa ja joko hypotensiota tai sinustakykardiaa). Kliinisissä tutkimuksissa käytettyjä annoksia suuremmat Lemtrada-annokset voivat suurentaa infuusioon ja immuunijärjestelmään liittyvien haittavaikutusten voimakkuutta ja/tai pidentää niiden kestoa.
Alemtutsumabin yliannostukseen ei tunneta vasta-ainetta. Hoitona on lääkevalmisteen annon keskeytys ja tukihoitotoimenpiteet.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosupressantit, monoklonaaliset vasta-aineet, ATC-koodi: L04AG06.
Vaikutusmekanismi
Alemtutsumabi on geeniteknologisesti valmistettu ihmisen monoklonaalinen vasta-aine, joka kohdistuu solun pinnalla oleviin 21–28 kD:n painoisiin CD52-pintaglykoproteiineihin. Alemtutsumabi on IgG1 kappa ‑vasta-aine, jossa on ihmisen variaabelirakenteita ja vakioalueita sekä ns. Complementary Determining -alueita rotan monoklonaalisesta vasta-aineesta. Vasta-aineen likimääräinen molekyylipaino on 150 kD.
Alemtutsumabi sitoutuu solun CD52-pinta-antigeeniin, jota on suuria pitoisuuksia T (CD3+)- ja B (CD19+) ‑lymfosyyteissä sekä pienempiä pitoisuuksia luonnollisissa tappajasoluissa, monosyyteissä ja makrofageissa. CD52:a esiintyy vähän tai ei lainkaan neutrofiileissä, plasmasoluissa tai luuytimen kantasoluissa. Alemtutsumabi vaikuttaa sekä vasta-aineriippuvaisen että komplementtivälitteisen solujen hajottamisen kautta sitouduttuaan T- ja B-lymfosyyttien pintaan.
Mekanismia, jolla Lemtrada vaikuttaa MS-taudissa, ei ole täysin selvitetty. Tutkimukset kuitenkin viittaavat immuunivastetta muuntaviin (immunomodulatorisiin) vaikutuksiin lymfosyyttikadon ja ‑populaation uudelleen muodostumisen kautta, sisältäen:
- muutokset joidenkin lymfosyyttien alatyyppien määrässä, suhteellisessa määrässä ja ominaisuuksissa hoidon jälkeen
- suurentunut regulatoristen T-solualatyyppien esiintyminen
- suurentunut muisti-T- ja B-lymfosyyttien esiintyminen
- ohimenevät vaikutukset luontaisen immuniteetin osa-alueisiin (ts. neutrofiileihin, makrofageihin, luonnollisiin tappajasoluihin).
Lemtrada-valmisteen aiheuttama verenkierrossa olevien B- ja T-solujen määrän väheneminen ja siitä seuraava solupopulaatioiden uudelleenmuodostuminen voi pienentää pahenemisvaiheen mahdollisuutta, mikä lopulta viivästyttää sairauden etenemistä.
Farmakodynaamiset vaikutukset
Lemtrada tuhoaa jokaisen hoitojakson jälkeen verenkierrossa olevia T- ja B-lymfosyytteja, joiden alimmat havaitut arvot esiintyvät kuukausi hoitojakson jälkeen (varhaisin hoidon jälkeinen aikapiste vaiheen 3 tutkimuksissa). Lymfosyyttipopulaatiot muodostuvat uudelleen ajan myötä, ja B-solujen palautuminen tapahtuu yleensä 6 kuukauden sisällä. CD3+- ja CD4+-lymfosyyttimäärät nousevat hitaammin kohti normaaliarvoja, mutta yleensä ne eivät palaa lähtötilanteen tasolle ennen 12 kuukautta hoidon jälkeen. Noin 40 %:lla potilaista lymfosyyttien kokonaismäärä saavutti viitevälin alarajan (LLN) 6 kuukautta kunkin hoitojakson jälkeen ja noin 80 %:lla potilaista lymfosyyttien kokonaismäärä saavutti LLN-tason 12 kuukautta kunkin hoitojakson jälkeen.
Lemtrada vaikuttaa vain ohimenevästi neutrofiileihin, monosyytteihin, eosinofiileihin, basofiileihin ja luonnollisiin tappajasoluihin.
Kliininen teho ja turvallisuus
Alemtutsumabin turvallisuutta ja tehoa MS-taudin hoidossa arvioitiin 3 satunnaistetussa, arvioijasokkoutetussa, aktiivista vertailuvalmistetta käyttävässä kliinisessä tutkimuksessa ja yhdessä kontrolloimattomassa, arvioijasokkoutetussa jatkotutkimuksessa, joihin osallistui aaltomaista MS-tautia (RRMS) sairastavia potilaita.
Tutkimusasetelma/-populaatio tutkimuksissa 1, 2, 3 ja 4 on esitetty taulukossa 2.
| Taulukko 2: Tutkimusasetelma ja lähtötilanteen ominaisuudet tutkimuksissa 1, 2, 3 ja 4 | |||
| Tutkimus 1 | Tutkimus 2 | Tutkimus 3 | |
| Tutkimuksen nimi | CAMMS323 (CARE-MS I) | CAMMS32400507 (CARE-MS II) | CAMMS223 |
| Tutkimusasetelma | Kontrolloitu, satunnaistettu, arvioijasokkoutettu | Kontrolloitu, satunnaistettu, arvioija- ja annossokkoutettu | Kontrolloitu, satunnaistettu, arvioijasokkoutettu |
| Sairaushistoria | MS-potilaat, joilla on aktiivinen MS-tauti, määritelmänä vähintään 2 pahenemisvaihetta edeltävien 2 vuoden aikana. | Potilaat, joilla on aktiivinen MS-tauti, määritelmänä vähintään 2 pahenemisvaihetta edeltävien 2 vuoden aikana ja vähintään 1 varjoaineella tehostuva leesio | |
| Kesto | 2 vuotta | 3 vuotta‡ | |
| Tutkimuspopulaatio | Aiemmin hoitoa saamattomat potilaat | Potilaat, joilla esiintyi riittämätön vaste ennen hoitoa* | Aiemmin hoitoa saamattomat potilaat |
| Lähtötilanteen ominaisuudet | |||
| Keskimääräinen ikä (vuotta) | 33 | 35 | 32 |
| Sairauden keskimääräinen / mediaanikesto | 2/1,6 vuotta | 4,5/3,8 vuotta | 1,5/1,3 vuotta |
| Aiemman MS-hoidon keskimääräinen kesto (käytetty ≥ 1 lääkettä) | Ei aiempaa hoitoa | 36 kuukautta | Ei aiempaa hoitoa |
| %, jotka saivat ≥ 2 aiempaa MS-hoitoa | Ei oleellinen | 28 % | Ei oleellinen |
| Keskimääräinen EDSS-pistemäärä aloitustilanteessa | 2,0 | 2,7 | 1,9 |
| Tutkimus 4 | |||
| Tutkimuksen nimi | CAMMS03409 | ||
| Tutkimusasetelma | Kontrolloimaton, arvioijasokkoutettu jatkotutkimus | ||
| Tutkimuspopulaatio | Potilaat, jotka osallistuivat CAMMS223-, CAMMS323- tai CAMMS32400507-tutkimuksiin (ks. lähtötilanteen ominaisuudet yllä) | ||
| Jatkotutkimuksen kesto | 4 vuotta | ||
* Tarkoittaa potilaita, joilla esiintyi ainakin yksi pahenemisvaihe beetainterferoni- tai glatirameeriasetaattihoidon aikana, kun potilas oli saanut lääkehoitoa vähintään 6 kuukautta.
‡ Tutkimuksen ensisijainen päätetapahtuma arvioitiin kolmen vuoden kohdalla. Lisäseurannasta saatiin tietoja ajalta, jonka mediaani oli 4,8 vuotta (enimmäisaika 6,7).
Tulokset tutkimuksista 1 ja 2 on esitetty taulukossa 3.
| Taulukko 3: Tärkeät kliiniset ja magneettikuvaukseen liittyvät päätetapahtumat tutkimuksissa 1 ja 2 | ||||
| Tutkimus 1 | Tutkimus 2 | |||
| Tutkimuksen nimi | CAMMS323 (CARE-MS I) | CAMMS32400507 (CARE-MS II) | ||
| Kliiniset päätetapahtumat | Lemtrada 12 mg (N = 376) | SC IFNB-1a (N = 187) | Lemtrada 12 mg (N = 426) | SC IFNB-1a (N = 202) |
Pahenemisvaiheiden määrä1 Vuosittainen pahenemisvaiheiden määrä (ARR) (95 % CI) | 0,18 (0,13, 0,23) | 0,39 (0,29, 0,53) | 0,26 (0,21, 0,33) | 0,52 (0,41, 0,66) |
| Esiintyvyyssuhde (95 % CI) Riskin väheneminen (%) | 0,45 (0,32, 0,63) 54,9 (p < 0,0001) | 0,51 (0,39, 0,65) 49,4 (p < 0,0001) | ||
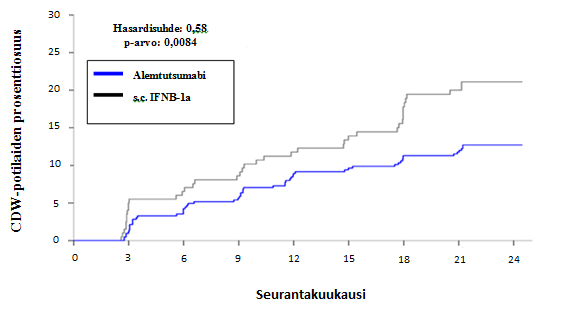
Toimintakyvyn heikkeneminen1 Vahvistettu toimintakyvyn heikkeneminen [CDW]2 Potilaat, joilla 6 kuukautta vahvistettu toimintakyvyn heikkeneminen (95 % CI) | 8,0 % (5,7, 11,2) | 11,1 % (7,3, 16,7) | 12,7 % (9,9, 16,3) | 21,1 % (15,9, 27,7) |
| Hasardisuhde (95 % CI) | 0,70 (0,40, 1,23) (p = 0,22) | 0,58 (0,38, 0,87) (p = 0,0084) | ||
| Pahenemisvaiheettomat potilaat vuonna 2 (95 % CI) | 77,6 % (72,9, 81,6) (p < 0,0001) | 58,7 % (51,1, 65,5) | 65,4 % (60,6, 69,7) (p < 0,0001) | 46,7 % (39,5, 53,5) |
Muutos aloitustilanteen EDSS-pisteissä vuonna 23 (95 % CI) | –0,14 (‑0,25, ‑0,02) (p = 0,42) | -0,14 (-0,29, 0,01) | –0,17 (‑0,29, ‑0,05) (p < 0,0001) | 0,24 (0,07, 0,41) |
| Magneettikuvauksen päätetapahtumat (0–2 vuotta) | ||||
| Mediaanimuutos (%) T2-leesioiden tilavuudessa | –9,3 (–19,6, –0,2) (p = 0,31) | -6,5 (-20,7, 2,5) | –1,3 (p = 0,14) | -1,2 |
| Potilaat, joilla on uusia tai suurenevia T2-leesioita vuonna 2 | 48,5 % (p = 0,035) | 57,6 % | 46,2 % (p <0,0001) | 67,9 % |
| Potilaat, joilla on gadoliniumilla tehostuvia leesioita vuonna 2 | 15,4 % (p = 0,001) | 27,0 % | 18,5 % (p < 0,0001) | 34,2 % |
| Potilaat, joilla on uusia T1- hypointensiivisiä leesioita vuonna 2 | 24,0 % (p = 0,055) | 31,4 % | 19,9 % (p < 0,0001) | 38,0 % |
| Mediaanimuutos (%) aivokudoksen tilavuusosuudessa | –0,867 (p < 0,0001) | –1,488 | –0,615 (p = 0,012) | -0,810 |
| 1 Yhdistetyt päätetapahtumat: vuosittainen pahenemisvaiheiden määrä (ARR) ja vahvistettu toimintakyvyn heikkeneminen (CDW). Tutkimus oli onnistunut, jos vähintään yksi kahdesta yhdistetyistä päätetapahtumista täyttyi. 2 Vahvistetuksi toimintakyvyn heikkenemiseksi (CDW) katsottiin vähintään 1 pisteen nousu EDSS-asteikolla aloitustilanteen EDSS-pisteissä ≥ 1,0 (1,5 pisteen nousu potilailla, joiden EDSS-pisteet aloitustilanteessa olivat 0), mikä säilyi 6 kuukautta. 3 Arvioitiin käyttämällä toistettujen mittausten sekamallia. | ||||
Kuva 1: Aika 6 kuukauden vahvistettuun toimintakyvyn heikkenemiseen tutkimuksessa 2
Pahenemisvaiheen vaikeusaste
Vaikutuksessa pahenemisvaiheiden määrään, tutkimuksen 1 (CAMMS323) toissijaiset analyysit osoittivat, että 12 mg:n Lemtrada-valmisteen vuorokausiannos johti merkitsevästi pienempään vaikeiden pahenemisvaiheiden määrään (61 %:n vähenemä, p = 0,0056) ja merkitsevästi pienempään steroidihoitoon johtaneiden pahenemisvaiheiden määrään (58 %:n vähenemä, p < 0,0001) verrattuna IFNB-1a:an.
Tutkimuksen 2 (CAMMS32400507) toissijaiset analyysit osoittivat, että 12 mg:n Lemtrada-valmisteen vuorokausiannos johti merkitsevästi pienempään vaikeiden pahenemisvaiheiden määrään (48 %:n vähenemä, p = 0,0121) ja merkitsevästi pienempään steroidihoitoon (56 %:n vähenemä, p < 0,0001) tai sairaalahoitoon (55 % vähenemä, p = 0,0045) johtaneiden pahenemisvaiheiden määrään verrattuna IFNB-1a:an.
Vahvistettu toimintakyvyn paraneminen (Confirmed disability improvement, CDI)
CDI:n alkamisajankohdaksi katsottiin vähintään 6 kuukautta pysyvä yhden pisteen lasku EDSS-asteikolla aloitustilanteessa, jossa EDSS-pistemäärä oli ≥ 2. CDI on pitkäkestoisen toimintakyvyn paranemisen mittari. 29 % Lemtrada-valmisteella hoidetuista potilaista ja 13 % ihonalaisella IFNB-1a:lla hoidetuista potilaista saavutti tämän päätetapahtuman tutkimuksessa 2. Ero oli tilastollisesti merkitsevä (p = 0,0002).
Tutkimuksessa 3 (faasin 2 tutkimus CAMMS223) arvioitiin Lemtrada-valmisteen turvallisuutta ja tehoa aaltomaista MS-tautia (RRMS) sairastavilla potilailla 3 vuoden aikana. Potilailla oli EDSS-pistemäärä 0–3,0, ainakin 2 kliinistä pahenemisjaksoa 2 edellisen vuoden aikana ja vähintään yksi gadoliniumilla tehostuva leesio tutkimuksen alkamisvaiheessa. Potilaat eivät olleet saaneet aiempaa hoitoa MS-tautiin. Potilaita hoidettiin Lemtrada-valmisteella 12 mg/vrk (N = 108) tai 24 mg/vrk (N = 108) annettuna kerran päivässä 5 päivänä kuukautena 0 ja 3 päivänä kuukautena 12 tai ihonalaisen IFNB-1a:n 44 µg:n annoksella (N = 107) annettuna 3 kertaa viikossa 3 vuoden ajan. 46 potilasta sai kolmannen Lemtrada-hoitojakson, jolloin sitä annettiin 12 mg/vrk tai 24 mg/vrk 3 päivän ajan kuukautena 24.
Kolmen vuoden kohdalla Lemtrada pienensi 6 kuukauden ajan vahvistetun toimintakyvyn heikkenemisen riskiä 76 % (hasardisuhde 0,24 [95 % CI: 0,110; 0,545], p < 0,0006) ja vähensi vuosittaista pahenemisvaiheiden määrää 67 % (esiintyvyyssuhde 0,33 [95 % CI: 0,196; 0,552], p <0,0001) verrattuna ihonalaiseen IFNB‑1a:han. Lemtrada 12 mg/vrk johti merkitsevästi alhaisempiin EDSS-pisteisiin (parani verrattuna aloitustilanteeseen) kahden vuoden seurannassa verrattuna IFNB-1a:han (p < 0,0001).
Aaltomaista MS-tautia sairastavien potilaiden alaryhmässä, jossa potilailla oli ollut vähintään 2 pahenemisvaihetta edeltävän vuoden aikana ja vähintään yksi gadoliniumilla tehostuva T1-leesio lähtötilanteessa, vuosittainen pahenemisvaiheiden määrä oli 0,26 (95 % CI:0,20; 0,34) Lemtrada-valmistetta saaneiden ryhmässä (n = 205) ja 0,51 (95 % CI: 0,40; 0,64) IFNB‑1a-ryhmässä (n = 102) (p < 0,0001). Tämä analyysi sisältää vain tiedot vaiheen 3 tutkimuksista (CAMMS324 ja CAMMS323) vaiheiden 2 ja 3 tutkimusten magneettikuvauksissa käytettyjen algoritmien eroavaisuuksien vuoksi. Nämä tulokset on saatu post hoc ‑analyysistä ja niihin on suhtauduttava varauksella.
Pitkäaikaista tehokkuutta koskevat tiedot
Monikeskustutkimus 4 oli vaiheen 3 avoin ja arvioijasokkoutettu tehoa ja turvallisuutta mittaava jatkotutkimus aaltomaista MS-tautia (RRMS) sairastavilla potilailla, jotka osallistuivat tutkimuksiin 1, 2 tai 3 (ennen vaiheen 3 ja 2 tutkimuksia). Tutkimuksessa arvioitiin Lemtrada-valmisteen pitkäaikaista tehoa ja turvallisuutta. Tutkimus antaa tietoa tehosta ja turvallisuudesta kuuden (mediaani) vuoden ajalta tutkimuksissa 1 ja 2 aloittamisen jälkeen. Jatkotutkimuksen (tutkimus 4) potilailla oli mahdollisuus saada tarpeen mukaan Lemtrada-valmisteen lisähoitojaksoja, kun potilaalla dokumentoitiin taudin aktivoituminen uudelleen. Taudin aktivoitumiseksi uudelleen määriteltiin vähintään yksi MS-taudin pahenemisvaihe ja/tai vähintään 2 uutta tai kasvavaa magneettikuvauksella todettua aivojen tai selkäytimen leesiota. Lemtrada-valmisteen lisähoitojakso(i)lla annettiin 12 mg/vrk kolmena peräkkäisenä päivänä (36 mg:n kokonaisannos) vähintään 12 kuukautta aloitushoitojakson jälkeen.
91,8 % Lemtrada 12 mg ‑valmisteella tutkimuksissa 1 ja 2 hoidetusta potilaista osallistui tutkimukseen 4. 82,7 % näistä potilaista oli tutkimuksessa mukana loppuun asti. Noin puolet (51,2 %) potilaista, jotka saivat aluksi 12 mg Lemtrada-valmistetta vuorokaudessa tutkimuksessa 1 tai 2 ja jotka osallistuivat tutkimukseen 4, saivat vain kaksi aloitushoitojaksoa Lemtrada-valmistetta, eivätkä he saaneet muita sairauteen vaikuttavia hoitoja kuuden vuoden seurannan aikana.
46,6 % potilaista, jotka saivat aluksi Lemtrada-valmistetta 12 mg/vrk tutkimuksessa 1 tai 2, saivat lisähoitojaksoja, kun potilaalla esiintyi dokumentoituja MS-taudin aktiivisuuden merkkejä (pahenemisvaihe ja/tai toteaminen magneettikuvauksella) ja hoitava lääkäri oli tehnyt päätöksen hoidon uusimisesta. Tutkimuksen sisäänottovaiheessa ei havaittu ominaisuuksia, joilla tunnistettaisiin potilaat, jotka myöhemmin saisivat yhden tai useamman lisähoitojakson.
6 vuotta Lemtrada-hoidon aloituksen jälkeen seurannassa jatkaneilla potilailla todetut MS-taudin pahenemisvaiheet, aivoihin muodostuneet leesiot magneettikuvissa ja aivojen tilavuuden pieneneminen olivat yhtäpitäviä tutkimuksissa 1 ja 2 todettujen Lemtrada-hoidon vaikutusten kanssa ja potilaiden tila oli myös etupäässä vakaa tai heidän toimintakykyä mittaavat pisteet olivat parantuneet. Tutkimuksissa 1 ja 2 sekä näiden seurantatutkimuksessa 4 Lemtrada-hoitoa alun perin saaneiden potilaiden vuosittaisten pahenemisvaiheiden määrät olivat 0,17 ja 0,23, vahvistettu toimintakyvyn heikkeneminen oli 22,3 % ja 29,7 % vahvistetun toimintakyvyn paranemisen ollessa 32,7 % ja 42,5 % vastaavasti. Tutkimuksen 4 jokaisena vuotena kummassakin aiemmassa tutkimuksissa mukana olleiden potilaiden riski muodostaa uusia T2-soluja (27,4 %–33,2 %) tai gadoliniumilla tehostuvia leesioita (9,4 %–13,5 %) oli pieni ja vuosittainen aivokudoksen tilavuusosuuden muutoksen mediaani vaihteli välillä 0,19 % – -0,09 %.
Yhden tai kaksi Lemtrada-lisähoitojaksoa saaneiden potilaiden joukossa havaittiin parantumista pahenemisvaiheiden määrässä, magneettikuvauksella todettavassa tautiaktiivisuudessa ja toimintakykyä mittaavien pisteiden keskiarvossa ensimmäisen ja toisen Lemtrada-lisähoitojakson jälkeen (jaksot 3 ja 4), kun tuloksia verrattiin edeltävään vuoteen. Näiden potilaiden vuosittaisten pahenemisvaiheiden määrä laski 3. hoitojaksoa edeltävän vuoden arvosta 0,79 seuraavan vuoden arvoon 0,18 ja EDSS-pisteiden keskiarvo 2,89:stä 2,69:ään. Uusia tai suurentuneita T2-leesioita saaneiden potilaiden prosenttiosuus laski 3. hoitojaksoa edeltävän vuoden arvosta 50,8 % seuraavan vuoden arvoon 35,9 % ja uusien gadoliniumilla tehostuvien leesioiden määrät 32,2 %:sta 11,9 %:iin. Samanlaista parannusta nähtiin myös vuosittaisten pahenemisvaiheiden määrissä, EDSS-pisteiden keskiarvossa ja T2- ja gadoliniumilla tehostuvissa leesioissa 4. hoitojakson jälkeen verrattaessa edeltävään vuoteen. Nämä paremmat arvot pysyivät myöhemmin, mutta varmoja johtopäätöksiä pitkäaikaisvaikutuksista (esim. 3 tai 4 vuotta lisähoitojaksojen jälkeen) ei voida tehdä, koska useat potilaat lopettivat tutkimuksen ennen näiden aikapisteiden saavuttamista.
Viiden tai sitä useamman hoitojakson hyötyjä ja riskejä ei ole määritetty.
Immunogeenisuus
Kuten kaikkien terapeuttisten proteiinien kohdalla, immunogeenisuuden mahdollisuus on olemassa. Tiedot heijastavat niiden potilaiden prosenttiosuutta, joiden testituloksista löytyi alemtutsumabin vasta-aineita ELISA-analyysissä, mikä vahvistettiin kilpailevan sitoutumisen kokeella. Positiiviset näytteet arvioitiin lisäksi in vitro ‑estovaikutuksen merkkien varalta käyttämällä virtaussytometria-analyysiä. MS-tautia koskeviin kliinisiin tutkimuksiin osallistuneilta potilailta otettiin seeruminäytteet 1, 3 ja 12 kuukautta kunkin hoitojakson jälkeen alemtutsumabi-vasta-aineiden määritystä varten. Noin 85 % Lemtrada-valmistetta saaneista potilaista antoi positiivisen alemtutsumabi-vasta-ainenäytteen tutkimuksen aikana, ja näistä potilaista ≥ 90%:lla esiintyi myös vasta-aineita, jotka estivät alemtutsumabin sitoutumista in vitro. Potilaille, joille kehittyi alemtutsumabi-vasta-aineita, kehittyi niitä 15 kuukauteen mennessä hoidon aloituksen jälkeen. Kahden hoitojakson aikana alemtutsumabin vasta-aineiden tai estävien alemtutsumabin vasta-aineiden esiintymisen ei katsottu liittyvän tehon laskuun, farmakodynamiikan muuttumiseen tai haittavaikutusten, kuten infuusioon liittyvien reaktioiden, esiintymiseen. Osalla potilaista havaittiin suuri alemtutsumabivasta-ainetiitteri, joka liittyi epätäydelliseen lymfosyyttikatoon kolmannen tai neljännen hoitojakson jälkeen. Alemtutsumabivasta-aineilla ei kuitenkaan ollut selvää vaikutusta Lemtrada-valmisteen kliiniseen tehoon tai turvallisuusprofiiliin.
Vasta-aineiden esiintyvyys riippuu suuresti analyysin herkkyydestä ja spesifisyydestä. Lisäksi vasta-aineen (mukaan lukien estävän vasta-aineen) havaittuun esiintyvyyteen analyysissä voivat vaikuttaa useat tekijät, kuten analyysin metodologia, näytteen käsittely, näytteenoton ajoitus, samanaikainen lääkitys ja taustasairaus. Näistä syistä Lemtrada-vasta-aineiden esiintyvyyden vertailu muiden valmisteiden vasta-aineiden esiintyvyyteen voi olla harhaanjohtavaa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset alemtutsumabin käytöstä alle 10-vuotiaiden lasten multippeliskleroosin hoidossa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Lemtrada-valmisteen käytöstä yhden tai useamman pediatrisen RRMS-potilasryhmän hoidossa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Alemtutsumabin farmakokinetiikka arvioitiin yhteensä 216 aaltomaista MS-tautia sairastavalla potilaalla, jotka saivat laskimonsisäisiä infuusioita joko 12 mg/vrk tai 24 mg/vrk viitenä peräkkäisenä päivänä ja sen jälkeen kolmena peräkkäisenä päivänä 12 kuukautta aloitushoitojakson jälkeen. Seerumin lääkeainepitoisuudet suurenivat hoitojakson jokaisen peräkkäisen annoksen myötä ja suurimmat havaitut pitoisuudet esiintyivät hoitojakson viimeisen infuusion jälkeen. 12 mg:n vuorokausiannos sai aikaan keskimäärin 3014 ng/ml suuruisen Cmax-arvon aloitushoitojakson 5. päivänä ja 2276 ng/ml suuruisen arvon toisen hoitojakson 3. päivänä. Alfa-puoliintumisaika oli likimäärin 4–5 päivää, mikä oli samanveroinen eri hoitojaksoilla ja johti pieniin tai havaitsemattomiin seerumipitoisuuksiin noin 30 päivän sisällä kunkin hoitojakson jälkeen.
Alemtutsumabi on proteiini, jonka ennakoitu metabolinen reitti on hajoaminen pieniksi peptideiksi ja yksittäisiksi aminohapoiksi laajasti esiintyvien proteolyyttisten entsyymien vaikutuksesta. Klassisia biotransformaatiotutkimuksia ei ole tehty.
Saatavilla olevien tietojen perusteella johtopäätöksiä ei voida tehdä rodun tai sukupuolen vaikutuksista alemtutsumabin farmakokinetiikkaan. Alemtutsumabin farmakokinetiikkaa ei ole tutkittu 55-vuotiailla ja sitä vanhemmilla potilailla.
Prekliiniset tiedot turvallisuudesta
Karsinogeneesi ja mutageneesi
Alemtutsumabin karsinogeenisuus- tai mutageenisuuspotentiaalia ei ole tutkittu.
Fertiliteetti ja suvunjatkamiskyky
Alemtutsumabin laskimoon annettu hoito 10 mg/kg/vrk yltävillä annoksilla annettuna 5 peräkkäisenä päivänä (AUC-arvo 7,1-kertainen verrattuna ihmisen altistukseen suositellulla päiväannoksella) ei vaikuttanut huCD52-siirtogeenisten uroshiirten hedelmällisyyteen ja suvunjatkamiskykyyn. Normaalien siittiöiden määrä pieneni merkitsevästi (< 10 %) suhteessa kontrolleihin ja epänormaalien siittiöiden prosenttimäärä (irronneet päät tai ei päitä) suureni merkitsevästi (jopa 3 %). Nämä muutokset eivät kuitenkaan vaikuttaneet hedelmällisyyteen ja siten ne katsottiin haitattomiksi.
Naarashiirillä, jotka saivat alemtutsumabia enintään 10 mg/kg/vrk laskimoinfuusiona (AUC-arvo 4,7‑kertainen verrattuna ihmisten altistukseen suositellulla vuorokausiannoksella) 5 peräkkäisenä päivänä ennen siirtämistä yhteen villin tyypin uroshiiren kanssa, keskimääräinen keltarauhasen ja implantaatiokohtien määrä/hiiri väheni merkittävästi verrattuna vehikkelillä käsiteltyihin eläimiin. Pienentynyt tiineysajan painonnousu suhteessa vehikkeli-kontrolleihin havaittiin tiineillä hiirillä, jotka saivat jopa 10 mg/kg/vrk alemtutsumabia.
Tiineinä olevien hiirten lisääntymistoksisuustutkimuksessa hiiret saivat alemtutsumabia annettuna laskimoinfuusiona enintään 10 mg/kg/vrk (AUC-arvo 2,4-kertainen verrattuna suositeltuun ihmisten altistukseen 12 mg vuorokausiannoksella) viitenä peräkkäisenä päivänä tiineyden aikana, mikä aiheutti lisääntymisen niiden emojen määrässä, joiden kaikki sikiöt olivat kuolleet tai resorboituneet, sekä samanaikaisesti pienenemisen niiden emojen määrässä, joilla oli elinkykyisiä sikiöitä. Ulkoisia, pehmytkudoksen tai luuston epämuodostumia tai muutoksia ei havaittu 10 mg/kg/vrk yltävillä annoksilla.
Istukan läpi kulkeutumista ja mahdollista alemtutsumabin farmakologista aktiviteettia havaittiin hiirillä raskauden aikana ja poikimisen jälkeen. Hiirten tutkimuksissa alemtutsumabille altistuneilla poikasilla havaittiin lymfosyyttimäärän muutoksia tiineyden aikana annoksilla 3 mg/kg/vrk annettuna viitenä peräkkäisenä päivänä (AUC-arvo 0,6-kertainen verrattuna ihmisen altistukseen suositellulla 12 mg:n suuruisella päiväannoksella). Alemtutsumabiannokset, jotka ylsivät enintään tasolle 10 mg/kg/vrk, eivät vaikuttaneet poikasten kognitiiviseen, fyysiseen tai seksuaaliseen kehitykseen imetyksen aikana.
Farmaseuttiset tiedot
Apuaineet
Dinatriumfosfaattidihydraatti (E339), dinatriumedetaattidihydraatti, kaliumkloridi (E508), kaliumdivetyfosfaatti (E340), polysorbaatti 80 (E433), natriumkloridi, injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Konsentraatti
4 vuotta
Laimennettu liuos
Kemiallinen ja fyysinen käytönaikainen stabiilius on osoitettu 8 tunnin ajan lämpötilassa 2°C – 8°C.
Mikrobiologiselta kannalta valmiste olisi käytettävä heti. Jos valmistetta ei käytetä heti, säilytysaika ja -olosuhteet ovat käyttäjän vastuulla. Säilytysaika ei tavallisesti saa ylittää 8 tuntia 2°C – 8°C:ssa valolta suojattuna.
Säilytys
Konsentraatti
Säilytä jääkaapissa (2°C – 8°C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LEMTRADA infuusiokonsentraatti, liuosta varten
12 mg (L:ei) 1 kpl (10 mg/ml) (8000,75 €)
PF-selosteen tieto
Lemtrada toimitetaan kirkkaassa 2 ml:n injektiopullossa, jossa on butyylikuminen tulppa ja alumiininen tiiviste sekä muovinen napsautuskorkki.
Pakkauskoko: laatikko, jossa on 1 injektiopullo.
Valmisteen kuvaus:
Kirkas, väritön tai hieman kellertävä konsentraatti, jonka pH on 7,0–7,4.
Käyttö- ja käsittelyohjeet
Injektiopullon sisältö on tarkistettava silmämääräisesti hiukkasten ja värimuutosten varalta ennen antoa. Valmistetta ei saa käyttää, jos siinä näkyy hiukkasia tai konsentraatissa on värimuutoksia.
Injektiopulloja ei saa ravistaa ennen käyttöä.
Laskimoon antoa varten ota 1,2 ml Lemtrada-valmistetta injektiopullosta ruiskuun käyttämällä aseptista tekniikkaa. Injektoi se 100 ml:aan natriumkloridin 9 mg/ml (0,9 %) infuusionesteeseen tai glukoosin (5 %) infuusionesteeseen. Tätä lääkevalmistetta ei saa laimentaa muilla liuottimilla. Pussia on käänneltävä varovasti, jotta liuos sekoittuu.
Valmistetun liuoksen steriiliys on varmistettava huolellisesti. On suositeltavaa, että laimennettu valmiste annetaan välittömästi. Injektiopullot ovat kertakäyttöisiä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LEMTRADA infuusiokonsentraatti, liuosta varten
12 mg 1 kpl
- Ei korvausta.
ATC-koodi
L04AG06
Valmisteyhteenvedon muuttamispäivämäärä
27.10.2025
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi