
EYLEA injektioneste, liuos 40 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Potilasoppaan äänitiedosto
Terveydenhuollon ammattilainen
EYLEA - lasiaisinjektion antaminen
EYLEA Lääkkeen määrääjän opas
Vaikuttavat aineet ja niiden määrät
1 ml injektionestettä, liuosta, sisältää 40 mg afliberseptiä*.
Yhden injektiopullon sisältämä kokonaismäärä on vähintään 0,1 ml, mikä vastaa vähintään 4 mg:aa afliberseptiä. Tästä saadaan annettavaksi yksi 0,05 ml:n annos, jossa on 2 mg afliberseptiä.
*Fuusioproteiini, joka sisältää osia ihmisen verisuonten endoteelin kasvutekijän (VEGF) reseptoreiden 1 ja 2 solunulkoisista domeeneista yhdistettynä ihmisen IgG1:n Fc-osaan. Se on tuotettu kiinanhamsterin munasarja (CHO) K1-soluissa yhdistelmä-DNA-tekniikalla.
Apuaine, jonka vaikutus tunnetaan
1 ml injektionestettä, liuosta, sisältää 0,3 mg polysorbaatti 20:tä (E 432).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektio)
Kliiniset tiedot
Käyttöaiheet
Eylea on tarkoitettu aikuisille
- neovaskulaarisen (kostean) silmänpohjan ikärappeuman (AMD) hoitoon (ks. kohta Farmakodynamiikka)
- verkkokalvon laskimotukoksesta (verkkokalvon keskuslaskimotukos (CRVO) tai haaralaskimotukos (BRVO)) johtuvan makulaturvotuksen aiheuttaman näkökyvyn heikkenemisen hoitoon (ks. kohta Farmakodynamiikka)
- diabeettisesta makulaturvotuksesta (DME) johtuvan näön heikkenemisen hoitoon (ks. kohta Farmakodynamiikka)
- likitaitteisuuden aiheuttaman suonikalvon uudissuonittumisesta (myooppinen CNV) johtuvan näön heikkenemisen hoitoon (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista.
Annostus ja antotapa
Eylea on tarkoitettu vain silmän lasiaiseen injektoitavaksi.
Eylea-valmistetta saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista.
Annostus
Kostea silmänpohjan ikärappeuma
Eylea-valmisteen suositeltu annos on 2 mg afliberseptiä, joka vastaa 0,05 ml:aa.
Eylea-hoito aloitetaan antamalla yksi injektio kuukaudessa kolmen kuukauden ajan. Tämän jälkeen hoitoväliä pidennetään kahteen kuukauteen.
Lääkärin näkö- ja/tai anatomiavasteisiin perustuvalla arviolla hoitoväliä voidaan pitää kahden kuukauden pituisena tai pidentää edelleen treat-and-extend-hoito-ohjelmalla, jossa injektioväliä pidennetään asteittain 2 tai 4 viikon jaksoissa vakaiden näkö- ja/tai anatomiavasteiden ylläpitämiseksi. Jos näkö- ja/tai anatomiavasteet huonontuvat, tulee hoitoväliä vastaavasti lyhentää.
Seuranta injektioiden välillä ei ole tarpeen. Lääkärin arvioon perustuen seurantakäyntejä voi olla useammin kuin injektiokertoja.
Yli neljän kuukauden tai alle neljän viikon pituisia hoitovälejä injektioiden välillä ei ole tutkittu (ks. kohta Farmakodynamiikka).
Verkkokalvon laskimotukoksesta (verkkokalvon haaralaskimotukos tai keskuslaskimotukos) aiheutuva makulaarinen edeema
Suositeltu Eylea-annos on 2 mg afliberseptiä, joka vastaa 0,05 ml:aa.
Ensimmäisen injektion jälkeen hoito annetaan kuukausittain. Kahden annoksen välisen jakson on oltava vähintään kuukausi.
Jos näkö- ja anatomiavasteista ilmenee, ettei potilas hyödy jatkuvasta hoidosta, tulee hoito lopettaa.
Kuukausittaista hoitoa jatketaan, kunnes suurin mahdollinen näöntarkkuus saavutetaan ja/tai sairauden aktiivisuudesta ei ole merkkejä. Voidaan tarvita kolme tai useampi peräkkäistä kuukausittaista injektiota.
Hoitoa voidaan tuolloin jatkaa asteittain pitenevin hoitovälein vakaiden näkö- ja/tai anatomiavasteiden ylläpitämiseksi. Hoitovälien pituudesta ei ole kuitenkaan riittävästi tietoa. Jos näkö- ja/tai anatomiavasteet huonontuvat, voidaan hoitoväliä lyhentää vastaavasti.
Hoitava lääkäri päättää arviointiaikataulusta ja hoito-ohjelmasta yksilöllisesti potilaan hoitovasteen perusteella.
Sairauden aktiivisuuden seuranta voi sisältää kliinisen tarkastuksen, toiminnallisen kokeen tai kuvantamistekniikoita (esim. OCT (optical coherence tomography, valokerroskuvaus) tai fluoreseiiniangiografia).
Diabeettinen makulaturvotus
Suositeltu Eylea-annos on 2 mg afliberseptiä, joka vastaa 0,05 ml:aa.
Eylea-hoito aloitetaan yhdellä injektiolla kuukaudessa viiden peräkkäisen kuukauden ajan, minkä jälkeen annetaan yksi injektio joka toinen kuukausi.
Lääkärin näkö- ja/tai anatomiavasteisiin perustuvan arvion mukaisesti hoitovälinä voidaan pitää 2 kuukautta tai hoitoväliä voidaan säätää yksilöllisesti esim. treat-and-extend-annosteluohjelman mukaisesti vakaiden näkö- ja/tai anatomiavasteiden ylläpitämiseksi. Annosteluväliä pidennetään tavallisesti kaksi viikkoa kerrallaan. Yli neljän kuukauden hoitoväleistä on rajoitetusti tietoa saatavilla. Jos näkö- ja/tai anatomiavasteet huonontuvat, tulee hoitoväliä lyhentää. Alle 4 viikon hoitovälejä ei ole tutkittu (ks. kohta Farmakodynamiikka).
Hoitavan lääkärin pitää päättää hoitovasteiden arviointiaikataulusta.
Jos näkö- ja anatomiavasteissa ei ilmene paranemista hoidon jatkumisesta huolimatta, Eylea-hoito pitää lopettaa.
Likitaitteisuuden aiheuttama suonikalvon uudissuonittuminen
Suositeltu Eylea-annos on yksi silmän lasiaiseen annettava, 2 mg afliberseptiä sisältävä injektio, joka vastaa 0,05 ml:aa.
Lisäannoksia voidaan antaa, jos näkö- ja/tai anatomiavasteista ilmenee, että sairaus ei häviä. Uusiutunutta tautia pitää hoitaa taudin uutena ilmentymänä.
Hoitavan lääkärin pitää päättää hoitovasteiden arviointiaikatauluista
Kahden annoksen välisen jakson on oltava vähintään kuukausi.
Erityisryhmät
Maksan ja/tai munuaisten vajaatoiminta
Eylea-valmisteesta ei ole tehty erityisiä tutkimuksia potilaille, joilla on maksan ja/tai munuaisten vajaatoiminta.
Saatavilla olevat tiedot eivät viittaa tarpeeseen muuttaa näiden potilaiden Eylea-annosta (ks. kohta Farmakokinetiikka).
Iäkkäät
Erityisiä varotoimia ei tarvita. Yli 75-vuotiaista diabeettista makulaturvotusta sairastavista potilaista on rajallisesti kokemusta.
Pediatriset potilaat
Eylea-valmisteen turvallisuutta ja tehoa lasten ja nuorten hoidossa ei ole varmistettu. Ei ole asianmukaista käyttää Eylea-valmistetta pediatrisille potilaille neovaskulaarisen (kostean) silmänpohjan ikärappeuman (AMD), verkkokalvon keskuslaskimotukoksen (CRVO), haaralaskimotukoksen (BRVO), diabeettisen makulaturvotuksen (DME) ja likitaitteisuuden aiheuttaman suonikalvon uudissuonittumisen (myooppinen CNV) hoidossa.
Antotapa
Lasiaiseen annettavat injektiot on annettava lääketieteellisten standardien sekä soveltuvien ohjeiden mukaisesti. Injektiot saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista. Yleisesti ottaen on varmistettava riittävä puudutus ja aseptiikka, mukaan lukien topikaalinen laajakirjoinen mikrobisidi (esim. jodattu povidoni silmää ympäröivälle iholle, silmäluomelle ja silmän pinnalle). On suositeltavaa käyttää kirurgian edellyttämää käsien desinfiointia, steriilejä käsineitä, steriiliä liinaa ja steriiliä luomenlevitintä (tai vastaavaa).
Injektioneula pistetään 3,5–4,0 mm limbuksesta posteriorisesti lasiaiseen, vältetään horisontaalista meridiaania ja tähdätään silmämunan keskikohtaan ja annettava 0,05 ml:n määrä injisoidaan. Kovakalvon pistoskohtaa vaihdetaan seuraavissa injektioissa.
Potilaita on seurattava silmänpaineen kohoamisen varalta välittömästi lasiaisinjektion jälkeen. Sopiva seuranta voi sisältää näköhermon pään perfuusion tarkistuksen tai tonometrian. Steriilejä parasenteesivälineitä on tarvittaessa oltava saatavilla.
Lasiaisinjektion jälkeen potilaita on neuvottava viipymättä ilmoittamaan kaikista endoftalmiittiin viittaavista oireista (esim. silmäkipu, silmän punoitus, valoherkkyys, näön sumeneminen).
Yhtä injektiopulloa saa käyttää vain yhden silmän hoitamiseen. Useiden annosten ottaminen yhdestä injektiopullosta voi lisätä kontaminaation riskiä ja sen seurauksena infektion riskiä.
Injektiopullo sisältää enemmän kuin suositellun 2 mg:n afliberseptiannoksen (vastaa 0,05 ml:aa injektionestettä). Injektiopullosta saatava kokonaismäärä, on se määrä injektionestettä, joka injektiopullosta voidaan saada ja sitä ei pidä käyttää kokonaan. Eylea injektiopullosta saatava kokonaismäärä on vähintään 0,1 ml. Ylimäärä on poistettava ennen suositellun annoksen injisointia (ks. kohta Käyttö- ja käsittelyohjeet). Injektiopullon kokonaismäärän injisointi voi johtaa yliannostukseen. Poista ilmakuplat ja ylimääräinen lääkevalmiste ruiskusta painamalla mäntää hitaasti niin, että männän litteä reuna kohdistuu ruiskussa olevaan 0,05 ml:n viivaan (vastaa 0,05 ml:aa eli 2 mg:aa afliberseptiä) (ks. kohdat Yliannostus ja Käyttö- ja käsittelyohjeet)
Injektion jälkeen käyttämätön valmiste on hävitettävä.
Lääkevalmisteen käsittely ennen sen antamista, ks. kohta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle afliberseptille tai kohdassa Apuaineet mainituille apuaineille.
Aktiivinen tai epäilty silmän tai silmänympärysalueen infektio.
Aktiivinen vakava silmänsisäinen inflammaatio.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Lasiaisinjektioihin liittyvät reaktiot
Lasiaisinjektioihin, mukaan lukien Eylea-injektiot, on liittynyt seuraavia: endoftalmiitti, silmänsisäinen tulehdusreaktio, verkkokalvoreiästä aiheutunut verkkokalvon irtauma, verkkokalvon repeämä ja hoidosta johtuva traumaperäinen kaihi (ks. kohta Haittavaikutukset). Oikeanlaisia aseptisia injektointitekniikoita on aina käytettävä injektoitaessa Eylea-valmistetta. Lisäksi potilaita tulisi seurata injektion jälkeisellä viikolla, jotta infektiotilanteessa hoito voitaisiin aloittaa nopeasti. Potilaita on kehotettava ilmoittamaan viipymättä kaikista endoftalmiittiin tai yllä mainittuihin tapahtumiin viittaavista oireista.
Injektiopullo sisältää enemmän kuin suositellun 2 mg:n annoksen afliberseptiä (vastaa 0,05 ml:aa). Ylimääräinen lääkevalmiste on poistettava ennen antoa (ks. kohdat Annostus ja antotapa ja Käyttö- ja käsittelyohjeet). Silmänpaineen kohoamista on havaittu 60 minuutin sisällä lasiaiseen annetusta injektiosta, myös Eylea-valmisteen yhteydessä (ks. kohta Haittavaikutukset). Erityistä varovaisuutta tarvitaan sellaisten potilaiden kohdalla, joilla on huonosti hallinnassa oleva glaukooma (Eylea-valmistetta ei saa injektoida, kun silmänpaine on ≥ 30 mmHg). Siksi, kaikissa tapauksissa silmänpainetta ja näköhermon pään perfuusiota on seurattava ja hoidettava asianmukaisesti.
Immunogeenisuus
Eylea-valmisteen käyttöön voi liittyä immunogeenisuutta, koska kyseessä on terapeuttinen proteiini (ks. kohta Haittavaikutukset). Potilaita on kehotettava ilmoittamaan, mikäli heillä ilmenee silmänsisäisen tulehduksen oireita kuten kipua, valonarkuutta tai punoitusta, mikä voi olla yliherkkyydestä johtuva kliininen oire.
Systeemiset vaikutukset
Systeemisiä haittatapahtumia, mukaan lukien silmän ulkopuolisia verenvuotoja ja valtimoiden tromboemboliatapahtumia, on raportoitu verisuonikasvutekijän (VEGF) estäjien lasiaisinjektioiden jälkeen, ja on teoreettinen mahdollisuus, että ne liittyvät VEGF-estoon. Sellaisten potilaiden hoidon turvallisuudesta, joilla on verkkokalvon keskuslaskimotukos (CRVO), haaralaskimotukos (BRVO), diabeettinen makulaturvotus (DME) tai likitaitteisuuden aiheuttama suonikalvon uudissuonittuminen (myooppinen CNV) sekä joilla on aiemmin ollut aivohalvaus tai ohimeneviä iskeemisiä kohtauksia tai sydäninfarkti edellisen 6 kuukauden aikana, on rajallisesti tietoa. Näiden potilaiden hoidossa on noudatettava varovaisuutta.
Muuta
Kuten muidenkin silmänpohjan ikärappeuman, verkkokalvon keskuslaskimotukoksen, haaralaskimotukoksen, diabeettisen makulaturvotuksen ja likitaitteisuuden aiheuttaman suonikalvon uudissuonittumisen anti-VEGF (verisuonikasvutekijän estäjä) -hoitojen kohdalla, seuraavat koskevat myös Eylea-hoitoa:
- Molempiin silmiin samanaikaisesti annettavan Eylea-hoidon turvallisuutta ja tehoa ei ole tutkittu systemaattisesti (ks. kohta Farmakodynamiikka). Jos molemminpuolinen hoito toteutetaan samanaikaisesti, tämä voi aiheuttaa suuremman systeemisen altistuksen ja lisätä systeemisten haittavaikutusten riskiä.
-
Muiden anti-VEGF-hoitojen samanaikainen käyttö
Tietoa ei ole saatavilla Eylea-valmisteen käytöstä muiden anti-VEGF-valmisteiden kanssa (systeeminen tai paikallinen). - Kostean silmänpohjan ikärappeuman anti-VEGF-hoidon jälkeen verkkokalvon pigmenttiepiteelin repeytymän kehittymiseen liittyviä riskitekijöitä ovat laaja ja/tai korkea verkkokalvon pigmenttiepiteelin irtoaminen. Aloitettaessa Eylea-hoitoa on noudatettava varovaisuutta, jos potilaalla on näitä verkkokalvon pigmenttiepiteelin repeytymien riskitekijöitä.
- Hoitoa ei saa antaa potilaille, joilla on repeytymisestä johtuva verkkokalvon irtauma tai 3. tai 4. asteen makulareikiä.
- Verkkokalvon repeytymisen yhteydessä annosta ei tule antaa ja hoitoa täytyy siirtää, kunnes repeytymä on riittävästi korjautunut
-
Seuraavissa tapauksissa annosta ei tule antaa ja hoitoa ei tule aloittaa uudelleen ennen seuraavaa suunniteltua hoitoajankohtaa:
- paras korjattu näöntarkkuus (BCVA) on laskenut ≥ 30 kirjainta verrattuna edelliseen näöntarkkuuden mittaamiseen
- verkkokalvon alainen verenvuoto keskeisen näön alueella, jos verenvuodon laajuus on ≥ 50 % leesion kokonaispinta-alasta
- Annosta ei tule antaa 28 vuorokautta ennen tai jälkeen suoritetun tai suunnitellun silmäleikkauksen
- Eylea-valmistetta ei pidä käyttää raskauden aikana, ellei hoidon mahdollinen hyöty ole sikiölle aiheutuvaa mahdollista riskiä suurempi (ks. kohta Raskaus ja imetys).
- Hedelmällisessä iässä olevien naisten on käytettävä tehokasta ehkäisyä hoidon aikana sekä vähintään 3 kuukautta viimeisen lasiaiseen annettavan aflibersepti-injektion jälkeen (ks. kohta Raskaus ja imetys).
- Kokemusta on rajallisesti sellaisten potilaiden hoidosta, joilla on iskeeminen verkkokalvon keskuslaskimotukos tai haaralaskimotukos. Tätä lääkehoitoa ei suositella potilaille, joilla on kliinisiä merkkejä palautumattomasta iskeemisestä näön menetyksestä.
Potilasryhmät, joista on rajallisesti tietoa
Hoidosta potilailla, joilla on tyypin 1 diabeteksesta johtuva diabeettinen makulaturvotus, tai diabeetikoilla, joiden HbA1c on yli 12 % tai jotka sairastavat proliferatiivista diabeettista retinopatiaa, on vain rajallisesti tietoa.
Eylea-valmistetta ei ole tutkittu potilailla, joilla on aktiivisia systeemisiä tulehduksia, eikä potilailla, joilla on samanaikaisesti silmäsairauksia, kuten verkkokalvon irtauma tai makulareikä. Eylea-hoidosta ei myöskään ole kokemusta diabeetikoilla, joiden verenpaine ei ole hoitotasapainossa. Tällaisten potilaiden hoidossa lääkärin on otettava huomioon tietojen puutteellisuus.
Eylea-valmisteen käytöstä likitaitteisuuden aiheuttaman suonikalvon uudissuonittumisen hoitoon ei ole kokemusta ei-aasialaista syntyperää olevilla potilailla, likitaitteisuuden aiheuttamaan suonikalvon uudissuonittumiseen aiemmin hoitoa saaneilla potilailla eikä potilailla, joilla on makulan keskiosan ulkopuolella olevia vaurioita (ekstrafoveaalisia leesioita).
Tietoa apuaineista
Tämä lääkevalmiste sisältää
- alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”
- 0,015 mg polysorbaatti 20:tä per 0,05 ml:n annos, joka vastaa 0,3 mg/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Verteporfiinilla annettavan fotodynaamisen hoidon käyttöä Eylea-hoidon lisänä ei ole tutkittu, ja siksi turvallisuutta ei tunneta.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Hedelmällisessä iässä olevien naisten on käytettävä tehokasta ehkäisyä hoidon aikana sekä vähintään 3 kuukautta viimeisen lasiaiseen annettavan aflibersepti-injektion jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Ei ole olemassa tietoja afliberseptin käytöstä raskaana oleville naisille.
Eläinkokeissa on havaittu alkio-sikiötoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikka systeeminen altistus lasiaiseen injektoinnin jälkeen on erittäin vähäistä, Eylea-valmistetta ei pidä käyttää raskauden aikana, ellei mahdollinen hyöty ole sikiölle aiheutuvaa mahdollista riskiä suurempi.
Imetys
Hyvin niukkojen ihmisistä saatujen tietojen perusteella aflibersepti saattaa erittyä ihmisen äidinmaitoon vähäisissä määrin. Aflibersepti on suuri proteiinimolekyyli, ja vauvaan imetyksen yhteydessä imeytyvän lääkkeen määrän odotetaan olevan vähäinen. Afliberseptin vaikutuksia imetettävään vauvaan ei tunneta.
Varotoimena imetystä ei suositella Eylean käytön aikana.
Hedelmällisyys
Korkeaa systeemistä altistusta tutkineiden eläinkokeiden tulokset osoittavat, että aflibersepti voi heikentää miesten ja naisten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Sellaisia vaikutuksia ei odoteta silmäkäytön jälkeen, koska systeeminen altistus on erittäin vähäistä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Eylea-injektiolla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn johtuen lasiaiseen annetun injektion tai injektioon liittyvien silmätutkimusten aiheuttamista väliaikaisista näköhäiriöistä. Potilaat eivät saa ajaa tai käyttää koneita, ennen kuin heidän näkökykynsä on palautunut riittävästi.
Haittavaikutukset
Yhteenveto turvallisuudesta
Kahdeksassa vaiheen III tutkimuksessa turvallisuusryhmän muodosti 3 102 potilasta. Näistä potilaista 2 501 potilasta hoidettiin suositellulla 2 mg:n annoksella.
Injektointitoimenpiteeseen liittyviä vakavia okulaarisia haittavaikutuksia tutkittavassa silmässä esiintyi harvemmin kuin yhdessä 1 900 lasiaiseen annetusta Eylea-injektiosta. Vakavia haittavaikutuksia olivat sokeutuminen, endoftalmiitti, verkkokalvon irtauma, traumaperäinen kaihi, kaihi, lasiaisverenvuoto, lasiaisirtauma ja silmänpaineen kohoaminen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yleisimpiä haittavaikutuksia (vähintään 5 %:lla Eylea-valmisteella hoidetuista potilaista) olivat sidekalvon verenvuoto (25 %), verkkokalvon verenvuoto (11 %), näöntarkkuuden heikkeneminen (11 %), silmäkipu (10 %), kaihi (8 %), kohonnut silmänpaine (8 %), lasiaisirtauma (7 %) ja lasiaiskellujat (7 %).
Haittavaikutustaulukko
Alla kuvatut turvallisuustiedot sisältävät kaikki sellaiset haittavaikutukset silmänpohjan ikärappeuman kostean muodon, verkkokalvon keskuslaskimotukoksen, haaralaskimotukoksen, diabeettisen makulaturvotuksen ja likitaitteisuuden aiheuttaman suonikalvon uudissuonittumisen käyttöaiheiden kahdeksasta vaiheen III tutkimuksista, joilla oli kohtuullinen mahdollisuus syy-seuraus-suhteesta injektointitoimenpiteeseen tai lääkevalmisteen käyttöön.
Haittavaikutukset on lueteltu elinjärjestelmäluokan ja esiintymistiheyden mukaan seuraavasti:
hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Todetut haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Kaikki vaiheen III tutkimuksissa hoidosta johtuvat potilailla raportoidut haittavaikutukset (yhdistetyt tiedot vaiheen III tutkimuksista kostean silmänpohjan ikärappeuman, verkkokalvon keskuslaskimotukoksen, haaralaskimotukoksen, diabeettisen makulaturvotuksen ja likitaitteisuuden aiheuttaman suonikalvon uudissuonittumisen käyttöaiheissa) tai markkinoille tulon jälkeisessä seurannassa havaitut haittavaikutukset
| Elinjärjestelmäluokka | Esiintymistiheys | Haittavaikutus |
| Immuunijärjestelmä | Melko harvinainen | Yliherkkyys*** |
| Silmät | Hyvin yleinen | Näöntarkkuuden heikkeneminen, verkkokalvon verenvuoto, sidekalvon verenvuoto, silmäkipu |
| Yleinen | Verkkokalvon pigmenttiepiteelin repeämä*, verkkokalvon pigmenttiepiteelin irtauma, verkkokalvon rappeutuminen, lasiaisen verenvuoto, kaihi, kortikaalinen kaihi, tumakaihi, subkapsulaarinen kaihi, sarveiskalvon eroosio, sarveiskalvon naarmu, kohonnut silmänpaine, näön hämärtyminen, lasiaiskellujat, lasiaisen irtauma, injektiokohdan kipu, vierasainetuntemus silmissä, lisääntynyt kyynelnesteen eritys, silmäluomen turvotus, injektiokohdan verenvuoto, pisteinen sarveiskalvotulehdus, sidekalvon verekkyys, silmän verekkyys | |
| Melko harvinainen | Endoftalmiitti**, verkkokalvon irtauma, verkkokalvon repeämä, iriitti, uveiitti, iridosykliitti, mykiön samentuma, sarveiskalvon epiteelin vaurio, injektiokohdan ärsytys, epänormaali tunne silmässä, silmäluomen ärsytys, etukammion punoitus, sarveiskalvon turvotus | |
| Harvinainen | Sokeutuminen, traumaperäinen kaihi, lasiaistulehdus, silmän etukammion märkäsakka | |
| Tuntematon | Skleriitti**** |
* Tilat, joiden tiedetään liittyvän kosteaan silmänpohjan ikärappeumaan. Havaittiin vain kostean silmänpohjan ikärappeuman tutkimuksissa.
** viljelypositiivinen ja viljelynegatiivinen endoftalmiitti
*** markkinoille tulon jälkeisenä aikana on raportoitu yliherkkyyttä, johon liittyy ihottumaa, kutinaa, nokkosihottumaa sekä yksittäisiä vaikeita anafylaktisia/anafylaktoidisia reaktioita.
Kuvaus valikoiduista haittavaikutuksista
Silmänpohjan kostean ikärappeuman (AMD) vaiheen III tutkimuksissa havaittiin sidekalvon verenvuodon kohonnut insidenssi potilailla, jotka saivat antitromboottista lääkettä. Insidenssin kohoaminen oli vastaavaa ranibitsumabilla ja Eylea-valmisteella hoidetuilla potilailla.
Valtimon tromboemboliatapahtumat ovat haittavaikutuksia, jotka mahdollisesti liittyvät systeemiseen verisuonikasvutekijän (VEGF) estoon. Verisuonikasvutekijän estäjien lasiaisen sisäisen käytön jälkeen on olemassa teoreettinen riski valtimon tromboemboliatapahtumiin, mukaan lukien aivohalvaus ja sydäninfarkti.
Eylea-valmisteen kliinisissä tutkimuksissa todettiin matala insidenssi valtimoiden tromboembolisten tapahtumien suhteen kaikissa käyttöaiheissa (AMD, DME, RVO ja myooppinen CNV). Eri käyttöaiheissa ei todettu merkittävää eroa aflibersepti- ja vertailuryhmän välillä.
Kuten kaikkien terapeuttisten proteiinien kohdalla, myös Eylea-valmisteeseen liittyy immunogeenisuuden mahdollisuus.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa käytettiin enintään 4 mg:n annoksia kuukauden välein ja yksittäisiä yliannostuksia 8 mg:n annoksella tapahtui.
Yliannostus ja suuri injektiomäärä voivat nostaa silmänpainetta. Siksi yliannostustapauksissa silmänpainetta on seurattava ja hoitavan lääkärin suosituksen mukaan aloitettava sopiva hoito. (ks. kohta Käyttö- ja käsittelyohjeet)
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: silmälääkkeet/antineovaskularisaatioaineet ATC-koodi: S01LA05
Aflibersepti on rekombinantti fuusioproteiini, joka sisältää osia ihmisen VEGF-reseptorien 1 ja 2 solunulkoisista domeeneista yhdistettynä ihmisen IgG1:n Fc-osaan.
Aflibersepti on tuotettu kiinanhamsterin munasarja (CHO) K1-soluissa yhdistelmä-DNA-tekniikalla.
Aflibersepti toimii liukoisena syöttireseptorina, joka sitoo VEGF-A:ta ja PlGF:ä suuremmalla affiniteetilla kuin näiden luonnolliset reseptorit, ja siten se estää näihin kuuluvien VEGF-reseptoreiden sitoutumista ja aktivaatiota.
Vaikutusmekanismi
Verisuonikasvutekijä A (VEGF-A) ja plasentaalinen kasvutekijä (PlGF) kuuluvat angiogeenisiin kasvutekijöihin (VEGF), jotka toimivat endoteelisolujen potentteina mitogeenisinä, kemotaktisina ja vaskulaarista permeabiliteettia lisäävinä tekijöinä. VEGF toimii kahden endoteelisolujen pinnalla esiintyvän reseptorityrosiinikinaasin, VEGFR-1:n ja VEGFR-2:n, välityksellä. PlGF sitoutuu vain VEGFR-1:een, jota esiintyy myös leukosyyttien pinnalla. Jos VEGF-A aktivoi näitä reseptoreita liikaa, se voi aiheuttaa patologista neovaskularisaatiota ja liiallista vaskulaarista permeabiliteettia.
PlGF voi tehostaa VEGF-A:n vaikutusta näissä prosesseissa ja sen tiedetään myös edistävän leukosyyttien infiltraatiota ja vaskulaarista inflammaatiota.
Farmakodynaamiset vaikutukset
Kostea silmänpohjan ikärappeuma
Kosteaan silmänpohjan ikärappeumaan liittyy patologinen suonikalvon verisuonten uudismuodostus (CNV). Suonikalvon verisuonten uudismuodostuksen (CNV) aiheuttama veren ja nesteen vuoto voi aiheuttaa verkkokalvon paksuuntumista tai turvotusta ja/tai verkkokalvon alaista/sisäistä verenvuotoa, johtaen näöntarkkuuden heikkenemiseen.
Eylea-valmisteella hoidetuilla (yksi injektio kuukaudessa kolmena peräkkäisenä kuukautena ja sen jälkeen yksi injektio kahden kuukauden välein) potilailla keskeinen verkkokalvon paksuus [CRT] pieneni pian hoidon aloittamisen jälkeen, ja suonikalvon verisuonten uudismuodostusleesion keskimääräinen koko pieneni. Tämä on yhdenmukaista kuukausittaisella 0,5 mg:n ranibitsumabiannoksella saatujen tulosten kanssa.
VIEW1-tutkimuksessa CRT väheni keskimäärin optisella koherenssitomografialla (OCT) mitattuna (-130 mikrometriä 2 mg:n Eylea-ryhmässä, jossa hoitoväli oli kaksi kuukautta, ja -129 mikrometriä 0,5 mg:n ranibitsumabiryhmässä, jossa hoitoväli oli kuukausi, kun paksuus mitattiin viikolla 52). Viikolla 52 VIEW2-tutkimuksessa CRT pieneni OCT:llä mitattuna (-149 mikrometriä 2 mg:n Eylea-ryhmässä, jossa hoitoväli oli kaksi kuukautta, ja -139 mikrometriä 0,5 mg:n ranibitsumabiryhmässä, jossa hoitoväli oli kuukausi). Suonikalvon verisuonten uudismuodostuksen pinta-ala ja CRT:n pieneneminen säilyivät yleisesti tutkimusten toisen vuoden aikana.
ALTAIR-tutkimus suoritettiin japanilaisilla potilailla, jotka eivät olleet saaneet aiempaa hoitoa kosteaan silmänpohjan ikärappeumaan. Tutkimus osoitti samankaltaisia tuloksia kuin VIEW- tutkimukset käyttäen aluksi kolmea kuukausittaista 2 mg Eylea-injektiota, jonka jälkeen annettiin yksi injektio 2 kuukauden kuluttua. Tämän jälkeen jatkettiin treat-and-extend-hoito-ohjelmalla (ohjelma, jossa injektioväliä voidaan pidentää asteittain silmän hoitovasteen perusteella) vaihtelevin hoitovälein (2 tai 4 viikon muutokset) korkeintaan 16 viikon hoitoväliin etukäteen määriteltyjen kriteerien perusteella. Viikolla 52 verkkokalvon keskeisen alueen paksuus (CRT) pieneni OCT:llä mitattuna (-134,4 mikrometriä ryhmässä, jossa hoitovälin muutokset olivat 2 viikkoa ja -126,1 mikrometriä ryhmässä, jossa hoitovälin muutokset olivat 4 viikkoa). Viikolla 52 niiden potilaiden määrä, joilla ei ollut nestettä OCT:llä mitattuna, oli 68,3 % ryhmässä, jossa hoitovälin muutokset olivat 2 viikkoa ja 69,1 % ryhmässä, jossa hoitovälin muutokset olivat 4 viikkoa. ALTAIR-tutkimuksen toisena vuonna CRT:n pieneneminen yleisesti säilyi molemmissa tutkimushaaroissa.
ARIES-tutkimus suunniteltiin tutkimaan Eylea 2 mg treat-and-extend-hoito-ohjelman, joka aloitettiin heti kolmen kuukausittain annettavan injektion ja sen jälkeen kahden kuukauden kuluttua annettavan lisäinjektion jälkeen, vertailukelpoisuutta treat-and-extend-hoito-ohjelmaan, joka aloitettiin vuoden kestäneen hoidon jälkeen. Potilailla, jotka tarvitsivat tutkimuksen aikana vähintään kerran hoitoa useammin kuin 8 viikon välein, verkkokalvon keskeisen alueen paksuus (CRT) pysyi suurempana, mutta keskimääräinen CRT:n pieneneminen lähtötasosta viikolle 104 oli -160,4 mikrometriä, samoin kuin potilailla, joita hoidettiin 8 viikon välein tai harvemmin.
Verkkokalvon keskuslaskimotukoksesta ja haaralaskimotukoksesta aiheutuva makulaarinen edeema
Verkkokalvon keskuslaskimotukoksen ja haaralaskimotukoksen yhteydessä ilmenee verkkokalvon iskemiaa, joka johtaa verisuonikasvutekijän (VEGF) vapautumiseen. VEGF:n vaikutuksesta tiiviit liitokset heikentyvät ja endoteelisolujen proliferaatio stimuloituu. Verisuonikasvutekijän vaikutuksen tehostumiseen (up-regulation) liittyy veri-verkkokalvoesteen heikkenemistä, verisuonten lisääntynyttä läpäisevyyttä, verkkokalvon edeemaa ja verisuonten uudismuodostusta.
Potilailla, jotka ovat saaneet 6 perättäistä 2 mg Eylea-injektiota kerran kuukaudessa ilmeni yhdenmukainen, nopea ja selkeä morfologinen vaste (CRT:n pieneneminen OCT:llä mitattuna). Viikolla 24 CRT pieneni merkitsevästi enemmän kuin vertailuryhmässä kaikissa kolmessa tutkimuksessa (COPERNICUS CRVO-tutkimuksessa: -457 mikrometriä vs. -145 mikrometriä; GALILEO CRVO-tutkimuksessa -449 mikrometriä vs. -169 mikrometriä; VIBRANT BRVO-tutkimuksessa-280 mikrometriä vs. -128 mikrometriä). CRT:n pieneneminen lähtötilanteesta säilyi jokaisen tutkimuksen loppuun asti, viikolle 100 COPERNICUS-tutkimuksessa, viikolle 76 GALILEO-tutkimuksessa ja viikolle 52 VIBRANT-tutkimuksessa.
Diabeettinen makulaturvotus
Diabeettinen makulaturvotus on seurausta diabeettisesta retinopatiasta ja jonka merkkejä ovat lisääntynyt verisuonien läpäisevyys ja verkkokalvon hiussuonien vaurioituminen, mikä voi johtaa näöntarkkuuden heikkenemiseen.
Potilailla, jotka saivat Eylea-hoitoa, ja joista suurin osa luokiteltiin tyypin II diabetesta sairastaviksi, havaittu morfologinen vaste (CRT, DRSS-aste [diabeettisen retinopatian vaikeusaste]) oli nopea ja selkeä.
VIVIDDME- ja VISTADME-tutkimuksissa havaittiin tilastollisesti merkitsevästi suurempi CRT:n pieneneminen lähtötasosta Eylea-hoitoa saaneilla potilailla verrattuna laserkontrolliryhmään viikolla 52, -192,4 ja -183,1 mikrometriä Eylea 2Q8 -ryhmissä ja -66,2 ja -73,3 mikrometriä kontrolliryhmissä. Viikolla 100 pieneneminen säilyi siten, että se oli -195,8 ja -191,1 mikrometriä 2Q8 Eylea -ryhmissä ja -85,7 ja -83,9 mikrometriä kontrolliryhmissä VIVIDDME ja VISTADME-tutkimuksissa, tässä järjestyksessä.
DRSS:ssa havaittu ≥ 2 askeleen paraneminen määritettiin etukäteen määrätyllä tavalla VIVIDDME- ja VISTADME-tutkimuksissa. DRSS-pistemäärä oli luokiteltavissa 73,7 %:lla potilaista VIVID DME- tutkimuksessa ja 98,3 %:lla potilaista VISTADME-tutkimuksessa. Viikolla 52 ilmeni DRSS:ssa ≥ 2 askeleen paraneminen 27,7 %:ssa ja 29,1 %:ssa Eylea 2Q8-ryhmistä ja 7,5 %:ssa ja 14,3 %:ssa kontrolliryhmistä. Viikolla 100 vastaavat prosenttiosuudet olivat 32,6 % ja 37,1 % Eylea 2Q8-ryhmistä ja 8,2 % ja 15,6 % kontrolliryhmistä.
VIOLET-tutkimuksessa verrattiin Eylea 2 mg:n kolmea erilaista annosteluohjelmaa diabeettisen makulaturvotuksen hoidossa vähintään vuoden kiinteän annosvälin hoidon jälkeen (viiden kuukausittaisen annoksen jälkeen annostelua jatkettiin kahden kuukauden välein). Tutkimuksen viikolla 52 ja viikolla 100 (eli toisen ja kolmannen hoitovuoden aikana) CRT:n keskimääräiset muutokset olivat kliinisesti samanlaiset treat-and-extend (2T&E), pro re nata (2PRN) ja 2Q8 -ryhmissä, keskimääräisten muutosten ollessa vastaavasti -2,1, 2,2 ja -18,8 mikrometriä viikolla 52 ja 2,3, -13,9 ja -15,5 mikrometriä viikolla 100.
Likitaitteisuuden aiheuttama suonikalvon uudissuonittuminen
Likitaitteisuuden aiheuttama suonikalvon uudissuonittuminen (myooppinen CNV) on yleinen näön menettämisen syy aikuisilla, joilla on patologinen myopia. Siinä syntyy haavojen paranemismekanismi Bruchin kalvon repeymien seurauksena; tämä onkin patologisessa myopiassa kaikkein eniten näköä uhkaava tapahtuma.
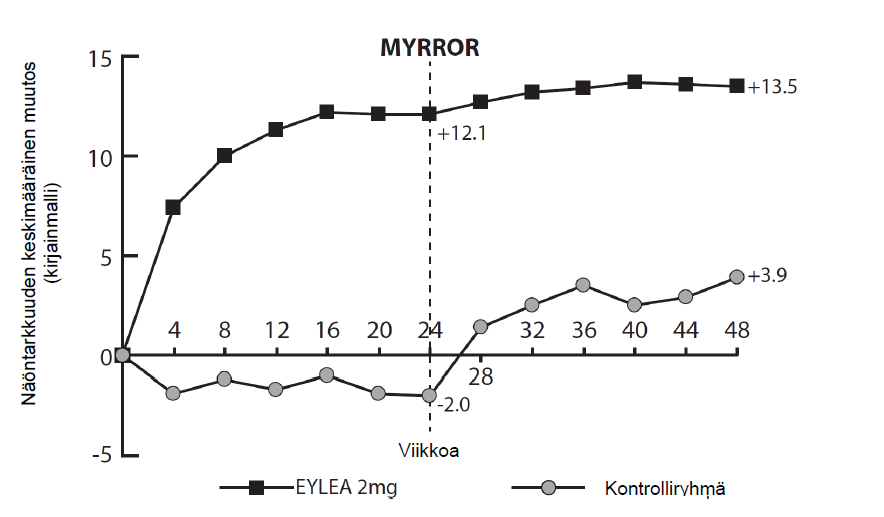
Potilailla, joita hoidettiin Eylea-valmisteella MYRROR-tutkimuksessa (yksi injektio annettiin hoidon alussa; jos sairaus ei hävinnyt tai uusiutui, annettiin lisäinjektioita), CRT pieneni pian hoidon aloittamisen jälkeen Eylea-valmisteella viikolla 24 (-79 mikrometriä Eylea 2 mg -hoitoryhmässä ja -4 mikrometriä kontrolliryhmässä) vaikutuksen kestäessä viikolle 48 asti. Lisäksi keskimääräisen CNV-leesion koko pieneni.
Kliininen teho ja turvallisuus
Kostea silmänpohjan ikärappeuma
Eylea-valmisteen turvallisuutta ja tehoa arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, aktiivisesti kontrolloidussa monikeskustutkimuksessa potilailla, joilla oli kostea silmänpohjan ikärappeuma (VIEW1 ja VIEW2). Tutkimuksissa yhteensä 2 412 potilasta hoidettiin ja hoidon teho arvioitiin. Näistä 1 817 potilasta sai Eylea-valmistetta. Potilaiden iät vaihtelivat 49 ja 99 vuoden välillä keski-iän ollen 76 vuotta. Näissä kliinisissä tutkimuksissa noin 89 % (1 616/1 817) randomisoiduista Eylea-hoitoa saaneista potilaista olivat iältään 65 vuotta tai vanhempia ja noin 63 % (1 139/1 817)
75 vuotta tai vanhempia. Kummassakin tutkimuksessa potilaat määrättiin satunnaisesti yhteen neljästä annosryhmästä suhteessa 1:1:1:1:
- Eylea-valmistetta annettiin 2 mg 8 viikon välein kolmen kuukausittaisen alkuannoksen jälkeen (Eylea 2Q8);
- Eylea-valmistetta annettiin 2 mg neljän viikon välein (Eylea 2Q4);
- Eylea-valmistetta annettiin 0,5 mg neljän viikon välein (Eylea 0.5Q4); ja
- ranibitsumabia annettiin 0,5 mg neljän viikon välein (ranibitsumabi 0.5Q4).
Tutkimusten toisena vuotena potilaat saivat samaa annosvahvuutta, jota heidät oli alun perin satunnaistettu saamaan, mutta annosaikataulua muutettiin visuaalisten ja anatomisten tulosten arvion mukaan. Tutkimussuunnitelmassa määritetty annosten enimmäisväli oli 12 viikkoa.
Molemmissa tutkimuksissa ensisijainen tehon päätetapahtuma oli niiden potilaiden suhteellinen määrä tutkimussuunnitelmassa määritetystä joukosta, joiden näkökyky säilyi (määritelmänä alle 15 kirjaimen näkötarkkuuden menetys) viikolla 52 verrattuna lähtötasoon.
VIEW1-tutkimuksessa viikolla 52 Eylea 2Q8 -ryhmän potilaista 95,1 %:lla säilyi näkö ja 94,4 %:lla potilaista ranibitsumabi 0.5Q4 -ryhmässä. VIEW2-tutkimuksessa viikolla 52 Eylea 2Q8 -ryhmän potilaista 95,6 %:lla säilyi näkö ja 94,4 %:lla potilaista ranibitsumabi 0.5Q4 -ryhmässä. Molemmissa tutkimuksissa Eylea-hoidon osoitettiin olevan vähintään yhtä hyvä (non-inferior) ja kliinisesti vastaava kuin ranibitsumabi 0.5Q4 hoito.
Tarkemmat tulokset molempien tutkimusten yhdistetystä analyysistä on esitetty seuraavassa taulukossa 2 ja kuvassa 1.
Taulukko 2: Tehoa kuvaavat tulokset viikolla 52 (ensisijainen analyysi) ja viikolla 96; yhdistetyt tiedot VIEW1- ja VIEW2-tutkimuksistaB)
| Tehoa kuvaava tulos | Eylea 2Q8 E) (Eylea 2 mg 8 viikon välein 3 kuukausittaisen alkuannoksen jälkeen) (n = 607) | Ranibitsumabi 0.5Q4 (ranibitsumabi 0,5 mg 4 viikon välein) (n = 595) | ||
| Injektioiden keskimääräinen lukumäärä lähtötilanteesta | Viikko 52 | Viikko 96 | Viikko 52 | Viikko 96 |
| Injektioiden keskimääräinen lukumäärä lähtötilanteesta | 7,6 | 11,2 | 12,3 | 16,5 |
| Injektioiden keskimääräinen lukumäärä viikoilla 52-96 | 4,2 | 4,7 | ||
| Niiden potilaiden osuus, joiden näöntarkkuus säilyi (<15 kirjainta BCVA:nA) menetystä) (tutkimussuunnitelman mukaisesti) | 95,33 %B) | 92,42 % | 94,42 % B) | 91,60 % |
EroC) (95 % CI)D) | 0,9 % (–1,7, 3,5)F) | 0,8 % (–2,3, 3,8)F) | ||
Keskimääräinen BCVA:n muutos mitattuna ETDRS -kirjainpisteinäA) lähtötasosta | 8,40 | 7,62 | 8,74 | 7,89 |
| Ero keskimääräisenä muutoksena, LS A) (ETDRS-kirjaimet)C) (95 % CI)D) | –0,32 (–1,87, 1,23) | –0,25 (–1,98, 1,49) | ||
| Potilaiden osuus, joiden näöntarkkuus parani lähtötasosta vähintään 15 kirjainta | 30,97 % | 33,44 % | 32,44 % | 31,60 % |
EroC) (95 % CI)D) | –1,5 % (–6,8, 3,8) | 1,8 % (–3,5, 7,1) | ||
A) BCVA: paras korjattu näöntarkkuus.
ETDRS: Early Treatment Diabetic Retinopathy Study.
LS: ANCOVA:sta johdetut pienimmät neliösummat.
PPS: Per Protocol Set
B) Täysi analyysijoukko (FAS), viimeinen havainto eteenpäin (LOCF) kaikista analyyseistä, paitsi niiden potilaiden osuudesta, joiden näöntarkkuus säilyi viikolla 52, mikä on tutkimussuunnitelman mukaista.
C) Ero on Eylea-ryhmän arvo miinus ranibitsumabiryhmän arvo. Positiivinen arvo tarkoittaa Eylea-valmisteen hyötyä.
D) Luottamusväli (CI) laskettuna normaalilla approksimaatiolla.
E) Kun hoito aloitettiin kolmella kuukausittaisella annoksella.
F) Luottamusväli, joka on kokonaan –10 %:n yläpuolella, osoittaa Eylea-valmisteen ei-huonommuuden verrattuna ranibitsumabiin.
Kuva 1. Näkötarkkuuden keskimääräinen muutos
Näkötarkkuuden keskimääräinen muutos lähtötasosta viikkoon 96 mennessä (yhdistetyt tiedot View1- ja View2-tutkimuksista)
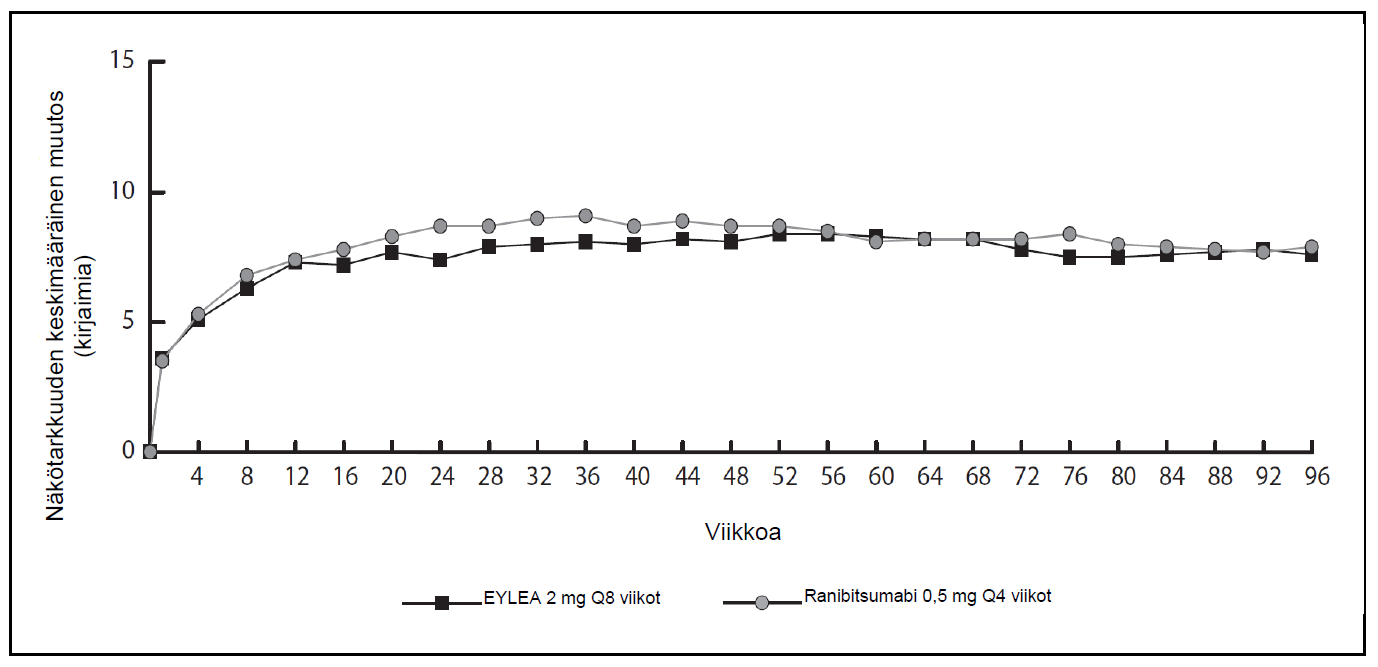
VIEW1- ja VIEW2-tutkimusten tulosten yhdistetyssä analyysissä Eylea-valmisteella todettiin kliinisesti merkittäviä muutoksia lähtötasosta esimääritetyssä toissijaisessa tehon päätetapahtumassa NEI VFQ-25 (National Eye Institute Visual Function Questionnaire) -kyselyssä, ilman kliinisesti merkittävää eroa ranibitsumabiin. Näiden muutosten suuruus oli samanlainen kuin julkaistuissa tutkimuksissa, ja vastasi 15 kirjaimen parannusta parhaassa korjatussa näöntarkkuudessa (BCVA).
Tutkimusten toisen vuoden aikana, teho säilyi yleisesti viimeiseen arviointiin viikolle 96 asti. Potilaista 2-4 % tarvitsi kaikki injektiot kuukauden välein ja kolmannes potilaista tarvitsi vähintään yhden injektion kuukauden kuluttua edellisestä injektiosta.
Suonikalvon verisuonten uudismuodostusalueen keskimääräisen pinta-alan pieneneminen oli selvä kaikissa annosryhmissä molemmissa tutkimuksissa.
Tehoa kuvaavat tulokset kaikissa arvioitavissa alaryhmissä (esim. ikä, sukupuoli, rotu, lähtötason näöntarkkuus, leesiotyyppi, leesion koko) molemmissa tutkimuksissa ja yhdistetyssä analyysissä vastasivat yleisen väestön tuloksia.
ALTAIR oli 96 viikkoa kestänyt satunnaistettu, avoin, monikeskustutkimus 247 japanilaisella potilaalla, jotka eivät ole saaneet aiempaa hoitoa kosteaan silmänpohjan ikärappeumaan. Tutkimus on suunniteltu arvioimaan Eylea-valmisteen tehoa ja turvallisuutta treat-and-extend-hoito-ohjelmalla kahdella eri hoitovälin muutoksella (2 tai 4 viikon muutokset).
Kaikki potilaat saivat kerran kuukaudessa Eylea 2 mg -injektion kolmen kuukauden ajan, jonka jälkeen annettiin vielä yksi injektio 2 kuukauden kuluttua. Viikolla 16 potilaat satunnaistettiin suhteessa 1:1 kahteen hoitoryhmään: 1) Eylea treat-and-extend-hoito 2 viikon hoitovälin muutoksina
2) Eylea treat-and-extend-hoito 4 viikon hoitovälin muutoksina. Päätös hoitovälin pidentämisestä tai lyhentämisestä perustui protokollassa määriteltyihin näkö- ja/tai anatomiavasteisiin. Molemmissa hoitoryhmissä enimmäishoitoväli oli 16 viikkoa.
Ensisijainen tehon päätetapahtuma oli keskimääräinen muutos parhaassa korjatussa näöntarkkuudessa (BCVA) lähtötasosta viikolle 52. Toissijaiset tehon päätetapahtumat olivat niiden potilaiden osuus, joiden näkökyky ei heikentynyt ≥ 15 kirjainta ja niiden potilaiden osuus, joiden näkökyky parani vähintään 15 kirjainta (BCVA) viikolla 52 verrattuna lähtötasoon.
Viikolla 52 tutkimushaarassa, jossa hoitoa pidennettiin 2 viikon hoitovälin lisäyksinä, potilaiden näkökyky parani keskimäärin 9,0 kirjainta lähtötasosta verrattuna 8,4 kirjaimeen tutkimushaarassa, jossa hoitoa pidennettiin 4 viikon hoitovälin lisäyksinä [LS (pienimpien neliösummien keskiarvo) kirjaimina (95 % CI): -0,4 (-3,8,3,0), ANCOVA]. Niiden potilaiden osuus, joiden näkökyky ei heikentynyt ≥ 15 kirjainta oli samankaltainen molemmissa ryhmissä (96,7 % ryhmä, jossa hoitovälin muutokset tehtiin 2 viikon välein ja 95,9 % ryhmä, jossa hoitovälin muutokset tehtiin 4 viikon välein). Niiden potilaiden osuus, joiden näkökyky parani ≥ 15 kirjainta viikolla 52 oli 32,5 % ryhmässä, jossa hoitovälin muutokset tehtiin 2 viikon välein ja 30,9 % ryhmässä, jossa hoitovälin muutokset tehtiin 4 viikon välein. Niiden potilaiden suhteellinen määrä, joiden hoitoväliä pidennettiin 12 viikkoon tai pidempään, oli 42,3 % ryhmässä, jossa hoitovälin muutokset tehtiin 2 viikon välein ja 49,6 % ryhmässä, jossa hoitovälin muutokset tehtiin 4 viikon välein. Lisäksi ryhmässä, jossa hoitovälin muutokset tehtiin 4 viikon välein, hoitoväliä pidennettiin 16 viikkoon 40,7 % potilaalla. Viimeisellä käynnillä korkeintaan viikolla 52, seuraava injektio oli suunniteltu annettavaksi 12 viikon kuluttua tai myöhemmin 56,8 % potilaista ryhmässä, jossa hoitovälin muutokset tehtiin 2 viikon välein ja 57,8 % potilaista ryhmässä, jossa hoitovälin muutokset tehtiin 4 viikon välein.
Tutkimuksen toisen vuoden aikana teho säilyi yleisesti viikon 96 viimeiseen arviointiin asti. Näkökyky parani keskimäärin 7,6 kirjainta lähtötasosta ryhmässä, jossa hoitovälin muutokset olivat 2 viikkoa ja 6,1 kirjainta ryhmässä, jossa hoitovälin muutokset olivat 4 viikkoa. Niiden potilaidenmäärä, joiden hoitoväliä pidennettiin 12 viikkoon tai pidempään, oli 56,9 % ryhmässä, jossa hoitovälin muutokset olivat 2 viikkoa ja 60,2 % ryhmässä, jossa hoitovälin muutokset olivat 4 viikkoa. Viimeisellä käynnillä ennen viikkoa 96 seuraava injektio oli suunniteltu annettavaksi 12 viikon kuluttua tai myöhemmin 64,9 % potilaista ryhmässä, jossa hoitovälin muutokset tehtiin 2 viikon välein ja 61,2 % potilaista ryhmässä, jossa hoitovälin muutokset tehtiin 4 viikon välein. Hoidon toisen vuoden aikana potilaat saivat keskimäärin 3,6 injektiota ryhmässä, jossa hoitovälin muutokset tehtiin 2 viikon välein ja vastaavasti 3,7 injektiota ryhmässä, jossa hoitovälin muutokset tehtiin 4 viikon välein. Kahden vuoden hoitojakson aikana potilaat saivat keskimäärin 10,4 injektiota.
Turvallisuusprofiili silmässä ja systeemisesti oli samankaltainen kuin turvallisuustiedot, jotka havaittiin VIEW1- ja VIEW2- avaintutkimuksissa.
ARIES oli 104 viikkoa kestänyt satunnaistettu, avoin, aktiivisesti kontrolloitu monikeskustutkimus 269 potilaalla, jotka eivät olleet saaneet aiempaa hoitoa kosteaan silmänpohjan ikärappeumaan. Tutkimus oli suunniteltu arvioimaan tehon sekä turvallisuuden vertailukelpoisuutta treat-and-extend-hoito-ohjelmassa, joka aloitettiin kolmen peräkkäisen kuukausittain annettavan annoksen ja näitä seuraavan kahden kuukauden hoitovälin annoksen jälkeen verrattuna treat-and-extend-hoito-ohjelmaan, joka aloitettiin ensimmäisen hoitovuoden jälkeen.
ARIES-tutkimuksessa tutkittiin myös niiden potilaiden prosentuaalista osuutta, jotka tutkijan päätökseen perustuen tarvitsivat hoitoa useammin kuin joka 8. viikko. Tutkimuksen aikana 269 potilaasta 62 potilasta sai vähintään kerran annoksen useammin. Nämä potilaat jatkoivat tutkimuksessa ja saivat hoitoa tutkijan parhaan kliinisen arvion mukaisesti. He eivät kuitenkaan saaneet hoitoa useammin kuin joka 4. viikko ja heidän hoitoväliä voitiin pidentää myöhemmin uudelleen. Keskimääräinen hoitoväli oli 6,1 viikkoa, sen jälkeen kun päätös lyhyemmästä hoitovälistä oli tehty. Viikolla 104 korjattu näöntarkkuus (BCVA) oli pienempi potilailla, jotka tarvitsivat intensiivisempää hoitoa vähintään kerran tutkimuksen aikana verrattaessa potilaisiin, jotka eivät sitä tarvinneet. Keskimääräinen BCVA:n muutos lähtötasosta tutkimuksen loppuun oli +2,3 ± 15,6 kirjainta. Potilaista, joita hoidettiin useammin, 85,5 prosentilla näkökyky säilyi eli menetys vähemmän kuin 15 kirjainta ja 19,4 prosentilla parannus 15 kirjainta tai enemmän. Turvallisuusprofiili useammin kuin 8 viikon hoitovälillä hoidetuilla potilailla oli verrattavissa turvallisuustietoihin, jotka havaittiin VIEW1- ja VIEW2- tutkimuksissa.
Verkkokalvon keskuslaskimotukoksesta johtuva makulaarinen edeema
Eylea-valmisteen turvallisuutta ja tehoa arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa potilailla, joilla oli verkkokalvon keskuslaskimotukoksesta aiheutuva makulaarinen edeema (COPERNICUS ja GALILEO). Yhteensä 358 potilaista hoidettiin ja hoidon teho arvioitiin. Näistä 217 potilasta sai Eylea-valmistetta. Potilaiden iät vaihtelivat 22 ja 89 vuoden välillä keski-iän ollen 64 vuotta. CRVO-tutkimuksissa noin 52 % (112/217) randomisoiduista Eylea-hoitoa saaneista potilaista olivat iältään 65 vuotta tai vanhempia ja noin 18 % (38/217) 75 vuotta tai vanhempia. Molemmissa tutkimuksissa potilaat määrättiin satunnaisesti suhteessa 3:2 joko 2 mg Eylea-valmistetta kerran 4 viikossa saavaan ryhmään (2Q4) tai yhden lumelääkeinjektion kerran 4 viikossa (yhteensä 6 injektiota) saavaan kontrolliryhmään.
Kun potilaat olivat saaneet yhteensä 6 injektiota kuukauden välein, heille annettiin hoitoa vain, jos ennalta määritellyt hoitokriteerit täyttyivät. Poikkeuksen muodostivat GALILEO-tutkimuksen kontrolliryhmän potilaat, jotka saivat lumelääkettä (kontrollista kontrolliin) viikolle 52 saakka. Tästä lähtien kaikki potilaat hoidettiin ennalta määriteltyjen kriteereiden mukaan.
Molemmissa tutkimuksissa ensisijainen tehon päätetapahtuma oli niiden potilaiden suhteellinen määrä, joiden näkökyky parani vähintään 15 kirjainta (BCVA) viikolla 24 verrattuna lähtötasoon. Toissijainen tehomuuttuja oli näöntarkkuuden muutos viikolla 24 verrattuna lähtötasoon.
Ero hoitoryhmien välillä oli molemmissa tutkimuksissa tilastollisesti merkitsevä Eylea-valmisteen eduksi. Maksimaalisen näöntarkkuuden paraneminen saavutettiin 3 kuukaudessa ja tästä seurannut näöntarkkuuden ja CRT:n stabiloituminen saavutettiin 6 kuukauden aikana. Tilastollisesti merkitsevä ero säilyi viikolle 52 saakka.
Molempien tutkimusten analyysin yksityiskohtaiset tulokset on esitetty seuraavassa taulukossa 3 ja kuvassa 2.
Taulukko 3: Tehoa kuvaavat tulokset viikoilla 24, 52 ja 76/100 (täysi analyysijoukko, viimeinen havainto eteenpäin (LOCFC)) COPERNICUS- ja GALILEO- tutkimuksissa.
| Tehoa kuvaava tulos | COPERNICUS | GALILEO | ||||||||||
| 24 viikkoa | 52 viikkoa | 100 viikkoa | 24 viikkoa | 52 viikkoa | 76 viikkoa | |||||||
Eylea (n=114) | Kontrolli (n = 73) | Eylea (n=114) | KontrolliE) (n = 73) | EyleaF) 2 mg (n = 114) | KontrolliE,F) (n = 73) | Eylea (n=103) | Kontrolli (n = 68) | Eylea (n=103) | Kontrolli (n = 68) | EyleaG) (n = 103) | KontrolliG) (n = 68) | |
Niiden potilaiden osuus, joiden näöntarkkuus parani lähtötasosta vähintään 15 kirjainta | 56 % | 12 % | 55 % | 30 % | 49,1 % | 23,3 % | 60 % | 22 % | 60 % | 32 % | 57,3 % | 29,4 % |
Painotettu eroA,B) (95 % CI) p-arvo | 44,8 % (33,0, 56,6) p < 0,0001 | 25,9 % (11,8, 40,1) p = 0,0006 | 26,7 % (13,1, 40,3) p=0,0003 | 38,3 % (24,4, 52,1) p < 0,0001 | 27,9 % (13,0, 42,7) p = 0,0004 | 28,0 % (13,3, 42,6) p=0,0004 | ||||||
Keskimääräinen BVCAC):n muutos mitattuna ETDRSC) -kirjainpisteinä lähtötasosta (SD) | 17,3 (12,8) | -4,0 (18,0) | 16,2 (17,4) | 3,8 (17,1) | 13,0 (17,7) | 1,5 (17,7) | 18,0 (12,2) | 3,3 (14,1) | 16,9 (14,8) | 3,8 (18,1) | 13,7 (17,8) | 6,2 (17,7) |
Ero keskimääräisenä muutoksen, LSA,C,D) (95 % CI) p-arvo | 21,7 (17,4; 26,0) p < 0,0001 | 12,7 (7,7; 17,7) p < 0,0001 | 11,8 ( 6,7; 17,0) p < 0,0001 | 14,7
p < 0,0001 | 13,2 (8,2; 18,2)
p < 0,0001 | 7,6
p=0,0070 | ||||||
A) Ero on Eylea 2 mg Q4 miinus kontrolli
B) Ero ja luottamusväli (CI) laskettiin käyttäen Cochran-Mantel-Haenszelin testiä (CMH) sopeutettuna tietylle alueelle (Amerikka vs. muu maailma COPERNICUS-tutkimuksessa ja Eurooppa vs. Aasia/Tyynenmeren alue GALILEO-tutkimuksessa) ja lähtötason BCVA-kategoriaa (> 20/200 ja ≤ 20/200)
C) BCVA: Paras korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study
LOCF: viimeinen havainto eteenpäin
SD: Standard deviation
LS: pienimpien neliösummien keskiarvo perustuen ANCOVA-malliin
D) Pienimpien neliösummien keskiarvojen ero ja luottamusväli perustuen ANCOVA-malliin, jossa tekijöinä hoitoryhmä, alue (Amerikka vs. muu maailma COPERNICUS- tutkimuksessa ja Eurooppa vs. Aasia/Tyynenmeren alue GALILEO-tutkimuksessa) ja lähtötason BCVA-kategoria (> 20/200 ja ≤ 20/200)
E) COPERNICUS-tutkimuksessa kontrolliryhmän potilaat saattoivat tarvittaessa saada Eylea-valmistetta 4 viikon välein viikoilla 24–52; potilailla oli käynnit 4 viikon välein
F) COPERNICUS-tutkimuksessa sekä kontrolliryhmä että Eylea 2 mg -potilaat saivat tarvittaessa 2 mg Eylea-valmistetta 4 viikon välein viikoilla 52–96; potilailla oli pakolliset käynnit neljännesvuosittain, mutta tarvittaessa käynti on saattanut olla jopa 4 viikon välein
G) GALILEO-tutkimuksessa sekä kontrolliryhmä että Eylea 2 mg –potilaat saivat tarvittaessa 2 mg Eylea-valmistetta 8 viikon välein viikoilla 52–68; potilailla oli pakolliset käynnit 8 viikon välein.
Kuva 2: Keskimääräinen näöntarkkuuden muutos lähtötasosta viikolle 76/100 hoitoryhmittäin COPERNICUS- ja GALILEO-tutkimuksissa (täysi analyysijoukko)
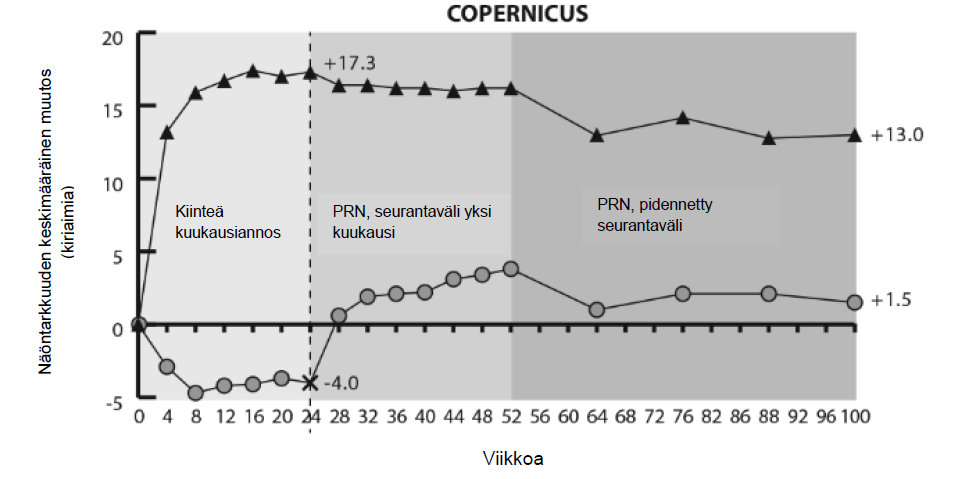
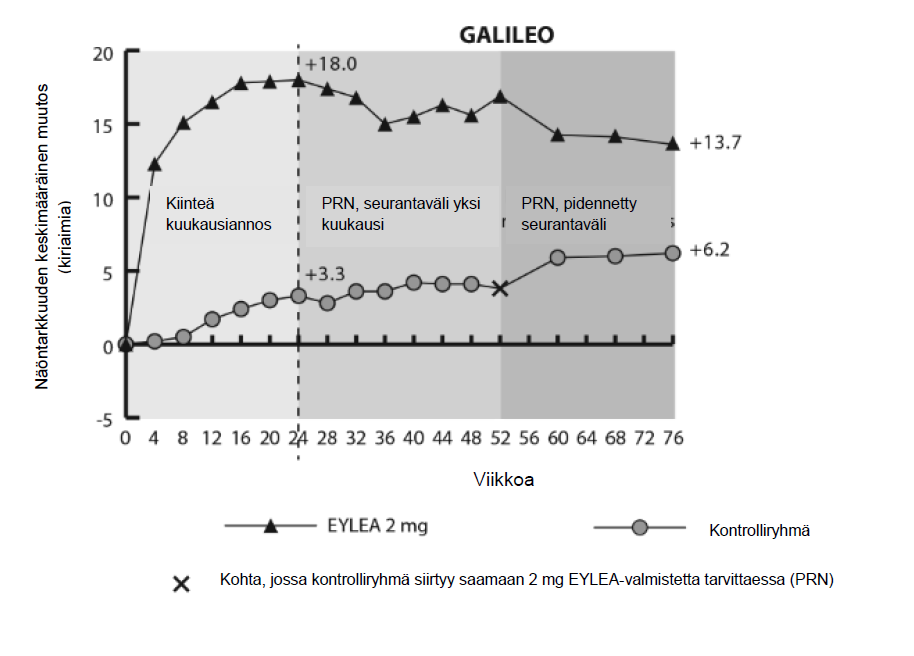
GALILEO-tutkimuksessa perfusoituneiden potilaiden osuus lähtötilanteessa oli 86,4 % (n = 89) Eylea- tutkimusryhmässä ja 79,4 % (n=54) kontrolliryhmässä. Viikolla 24 osuus oli 91,8 % (n = 89) Eylea- ryhmässä ja 85,5 % (n=47) kontrolliryhmässä. Nämä osuudet pysyivät samansuuruisina viikolle 76 asti; 84,3 % (n=75) Eylea-ryhmässä ja 84,0 % (n=42) kontrolliryhmässä.
COPERNICUS-tutkimuksessa perfusoituneiden potilaiden osuus lähtötilanteessa oli 67,5 % (n = 77) Eylea-ryhmässä ja 68,5 % (n = 50) vertailuryhmässä. Viikolla 24 osuus oli 87,4 % (n = 90) Eylea- ryhmässä ja 58,6 % (n = 34) vertailuryhmässä. Nämä osuudet pysyivät samansuuruisina viikolle 100 asti 76,8 % (n = 76) potilaista Eylea-ryhmässä ja 78 % (n = 39) vertailuryhmässä. Lumeryhmän potilaat hoidettiin viikosta 24 lähtien Eylea-valmisteella ennalta sovittujen kriteerien mukaisesti.
Eylea-hoidon hyödyllinen vaikutus näkökykyyn oli samanlainen lähtötilanteen perfusoituneiden ja ei- perfusoituneiden potilaiden alaryhmissä. Hoidon tehoa kuvaavat tulokset muissa arvioitavissa alaryhmissä (esim. ikä, sukupuoli, rotu, lähtötason näöntarkkuus, verkkokalvon keskuslaskimotukoksen kesto) vastasivat molemmissa tutkimuksissa normaalin väestön tuloksia.
GALILEO ja COPERNICUS -tutkimusten yhdistetyssä tulosanalyysissä, Eylea-valmisteella osoitettiin kliinisesti merkittäviä muutoksia lähtötasoon verrattuna ennalta sovittuun toissijaiseen päätetapahtumaan, National Eye Institute Visual Function Questionnaire (NEI VFQ-25) -kyselyn pisteisiin. Näiden muutosten suuruus oli samanlainen kuin julkaistuissa tutkimuksissa, vastaten 15 kirjaimen korjattua näöntarkkuuden paranemista (BCVA).
Verkkokalvon haaralaskimotukoksesta johtuva makulaarinen edeema
Eylea-valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa kaksoissokkoutetussa aktiivisesti kontrolloidussa monikeskustutkimuksessa potilailla, joilla oli verkkokalvon haaralaskimotukoksesta johtuva makulaarinen edeema (VIBRANT) mukaan lukien hemiretinaalinen laskimotukos. Yhteensä 181 potilasta hoidettiin ja oli arvioitavissa tehon suhteen. Potilaista 91 sai Eylea-valmistetta. Potilaiden iät vaihtelivat 42 ja 94 vuoden välillä keski-iän ollen 65 vuotta. BRVO-tutkimuksessa noin 58 % (53/91) randomisoiduista Eylea-hoitoa saaneista potilaista olivat iältään 65 vuotta tai vanhempia ja noin 23 % (21/91) 75 vuotta tai vanhempia. Tutkimuksessa potilaat satunnaistettiin suhteessa 1:1 seuraaviin ryhmiin: 2 mg Eylea-valmistetta 6 injektiota kuukauden välein ja sen jälkeen annosteltuna kahdeksan viikon välein tai laserhoitoon annettuna lähtötilanteessa (laserkontrolliryhmä).
Laserkontrolliryhmässä potilaiden oli mahdollista saada tarvittaessa lisälaserhoito (ns. rescue- laserhoito) viikolta 12 alkaen. Laserhoitojen välisen ajan tuli olla vähintään 12 viikkoa. Viikolta 24 alkaen potilaiden laserkontrolliryhmässä oli mahdollista saada lisähoitona (ns. rescue-hoito) Eylea- valmistetta 2 mg, tarvittaessa, neljän viikon välein 3 kuukauden ajan ja sen jälkeen 8 viikon välein, ennalta määrättyjen kriteerien mukaisesti.
VIBRANT-tutkimuksessa ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saivat vähintään 15 kirjainta viikolla 24 verrattuna lähtötasoon mitattuna BCVA (paras korjattu näöntarkkuus). Viikolla 24 Eylea-ryhmä oli parempi verrattuna laserkontrolliryhmään.
Muutos näöntarkkuudessa viikolla 24 verrattuna lähtötasoon oli toissijainen päätetapahtuma VIBRANT-tutkimuksessa. Ero hoitoryhmien välillä oli tilastollisesti merkitsevä Eylea-ryhmän eduksi. Näöntarkkuuden paraneminen tapahtui nopeasti ja huippu saavutettiin kolmen kuukauden kohdalla ja sen jälkeinen vaikutus säilyi 12 kuukauden kohtaan asti.
Laserkontrolliryhmässä 67 potilasta sai ns. rescue-hoitona Eylea-valmistetta viikolta 24 alkaen (Aktiivinen kontrolliryhmä/Eylea 2 mg ryhmä), minkä seurauksena näöntarkkuus parani noin 5 kirjainta viikkojen 24–52 aikana.
Yksityiskohtaiset tulokset VIBRANT-tutkimuksen analyysistä on esitetty seuraavassa taulukossa 4 ja kuvassa 3.
Taulukko 4: Tehoa kuvaavat tulokset viikolla 24 ja viikolla 52 (täysi analyysijoukko ja LOCF) VIBRANT-tutkimuksessa
| Tehoa kuvaavat tulokset | VIBRANT | |||
| Viikko 24 | Viikko 52 | |||
| Eylea 2mg Q4 (N = 91) | Active Control (laser) (N = 90) | Eylea 2mg Q8 (N = 91) D) | Active Control (laser)/Eylea 2mgE) (N = 90) | |
Niiden potilaiden osuus, joiden näöntarkkuus parani lähtötasosta vähintään 15 kirjainta BCVA:sta | 52,7% | 26,7% | 57,1% | 41,1% |
Painotettu ero A,B (%) (95% CI) p-arvo | 26,6% (13,0; 40,1) p=0,0003 | 16,2% (2,0; 30,5) p=0,0296 | ||
Keskimääräinen BCVA:n muutos mitattuna ETDRS- kirjainpisteinä lähtötasosta (SD) | 17,0 (11,9) | 6,9 (12,9) | 17,1 (13,1) | 12,2 (11,9) |
Ero keskimääräisenä muutoksena LS A,C (95% CI) p-arvo | 10,5 (7,1; 14,0) p<0,0001 | 5,2 (1,7; 8,7) p=0,0035F) | ||
A) Ero on Eylea 2 mg Q4 viikkoa -arvo miinus laserkontrolliarvo
B) Ero ja 95% CI laskettiin käyttäen Mantel-Haenszel painotuskaaviota sopeutettuna tietylle alueelle (Pohjois-Amerikka vs. Japani) ja lähtötason BCVA-kategoria (> 20/200 ja ≤ 20/200)
C) Ero keskimääräisenä muutoksena LS ja 95% CI perustuen ANCOVA-malliin, jossa hoitoryhmä, lähtötason BCVA-kategoria (> 20/200 and ≤ 20/200) ja alue (Pohjois-Amerikka vs. Japani) oli määritetty tekijöiksi ja lähtötason BCVA oli kovariaatti.
D) Viikolta 24 alkaen hoitoväliä pidennettiin kaikilla potilailla Eylea-hoitoryhmässä neljästä viikosta kahdeksaan viikkoon viikolle 48 asti.
E) Viikolta 24 alkaen tutkittavien laserkontrolliryhmässä oli mahdollista saada Eylea-hoitoa ns. rescue- hoitona, jos vähintään yksi ennalta määritelty hoitokriteeri täyttyi. Kaikkiaan 67 tutkittavaa tässä hoitoryhmässä sai Eylea-hoitoa. Hoito-ohjelmassa 2 mg Eylea-valmistetta annettiin neljän viikon välein kolme kertaa minkä jälkeen injektioväli oli 8 viikkoa.
F) Nominaalinen p-arvo
Kuva 3: Keskimääräinen BCVA:n muutos mitattuna ETDRS-kirjainpisteinä lähtötasosta viikolla 52 VIBRANT-tutkimuksessa.
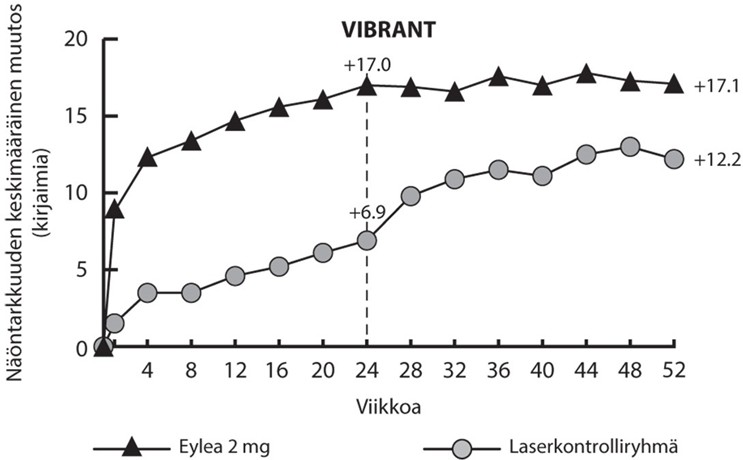
Lähtötilanteessa perfusoituneiden potilaiden osuus Eylea-ryhmässä oli 60 % ja laserkontrolliryhmässä 68 %. Viikolla 24 nämä osuudet olivat 80 % ja 67 %. Eylea-ryhmässä perfusoituneiden potilaiden osuus säilyi viikolle 52 asti. Laserkontrolliryhmässä, jossa potilaiden oli mahdollista saada ns. rescue- Eylea-hoitoa viikolta 24 alkaen, perfusoituneiden potilaiden osuus kasvoi 78 %:iin viikolle 52 mennessä.
Diabeettinen makulaturvotus
Eylea-valmisteen turvallisuutta ja tehoa arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, aktiivisesti kontrolloidussa monikeskustutkimuksessa potilailla, joilla oli diabeettinen makulaturvotus (VIVIDDME ja VISTADME). Yhteensä 862 potilasta hoidettiin ja voitiin arvioida tehon osalta, potilaista 576 satunnaistettiin Eylea-ryhmiin. Potilaiden iät vaihtelivat 23 ja 87 vuoden välillä keski-iän ollen 63 vuotta. DME-tutkimuksissa noin 47 % (268/576) randomisoiduista Eylea-hoitoa saaneista potilaista olivat iältään 65 vuotta tai vanhempia ja noin 9 % (52/576) 75 vuotta tai vanhempia. Suurimmalla osalla potilaista kummassakin tutkimuksessa oli tyypin 2 diabetes. Kummassakin tutkimuksessa potilaat satunnaistettiin yhteen kolmesta annosryhmästä suhteessa 1:1:1:
- Eylea-valmistetta annettiin 2 mg 8 viikon välein viiden kuukausittaisen alkuannoksen jälkeen (Eylea 2Q8);
- Eylea-valmistetta annettiin 2 mg 4 viikon välein (Eylea 2Q4) ja
- silmänpohjan laserfotokoagulaatio (aktiivinen kontrolli).
Viikolta 24 alkaen potilaat, joiden näkökyvyn heikkeneminen oli saavuttanut ennalta määritetyn arvon, olivat oikeutettuja saamaan lisähoitoa: Eylea-ryhmän potilaille voitiin antaa laserhoitoa ja kontrolliryhmän potilaille Eylea-valmistetta.
Molemmissa tutkimuksissa ensisijainen tehon päätetapahtuma oli parhaan korjatun näöntarkkuuden (BCVA) keskimääräinen muutos viikolla 52 verrattuna lähtötasoon. Sekä Eylea 2Q8 että Eylea 2Q4
-ryhmien tehon osoitettiin olevan tilastollisesti merkitsevästi parempi kuin kontrolliryhmässä. Tämä hyöty säilyi viikolle 100 asti.
Tarkemmat tulokset VIVIDDME- ja VISTADME-tutkimusten analyysistä on esitetty seuraavassa taulukossa 5 ja kuvassa 4.
Taulukko 5: Tehoa kuvaavat tulokset viikolla 52 ja viikolla 100 (täysi analyysijoukko ja LOCF) VIVIDDME- ja VISTADME-tutkimuksissa
| Tehoa kuvaava tulos | VIVIDDME | VISTADME | ||||||||||
| Viikko 52 | Viikko 100 | Viikko 52 | Viikko 100 | |||||||||
Eylea 2 mg Q8 A (N = 135) | Eylea 2 mg Q4 (N = 136) | Aktiivinen kontrolli (laser) (N = 132) | Eylea 2 mg Q8 A (N = 135) | Eylea 2 mg Q4 (N = 136) | Aktiivinen kontrolli (laser) (N = 132) | Eylea 2 mg Q8 A (N = 151) | Eylea 2 mg Q4 (N = 154) | Aktiivinen kontrolli (laser) (N = 154) | Eylea 2 mg Q8 A (N = 151) | Eylea 2 mg Q4 (N = 154) | Aktiivinen kontrolli (laser) (N = 154) | |
Keskimääräinen BCVA:n muutos mitattuna ETDRS- kirjainpisteinäE lähtötasosta | 10,7 | 10,5 | 1,2 | 9,4 | 11,4 | 0,7 | 10,7 | 12,5 | 0,2 | 11,1 | 11,5 | 0,9 |
Ero keskimääräi- senä muutoksena, LS B,C,E (97,5 % CI) | 9,1 (6,4, 11,8) | 9,3 (6,5, 12,0) | 8,2 (5,2; 11,3) | 10,7 (7,6; 13,8) | 10,45 (7,7, 13,2) | 12,19 (9,4, 15,0) | 10,1 (7,0; 13,3) | 10,6 (7,1; 14,2) | ||||
Niiden potilaiden osuus, joiden BCVA-arvoE parani lähtötasosta vähintään 15 kirjainta | 33 % | 32 % | 9 % | 31,1 % | 38,2 % | 12,1 % | 31 % | 42 % | 8 % | 33,1 % | 38,3 % | 13,0 % |
Sopeutettu ero D,C,E (97,5 % CI) | 24 % (13,5, 34,9) | 23 % (12,6, 33,9) | 19,0 % (8,0; 29,9) | 26,1 % (14,8; 37,5) | 23 % (13,5, 33,1) | 34 % (24,1, 44,4) | 20,1 % (9,6; 30,6) | 25,8 % (15,1; 36,6) | ||||
A Kun hoito aloitettiin viidellä kuukausittaisella injektiolla
B Keskimääräinen LS ja CI perustuen ANCOVA-malliin, jossa lähtötason BCVA-mittaus on kovariaatti ja tekijä hoitoryhmälle. Lisäksi alue (Eurooppa/Australia vs. Japani) oli lisätty tekijänä VIVIDDME-tutkimukselle ja MI:n ja/tai CVA:n historia tekijänä VISTADME-tutkimukselle.
C Ero on Eylea-ryhmän arvo miinus aktiivisen kontrolliryhmän (laser) arvo.
D Ero ja luottamusväli (CI) sekä tilastollinen testi laskettiin käyttäen Mantel-Haenszelin painotuskaaviota sopeutettuna tietylle alueelle (Eurooppa/Australia vs. Japani) VIVIDDME- tutkimuksessa ja MI:n tai CVA:n lääketieteellinen historia VISTADME-tutkimukselle
E BCVA: Paras korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study LOCF: viimeinen havainto eteenpäin
LS: pienimpien neliösummien keskiarvo perustuen ANCOVA-malliin
CI: luottamusväli
Kuva 4: Keskimääräinen BCVA:n muutos mitattuna ETDRS-kirjainpisteinä lähtötasosta viikolla 100 VIVIDDME- ja VISTADME-tutkimuksissa
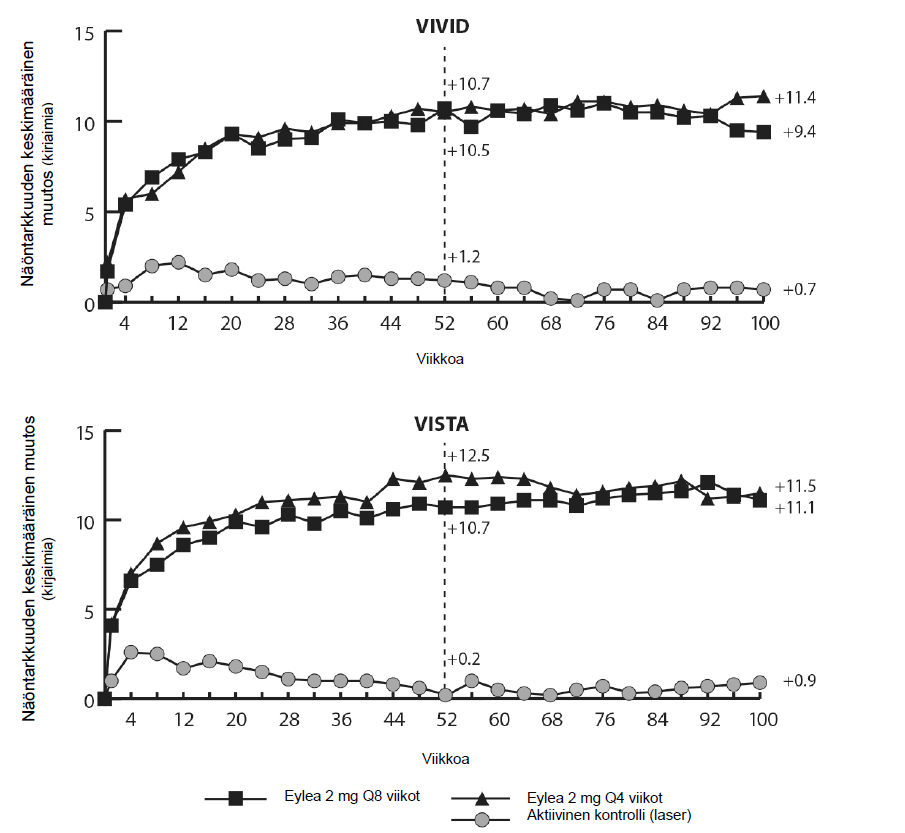
Hoitovasteet arvioitavissa alaryhmissä (esim. ikä, sukupuoli, rotu, lähtötason HbA1c, lähtötason näöntarkkuus, ennen anti-VEGF-hoitoa) molemmissa tutkimuksissa erikseen ja yhdistetyssä analyysissä vastasivat yleisesti ottaen muita saatuja tuloksia.
VIVIDDME- ja VISTADME-tutkimuksissa 36 (9 %) ja 197 (43 %) potilasta oli saanut anti-VEGF-hoitoa, jota oli seurannut 3 kuukauden pituinen tai pidempi lääkkeetön (washout) jakso. Hoitovasteet niiden potilaiden alaryhmissä, joita oli aiemmin hoidettu VEGF:n estäjillä, olivat samanlaiset kuin potilailla, jotka eivät olleet saaneet VEGF:n estäjiä ennen tutkimukseen osallistumista.
Potilailla, joiden molemmat silmät olivat sairastuneet, oli mahdollisuus saada anti-VEGF-hoitoa myös toiseen silmään, jos lääkäri piti sitä tarpeellisena. VISTADME-tutkimuksessa 217 (70,7 %) Eylea- potilasta sai molemminpuolisia Eylea-injektioita viikolle 100 asti; VIVIDDME-tutkimuksessa 97 (35,8 %) Eylea-potilasta sai toiseen silmään erilaista anti-VEGF-hoitoa.
Riippumaton vertaileva tutkimus (DRCR.net Protocol T) toteutti joustavaa hoito-ohjelmaa, joka perustui tiukkoihin OCT- ja uudelleenhoitokriteereihin. Afliberseptihoitoryhmässä (n=224) viikolla 52 hoito- ohjelman mukaisesti potilaat saivat keskimäärin 9,2 injektiota, vastaten Eylea-annosten lukumäärää VIVIDDME ja VISTADME -tutkimusten 2Q8-ryhmissä. Kokonaisteho afliberseptihoitoryhmässä Protocol T -tutkimuksessa oli verrattavissa Eylea 2Q8 -hoitoryhmään VIVIDDME ja VISTADME-tutkimuksissa. Protocol T -tutkimuksessa saavutettiin keskimäärin 13,3 kirjaimen parannus ja 42 % potilaista saavutti vähintään 15 kirjaimen parannuksen näöntarkkuudessa lähtötasoon verrattuna. Turvallisuustulokset osoittivat, että okulaaristen ja muiden haittatapahtumien (mukaan lukien aterotromboottiset tapahtumat) kokonaisilmaantuvuus oli vastaavalla tasolla kaikissa hoitoryhmissä kussakin tutkimuksessa ja tutkimusten välillä.
VIOLET oli 100 viikkoa kestänyt, satunnaistettu, avoin, aktiivisesti kontrolloitu monikeskustutkimus, jossa verrattiin Eylea 2 mg:n kolmea erilaista annosteluohjelmaa diabeettisen makulaturvotuksen hoidossa vähintään vuoden kiinteän annosvälin hoidon jälkeen (viiden kuukausittaisen annoksen jälkeen annostelua jatkettiin kahden kuukauden välein). Tutkimuksessa arvioitiin Eylea 2 mg:n annostelun non-inferioriteettiä annosteltuna treat-and-extend-ohjelman mukaisesti (2T&E-annosteluohjelmassa injektiovälit pidettiin vähintään 8 viikossa ja pidennettiin vähitellen kliinisten ja anatomisten hoitovasteiden perusteella) ja tarvittaessa annosteltuna (2PRN-annosteluohjelmassa potilaita tarkkailtiin 4 viikon välein ja injisoitiin tarvittaessa kliinisten ja anatomisten hoitovasteiden perusteella) verrattuna Eylea 2 mg:n annokseen 8 viikon välein (2Q8) toisen ja kolmannen hoitovuoden aikana.
Tehon ensisijainen päätetapahtuma (BCVA:n muutos lähtötasosta viikolle 52) oli 0,5 ± 6,7 kirjainta 2T&E-ryhmässä ja 1,7 ± 6,8 kirjainta 2PRN-ryhmässä verrattuna 0,4 ± 6,7 kirjaimeen 2Q8-ryhmässä, jolloin saavutettiin tilastollinen non-inferioriteetti (p < 0,0001 kummassakin vertailussa; vertailukelpoisuuden marginaali 4 kirjainta). BCVA:n muutokset lähtötasosta viikolle 100 olivat yhdenmukaisia viikon 52 tulosten kanssa: -0,1 ± 9,1 kirjainta 2T&E-ryhmässä ja 1,8 ± 9,0 kirjainta 2PRN-ryhmässä verrattuna 0,1 ± 7,2 kirjaimeen 2Q8-ryhmässä. Injektioiden keskimääräinen määrä 100 viikon aikana oli 12,3 2Q8fix-ryhmässä, 10,0 2T&E-ryhmässä ja 11,5 2PRN-ryhmässä.
Silmiin liittyvä ja systeeminen turvallisuusprofiili oli samankaltainen kuin VIVID- ja VISTA- avaintutkimuksissa.
2T&E-ryhmässä injektiovälien pidennykset ja lyhennykset tehtiin tutkijan harkinnan mukaan. Tutkimuksessa suositeltiin kahden viikon pidennyksiä.
Likitaitteisuuden aiheuttaman suonikalvon uudissuonittuminen
Eylea-valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa lumekontrolloidussa monikeskustutkimuksessa aiemmin hoitamattomilla aasialaisilla potilailla, jotka sairastivat likitaitteisuuden aiheuttamaa suonikalvon uudissuonittumista. Yhteensä 121 potilasta sai hoitoa ja hoidon teho arvioitiin (90 potilasta sai Eylea-valmistetta). Potilaiden iät vaihtelivat 27 ja 83 vuoden välillä keski-iän ollen 58 vuotta. Myooppisen CNV:n tutkimuksessa noin 36 % (33/91) randomisoiduista Eylea-hoitoa saaneista potilaista olivat iältään 65 vuotta tai vanhempia ja noin 10 % (9/91) 75 vuotta tai vanhempia.
Potilaat jaettiin satunnaisesti suhteessa 3:1 saamaan joko 2 mg Eylea-valmistetta lasiaisensisäisesti tai lumeinjektioita; injektio annettiin kerran tutkimuksen alussa, ja jos sairaus ei hävinnyt tai uusiutui, annettiin lisäinjektio kerran kuukaudessa viikolle 24 asti, jolloin ensisijainen päätetapahtuma arvioitiin. Viikolla 24 potilaat, jotka oli aluksi satunnaistettu saamaan lumevalmistetta, olivat kelpoisia saamaan ensimmäisen Eylea-annoksen. Tämän jälkeen molempien ryhmien potilaat olivat edelleen kelpoisia saamaan lisäinjektioita, jos sairaus ei hävinnyt tai uusiutui.
Ero hoitoryhmien välillä oli tilastollisesti merkitsevä Eylea-valmisteen eduksi ensisijaisen päätetapahtuman suhteen (BCVA-arvon muutos) ja tehon toissijaisen päätetapahtuman suhteen (niiden potilaiden osuus, jotka saivat 15 kirjainta BCVA-testissä) viikolla 24 verrattuna lähtötilanteeseen. Molempien päätetapahtumien erot säilyivät viikolle 48.
Yksityiskohtaiset tulokset MYRROR-tutkimuksen analyysista on esitetty seuraavassa taulukossa 6 ja kuvassa 5.
Taulukko 6: Tehoa kuvaavat tulokset viikolla 24 (ensisijainen analyysi) ja viikolla 48 MYRROR- tutkimuksessa (täysi analyysijoukko ja LOCFA))
| Tehoa kuvaavat tulokset | MYRROR | |||
| 24 viikkoa | 48 viikkoa | |||
| Eylea 2 mg (N = 90) | Lumelääke (N = 31) | Eylea 2 mg (N = 90) | Lumelääke/ Eylea 2 mg (N = 31) | |
| Keskimääräinen BCVA B):n muutos mitattuna ETDRS-kirjainpisteinä lähtötasosta (SD) B) | 12,1 (8,3) | -2,0 (9,7) | 13,5 (8,8) | 3,9 (14,3) |
Ero keskimääräisenä muutoksena, LS C,D,E) (95% CI) | 14,1 (10,8; 17,4) | 9,5 (5,4; 13,7) | ||
| Niiden potilaiden osuus, jotka saivat vähintään 15 kirjainta lähtötilanteesta | 38,9 % | 9,7 % | 50,0 % | 29,0 % |
| Painotettu ero D,F) (95 %:n CI) | 29,2 % (14,4; 44,0) | 21,0 % (1,9; 40,1) | ||
A) LOCF: viimeinen havainto eteenpäin
B) BCVA: Paras korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study
SD: keskihajonta (Standard Deviation)
C) LS-keskiarvo: Pienimpien summien keskiarvot perustuen ANCOVA-malliin
D) CI: Luottamusväli
E) Pienimpien neliösummien keskiarvojen ero ja 95 %:n luottamusväli perustuen ANCOVA-malliin, jossa tekijöinä hoitoryhmä ja maa (maiden määritelmät) ja lähtötason BCVA oli kovariaatti.
F) Ero ja 95 %:n CI lasketaan käyttäen Cochran-Mantel-Haenszel (CMH) -testiä maan mukaan sopeutettuna (maiden määritelmät)
Kuva 5: Keskimääräinen muutos näöntarkkuudessa lähtötilanteesta viikolle 48 hoitoryhmittäin MYRROR-tutkimuksessa (täysi analyysijoukko, LOCF)
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Eylea- valmisteen käytöstä kaikkien pediatristen potilasryhmien kostean silmänpohjan ikärappeuman, verkkokalvon keskuslaskimotukoksen, haaralaskimotukoksen, diabeettisen makulaturvotuksen ja likitaitteisuuden aiheuttaman suonikalvon uudissuonittumisen hoidossa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Eylea injektoidaan suoraan lasiaiseen, jotta se aiheuttaa paikallisia vaikutuksia silmässä. Imeytyminen/jakautuminen
Aflibersepti imeytyy hitaasti silmästä systeemiseen verenkiertoon lasiaiseen antamisen jälkeen, ja sitä havaitaan systeemisessä verenkierrossa lähinnä inaktiivisena, stabiilina kompleksina VEGF:n kanssa. Kuitenkin vain ”vapaa aflibersepti” voi sitoa endogeenistä VEGF:ää.
Kuuden neovaskulaarista kosteaa silmänpohjan ikärappeumaa sairastavan potilaan farmakokineettisessä alatutkimuksessa, jossa näytteitä otettiin usein, vapaan afliberseptin enimmäispitoisuudet plasmassa (systeeminen Cmax) olivat pieniä, keskiarvo noin 0,02 mikrogrammaa/ml (vaihteluväli 0–0,054) 1–3 päivän sisällä lasiaiseen annetusta 2 mg:n injektiosta. Plasmapitoisuudet eivät olleet mitattavissa kaksi viikkoa annoksen antamisen jälkeen lähes kaikilla potilailla. Aflibersepti ei kerry plasmaan, kun sitä annetaan lasiaiseen neljän viikon välein.
Vapaan afliberseptin keskimääräinen enimmäispitoisuus plasmassa on noin 50–500 kertaa pienempi kuin pitoisuus, jota tarvitaan estämään systeemisen VEGF:n biologinen aktiivisuus 50 %:lla eläintutkimuksissa, joissa havaittiin verenpaineen muutoksia, kun vapaan kiertävän afliberseptin saavutetut pitoisuudet olivat noin 10 mikrogrammaa/ml, ja palasivat lähtötasolle, kun pitoisuudet putosivat noin 1 mikrogrammaan/ml. Terveillä vapaaehtoisilla on arvioitu, että afliberseptin 2 mg:n intravitreaalisen annostelun jälkeen, vapaan afliberseptin keskimääräinen plasman enimmäispitoisuus oli yli 100 kertaa pienempi kuin pitoisuus, jota tarvitaan sitomaan systeemistä VEGF:ää (2,91 mikrogrammaa/ml) puolimaksimaalisesti. Siksi systeemiset farmakodynaamiset vaikutukset, kuten verenpaineen muutokset, ovat epätodennäköisiä.
Farmakokineettisissä alatutkimuksissa verkkokalvon keskuslaskimotukosta, haaralaskimotukosta, diabeettista makulaturvotusta tai likitaitteisuuden aiheuttamaa suonikalvon uudissuonittumista sairastavilla potilailla vapaan afliberseptin keskimääräinen Cmax plasmassa oli välillä 0,03–0,05 mikrogrammaa/ml yksittäisten arvojen ollessa alle 0,14 mikrogrammaa/ml. Vapaan afliberseptin plasmapitoisuudet pienenivät alle mitattavien arvojen tai lähelle ala-arvoa tavallisesti viikon kuluessa; kaikilla potilailla pitoisuudet laskivat alle havaitsemisrajan ennen seuraavaa hoitokertaa neljän viikon jälkeen.
Eliminaatio
Koska Eylea on proteiinipohjainen lääkeaine, metaboliatutkimuksia ei ole suoritettu.
Vapaa aflibersepti sitoutuu verisuonikasvutekijään ja muodostaa stabiilin, reagoimattoman kompleksin. Kuten muidenkin suurten proteiinien kohdalla, proteolyyttisen katabolian odotetaan eliminoivan sekä vapaan että sitoutuneen afliberseptin.
Munuaisten vajaatoiminta
Eylea-valmisteesta ei ole tehty erityisiä tutkimuksia potilaille, joilla on munuaisten vajaatoiminta.
VIEW2-tutkimuksen potilaiden, joista 40 %:lla oli munuaisten vajaatoiminta (24 %:lla lievä, 15 %:lla keskivaikea ja 1 %:lla vaikea), farmakokineettinen analyysi ei paljastanut eroja aktiivisen lääkkeen plasmapitoisuuksissa, kun valmistetta annettiin lasiaiseen 4 tai 8 viikon välein.
Samanlaisia tuloksia havaittiin verkkokalvon keskuslaskimotukosta sairastavilla potilailla GALILEO- tutkimuksessa, diabeettista makulaturvotusta sairastavilla potilailla VIVIDDME-tutkimuksessa ja likitaitteisuuden aiheuttamaa suonikalvon uudissuonittumista sairastavilla potilailla MYRROR- tutkimuksessa.
Prekliiniset tiedot turvallisuudesta
Ei-kliinisissä tutkimuksissa toistuvan annoksen aiheuttamaa toksisuutta on todettu systeemisissä altistuksissa vain silloin, kun on käytetty annosta, joka ylittää suurimman ihmisille käytettävän kliinisen annoksen lasiaiseen, viitaten vain vähäiseen kliiniseen merkitykseen.
Lasiaiseen annetulla afliberseptillä hoidettujen apinoiden nenän kuorikoissa havaittiin hengitysepiteelin eroosiota ja haavaumia systeemisillä altistuksilla, jotka olivat ihmisen enimmäisaltistusta suurempia. Systeeminen altistus, joka perustui vapaan afliberseptin Cmax- ja AUC- arvoihin, oli noin 200 ja 700 kertaa korkeampi verrattuna ihmisillä havaittuihin vastaaviin arvoihin lasiaiseen annetun 2 mg:n annoksen jälkeen. NOAEL-pitoisuudella (No Observed Adverse Effect Level) 0,5 mg / silmä apinoiden systeeminen altistus oli 42 kertaa suurempi Cmax-arvon perusteella ja 56 kertaa suurempi AUC-arvon perusteella.
Afliberseptin mutageenisistä tai karsinogeenisistä ominaisuuksista ei ole tehty tutkimuksia.
Afliberseptin osoitettiin vaikuttavan kohdunsisäiseen kehitykseen kaniinien alkio-sikiö- kehitystutkimuksessa, kun ainetta annettiin laskimoon (3–60 mg/kg) ja ihon alle (0,1–1 mg/kg). Emon NOAEL-tasot olivat 3 mg/kg laskimoon annettuna ja 1 mg/kg ihon alle annettuna. Kehitykseen vaikuttavaa NOAEL-tasoa ei määritetty. Annoksella 0,1 mg/kg systeeminen altistus vapaalle afliberseptille oli noin 17 kertaa korkeampi Cmax-arvon perusteella ja 10 kertaa korkeampi AUC-arvon perusteella kuin ihmisillä havaitut arvot lasiaiseen annetun 2 mg:n annoksen jälkeen.
Vaikutukset miesten ja naisten hedelmällisyyteen arvioitiin osana 6 kuukauden tutkimusta apinoilla, joille annettiin laskimoon afliberseptiannos 3–30 mg/kg. Puuttuvat tai epäsäännölliset kuukautiset liittyivät naisten lisääntymishormonipitoisuuksien muutoksiin, ja siemennesteen morfologian ja siittiöiden liikkuvuuden muutoksia havaittiin kaikilla annostasoilla. 3 mg:n/kg laskimoon annetun annoksen kohdalla vapaan afliberseptin systeeminen altistus oli noin 4 900 kertaa suurempi Cmax-arvon perusteella ja 1 500 kertaa suurempi AUC-arvon perusteella kuin ihmisillä havaittu altistus lasiaiseen annetun 2 mg:n annoksen jälkeen. Kaikki muutokset olivat palautuvia.
Farmaseuttiset tiedot
Apuaineet
Polysorbaatti 20 (E 432)
Natriumdivetyfosfaatti, monohydraatti (pH:n säätöön)
Dinatriumvetyfosfaatti, heptahydraatti (pH:n säätöön)
Natriumkloridi
Sakkaroosi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
2 vuotta
Säilytys
Säilytä jääkaapissa (2°C - 8°C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Avaamaton injektiopullo voidaan säilyttää poissa jääkaapista alle 25 °C:ssa enintään 24 tunnin ajan. Injektiopullon avaamisen jälkeen on jatkettava aseptisissa olosuhteissa.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
EYLEA injektioneste, liuos
40 mg/ml (L:ei) 1 kpl (0,1 ml) (1090,58 €)
PF-selosteen tieto
Liuos injektiopullossa (tyypin I lasia), jossa on tulppa (elastomeerikumia) ja 18 G:n suodatinneula. Yhden injektiopullon sisältämä kokonaismäärä on vähintään 0,1 ml. Pakkauskoko: 1 injektiopullo + 1 suodatinneula
Valmisteen kuvaus:
Liuos on kirkas, väritön tai vaaleankeltainen iso-osmoottinen liuos.
Käyttö- ja käsittelyohjeet
Injektiopullo on kertakäyttöinen ja yhden silmän hoitoon. Injektiopullo sisältää enemmän kuin suositellun 2 mg:n afliberseptiannoksen (vastaa 0,05 ml:aa injektionestettä). Ylimäärä on poistettava ennen antoa.
Ennen lääkkeen antamista, liuos on tarkastettava silmämääräisesti mahdollisten hiukkasten ja/tai värimuutosten ja/tai minkä tahansa valmisteen fysikaalisen ulkonäön muutoksen havaitsemiseksi. Mikäli tällaista havaitaan, tulee lääkevalmiste hävittää.
Suodatinneula:
Blunt Fill -suodatinneula, ei pistämiseen ihon läpi.
Blunt Fill -suodatinneulaa ei saa autoklavoida.
Suodatinneula on pyrogeeniton. Älä käytä sitä, jos sen pakkaus on vahingoittunut.
Hävitä Blunt Fill -suodatinneula terävälle lääkejätteelle tarkoitettuun keräysastiaan.
Varoitus: Suodatinneulan uudeelleenkäyttäminen voi aiheuttaa infektion tai muun sairauden/vamman.
Lasiaiseen annettavaan injektioon on käytettävä 30 G x ½ tuuman kokoista injektioneulaa.
Injektiopullon käyttöohjeet:
| 1. | Poista muovikorkki ja desinfioi injektiopullon kumitulpan ulkopuoli. |  |
| 2. | Liitä pakkauksessa tuleva 18 G:n, 5 mikronin suodatinneula 1 ml:n steriiliin Luer-lock-ruiskuun. |  |
| 3. | Työnnä suodatinneulaa injektiopullon tulpan keskiosaan, kunnes neula on kokonaan työntynyt injektiopulloon ja kärki koskee injektiopullon pohjaan tai pohjan reunaan. | |
| 4. | Käytä aseptista tekniikkaa ja vedä Eylea-injektiopullon koko sisältö ruiskuun samalla, kun pidät injektiopulloa pystysuorassa ja hieman kallistetussa asennossa. Tämä asento helpottaa koko sisällön vetämistä ruiskuun. Estääksesi ilman pääsyn ruiskuun, varmista että suodatinneulan kärki on nesteen pinnan alla. Pidä pullo kallellaan, kun vedät nestettä ruiskuun, niin että suodatinneulan kärki on nesteen pinnan alapuolella. | |
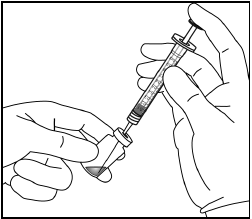 | 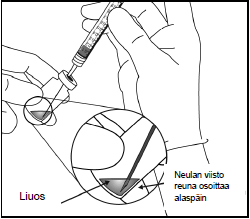 | |
| 5. | Varmista, että mäntä on vedetty tarpeeksi taakse, kun tyhjennät injektiopulloa. Näin suodatinneula tyhjenee varmasti kokonaan. | |
| 6. | Poista suodatinneula ja hävitä se asianmukaisesti. Huomautus: suodatinneulaa ei saa käyttää lasiaisinjektioon. | |
| 7. | Käytä aseptista tekniikkaa ja kiinnitä 30 G:n x ½ tuuman injektioneula tiukasti Luer-lock-ruiskun kärkeen. | 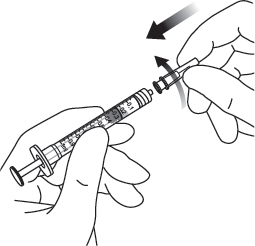 |
| 8. | Pidä ruiskua niin, että neula osoittaa ylöspäin, ja tarkista ruisku kuplien varalta. Jos ruiskussa näkyy kuplia, naputa ruiskua varovasti sormella, kunnes kuplat nousevat pinnalle. | 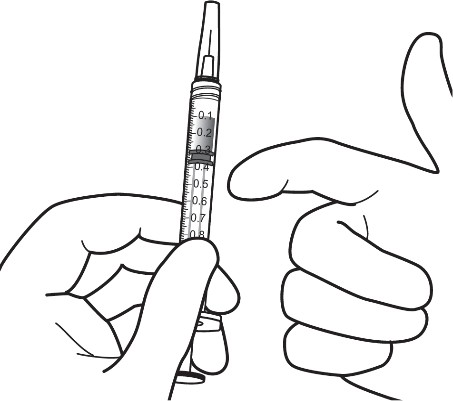 |
| 9. | Poista kaikki kuplat ja ylimääräinen lääkevalmiste painamalla mäntää hitaasti niin, että männän kärki kohdistuu ruiskussa olevaan 0,05 ml:n annosviivaan. | |
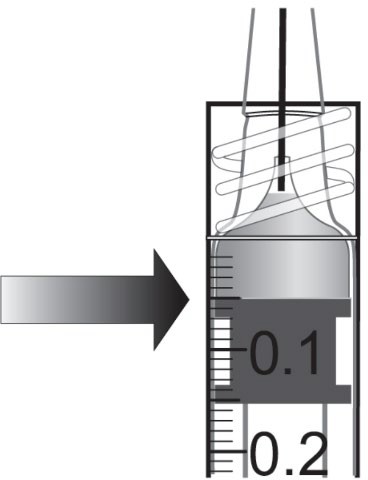 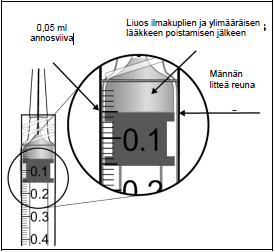 | ||
| 10. | Injektiopullo on tarkoitettu vain kertakäyttöön. Useiden annosten ottaminen yhdestä injektiopullosta voi lisätä kontaminaation riskiä ja sen seurauksena infektion riskiä. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti. | |
Korvattavuus
EYLEA injektioneste, liuos
40 mg/ml 1 kpl
- Ei korvausta.
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
S01LA05
Valmisteyhteenvedon muuttamispäivämäärä
25.06.2025
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi