
XARELTO tabletti, kalvopäällysteinen 2,5 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Terveydenhuollon ammattilainen
Lääkkeen määrääjän opas antaa ohjeita XARELTO-valmisteen käyttöön vuotoriskin pienentämiseksi XARELTO-hoidon aikana.
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 2,5 mg rivaroksabaania.
Apuaine, jonka vaikutus tunnetaan: Yksi kalvopäällysteinen tabletti sisältää 33,92 mg laktoosia (monohydraattina), ks. kohta Varoitukset ja käyttöön liittyvät varotoimet
Täydellinen apuaineluettelo, ks. kohta Apuaineet
Lääkemuoto
Tabletti, kalvopäällysteinen
Kliiniset tiedot
Käyttöaiheet
Samanaikaisesti pelkän asetyylisalisyylihapon (ASA) tai asetyylisalisyylihapon ja joko klopidogreelin tai tiklopidiinin yhdistelmän kanssa aterotromboottisten tapahtumien ehkäisyyn aikuisille potilaille akuutin sepelvaltimotautikohtauksen jälkeen sydämen biomarkkerien ollessa koholla (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet sekä Farmakodynamiikka).
Samanaikaisesti asetyylisalisyylihapon (ASA) kanssa aterotromboottisten tapahtumien ehkäisyyn sepelvaltimotautia tai oireista ääreisvaltimotautia sairastaville aikuisille potilaille, joilla on suuri iskeemisen tapahtuman riski.
Annostus ja antotapa
Annostus
Suositeltu annos on 2,5 mg kaksi kertaa päivässä.
- Akuutti sepelvaltimotautikohtaus
Xarelto-valmistetta 2,5 mg kaksi kertaa päivässä otettavien potilaiden tulee lisäksi ottaa päivittäin 75–100 mg:n annos asetyylisalisyylihappoa tai 75–100 mg:n annos asetyylisalisyylihappoa yhdistettynä 75 mg:n annokseen klopidogreelia tai normaaliin päiväannokseen tiklopidiinia.
Hoidon aikana tilannetta tulee seurata säännöllisesti ja arvioida iskeemisten tapahtumien riskiä suhteessa verenvuotoriskiin. Päätös hoidon jatkamisesta 12 kuukautta pidempään tulee tehdä tapauskohtaisesti, sillä enintään 24 kuukautta kestäneestä hoidosta on rajallisesti kokemuksia (ks. kohta Farmakodynamiikka).
Xarelto-hoito tulee aloittaa mahdollisimman pian akuutin sepelvaltimotautikohtauksen stabiloinnin jälkeen (mukaan lukien revaskularisaatiotoimenpiteet), kuitenkin aikaisintaan 24 tuntia sairaalaan tulon jälkeen hetkellä, jolloin parenteraalinen antikoagulaatiohoito tavallisesti lopetettaisiin.
- Sepel-/ääreisvaltimotauti
Xarelto-valmistetta 2,5 mg kaksi kertaa päivässä ottavien potilaiden tulee lisäksi ottaa päivittäin 75–100 mg:n annos asetyylisalisyylihappoa.
Oireisen ääreisvaltimotaudin takia onnistuneesti tehdyn alaraajan revaskularisaatiotoimenpiteen (kirurgisen tai endovaskulaarisen, hybriditoimenpiteet mukaan lukien) jälkeen hoidon saa aloittaa vasta, kun hemostaasi on saavutettu (ks. kohta Farmkodynamiikka).
Hoidon kesto on määritettävä yksilöllisesti säännöllisen potilaan arvioinnin perusteella, jossa on huomioitava tromboottisten tapahtumien riski suhteessa verenvuotoriskiin.
- Akuutti sepelvaltimokohtaus, sepel-/ääreisvaltimotauti
Samanaikainen antitromboottinen hoito
Jos potilas tarvitsee akuutin tromboottisen tapahtuman tai verisuonitoimenpiteen yhteydessä kaksinkertaista antitromboottista hoitoa, Xarelto-valmisteen 2,5 mg kahdesti päivässä otettavan hoidon jatkamista on arvioitava uudelleen tromboottisen tapahtuman tai verisuonitoimenpiteen sekä antitromboottisen hoito-ohjelman mukaan.
Xarelto-valmisteen turvallisuutta ja tehoa 2,5 mg kaksi kertaa päivässä otettuna yhdessä kaksinkertaisen antitromboottisen hoidon kanssa, on tutkittu
- potilailla, joilla on äskettäin ollut akuutti sepelvaltimotautikohtaus, yhdessä asetyylisalisyylihapon ja klopidogreelin/tiklopidiinin kanssa (ks. kohta Käyttöaiheet), sekä
- potilailla, joille on äskettäin tehty alaraajan revaskularisaatiotoimenpide oireisen ääreisvaltimotaudin takia, yhdessä asetyylisalisyylihapon ja tarvittaessa lyhytkestoisen klopidogreelihoidon kanssa (ks. kohdat Varoitukset ja käyttöön liityvät varotoimet ja Farmakodynamiikka).
Annoksen unohtaminen
Jos yksi annos jää ottamatta, potilaan tulee ottaa seuraava normaali annos normaalin aikataulun mukaan. Annosta ei tule kaksinkertaistaa ottamatta jääneen annoksen korvaamiseksi.
Siirtyminen K-vitamiinien antagonisteista (VKA) Xarelto-valmisteeseen
Kun potilaat siirtyvät VKA-hoidosta Xarelto-hoitoon, INR-arvot (International Normalized Ratio) voivat kohota virheellisesti Xarelto-valmisteen ottamisen jälkeen. INR-arvoa ei tule käyttää, koska se ei ole luotettava Xarelto-valmisteen antikoagulatiivisen vaikutuksen mittaamiseen (ks. kohta Yhteisvaikutukset).
Siirtyminen Xarelto-valmisteesta K-vitamiinien antagonisteihin (VKA)
Antikoagulaatio saattaa olla riittämätöntä siirtymäjakson aikana, kun siirrytään Xarelto-hoidosta VKA-hoitoon. Jatkuva ja riittävä antikoagulaatio on varmistettava aina siirryttäessä toiseen antikoagulanttiin. On huomattava, että Xarelto saattaa suurentaa INR-arvoa.
Xarelto-hoidosta VKA-hoitoon siirtyville potilaille tulee antaa samanaikaisesti VKA-hoitoa, kunnes INR on ≥ 2,0. Siirtymäjakson kahtena ensimmäisenä päivänä tulee käyttää VKA:n tavanomaista aloitusannosta ja sen jälkeen INR-testien mukaista VKA-annosta. Potilaiden saadessa samanaikaisesti sekä Xarelto-hoitoa että VKA-hoitoa, INR-arvo tulee testata aikaisintaan 24 tunnin kuluttua edellisestä annoksesta, mutta ennen seuraavaa Xarelto-annosta. Jos Xarelto-hoito keskeytetään, INR-testi voidaan tehdä luotettavasti aikaisintaan 24 tunnin kuluttua viimeisestä annoksesta (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Siirtyminen parenteraalisista antikoagulanteista Xarelto-valmisteeseen
Jos potilas saa parenteraalista antikoagulanttia, sen käyttö tulee lopettaa ja Xarelto-hoito aloittaa 0–2 tuntia ennen lopetettavan parenteraalisen lääkevalmisteen (esim. pienimolekyyliset hepariinit) seuraavaa annosteluajankohtaa tai jatkuvasti annetun parenteraalisen lääkevalmisteen (esim. laskimoon annettu fraktioimaton hepariini) keskeyttämisajankohtana.
Siirtyminen Xarelto-valmisteesta parenteraalisiin antikoagulantteihin
Parenteraalisen antikoagulantin ensimmäinen annos annetaan, kun seuraava Xarelto-annos otettaisiin.
Erityisryhmät
Munuaisten vajaatoiminta
Tähän mennessä saadut kliiniset tiedot vakavaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma 15–29 ml/min) sairastavista potilaista osoittavat, että plasman rivaroksabaanipitoisuus on merkittävästi lisääntynyt. Tämän vuoksi Xarelto-valmistetta tulee käyttää harkiten näillä potilailla. Käyttöä ei suositella potilaille, joiden kreatiniinipuhdistuma on < 15 ml/min (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet sekä Farmakokinetiikka).
Annoksen sovittaminen ei ole tarpeen lievää munuaisten vajaatoimintaa (kreatiniinipuhdistuma 50–80 ml/min) tai kohtalaista munuaisten vajaatoimintaa (kreatiniinipuhdistuma 30–49 ml/min) sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Xarelto on vasta-aiheinen potilailla, joiden maksasairauteen liittyy hyytymishäiriö ja kliinisesti merkittävä verenvuotoriski mukaan lukien Child Pugh -luokkien B ja C kirroosipotilaat (ks. kohdat Vasta-aiheet ja Farmakokinetiikka).
Iäkkäät potilaat
Annoksen muuttaminen ei ole tarpeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet sekä Farmakokinetiikka).
Verenvuodon riski kasvaa iän myötä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Paino
Annoksen muuttaminen ei ole tarpeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet sekä Farmakokinetiikka).
Sukupuoli
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Xarelto-valmisteen 2,5 mg tablettien turvallisuutta ja tehoa alle 18-vuotiaiden lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla. Tämän vuoksi Xarelto-valmisteen 2,5 mg tablettien käyttöä alle 18-vuotiaille lapsille ei suositella.
Antotapa
Xarelto on tarkoitetttu otettavaksi suun kautta.
Tabletit voidaan ottaa ruuan kanssa tai ilman (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Tablettien murskaaminen
Sellaisille potilaille, jotka eivät pysty nielemään kokonaisia tabletteja, Xarelto-tabletti voidaan murskata ja sekoittaa veteen tai omenasoseeseen juuri ennen sen antamista suun kautta.
Murskattu tabletti voidaan myös antaa mahaletkun kautta. (ks. kohdat Farmakokinetiikka ja Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiivinen kliinisesti merkitsevä verenvuoto.
Leesio tai sairaus, jos sen katsotaan olevan merkittävän verenvuodon riski. Näitä voivat olla nykyinen tai äskettäinen maha-suolikanavan haavauma; pahanlaatuiset kasvaimet, joiden vuotoriski on suuri; äskettäinen aivo- tai selkäydinvaurio; äskettäinen aivo-, selkäydin- tai silmäleikkaus; äskettäinen kallonsisäinen verenvuoto; todetut tai epäillyt ruokatorven laskimonlaajentumat; valtimo-laskimoepämuodostumat; valtimonpullistumat tai merkittävät selkärangan- tai aivojensisäiset verisuonipoikkeavuudet.
Samanaikaisesti käytetty muu antikoagulantti, esim. fraktioimaton hepariini, pienimolekyyliset hepariinit (enoksapariini, daltepariini jne.), hepariinijohdokset (fondaparinuuksi jne.), oraaliset antikoagulantit (varfariini, dabigatraanieteksilaatti, apiksabaani jne.), paitsi kun antikoagulanttihoitoa vaihdetaan tietyissä tilanteissa (ks. kohta Annostus ja antotapa) tai kun fraktioimatonta hepariinia annetaan annoksena, jonka tarkoituksena on pitää keskuslaskimo- tai -valtimokatetri avoimena (ks. kohta Yhteisvaikutukset).
Akuutin sepelvaltimotautikohtauksen samanaikainen antitromboottinen hoito potilailla, joilla on ollut aiemmin aivohalvaus tai ohimenevä aivoverenkiertohäiriö (TIA) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sepel-/ääreisvaltimotaudin samanaikainen hoito asetyylisalisyylihapolla potilailla, joilla on ollut aiemmin verenvuodosta aiheutuva tai lakunaarinen aivohalvaus tai mikä tahansa aivohalvaus viimeisen kuukauden aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksasairaus, johon liittyy hyytymishäiriö ja kliinisesti merkittävä verenvuotoriski mukaan lukien Child Pugh -luokkien B ja C kirroosipotilaat (ks. kohta Farmakokinetiikka).
Raskaus ja imetys (ks. kohta Fertiliteetti, raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Kaksi kertaa päivässä otettavan Xarelto 2,5 mg -valmisteen tehoa ja turvallisuutta on tutkittu akuutin sepelvaltimotautikohtauksen hoidossa, jolloin Xarelto-valmistetta on annettu yhdessä antitromboottisista lääkeaineista joko pelkän asetyylisalisyylihapon kanssa tai jaasetyylisalisyylihapon ja klopidogreelin//tiklopidiinin yhdistelmän kanssa.
Sepel-/ääreisvaltimotautia sairastavilla potilailla, joilla on korkea iskeemisten tapahtumien riski, kaksi kertaa päivässä otettavan Xarelto 2,5 mg -valmisteen tehoa ja turvallisuutta on tutkittu yhdessä asetyylisalisyylihapon kanssa.
Potilailla, joille on äskettäin tehty alaraajan revaskularisaatiotoimenpide oireisen ääreisvaltimotaudin takia, kaksi kertaa päivässä otettavan Xarelto 2,5 mg -valmisteen tehoa ja turvallisuutta on tutkittu yhdessä pelkän antitromboottisen asetyylisalisyylihapon kanssa tai asetyylisalisyylihapon ja lyhytkestoisen klopidogreelihoidon kanssa. Jos kaksinkertainen antitromboottinen hoito klopidogreelilla on tarpeen, sen tulee olla lyhytkestoista. Pitkäkestoista kaksinkertaista antitromboottista hoitoa tulee välttää (ks. kohta Farmakodynamiikka).
Samanaikaista käyttöä muiden antitromboottisten lääkeaineiden, kuten prasugreelin tai tikagrelorin, kanssa ei ole tutkittu, eikä sitä suositella.
Hoitojakson ajan suositellaan antikoagulaatiohoitokäytäntöjen mukaista kliinistä seurantaa.
Verenvuotoriski
Kuten muitakin antikoagulantteja käytettäessä, myös Xarelto-valmistetta käytettäessä potilaita on seurattava verenvuodon mahdollisten merkkien havaitsemiseksi. Xarelto-valmistetta suositellaan käytettäväksi varoen tilanteissa, joissa verenvuotoriski on tavallista suurempi. Jos potilaalla ilmenee vakava verenvuoto, valmisteen käyttö tulee lopettaa.
Kliinisissä tutkimuksissa limakalvoverenvuotoja (nenä, ien, gastrointestinaalikanava ja virtsateiden alue, mukaan lukien epänormaali emätinverenvuoto tai lisääntynyt kuukautisvuoto) ja anemiaa havaittiin useammin yksin- tai kaksinkertaisen antitromboottisen lääkityksen lisäksi annettavan pitkäkestoisen rivaroksabaanihoidon aikana. Riittävän kliinisen seurannan lisäksi voidaan harkita hemoglobiinin tai hematokriitin määrittämistä piilevän verenvuodon havaitsemiseksi ja näkyvän verenvuodon kliinisen merkityksen selvittämiseksi.
Useissa potilaiden alaryhmissä on tavallista suurempi verenvuotoriski seuraavassa esitetyn mukaisesti. Tämän vuoksi arvioitaessa Xarelto-valmisteen käyttöä samanaikaisesti kaksinkertaisen antitromboottisen hoidon kanssa potilaille, joilla tiedetään olevan kohonnut verenvuotoriski, on huomioitava aterotromboottisten tapahtumien ehkäisy. Lisäksi näitä potilaita tulee hoidon aloittamisen jälkeen tarkkailla huolellisesti verenvuotokomplikaatioiden ja anemian merkkien ja oireiden varalta (ks. kohta Haittavaikutukset).
Mikäli hemoglobiini tai verenpaine laskee tuntemattomasta syystä, mahdollinen vuotokohta on selvitettävä.
Vaikka rivaroksabaanihoidon yhteydessä ei tarvita rutiininomaista seurantaa, rivaroksabaanipitoisuuksien mittaamisesta kalibroidulla antifaktori Xa-aktiivisuustestillä saattaa olla hyötyä erikoistilanteissa, joissa tieto rivaroksabaanin antikoagulaatiovaikutuksesta voi auttaa tekemään kliinisiä hoitopäätöksiä esimerkiksi yliannostuksen tai hätäleikkauksen yhteydessä (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Vakavaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastavilla potilailla plasman rivaroksabaanipitoisuus saattaa suurentua merkitsevästi (keskimäärin 1,6-kertaiseksi) ja johtaa verenvuotoriskin lisääntymiseen. Xarelto-valmistetta tulee käyttää harkiten potilailla, joiden kreatiniinipuhdistuma on 15–29 ml/min. Käyttöä ei suositella potilaille, joiden kreatiniinipuhdistuma on < 15 ml/min (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Xarelto-valmistetta tulee käyttää harkiten kohtalaista munuaisten vajaatoimintaa (kreatiniinipuhdistuma 30–49 ml/min) sairastavilla potilailla, jotka saavat samanaikaisesti muita lääkevalmisteita, jotka lisäävät rivaroksabaanin pitoisuutta plasmassa (ks. kohta Yhteisvaikutukset).
Yhteisvaikutus muiden lääkevalmisteiden kanssa
Xarelto-valmisteen käyttöä ei suositella potilaille, jotka saavat samanaikaista systeemistä hoitoa atsoliryhmän sienilääkkeillä (kuten ketokonatsoli, itrakonatsoli, vorikonatsoli ja posakonatsoli) tai HIV-proteaasin estäjillä (esim. ritonaviiri). Nämä vaikuttavat aineet ovat voimakkaita CYP3A4- ja P-gp-estäjiä, minkä vuoksi ne saattavat lisätä plasman rivaroksabaanipitoisuutta kliinisesti merkittävästi (keskimäärin 2,6-kertaiseksi), mikä voi johtaa suurempaan verenvuotoriskiin (ks. kohta Yhteisvaikutukset).
Erityistä varovaisuutta tulee noudattaa, jos potilaat saavat samanaikaista hoitoa hemostaasiin vaikuttavilla lääkkeillä, kuten steroideihin kuulumattomilla tulehduskipulääkkeillä (NSAID), asetyylisalisyylihapolla tai trombosyyttiaggregaation estäjillä tai selektiivisillä serotoniinin takaisinoton estäjillä (SSRI-lääkkeet) tai serotoniinin ja noradrenaliinin takaisinoton estäjillä (SNRI-lääkkeet). Potilaille, joilla on haavaisen gastrointestinaalisairauden vaara, voidaan harkita asianmukaista ennaltaehkäisevää hoitoa (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka).
Potilaiden, jotka saavat Xarelto-valmistetta ja antitromboottisia lääkkeitä, tulee saada samanaikaisesti steroideihin kuulumattomia tulehduskipulääkkeitä vain siinä tapauksessa, että edut ovat verenvuotoriskiä suuremmat.
Muut verenvuodon riskitekijät
Muiden antitromboottisten lääkeaineiden tavoin rivaroksabaania ei suositella potilaille, joilla on lisääntynyt verenvuotoriski, kuten
- synnynnäisiä tai hankinnaisia verenvuotohäiriöitä
- vakava valtimoperäinen hypertensio, joka ei ole hoitotasapainossa
- muu ruoansulatuskanavan sairaus (ilman aktiivista haavaumaa), johon voi liittyä verenvuotoja (esim. tulehduksellinen suolistosairaus, esofagiitti, gastriitti ja ruokatorven refluksitauti)
- vaskulaarinen retinopatia
- keuhkoputkien laajentuma tai aiempi keuhkoverenvuoto.
Sitä tulee käyttää varoen akuuttia sepelvaltimotautia ja sepel-/ääreisvaltimotautia sairastavilla potilailla,
- jotka ovat ≥ 75-vuotiaita ja saavat samanaikaisesti pelkkää asetyylisalisyylihappoa tai asetyylisalisyylihappoa yhdessä joko klopidogreelin tai tiklopidiinin kanssa
- joiden paino on alhainen (< 60 kg) ja jotka saavat samanaikaisesti pelkkää asetyylisalisyylihappoa tai asetyylisalisyylihappoa yhdessä joko klopidogreelin tai tiklopidiinin kanssa. Hoidon yksilöllinen hyöty-riskiarvio on tehtävä säännöllisin väliajoin
- Tutkimustiedot osoittavat, että sepelvaltimotautipotilaat, joilla on vaikea-asteinen, oireinen sydämen vajaatoiminta saattavat hyötyä rivaroksabaanihoidosta muita vähemmän (ks. kohta Farmakodynamiikka).
Syöpäpotilaat
Potilailla, joilla on pahanlaatuinen sairaus voi samanaikaisesti olla suurempi verenvuotojen ja verisuonitukosten riski. Aktiivista syöpää sairastavien potilaiden antitromboottisen hoidon hyötyä ja verenvuotoriskiä on punnittava yksilöllisesti riippuen kasvaimen sijainnista, antineoplastisesta hoidosta ja sairauden vaiheesta. Maha-suolikanavassa tai virtsa- ja sukupuoliteissä sijaitseviin tuumoreihin on liittynyt lisääntynyt verenvuotoriski rivaroksabaanihoidon aikana.
Rivaroksabaanin käyttö on vasta-aiheinen potilailla, joilla on pahanlaatuisia kasvaimia, joiden vuotoriski on suuri (ks. kohta Vasta-aiheet).
Potilaat, joilla on sydämen tekoläppä
Rivaroksabaania ei pidä antaa tromboosin estolääkityksenä potilaille, joille on äskettäin asennettu katetrin avulla aorttaläppäproteesi (transcatheter aortic valve implantation, TAVI). Xarelto-valmisteen tehoa ja turvallisuutta ei ole tutkittu potilailla, joilla on sydämen tekoläppä. Tämän vuoksi ei ole tietoa siitä, että Xarelto takaisi riittävän antikoagulaation tässä potilasryhmässä. Xarelto-hoitoa ei suositella näille potilaille.
Fosfolipidivasta-aineoireyhtymää sairastavat potilaat
Suun kautta otettavia suoravaikutteisia antikoagulantteja, jotka sisältävät rivaroksabaania, ei suositella potilaille, joilla on ollut verisuonitukos ja joilla on diagnosoitu fosfolipidivasta-aineoireyhtymä. Erityisesti potilailla, joilla on positiivinen tulos kaikissa kolmessa testissä (lupusantikoagulantti, kardiolipiinivasta-aineet ja beeta-2-glykoproteiini I vasta-aineet), hoito suun kautta otettavilla suoravaikutteisilla antikoagulanteilla saattaa aiheuttaa uusiutuvia verisuonitukoksia useammin kuin K-vitamiinin antagonistihoito.
Potilaat, joilla on ollut aiemmin aivohalvaus tai ohimenevä aivoverenkiertohäiriö
Akuutin sepelvaltimotautikohtauksen saaneet potilaat
Xarelto 2,5 mg on vasta-aiheinen akuutin sepelvaltimotaudin hoitoon potilailla, joilla on ollut aiemmin aivohalvaus tai ohimenevä aivoverenkiertohäiriö (ks. kohta Vasta-aiheet). Vain harvoja akuuttia sepelvaltimotautia sairastavia potilaita, joilla on ollut aiemmin aivohalvaus tai ohimenevä aivoverenkiertohäiriö, on tutkittu, mutta saatavilla olevista rajallisista tehokkuustiedoista ilmenee, että nämä potilaat eivät hyödy hoidosta.
Potilaat, joilla on sepel-/ääreisvaltimotauti
Sepel-/ääreisvaltimotautia sairastavia potilaita, joilla oli ollut verenvuodosta aiheutuva tai lakunaarinen aivohalvaus tai joilla oli ollut iskeeminen tai ei-lakunaarinen aivohalvaus viimeisen kuukauden aikana, ei ole tutkittu (ks. kohta Vasta-aiheet). Potilaita, joille oli äskettäin tehty alaraajan revaskularisaatiotoimenpide oireisen ääreisvaltimotaudin takia ja joilla oli ollut aiemmin aivohalvaus tai ohimenevä aivoverenkiertohäiriö, ei tutkittu. Xarelto 2,5 mg ‑valmisteen käyttöä on vältettävä tällaisilla potilailla, jotka saavat kaksinkertaista antitromboottista hoitoa.
Spinaali-/epiduraalipuudutus tai -punktio
Potilailla, jotka saavat antitromboottista lääkitystä tromboembolisten komplikaatioiden ehkäisyyn, on olemassa pitkäaikaiseen tai pysyvään halvaukseen johtavan spinaali-/epiduraalihematooman riski käytettäessä spinaali-/epiduraalipuudutusta tai -punktiota. Näiden tapahtumien riskiä saattaa lisätä postoperatiivinen kestoepiduraalikatetrien käyttö tai muiden hemostaasiin vaikuttavien lääkevalmisteiden samanaikainen käyttö. Riskiä voi myös lisätä traumaattinen tai toistuva epiduraali- tai spinaalipunktio. Potilaita on seurattava tiheästi neurologisen tilan huonontumista osoittavien oireiden ja merkkien toteamiseksi (esim. alaraajojen puutuminen tai heikkous sekä suolen tai rakon toimintahäiriöt). Jos neurologisia oireita huomataan, kiireellinen diagnoosi ja hoito ovat välttämättömiä. Lääkärin on ennen selkäydinkanavaan kohdistuvaa toimenpidettä arvioitava mahdollinen hyöty ja riski potilailla, jotka ovat saaneet tai tulevat saamaan hyytymisenestolääkitystä tromboosiprofylaksina. Tällaisissa tilanteissa Xarelto 2,5 mg -tablettien käytöstä antitromboottisten lääkkeiden kanssa kanssa ei ole kliinisiä kokemuksia. Trombosyyttiaggregaation estäjien käyttö on lopetettava valmistajan antamien ohjeiden mukaisesti.
Spinaali-/epiduraalipuudutuksen tai -punktion ja samanaikaiseen rivaroksabaanin käyttöön liittyvän mahdollisen verenvuotoriskin pienentämiseksi on otettava huomioon rivaroksabaanin farmakokineettiset ominaisuudet. Epiduraalikatetrin asetus tai poisto ja lannepunktio on parasta ajoittaa hetkeen, jolloin rivaroksabaanin antikoagulanttivaikutuksen arvellaan olevan vähäinen (ks. kohta Farmakokinetiikka). Yksittäisen potilaan kohdalla riittävän pienen antikoagulanttivaikutuksen tarkka ajankohta ei kuitenkaan ole tiedossa.
Annossuositukset ennen invasiivisia ja kirurgisia toimenpiteitä sekä niiden jälkeen
Jos invasiivinen tai kirurginen toimenpide on tarpeen, tulee Xarelto 2,5 mg -tablettien käyttö keskeyttää, mikäli mahdollista, vähintään 12 tuntia ennen toimenpidettä ja lääkärin kliiniseen harkintaan perustuen. Jos potilaalle tehdään elektiivinen leikkaus ja antitromboottinen vaikutus halutaan välttää, trombosyyttiaggregaation estäjien käyttö on lopetettava valmistajan antamien ohjeiden mukaisesti.
Jos toimenpidettä ei voida viivästyttää, lisääntynyttä verenvuotoriskiä on arvioitava suhteessa toimenpiteen kiireellisyyteen.
Xarelto-hoito tulee aloittaa uudelleen mahdollisimman pian invasiivisen tai kirurgisen toimenpiteen jälkeen edellyttäen, että kliininen tilanne sallii sen ja riittävä hemostaasi on saavutettu hoitavan lääkärin arvion mukaan (ks. kohta Farmakokinetiikka).
Iäkkäät potilaat
Korkea ikä voi suurentaa verenvuotovaaraa (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Dermatologiset reaktiot
Valmisteen markkinoille tulon jälkeen rivaroxabaanin käytön yhteydessä on raportoitu vakavia ihoreaktioita, mukaan lukien Stevens-Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysi ja DRESS eli yleisoireinen eosinofiilinen oireyhtymä (ks. kohta Haittavaikutukset). Ihoreaktioiden riski näyttää olevan suurimmillaan hoidon alussa: oireet alkavat useimmiten ensimmäisten hoitoviikkojen aikana. Rivaroksabaanin käyttö tulisi lopettaa heti, jos havaitaan vakavaa ihottumaa (esim. jos ihottuma leviää tai pahenee ja/tai syntyy rakkuloita) tai jos ilmenee muita yliherkkyysoireita yhdessä limakalvomuutosten kanssa.
Tietoja apuaineista
Xarelto sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasin puutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
CYP3A4:n ja P-gp:n estäjät
Kun rivaroksabaania annettiin samanaikaisesti ketokonatsolin (400 mg kerran päivässä) tai ritonaviirin (600 mg kahdesti päivässä) kanssa, rivaroksabaanin keskimääräinen AUC-arvo nousi 2,6- / 2,5-kertaiseksi ja rivaroksabaanin keskimääräinen Cmax nousi 1,7- / 1,6-kertaiseksi tehostaen merkitsevästi farmakodynaamisia vaikutuksia, mikä saattaa johtaa suurempaan verenvuotoriskiin. Tämän vuoksi Xarelto-valmisteen käyttöä ei suositella potilaille, jotka saavat samanaikaista systeemistä hoitoa atsoliryhmän sienilääkkeillä, kuten ketokonatsoli, itrakonatsoli, vorikonatsoli tai posakonatsoli, tai HIV-proteaasin estäjillä. Nämä vaikuttavat aineet ovat voimakkaita sekä CYP3A4:n että P-gp:n estäjiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Voimakkaasti vain toista rivaroksabaanin eliminaatioreiteistä, joko CYP3A4:ää tai P-gp:tä, estävien vaikuttavien aineiden odotetaan lisäävän rivaroksabaanin pitoisuutta plasmassa vähäisesti. Esimerkiksi klaritromysiini (500 mg kahdesti päivässä), jota pidetään voimakkaana CYP3A4:n estäjänä ja kohtalaisena P-gp:n estäjänä, nosti rivaroksabaanin keskimääräisen AUC-arvon 1,5-kertaiseksi ja Cmax-arvon 1,4-kertaiseksi. Yhteisvaikutus klaritromysiinin kanssa ei todennäköisesti ole kliinisesti merkittävä suurimmalle osalle potilaista, mutta se saattaa olla merkitsevä suuren riskin potilaille. (Munuaisten vajaatoimintaa sairastavat: ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
CYP3A4:ää ja P-gp:tä kohtalaisesti estävä erytromysiini (500 mg kolmesti päivässä) nosti rivaroksabaanin keskimääräiset AUC- ja Cmax-arvot 1,3-kertaisiksi. Yhteisvaikutus erytromysiinin kanssa ei todennäköisesti ole kliinisesti merkittävä suurimmalle osalle potilaista, mutta se saattaa olla merkitsevä suuren riskin potilaille. Lievää munuaisten vajaatoimintaa sairastavilla potilailla erytromysiini (500 mg kolmesti päivässä) nosti rivaroksabaanin keskimääräisen AUC-arvon 1,8-kertaiseksi ja Cmax-arvon 1,6-kertaiseksi verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Kohtalaista munuaisten vajaatoimintaa sairastavilla potilailla erytromysiini nosti rivaroksabaanin keskimääräisen AUC-arvon 2,0-kertaiseksi ja Cmax-arvon 1,6-kertaiseksi verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Erytromysiini voi suurentaa munuaisten vajaatoiminnan vaikutusta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Flukonatsoli (400 mg kerran päivässä), jota pidetään kohtalaisen voimakkaana CYP3A4:n estäjänä, nosti rivaroksabaanin keskimääräisen AUC-arvon 1,4-kertaiseksi ja Cmax-arvon 1,3-kertaiseksi. Yhteisvaikutus flukonatsolin kanssa ei todennäköisesti ole kliinisesti merkittävä suurimmalle osalle potilaista, mutta se saattaa olla merkitsevä suuren riskin potilaille. (Munuaisten vajaatoimintaa sairastavat: ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Rivaroksabaanin ja dronedaronin yhteiskäyttöä pitää välttää, koska kliinistä tietoa yhteiskäytöstä dronedaronin kanssa on rajoitetusti.
Hyytymisenestolääkkeet
Kun enoksapariinia (40 mg:n kerta-annos) annettiin yhdessä rivaroksabaanin (10 mg:n kerta-annos) kanssa, havaittiin additiivinen vaikutus antifaktori Xa -aktiivisuuteen, mutta ei muita vaikutuksia verenhyytymistutkimuksiin (PT, aPTT). Enoksapariini ei vaikuttanut rivaroksabaanin farmakokinetiikkaan.
Lisääntyneen verenvuotoriskin vuoksi on noudatettava varovaisuutta, jos potilaita hoidetaan samanaikaisesti muilla hyytymisenestoaineilla (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
NSAID:t / trombosyyttiaggregaation estäjät
Kun rivaroksabaania (15 mg) ja 500 mg naprokseenia annettiin samanaikaisesti, verenvuodon keston ei havaittu pidentyneen kliinisesti merkittävällä tavalla. Joillakin yksilöillä farmakodynaaminen vaste saattaa kuitenkin tehostua.
Kun rivaroksabaania annettiin samanaikaisesti 500 mg:n asetyylisalisyylihappoannoksen kanssa, kliinisesti merkittäviä farmakokineettisiä tai farmakodynaamisia yhteisvaikutuksia ei todettu.
Klopidogreelin (300 mg:n kyllästysannos ja sen jälkeen 75 mg:n ylläpitoannos) ei todettu aiheuttavan farmakokineettistä yhteisvaikutusta rivaroksabaanin (15 mg) kanssa, mutta verenvuodon kestossa todettiin eräässä potilasalaryhmässä relevantti pidentyminen, joka ei korreloinut verihiutaleiden aggregaatioon eikä P-selektiinin tai GPIIb/IIIa-reseptorin tasoihin.
Varovaisuutta on noudatettava, jos potilaat saavat samanaikaista hoitoa NSAID-lääkkeillä (mukaan lukien asetyylisalisyylihappo) ja verihiutaleaggregaation estäjillä, sillä nämä lääkkeet lisäävät tyypillisesti verenvuotoriskiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
SSRI-/SNRI-lääkkeet
Kuten muitakin antikoagulantteja käytettäessä potilailla saattaa olla suurentunut verenvuotoriski samanaikaisen SSRI- tai SNRI-lääkkeiden käytön yhteydessä, johtuen kyseisten lääkkeiden raportoidusta vaikutuksesta verihiutaleisiin. Kun näitä lääkkeitä käytettiin samanaikaisesti rivaroksabaanin kliinisessä ohjelmassa, kaikissa hoitoryhmissä havaittiin merkittävien tai muiden kuin suurten kliinisesti merkittävien verenvuotojen korkeampi ilmaantuvuus.
Varfariini
Potilaiden siirtäminen K-vitamiinin antagonisti varfariinista (INR 2,0–3,0) rivaroksabaaniin (20 mg) tai rivaroksabaanista (20 mg) varfariiniin (INR 2,0–3,0) johti protrombiiniajan / INR-arvon (Neoplastin) lisääntymiseen enemmän kuin additiivisesti (yksilöllinen INR-arvo voi nousta jopa arvoon 12), kun puolestaan vaikutukset aPTT-arvoon, faktori Xa -aktiivisuuden estymiseen ja endogeenisen trombiinin potentiaaliin olivat additiivisia.
Jos rivaroksabaanin farmakodynaamisten vaikutusten testaaminen on tarpeen siirtymäjakson aikana, antifaktori Xa -aktiivisuutta, PiCT:tä ja Heptestiä voidaan käyttää, sillä varfariini ei vaikuttanut näihin testeihin. Neljäntenä päivänä viimeisen varfariiniannoksen jälkeen kaikki testit (mukaan lukien PT, aPTT, antifaktori Xa -aktiivisuuden estäminen ja ETP) heijastivat vain rivaroksabaanin vaikutusta.
Jos varfariinin farmakodynaamisia vaikutuksia halutaan testata siirtymäjakson aikana, INR voidaan mitata rivaroksabaanin Ctrough-vaiheessa (24 tunnin kuluttua edellisestä rivaroksabaaniannoksesta), sillä tässä vaiheessa rivaroksabaani vaikuttaa vain minimaalisesti INR-testiin.
Varfariinin ja rivaroksabaanin välillä ei havaittu farmakokineettisiä yhteisvaikutuksia.
CYP3A4:n indusoijat
Kun rivaroksabaania annettiin samanaikaisesti voimakkaan CYP3A4:n indusoijan rifampisiinin kanssa, rivaroksabaanin keskimääräinen AUC-arvo laski noin 50 % ja sen farmakodynaamiset vaikutukset vähenivät vastaavasti. Rivaroksabaanin samanaikainen käyttö muiden voimakkaiden CYP3A4:n indusoijien (esim. fenytoiini, karbamatsepiini, fenobarbitaali tai mäkikuismauute (Hypericum perforatum)) kanssa saattaa myös pienentää rivaroksabaanin pitoisuutta plasmassa. Siksi voimakkaiden CYP3A4:n indusoijien antamista samanaikaisesti tulee välttää, ellei potilasta seurata tarkasti tromboosin merkkien ja oireiden varalta.
Muut samanaikaiset hoidot
Kliinisesti merkittäviä farmakokineettisiä tai farmakodynaamisia yhteisvaikutuksia ei todettu, kun rivaroksabaania annettiin samanaikaisesti midatsolaamin (CYP3A4:n substraatti), digoksiinin (P-gp:n substraatti), atorvastatiinin (CYP3A4:n ja P-gp:n substraatti) tai omepratsolin (protonipumpun estäjän) kanssa. Rivaroksabaani ei estä eikä indusoi mitään tärkeitä CYP-isoformeja, kuten CYP3A4:ää.
Kliinisesti merkityksellisiä yhteisvaikutuksia ruoan kanssa ei todettu (ks. kohta Annostus ja antotapa).
Laboratorioparametrit
Vaikutus hyytymisparametreihin (esim. PT, aPTT, Heptest) on odotetusti rivaroksabaanin vaikutusmekanismin mukainen (ks. kohta Farmakodynamiikka).
Raskaus ja imetys
Raskaus
Xarelto-valmisteen turvallisuutta ja tehoa raskaana olevilla naisilla ei ole varmistettu. Eläintutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Xarelto on vasta-aiheinen raskauden aikana mahdollisen lisääntymistoksisuuden ja verenvuodon olennaisen riskin vuoksi ja koska rivaroksabaanin on osoitettu läpäisevän istukan (ks. kohta Vasta-aiheet).
Hedelmällisessä iässä olevien naisten tulee välttää raskaaksi tulemista rivaroksabaanihoidon aikana.
Imetys
Xarelto-valmisteen turvallisuutta ja tehoa imettävillä naisilla ei ole varmistettu. Eläintutkimukset osoittavat rivaroksabaanin erittyvän maitoon. Sen vuoksi Xarelto on vasta-aiheinen imetyksen aikana (ks. kohta Vasta-aiheet). On päätettävä joko imettämisen lopettamisesta tai hoidon keskeyttämisestä/hoidosta luopumisesta.
Hedelmällisyys
Rivaroksabaanilla ei ole tehty erityisiä tutkimuksia, joissa olisi arvioitu vaikutuksia ihmisen fertiliteettiin. Uros- ja naarasrotilla tehdyssä tutkimuksessa ei havaittu vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Xarelto-valmisteella on vähäinen vaikutus ajokykyyn ja koneiden käyttökykyyn. Pyörtyilyn (melko harvinainen) ja huimauksen (yleinen) kaltaisia haittavaikutuksia on raportoitu (ks. kohta Haittavaikutukset). Potilaiden, joilla esiintyy näitä haittavaikutuksia, ei tule ajaa eikä käyttää koneita.
Haittavaikutukset
Yhteenveto turvallisuudesta
Rivaroksabaanin turvallisuutta on arvioitu 13:ssa vaiheen III avaintutkimuksessa (ks. taulukko 1).
Rivaroksabaania annettiin yhteensä 69 608 aikuispotilaalle 19:ssä vaiheen III tutkimuksessa ja 488 pediatriselle potilaalle kahdessa vaiheen II ja kahdessa vaiheen III tutkimuksessa
Taulukko 1: Tutkittujen potilaiden määrä, kokonaisvuorokausiannos ja suurin hoidon kesto aikuisille ja lapsille tehdyissä vaiheen III tutkimuksissa
| Käyttöaihe | Potilaiden lukumäärä * | Kokonaisvuorokausiannos | Suurin hoidon kesto |
| Laskimotromboembolioiden (VTE) ehkäisy aikuisille potilaille, joille tehdään elektiivinen lonkka- tai polviproteesileikkaus | 6 097 | 10 mg | 39 päivää |
| Sairaalahoitopotilaiden VTE:n ehkäisy | 3 997 | 10 mg | 39 päivää |
| Syvän laskimotukoksen (SLT), keuhkoembolian (KE) hoito ja uusiutumisen ehkäisy | 6 790 | Päivä 1–21: 30 mg Päivä 22 ja sen jälkeen 20 mg | 21 kuukautta |
| VTE:n hoito ja VTE:n uusiutumisen ehkäisy täysiaikaisille vastasyntyneille ja alle 18 vuoden ikäisille lapsille tavanomaisen antikoagulaatiohoidon aloittamisen jälkeen | 329 | Kehon painoon mukautettu annos, jolla saavutettava altistus on samankaltainen kuin aikuisilla, jotka saavat SLT:n hoitoon 20 mg rivaroksabaania kerran päivässä | 12 kuukautta |
| Aivohalvauksen ja systeemisen embolian ehkäisy potilailla, joilla on ei-valvulaarinen eteisvärinä | 7 750 | 20 mg | 41 kuukautta |
| Aterotromboottisten tapahtumien ehkäisy akuutin sepelvaltimotautikohtauksen jälkeen | 10 225 | samanaikaisesti 5 mg asetyylisalisyylihapon kanssa tai 10 mg asetyylisalisyylihapon ja klopidogreelin tai tiklopidiinin yhdistelmän kanssa | 31 kuukautta |
Aterotromboottisten tapahtumien ehkäisy sepel /ääreisvaltimotautia sairastavilla | 18 244 | 5 mg samanaikaisesti asetyylisalisyylihapon kanssa tai 10 mg pelkästään | 47 kuukautta |
| 3 256** | 5 mg samanaikaisesti asetyylisalisyylihapon kanssa | 42 kuukautta |
*Vähintään yhdelle rivaroksabaaniannokselle altistuneet potilaat
** Tiedot VOYAGER PAD -tutkimuksesta
Yleisimmin raportoidut haittavaikutukset rivaroksabaania saavilla potilailla olivat verenvuodot (taulukko 2) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja ”Kuvaus valituista haittavaikutuksista”). Yleisimmin raportoituja verenvuotoja olivat nenäverenvuoto (4,5 %) ja ruoansulatuskanavan verenvuoto (3,8 %).
Taulukko 2. Verenvuoto- ja anemiatapahtumien ilmaantuvuus rivaroksabaanille altistuneilla potilailla kaikissa päätökseen saatetuissa, aikuisille ja lapsille tehdyissä vaiheen III tutkimuksissa:
Käyttöaihe | Jokin verenvuoto | Anemia |
VTE:n ehkäisy aikuisilla potilailla, joille tehdään elektiivinen lonkka- tai polviproteesileikkaus | 6,8 % potilaista | 5,9 % potilaista |
Sairaalahoitopotilaiden VTE:n ehkäisy | 12,6 % potilaista | 2,1 % potilaista |
SLT:n, KE:n hoito ja uusiutumisen ehkäisy | 23 % potilaista | 1,6 % potilaista |
| VTE:n hoito ja VTE:n uusiutumisen ehkäisy täysiaikaisilla vastasyntyneillä ja alle 18 vuoden ikäisillä lapsilla tavanomaisen antikoagulaatiohoidon aloittamisen jälkeen | 39,5 % potilaista | 4,6 % potilaista |
Aivohalvauksen ja systeemisen embolian ehkäisy potilailla, joilla on ei-valvulaarinen eteisvärinä | 28 /100 potilasvuotta | 2,5 /100 potilasvuotta |
Aterotromboottisten tapahtumien ehkäisy akuutin sepelvaltimotautikohtauksen jälkeen | 22 / 100 potilasvuotta | 1,4 / 100 potilasvuotta |
| Aterotromboottisten tapahtumien ehkäisy sepel /ääreisvaltimotautia sairastavilla | 6,7 / 100 potilasvuotta | 0,15 / 100 potilasvuotta** |
| 8,38 / 100 potilasvuotta# | 0,74 / 100 potilasvuotta*** # |
* Kaikissa rivaroksabaanitutkimuksissa kerättiin, raportoitiin ja arvioitiin kaikki verenvuototapahtumat.
** COMPASS-tutkimuksessa anemian esiintyvyys oli alhaista kun käytössä oli valikoiva haittatapahtumien keräystapa.
*** Käytössä oli valikoiva haittatapahtumien keräystapa.
# Tiedot VOYAGER PAD -tutkimuksesta
Luettelo haittavaikutuksista taulukon muodossa
Aikuispotilailla ja pediatrisilla potilailla Xarelto-valmisteen yhteydessä raportoitujen haittavaikutusten esiintymistiheydet luetellaan alla olevassa taulukossa 3 elinjärjestelmän (MedDRA) ja esiintyvyyden mukaan.
Esiintyvyys on määritetty seuraavalla tavalla:
hyvin yleinen (≥ 1/10)
yleinen (≥ 1/100, < 1/10)
melko harvinainen (≥ 1/1 000, < 1/100)
harvinainen (≥ 1/10 000, < 1/1 000)
hyvin harvinainen (< 1/10 000)
tuntematon (koska saatavissa oleva tieto ei riitä arviointiin)
Taulukko 3: Kaikki haittavaikutukset, jotka on raportoitu aikuispotilaille vaiheen III kliinisissä tutkimuksissa tai valmisteen markkinoille tulon jälkeen* sekä pediatrisille potilaille kahdessa vaiheen II tutkimuksessa ja kahdessa vaiheen III tutkimuksessa
| Veri ja imukudos | |
| yleinen | Anemia (ml. vastaavat laboratorioparametrit) |
| melko harvinainen | Trombosytoosi (ml. verihiutaleiden määrän lisääntyminen)A, trombosytopenia |
| Immuunijärjestelmä | |
| melko harvinainen | Allerginen reaktio, allerginen ihottuma, angioedeema ja allerginen edeema |
| hyvin harvinainen | Anafylaktiset reaktiot, ml. anafylaktinen sokki |
| Hermosto | |
| yleinen | Huimaus, päänsärky |
| melko harvinainen | Aivoverenvuoto ja kallonsisäinen verenvuoto, pyörtyminen |
| Silmät | |
| yleinen | Silmäverenvuoto (ml. sidekalvon verenvuoto) |
| Sydän | |
| melko harvinainen | Takykardia |
| Verisuonisto | |
| yleinen | Hypotensio, hematooma |
| Hengityselimet, rintakehä ja välikarsina | |
| yleinen | Nenäverenvuoto, veriyskä |
| hyvin harvinainen | Eosinofiilinen keuhkokuume |
| Ruoansulatuselimistö | |
| yleinen | Ienverenvuoto, ruoansulatuskanavan verenvuoto (ml. peräsuolen verenvuoto), maha-, suolisto- ja vatsakivut, dyspepsia, pahoinvointi, ummetusA, ripuli, oksenteluA |
| melko harvinainen | Suun kuivuminen |
| Maksa ja sappi | |
| yleinen | Transaminaasipitoisuuksien suureneminen |
| melko harvinainen | Maksan vajaatoiminta; bilirubiinin, veren alkalisen fosfataasinA, GGT:nA pitoisuuden suureneminen |
| harvinainen | Keltaisuus, konjugoituneen bilirubiinin pitoisuuden suureneminen (johon voi liittyä ALAT-arvon samanaikainen suureneminen), kolestaasi, hepatiitti (ml. heptosellulaarinen vaurio) |
| Iho ja ihonalainen kudos | |
| yleinen | Kutina (ml. harvinaiset yleisen kutinan tapaukset), ihottuma, ekkymoosi, iho- ja ihonalainen verenvuoto |
| melko harvinainen | Urtikaria |
| hyvin harvinainen | Stevens-Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysi, DRESS eli yleisoireinen eosinofiilinen oireyhtymä |
| Luusto, lihakset ja sidekudos | |
| yleinen | RaajakipuA |
| melko harvinainen | Hemartroosi |
| harvinainen | Lihasverenvuoto |
| tuntematon | Verenvuodon aiheuttama lihasaitio-oireyhtymä |
| Munuaiset ja virtsatiet | |
| yleinen | Urogenitaalikanavan verenvuoto (ml. hematuria ja menorragiaB), munuaisten vajaatoiminta (ml. veren kreatiniinipitoisuuden lisääntyminen, veren ureapitoisuuden lisääntyminen) |
| tuntematon | Munuaisten toimintahäiriö / akuutti munuaisten toimintahäiriö, joka aiheutuu verenvuodon aiheuttamasta hypoperfuusiosta, antikoagulanttiin liittyvä nefropatia |
| Yleisoireet ja antopaikassa todettavat haitat | |
| yleinen | KuumeA, perifeerinen ödeema, yleinen voiman ja energian väheneminen (ml. väsymys ja astenia) |
| melko harvinainen | Huonovointisuus (ml. kuvotus) |
| harvinainen | Paikallinen ödeemaA |
| Tutkimukset | |
| melko harvinainen | LDH:nA, lipaasinA, amylaasinA pitoisuuden suureneminen |
| Vammat ja myrkytykset | |
| yleinen | Toimenpiteen jälkeinen verenvuoto (ml. postoperatiivinen anemia ja haavaverenvuoto), kontuusio, haavaeriteA |
| harvinainen | Vaskulaarinen pseudoaneurysmaC |
A: Havaittu VTE:n ehkäisyhoidossa aikuisilla, joille oli tehty elektiivinen lonkka- tai polviproteesileikkaus
B: havaittu hyvin yleisenä SLT:n ja KE:n hoidossa ja uusiutumisen ehkäisyssä < 55-vuotiailla naisilla
C: havaittu melko harvinaisena aterotromboottisten tapahtumien preventiossa akuutin sepelvaltimotautikohtauksen jälkeen (perkutaanisen sepelvaltimotoimenpiteen yhteydessä)
* Valituissa vaiheen III tutkimuksissa käytössä oli ennalta asetettu valikoiva haittatapahtumien keräystapa. Näiden tutkimusten analyyseissa haittavaikutusten esiintyvyys ei kasvanut eikä uusia haittavaikutuksia havaittu,
Kuvaus valituista haittavaikutuksista
Farmakologisesta vaikutusmekanismista johtuen Xarelto-valmisteen käyttöön saattaa liittyä lisääntynyt piilevän tai avoimen verenvuodon riski mistä tahansa kudoksesta tai elimestä, mikä saattaa johtaa verenvuodon aiheuttamaan anemiaan. Merkit, oireet ja vakavuus (mukaan lukien kuolema) vaihtelevat verenvuodon paikan ja määrän tai laajuuden ja/tai anemian mukaan (ks. kohta Yliannostus: "Verenvuodon tyrehdyttäminen"). Kliinisissä tutkimuksissa limakalvoverenvuotoja (nenä, ien, gastrointestinaalikanava, urogenitaalialue , mukaan lukien epänormaali emätinverenvuoto tai lisääntynyt kuukautisvuoto) ja anemiaa havaittiin pitkäkestoisen rivaroksabaanihoidon aikana useammin kuin VKA-hoidon aikana. Sen vuoksi asianmukaisen kliinisen seurannan lisäksi hemoglobiinin/hematokriitin määrittämisestä voi olla hyötyä piilevän verenvuodon havaitsemisessa ja näkyvän verenvuodon kliinisen merkityksen selvittämisessä, mikäli em. määrityksiä pidetään tarkoituksenmukaisina. Verenvuotoriski voi olla tavallista suurempi tietyissä potilasryhmissä, kuten esimerkiksi potilailla, joilla on hyvin korkea hoitoresistentti verenpaine ja/tai jotka saavat samanaikaista hemostaasiin vaikuttavaa hoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ”Verenvuotoriski”). Kuukautisvuoto saattaa olla tavallista runsaampaa ja/tai kestää pidempään.
Verenvuotokomplikaatioiden oireita voivat olla heikkous, kalpeus, huimaus, päänsärky tai selittämätön turvotus, dyspnea ja selittämätön sokki. Joissakin tapauksissa anemian seurauksena on havaittu sydänlihasiskemian oireita, kuten rintakipua tai angina pectorista.
Tunnettuja vakavan verenvuodon aiheuttamia komplikaatioita, kuten lihasaitio-oireyhtymää ja hypoperfuusiosta johtuvaa munuaisten toimintahäiriötä, tai antikoagulanttiin liittyvää nefropatiaa on raportoitu Xarelto-valmisteen yhteydessä. Sen vuoksi verenvuodon mahdollisuus on otettava huomioon arvioitaessa hyytymisenestohoitoa saaneen potilaan vointia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Harvinaisia yliannostapauksia enintään 1960 mg:aan saakka on raportoitu. Yliannostustapauksessa potilasta on seurattava huolellisesti verenvuotokomplikaatioita tai muiden haittavaikutuksien havaitsemiseksi (ks. kohta ”Verenvuodon tyrehdyttäminen”). Vähäisen imeytymisen vuoksi 50 mg:n tai sen ylittävillä rivaroksabaanin supraterapeuttisilla annoksilla on odotettavissa maksimaalinen vaikutus ilman keskimääräisen plasmapitoisuuden lisääntymistä.
Rivaroksabaanin farmakodynaamisen vaikutuksen kumoamiseen ei ole käytettävissä erityistä vastalääkettä.
Rivaroksabaanin yliannostuksen yhteydessä voidaan imeytymisen vähentämiseksi harkita lääkehiilen käyttöä.
Verenvuodon tyrehdyttäminen
Jos rivaroksabaania saavalla potilaalla ilmenee verenvuotokomplikaatio, seuraavaa rivaroksabaanin antoa pitää lykätä tai hoito on tarvittaessa keskeytettävä. Rivaroksabaanin puoliintumisaika on noin 5–13 tuntia (ks. kohta Farmakokinetiikka).
Verenvuodon tyrehdyttämistoimenpiteet valitaan potilaskohtaisesti verenvuodon vaikeusasteen ja vuotokohdan mukaan. Asianmukaista oireiden hoitoa, johon kuuluu esim. mekaaninen kompressio (esim. vakavassa nenäverenvuodossa), kirurginen hemostaasi ja verenvuodon tyrehdytystoimenpiteet, nestehoito ja hemodynaaminen tuki, sekä verivalmisteet (pakatut punasolut tai jääplasma, riippuen anemiasta tai koagulopatiasta) tai trombosyyttien anto, käytetään tarpeen mukaan.
Jos verenvuotoa ei saada tyrehtymään edellä mainituin toimenpitein, voidaan harkita tiettyjen hyytymistekijävalmisteiden, kuten protrombiinikompleksikonsentraatin (PCC), aktivoidun protrombiinikompleksikonsentraatin (APCC) tai rekombinantti tekijä VIIa:n (r-FVIIa) antamista. Tällä hetkellä on kuitenkin hyvin vähän kokemusta näiden lääkevalmisteiden käytöstä rivaroksabaania saavilla henkilöillä. Suositus perustuu myös rajalliseen ei-kliiniseen aineistoon. Riippuen verenvuodon korjaantumisesta voidaan harkita rekombinantti tekijä VIIa:n uudelleen antamista ja annoksen säätämistä. Merkittävien verenvuotojen yhteydessä tulee harkita veren hyytymiseen erikoistuneen lääkärin konsultointia mahdollisuuksien mukaan. (ks. kohta Farmakodynamiikka).
Protamiinisulfaatin ja K-vitamiinin ei oleteta vaikuttavan rivaroksabaanin verenhyytymistä estävään vaikutukseen. Kokemuksia (traneksaamihapon käytöstä on vain vähän- ja aminokapronihaponja aprotiinin käytöstä ei lainkaan rivaroksabaania saavilla henkilöillä. Systeemiseen hemostaasiin vaikuttavia lääkeaineen (desmopressiini) hyödylle ei ole tieteellisiä todisteita eikä käytöstä ole kokemuksia rivaroksabaania saavilla henkilöillä. Koska rivaroksabaani sitoutuu voimakkaasti plasman proteiineihin, sen ei oleteta olevan dialysoitavissa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antitromboottiset lääkeaineet, suorat hyytymistekijä Xa:n estäjät, ATC-koodi: B01AF01.
Vaikutusmekanismi
Rivaroksabaani on suun kautta annosteltava hyvin selektiivinen hyytymistekijä Xa:n suora estäjä. Hyytymistekijä Xa:n estäminen keskeyttää veren hyytymisjärjestelmän ulkoisen ja sisäisen aktivaatioreitin estäen sekä trombiinin muodostumisen että trombin kehittymisen. Rivaroksabaani ei estä trombiinia (aktivoitu hyytymistekijä II) eikä vaikutuksia verihiutaleisiin ole osoitettu.
Farmakodynaamiset vaikutukset
Ihmisillä hyytymistekijä Xa:n vaikutuksen on havaittu estyvän annosriippuvaisesti. Rivaroksabaani vaikuttaa protrombiiniaikaan (PT) annosriippuvaisesti korreloiden läheisesti plasmapitoisuuksien kanssa (r-arvo on 0,98), kun määrityksessä käytetään Neoplastin-reagenssia. Muilla reagensseilla voidaan saada erilaisia tuloksia. PT tulee lukea sekunteina, sillä INR on kalibroitu ja validoitu ainoastaan kumariineille eikä sitä voi käyttää muilla hyytymisenestolääkeaineilla.
Rivaroksabaanin farmakodynaamisten vaikutusten palautumista terveillä aikuisilla (n=22) tarkastelleessa kliinisessä farmakologisessa tutkimuksessa arvioitiin kahden erityyppisen protrombiinikompleksikonsentraatin (PCC) kerta-annosten (50 IU/kg) vaikutuksia. Tutkimuksessa käytetyt protrombiinikompleksikonsentraatit olivat kolmea hyytymistekijää sisältävä PCC (hyytymistekijät II, IX ja X) ja neljää hyytymistekijää sisältävä PCC (tekijät II, VII, IX ja X). Kolmen hyytymistekijän PCC lyhensi Neoplastin-reagenssia käytettäessä keskimääräisiä protrombiiniaikoja (PT) noin 1,0 sekuntia 30 minuutin kuluessa ja neljän hyytymistekijän PCC noin 3,5 sekuntia. Kolmen hyytymistekijän PCC:llä oli kuitenkin suurempi ja nopeampi kokonaisvaikutus endogeenisen trombiinin tuotannossa ilmenneiden muutosten palautumiseen kuin neljän hyytymistekijän PCC:llä (ks. kohta Yliannostus).
Myös aktivoitu partiaalinen tromboplastiiniaika (aPTT) ja Heptest pidentyvät annosriippuvaisesti. Niitä ei kuitenkaan suositella rivaroksabaanin farmakodynaamisen vaikutuksen määritykseen. Rutiininomainen koagulaatioparametrien tarkkailu ei ole tarpeen rivaroksabaanihoidon aikana. Tarvittaessa rivaroksabaanipitoisuus voidaan kuitenkin mitata kalibroiduilla kvantitatiivisilla antifaktori Xa -testeillä (ks. kohta Farmakokinetiikka).
Kliininen teho ja turvallisuus
Akuuttisepelvaltimotautikohtaus
Rivaroksabaanin kliininen tutkimusohjelma tässä käyttöaiheessa suunniteltiin osoittamaan rivaroksabaanin-valmisteen teho kardiovaskulaarisista syistä johtuvan kuoleman, sydäninfarktin ja aivohalvauksen ehkäisyssä potilailla, joilla on äskettäin ollut akuutti sepelvaltimotautikohtaus (ST-nousuinfarkti [STEMI], sydäninfarkti ilman ST-nousua [NSTEMI] tai epästabiili angina [UA]). Keskeisessä kaksoissokkoutetussa ATLAS ACS 2 TIMI 51 -tutkimuksessa 15 526 potilasta jaettiin satunnaistetusti suhteessa 1:1:1 kolmeen eri hoitoryhmään: rivaroksabaanin 2,5 mg suun kautta kahdesti päivässä, 5 mg suun kautta kahdesti päivässä tai lumelääke kahdesti päivässä samanaikaisesti pelkän asetyylisalisyylihapon kanssa tai asetyylisalisyylihapon ja tienopyridiinin (klopidogreelin tai tiklopidiinin) yhdistelmän kanssa. Akuutin sepelvaltimotautikohtauksen saaneilla alle 55-vuotiailla potilailla tuli olla joko diabetes mellitus tai aiemmin sairastettu sydäninfarkti. Keskimääräinen hoitoaika oli 13 kuukautta ja hoidon kesto oli yhteensä lähes 3 vuotta. Potilaista 93,2 % sai samanaikaisesti asetyylisalisyylihapon ja tienopyridiinin yhdistelmää ja 6,8 % vain asetyylisalisyylihappoa. Kaksinkertaista antitromboottista lääkitystä saavista potilaista 98,8 % sai klopidogreelia, 0,9 % sai tiklopidiinia ja 0,3 % sai prasugreeliä. Potilaat saivat ensimmäisen rivaroksabaaniannoksen vähintään 24 tunnin ja enintään 7 päivän (keskiarvo 4,7 päivää) kuluttua sairaalaan tulosta, kuitenkin mahdollisimman pian akuutin sepelvaltimotautikohtauksen stabiloinnin (mukaan lukien revaskularisaatiotoimenpiteet) jälkeen hetkellä, jolloin parenteraalinen antikoagulaatiohoito tavallisesti lopetettaisiin.
Sekä annoksella 2,5 mg kahdesti päivässä että annoksella 5 mg kahdesti päivässä rivaroksabaani vähensi entisestään kardiovaskulaaristen tapahtumien määrää, kun taustalla oli normaali antitromboottinen lääkitys. Annos 2,5 mg kahdesti päivässä vähensi kuolleisuutta, ja on todisteita siitä, että pienempi annos aiheutti pienemmän verenvuotoriskin. Tämän vuoksi suositellaan annettavan 2,5 mg rivaroksabaania kahdesti päivässä samanaikaisesti vain asetyylisalisyylihapon tai asetyylisalisyylihapon ja joko klopidogreelin tai tiklopidiinin yhdistelmän kanssa ateromboottisten tapahtumien ehkäisemiseksi akuutin sepelvaltimotautikohtauksen jälkeen aikuisille potilaille, joiden sydämen biomarkkerit ovat koholla.
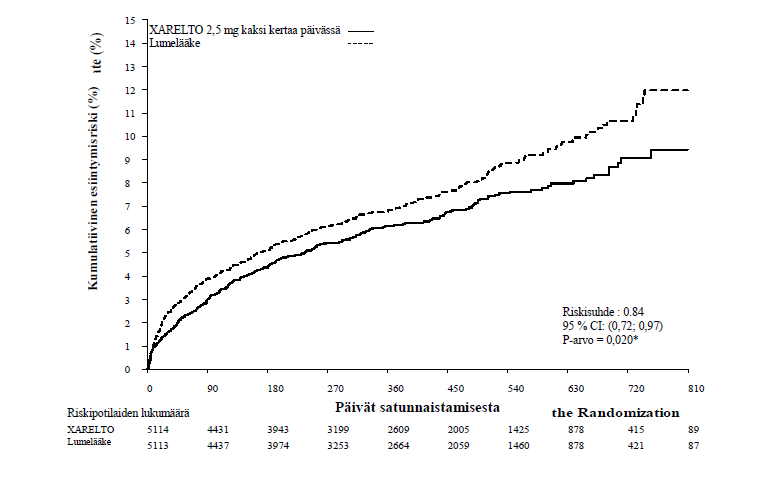
Lumelääkkeeseen verrattuna rivaroksabaani vähensi merkitsevästi tehokkuuden ensisijaisen yhdistetyn päätetapahtuman eli kardiovaskulaarisista syistä johtuvan kuoleman, sydäninfarktin ja aivohalvauksen esiintyvyyttä. Tulokseen vaikutti erityisesti kardiovaskulaarisista syistä johtuvan kuoleman ja sydäninfarktin väheneminen. Vaikutus todettiin varhaisessa vaiheessa ja se säilyi vakaana koko hoitojakson ajan (ks. taulukko 4 ja kuva 1). Myös tärkeimmän toissijaisen päätätapahtuman (kuolema, sydäninfarkti tai aivohalvaus) esiintyvyys väheni merkitsevästi. Retrospektiivinen lisäanalyysi osoitti nimellisesti merkitsevän laskun stenttitromboosin esiintyvysluvuissa lumelääkkeeseen verrattuna (ks. taulukko 4). Turvallisuuteen liittyvän tärkeimmän päätetapahtuman (ei ohitusleikkaukseen liittyvä, TIMI:n mukaan suuri verenvuoto) esiintyvyys oli rivaroksabaanihoitoa saaneilla potilailla suurempi kuin lumelääkettä saaneilla (ks. taulukko 6). Esiintyvyysluvut jakautuivat kuitenkin tasaisesti rivaroksabaanin ja lumelääkkeen välillä koskien kuolemaan johtaneiden verenvuotojen komponentteja, laskimonsisäistä inotrooppista lääkehoitoa vaativaa hypotensiota ja verenvuodon vaatimia kirurgisia toimenpiteitä.
Taulukossa 5 on esitetty tehoa koskevat tulokset potilailla, joille tehtiin perkutaaninen sepelvaltimotoimenpide. Turvallisuustulokset olivat tässä alaryhmässä verrattavissa turvallisuutta koskeviin kokonaistuloksiin.
Tutkimusjoukosta 80 %:lla oli kohonneet biomarkkeriarvot (troponiini tai CK-MB) mutta ei aiempaa aivohalvausta /ohimenevää aivoverenkiertohäiriötä. Myös tämän potilasjoukon tulokset olivat yhdenmukaiset tehoa ja turvallisuutta koskevien kokonaistulosten kanssa.
Taulukko 4: Vaiheen III ATLAS ACS 2 TIMI 51 -tutkimuksen tehoa koskevat tulokset
| Tutkimuspopulaatio | Potilaat, joilla on ollut äskettäin sepelvaltimotautikohtausa) | |
| Hoitoannos | Rivaroksabaani 2,5 mg, kahdesti päivässä, N=5 114 | Lumelääke N=5 113 n (%) |
| Kardiovaskulaarisista syistä johtuva kuolema, sydäninfarkti tai aivohalvaus | 313 (6,1 %) 0,84 (0,72; 0,97) p = 0,020* | 376 (7,4 %) |
| Kaikki kuolemansyyt, sydäninfarkti tai aivohalvaus | 320 (6,3 %) 0,83 (0,72; 0,97) p = 0,016* | 386 (7,5 %) |
| Kardiovaskulaarisista syistä johtuva kuolema | 94 (1,8 %) 0,66 (0,51; 0,86) p = 0,002** | 143 (2,8 %) |
| Kaikki kuolemansyyt | 103 (2,0 %) 0,68 (0,53; 0,87) p = 0,002** | 153 (3,0 %) |
| Sydäninfarkti | 205 (4,0 %) 0,90 (0,75; 1,09) p = 0,270 | 229 (4,5 %) |
| Aivohalvaus | 46 (0,9 %) 1,13 (0,74; 1,73) p = 0,562 | 41 (0,8 %) |
| Stenttitromboosi | 61 (1,2 %) 0,70 (0,51; 0,97) p = 0,033** | 87 (1,7 %) |
a) modifioidun hoitoaikeen mukainen analyysijoukko (stenttitromboosia koskeva hoitoaikeen mukainen kokonaisanalyysijoukko)
b) verrattuna lumelääkkeeseen; Logrank-testin p-arvo
* tilastollisesti parempi
** nimellisesti merkitsevä
Taulukko 5: Vaiheen III ATLAS ACS 2 TIMI 51 -tutkimuksen tehoa koskevat tulokset potilailla, joille tehtiin perkutaaninen sepelvaltimotoimenpide
| Tutkimuspopulaatio | Potilaat, joilla on ollut äskettäin sepelvaltimotautikohtaus ja joille tehtiin perkutaaninen sepelvaltimotoimenpidea) | |
| Hoitoannos | Rivaroksabaani 2,5 mg, kahdesti päivässä, N=3114 | Lumelääke N=3096 n (%) |
| Kardiovaskulaarisista syistä johtuva kuolema, sydäninfarkti tai aivohalvaus | 153 (4,9 %) 0,94 (0,75, 1,17) p = 0,572 | 165 (5,3 %) |
| Kardiovaskulaarisista syistä johtuva kuolema | 24 (0,8 %) 0,54 (0,33, 0,89) p = 0,013** | 45 (1,5 %) |
| Kaikki kuolemansyyt | 31 (1,0 %) 0,64 (0,41, 1,01) p = 0,053 | 49 (1,6 %) |
| Sydäninfarkti | 115 (3,7 %) 1,03 (0,79, 1,33) p = 0,829 | 113 (3,6 %) |
| Aivohalvaus | 27 (0,9 %) 1,30 (0,74, 2,31) p = 0,360 | 21 (0,7 %) |
| Stenttitromboosi | 47 (1,5 %) 0,66 (0,46, 0,95) p = 0,026** | 71 (2,3 %) |
a) modifioidun hoitoaikeen mukainen analyysijoukko (stenttitromboosia koskeva hoitoaikeen mukainen kokonaisanalyysijoukko)
b) verrattuna lumelääkkeeseen; Logrank-testin p-arvo
** nimellisesti merkitsevä
Taulukko 6: Vaiheen III ATLAS ACS 2 TIMI 51 -tutkimuksen turvallisuutta koskevat tulokset
| Tutkimuspopulaatio | Potilaat, joilla on ollut äskettäin sepelvaltimotautikohtausa) | |
| Hoitoannos | Rivaroksabaani 2,5 mg, kahdesti päivässä, N=5115 n (%) Riskisuhde (95 % CI) p-arvo b) | Lumelääke N=5125 n (%) |
| Ei ohitusleikkaukseen liittyvä, TIMI:n mukaan suuri verenvuoto | 65 (1,3 %) 3,46 (2,08, 5,77) p = < 0,001* | 19 (0,4 %) |
| Kuolemaan johtanut verenvuoto | 6 (0,1 %) 0,67 (0,24, 1,89) p = 0,450 | 9 (0,2 %) |
| Symtomaattinen kallonsisäinen verenvuoto | 14 (0,3 %) 2,83 (1,02, 7,86) p = 0,037 | 5 (0,1 %) |
| Hypotensio, joka vaatii laskimonsisäistä inotrooppista lääkehoitoa | 3 (0,1 %) | 3 (0,1 %) |
| Verenvuodon vaatima kirurginen toimenpide | 7 (0,1 %) | 9 (0,2 %) |
| Verensiirto, jossa annetaan 4 yksikköä tai enemmän verta 48 tunnin kuluessa | 19 (0,4 %) | 6 (0,1 %) |
a) turvallisuuspopulaatio, lääkehoidon aikana
b) verrattuna lumelääkkeeseen; Logrank-testin p-arvo
** tilastollisesti merkitsevä
Kuva 1: Aika tehon ensisijaisen päätetapahtuman (kardiovaskulaarisista syistä johtuva kuolema, sydäninfarkti tai aivohalvaus) ensimmäiseen tapahtumaan
Sepel‑/ääreisvaltimotauti
Vaiheen III COMPASS-tutkimus (27 395 potilasta, 78,0 % miehiä, 22,0 % naisia) osoitti rivaroksabaanin tehon ja turvallisuuden kardiovaskulaarisista syistä johtuvan kuoleman, sydäninfarktin ja aivohalvauksen yhdistelmäpäätetapahtuman ehkäisyssä sepelvaltimotautipotilailla tai oireista ääreisvaltimotautia sairastavilla potilailla, joilla on suuri iskeemisten tapahtumien riski. Potilaiden seuranta-ajan mediaani oli 23 kuukautta ja pisin seuranta-aika 3,9 vuotta.
Tutkittavat, joilla ei ollut jatkuvaa tarvetta protonipumpun estäjä ‑hoidolle, satunnaistettiin pantopratsoli- tai lumelääkeryhmiin. Kaikki potilaat satunnaistettiin sitten suhteessa 1:1:1 hoitoryhmiin 2,5 mg rivaroksabaania kaksi kertaa päivässä/100 mg asetyylisalisyylihappoa kerran päivässä; 5 mg rivaroksabaania kaksi kertaa päivässä; tai pelkästään 100 mg asetyylisalisyylihappoa kerran päivässä, ja niitä vastaaviin lumelääkeannoksiin.
Sepelvaltimotautipotilailla oli monen suonen sepelvaltimotauti ja/tai taustalla oli aiempi sydäninfarkti. Alle 65‑vuotiailla potilailla vaadittiin kahden tai useamman elimen valtimopuustossa todettu ateroskleroosi tai vähintään kaksi muuta kardiovaskulaarista riskitekijää.
Ääreisvaltimotautipotilaille oli tehty aiempia toimenpiteitä, kuten ohitusleikkaus, perkutaaninen transluminaalinen angioplastia tai raajan tai jalkaterän amputaatio, kun syynä oli valtimosairaus tai katkokävely, jossa nilkan ja käsivarren verenpainesuhde oli < 0,90 ja/tai merkittävä ääreisvaltimon ahtauma, aiempi kaulavaltimon revaskularisaatio tai oireeton kaulavaltimon ahtauma ≥ 50 %.
Poissulkuperusteisiin kuuluivat tarve kaksinkertaiseen antitromboottiseen hoitoon, muuhun antitromboottiseen hoitoon asetyylisalisyylihappoa lukuun ottamatta tai suun kautta otettavaan antikoagulanttihoitoon. Pois suljettiin myös potilaat, joilla oli korkea verenvuotoriski, joilla oli sydämen vajaatoiminta, jossa ejektiofraktio oli < 30 % tai New York Heart Association ‑järjestön mukaan luokitus III–IV, tai joilla oli ollut iskeeminen, ei-lakunaarinen aivohalvaus 1 kuukauden sisällä tai verenvuodosta aiheutuva tai lakunaarinen aivohalvaus milloin tahansa menneisyydessä.
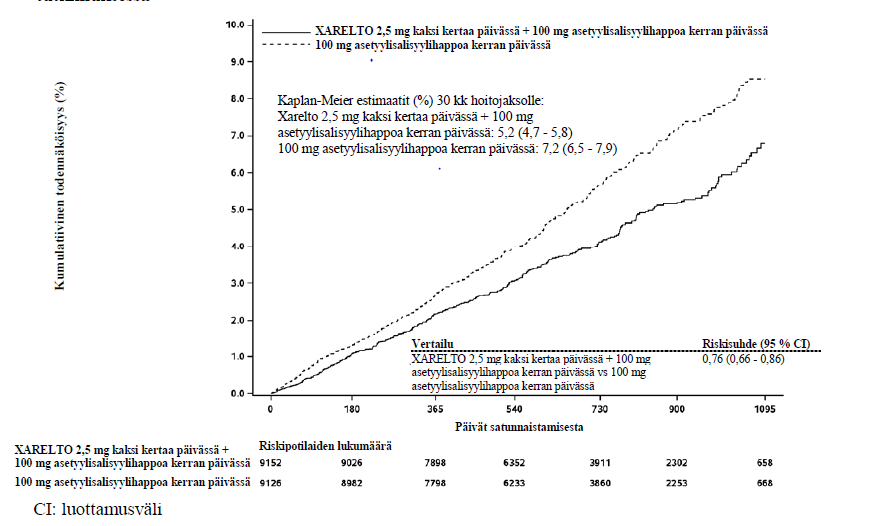
Rivaroksabaani 2,5 mg kaksi kertaa päivässä yhdessä 100 mg asetyylisalisyylihappoa kerran päivässä annettuna osoittautui pelkkää 100 mg asetyylisalisyylihappoa kerran päivässä ‑annostusta paremmaksi ensisijaisen yhdistelmäpäätetapahtuman; kardiovaskulaarisista syistä johtuvan kuoleman, sydäninfarktin sekä aivohalvauksen ehkäisemisessä. (ks. taulukko 7 ja kuva 2).
Turvallisuuden ensisijaisessa päätetapahtumassa (modifioidut ISTH:n merkittävät verenvuototapahtumat) havaittiin merkittävää kasvua potilailla, jotka saivat rivaroksabaani 2,5 mg kaksi kertaa päivässä yhdessä 100 mg asetyylisalisyylihappoa kerran päivässä verrattuna potilaisiin, jotka saivat vain 100 mg asetyylisalisyylihappoa (ks. taulukko 8).
Tehon ensisijaisessa päätetapahtumassa rivaroksabaani 2,5 mg kahdesti päivässä ja 100 mg asetyylisalisyylihappoa kerran päivässä hoito-ohjelman havaittu hyöty pelkän asetyylisalisyylihapon 100 mg:n päivittäisannoksen antamiseen verrattuna oli riskisuhteena ilmaistuna 0,89 (95 % CI 0,7–1,1) ≥ 75 vuotiailla potilailla (esiintyvyys: 6,3 % vs. 7,0 %) ja 0,70 (95 % CI 0,6–0,8) < 75 vuotiailla potilailla (3,6 % vs. 5,0 %). Modifioidun ISTH:n merkittävän verenvuodon kohdalla havaittu riskin kasvu oli riskisuhteena ilmaistuna 2,12 (95 % CI 1,5–3,0) ≥ 75 vuotiailla potilailla (5,2 % vs. 2,5 %) ja 1,53 (95 % CI 1,2–1,9) < 75 vuotiailla potilailla (2,6 % vs. 1,7 %).
Kerran päivässä otetusta 40 mg:n pantopratsoliannoksesta antitromboottiseen tutkimuslääkkeeseen yhdistettynä ei ollut hyötyä, kun tarkasteltiin ruoansulatuskanavan yläosan tapahtumia (yhdistetty päätetapahtuma, johon sisältyi verenvuoto, haavauma, tukos tai perforaatio ruoansulatuskanavan yläosassa) ja niiden ehkäisyä potilailla, joilla ei ollut kliinistä tarvetta käyttää protonipumpun estäjää. Ruoansulatuskanavan yläosan tapahtumien esiintyvyys oli 0,39 sataa potilasvuotta kohti 40 mg pantopratsolia kerran päivässä käyttäneiden ryhmässä ja 0,44 sataa potilasvuotta kohti lumelääkettä kerran päivässä käyttäneiden ryhmässä.
Taulukko 7: Vaiheen III COMPASS-tutkimuksen tehoa koskevat tulokset
Tutkimuspopulaatio | Sepel‑/ääreisvaltimotautia sairastavat potilaat a) | |||||
Hoitoannos | Rivaroksabaani 2,5 mg kahdesti päivässä ja 100 mg asetyylisalisyylihappoa kerran päivässä N=9 152 | 100 mg asetyylisalisyylihappoa kerran päivässä |
| |||
| Potilaat, joilla tapahtumia | KM‑% | Potilaat, joilla tapahtumia | KM‑% | Riskisuhde | p‑arvo b) |
| ||||||
Aivohalvaus, sydäninfarkti tai kardiovaskulaarisista syistä johtuva kuolema | 379 (4,1 %) | 5,20 % | 496 (5,4 %) | 7,17 % | 0,76 | p = 0,00004* |
| 83 (0,9 %) | 1,17 % | 142 (1,6 %) | 2,23 % | 0,58 | p = 0,00006 |
| 178 (1,9 %) | 2,46 % | 205 (2,2 %) | 2,94 % | 0,86 | p = 0,14458 |
| 160 (1,7 %) | 2,19 % | 203 (2,2 %) | 2,88 % | 0,78 | p = 0,02053 |
| ||||||
Kaikki kuolemansyyt | 313 (3.4%) | 4,50 % | 378 (4,1 %) | 5,57 % | 0,82 |
|
Akuutti raajan iskemia | 22 (0,2 %) | 0,27 % | 40 (0,4 %) | 0,60 % | 0,55 |
|
a) hoitoaikeen mukainen analyysijoukko, ensisijaiset analyysit b) verrattuna asetyylisalisyylihappo 100 mg ‑annostukseen; Log-Rank-testin p‑arvo * Ensisijaisen tehon päätetapahtuman alenema oli tilastollisesti korkeampi. CI: luottamusväli; KM‑%: kumulatiivisen esiintyvyysriskin 900 päivän kohdalla lasketut Kaplan-Meier-estimaatit | ||||||
Taulukko 8: Vaiheen III COMPASS-tutkimuksen turvallisuutta koskevat tulokset
Tutkimuspopulaatio | Sepel‑/ääreisvaltimotautia sairastavat potilaat a) | |||
Hoitoannos | Rivaroksabaani 2,5 mg kaksi kertaa päivässä ja 100 mg asetyylisalisyylihap- | 100 mg asetyylisalisyylihappoa kerran päivässä | Riskisuhde (95 % CI)
| |
Modifioitu ISTH:n merkittävä verenvuoto | 288 (3,9 %) | 170 (2,5 %) | 1,70 (1,40; 2,05) | |
| 15 (0,2 %) | 10 (0,2 %) | 1,49 (0,67; 3,33) | |
| 63 (0,9 %) | 49 (0,7 %) | 1,28 (0,88; 1,86) | |
| 10 (0,1 %) | 8 (0,1 %) | 1,24 (0,49; 3,14) | |
| 208 (2,9 %) | 109 (1,6 %) | 1,91 (1,51; 2,41) | |
| 172 (2,3 %) | 90 (1,3 %) | 1,91 (1,48; 2,46) | |
| 36 (0,5 %) | 21 (0,3 %) | 1,70 (0,99; 2,92) | |
merkittävä maha-suolikanavan verenvuoto | 140 (2,0 %) | 65 (1,1 %) | 2,15 (1,60; 2,89) | |
Merkittävä kallonsisäinen verenvuoto | 28 (0,4 %) | 24 (0,3 %) | 1,16 (0,67; 2,00) | |
a) hoitoaikeen mukainen analyysijoukko, ensisijaiset analyysit b) verrattuna asetyylisalisyylihappo 100 mg:aan; Log-Rank-testin p‑arvo CI: luottamusväli; kum. riski: kumulatiivinen esiintyvyysriski 30 kuukauden kohdalla (Kaplan-Meier-estimaatit); ISTH: International Society on Thrombosis and Haemostasis (kansainvälinen tromboosi- ja hemostaasiseura) |
| |||
Kuva 2: Aika tehon ensisijaisen päätetapahtuman (aivohalvaus, sydäninfarkti tai kardiovaskulaarisista syistä johtuva kuolema) ensimmäiseen tapahtumaan COMPASS-tutkimuksessa
Kaplan-Meier estimaatit (%) 30 kk hoitojaksolle: Xarelto 2,5 mg kaksi kertaa päivässä + 100 mg asetyylisalisyylihappoa kerran päivässä: 5,2 (4,7 - 5,8) 100 mg asetyylisalisyylihappoa kerran päivässä: 7,2 (6,5 - 7,9)
Oireisen ääreisvaltimotaudin takia äskettäin tehty alaraajan revaskularisaatiotoimenpide
VOYAGER PAD -tutkimus oli vaiheen III kaksoissokkoutettu avaintutkimus, jossa 6 564 potilasta, joille oli äskettäin tehty onnistuneesti alaraajan revaskularisaatiotoimenpide (kirurginen tai endovaskulaarinen, hybriditoimenpiteet mukaan lukien) oireisen ääreisvaltimotaudin takia, satunnaistettiin suhteessa 1:1 kahteen antitromboottista hoitoa saavaan ryhmään: rivaroksabaania 2,5 mg kaksi kertaa päivässä ja asetyylisalisyylihappoa 100 mg kerran päivässä tai asetyylisalisyylihappoa 100 mg kerran päivässä. Potilaat saivat käyttää lisäksi klopidogreelia vakioannoksena kerran päivässä enintään 6 kuukauden ajan. Tutkimuksen tavoitteena oli osoittaa rivaroksabaanin ja asetyylisalisyylihapon yhdistelmän teho ja turvallisuus sydäninfarktin, iskeemisen aivohalvauksen, kardiovaskulaarisista syistä johtuvan kuoleman, akuutin raajan iskemian tai vaskulaarisita syistä johtuvan suuren amputaation ehkäisyssä potilailla, joille oli äskettäin tehty onnistuneesti alaraajan revaskularisaatiotoimenpide oireisen ääreisvaltimotaudin takia. Tutkimukseen otetut potilaat olivat vähintään 50-vuotiaita ja heillä oli dokumentoitu keskivaikea tai vaikea oireinen alaraajan ateroskleroottinen ääreisvaltimotauti, joka oli todennettu kaikilla seuraavilla tavoilla: kliinisesti (toiminnallisia rajoituksia), anatomisesti (kuvantamistutkimuksissa näyttöä distaalisen tai ulomman lonkkavaltimon ääreisvaltimotaudista) ja hemodynaamisesti (nilkka-olkavarsipainesuhde [ABI] ≤ 0,80 tai varvas-olkavarsipainesuhde [TBI] ≤ 0,60 potilailla, joille ei ollut tehty aiempia raajan revaskularisaatiotoimenpiteitä, tai ABI ≤ 0,85 tai TBI ≤ 0,65 potilailla, joille oli tehty aiemmin jokin raajan revaskularisaatiotoimenpide). Tutkimukseen ei otettu potilaita, jotka tarvitsivat kaksinkertaista antitromboottista hoitoa yli 6 kuukauden ajan tai mitä tahansa muuta antitromboottista hoitoa kuin asetyylisalisyylihappoa ja klopidogreelia, tai oraalista antokoagulanttihoitoa, potilaita, joilla oli aiemmin ollut kallonsisäinen verenvuoto, aivohalvaus tai ohimenevä aivoverenkiertohäiriö, eikä potilaita, joiden eGFR oli < 15 ml/min.
Seuranta kesti keskimäärin 24 kuukautta ja enintään 4,1 vuotta. Tutkimukseen otettujen potilaiden keskimääräinen ikä oli 67 vuotta, ja 17 % potilaista oli yli 75-vuotiaita. Mediaaniaika indeksirevaskularisaatiotoimenpiteen ja tutkimushoidon aloittamisen välillä oli kokonaispopulaatiossa 5 päivää (6 päivää kirurgisen revaskularisaatiotoimenpiteen jälkeen ja 4 päivää endovaskulaarisen revaskularisaatiotoimenpiteen jälkeen, hybriditoimenpiteet mukaan lukien). Yhteensä 53,0 % potilaista sai taustalla lyhytkestoista klopidogreelihoitoa, jonka mediaanikesto oli 31 päivää. Tutkimussuunnitelman mukaan tutkimushoito voitiin aloittaa mahdollisimman pian mutta kuitenkin viimeistään 10 päivää onnistuneen, tutkimukseenottokriteerit täyttävän revaskularisaatiotoimenpiteen jälkeen, kun hemostaasi oli saavutettu.
Hoito rivaroksabaanilla 2,5 mg kaksi kertaa päivässä ja asetyylisalisyylihapolla 100 mg kerran päivässä vähensi pelkkää asetyylisalisyylihappoa paremmin ensisijaista yhdistelmäpäätetapahtumaa eli sydäninfarkteja, iskeemisiä aivohalvauksia, kardiovaskulaarisista syistä johtuvia kuolemia, akuuttia raajan iskemiaa ja vaskulaarisista syistä johtuvia suuria amputaatioita (ks. taulukko 9). Ensisijaisen turvallisuuspäätetapahtuman eli TIMI:n mukaisten merkittävien verenvuototapahtumien yleisyys oli suurempi rivaroksabaania ja asetyylisalisyylihappoa saaneilla potilailla, mutta kuolemaan johtaneiden verenvuotojen ja kallonsisäisten verenvuotojen yleisyys ei lisääntynyt (ks. taulukko 10).
Toissijaiset tehon päätetapahtumat testattiin ennalta määrätyssä hierarkkisessa järjestyksessä (ks. taulukko 9).
Taulukko 9: Vaiheen III VOYAGER PAD -tutkimuksen tehoa koskevat tulokset
| Tutkimuspopulaatio | Potilaat, joille oli oireisen ääreisvaltimotaudin takia tehty äskettäin alaraajan revaskularisaatiotoimenpidea) | ||
| Hoitoannos | Rivaroksabaani 2,5 mg kaksi kertaa päivässä ja 100 mg asetyylisalisyylihap- poa kerran päivässä, N = 3 286 n (kum. riski‑%)c) | 100 mg asetyylisalisyylihappoa kerran päivässä n (kum. riski‑%)c) | Riskisuhde (95 % CI)d) |
| Ensisijainen tehon päätetapahtumab) | 508 (15,5 %) | 584 (17,8 %) | 0,85 (0,76; 0,96) p = 0,0043e)* |
- sydäninfarkti | 131 (4,0 %) | 148 (4,5 %) | 0,88 (0,70; 1,12) |
- iskeeminen aivohalvaus | 71 (2,2 %) | 82 (2,5 %) | 0,87 (0,63; 1,19) |
- kardiovaskulaarisista syistä johtuva kuolema | 199 (6,1 %) | 174 (5,3 %) | 1,14 (0,93; 1,40) |
- akuutti raajan iskemiaf) | 155 (4,7 %) | 227 (6,9 %) | 0,67 (0,55; 0,82) |
- vaskulaarisista syistä johtuva suuri amputaatio | 103 (3,1 %) | 115 (3,5 %) | 0,89 (0,68; 1,16) |
| Toissijainen tehon päätetapahtuma | |||
Suunnittelematon indeksiraajan revaskularisaatio raajan iskemian uusiutumisen takia | 584 (17,8 %) | 655 (20,0 %) | 0,88 (0,79; 0,99) p = 0,0140e)* |
Sairaalahoito sepelvaltimon tai ääreisvaltimon tromboosin takia (kumpi tahansa alaraaja) | 262 (8,0 %) | 356 (10,9 %) | 0,72 (0,62; 0,85) p < 0,0001e)* |
Kaikki kuolemansyyt | 321 (9,8 %) | 297 (9,1 %) | 1,08 (0,92; 1,27) |
Laskimotromboemboliatapahtumat | 25 (0,8 %) | 41 (1,3 %) | 0,61 (0,37; 1,00) |
a) Hoitoaikeen mukainen analyysijoukko, ensisijaiset analyysit; ICAC:n arvioima.
b) Yhdistelmä seuraavista: sydäninfarkti, iskeeminen aivohalvaus, kardiovaskulaarisista syistä johtuva kuolema (kardiovaskulaarisista syistä johtuva kuolema ja tuntematon kuolemansyy), akuutti raajan iskemia ja vaskulaarisista syistä johtuva suuri amputaatio.
c) Vain analysoitavan päätetapahtuman ensimmäinen esiintymiskerta otettiin huomioon kyseisen tutkittavan tiedoissa.
d) Riskisuhde (95 % CI) perustuu Coxin suhteellisen vaaran malliin, jossa osituksen ainoana kovariaattina on käytetty toimenpiteen tyyppiä ja klopidogreelin käyttöä hoidon yhteydessä.
e) Yksitahoinen p-arvo perustuu log-rank-testiin, jossa osituksen faktorina on käytetty toimenpiteen tyyppiä ja klopidogreelin käyttöä hoidon yhteydessä.
f) Akuutti raajan iskemia määritellään raajaperfuusion äkilliseksi ja merkittäväksi huononemiseksi, johon joko liittyy pulssivajauksen kehittyminen tai hoitotoimenpiteen tarve (eli trombolyysin, trombektomian tai kiireellisen revaskularisaation tarve) ja joka vaatii sairaalahoitoa.
* Tilastollisesti suurempi tehon päätetapahtuman vähenemä.
CI: luottamusväli; ICAC: Independent Clinical Adjudication Committee (kansainvälinen kliininen arviointilautakunta)
Taulukko 10: Vaiheen III VOYAGER PAD -tutkimuksen turvallisuutta koskevat tulokset
| Tutkimuspopulaatio | Potilaat, joille oli oireisen ääreisvaltimotaudin takia tehty äskettäin alaraajan revaskularisaatiotoimenpidea) | ||
| Hoitoannos | Rivaroksabaani 2,5 mg kaksi kertaa päivässä ja 100 mg asetyylisalisyylihap- poa kerran päivässä, N = 3 256 n (kum. riski‑%)b) | 100 mg asetyylisalisyylihappoa kerran päivässä n (kum. riski‑%)b) | Riskisuhde (95 % CI)c)
p-arvod) |
TIMI:n mukaiset merkittävät verenvuodot (CABG/ei-CABG) | 62 (1,9 %) | 44 (1,4 %) | 1,43 (0,97; 2,10) p = 0,0695 |
- kuolemaan johtanut verenvuoto | 6 (0,2 %) | 6 (0,2 %) | 1,02 (0,33; 3,15) |
- kallonsisäinen verenvuoto | 13 (0,4 %) | 17 (0,5 %) | 0,78 (0,38; 1,61) |
- runsas verenvuoto, johon liittyi Hb-arvon lasku ≥ 5g/dl / hematokriitin lasku ≥ 15 % | 46 (1,4 %) | 24 (0,7 %) | 1,94 (1,18; 3,17) |
| ISTH:n merkittävä verenvuoto | 140 (4,3 %) | 100 (3,1 %) | 1,42 (1,10; 1,84) p = 0,0068 |
- kuolemaan johtanut verenvuoto | 6 (0,2 %) | 8 (0,2 %) | 0,76 (0,26; 2,19) |
- ei kuolemaan johtanut verenvuoto kriittisessä elimessä | 29 (0,9 %) | 26 (0,8 %) | 1,14 (0,67; 1,93) |
| ISTH:n kliinisesti relevantti ei-merkittävä verenvuoto | 246 (7,6 %) | 139 (4,3 %) | 1,81 (1,47; 2,23) |
a) Turvallisuusanalyysijoukko (kaikki satunnaistetut tutkittavat, jotka saivat vähintään yhden annoksen tutkimuslääkettä), ICAC: Independent Clinical Adjudication Committee (kansainvälinen kliininen arviointilautakunta).
b) n = tutkittavat, joilla esiintyi tapahtumia, N = tutkittavat, joita riski koski, % = 100 * n/N, n/100 potilasvuotta = tapahtumia saaneiden tutkittavien ja kumulatiivisen riskin keston suhde.
c) Riskisuhde (95 % CI) perustuu Coxin suhteellisen vaaran malliin, jossa osituksen ainoana kovariaattina on käytetty toimenpiteen tyyppiä ja klopidogreelin käyttöä hoidon yhteydessä.
d) Kaksitahoinen p-arvo perustuu log-rank-testiin, jossa osituksen faktorina on käytetty toimenpiteen tyyppiä ja klopidogreelin käyttöä hoidon yhteydessä.
Sepelvaltimotauti, johon liittyy sydämen vajaatoiminta
COMMANDER HF ‑tutkimuksessa oli mukana 5 022 potilasta, joilla oli sydämen vajaatoiminta ja merkittävä sepelvaltimotauti, ja jotka olivat olleet sairaalahoidossa pahentuneen sydämen vajaatoiminnan vuoksi. Potilaat satunnaistettiin kahteen hoitoryhmään: rivaroksabaani 2,5 mg kaksi kertaa päivässä (N = 2 507) tai kaltaistettu lumelääke (N = 2 515). Tutkimushoidon kokonaiskeston mediaani oli 504 päivää.
Potilailla edellytettiin olleen oireinen sydämen vajaatoiminta vähintään 3 kuukauden ajan ja vasemman kammion ejektiofraktio (LVEF) ≤ 40 % tutkimukseen mukaan tuloa edeltävän vuoden aikana. Lähtötilanteessa ejektiofraktion mediaani oli 34 % (kvartiilivälin pituus: 28–38 %), ja 53 %:lla tutkittavista NYHA-luokka oli III tai IV.
Tehon ensisijainen analyysi (yhdistetty päätetapahtuma, johon sisältyi kokonaiskuolleisuus, sydäninfarkti tai aivohalvaus) ei osoittanut tilastollisesti merkitsevää eroa 2,5 mg rivaroksabaania kaksi kertaa päivässä saaneen ryhmän ja lumelääkettä saaneen ryhmän välillä (HR 0,94; 95 %:n luottamusväli 0,84–1,05; p = 0,270). Kokonaiskuolleisuuden osalta rivaroksabaanin ja lumelääkkeen välillä ei ollut eroa tapahtumien lukumäärässä (tapahtumien määrä 100 potilasvuotta kohden 11,41 vs. 11,63; HR 0,98; 95 %:n luottamusväli: 0,87–1,10; p = 0,743). Sydäninfarktin osalta tapahtumien lukumäärä 100 potilasvuotta kohden (rivaroksabaani vs. lumelääke) oli 2,08 vs. 2,52 (HR 0,83; 95 %:n luottamusväli: 0,63–1,08; p = 0,165), ja aivohalvauksen osalta tapahtumien lukumäärä 100 potilasvuotta kohden oli 1,08 vs. 1,62 (HR 0,66; 95 %:n luottamusväli: 0,47–0,95; p = 0,023). Turvallisuutta koskeva ensisijainen yhdistetty päätetapahtuma (eli kuolemaan johtaneet verenvuodot tai verenvuodot kriittiseen tilaan, joihin liittyi pysyvän vammautumisen mahdollisuus) todettiin 18 (0,7 %) potilaalla 2,5 mg rivaroksabaania kaksi kertaa päivässä saaneessa ryhmässä ja 23 (0,9 %) potilaalla lumelääkettä saaneessa ryhmässä (HR 0,80; 95 %:n luottamusväli 0,43–1,49; p = 0,484). ISTH:n merkittävät verenvuototapahtumat lisääntyivät tilastollisesti merkitsevästi rivaroksabaaniryhmässä verrattuna lumeryhmään (tapahtumien lukumäärä 100 potilasvuotta kohden: 2,04 vs. 1,21, HR 1,68; 95 %:n luottamusväli: 1,18–2,39; p = 0,003).
COMPASS-tutkimuksessa lievää ja keskivaikeaa sydämen vajaatoimintaa sairastavien potilaiden alaryhmässä hoidon vaikutus oli samankaltainen kuin koko tutkimuspotilasjoukossa (ks. kohta Sepel-/ääreisvaltimotauti).
Potilaat, joilla on suuririskinen fosfolipidivasta-aineoireyhtymä, jossa kaikki kolme vasta-ainetestiä ovat positiiviset
Tutkijalähtöisessä, satunnaistetussa, avoimessa monikeskustutkimuksessa, jossa käytettiin sokkoutettua päätetapahtumien arviointia, rivaroksabaania verrattiin varfariiniin fosfolipidivasta-aineoireyhtymää sairastavilla potilailla, joilla oli ollut verisuonitukos ja joilla oli korkea tromboembolisten tapahtumien riski (positiivinen tulos kaikissa kolmessa fosfolipidivasta-ainetestissä: lupusantikoagulantti, kardiolipiinivasta-aineet ja beeta-2-glykoproteiini I -vasta-aineet). Tutkimukseen osallistui 120 potilasta, ja se keskeytettiin ennenaikaisesti, koska rivaroksabaania saaneilla potilailla oli enemmän tapahtumia. Seuranta kesti keskimäärin 569 päivää. 59:lle satunnaistetulle potilaalle annettiin 20 mg rivaroksabaania (15 mg potilaille, joilla kreatiniinipuhdistuma oli < 50 ml/min), ja 61:lle potilaalle annettiin varfariinia (INR 2,0–3,0). Rivaroksabaaniryhmään satunnaistetuista potilaista 12 %:lle ilmeni tromboembolinen tapahtuma (4 iskeemistä aivohalvausta ja 3 sepelvaltimotukosta). Varfariiniryhmään satunnaistetuilla potilailla ei todettu päätetapahtumia. Merkittävää verenvuotoa esiintyi neljällä (7 %) rivaroksabaaniryhmän potilaalla ja kahdella ( %) varfariiniryhmän potilaalla.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteelle toimittaa tutkimustulokset Xarelto-valmisteen käytöstä kaikkien pediatristen potilasryhmien hoidossa laskimotukoksen ehkäisyssä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Rivaroksabaani imeytyy nopeasti ja sen huippupitoisuus (Cmax) saavutetaan 2–4 tunnin kuluttua tabletin ottamisesta.
Suun kautta otettu rivaroksabaani imeytyy lähes täydellisesti, ja biologinen hyötyosuus suun kautta otettuna on suuri (80–100 %) tablettiannoksen ollessa 2,5 mg ja10 mg riippumatta siitä, onko ihminen paastonnut tai ruokaillut. Ottaminen ruoan kanssa ei vaikuta rivaroksabaanin AUC- ja Cmax-arvoihin annoksen ollessa 2,5 mg ja 10 mg. Rivaroksabaani 2,5 mg ja 10 mg tabletit voidaan ottaa ruoan kanssa tai ilman.
Rivaroksabaanin farmakokinetiikka on likimain lineaarinen noin 15 mg kerran päivässä annokseen saakka. Suurempana annoksena rivaroksabaanin liukeneminen rajoittaa imeytymistä johtaen pienempään biologiseen hyötyosuuteen. Imeytymisnopeus on pienempi suuremmalla annoksella. Tämä on merkittävämpää paastotilassa kuin ravitussa tilassa. Vaihtelevuus rivaroksabaanin farmakokinetiikassa on kohtalaista yksilöiden välisen variaation (CV %) ollessa 30–40 %.
Rivaroksabaanin imeytyminen riippuu sen vapautumiskohdasta ruoansulatuskanavassa. Annettaessa rivaroksabaanirakeita pohjukaissuoleen raportoitiin 29 %:n lasku AUC-arvossa ja 56 %:n lasku Cmax-arvossa verrattuna tablettien käyttöön. Altistus laskee vielä enemmän rivaroksabaanin vapautuessa ileumissa tai nousevassa paksusuolessa. Näin ollen on vältettävä rivaroksabaanin antamista mahalaukusta distaalisesti, koska se voi heikentää imeytymistä ja alentaa siten rivaroksabaanialtistusta.
Kokonaiseen tablettiin verrattava biologinen hyötyosuus (AUC ja Cmax) saavutettiin antamalla 20 mg rivaroksabaania suun kautta joko murskaamalla tabletti ja sekoittamalla se omenasoseeseen tai antamalla veteen sekoitettuna suspensiona mahaletkun kautta ja antamalla sen jälkeen nestemäinen ateria. Koska rivaroksabaanin farmakokineettinen profiili on ennustettavissa ja se on suhteessa annokseen, tämän tutkimuksen biologista hyötyosuutta koskevat tulokset ovat oletettavasti sovellettavissa alhaisempiin rivaroksabaaniannoksiin.
Jakautuminen
Ihmisellä sitoutuminen plasman proteiineihin on voimakasta, noin 92–95 %, seerumin albumiinin ollessa tärkein sitova komponentti. Jakautumistilavuus on kohtalainen Vss-arvon ollessa noin 50 litraa.
Biotransformaatio ja eliminaatio
Annetusta rivaroksabaaniannoksesta noin 2/3 eliminoituu metaboloitumalla niin, että puolet metaboliiteista eliminoituu munuaisten kautta ja puolet ulosteiden kautta. 1/3 annetusta annoksesta erittyy muuttumattomana vaikuttavana aineena suoraan virtsaan pääasiassa aktiivisen munuaiserityksen kautta.
Rivaroksabaani metaboloituu CYP3A4:n, CYP2J2:n ja CYP-entsyymeistä riippumattomien mekanismien kautta. Morfolinonirakenteen oksidatiivinen degradaatio ja aminosidosten hydrolyysi ovat keskeiset biotransformaation kohteet. In vitro -tutkimuksiin perustuen rivaroksabaani on kuljettajaproteiinien P-gp (P-glykoproteiini) ja Bcrp (breast cancer resistance protein) substraatti.
Rivaroksabaani esiintyy ihmisen plasmassa pääasiassa muuttumattomana yhdisteenä ilman merkittäviä tai aktiivisia metaboliitteja. Rivaroksabaanin systeeminen puhdistuma on noin 10 l/h, minkä vuoksi se voidaan luokitella aineeksi, jolla on vähäinen puhdistuma. Laskimonsisäisesti annetun 1 mg:n annoksen jälkeen eliminaation puoliintumisaika on noin 4,5 tuntia. Suun kautta annon jälkeen eliminaatio muuttuu imeytymisrajoitetuksi. Rivaroksabaanin eliminoitumisen terminaalinen puoliintumisaika plasmasta on 5–9 tuntia nuorilla henkilöillä ja 11–13 tuntia vanhemmilla henkilöillä.
Erityisryhmät
Sukupuoli
Mies- ja naispotilailla ei ollut kliinisesti merkittäviä eroja farmakokineettisissä ja farmakodynaamisissa ominaisuuksissa.
Iäkkäät potilaat
Ikääntyneillä potilailla oli suurempi plasmapitoisuus kuin nuoremmilla, ja keskimääräiset AUC-arvot olivat noin 1,5 kertaa suurempia pääasiassa vähentyneen (näennäisen) kokonais- ja munuaispuhdistuman vuoksi. Annoksen sovittaminen ei ole tarpeen.
Eri painoryhmät
Erittäin pienellä tai suurella kehon painolla (< 50 kg tai > 120 kg) oli rivaroksabaanin pitoisuuteen plasmassa vain pieni vaikutus (alle 25 %). Annoksen sovittaminen ei ole tarpeen.
Etnisten ryhmien väliset erot
Rivaroksabaanin farmakokineettisissä ja farmakodynaamisissa ominaisuuksissa ei todettu kliinisesti merkittäviä etnisten ryhmien välisiä eroja kaukaasialaisissa, afroamerikkalaisissa, latinalaisamerikkalaisissa, japanilaisissa tai kiinalaisissa potilaissa.
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa sairastavilla kirroosipotilailla (Child Pugh -luokka A) todettiin vain vähäisiä rivaroksabaanin farmakokinetiikan muutoksia (rivaroksabaanin AUC-arvo lisääntyi keskimäärin 1,2-kertaiseksi), mikä on lähes verrannollinen terveiden vapaaehtoisten verrokkiryhmään. Kohtalaista maksan vajaatoimintaa sairastavilla kirroosipotilailla (Child Pugh -luokka B) rivaroksabaanin AUC-arvo lisääntyi huomattavasti 2,3-kertaiseksi terveisiin vapaaehtoisiin verrattuna. Sitoutumaton AUC-arvo lisääntyi 2,6-kertaiseksi. Näillä potilailla rivaroksabaania myös eliminoitui vähemmän munuaisten kautta, mikä oli samankaltaista kohtalaista munuaisten vajaatoimintaa sairastavien potilaiden kanssa. Vakavaa maksan vajaatoimintaa sairastavista potilaista ei ole tietoja.
Kohtalaista maksan vajaatoimintaa sairastavilla potilailla tekijä Xa:n vaikutuksen estyminen lisääntyi 2,6-kertaiseksi terveisiin vapaaehtoisiin verrattuna; PT pidentyi vastaavasti 2,1-kertaiseksi. Kohtalaista maksan vajaatoimintaa sairastavat potilaat olivat herkempiä rivaroksabaanille, mikä johti jyrkempään PK/PD-suhteeseen pitoisuuden ja PT:n välillä.
Rivaroksabaani on vasta-aiheinen potilailla, joiden maksasairauteen liittyy hyytymishäiriö ja kliinisesti merkittävä verenvuotoriski mukaan lukien Child Pugh -luokkien B ja C kirroosipotilaat (ks. kohta Vasta-aiheet).
Munuaisten vajaatoiminta
Kreatiniinipuhdistuman mittauksiin perustuvien arvioiden mukaan rivaroksabaanialtistuksen lisääntyminen korreloi munuaistoiminnan heikentymiseen. Lievää (kreatiniinipuhdistuma 50–80 ml/min), kohtalaista (kreatiniinipuhdistuma 30–49 ml/min) ja vakavaa (kreatiniinipuhdistuma 15–29 ml/min) munuaisten vajaatoimintaa sairastavilla henkilöillä rivaroksabaanipitoisuus plasmassa (AUC) kasvoi 1,4-, 1,5- ja 1,6-kertaiseksi. Farmakodynaamisten vaikutusten vastaavat lisäykset olivat suuremmat. Lievää, kohtalaista ja vakavaa munuaisten vajaatoimintaa sairastavilla henkilöillä tekijä Xa:n vaikutuksen kokonaisestyminen lisääntyi kertoimella 1,5, 1,9 ja 2,0 terveisiin vapaaehtoisiin verrattuna. PT:n pidentyminen lisääntyi samoin kertoimella 1,3, 2,2 ja 2,4. Tietoa potilaista, joiden kreatiniinipuhdistuma on < 15 ml/min, ei ole.
Koska rivaroksabaani sitoutuu voimakkaasti plasman proteiineihin, sen ei oleteta olevan dialysoitavissa.
Käyttöä ei suositella potilaille, joiden kreatiniinipuhdistuma on < 15 ml/min. Rivaroksabaania tulee käyttää harkiten potilaille, joiden kreatiniinipuhdistuma on 15–29 ml/min (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Farmakokineettiset tiedot potilailla
Potilailla, joilla on sepelvaltimotauti ja jotka ovat saaneet rivaroksabaania aterotromboottisten tapahtumien ehkäisyyn annoksella 2,5 mg kahdesti päivässä, geometrinen keskikonsentraatio (90 % ennusteväli) 2–4 tuntia ja noin 12 tuntia annostelun jälkeen (vastaten karkeasti annosvälin maksimi- ja minimikonsentraatioita) oli 47 (13–123) ja 9,2 (4,4–18) mikrog/l.
Farmakokineettiset/farmakodynaamiset suhteet
Farmakokinetiikan/farmakodynamiikan (PK/PD) suhdetta plasman rivaroksabaanipitoisuuden ja useiden PD-päätepisteiden (tekijä Xa:n estyminen, PT, aPTT, Heptest) välillä on arvioitu useiden eri annosten (5–30 mg kahdesti päivässä) annon jälkeen. Rivaroksabaanin pitoisuuden ja tekijä Xa:n vaikutuksen suhdetta kuvattiin parhaiten Emax-mallilla. PT:n osalta lineaarinen leikkauspistemalli yleensä kuvasi tuloksia paremmin. Käytetyistä eri PT-reagensseista riippuen kulmakerroin vaihteli huomattavasti. Kun käytettiin Neoplastin PT:ta, lähtötason PT oli noin 13 s, ja kulmakerroin oli noin 3–4 s/(100 mikrog/l). PK/PD-analyysien tulokset vaiheessa II ja III olivat yhdenmukaiset terveiltä henkilöiltä saatujen tietojen kanssa.
Pediatriset potilaat
Turvallisuutta ja tehoa lasten ja enintään 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu akuutin sepelvaltimotautikohtauksen ja sepel-/ääreisvaltimotaudin käyttöaiheissa.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, yksittäisen altistuksen aiheuttamaa toksisuutta, fototoksisuutta, geenitoksisuutta, karsinogeenistä potentiaalia ja juveniilitoksisuutta koskevien tavanomaisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Toistuvan annoksen toksisuutta koskevissa tutkimuksissa havaitut vaikutukset johtuivat pääasiassa rivaroksabaanin liiallisesta farmakodynaamisesta vaikutuksesta. Rotilla todettiin kohonneita IgG- ja IgA-plasmatasoja kliinisesti merkittävällä altistumistasolla.
Rotilla ei havaittu vaikutuksia fertiliteettiin uros- tai naarasrotilla. Eläintutkimuksissa todettiin lisääntymistoksisuutta liittyen rivaroksabaanin farmakologiseen vaikutusmekanismiin (esim. verenvuotokomplikaatioita). Alkion ja sikiön toksisuutta (postimplantaation menetys, hidastunut/edistynyt luutuminen, multippelit vaaleanväriset läikät maksassa) ja yleisten epämuodostuminen lisääntynyttä esiintymistä sekä istukan muutoksia havaittiin kliinisesti merkittävissä plasmapitoisuuksissa. Rotilla tehdyssä pre- ja postnataalitutkimuksessa havaittiin jälkeläisten elinkyvyn heikkenemistä annoksilla, jotka olivat toksisia emoille.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin: mikrokiteinen selluloosa, kroskarmelloosinatrium, laktoosimonohydraatti, hypromelloosi (2910), natriumlauryylisulfaatti, magnesiumstearaatti.
Kalvopäällyste: makrogoli (3350), hypromelloosi (2910), titaanidioksidi (E 171), keltainen rautaoksidi (E 172).
Yhteensopimattomuudet
Ei oleellinen
Kestoaika
3 vuotta
Murskatut tabletit
Murskatut rivaroksabaanitabletit ovat stabiileja vedessä ja omenasoseessa enintään 4 tunnin ajan.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XARELTO tabletti, kalvopäällysteinen
2,5 mg (L:kyllä) 56 fol (38,64 €), 196 fol (125,24 €)
PF-selosteen tieto
PP/alumiiniläpipainopakkaukset 56, 168 kalvopäällystetyn tabletin pahvipakkauksissa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Vaaleankeltaisia, pyöreitä, kaksoiskuperia tabletteja (halkaisija 6 mm, kaarteen säde 9 mm), joiden toisella puolella on BAYER-risti ja toisella puolella ”2,5” ja kolmio.
Käyttö- ja käsittelyohjeet
Tablettien murskaaminen
Rivaroksabaanitabletit voidaan murskata ja suspendoida 50 ml:aan vettä ja antaa nenämahaletkun tai mahaletkun kautta. Ennen valmisteen antamista on tarkistettava letkun oikea sijainti mahassa. Valmisteen antamisen jälkeen letku on huuhdeltava vedellä. Rivaroksabaanin imeytyminen riippuu vaikuttavan aineen vapautumiskohdasta, joten rivaroksabaanin antamista mahalaukusta distaalisesti on vältettävä, koska se voi heikentää imeytymistä ja alentaa siten altistusta vaikuttavalle aineelle. Enteraalista ravintoa ei tarvita välittömästi 2,5 mg:n tablettien antamisen jälkeen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
XARELTO tabletti, kalvopäällysteinen
2,5 mg 56 fol, 196 fol
- Alempi erityiskorvaus (65 %). Krooninen sepelvaltimotauti ja krooniseen sepelvaltimotautiin liittyvä rasva-aineenvaihdunnan häiriö (206).
- Peruskorvaus (40 %).
ATC-koodi
B01AF01
Valmisteyhteenvedon muuttamispäivämäärä
27.06.2024
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi