
HIZENTRA injektioneste, liuos 200 mg/ml, injektioneste, liuos, esitäytetty ruisku 200 mg/ml
Vaikuttavat aineet ja niiden määrät
Ihmisen normaali immunoglobuliini (immunoglobulinum humanum normale) (SCIg)
Yksi millilitra sisältää:
Ihmisen normaalia immunoglobuliinia 200 mg
(puhtaus: vähintään 98 % on tyypin G immunoglobuliinia [IgG])
Injektiopullot
Jokainen 5 ml:n injektiopullo liuosta sisältää: 1 g:n ihmisen normaalia immunoglobuliinia
Jokainen 10 ml:n injektiopullo liuosta sisältää: 2 g ihmisen normaalia immunoglobuliinia
Jokainen 20 ml:n injektiopullo liuosta sisältää: 4 g ihmisen normaalia immunoglobuliinia
Jokainen 50 ml:n injektiopullo liuosta sisältää: 10 g normaalia immunoglobuliinia
Esitäytetyt ruiskut
Jokainen esitäytetty 5 ml:n ruisku liuosta sisältää: 1 g:n ihmisen normaalia immunoglobuliinia
Jokainen esitäytetty 10 ml:n ruisku liuosta sisältää: 2 g ihmisen normaalia immunoglobuliinia
Jokainen esitäytetty 20 ml:n ruisku liuosta sisältää: 4 g ihmisen normaalia immunoglobuliinia
Jokainen esitäytetty 50 ml:n ruisku liuosta sisältää: 10 g ihmisen normaalia immunoglobuliinia
IgG:n alaluokkien jakauma (likimääräiset osuudet):
IgG1 69 %
IgG2 26 %
IgG3 3 %
IgG4 2 %
Suurin IgA-pitoisuus on 50 mikrogrammaa/ml.
Valmistettu luovutetusta ihmisen plasmasta.
Apuaineet, joiden vaikutus tunnetaan
Hizentra sisältää noin 250 (210−290) mmol/l L-proliinia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste ihonalaiseen käyttöön, liuos.
Kliiniset tiedot
Käyttöaiheet
Korvaushoito aikuisille ja 0−18-vuotiaille lapsille ja nuorille seuraavissa tilanteissa:
- Primaari immuunipuutosoireyhtymä, johon liittyy heikentynyt vasta-ainemuodostus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
- Sekundaariset immuunipuutokset potilailla, joilla on vaikea-asteisia tai toistuvia infektioita, joille annettu mikrobilääkehoito ei tehoa ja joilla on joko todennettu spesifin vasta-aineen toimimattomuus (proven specific antibody failure, PSAF)* tai seerumin IgG-pitoisuus < 4 g/l.
*PSAF = IgG-vasta-ainetitteri pneumokokkipolysakkaridi- ja polypeptidiantigeenirokotteita vastaan ei nouse vähintään kaksinkertaiseksi.
Immunomodulatorinen hoito aikuisille ja 0−18-vuotiaille lapsille ja nuorille:
- Hizentra on tarkoitettu kroonista inflammatorista demyelinoivaa polyneuropatiaa (CIDP) sairastavien potilaiden ylläpitohoitoon, kun tila on vakautettu IVIg-hoidolla.
Ehto
Hoito on aloitettava ja sitä on seurattava immuunipuutostilojen hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
Annos ja annostusohjelma määräytyvät käyttöaiheen mukaan.
Hoito on aloitettava ja sitä on seurattava immuunipuutostilojen tai CIDP:n SCIg-hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus
Aikuiset ja 0–18-vuotiaat lapset
Korvaushoito
Lääkevalmiste annetaan ihon alle.
Korvaushoidossa annos saattaa olla tarpeen määritellä kullekin potilaalle yksilöllisesti kliinisen vasteen sekä seerumin pienimmän IgG-pitoisuuden mukaan. Seuraavat annostusohjeet ovat suuntaa-antavia.
Annostus toteutetaan siten, että IgG-minimipitoisuus (mitattuna ennen seuraavaa infuusiota) on vähintään 6 g/l tai potilasjoukon iänmukaisten normaalien viitearvojen puitteissa. Latausannos vähintään 0,2–0,5 g/kg (1,0–2,5 ml/kg) painokiloa kohti voi olla tarpeen. Tämä voi olla tarpeen jakaa usealle päivälle. Kun vakaan tilan IgG-pitoisuus on saavutettu, ylläpitoannoksia annetaan toistuvasti, jotta saavutetaan kumulatiivinen kuukausittainen annos, joka on suuruusluokkaa 0,4–0,8 g/kg (2,0–4,0 ml/kg) painokiloa kohti. Voi olla tarpeen injisoida jokainen kerta-annos eri anatomiseen kohtaan.
Minimipitoisuudet on mitattava ja arvioitava potilaan kliinisen vasteen mukaisesti. Annoksen ja/tai antovälin säätämistä voidaan harkita kliinisen vasteen perusteella (esim. infektiotaajuus) suuremman minimipitoisuuden saavuttamiseksi.
CIDP:n immunomodulatorinen hoito
Hizentra-hoito aloitetaan viikon kuluttua viimeisen IVIg-infuusion antamisesta. Suositeltu ihon alle annettava annos on 0,2–0,4 g/painokilo viikossa annettuna yhdellä tai kahdella kerralla yhtenä tai kahtena perättäisenä päivänä. Ihon alle annettava aloitusannos voidaan muuntaa aiemmasta IVIg-annoksesta (viikoittaisena annoksena laskettuna) suhteessa 1:1.
Esimerkki: IVIg-annos 1 g/kg kerran kolmessa viikossa on muunnettuna viikoittainen Hizentra-annos 0,33 g/kg.
Viikoittainen annos voidaan jakaa pienempiin annoksiin haluttujen antokertojen määrän mukaisesti. Hizentra-annos kaksinkertaistetaan, jos annos annetaan kahden viikon välein.
Annosta voi olla tarpeen säätää halutun kliinisen vasteen saavuttamiseksi. Potilaan yksilöllinen kliininen vaste on pääasiallinen annoksen säätämiskriteeri. Jos kliininen tila huononee, annos voidaan suurentaa suurimpaan suositeltuun viikoittaiseen annokseen 0,4 g/kg. Hizentralla tapahtuvaa CIDP-potilaiden ylläpitohoitoa on tutkittu enintään 18 kuukauden ajan. Potilaan vaste ja osoitettu jatkohoidon tarve määrittävät sen, voiko hoitoa yksilöllisesti jatkaa pidempään kuin 18 kuukautta.
Hizentran teho lumelääkkeeseen verrattuna on osoitettu laskimoon annettavista immunoglobuliineista (IVIg) siirtymisen jälkeen. Hizentraa ja IVIg-hoitoa suoraan vertailevia tietoja ei ole saatavissa. Ks. myös kohta Farmakodynamiikka.
Pediatriset potilaat
Annostus lapsille ja nuorille (0–18 vuotta) ei eroa aikuisten annostuksesta, koska käyttöaihekohtainen annostus ilmoitetaan suhteessa painoon ja sitä säädetään kliinisen hoitotuloksen mukaan korvaushoidon käyttöaiheissa.
Hizentraa arvioitiin 68:lla iältään 2 – < 12‑vuotiaalla pediatrisella tutkimushenkilöllä ja 57:llä iältään 12 – < 18-vuotiaalla nuorella, joilla oli primaari immuunipuutos (PID). Erityiset pediatriset annosvaatimukset eivät olleet tarpeen halutun seerumin IgG-pitoisuuden saavuttamiseksi. Hizentraa ei ole arvioitu pediatrisilla potilailla tehdyissä kliinisissä tutkimuksissa alle 18-vuotiailla CIDP-potilailla.
Iäkkäät
Koska annos ilmoitetaan suhteessa painoon ja sitä säädetään kliinisen hoitotuloksen mukaan edellä mainituin edellytyksin, iäkkäille annettavan annoksen ei katsota eroavan 18–65-vuotiaiden annostuksesta.
Hizentraa arvioitiin kliinisissä tutkimuksissa 13:llä yli 65‑vuotiaalla tutkimushenkilöllä, joilla oli PID; erityiset annosmuutokset eivät olleet tarpeen halutun seerumin IgG-pitoisuuden saavuttamiseksi.
Hizentraa arvioitiin kliinisissä tutkimuksissa 61:llä yli 65‑vuotiaalla tutkimushenkilöllä, joilla oli CIDP; erityiset annosmuutokset eivät olleet tarpeen halutun kliinisen hoitotuloksen saavuttamiseksi.
Antotapa
Vain ihon alle.
Kotihoito
Kotihoidossa ihon alle annettavan infuusiohoidon saa aloittaa ja sitä saa valvoa vain terveydenhuoltohenkilö, jolla on kokemusta potilaiden kotihoidon ohjauksesta. Terveydenhuollon ammattilaisen on valittava sopiva infuusiomenetelmä (laiteavusteinen infuusio tai manuaalinen bolusinjektio) potilaan sairauden yksilöllisen tilanteen sekä mieltymysten perusteella. Infuusiohoidossa voidaan käyttää immunoglobuliinien ihonalaiseen annosteluun soveltuvia infuusiolaitteita. Infuusiolaitteiden käyttö, hoitopäiväkirjan täyttäminen, vaikeiden haittavaikutusten tunnistaminen ja toimenpiteet niiden yhteydessä on neuvottava ja opetettava potilaalle tai häntä hoitavalle henkilölle.
Hizentra voidaan infusoida esim. vatsaan, reiteen, olkavarteen ja/tai lonkan sivuun.
Samanaikaisesti voidaan käyttää useampaa kuin yhtä infuusiolaitetta. Kuhunkin kohtaan infusoitavan valmisteen määrä voi vaihdella. Imeväisillä ja lapsilla infuusiokohtaa voidaan vaihtaa 5–15 ml:n välein. Aikuisille voidaan antaa enintään 50 ml/kohta. Infuusiokohtien määrälle ei ole ylärajaa. Infuusiokohtien on sijaittava vähintään 5 cm:n etäisyydellä toisistaan.
Infuusionopeus
Hizentra voidaan infusoida
- infuusiolaitteella tai
- manuaalisesti bolusinjektiona ruiskulla.
Infuusion suositeltu aloitusnopeus riippuu potilaan yksilöllisestä tarpeesta.
Laiteavusteinen infuusio
Infuusion suositeltu aloitusnopeus ei saa ylittää nopeutta 20 ml/tunti/kohta.
Infuusionopeus voidaan sen jälkeen suurentaa asteittain nopeuteen 35 ml/tunti/kohta seuraavien kahden infuusion yhteydessä, jos potilas sietää hoidon hyvin (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos potilas sietää aloitusinfuusiot täydellä annoksella per infuusiokohta ja enimmäisnopeudella, voidaan seuraavien infuusioiden infuusionopeuden suurentamista harkita tämän jälkeen potilaan tuntemuksen ja terveydenhuoltohenkilön arvion mukaan.
Manuaalinen bolusinjektio
Infuusion suositeltu aloitusnopeus ei saa ylittää nopeutta 0,5 ml/min/kohta (30 ml/tunti/kohta).
Jos tämä on hyvin siedetty (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet), infuusionopeus voidaan suurentaa nopeuteen 2,0 ml/min/kohta (120 ml/tunti/kohta). Jos potilas sietää aloitusinfuusiot täydellä annoksella per infuusiokohta ja enimmäisnopeudella, voidaan seuraavien infuusioiden infuusionopeuden suurentamista harkita tämän jälkeen potilaan tuntemuksen ja terveydenhuoltohenkilön arvion mukaan.
Potilas voi tarvita suurten infuusionopeuksien infusoimiseksi 24 gaugen tai suuremman (eli gauge-numeroltaan pienemmän) neulan. Tätä pienempien neulojen (eli gauge-numeroltaan suurempien) käyttö voi vaikeuttaa Hizentran antamista manuaalisena bolusinjektiona. Yhtä ruiskua kohti saa käyttää vain yhtä infuusiokohtaa. Jos tarvitaan ruiskulla annettava Hizentra-lisäannos, on käytettävä uutta steriiliä injektioneulaa ja vaihdettava infuusiokohtaa.
Jos Hizentra- esitäytettyä ruiskua käytetään manuaalisen bolusinjektion antamiseen, on suositeltavaa käyttää 5 ml:n, 10 ml:n tai 20 ml:n esitäytettyä ruiskua.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaat, joilla on tyypin I tai II hyperprolinemia.
Hizentraa ei saa antaa suonensisäisesti.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Hizentra on tarkoitettu annettavaksi vain ihon alle. Jos Hizentra annetaan vahingossa verisuoneen, potilaalle voi kehittyä sokki.
Kohdassa Annostus ja antotapa mainittua infuusion suositusnopeutta on noudatettava. Potilaita on seurattava ja tarkkailtava huolellisesti koko infuusion ajan haittavaikutusten havaitsemiseksi.
Tiettyjä haittavaikutuksia saattaa esiintyä useammin potilailla, jotka saavat ihmisen normaalia immunoglobuliinia ensimmäistä kertaa tai harvoissa tapauksissa, kun ihmisen normaalia immunoglobuliinia sisältävä valmiste vaihdetaan toiseen tai kun hoito on keskeytetty yli kahdeksaksi viikoksi.
Mahdolliset komplikaatiot voidaan usein välttää varmistamalla, että:
- potilas ei ole herkistynyt ihmisen normaalille immunoglobuliinille antamalla valmiste aluksi hitaana injektiona (ks. kohta Annostus ja antotapa)
- potilasta seurataan tarkkaan koko infuusion ajan oireiden havaitsemiseksi varsinkin, jos potilas ei ole aiemmin saanut ihmisen normaalia immunoglobuliinia, jos potilas siirtyy toisesta valmisteesta tämän valmisteen käyttöön tai jos edellisestä infuusiosta on kulunut pitkä aika. Potilasta on tällöin seurattava ensimmäisen infuusion aikana ja tunnin ajan sen jälkeen, jotta mahdolliset haittavaikutukset voidaan havaita. Kaikkia muita potilaita on seurattava vähintään 20 minuutin ajan annon jälkeen.
Allergisia tai anafylaktisia reaktioita epäiltäessä injektion antaminen on lopetettava välittömästi. Sokin yhteydessä on annettava asianmukaista lääkärinhoitoa.
Yliherkkyys
Todelliset allergiset reaktiot ovat harvinaisia. Niitä voi ilmetä erityisesti potilailla, joilla on IgA-vasta-aineita, ja näiden potilaiden hoidossa on oltava erityisen varovainen. Jos potilaalla on IgA-vasta-aineita ja hoito ihon alle annettavilla IgG-valmisteilla on ainoa vaihtoehto, potilaalle voidaan antaa Hizentra-hoitoa vain lääkärin tarkassa valvonnassa.
Ihmisen normaali immunoglobuliini voi aiheuttaa harvoissa tapauksissa verenpaineen laskun, johon liittyy anafylaktinen reaktio, myös potilaille, jotka ovat aikaisemmin sietäneet hoidon ihmisen normaalilla immunoglobuliinilla.
Tromboembolia
Immunoglobuliinien käytön yhteydessä on todettu valtimon ja laskimon tromboemboliatapahtumia, kuten sydäninfarkteja, aivohalvauksia, syviä laskimotrombooseja ja keuhkoembolioita. Varovaisuus on tarpeen, jos potilaalla on ennestään tromboositapahtumien riskitekijöitä (kuten korkea ikä, verenpainetauti, diabetes mellitus ja anamneesissa verisuonisairaus tai tromboottisia episodeja, hankinnainen tai perinnöllinen tromboositaipumus, pitkäaikainen immobilisaatio, vaikea hypovolemia, veren viskoosiutta lisäävä sairaus). Potilaille on kerrottava tromboemboliatapahtumien ensi oireista, joita ovat hengästyminen, raajan kipu ja turvotus, fokaaliset neurologiset oireet ja rintakipu, ja heitä on kehotettava ottamaan heti yhteyttä lääkäriin, jos oireita ilmenee. Riittävästä nesteytyksestä on syytä huolehtia ennen immunoglobuliinien käyttöä.
Aseptinen meningiitti ‑oireyhtymä (AMS)
Laskimoon annosteltavia immunoglobuliineja (IVIg) käytettäessä ja SCIg-hoidon yhteydessä on raportoitu AMS-tapauksia. Oireyhtymä alkaa yleensä, kun immunoglobuliinihoidosta on kulunut muutamasta tunnista kahteen vuorokautta. Tyypillisiä AMS-oireita ovat kova päänsärky, niskajäykkyys, uneliaisuus, kuume, valonarkuus, pahoinvointi ja oksentelu. Jos potilaalla on AMS-tyyppisiä merkkejä tai oireita, hänelle on tehtävä perusteellinen neurologinen tutkimus ja otettava aivo-selkäydinnestenäyte muiden aivokalvotulehduksen syiden pois sulkemiseksi. Kun immunoglobuliinihoito keskeytetään, AMS voi korjautua muutamassa päivässä ilman seurauksia.
Tietoa turvallisuudesta tarttuvien taudinaiheuttajien suhteen
Ihmisen verestä tai plasmasta valmistettujen lääkevalmisteiden välityksellä tarttuvien infektioiden ehkäisemiseksi vakiotoimenpiteisiin sisältyy mm. luovuttajien valinta, jokaisen luovutetun erän sekä plasman kokoomaerien testaus erityisten infektioiden merkkiaineiden toteamiseksi sekä tehokkaiden valmistusmenetelmien käyttö virusten inaktivoimiseksi tai poistamiseksi. Näistä toimenpiteistä huolimatta ei ihmisverestä tai ‑plasmasta valmistettujen lääkevalmisteiden antoon liittyvää infektion aiheuttajien siirtymisen mahdollisuutta voida sulkea kokonaan pois. Tämä koskee myös kaikkia tuntemattomia tai vasta kehittyviä viruksia tai muita patogeeneja.
Käytettyjä menetelmiä pidetään tehokkaina vaipallisia viruksia, kuten HIV, HBV ja HCV, sekä vaipattomia HA-viruksia ja parvoviruksia B19 vastaan.
Kliininen kokemus viittaa siihen, että hepatiitti A ja parvovirus B19 eivät siirry immunoglobuliinien annon yhteydessä ja oletetaan, että valmisteiden sisältämillä vasta-aineilla on merkittävä viruksilta suojaava vaikutus.
Vaikutukset serologisten tutkimusten tuloksiin
Erilaisten passiivisesti siirtyneiden vasta-aineiden määrä saattaa lisääntyä potilaan veressä tilapäisesti immunoglobuliini-infuusion jälkeen, mikä voi aiheuttaa harhaanjohtavia positiivisia tuloksia serologisissa määrityksissä.
Punasoluantigeeneihin, kuten A, B, ja D, kohdistuvien vasta-aineiden passiivinen siirtyminen voi häiritä joitakin punasolujen allovasta-ainemäärityksiä (esim. Coombsin koe).
Natriumpitoisuus
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per injektiopullo/ruisku eli sen voidaan sanoa olevan ”natriumiton”.
Pediatriset potilaat
Samat varoitukset ja varotoimet pätevät myös pediatrisiin potilaisiin.
Iäkkäät
Samat varoitukset ja varotoimet pätevät myös iäkkäisiin.
Yhteisvaikutukset
Eläviä heikennettyjä viruksia sisältävät rokotteet
Immunoglobuliinin anto voi heikentää eläviä, heikennettyjä viruksia sisältävien rokotteiden, kuten tuhkarokko-, vihurirokko-, sikotauti- ja vesirokkorokotteiden, tehoa vähintään kuudesta viikosta jopa kolmeen kuukauteen asti. Tämän lääkevalmisteen annon jälkeen on odotettava kolme kuukautta ennen rokottamista eläviä heikennettyjä viruksia sisältävällä rokotteella. Tuhkarokkorokotteen yhteydessä tämä heikkeneminen voi kestää vuoden. Sen vuoksi potilailta, joille on tarkoitus antaa tuhkarokkorokotus, on määritettävä vasta-aineet.
Pediatriset potilaat
Samoja yhteisvaikutuksia voi ilmetä myös pediatrisilla potilailla.
Iäkkäät
Samoja yhteisvaikutuksia voi ilmetä myös iäkkäillä.
Raskaus ja imetys
Raskaus
Kliinisistä tutkimuksista saatuja tietoja ihmisen normaalin immunoglobuliinin käytöstä raskauden aikana on vähän. Sen vuoksi Hizentraa saa antaa vain varoen raskaana oleville naisille. Immunoglobuliineista saatu kliininen kokemus viittaa siihen, että niistä ei ole odotettavissa haitallisia vaikutuksia raskauden kulkuun, sikiölle tai vastasyntyneelle.
Raskaana olevan naisen jatkuva hoito varmistaa, että vastasyntyneellä on asianmukainen passiivinen immuniteetti.
Imetys
Prospektiivisista kliinisistä tutkimuksista saatuja tietoja ihmisen normaalin immunoglobuliinin käytöstä imetyksen aikana on vähän. Sen vuoksi Hizentraa saa antaa vain varoen imettäville äideille. Immunoglobuliineista saatu kliininen kokemus viittaa kuitenkin siihen, että niistä ei ole odotettavissa haitallisia vaikutuksia vastasyntyneelle. Immunoglobuliinit erittyvät äidinmaitoon ja saattavat edistää suojaavien vasta-aineiden välittymistä vastasyntyneelle.
Hedelmällisyys
Immunoglobuliineista saatu kliininen kokemus viittaa siihen, että niistä ei ole odotettavissa haitallisia vaikutuksia hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Hizentra-valmisteella on vähäinen vaikutus (esim. huimaus) ajokykyyn ja koneidenkäyttökykyyn (ks. kohta Haittavaikutukset). Jos potilaalla esiintyy haittavaikutuksia hoidon aikana, hänen on odotettava näiden häviämistä ennen autolla ajoa tai koneiden käyttöä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Haittavaikutuksina saattaa toisinaan ilmetä esimerkiksi vilunväristyksiä, päänsärkyä, kuumetta, oksentelua, allergisia reaktioita, pahoinvointia, nivelsärkyä, matalaa verenpainetta ja kohtalaista alaselkäsärkyä.
Ihmisen normaali immunoglobuliini voi aiheuttaa harvoissa tapauksissa äkillisen verenpaineen laskun, ja yksittäisissä tapauksissa anafylaktisen sokin, myös potilaalle, jolla ei ole aikaisempien antokertojen aikana ilmennyt yliherkkyyttä.
Paikallisia reaktioita infuusiokohdissa: turvotus, arkuus, punoitus, kovettumat, paikallinen kuumotus, kutina, mustelmat ja ihottuma.
Tartuntatauteja koskevat turvallisuustiedot, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Haittavaikutustaulukko
Haittavaikutukset on kerätty Hizentralla tehdyistä kliinisistä tutkimuksista: seitsemästä vaiheen III tutkimuksesta potilailla, joilla on primaari immuunipuutos (n = 231), kahdesta vaiheen IV tutkimuksesta potilailla, joilla oli PID (n = 74), yhdestä vaiheen III tutkimuksesta (n = 115) ja yhdestä jatkotutkimuksesta (n = 82) CIDP-potilailla (yhteensä 502 potilasta; 26 646 infuusiota). Yhteenveto näissä kliinisissä tutkimuksissa raportoiduista haittavaikutuksista on esitetty seuraavassa taulukossa MedDRA-elinluokitusjärjestelmän (SOC ja Preferred Term) ja esiintymistiheyden mukaisesti luokiteltuina.
Esiintymistiheys potilasta tai infuusiota kohti on määritetty seuraavien kriteerien mukaisesti: hyvin yleinen (≥ 1/10), yleinen ≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000). Myyntiluvan saamisen jälkeen spontaanisti raportoitujen haittavaikutusten esiintymistiheys on luokitukseltaan tuntematon.
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Hizentraan liittyvien haittavaikutusten esiintymistiheys potilasta tai infuusiota kohti kliinisissä tutkimuksissa ja myyntiluvan saamisen jälkeisessä seurannassa
Elinjärjestelmä (SOC, MedDRA) | Haittavaikutus (MedDRA Preferred Term, PT) | Esiintymistiheys potilasta kohti | Esiintymistiheysinfuusiota kohti |
Immuunijärjestelmä | Yliherkkyys | Melko harvinainen | Harvinainen |
Anafylaktinen reaktio | Tuntematon | Tuntematon | |
Hermosto | Päänsärky | Hyvin yleinen | Melko harvinainen |
Heitehuimaus, migreeni | Yleinen | Harvinainen | |
Vapina (myös psykomotorinen hyperaktiivisuus) | Melko harvinainen | Harvinainen | |
Aseptinen meningiitti | Melko harvinainen | Hyvin harvinainen | |
Polttava tunne | Tuntematon | Tuntematon | |
Sydän | Takykardia | Melko harvinainen | Hyvin harvinainen |
Verisuonisto | Kohonnut verenpaine | Yleinen | Harvinainen |
Kasvojen punoitus | Melko harvinainen | Harvinainen | |
Emboliset ja tromboottiset tapahtumat | Tuntematon | Tuntematon | |
Ruoansulatuselimistö | Ripuli, vatsakipu | Yleinen | Melko harvinainen |
Pahoinvointi, oksentelu | Yleinen | Harvinainen | |
Iho ja ihonalainen kudos | Ihottuma | Hyvin yleinen | Melko harvinainen |
Kutina, nokkosihottuma | Yleinen | Harvinainen | |
Luusto, lihakset ja sidekudos | Tuki- ja liikuntaelimistön kipu, nivelkipu | Yleinen | Melko harvinainen |
Lihaskouristus, lihasheikkous | Melko harvinainen | Harvinainen | |
Yleisoireet ja antopaikassa todettavat haitat | Infuusiokohdan reaktio | Hyvin yleinen | Hyvin yleinen |
Väsymys (myös huonovointisuus), kuume | Yleinen | Melko harvinainen | |
Rintakipu, influenssan kaltainen sairaus, kipu | Yleinen | Harvinainen | |
Vilunväristykset (myös hypotermia) | Melko harvinainen | Harvinainen | |
Infuusiokohdan haavauma | Tuntematon | Tuntematon | |
Tutkimukset | Suurentunut veren kreatiniiniarvo | Melko harvinainen | Harvinainen |
Pediatriset potilaat
Kaiken kaikkiaan turvallisuusprofiili oli kliinisten Hizentra-tutkimusten pediatrisilla ja aikuisilla PID-potilailla samankaltainen.
Hizentraa ei arvioitu kliinisissä tutkimuksissa alle 18‑vuotiailla pediatrisilla CIDP-potilailla.
Iäkkäät
Samoja haittavaikutuksia voi ilmetä iäkkäillä. Kliinisistä tutkimuksista saatujen tietojen perusteella ≥ 65-vuotiaiden potilaiden turvallisuusprofiili ei eronnut nuorempien potilaiden turvallisuusprofiilista.
Hizentran markkinoilletulon jälkeen ≥ 65-vuotiaista potilaista saatujen tietojen perusteella turvallisuusprofiili on kyseisessä ikäryhmässä kaiken kaikkiaan samanlainen kuin nuoremmilla potilailla.
Kohdassa Varoitukset ja käyttöön liittyvät varotoimet on kerrottu tarkemmin riskitekijöistä ja seurantasuosituksista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostuksen seurauksia ei tunneta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunoseerumit ja immunoglobuliinit: immunoglobuliinit, ihmisen normaali immunoglobuliini, immunoglobuliini im, ATC-koodi: J06BA01.
Ihmisen normaali immunoglobuliini sisältää pääasiassa immunoglobuliini G:tä (IgG) sekä laajan kirjon vasta-aineita taudinaiheuttajia vastaan.
Ihmisen normaali immunoglobuliini sisältää normaaliväestössä esiintyviä IgG-vasta-aineita. Se valmistetaan yleensä vähintään 1000 luovuttajan plasmapoolista. Sen immunoglobuliini G ‑alaluokkien jakauma vastaa läheisesti vastaavaa jakaumaa ihmisen luonnollisessa plasmassa.
Vaikutusmekanismi
Immuunipuutoksessa riittävät Hizentra-annokset voivat palauttaa poikkeavan pienet immunoglobuliini G ‑vasta-ainepitoisuudet normaalialueelle ja siten auttaa torjumaan infektioita.
Vaikutusmekanismi muissa käyttöaiheissa kuin korvaushoidossa ei ole täysin selvillä, mutta mekanismilla on immunomodulatorisia vaikutuksia.
PID
Euroopassa tehdyssä keskeisessä prospektiivisessa, avoimessa, yhden hoitoryhmän sisältäneessä monikeskustutkimuksessa yhteensä 51:tä iältään 3–60-vuotiasta tutkimushenkilöä, joilla oli primaari immuunipuutostauti, hoidettiin Hizentralla enintään 41 viikon ajan. Keskimääräinen viikoittainen annos oli 0,12 g/kg painokiloa kohti. Näin saavutettiin pitkäkestoisesti IgG-minimipitoisuudeksi keskimäärin 7,99–8,25 g/l koko hoitojakson ajaksi. Tutkimushenkilöt saivat yhteensä 1831 viikoittaista Hizentra-infuusiota.
Yhdysvalloissa tehdyssä prospektiivisessa, avoimessa, yhden hoitoryhmän sisältäneessä monikeskustutkimuksessa yhteensä 49:ää iältään 5–72-vuotiasta tutkimushenkilöä, joilla oli primaari immuunipuutostauti, hoidettiin Hizentralla enintään 15 kuukauden ajan. Keskimääräinen viikoittainen annos oli 0,23 g/kg painokiloa kohti. Näin saavutettiin pitkäkestoisesti IgG-minimipitoisuudeksi keskimäärin 12,53 g/l koko hoitojakson ajaksi. Tutkimushenkilöt saivat yhteensä 2264 viikoittaista Hizentra-infuusiota.
Hizentra-hoitoa saaneilla tutkimushenkilöillä ei raportoitu kliinisten tutkimusten tehokkaan hoidon aikana vakavia bakteeri-infektioita.
Suurten infuusionopeuksien turvallisuutta ja siedettävyyttä manuaalisen bolusinjektion ja pumppuavusteisen annostelun yhteydessä arvioitiin avoimessa, satunnaistamattomassa vaiheen IV HILO (Hizentra Label Optimization) -monikeskustutkimuksessa, jossa käytettiin rinnakkaisryhmiä ja johon otettiin mukaan 49 iältään 2–75-vuotiasta tutkimushenkilöä, joilla oli PID. Tutkimushenkilöt saivat Hizentra-hoitoa vähintään 12 viikon ajan (11 iältään 2 – < 18‑vuotiasta pediatrista potilasta, 35 iältään 18–65‑vuotiasta aikuispotilasta ja 3 iältään > 65‑vuotiasta iäkästä potilasta). Ensimmäinen potilasryhmä sai Hizentraa manuaalisena bolusinjektiona (n = 16) 2–7 infuusiota viikossa infuusionopeuksilla 30, 60 ja 120 ml/tunti/kohta (ks. kohta Annostus ja antotapa). Toinen potilasryhmä sai Hizentra-infuusioita pumppuavusteisesti (n = 18) viikoittain infuusionopeuksilla 25, 50, 75 ja 100 ml/tunti/kohta. Kolmannessa ryhmässä arvioitiin lisäksi viikoittaisten Hizentra-annosten pumppuavusteista annostelua infuusiotilavuuksilla 25, 40 ja 50 ml/kohta (n = 15). Kutakin infuusioparametria käytettiin kussakin kolmesta ryhmästä 4 viikon ajan, minkä jälkeen voitiin ottaa käyttöön järjestyksessä seuraavaksi suurempi infuusioparametri niille tutkimushenkilöille, joille validien infuusioiden vaadittu minimimäärä oli saatu onnistuneesti infusoitua.
Ensisijainen päätetapahtuma oli niiden tutkimushenkilöiden prosentuaalinen osuus, joilla saavutettiin vaste suurempaan infuusioparametriin.
| Ryhmä | Infuusioparametri ja hoitoon vastanneet (%) | |||
| 30 ml/tunti/kohta | 60 ml/tunti/kohta | 120 ml/tunti/kohta | - |
| 100,0 % | 100,0 % | 87,5 % | - | |
| 2. pumppuavusteiset virtausnopeudet | 25 ml/tunti/kohta | 50 ml/tunti/kohta | 75 ml/tunti/kohta | 100 ml/tunti/kohta |
| 77,8 % | 77,8 % | 66,7 % | 61,1 % | |
| 3. pumppuavusteiset tilavuudet | 25 ml/kohta | 40 ml/kohta | 50 ml/kohta | - |
| 86,7 % | 73,3 % | 73,3 % | - | |
Hoitoon vastannut: pumppuavusteisen annostelun ryhmässä tutkimushenkilö, joka suoritti ≥ 3 validia infuusiota 4:stä infuusiosta yhtä infuusioparametria kohti; manuaalisen bolusinjektion ryhmässä tutkimushenkilö, joka suoritti ≥ 60 % valideja infuusioita infuusioparametriä kohti. Infuusiota pidettiin validina, jos ≥ 95 % suunnitellusta virtausnopeudesta/tilavuus per ≥1 infuusiokohta saavutettiin.
Kaiken kaikkiaan niiden infuusioiden lukumäärä, joissa ei ilmennyt vaikeita paikallisia reaktioita, verrattuna infuusioiden kokonaismäärään (siedettävyys) oli ≥ 0,98 kaikissa ryhmissä kaikkien infuusioparametrien suhteen. Yhdelläkään tutkimushenkilöllä ei havaittu kliinisesti merkityksellisiä eroja seerumin pienimmässä IgG-pitoisuudessa (IgG trough) lähtötilanteen 1. päivän ja tutkimuksen lopun välillä.
CIDP
Hizentran turvallisuutta, tehoa ja siedettävyyttä CIDP-potilaille arvioitiin vaiheen III PATH-tutkimuksessa [Polyneuropathy and Treatment with Hizentra]. Tutkimus oli kaksoissokkoutettu, satunnaistettu, lumelääkekontrolloitu ja rinnakkaisryhmillä suoritettu monikeskustutkimus. Aiemmin IVIg-hoitoa saaneet 172 aikuista, joilla oli varmistettu tai todennäköinen CIDP ja jotka saivat vasteen IVIg-hoitoon, satunnaistettiin saamaan viikoittain Hizentraa 0,2 g/kg painokilo, 0,4 g/kg painokilo tai lumelääkettä, ja heitä seurattiin seuraavien 24 viikon ajan. Altistuksen keskimääräinen kesto Hizentraa 0,2 g/kg painokilo saaneiden ryhmässä oli 118,9 vuorokautta (maksimialtistus oli enintään 167 vuorokautta) ja Hizentraa 0,4 g/kg painokilo saaneiden ryhmässä 129 vuorokautta (maksimialtistus oli enintään 166 vuorokautta). Tutkimushenkilöillä oli tavallisesti yhtä aikaa käytössä 4 infuusiokohtaa (enintään 8 kohtaa yhtä aikaa). Kaiken kaikkiaan lumelääkeryhmässä 57 tutkimushenkilöä sai 1 514 infuusiota, 0,2 g/kg painokiloa Hizentraa saavien ryhmässä 57 tutkimushenkilöä sai 2 007 infuusiota ja 0,4 g/kg painokilo Hizentraa saaneiden ryhmässä 58 tutkimushenkilöä sai 2 218 infuusiota (yhteensä 5 739 infuusiota).
Tehon ensisijainen päätetapahtuma oli niiden tutkimushenkilöiden prosentuaalinen määrä, jotka saivat CIDP-relapsin (määritelty korjatun Inflammatory Neuropathy Cause and Treatment [INCAT] ‑pistemäärän ≥ 1 pisteen suurenemana lähtötilanteeseen verrattuna) tai jotka keskeyttivät tutkimuksen mistä tahansa muusta syystä Hizentra-hoitojakson aikana.
Molemmat Hizentra-annokset olivat lumelääkettä parempia ensisijaisen päätetapahtuman suhteen. Lumelääkkeeseen verrattuna tilastollisesti merkitsevästi pienempi prosenttiosuus Hizentralla hoidetuista tutkimushenkilöistä (32,8 % annoksella 0,4 g/kg painokilo hoidetuista (p < 0,001), 38,6 % annoksella 0,2 g/kg painokilo hoidetuista (p = 0,007) ja 63,2 % lumelääkkeellä hoidetuista) sai CIDP-relapsin tai keskeytti tutkimuksen muista syistä. Kun vain relapsi huomioidaan, CIDP-relapsin saaneiden osuus oli 19,0 % Hizentraa annoksella 0,4 g/kg painokilo saaneilla (p < 0,001), 33,3 % Hizentraa annoksella 0,2 g/kg painokilo saaneilla (p = 0,012) ja 56,1 % lumelääkettä saaneilla. Vastaavasti enintään 24 viikon hoitojakson aikana Hizentra esti relapsin 81 %:lla tutkimushenkilöistä annosta 0,4 g/kg painokilo saaneiden ryhmässä ja 67 %:lla tutkimushenkilöistä annosta 0,2 g/kg painokilo saaneiden ryhmässä, lumelääkettä saaneiden ryhmässä puolestaan 44 % tutkimushenkilöistä välttyi relapseilta.
CIDP-relapsiin kulunut aika (Kuva 1) arvioitiin ja vastaavat Kaplan-Meier -estimaatteihin perustuvat CIDP-relapsin todennäköisyydet olivat lumelääkkeellä 58,8 %, Hizentra-annoksella 0,2 g/kg painokilo 35,0 % ja Hizentra-annoksella 0,4 g/kg painokilo 22,4 %. Riskisuhde lumelääkkeeseen verrattuna (95 %:n luottamusväli) oli pienemmällä annoksella 0,48 (0,27; 0,85) ja suuremmalla annoksella 0,25 (0,12; 0,49).
Hizentra-ryhmien 0,2 g/kg painokilo ja 0,4 g/kg painokilo välillä havaittu ero ei saavuttanut tilastollista merkitsevyyttä.
Kuva 1. CIDP-relapsiin kuluvan ajan Kaplan-Meier -kuvaaja
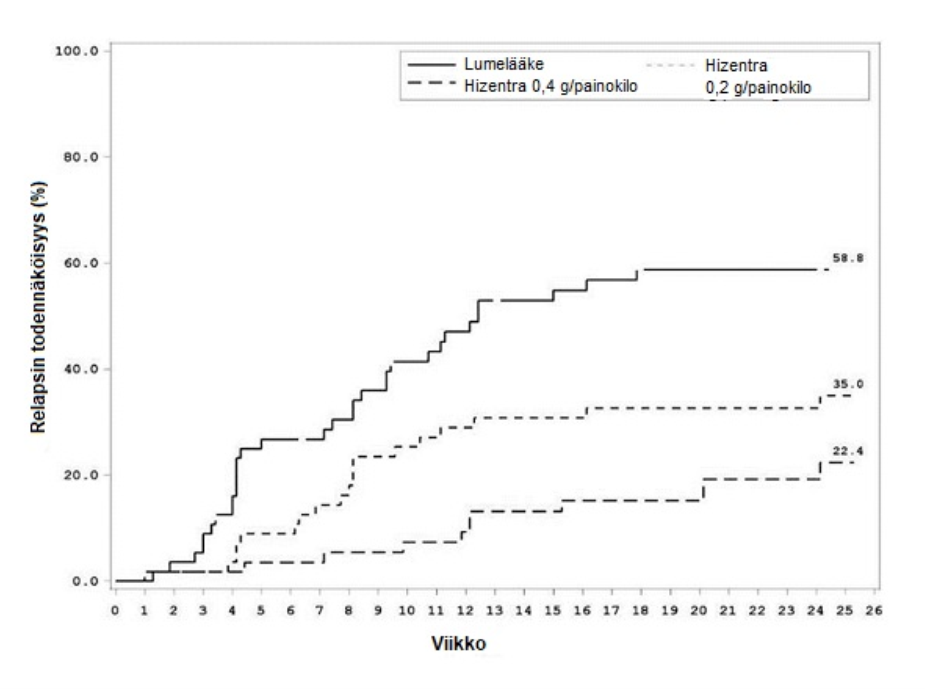
Tehoa kuvaavat pistemäärät (INCAT-pistemäärä, keskimääräinen puristusvoima ja Medical Research Council -yhteispistemäärä) pysyivät vakaina molempien Hizentra-annosryhmien tutkimushenkilöillä, mutta lumelääkeryhmän tutkimushenkilöiden pistemäärät huononivat. Suurta Hizentra-annosta saaneessa ryhmässä tutkimushenkilöiden Rasch-built Overall Disability Scale (R-ODS) ‑sadannespistemäärä pysyi vakaana. Molemmissa Hizentra-annosryhmissä tutkimushenkilöiden elektrofysiologiset parametrit pysyivät vakaina.
Useassa eri tutkimuskeskuksessa tehtyyn 48 viikon avoimeen vaiheen III jatkotutkimukseen otettiin mukaan 82 CIDP‑potilasta PATH-tutkimuksesta. Tässä jatkotutkimuksessa tutkittiin kahdella viikoittaisella annoksella (0,2 g/kg painokilo ja 0,4 g/kg painokilo) annetun Hizentra-ylläpitohoidon pitkäaikaista turvallisuutta ja tehoa. Tutkimusasetelman vuoksi sama tutkimushenkilö saattoi saada kumpaakin annosta tutkimuksen aikana; tehon arviointijakson aikana 72 tutkimushenkilöä sai annosta 0,4 g/kg painokilo ja 73 tutkimushenkilöä annosta 0,2 g/kg painokilo. Tehon arviointijakson keskimääräinen kesto oli 125,8 päivää (vaihteluväli: 1–330) 0,2 g/kg painokilo ‑ryhmässä ja 196,1 päivää (vaihteluväli: 1–330) 0,4 g/kg painokilo ‑ryhmässä. Niillä potilailla, jotka pysyivät keskeisessä PATH-tutkimuksessa mukana loppuun saakka ilman relapsia annoksella 0,4 g/kg painokilo ja jotka saivat aluksi tätä annosta jatkotutkimuksessa, relapseja ilmeni 5,6 %:lla (1/18 potilaalla). Kaikista niistä potilaista, joiden annos PATH-jatkotutkimuksessa oli 0,4 g/kg painokilo, relapsin sai 9,7 % (7/72 potilasta). Potilailla, jotka pysyivät PATH-tutkimuksessa mukana loppuun saakka ilman relapsia annoksella 0,2 g/kg painokilo ja jotka saivat tätä annosta aluksi jatkotutkimuksessa, relapseja ilmeni 50 %:lla (3/6 potilaalla). Niistä kaikista potilaista, jotka saivat annosta 0,2 g/kg painokilo jatkotutkimuksessa, relapsin sai 47,9 % (35/73 potilasta). PATH-tutkimuksessa loppuun asti pysyneiden, kumpaa tahansa annosta saaneiden ja jatkotutkimukseen siirtyneiden potilaiden annosta voitiin pienentää määrästä 0,4 g/kg painokilo määrään 0,2 g/kg painokilo 67,9 %:lla tutkimushenkilöistä (19/28 potilaalla) ilman relapsia. Kaikki 9 relapsin saanutta toipuivat 4 viikossa saatuaan hoitoa annoksella 0,4 g/kg painokilo. Puristusvoima, Medical Research Council (MRC) ‑yhteispistemäärä ja R‑ODS‑sadannespistemäärä pysyivät vakaina verrattuna lähtötilanteeseen potilailla, jotka eivät olleet koskaan saaneet relapsia jatkotutkimuksessa.
Pediatriset potilaat
Hizentran turvallisuus ja tehokkuus on osoitettu 2–18‑vuotiailla pediatrisilla henkilöillä. Hizentraa on arvioitu 68:lla iältään 2 – < 12‑vuotiaalla pediatrisella henkilöllä, joilla on PID, ja 57:llä iältään 12 – < 18‑vuotiaalla pediatrisella henkilöllä. Farmakokinetiikassa tai turvallisuus- ja tehoprofiileissa ei ollut eroa verrattuna aikuisiin tutkimushenkilöihin. Erityiset pediatriset annosmuutokset eivät olleet tarpeen halutun seerumin IgG‑pitoisuuden saavuttamiseksi. Farmakodynamiikassa ei todettu eroa aikuisten ja pediatristen primaaria immuunipuutostautia sairastavien tutkimuspotilaiden välillä.
Hizentraa ei ole arvioitu pediatrisilla potilailla tehdyissä kliinisissä tutkimuksissa alle 18-vuotiailla CIDP-potilailla.
Iäkkäät
Kaiken kaikkiaan turvallisuudessa tai tehossa ei havaittu eroa yli 65‑vuotiaiden PID-potilaiden ja 18–65‑vuotiaiden PID-potilaiden välillä. Kliinisissä tutkimuksissa Hizentraa arvioitiin 13 yli 65‑vuotiaalla PID‑potilaalla.
Kaiken kaikkiaan turvallisuudessa tai tehossa ei havaittu eroa yli 65‑vuotiaiden CIDP-potilaiden ja 18–65‑vuotiaiden CIDP-potilaiden välillä. CIDP-potilailla tehdyissä kliinisissä tutkimuksissa Hizentra-hoitoa sai 61 yli 65‑vuotiasta henkilöä.
Farmakokinetiikka
Imeytyminen ja jakautuminen
Kun Hizentra annetaan ihon alle, seerumin huippupitoisuudet saavutetaan noin 2 päivän kuluttua.
Eliminaatio
IgG ja IgG-kompleksit pilkkoutuvat retikuloendoteliaalijärjestelmän soluissa.
PID
Tutkimushenkilöt saavuttivat Hizentralla tehdyssä kliinisessä vaiheen III tutkimuksessa (n = 46) minimipitoisuudet (mediaani 8,1 g/l) pitkäkestoisesti 29 viikon aikana, kun he saivat viikoittain annoksia 0,06–0,24 g painokiloa kohti (mediaani).
Empiirisillä populaatiofarmakokineettisillä malleilla tehtyjen simulaatioiden perusteella IgG-altistus (AUC0-14vrk, Cmin 14vrk) voi olla samaa luokkaa, jos Hizentraa annetaan ihon alle kahden viikon välein käyttäen kaksi kertaa viikoittaisten ylläpitoannosten suuruisia annoksia.
Lisäksi näiden simulaatioiden perusteella IgG:n minimipitoisuudet seerumissa voivat olla samaa luokkaa, jos Hizentran viikoittainen ylläpitoannos annetaan vastaavina määrinä useammin kuin kerran viikossa (esim. 2 kertaa viikossa, 3 kertaa viikossa, 5 kertaa viikossa tai päivittäin).
Kahden tai kolmen unohtuneen päivittäisen annoksen simulaatiossa seerumin IgG-pitoisuuden pienenemä (mediaani) oli ≤ 4 % jatkuvaan päivittäiseen annostukseen verrattuna. Kun unohtuneet annokset korvattiin päivittäisen annostuksen alettua uudelleen, pitoisuusprofiili (mediaani) palasi ennalleen 2–3 päivässä. Jos unohtuneita annoksia ei kuitenkaan korvattu annostuksen alettua uudelleen, IgG:n minimipitoisuuksien palaaminen vakaaseen tilaan kesti 5–6 viikkoa.
Pediatriset potilaat
Aikuisten ja pediatristen PID‑tutkimuspotilaiden farmakokineettisten parametrien välillä ei havaittu eroja.
Iäkkäät
Yli 65‑vuotiaiden ja 18–65‑vuotiaiden PID‑potilaiden farmakokineettisten parametrien välillä ei kaiken kaikkiaan havaittu eroja.
CIDP
PATH-tutkimuksessa tutkimushenkilöt (n = 172) saavuttivat pysyvän jäännöspitoisuuden 24 viikon jakson aikana, kun he saivat viikoittain annosta 0,2 g/kg painokilo tai 0,4 g/kg painokilo. Keskimääräinen (keskihajonta) IgG-jäännöspitoisuus Hizentra-hoidon jälkeen oli 20,4 (3,24) g/l annoksella 0,4 g/kg painokilo ja 15,4 (3,06) g/l annoksella 0,2 g/kg painokilo. PATH-tutkimuksessa populaatiofarmakokineettisillä malleilla tehdyt simulaatiot viittaavat siihen, että vastaava IgG-altistus (Cmax, AUC0-14 vrk, Cmin, 14 vrk) saavutetaan, kun kaksinkertaista Hizentran viikkoannosta annetaan CIDP-potilaille kahden viikon välein. Simulaatiot viittaavat lisäksi siihen, että CIDP-potilasryhmässä saavutetaan vastaava IgG-altistus, kun Hizentran viikoittainen ylläpitoannos jaetaan useaan, tiheämmin annettavaan annokseen (2–7 kertaa viikossa).
Pediatriset potilaat
Hizentraa ei ole arvioitu pediatrisilla potilailla tehdyissä kliinisissä tutkimuksissa alle 18-vuotiailla CIDP-potilailla.
Iäkkäät
Yli 65‑vuotiaiden ja 18–65‑vuotiaiden CIDP‑potilaiden farmakokineettisten parametrien välillä ei kaiken kaikkiaan havaittu eroja.
Prekliiniset tiedot turvallisuudesta
Immunoglobuliinit ovat ihmisen elimistöön normaalisti kuuluvia aineita. L-proliini on fysiologinen, ei välttämätön aminohappo.
Hizentran turvallisuutta on arvioitu useissa prekliinisissä tutkimuksissa, etenkin apuaineen L-proliinin suhteen. Farmakologista turvallisuutta ja toksisuutta koskevien ei-kliinisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
L-proliini
polysorbaatti 80
injektionesteisiin käytettävä vesi
kloorivetyhappo (pH:n säätöön)
natriumhydroksidi (pH:n säätöön).
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
30 kuukautta.
Kun injektiopullo tai muovikoteloon pakattu esitäytetty ruisku on avattu, liuos on käytettävä välittömästi.
Säilytys
Säilytä alle 25 °C.
Ei saa jäätyä.
Pidä injektiopullo tai muovikoteloon pakattu esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Avatun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
HIZENTRA injektioneste, liuos
200 mg/ml (L:ei) 5 ml (1 g/5 ml) (90,11 €), 10 ml (2 g/10 ml) (173,48 €), 20 ml (4 g/20 ml) (330,94 €), 50 ml (10 g/50 ml) (796,86 €), 10 x 10 ml (2 g/10 ml) (1549,32 €), 10 x 20 ml (2967,49 €)
HIZENTRA injektioneste, liuos, esitäytetty ruisku
200 mg/ml (L:ei) 10 ml (173,48 €), 20 ml (330,94 €), 50 ml (796,86 €)
PF-selosteen tieto
Injektiopullot
5, 10 tai 20 ml liuosta injektiopullossa (tyypin I lasia) ja 50 ml liuosta injektiopullossa (tyypin II lasia), joka on suljettu (halobutyyli) tulpalla, (alumiinipuriste) korkilla ja irti napsautettavalla muovilevyllä.
Pakkauskoot 1, 10 tai 20 injektiopulloa:
1 g / 5 ml
2 g / 10 ml
4 g / 20 ml
10 g / 50 ml
Esitäytetyt ruiskut
5, 10, 20 tai 50 ml liuosta esitäytetyssä ruiskussa (syklo-olefiini-kopolymeeri (COC)), joka on läpipainopakkauksessa, jossa on hapenpoistajapakkaus (oxygen absorber).
Pakkauskoot 1 esitäytetyssä ruiskussa:
1 g / 5 ml
2 g / 10 ml
4 g / 20 ml
10 g / 50 ml
Pakkauskoot 10 esitäytettyä ruiskua:
1 g / 5 ml
2 g / 10 ml
4 g / 20 ml
10 g / 50 ml
Pakkauskoot 20 esitäytettyä ruiskua:
2 g / 10 ml
4 g / 20 ml
Pakkaus ei sisällä alkoholipyyhkeitä, neuloja eikä muita välineitä tai tarvikkeita.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Liuos on kirkas ja vaaleankeltainen tai vaaleanruskea.
Hizentran osmolaalisuus on noin 380 mOsmol/kg.
Käyttö- ja käsittelyohjeet
Hizentra on käyttövalmis liuos yhteen käyttökertaan tarkoitetuissa injektiopulloissa tai yhteen käyttökertaan tarkoitetuissa esitäytetyissä ruiskuissa. Hizentra on käytettävä/infusoitava mahdollisimman pian injektiopullon tai esitäytetyn ruiskun muovikotelon aukaisemisen jälkeen. Hizentra-valmistetta ei saa käyttää, jos injektiopullo tai esitäytetyn ruiskun muovikotelo on avattu tai jos se on viallinen.
Lääkevalmisteen on annettava lämmetä huoneen- tai kehonlämpöiseksi ennen käyttöä.
Liuoksen on oltava kirkasta ja vaaleankeltaista tai vaaleanruskeaa.
Sameita tai sakkautuneita liuoksia ei saa käyttää.
Käyttämätön lääkevalmiste, jäte ja hapenpoistajapakkaus on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
HIZENTRA injektioneste, liuos
200 mg/ml 5 ml, 10 ml, 20 ml, 50 ml, 10 x 10 ml, 10 x 20 ml
HIZENTRA injektioneste, liuos, esitäytetty ruisku
200 mg/ml 10 ml, 20 ml, 50 ml
- Ylempi erityiskorvaus (100 %). Gammaglobuliinin puutostila (120), Itsenäinen verihiutaleiden tai granulosyyttien niukkuus (129).
- Peruskorvaus (40 %).
ATC-koodi
J06BA01
Valmisteyhteenvedon muuttamispäivämäärä
25.09.2025
Yhteystiedot
 CSL BEHRING AB
CSL BEHRING AB Box 712
182 17 Danderyd
Ruotsi
+46 (0) 8 544 966 70
www.cslbehring.fi
info@cslbehring.se