
XGEVA injektioneste, liuos 120 mg, injektioneste, liuos, esitäytetty ruisku 120 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Leuan luukuolion riskin minimoiminen
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 120 mg denosumabia 1,7 millilitrassa liuosta (70 mg/ml).
Yksi esitäytetty ruisku sisältää 120 mg denosumabia 1,0 millilitrassa liuosta (120 mg/ml).
Denosumabi on ihmisen monoklonaalinen IgG2-vasta-aine, joka tuotetaan yhdistelmä‑DNA‑menetelmällä nisäkässolulinjassa (kiinanhamsterin munasarjasoluissa).
Apuaine, joiden vaikutus tunnetaan
1,7 ml liuosta sisältää 78 mg sorbitolia (E420).
1,0 ml liuosta sisältää 37 mg sorbitolia (E420) ja 6,1 mg L‑fenyylialaniinia.
1,7 ml liuosta sisältää 0,17 mg polysorbaatti 20:tä.
1,0 ml liuosta sisältää 0,1 mg polysorbaatti 20:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
XGEVA 120 mg injektioneste, liuos
Injektioneste, liuos (injektioneste).
XGEVA 120 mg injektioneste, liuos, esitäytetty ruisku
Injektioneste, liuos (injektioneste).
Kliiniset tiedot
Käyttöaiheet
Luustotapahtumien (patologisten murtumien, luuston sädehoidon, selkäydinkompression tai luuston kirurgisten toimenpiteiden) ehkäiseminen aikuispotilailla, joilla on pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä (ks. kohta Farmakodynamiikka).
Luun jättisolukasvaimen hoito aikuisilla ja nuorilla, joiden luuston kasvu on päättynyt, kun poistoleikkaus ei ole mahdollinen tai se johtaisi todennäköisesti vaikeaan toimintakyvyn heikkenemiseen.
Ehto
Valmiste annetaan terveydenhuollon ammattilaisen vastuulla.
Annostus ja antotapa
Xgeva annetaan terveydenhuollon ammattilaisen vastuulla.
Annostus
Kaikki potilaat tarvitsevat kalsiumlisää vähintään 500 mg/vrk ja D-vitamiinilisää vähintään 400 IU/vrk, ellei hyperkalsemiaa esiinny (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Xgeva-hoitoa saaville potilaille on annettava pakkausseloste ja potilaan muistutuskortti.
Luustotapahtumien ehkäisy aikuispotilailla, joilla on pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä
Suositeltu annos on 120 mg, joka annetaan 4 viikon välein kertainjektiona ihon alle reiteen, vatsaan tai olkavarteen.
Luun jättisolukasvain
Suositeltu Xgeva-annos on 120 mg kertainjektiona ihon alle reiteen, vatsaan tai olkavarteen 4 viikon välein ja lisäksi 120 mg:n annokset hoidon 8. ja 15. päivänä ensimmäisen hoitokuukauden aikana.
Toisen vaiheen tutkimukseen osallistuneet potilaat, joille tehtiin luun jättisolukasvaimen täydellinen poisto, saivat hoitoa vielä 6 kuukauden ajan leikkauksen jälkeen tutkimussuunnitelman mukaisesti.
Luun jättisolukasvainta hoidettaessa hoidon hyöty potilaalle on arvioitava uudelleen säännöllisin välein. Hoidon keskeyttämisen tai lopettamisen vaikutusta potilaisiin, joiden tauti pysyy hallinnassa Xgeva-hoidolla, ei ole arvioitu. Rajalliset tiedot näistä potilaista eivät kuitenkaan viittaa siihen, että hoidon lopettaminen aiheuttaisi oireiden tilapäistä vaikeutumista (rebound).
Munuaisten vajaatoiminta
Munuaisten vajaatoiminta ei vaadi annostuksen muuttamista (ks. kohdasta Varoitukset ja käyttöön liittyvät varotoimet kalsiumarvon seurantaa koskevat suositukset ja kohdat Haittavaikutukset ja Farmakokinetiikka).
Maksan vajaatoiminta
Denosumabin turvallisuutta ja tehoa ei ole tutkittu maksan vajaatoiminnan yhteydessä (ks. kohta Farmakokinetiikka).
Iäkkäät (≥ 65-vuotiaat) potilaat
Iäkkäiden potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Xgevan turvallisuutta ja tehoa ei ole varmistettu alle 18‑vuotiaiden lasten hoidossa, lukuun ottamatta 12–17‑vuotiaita nuoria, joilla on luun jättisolukasvain ja joiden luuston kasvu on päättynyt.
Xgevaa ei suositella alle 18‑vuotiaille lapsille, lukuun ottamatta 12–17‑vuotiaita nuoria, joilla on luun jättisolukasvain ja joiden luuston kasvu on päättynyt (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Luun jättisolukasvaimen hoito nuorilla, joiden luuston kasvu on päättynyt, kun poistoleikkaus ei ole mahdollinen tai se johtaisi todennäköisesti vaikeaan toimintakyvyn heikkenemiseen: annostus on sama kuin aikuispotilailla.
Eläinkokeissa RANK:n tai RANK-ligandin (RANKL) estoon on liitetty luuston kasvun vähenemistä ja hampaiden puhkeamattomuutta. Nämä muutokset korjautuivat osittain, kun RANK-ligandia estävä vaikutus päättyi (ks. kohta Prekliiniset tiedot turvallisuudesta).
Antotapa
Ihon alle.
Xgeva 120 mg/1,7 ml injektioneste kertakäyttöisessä injektiopullossa:
Vain terveydenhuollon ammattilainen saa antaa 120 mg/1,7 ml injektionesteen injektiopullosta.
Xgeva 120 mg/1,0 ml injektioneste esitäytetyssä ruiskussa:
Potilas tai potilaasta huolehtiva henkilö voi antaa 120 mg/1,0 ml injektionesteen esitäytetyllä ruiskulla, kun terveydenhuollon ammattilainen on opettanut hänelle oikean injektiotekniikan. Esitäytetyn Xgeva‑ruiskun ensimmäisen itsenäisen antokerran on tapahduttava terveydenhuollon ammattilaisen valvonnassa.
Käyttö-, käsittely- ja hävittämisohjeet, ks. kohta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Vaikea hoitamaton hypokalsemia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Huonosti parantuneet haavat hammas- tai suukirurgisen toimenpiteen jälkeen.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Kalsium- ja D-vitamiinilisä
Kaikki potilaat tarvitsevat kalsium- ja D-vitamiinilisää, ellei hyperkalsemiaa esiinny (ks. kohta Annostus ja antotapa).
Hypokalsemia
Jo olemassa oleva hypokalsemia on korjattava ennen Xgeva-hoidon aloittamista. Hypokalsemiaa voi esiintyä milloin tahansa Xgeva-hoidon aikana. Kalsiumarvoja pitäisi seurata 1) ennen ensimmäistä Xgeva-annosta, 2) kahden viikon kuluessa ensimmäisestä annoksesta, 3) jos havaitaan hypokalsemiaan viittaavia oireita (ks. tietoa oireista kohdasta Haittavaikutukset). Kalsiumarvojen seurantaa on harkittava hoidon aikana myös, jos potilaalla on hypokalsemian riskitekijöitä tai jos se on muuten aiheellista potilaan kliinisen tilan perusteella.
Potilaita on kehotettava ilmoittamaan hypokalsemiaan viittaavista oireista. Jos Xgeva-hoidon aikana esiintyy hypokalsemiaa, ylimääräinen kalsiumlisä ja kalsiumarvojen lisäseuranta saattavat olla tarpeen.
Markkinoille tulon jälkeen on raportoitu vaikeaa (myös kuolemaan johtanutta) oireista hypokalsemiaa (ks. kohta Haittavaikutukset). Useimmat tapaukset ilmaantuivat ensimmäisinä viikkoina hoidon aloittamisen jälkeen, mutta hypokalsemiaa voi esiintyä myöhemminkin.
Munuaisten vajaatoiminta
Hypokalsemian riski on suurempi potilailla, joilla on vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma < 30 ml/min) tai jotka ovat dialyysihoidossa. Hypokalsemian ja siihen liittyvän lisäkilpirauhashormonipitoisuuksien suurenemisen riski on sitä suurempi mitä vaikea-asteisempi munuaisten vajaatoiminta on. Kalsiumarvojen säännöllinen seuranta on näissä tapauksissa erityisen tärkeää.
Leuan luukuolio
Leuan luukuoliota on raportoitu yleisesti Xgeva-hoitoa saavilla potilailla (ks. kohta Haittavaikutukset).
Hoidon aloittamista tai uutta hoitojaksoa on lykättävä, jos potilaalla on huonosti paranevia avoimia pehmytkudosvaurioita suussa. Hammastarkastusta ja ehkäisevää hammashoitoa sekä yksilöllistä riski‑hyötyarviota suositellaan ennen denosumabihoidon aloittamista.
Seuraavat riskitekijät on otettava huomioon leuan luukuolion riskiä arvioitaessa:
- luun hajoamista estävän lääkevalmisteen voimakkuus (erittäin voimakkaiden lääkeaineiden käyttöön liittyy suurempi riski), antotapa (parenteraaliseen hoitoon liittyy suurempi riski) ja luun hajoamista estävän lääkityksen kumulatiivinen annos.
- syöpä, muut samanaikaiset sairaudet (esim. anemia, hyytymishäiriöt, infektio), tupakointi.
- muu samanaikainen hoito: kortikosteroidit, solunsalpaajat, angiogeneesin estäjät, pään ja kaulan alueen sädehoito.
- huono suuhygienia, hampaan kiinnityskudoksen sairaus, huonosti istuvat hammasproteesit, olemassa oleva hammassairaus, invasiiviset hammastoimenpiteet (esim. hampaanpoistot).
Kaikkia potilaita on kehotettava huolehtimaan hyvin suuhygieniasta, käymään säännöllisesti hammastarkastuksessa ja ilmoittamaan heti, jos denosumabihoidon aikana esiintyy suuoireita, kuten hampaiden heilumista, kipua tai turvotusta tai huonosti paranevia haavoja tai eritevuotoa. Hoidon aikana invasiivisia hammastoimenpiteitä on tehtävä vain huolellisen harkinnan jälkeen, eikä niitä pidä ajoittaa lähelle Xgeva-annoksen antamista.
Jos potilaalle kehittyy leuan luukuolio, hoitavan lääkärin on tehtävä hoitosuunnitelma yhteistyössä leuan luukuolion hoitoon perehtyneen hammaslääkärin tai suukirurgin kanssa. Xgeva-hoidon keskeyttämistä on harkittava, kunnes tila paranee ja riskitekijät vähenevät, mikäli mahdollista.
Korvakäytävän osteonekroosi
Denosumabin käytön yhteydessä on raportoitu korvakäytävän osteonekroosia. Korvakäytävän osteonekroosin mahdollisia riskitekijöitä ovat steroidien käyttö ja solunsalpaajahoito ja/tai paikalliset riskitekijät, kuten infektio tai vamma. Korvakäytävän osteonekroosin mahdollisuus on otettava huomioon, jos denosumabihoitoa saavalla potilaalla on korvaoireita, krooniset korvatulehdukset mukaan lukien.
Epätyypilliset reisiluun murtumat
Denosumabihoitoa saavilla potilailla on raportoitu epätyypillisiä reisiluun murtumia (ks. kohta Haittavaikutukset). Subtrokanteeriselle alueelle tai reisiluun varteen voi syntyä epätyypillisiä murtumia vain vähäisen trauman seurauksena tai ilman traumaa. Näihin tapahtumiin liittyy spesifisiä radiologisia löydöksiä. Epätyypillisiä reisiluun murtumia on raportoitu myös potilailla, joilla on samanaikaisesti tiettyjä muita sairauksia tai häiriöitä (esim. D-vitamiinin puutos, nivelreuma, hypofosfatasia), ja myös joidenkin lääkkeiden (esim. bisfosfonaattien, glukokortikoidien, protonipumpun estäjien) käytön yhteydessä. Näitä tapahtumia on esiintynyt myös potilailla, jotka eivät ole saaneet luun hajoamista vähentäviä lääkkeitä. Bisfosfonaatteja saaneilla potilailla raportoidut samanlaiset murtumat ovat olleet usein molemminpuolisia. Siksi myös vastakkainen reisiluu on tutkittava, jos denosumabihoitoa saavalla potilaalla todetaan reisiluun varren murtuma. Xgeva-hoidon keskeyttämistä on harkittava, jos potilaalla epäillään epätyypillistä reisiluun murtumaa, kunnes potilaasta on tehty yksilöllinen riski‑hyötyarvio. Denosumabihoitoa saavia potilaita on kehotettava kertomaan lääkärille uudesta tai epätavallisesta reiden, lonkan tai nivustaipeen kivusta. Jos tällaisia oireita ilmaantuu, potilas on tutkittava mahdollisen epätäydellisen reisiluun murtuman havaitsemiseksi.
Hyperkalsemia hoidon päättymisen jälkeen potilailla, joilla on luun jättisolukasvain tai joilla luuston kasvu on kesken
Kun Xgeva‑hoitoa on annettu potilaille, joilla on luun jättisolukasvain, kliinisesti merkittävää hyperkalsemiaa, joka on vaatinut sairaalahoitoa ja jonka komplikaationa on akuutti munuaisvaurio, on raportoitu viikkojen–kuukausien kuluttua hoidon päättymisestä.
Kun hoito on päättynyt, tarkkaile, esiintyykö potilaalla hyperkalsemian oireita, harkitse seerumin kalsiumarvojen seuraamista säännöllisesti ja arvioi potilaan kalsium- ja D‑vitamiinilisien tarve uudelleen (ks. kohta Haittavaikutukset).
Xgevaa ei suositella potilaille, joiden luuston kasvu ei ole päättynyt (ks. kohta Annostus ja antotapa). Kliinisesti merkittävää hyperkalsemiaa on raportoitu myös näillä potilailla viikkojen–kuukausien kuluttua hoidon päättymisestä.
Muut
Xgeva-hoitoa saaville potilaille ei saa antaa samanaikaisesti denosumabia sisältäviä muita lääkkeitä (osteoporoosin hoitoon).
Bisfosfonaattihoitoa ei saa antaa samanaikaisesti Xgeva-hoidon kanssa.
Luun jättisolukasvaimen muuttuminen pahanlaatuiseksi tai eteneminen etäpesäkkeitä lähettäväksi taudiksi on harvinainen tapahtuma ja luun jättisolukasvaimeen liittyvä tunnettu riski. Potilaiden tilaa on seurattava pahanlaatuisuuteen viittaavien radiologisten löydösten, uusien kirkastumien tai osteolyysin havaitsemiseksi. Käytettävissä olevat kliiniset tiedot eivät viittaa siihen, että luun jättisolukasvaimen pahanlaatuistumisen riski olisi suurentunut Xgeva-hoitoa saaneilla potilailla.
Apuaineita koskevat varoitukset
Tämä lääkevalmiste sisältää sorbitolia. Sorbitolia (tai fruktoosia) sisältävien muiden valmisteiden samanaikaisen annon sekä ravinnosta saatavan sorbitolin (tai fruktoosin) additiivinen vaikutus on huomioitava.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 120 mg:n annos eli sen voidaan sanoa olevan "natriumiton".
Tämä lääkevalmiste sisältää 0,17 mg polysorbaatti 20:tä per 1,7 ml liuosta injektiopullossa.
Tämä lääkevalmiste sisältää 0,1 mg polysorbaatti 20:tä per 1,0 ml liuosta esitäytetyssä ruiskussa.
Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Potilaat, joilla on fenyyliketonuria (PKU)
Xgeva 120 mg/1,7 ml injektioneste kertakäyttöisessä injektiopullossa ei sisällä fenyylialaniinia. Jos potilaalla on fenyyliketonuria (PKU), Xgeva annetaan hänelle kertakäyttöisestä injektiopullosta, joka sisältää 120 mg 1,7 millilitrassa liuosta.
Xgeva 120 mg/1,0 ml injektioneste kerta-annoksen sisältävässä esitäytetyssä ruiskussa sisältää 6,1 mg fenyylialaniinia. Fenyylialaniini saattaa olla haitallista potilaille, joilla on fenyyliketonuria (PKU), harvinainen perinnöllinen sairaus, jossa kertyy fenyylialaniinia, koska elimistö ei kykene poistamaan sitä kunnolla.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Kliinisissä tutkimuksissa Xgevaa on annettu yhdessä tavanomaisten syöpähoitojen kanssa ja potilaille, jotka ovat saaneet aikaisemmin bisfosfonaatteja. Samanaikainen solunsalpaaja- ja/tai hormonihoito tai aikaisempi laskimoon annettu bisfosfonaattihoito eivät vaikuttaneet kliinisesti merkittävästi denosumabin jäännöspitoisuuksiin (trough) seerumissa eivätkä sen farmakodynamiikkaan (kreatiniiniin suhteutettu virtsan N-telopeptidi, uNTX/Cr).
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja denosumabin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Xgevaa ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä. Naisia on kehotettava välttämään raskaaksi tulemista Xgeva-hoidon aikana ja vähintään 5 kuukauden ajan hoidon jälkeen. Xgevan mahdolliset vaikutukset ovat todennäköisesti suurempia raskauden toisen ja kolmannen kolmanneksen aikana, sillä istukan läpi kulkeutuvien monoklonaalisten vasta-aineiden määrä suurenee lineaarisesti raskauden edetessä, ja istukan läpäisevä määrä on suurin kolmannen raskauskolmanneksen aikana.
Imetys
Ei tiedetä, erittyykö denosumabi ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Tutkimukset poistogeenisillä hiirillä viittaavat siihen, että RANKL:n puuttuminen tiineyden aikana voi häiritä maitorauhasen kehittymistä ja heikentää maidoneritystä synnytyksen jälkeen (ks. kohta Prekliiniset tiedot turvallisuudesta). On päätettävä lopetetaanko rintaruokinta vai lopetetaanko Xgeva-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Denosumabin vaikutuksesta ihmisen hedelmällisyyteen ei ole tutkimustietoa. Eläinkokeissa ei ole havaittu suoria tai epäsuoria hedelmällisyyteen kohdistuvia haittoja (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Xgevalla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Yleinen turvallisuusprofiili on yhdenmukainen Xgevan kaikissa hyväksytyissä käyttöaiheissa.
Hypokalsemiaa on raportoitu hyvin yleisesti Xgevan annon jälkeen, useimmiten kahden ensimmäisen viikon aikana. Hypokalsemia voi olla vaikeaa, ja siihen voi liittyä oireita (ks. kohta Haittavaikutukset – Tärkeimpien haittavaikutusten kuvaus). Seerumin kalsiumarvon lasku saatiin yleensä hallintaan antamalla kalsium- ja D-vitamiinilisää. Xgeva-hoidon yleisin haittavaikutus on lihas- ja luustokipu. Xgevaa käyttävillä potilailla on havaittu yleisesti leuan luukuoliota (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset – Tärkeimpien haittavaikutusten kuvaus).
Haittavaikutustaulukko
Haittavaikutusten yleisyysluokat on määritelty seuraavasti neljässä kolmannen vaiheen ja kahdessa toisen vaiheen kliinisessä tutkimuksessa raportoitujen ilmaantuvuuksien ja markkinoille tulon jälkeen saatujen kokemusten perusteella (ks. taulukko 1): hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1000, < 1/100), harvinainen (≥ 1/10 000, < 1/1000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyys- ja elinjärjestelmäluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Raportoidut haittavaikutukset potilailla, joilla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä, multippeli myelooma tai luun jättisolukasvain
| Elinjärjestelmä (MedDRA) | Yleisyys | Haittavaikutukset |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Yleinen | Uusi primaarinen syöpä1 |
| Immuunijärjestelmä | Harvinainen | Lääkeyliherkkyys1 |
| Harvinainen | Anafylaktinen reaktio1 | |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Hypokalsemia1, 2 |
| Yleinen | Hypofosfatemia | |
| Melko harvinainen | Hyperkalsemia hoidon päättymisen jälkeen potilailla, joilla on luun jättisolukasvain3 | |
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Hengenahdistus |
| Ruoansulatuselimistö | Hyvin yleinen | Ripuli |
| Yleinen | Hampaanpoisto | |
| Iho ja ihonalainen kudos | Yleinen | Runsas hikoilu |
| Melko harvinainen | Likenoidit lääkeihottumat1 | |
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Lihas- ja luustokipu1 |
| Yleinen | Leuan luukuolio1 | |
| Melko harvinainen | Epätyypillinen reisiluun murtuma1 | |
| Tuntematon | Korvakäytävän osteonekroosi3, 4 | |
| Yleisoireet ja antopaikassa todettavat haitat | Melko harvinainen | Injektiokohdan reaktiot5 |
1 Ks. kohta Tärkeimpien haittavaikutusten kuvaus. 2 Ks. kohta Muut erityisryhmät. 3 Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet. 4 Lääkeaineryhmälle tyypillinen vaikutus. | ||
Tärkeimpien haittavaikutusten kuvaus
Hypokalsemia
Luustotapahtumien ehkäisemistä koskevissa kliinisissä tutkimuksissa hypokalsemian ilmaantuvuus oli suurempi denosumabia kuin tsoledronihappoa saaneilla potilailla.
Hypokalsemian ilmaantuvuus oli suurin kolmannen vaiheen kliinisessä tutkimuksessa, johon osallistuneilla potilailla oli multippeli myelooma. Hypokalsemiaa raportoitiin 16,9 prosentilla Xgevaa ja 12,4 prosentilla tsoledronihappoa saaneista potilaista. Kolmannen asteen seerumin kalsiumpitoisuuden pieneneminen todettiin 1,4 prosentilla Xgevaa ja 0,6 prosentilla tsoledronihappoa saaneista potilaista. Neljännen asteen seerumin kalsiumpitoisuuden pieneneminen todettiin 0,4 prosentilla Xgevaa ja 0,1 prosentilla tsoledronihappoa saaneista potilaista.
Kolmessa vaikuttavalla vertailuaineella tehdyssä kolmannen vaiheen kliinisessä tutkimuksessa, joihin osallistuneilla potilailla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä, hypokalsemiaa raportoitiin 9,6 prosentilla Xgeva-hoitoa saaneista ja 5,0 prosentilla tsoledronihappoa saaneista potilaista.
Kolmannen asteen seerumin kalsiumpitoisuuden pieneneminen todettiin 2,5 prosentilla Xgevaa ja 1,2 prosentilla tsoledronihappoa saaneista potilaista. Neljännen asteen seerumin kalsiumpitoisuuden pieneneminen todettiin 0,6 prosentilla Xgevaa ja 0,2 prosentilla tsoledronihappoa saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kahdessa toisen vaiheen, yhden hoitohaaran kliinisessä tutkimuksessa, joihin osallistuneilla potilailla oli luun jättisolukasvain, hypokalsemiaa raportoitiin 5,7 prosentilla potilaista. Yksikään näistä haittatapahtumista ei ollut vakava.
Markkinoille tulon jälkeen on raportoitu vaikeaa (myös kuolemaan johtanutta) oireista hypokalsemiaa. Useimmat tapaukset ilmaantuivat ensimmäisinä viikkoina hoidon aloittamisen jälkeen. Vaikean oireisen hypokalsemian kliinisiä ilmenemismuotoja ovat olleet esimerkiksi pidentynyt QT-aika, tetania, kouristuskohtaukset ja psyykkisen tilan muutos (myös kooma) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kliinisissä tutkimuksissa hypokalsemian oireita olivat parestesiat tai lihasjäykkyys, lihasnykäykset, lihaskouristukset ja suonenvedot.
Leuan luukuolio
Kliinisissä tutkimuksissa leuan luukuolio yleistyi altistuksen pitkittyessä. Leuan luukuoliota on diagnosoitu myös Xgeva‑hoidon lopettamisen jälkeen. Suurin osa tapauksista ilmaantui 5 kuukauden kuluessa viimeisestä annoksesta. Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli aikaisemmin todettu leuan luukuolio tai leuan osteomyeliitti, suukirurgista hoitoa vaativa aktiivinen hampaiden tai leuan sairaus, parantumaton hammas‑ tai suukirurgisen toimenpiteen jälkitila tai suunnitteilla oleva invasiivinen hammastoimenpide.
Luustotapahtumien ehkäisemistä koskevissa kliinisissä tutkimuksissa leuan luukuolion ilmaantuvuus oli suurempi denosumabia kuin tsoledronihappoa saaneilla potilailla. Leuan luukuolion ilmaantuvuus oli suurin kolmannen vaiheen kliinisessä tutkimuksessa, johon osallistuneilla potilailla oli multippeli myelooma. Tämän tutkimuksen kaksoissokkovaiheessa leuan luukuolio varmistettiin 5,9 prosentilla Xgevaa saaneista (altistumisajan mediaani: 19,4 kuukautta, vaihteluväli: 1–52) ja 3,2 prosentilla tsoledronihappoa saaneista potilaista. Kaksoissokkovaiheen päättyessä varmistetun leuan luukuolion potilasvuosien mukaan korjattu ilmaantuvuus oli Xgeva‑ryhmässä (altistumisajan mediaani: 19,4 kuukautta, vaihteluväli: 1−52) ensimmäisen hoitovuoden aikana 2,0 sataa potilasvuotta kohti, toisen hoitovuoden aikana 5,0 ja sen jälkeen 4,5. Mediaaniaika leuan luukuolion ilmaantumiseen oli 18,7 kuukautta (vaihteluväli: 1−44).
Kolmessa vaikuttavalla vertailuaineella tehdyssä kolmannen vaiheen kliinisessä tutkimuksessa, joihin osallistuneilla potilailla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä, leuan luukuolio diagnosoitiin primaarisessa hoitovaiheessa 1,8 prosentilla Xgevaa saaneista (altistumisajan mediaani: 12,0 kuukautta, vaihteluväli: 0,1–40,5) ja 1,3 prosentilla tsoledronihappoa saaneista potilaista. Näiden tapausten kliiniset ominaisuudet olivat samanlaiset molemmissa hoitoryhmissä. Niistä potilaista, joilla leuan luukuolio oli varmistettu, suurimmalla osalla (81 % molemmissa hoitoryhmissä) oli huono suuhygienia, heiltä oli poistettu hammas ja/tai he käyttivät hammaslaitetta. Useimmat potilaat saivat tai olivat saaneet solunsalpaajahoitoa.
Tutkimuksiin, joihin osallistui rintasyöpää tai eturauhassyöpää sairastaneita potilaita, kuului myös Xgeva‑jatkohoitovaihe (koko altistumisajan mediaani: 14,9 kuukautta, vaihteluväli: 0,1–67,2). Leuan luukuolio varmistettiin 6,9 prosentilla rintasyöpä- ja eturauhassyöpäpotilaista jatkohoitovaiheen aikana.
Varmistetun leuan luukuolion potilasvuosien mukaan korjattu kokonaisilmaantuvuus oli ensimmäisen hoitovuoden aikana 1,1 sataa potilasvuotta kohti, toisen hoitovuoden aikana 3,7 ja sen jälkeen 4,6.Mediaaniaika leuan luukuolion ilmaantumiseen oli 20,6 kuukautta (vaihteluväli: 4–53).
Ruotsissa, Tanskassa ja Norjassa toteutettu satunnaistamaton, retrospektiivinen, havainnoiva tutkimus, johon osallistui 2 877 syöpäpotilasta, jotka saivat Xgevaa tai tsoledronihappoa, osoitti, että lääketieteellisesti varmistetun leuan luukuolion ilmaantuvuus oli viiden vuoden aikana 5,7 prosenttia (95 % CI: 4,4–7,3; seuranta-ajan mediaani 20 kuukautta [vaihteluväli: 0,2–60]) Xgevaa saaneessa potilaskohortissa ja 1,4 prosenttia (95 % CI: 0,8–2,3; seuranta-ajan mediaani 13 kuukautta [vaihteluväli: 0,1–60]) erillisessä, tsoledronihappoa saaneessa potilaskohortissa. Leuan luukuolion ilmaantuvuus viiden vuoden aikana potilailla, jotka vaihtoivat tsoledronihaposta Xgevaan, oli 6,6 prosenttia (95 % CI: 4,2–10,0; seuranta-ajan mediaani 13 kuukautta [vaihteluväli: 0,2–60]).
Kolmannen vaiheen tutkimuksessa potilailla, joilla oli metastasoitumaton eturauhassyöpä (joka ei ole Xgevan käyttöaihe) ja pitempi altistuksen kesto (enintään 7 vuotta), varmistetun leuan luukuolion potilasvuosien mukaan korjattu ilmaantuvuus oli ensimmäisen hoitovuoden aikana 1,1 sataa potilasvuotta kohti, toisen hoitovuoden aikana 3,0 ja sen jälkeen 7,1.
Pitkäaikaisessa toisen vaiheen avoimessa kliinisessä tutkimuksessa, johon osallistuneilla potilailla oli luun jättisolukasvain (tutkimus 6, ks. kohta Farmakodynamiikka), leuan luukuolio varmistettiin 6,8 prosentilla potilaista, joista yksi nuorella potilaalla (keskimäärin 34 annosta (mediaani); vaihteluväli 4–116). Tutkimuksen päättyessä potilaiden keskimääräinen osallistumisaika (mediaani) turvallisuuden varmistava seurantavaihe mukaan lukien oli 60,9 kuukautta (vaihteluväli: 0–112,6). Varmistetun leuan luukuolion potilasvuosien mukaan korjattu ilmaantuvuus oli kaikkiaan 1,5 sataa potilasvuotta kohti (ensimmäisen hoitovuoden aikana 0,2 sataa potilasvuotta kohti, toisen hoitovuoden aikana 1,5; kolmannen hoitovuoden aikana 1,8; neljännen hoitovuoden aikana 2,1; viidennen hoitovuoden aikana 1,4 ja sen jälkeen 2,2). Mediaaniaika leuan luukuolion ilmaantumiseen oli 41 kuukautta (vaihteluväli: 11–96).
Tutkimuksessa 7 jatkettiin tutkimuksessa 6 luun jättisolukasvaimen vuoksi hoidettujen potilaiden seurantaa vähintään viiden vuoden ajan. Leuan luukuolio raportoitiin kuudella (11,8 %) denosumabia saaneista 51 potilaasta. Heidän saamiensa denosumabiannosten lukumäärän mediaani oli 42. Näistä luukuoliotapauksista kolme varmistettiin kliinisesti.
Lääkkeestä johtuvat yliherkkyysreaktiot
Lääkkeen markkinoille tulon jälkeen Xgeva‑hoitoa saavilla potilailla on raportoitu yliherkkyysreaktioita, harvoin myös anafylaktisia reaktioita.
Epätyypilliset reisiluun murtumat
Kaiken kaikkiaan kliinisessä tutkimusohjelmassa Xgeva-hoitoa saaneilla potilailla raportoitiin epätyypillisiä reisiluun murtumia melko harvoin, ja riski kasvoi hoidon keston pidetessä. Murtumia on esiintynyt hoidon aikana ja jopa 9 kuukautta hoidon päättymisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Luun jättisolukasvaimia koskevassa kliinisessä tutkimusohjelmassa epätyypillisiä reisiluun murtumia on raportoitu yleisesti Xgeva-hoitoa saaneilla potilailla. Tutkimuksessa 6 varmistettujen epätyypillisten reisiluun murtumien ilmaantuvuus oli 0,95 % (5/526) potilailla, joilla oli luun jättisolukasvain. Jatkotutkimuksessa (tutkimus 7) varmistettujen epätyypillisten reisiluun murtumien ilmaantuvuus oli denosumabia saaneilla potilailla 3,9 % (2/51).
Lihas- ja luustokipu
Lääkkeen markkinoille tulon jälkeen Xgeva-hoitoa saaneilla potilailla on raportoitu lihas- ja luustokipua, joka on joissakin tapauksissa ollut vaikeaa. Kliinisissä tutkimuksissa lihas- ja luustokipua esiintyi hyvin yleisesti sekä denosumabiryhmässä että tsoledronihapporyhmässä. Tutkimushoidon keskeyttämiseen johtanut lihas- ja luustokipu oli melko harvinaista.
Uusi primaarinen syöpä
Neljässä vaikuttavalla vertailuaineella tehdyssä kolmannen vaiheen kliinisessä tutkimuksessa, joihin osallistuneilla potilailla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä, 54:llä Xgevaa saaneella potilaalla 3 691:stä (1,5 prosentilla) (altistumisajan mediaani: 13,8 kuukautta, vaihteluväli: 1,0–51,7) ja 33:lla tsoledronihappoa saaneella potilaalla 3 688:sta (0,9 prosentilla) (altistumisajan mediaani 12,9 kuukautta, vaihteluväli: 1,0–50,8) raportoitiin uusi primaarinen syöpä primaarisessa, kaksoissokkoutetussa hoitovaiheessa.
Kumulatiivinen ilmaantuvuus yhden vuoden jälkeen oli 1,1 % denosumabia saaneilla ja 0,6 % tsoledronihappoa saaneilla potilailla.
Yksittäisten syöpien tai syöpäryhmien yhteyttä hoitoon ei havaittu.
Uusien syöpien ilmaantuvuus tutkimuksen 6 potilailla, joilla oli luun jättisolukasvain, oli 3,8 % (20/526). Tässä huomioitiin sekä luustoon levinneet ja luukudokseen vaikuttavat että luuston ulkopuoliset syövät. Seurantatutkimuksessa (tutkimus 7) ilmaantuvuus denosumabia saaneilla potilailla oli 11,8 % (6/51).
Likenoidit lääkeihottumat
Lääkkeen markkinoille tulon jälkeen potilailla on raportoitu likenoideja lääkeihottumia (kuten punajäkälää muistuttavia reaktioita).
Pediatriset potilaat
Xgevaa annettiin avoimessa tutkimuksessa luun jättisolukasvaimen hoitoon 28 nuorelle, joiden luuston kasvu oli päättynyt. Näiden rajallisten tietojen perusteella haittatapahtumat näyttivät olevan samanlaisia kuin aikuisilla.
Markkinoille tulon jälkeen on pediatrisilla potilailla raportoitu kliinisesti merkittävää hyperkalsemiaa hoidon päättymisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muut erityisryhmät
Munuaisten vajaatoiminta
Kliinisessä tutkimuksessa potilailla, joilla ei ollut pitkälle edennyttä syöpää mutta joilla oli vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma < 30 ml/min) tai jotka olivat dialyysihoidossa, hypokalsemian riski oli suurempi, kun potilaat eivät saaneet kalsiumlisää. Hypokalsemian riski on Xgeva-hoidon aikana sitä suurempi mitä vaikea-asteisempi munuaisten vajaatoiminta on. Kliinisessä tutkimuksessa, johon osallistuneilla potilailla ei ollut pitkälle edennyttä syöpää, 19 prosentille vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastavista ja 63 prosentille dialyysihoidossa olevista potilaista kehittyi hypokalsemia, vaikka potilaat saivat kalsiumlisää. Kliinisesti merkittävän hypokalsemian kokonaisilmaantuvuus oli 9 %.
Xgeva-hoitoa saavilla potilailla, joilla on vaikea munuaisten vajaatoiminta tai jotka ovat dialyysihoidossa, on havaittu myös hypokalsemiaan liittyvää lisäkilpirauhashormonipitoisuuksien suurenemista. Kalsiumpitoisuuksien seuranta ja riittävä kalsiumin ja D-vitamiinin saanti on erityisen tärkeää potilaille, joiden munuaisten toiminta on heikentynyt (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä 1 haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta: www.fimea.fi.
Yliannostus
Kliinisissä tutkimuksissa ei ole saatu kokemuksia yliannostuksesta. Xgevaa on annettu kliinisissä tutkimuksissa enintään 180 mg neljän viikon välein ja 120 mg viikon välein kolmen viikon ajan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Luukudokseen vaikuttavat lääkkeet – muut luukudokseen vaikuttavat lääkkeet, ATC-koodi: M05BX04.
Vaikutusmekanismi
RANK-ligandi (RANKL) esiintyy solukalvon läpäisevänä tai liukoisena proteiinina. RANKL on välttämätön ainoan luuta hajottavan solutyypin, osteoklastien, muodostumiselle, toiminnalle ja elinkelpoisuudelle. RANKL:n stimuloima osteoklastien aktiivisuuden lisääntyminen on tärkeä välittäjä luustoetäpesäkkeisiin ja multippeliin myeloomaan liittyvässä luutuhossa. Denosumabi on ihmisen monoklonaalinen IgG2-vasta-aine, joka sitoutuu suurella affiniteetilla ja erittäin spesifisesti vaikutuskohteeseensa RANK-ligandiin ja estää näin RANKL:n ja RANK:n vuorovaikutuksen. Tämä vähentää osteoklastien lukumäärää ja aktiivisuutta ja sen myötä myös luun hajoamista ja syövän aiheuttamaa luutuhoa.
Luun jättisolukasvaimille ovat ominaisia RANK-ligandia ilmentävät neoplastiset stroomasolut ja RANK-reseptoria ilmentävät osteoklastien kaltaiset jättisolut. Luun jättisolukasvaimissa denosumabi sitoutuu RANK-ligandiin ja vähentää näin merkitsevästi osteoklastien kaltaisia jättisoluja tai hävittää ne kokonaan. Tämän seurauksena osteolyysi vähenee ja proliferatiivinen kasvaimen strooma korvautuu ei-proliferatiivisella, erilaistuneella tiiviillä uudella luulla.
Farmakodynaamiset vaikutukset
Toisen vaiheen kliinisissä tutkimuksissa, joihin osallistuneilla potilailla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä, Xgevaa annettiin ihon alle joko neljän viikon tai 12 viikon välein, mikä johti luun hajoamisen merkkiaineiden (uNTX/Cr, seerumin CTx) nopeaan vähenemiseen. uNTX/Cr-arvo laski viikon kuluessa keskimäärin 80 % (mediaani) aikaisemmasta bisfosfonaattihoidosta tai uNTX/Cr-lähtöarvosta riippumatta. Kolmannen vaiheen kliinisissä tutkimuksissa, joihin osallistuneilla potilailla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä, uNTX/Cr-arvon noin 80 prosentin alenema (mediaani) säilyi koko 49 viikkoa jatkuneen Xgeva-hoidon (120 mg neljän viikon välein) ajan.
Immunogeenisuus
Kliinisissä tutkimuksissa ei havaittu denosumabia neutraloivia vasta-aineita potilailla, joilla on pitkälle edennyt syöpä tai luun jättisolukasvain. Herkässä immunomäärityksessä alle prosentilla potilaista, jotka saivat denosumabia enintään kolmen vuoden ajan, todettiin ei-neutraloivia sitoutuvia vasta-aineita, eikä heillä havaittu farmakokinetiikan, toksisuuden eikä kliinisen vasteen muuttumista.
Kliininen teho ja turvallisuus potilailla, joilla oli kiinteiden kasvainten luustoetäpesäkkeitä
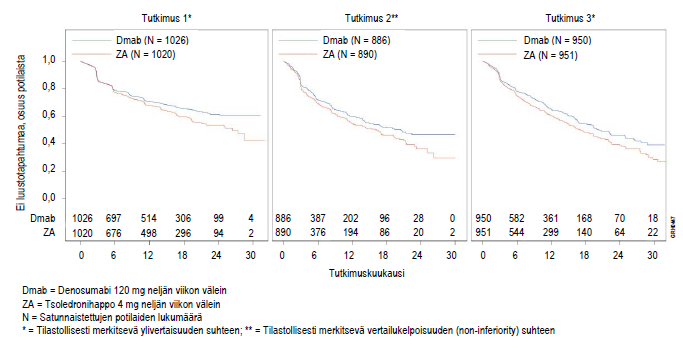
Xgevan (120 mg ihon alle neljän viikon välein) tehoa ja turvallisuutta verrattiin tsoledronihappoon (4 mg laskimoon neljän viikon välein; annosta säädettiin, jos munuaisten toiminta oli heikentynyt) kolmessa satunnaistetussa kaksoissokkoutetussa vaikuttavalla vertailuaineella tehdyssä tutkimuksessa. Tutkimukset tehtiin aikuispotilailla, jotka eivät olleet aikaisemmin saaneet laskimoon annettavia bisfosfonaatteja ja joilla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä: rintasyöpä (tutkimus 1), muita kiinteitä kasvaimia tai multippeli myelooma (tutkimus 2) tai kastraatioresistentti eturauhassyöpä (tutkimus 3). Turvallisuutta arvioitiin näissä vaikuttavalla vertailuaineella tehdyissä kliinisissä tutkimuksissa 5931 potilaalla. Näihin tutkimuksiin ei otettu mukaan potilaita, joilla oli aikaisemmin todettu leuan luukuolio tai leuan osteomyeliitti, suukirurgista hoitoa vaativa aktiivinen hampaiden tai leuan sairaus, parantumaton hammas- tai suukirurgisen toimenpiteen jälkitila tai suunnitteilla oleva invasiivinen hammastoimenpide. Ensisijaisella ja toissijaisella päätetapahtumalla arvioitiin yhden tai useamman luustotapahtuman esiintymistä. Tutkimuksissa, joissa Xgeva osoittautui tilastollisesti ylivertaiseksi tsoledronihappoon nähden, potilaille annettiin Xgevaa etukäteen määritellyssä avoimessa kahden vuoden jatkohoitovaiheessa. Luustotapahtumaksi katsottiin mikä tahansa seuraavista: patologinen murtuma (nikamamurtuma tai muu murtuma), luuston sädehoito (myös radioisotooppien käyttö), luuston kirurginen toimenpide tai selkäydinkompressio.
Xgeva pienensi yhden luustotapahtuman ja useampien (ensimmäisen ja sitä seuraavien) luustotapahtumien ilmaantumisen riskiä potilailla, joilla oli kiinteiden kasvainten luustoetäpesäkkeitä (ks. taulukko 2).
Taulukko 2. Tehoa kuvaavat tulokset potilailla, joilla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä
| Tutkimus 1 rintasyöpä | Tutkimus 2 muut kiinteät kasvaimet** tai multippeli myelooma | Tutkimus 3 eturauhassyöpä | Yhdistetyt tulokset pitkälle edennyt syöpä | |||||
| Xgeva | tsoledronihappo | Xgeva | tsoledronihappo | Xgeva | tsoledronihappo | Xgeva | tsoledronihappo | |
| N | 1026 | 1020 | 886 | 890 | 950 | 951 | 2862 | 2861 |
| Ensimmäinen luustotapahtuma | ||||||||
| Mediaaniaika (kk) | NR | 26,4 | 20,6 | 16,3 | 20,7 | 17,1 | 27,6 | 19,4 |
| Mediaaniaikojen ero (kk) | NA | 4,2 | 3,5 | 8,2 | ||||
| HR (95 % CI) / RRR (%) | 0,82 (0,71–0,95) / 18 | 0,84 (0,71–0,98) / 16 | 0,82 (0,71–0,95) / 18 | 0,83 (0,76–0,90) / 17 | ||||
| Vertailukelpoisuus (non-inferiority) / ylivertaisuus, p‑arvot | < 0,0001† /0,0101† | 0,0007† /0,0619† | 0,0002† / 0,0085† | < 0,0001 / < 0,0001 | ||||
| Osuus potilaista (%) | 30,7 | 36,5 | 31,4 | 36,3 | 35,9 | 40,6 | 32,6 | 37,8 |
| Ensimmäinen ja sitä seuraavat luustotapahtumat* | ||||||||
| Lukumäärä/potilas (keskiarvo) | 0,46 | 0,60 | 0,44 | 0,49 | 0,52 | 0,61 | 0,48 | 0,57 |
| Esiintymistiheyksien suhde (95 % CI) / RRR (%) | 0,77 (0,66–0,89) / 23 | 0,90 (0,77–1,04) / 10 | 0,82 (0,71–0,94) / 18 | 0,82 (0,75–0,89) / 18 | ||||
| Ylivertaisuus, p‑arvo | 0,0012† | 0,1447† | 0,0085† | < 0,0001 | ||||
| SMR / vuosi | 0,45 | 0,58 | 0,86 | 1,04 | 0,79 | 0,83 | 0,69 | 0,81 |
| Ensimmäinen luustotapahtuma tai syövästä johtuva hyperkalsemia (HCM) | ||||||||
| Mediaaniaika (kk) | NR | 25,2 | 19,0 | 14,4 | 20,3 | 17,1 | 26,6 | 19,4 |
| HR (95 % CI) / RRR (%) | 0,82 (0,70–0,95) / 18 | 0,83 (0,71–0,97) / 17 | 0,83 (0,72–0,96) / 17 | 0,83 (0,76–0,90) / 17 | ||||
| Ylivertaisuus, p‑arvo | 0,0074 | 0,0215 | 0,0134 | < 0,0001 | ||||
| Ensimmäinen luuston sädehoito | ||||||||
| Mediaaniaika (kk) | NR | NR | NR | NR | NR | 28,6 | NR | 33,2 |
| HR (95 % CI) / RRR (%) | 0,74 (0,59–0,94) / 26 | 0,78 (0,63–0,97) / 22 | 0,78 (0,66–0,94) / 22 | 0,77 (0,69–0,87) / 23 | ||||
| Ylivertaisuus, p‑arvo | 0,0121 | 0,0256 | 0,0071 | < 0,0001 | ||||
NR (not reached) = ei saavutettu, NA (not available) = ei saatavissa, HCM (hypercalcaemia of malignancy) = syövästä johtuva hyperkalsemia, SMR (skeletal morbidity rate) = luustotapahtumien ilmaantuvuus, HR (Hazard Ratio) = vaarasuhde; RRR (Relative Risk Reduction) = suhteellinen riskin väheneminen †Korjatut p-arvot on esitetty tutkimuksista 1, 2 ja 3 (päätetapahtumat ensimmäinen luustotapahtuma ja ensimmäinen ja sitä seuraavat luustotapahtumat); *Kaikki ajan myötä ilmaantuneet luustotapahtumat; vain tapahtumat, joiden välinen aika oli ≥ 21 vuorokautta, on laskettu. ** Mukaan lukien ei-pienisoluinen keuhkosyöpä, munuaissyöpä, kolorektaalisyöpä, pienisoluinen keuhkosyöpä, rakkosyöpä, pään ja kaulan alueen syöpä, ruoansulatuskanavan/urogenitaalialueen syöpä ja muut; pois lukien rintasyöpä ja eturauhassyöpä. | ||||||||
Kuva 1. Kaplan-Meierin kuvaajat – aika ensimmäisen tutkimuksenaikaisen luustotapahtuman ilmaantumiseen
Taudin eteneminen ja kokonaiselinaika potilailla, joilla oli kiinteiden kasvainten luustoetäpesäkkeitä
Taudin eteneminen oli samanlaista Xgeva- ja tsoledronihapporyhmissä kaikissa kolmessa tutkimuksessa ja etukäteen määritellyssä kaikkien tutkimusten yhdistetyssä analyysissä.
Tutkimuksissa 1, 2 ja 3 kokonaiselinaikojen välillä ei ollut eroa Xgeva- ja tsoledronihapporyhmien välillä potilailla, joilla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä: rintasyöpäpotilaat (vaarasuhde (HR) ja 95 % CI oli 0,95 [0,81–1,11]), eturauhassyöpäpotilaat (vaarasuhde ja 95 % CI oli 1,03 [0,91–1,17]) ja potilaat, joilla oli muita kiinteitä kasvaimia tai multippeli myelooma (vaarasuhde ja 95 % CI oli 0,95 [0,83–1,08]). Tutkimuksen 2 (potilaat, joilla oli muita kiinteitä kasvaimia tai multippeli myelooma) post hoc ‑analyysissä tarkasteltiin kokonaiselinaikaa stratifikaation mukaisissa kolmessa kasvaintyypissä (ei‑pienisoluinen keuhkosyöpä, multippeli myelooma ja muut). Kokonaiselinaika oli ei-pienisoluista keuhkosyöpää sairastaneilla pitempi Xgeva-ryhmässä (vaarasuhde [95 % CI] 0,79 [0,65–0,95]; n = 702) ja multippelia myeloomaa sairastaneilla pitempi tsoledronihapporyhmässä (vaarasuhde [95 % CI] 2,26 [1,13–4,50]; n = 180) ja muissa kasvaintyypeissä yhtä pitkä Xgeva- ja tsoledronihapporyhmässä (vaarasuhde [95 % CI] 1,08 [0,90–1,30]; n = 894). Taudin ennusteeseen vaikuttavia tekijöitä ja syöpälääkitystä ei otettu huomioon tässä tutkimuksessa. Tutkimusten 1, 2 ja 3 etukäteen määritellyssä yhdistetyssä analyysissä kokonaiselinaika oli samanlainen Xgeva- ja tsoledronihapporyhmissä (vaarasuhde ja 95 % CI 0,99 [0,91–1,07]).
Vaikutus kipuun
Aika kivun lievittymiseen (≥ 2 pisteen lasku lähtötasosta BPI-SF-kipuasteikon voimakkainta kipua kuvaavassa pistearvossa) oli samanlainen denosumabi- ja tsoledronihapporyhmissä jokaisessa tutkimuksessa ja yhdistetyissä analyyseissä. Yhdistetyn tutkimusaineiston post hoc –analyysissä mediaaniaika kivun pahenemiseen (voimakkaimman kivun pistearvo > 4) oli pitempi Xgeva-ryhmässä (198 vuorokautta) kuin tsoledronihapporyhmässä (143 vuorokautta) (p = 0,0002) potilailla, joilla ei ollut kipuja lähtötilanteessa tai kipu oli lievää.
Kliininen teho multippelia myeloomaa sairastavien potilaiden hoidossa
Kansainvälisessä, satunnaistetussa (1:1), vaikuttavalla vertailuaineella tehdyssä kaksoissokkotutkimuksessa Xgevaa verrattiin tsoledronihappoon hiljattain diagnosoitua multippelia myeloomaa sairastavien potilaiden hoidossa (tutkimus 4).
Tässä tutkimuksessa 1718:lle multippelia myeloomaa sairastavalle potilaalle, joilla oli vähintään yksi luuleesio, annettiin satunnaistetusti joko 120 mg Xgevaa ihon alle neljän viikon välein tai 4 mg tsoledronihappoa laskimoon neljän viikon välein (annosta säädettiin munuaisten toiminnan mukaan). Ensisijainen lopputulosmuuttuja oli ensimmäisen luustotapahtuman ilmaantumiseen kuluvan ajan vertailukelpoisuuden (non-inferiority) osoittaminen tsoledronihappoon verrattuna. Toissijaisia lopputulosmuuttujia olivat ensimmäisen luustotapahtuman ilmaantumiseen kuluvan ajan ylivertaisuus, ensimmäisen ja sitä seuraavien luustotapahtumien ilmaantumiseen kuluvan ajan ylivertaisuus ja kokonaiselinaika. Luustotapahtumaksi katsottiin mikä tahansa seuraavista: patologinen murtuma (nikamamurtuma tai muu murtuma), luuston sädehoito (myös radioisotooppien käyttö), luuston kirurginen toimenpide tai selkäydinkompressio.
Molemmissa tutkimushaaroissa 54,5 prosentille potilaista oli tarkoitus tehdä autologinen perifeerisen veren kantasolujen siirto, 95,8 % potilaista sai tai heille oli suunniteltu ensilinjan hoitona uudentyyppistä myeloomahoitoa (uudentyyppisiä hoitomuotoja ovat bortetsomibi, lenalidomidi ja talidomidi) ja 60,7 prosentilla potilaista oli aikaisemmin ollut luustotapahtuma. Molempien tutkimushaarojen potilaista 32,4 prosentilla oli diagnosointihetkellä ISS-ennusteluokan I tauti, 38,2 prosentilla oli ISS-luokan II tauti ja 29,3 prosentilla oli ISS-luokan III tauti.
Xgevaa annettiin keskimäärin 16 annosta (mediaani) ja tsoledronihappoa keskimäärin 15 annosta (mediaani).
Kuvassa 2 ja taulukossa 3 ovat tehoa kuvaavat tulokset tutkimuksesta 4.
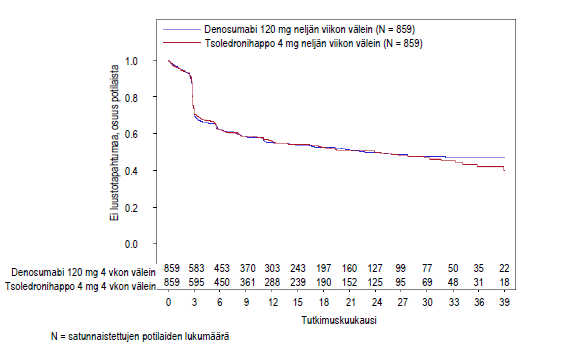
Kuva 2. Kaplan–Meierin kuvaaja – aika ensimmäisen tutkimuksenaikaisen luustotapahtuman ilmaantumiseen potilailla, joilla oli hiljattain diagnosoitu multippeli myelooma
Taulukko 3. Xgevan tehoa kuvaavat tulokset tsoledronihappoon verrattuna potilailla, joilla oli hiljattain diagnosoitu multippeli myelooma
Xgeva (N = 859) | Tsoledronihappo (N = 859) | |
| Ensimmäinen luustotapahtuma | ||
| Potilaita, joilla oli luustotapahtumia (%) | 376 (43,8) | 383 (44,6) |
| Mediaaniaika luustotapahtuman ilmaantumiseen (kk) | 22,8 (14,7–NE) | 23,98 (16,56–33,31) |
| Vaarasuhde (HR) (95 % CI) | 0,98 (0,85–1,14) | |
| Ensimmäinen ja sitä seuraavat luustotapahtumat | ||
| Tapahtumien lukumäärä / potilas (keskiarvo) | 0,66 | 0,66 |
| Esiintymistiheyksien suhde (RR) (95 % CI) | 1,01 (0,89–1,15) | |
| Luustotapahtumien ilmaantuvuus / vuosi | 0,61 | 0,62 |
| Ensimmäinen luustotapahtuma tai syövästä johtuva hyperkalsemia (HCM) | ||
| Mediaaniaika (kk) | 22,14 (14,26–NE) | 21,32 (13,86–29,7) |
| Vaarasuhde (HR) (95 % CI) | 0,98 (0,85–1,12) | |
| Ensimmäinen luuston sädehoito | ||
| Vaarasuhde (HR) (95 % CI) | 0,78 (0,53–1,14) | |
| Kokonaiselinaika | ||
| Vaarasuhde (HR) (95 % CI) | 0,90 (0,70–1,16) | |
| NE = ei arvioitavissa | ||
Kliininen teho ja turvallisuus luun jättisolukasvaimen hoidossa aikuisilla ja nuorilla, joiden luuston kasvu on päättynyt
Xgevan turvallisuutta ja tehoa luun jättisolukasvaimen hoidossa tutkittiin kahdessa toisen vaiheen avoimessa, yhden hoitohaaran tutkimuksessa (tutkimukset 5 ja 6) 554 potilaalla. Potilailla oli luun jättisolukasvain, jonka kirurginen poisto ei ollut mahdollista tai poistoleikkaus johtaisi vaikeaan toimintakyvyn heikkenemiseen. Lisäksi turvallisuuden seurantaa varten toteutettiin prospektiivinen avoin neljännen vaiheen monikeskustutkimus (tutkimus 7) tutkimuksen 6 päättäneillä potilailla. Potilaat saivat 120 mg Xgevaa ihon alle neljän viikon välein ja kyllästysannoksena 120 mg 8. ja 15. päivänä. Potilaat, jotka lopettivat Xgeva-hoidon, siirrettiin turvallisuuden seurantavaiheeseen vähintään 60 kuukauden ajaksi. Potilaille sai antaa Xgeva-hoitoa uudelleen turvallisuuden seurantavaiheessa (esimerkiksi taudin uusiutuessa), jos he olivat saavuttaneet vasteen Xgeva-hoitoon ensimmäisellä hoitokerralla.
Tutkimukseen 5 otettiin 37 aikuispotilasta, joilla oli histologisesti varmistettu leikkaukseen soveltumaton tai uusiutuva luun jättisolukasvain. Tutkimuksen ensisijainen lopputulosmuuttuja oli hoitovaste. Hoitovasteen kriteerinä oli jättisolujen häviäminen vähintään 90‑prosenttisesti lähtötasoon verrattuna (tai jättisolujen häviäminen kokonaan, kun niiden osuus kasvainsoluista oli alle 5 %). Ellei histopatologinen toteaminen ollut mahdollista, hoitovasteeksi luettiin radiologisissa mittauksissa todettu kohdeleesion etenemättömyys. Tehoa mittaavassa analyysissä oli mukana 35 potilasta, joista 85,7 prosentilla (95 % CI: 69,7–95,2) todettiin Xgeva‑hoitovaste. Kaikki histologisesti arvioidut 20 potilasta (100 %) täyttivät hoitovasteelle asetetut kriteerit. Muista 15 potilaasta 10:llä (67 %) ei havaittu kohdeleesion etenemistä radiologisissa mittauksissa.
Tutkimukseen 6 otettiin 535 aikuispotilasta tai nuorta potilasta, joiden luuston kasvu oli päättynyt. Potilailla oli luun jättisolukasvain. Potilaista 28 oli 12–17‑vuotiaita. Potilaat jaettiin kolmeen kohorttiin: kohorttiin 1 potilaat, joilla oli leikkaukseen soveltumaton tauti (esim. sakraaliset tai selkärangan leesiot tai useita leesioita, kuten keuhkometastaaseja), kohorttiin 2 potilaat, joilla oli leikkaukseen soveltuva tauti ja joiden suunniteltuun leikkaukseen liittyi vaikea toimintakyvyn heikkeneminen (esim. nivelresektio, raajan amputointi tai hemipelvektomia), ja kohorttiin 3 potilaat, jotka olivat aikaisemmin osallistuneet tutkimukseen 5 ja siirrettiin tähän tutkimukseen. Ensisijaisena tavoitteena oli arvioida denosumabin turvallisuus potilailla, joilla on luun jättisolukasvain. Tutkimuksen toissijaisiin lopputulosmuuttujiin kuului kohortissa 1 aika taudin etenemiseen (tutkijan arvion mukaan) ja kohortissa 2 osuus potilaista, joille ei ollut tehty leikkausta 6 kuukauden kuluttua.
Lopullisessa analyysissä tauti eteni 28:lla hoidetuista 260 potilaasta (10,8 %) kohortissa 1. Kohortissa 2 arvioitavissa olleista 238:sta Xgeva‑hoitoa saaneesta potilaasta 219:lle (92,0 %; 95 % CI: 87,8 %, 95,1 %) ei ollut tehty leikkausta 6 kuukauden kuluttua. Leikkausta ei tehty tutkimuksen aikana kaikkiaan 82 potilaalle (34,3 %) niistä 239 potilaasta, jotka kuuluivat kohorttiin 2 ja joiden kohdeleesio ei lähtötilanteessa eikä tutkimuksen aikana sijainnut keuhkoissa tai pehmytkudoksissa. Pääsääntöisesti tehoa kuvaavat tulokset olivat hyvin samankaltaisia aikuisilla ja nuorilla, joiden luuston kasvu on päättynyt.
Tutkimukseen 7 otettiin 85 aikuispotilasta, jotka olivat aiemmin osallistuneet tutkimukseen 6 ja päättäneet sen. Potilaille sai antaa denosumabia luun jättisolukasvaimen hoitoon, ja kaikkia potilaita seurattiin 5 vuoden ajan. Ensisijaisena tavoitteena oli arvioida denosumabin pitkän aikavälin turvallisuus potilailla, joilla on luun jättisolukasvain.
Vaikutus kipuun
Lopullisessa analyysissä, johon kohortit 1 ja 2 oli yhdistetty, raportoitiin voimakkaimman kivun kliinisesti merkittävää lievittymistä (≥ 2 pisteen lasku lähtötasosta) 30,8 prosentilla riskipotilaista (voimakkaimman kivun pistearvo lähtötilanteessa ≥ 2) viikon kuluessa hoidon aloittamisesta ja ≥ 50 prosentilla 5 viikon kuluessa. Tämä kivun lievittyminen säilyi myös kaikissa myöhemmissä arvioinneissa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Xgeva‑valmisteen käytöstä luustotapahtumien ehkäisemisessä kaikissa pediatrisissa potilasryhmissä, kun potilailla on luustoetäpesäkkeitä, ja luun jättisolukasvaimen hoidossa alle 12‑vuotiaiden pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Tutkimuksessa 6 Xgevaa on arvioitu luun jättisolukasvaimen hoidossa 28 nuoren potilaan alaryhmässä (ikäjakauma 13–17 vuotta). Potilaiden luuston kasvu oli päättynyt (vähintään yhden pitkän luun kasvu oli päättynyt, esim. olkaluun kasvulevy oli sulkeutunut) ja paino oli ≥ 45 kg. Tauti uusiutui ensimmäisen hoitovaiheen aikana yhdellä nuorella potilaalla, jolla oli leikkaukseen soveltumaton tauti (N = 14). 14 potilaasta 13:lla oli leikkaukseen soveltuva tauti ja suunniteltuun leikkaukseen liittyi vaikea toimintakyvyn heikkeneminen. Heistä kenellekään ei ollut tehty leikkausta 6 kuukauden kuluttua.
Farmakokinetiikka
Imeytyminen
Ihon alle annetun annoksen jälkeen hyötyosuus oli 62 %.
Biotransformaatio
Denosumabi koostuu yksinomaan aminohapoista ja hiilihydraateista, samoin kuin natiivi immunoglobuliini, eikä siten todennäköisesti eliminoidu maksametabolian kautta. Sen metabolian ja eliminoitumisen odotetaan noudattavan immunoglobuliinipuhdistuman reittiä, mikä johtaa pilkkoutumiseen pieniksi peptideiksi ja yksittäisiksi aminohapoiksi.
Eliminaatio
Levinnyttä syöpää sairastavilla potilailla, jotka saivat toistuvia 120 mg:n annoksia neljän viikon välein, seerumin denosumabipitoisuuksissa havaittiin noin kaksinkertainen kumuloituminen, ja vakaa tila saavutettiin kuuden kuukauden kuluttua ajasta riippumattoman farmakokinetiikan mukaisesti. Multippelia myeloomaa sairastavilla potilailla, jotka saivat 120 mg:n annoksia neljän viikon välein, jäännöspitoisuuksien mediaani vaihteli alle 8 % kuukausien 6 ja 12 välillä. Potilailla, joilla oli luun jättisolukasvain ja jotka saivat 120 mg:n annoksia neljän viikon välein ja kyllästysannoksen 8. ja 15. päivänä, vakaan tilan pitoisuus saavutettiin ensimmäisen hoitokuukauden aikana. Viikkojen 9 ja 49 välillä jäännöspitoisuuksien mediaani vaihteli alle 9 %. Potilailla, jotka lopettivat hoidon (120 mg neljän viikon välein), puoliintumisajan keskiarvo oli 28 vuorokautta (vaihteluväli 14–55 vuorokautta).
Populaatiofarmakokineettinen analyysi ei tuonut esiin kliinisesti merkittäviä vakaan tilan aikaisen systeemisen denosumabialtistuksen muutoksia suhteessa ikään (18–87 vuotta), etniseen taustaan (tutkitut olivat mustaihoisia, latinalaisamerikkalaisia, aasialaisia ja valkoihoisia), sukupuoleen tai kiinteän kasvaimen tyyppiin tai potilaan sairastamaan multippeliin myeloomaan. Painon noustessa systeeminen altistus pieneni ja päinvastoin. Näitä muutoksia ei kuitenkaan pidetty kliinisesti merkityksellisinä, sillä luun aineenvaihduntaa kuvaavien merkkiaineiden perusteella farmakodynaamiset vaikutukset olivat yhdenmukaiset hyvin eripainoisilla potilailla.
Lineaarisuus/ei-lineaarisuus
Denosumabin farmakokinetiikka oli epälineaarinen suhteessa annokseen laajalla annosalueella, mutta altistus suureni suunnilleen suhteessa annokseen 60 mg:n (tai 1 mg/kg) ja suurempia annoksia käytettäessä. Epälineaarisuus johtuu todennäköisesti saturoituvasta kohdemekanismin kautta välittyvästä (”target-mediated”) eliminoitumisreitistä, jolla on merkitystä pienillä pitoisuusalueilla.
Munuaisten vajaatoiminta
Denosumabitutkimuksissa (60 mg, n = 55 ja 120 mg, n = 32), joissa oli mukana eriasteista munuaisten vajaatoimintaa sairastavia ja myös dialyysihoidossa olevia potilaita, joilla ei ollut pitkälle edennyttä syöpää, munuaisten vajaatoiminnan asteen ei havaittu vaikuttavan denosumabin farmakokinetiikkaan. Munuaisten vajaatoiminta ei siis vaadi annoksen muuttamista. Munuaisten toimintaa ei tarvitse seurata Xgeva-hoidon aikana.
Maksan vajaatoiminta
Maksan vajaatoiminnasta ei ole tehty erillistä tutkimusta. Monoklonaaliset vasta-aineet eivät yleensä eliminoidu maksametabolian välityksellä. Maksan vajaatoiminnan ei oleteta vaikuttavan denosumabin farmakokinetiikkaan.
Iäkkäät potilaat
Hoidon turvallisuudessa ja tehossa ei havaittu yleisiä eroja iäkkäiden ja nuorempien potilaiden välillä. Xgevan kliinisissä vertailututkimuksissa, joihin osallistuneilla yli 65-vuotiailla potilailla oli pitkälle edennyt luustoon levinnyt tai luukudokseen vaikuttava syöpä, turvallisuus ja teho olivat samanlaiset vanhemmilla ja nuoremmilla potilailla. Iäkkäiden potilaiden annosta ei tarvitse muuttaa.
Pediatriset potilaat
Nuorilla potilailla (12–17‑vuotiailla), joiden luuston kasvu oli päättynyt, joilla oli luun jättisolukasvain (GCTB) ja jotka saivat 120 mg:n annoksen neljän viikon välein ja kyllästysannoksen 8. ja 15. päivänä, denosumabin farmakokinetiikka oli samankaltainen kuin aikuisilla GCTB-potilailla.
Prekliiniset tiedot turvallisuudesta
Koska eläimillä denosumabi on biologisesti aktiivinen vain kädellisillä, denosumabin farmakodynaamisia ominaisuuksia arvioitiin jyrsijämalleissa tutkimalla geenimuunneltuja (poistogeenisiä) hiiriä tai käyttämällä muita RANK/RANKL-reitin biologisia estäjiä, kuten OPG-Fc:tä ja RANK-Fc:tä.
Ihmisen estrogeenireseptoripositiivisen ja -negatiivisen rintasyövän, eturauhassyövän ja ei‑pienisoluisen keuhkosyövän luustometastaasien hiirimalleissa OPG-Fc vähensi osteolyyttisiä, osteoblastisia ja osteolyyttis-osteoblastisia leesioita, viivästytti de novo ‑luustoetäpesäkkeiden muodostumista ja hidasti luustokasvainten kasvua. Kun OPG-Fc yhdistettiin hormonihoitoon (tamoksifeeniin) rintasyöpämallissa tai solunsalpaajahoitoon (doketakseliin) eturauhassyöpä- ja keuhkosyöpämalleissa, sillä havaittiin olevan additiivinen luukasvainten kasvua estävä vaikutus. Hiirimallissa, jossa aiheutettiin maitorauhaskasvaimia, RANK-Fc vähensi hormonien aiheuttamaa maitorauhasen epiteelin proliferaatiota ja hidasti kasvaimen muodostumista.
Denosumabin genotoksisuutta ei ole tutkittu tavanomaisilla testeillä, koska ne eivät sovellu tämän molekyylin tutkimiseen. Denosumabin ominaisuuksien perusteella on kuitenkin epätodennäköistä, että sillä voisi olla genotoksisia vaikutuksia.
Denosumabin karsinogeenisuutta ei ole tutkittu pitkäaikaisissa eläinkokeissa.
Kun kerta-annoksen ja toistuvien annosten toksisuutta tutkittiin jaavanmakakeilla (cynomolgus‑apinoilla), denosumabiannokset, jotka saivat aikaan 2,7–15 kertaa suuremman systeemisen altistuksen kuin ihmisille suositeltu annos, eivät vaikuttaneet sydän- ja verisuonijärjestelmän fysiologiaan, urosten tai naaraiden hedelmällisyyteen eivätkä aiheuttaneet spesifistä kohde-elintoksisuutta.
Tutkimuksessa, jossa jaavanmakakeille annettiin denosumabia tiineyden ensimmäistä kolmannesta vastaavan jakson ajan, denosumabiannokset, jotka saivat aikaan 9 kertaa suuremman systeemisen altistuksen kuin ihmisille suositeltu annos, eivät aiheuttaneet emoon kohdistuneita toksisia vaikutuksia eivätkä sikiövaurioita tiineyden ensimmäistä kolmannesta vastaavan jakson aikana. Sikiöiden imusolmukkeita ei kuitenkaan tutkittu.
Toisessa tutkimuksessa, jossa jaavanmakakeille annettiin denosumabia koko tiineysajan annoksina, jotka saivat aikaan 12 kertaa suuremman systeemisen altistuksen kuin ihmisille suositeltu annos, havaittiin kuolleena syntyneiden poikasten määrän ja poikaskuolleisuuden lisääntymistä, luun poikkeavaa kasvua, joka heikensi luiden lujuutta, hematopoieesin vähenemistä sekä hampaiden asentovirheitä, perifeeristen imusolmukkeiden puuttumista ja neonataalisen kasvun hidastumista. Annostasoa, jolla ei ole lisääntymistoimintoihin kohdistuvia haitallisia vaikutuksia (NOAEL), ei määritetty. Kuuden kuukauden kuluttua syntymästä luustomuutokset olivat korjautumassa eikä vaikutuksia hampaiden puhkeamiseen ollut havaittavissa. Imusolmukemuutokset ja hampaiden asentovirheet olivat kuitenkin pysyneet ennallaan, ja yhdellä eläimellä havaittiin useiden kudosten vähäistä tai kohtalaista mineralisoitumista (yhteys hoitoon epävarma). Ennen poikimista ei havaittu viitteitä emoon kohdistuneista haitoista. Poikimisen aikana emoon kohdistuneita haittavaikutuksia esiintyi harvoin. Emojen maitorauhaset kehittyivät normaalisti.
Pitkään denosumabia saaneilla apinoilla tehdyissä luun laatua selvittävissä prekliinisissä tutkimuksissa luun aineenvaihdunnan hidastumiseen liittyi parantunut luun lujuus ja normaali luun histologia.
Geenimuunnelluilla uroshiirillä, jotka ilmensivät ihmisen RANK-ligandia (huRANKL) (siirtogeeninen knock-in ‑hiirimalli) ja joille aiheutettiin transkortikaalinen murtuma, denosumabi hidasti murtumakalluksen ruston hajoamista ja korvautumista uudisluulla verrokkiryhmään verrattuna, mutta luun biomekaaniseen lujuuteen sillä ei ollut haitallista vaikutusta.
Prekliinisissä tutkimuksissa poistogeeniset hiiret, joilta puuttui RANK tai RANKL, eivät erittäneet maitoa, koska maitorauhasen kypsyminen (rauhasen lobuloalveolaarinen kehitys tiineyden aikana) oli estynyt ja imusolmukkeiden muodostuminen oli heikentynyt. Vastasyntyneet poistogeeniset hiiret, joilta puuttui RANK tai RANKL, painoivat vähemmän, niiden luuston kasvu oli hidastunut, kasvulevyissä todettiin poikkeavuuksia eivätkä niiden hampaat puhjenneet. Luiden kasvun hidastumista, poikkeavia kasvulevyjä ja häiriöitä hampaiden puhkeamisessa todettiin myös tutkimuksissa, joissa vastasyntyneille rotille annettiin RANKL:n estäjiä. Nämä muutokset korjautuivat osittain, kun RANKL:n estäjän anto lopetettiin. Nuorilla kädellisillä todettiin poikkeavia kasvulevyjä, kun denosumabialtistus oli 2,7-kertainen (10 mg/kg) ja 15-kertainen (50 mg/kg) ihmisen altistukseen verrattuna. Denosumabihoito voi siis häiritä lapsen luuston kasvua, jos kasvulevyt ovat vielä avoimet, ja estää hampaiden puhkeamista.
Farmaseuttiset tiedot
Apuaineet
Injektiopullo
Etikkahappo, väkevä*
Natriumhydroksidi (pH:n säätöön)*
Sorbitoli (E420)
Polysorbaatti 20
Injektionesteisiin käytettävä vesi
* Asetaattipuskuri valmistetaan sekoittamalla etikkahappoa ja natriumhydroksidia.
Esitäytetty ruisku
Etikkahappo, väkevä*
Natriumhydroksidi (pH:n säätöön)*
Sorbitoli (E420)
L-fenyylialaniiniǂ
Polysorbaatti 20
Injektionesteisiin käytettävä vesi
* Asetaattipuskuri valmistetaan sekoittamalla etikkahappoa ja natriumhydroksidia.
ǂ Vain kerta-annoksen sisältävässä esitäytetyssä ruiskussa, joka sisältää 120 mg denosumabia 1,0 millilitrassa liuosta.
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Injektiopullo
4 vuotta.
Esitäytetty ruisku
3 vuotta.
Kun Xgeva on otettu pois jääkaapista, sitä voidaan säilyttää huoneenlämmössä (enintään 25 °C:ssa) enintään 30 vuorokautta alkuperäispakkauksessaan. Sitä ei saa laittaa takaisin jääkaappiin, vaan se on käytettävä näiden 30 vuorokauden kuluessa.
Säilytys
Säilytä jääkaapissa (2°C – 8°C).
Ei saa jäätyä.
Pidä injektiopullo tai esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XGEVA injektioneste, liuos
120 mg (L:ei) 1 kpl (1,7 ml (70 mg/ml)) (185,84 €)
XGEVA injektioneste, liuos, esitäytetty ruisku
120 mg (L:ei) 1 kpl (automaattinen turvamekanismi, 1 ml (120 mg/ml)) (258,62 €)
PF-selosteen tieto
Injektiopullo
1,7 ml liuosta kertakäyttöisessä injektiopullossa (tyypin I lasia), jossa on tulppa (fluoripolymeerilla pinnoitettua elastomeeria), suljin (alumiinia) ja suojakansi.
Pakkauksessa on yksi injektiopullo.
Esitäytetty ruisku
1 ml liuosta kertakäyttöisessä, tyypin I lasista valmistetussa esitäytetyssä ruiskussa, jossa on kumitulpalla (bromobutyylielastomeeri) varustettu mäntä ja ruostumattomasta teräksestä valmistettu 27 G:n neula. Ruiskussa on automaattinen turvamekanismi.
Pakkauksessa on yksi turvamekanismilla varustettu esitäytetty ruisku.
Valmisteen kuvaus:
Kirkas, väritön tai kellertävä liuos, joka saattaa sisältää hyvin pieniä määriä läpikuultavia tai valkoisia proteiinin kaltaisia hiukkasia.
Käyttö- ja käsittelyohjeet
- Pakkauskotelossa on pakkausseloste, jossa on tarkat käyttö- ja käsittelyohjeet.
- Xgeva‑liuos on tarkistettava silmämääräisesti ennen käyttöä. Liuos saattaa sisältää hyvin pieniä määriä läpikuultavia tai valkoisia proteiinin kaltaisia hiukkasia. Liuosta ei saa antaa injektiona, jos se on sameaa, sen väri on muuttunut, siinä on paljon hiukkasia tai siinä on vieraita hiukkasia.
- Älä ravista.
- Jotta pistos olisi miellyttävämpi, injektiopullon tai esitäytetyn ruiskun annetaan lämmetä huoneenlämpöiseksi (enintään 25 °C) ennen pistosta ja pistos annetaan hitaasti.
- Injektiopullon tai esitäytetyn ruiskun koko sisältö injisoidaan.
- Kun käytössä on injektiopullo, denosumabin pistämiseen suositellaan 27 G:n neulaa.
- Koko annos on otettava injektiopullosta kerralla. Tulppaa ei saa läpäistä toistamiseen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
XGEVA injektioneste, liuos
120 mg 1 kpl
XGEVA injektioneste, liuos, esitäytetty ruisku
120 mg 1 kpl
- Ylempi erityiskorvaus (100 %). Rintasyöpä (115), Eturauhassyöpä (116), Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117), Gynekologiset syövät (128), Pahanlaatuiset kasvaimet, joita ei ole edellä erikseen mainittu (130).
- Peruskorvaus (40 %).
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
M05BX04
Valmisteyhteenvedon muuttamispäivämäärä
12.03.2026
Yhteystiedot
Keilaranta 10, PL 86
02101 Espoo
09 5490 0500