
CONSTELLA kapseli, kova 290 mikrog
not_interestedSaatavuushäiriö
Ei saatavilla
CONSTELLA kapseli, kova
- 290 mikrog28 kpl20.07.2026 - 10.08.2026
Saatavilla
Saman valmisteen muut pakkaukset ja/tai vaihtokelpoiset valmisteet
CONSTELLA kapseli, kova
- 290 mikrog90 kpl
Vaikuttavat aineet ja niiden määrät
Yksi kapseli sisältää 290 mikrogrammaa linaklotidia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kova kapseli.
Kliiniset tiedot
Käyttöaiheet
Constella on tarkoitettu oireenmukaiseen hoitoon aikuisten keskivaikeaan tai vaikeaan ärtyvän suolen oireyhtymään, johon liittyy ummetusta (IBS‑C).
Annostus ja antotapa
Annostus
Suositeltu annos on yksi kapseli (290 mikrogrammaa) kerran vuorokaudessa.
Lääkäreiden tulee säännöllisesti arvioida jatkuvan hoidon tarvetta. Linaklotidin teho on osoitettu enintään 6 kuukautta kestäneissä kaksoissokkoutetuissa lumelääkekontrolloiduissa tutkimuksissa. Jos potilaan oireet eivät ole parantuneet neljän viikon hoidon jälkeen, potilas on tutkittava uudelleen ja hoidon jatkamisesta saatava hyöty ja riskit on arvioitava uudestaan.
Erityispotilaat
Munuaisten tai maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilaille, joilla on maksan tai munuaisten vajaatoimintaa (ks. kohta Farmakokinetiikka).
Iäkkäät potilaat
Vaikka iäkkäiden potilaiden annoksen muuttaminen ei ole tarpeen, hoitoa on seurattava huolellisesti ja lääkityksen tarve on arvioitava säännöllisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Constellan turvallisuutta ja tehoa alle 18-vuotiaiden lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Constella-valmistetta ei pidä käyttää lasten ja nuorten hoitoon (ks. kohta Farmakodynamiikka).
Antotapa
Suun kautta. Kapseli on otettava vähintään 30 minuuttia ennen ruokailua (ks. kohta Yhteisvaikutukset).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Potilaat, joilla on tunnettu tai epäilty vatsa‑suolikanavan mekaaninen tukos.
Varoitukset ja käyttöön liittyvät varotoimet
Constellaa saa käyttää sen jälkeen, kun elimelliset sairaudet on poissuljettu ja potilaalle on diagnosoitu keskivaikea tai vaikea IBS-C (ks. kohta Farmakodynamiikka).
Potilaiden tulee olla tietoisia ripulin ja alemman maha-suolikanavan verenvuodon mahdollisesta esiintymisestä hoidon aikana. Heitä on neuvottava kertomaan lääkärilleen, jos esiintyy vaikeaa tai pitkittynyttä ripulia tai alemman maha-suolikanavan verenvuotoa (ks. kohta Haittavaikutukset).
Jos pitkittynyttä (esimerkiksi yli yhden viikon kestävää) tai vaikeaa ripulia esiintyy, linaklotidin käytön väliaikaista keskeyttämistä on harkittava ja pyydettävä lääkärin ohjeita, kunnes ripulivaihe on päättynyt. Erityistä varovaisuutta on noudatettava sellaisten potilaiden kohdalla, joilla on alttius vesi‑ tai elektrolyyttitasapainon häiriöön (esim. iäkkäät sekä sydän‑ ja verisuonitauteja, diabetesta tai verenpainetautia sairastavat potilaat), ja heille on harkittava elektrolyyttiseurantaa.
Suolen puhkeamista on raportoitu sen jälkeen kun potilaat, joilla on sairaus, johon voi liittyä paikallinen tai diffuusi suolenseinämän heikkous, ovat käyttäneet linaklotidia. Potilaita on neuvottava hakeutumaan heti lääkärin hoitoon, jos heillä on vaikeaa, jatkuvaa tai pahenevaa vatsakipua; linaklotidin käyttö on keskeytettävä, jos tällaisia oireita ilmenee.
Linaklotidin käyttöä ei ole tutkittu suoliston kroonisia tulehdussairauksia, kuten Crohnin tautia tai haavaista koliittia sairastavien potilaiden hoidossa. Siksi Constellaa ei suositella annettavaksi näille potilaille.
Iäkkäät potilaat
Käytöstä iäkkäille potilaille on vain vähän tietoja (ks. kohta Farmakodynamiikka). Koska ripulivaaran on havaittu kliinisissä tutkimuksissa olevan suurempi (ks. kohta Haittavaikutukset), näitä potilaita on erityisesti tarkkailtava ja hoidon hyötyjen ja riskien suhdetta on arvioitava säännöllisesti huolella.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Linaklotidia havaitaan harvoin plasmassa suositeltujen kliinisten annosten antamisen jälkeen, ja in vitro ‑tutkimukset ovat osoittaneet, että linaklotidi ei ole sytokromi P450 ‑entsyymin substraatti eikä myöskään sen inhibiittori/induktori. Linaklotidilla ei ole vuorovaikutusta yleisten effluksi‑ ja sisäänkuljettajaproteiinien kanssa (ks. kohta Farmakokinetiikka).
Kliinisessä ruokayhteisvaikutustutkimuksessa terveille henkilöille annettiin terapeuttinen annos linaklotidia niin, että he olivat joko syöneet tai paastonneet. Linaklotidia ei havaittu plasmassa kummassakaan tapauksessa. Kun Constellaa otettiin syömisen jälkeen, tämä aiheutti tiheämpää ulostamista ja löysempiä ulosteita sekä enemmän ruoansulatuselimistöön liittyviä haittatapahtumia kuin silloin, kun lääkettä otettiin paastottaessa (ks. kohta Farmakodynamiikka.). Kapseli on otettava 30 minuuttia ennen ruokailua (ks. kohta Annostus ja antotapa).
Samanaikainen hoito protonipumpun estäjillä, laksatiiveilla tai ei-steroidisilla tulehduskipulääkkeillä (NSAID) saattaa lisätä ripulin vaaraa. Varovaisuutta on noudatettava, kun Constellaa annetaan samanaikaisesti tällaisten lääkkeiden kanssa.
Vaikean tai pitkittyneen ripulin esiintyminen saattaa vaikuttaa muiden suun kautta otettavien lääkevalmisteiden imeytymiseen. Suun kautta otettavien ehkäisyvalmisteiden teho saattaa vähentyä, ja muun ehkäisymenetelmän käyttöä suositellaan suun kautta otettavan ehkäisyvalmisteen tehon mahdollisen heikkenemisen estämiseksi (ks. suun kautta otettavan ehkäisyvalmisteen valmisteyhteenvetotietoja). Määrättäessä suolistosta imeytyviä kapean terapeuttisen indeksin omaavia lääkevalmisteita on noudatettava varovaisuutta, koska niiden teho saattaa vähentyä.
Raskaus ja imetys
Raskaus
Linaklotidin käytöstä raskaana oleville naisille on vain vähän tietoja. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Constellan käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Suun kautta otettu Constella imeytyy hyvin niukasti. Seitsemällä linaklotidihoitoa saavalla imettävällä naisella tehdyssä maidon lääkeainepitoisuutta selvittäneessä maidoneritystä koskevassa tutkimuksessa ei havaittu maidossa linaklotidia eikä sen aktiivista metaboliittia. Näin ollen imetys ei oletettavasti altista imeväistä linaklotidille ja Constellaa voi käyttää imetyksen aikana.
Linaklotidin tai sen metaboliitin vaikutuksia imettävien naisten maidontuotantoon ei ole tutkittu.
Hedelmällisyys
Eläimillä tehdyt tutkimukset osoittavat, että lääkkeellä ei ole vaikutusta uroksen tai naaraan hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Constellalla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Linaklotidia on annettu suun kautta 1 166 IBS‑C‑potilaalle kontrolloiduissa kliinisissä tutkimuksissa. Näistä potilaista 892 sai linaklotidin suositusannoksen, 290 mikrogrammaa vuorokaudessa. Kliinisen kehityssuunnitelman kokonaisaltistus oli yli 1 500 potilasvuotta. Yleisimmin ilmoitettu Constella‑hoidon yhteydessä ilmennyt haittavaikutus oli ripuli, joka oli vaikeusasteeltaan pääasiassa lievästä keskivaikeaan ja jota esiintyi alle 20 %:lla potilaista. Harvinaisissa ja vaikeammissa tapauksissa tämän seurauksena voi esiintyä dehydraatiota, hypokalemiaa, veren bikarbonaattipitoisuuden pienenemistä, heitehuimausta ja ortostaattista hypotensiota.
Muita yleisiä haittavaikutuksia (> 1 %) olivat vatsakipu, vatsan pingottuminen ja ilmavaivat.
Taulukoitu yhteenveto haittavaikutuksista
Kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä seurannassa ilmoitettiin seuraavia haittavaikutuksia käytettäessä suositusannosta, 290 mikrogrammaa vuorokaudessa (taulukko 1). Esiintyvyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000) ja hyvin harvinainen (< 1/10 000) sekä tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1 Kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä seurannassa ilmoitetut haittavaikutukset käytettäessä suositusannosta, 290 mikrogrammaa vuorokaudessa
MedDRA:n mukainen elinjärjestelmä-luokitus | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Tuntematon |
Infektiot |
| Viruksen aiheuttama maha‑suoli-tulehdus |
|
|
|
Immuunijärjestelmä |
|
|
|
| Anafylaktinen reaktio |
Aineenvaihdunta ja ravitsemus |
|
| Hypokalemia Dehydraatio Ruokahalun heikkeneminen |
|
|
Hermosto |
| Heitehuimaus |
|
|
|
Verisuonisto |
|
| Ortostaattinen hypotensio |
|
|
Ruoansulatuselimistö | Ripuli | Vatsakipu Ilmavaivat Vatsan pingottuminen | Ulosteinkontinenssi Ulostamispakko Alemman maha-suolikanavan verenvuoto, mukaan lukien pukamavuoto ja peräsuoliverenvuoto Pahoinvointi Oksentelu | Maha-suolikanavan puhkeaminen |
|
Iho ja ihonalainen kudos |
|
| Nokkosihottuma |
| Ihottuma |
Tutkimukset |
|
|
| Veren bikarbonaatti-pitoisuus pienentynyt |
|
Valikoitujen haittavaikutusten kuvaus
Yleisin haittavaikutuksista on ripuli, mikä sopii yhteen vaikuttavan aineen farmakologisen vaikutuksen kanssa. Hoidetuista potilaista 2 %:lla oli vaikea ripuli, ja 5 % keskeytti hoidon kliinisissä tutkimuksissa ripulin vuoksi.
Suurin osa ilmoitetuista ripulitapauksista oli lievistä (43 %) keskivaikeisiin (47 %); 2 %:lla hoidetuista potilaista oli vaikea ripuli. Noin puolet ripulikohtauksista alkoi hoidon ensimmäisen viikon aikana.
Ripuli parani seitsemän vuorokauden kuluessa noin kolmanneksella potilaista, mutta 80 potilaalla (50 %) esiintyi ripulia yli 28 vuorokauden ajan (vastaa 9,9 % osuutta kaikista linaklotidilla hoidetuista potilaista).
Potilaista 5 % keskeytti hoidon kliinisissä tutkimuksissa ripulin vuoksi. Ripulin takia hoidon keskeyttäneillä potilailla ripuli parani muutaman päivän kuluessa hoidon keskeyttämisestä.
Iäkkäillä (> 65‑vuotiaat), verenpainetautia ja diabetesta sairastavilla potilailla ilmoitettiin esiintyvän ripulia useammin kuin koko kliinisiin tutkimuksiin sisältyvän IBS‑C‑ryhmän potilailla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostus saattaa johtaa lääkevalmisteen tunnettujen farmakodynaamisten vaikutusten korostumisesta aiheutuviin oireisiin, pääasiassa ripuliin. Terveet vapaaehtoiset henkilöt saivat tutkimuksessa kerta-annoksen 2 897 mikrogrammaa (enintään kymmenkertaisesti enemmän kuin suositeltu terapeuttinen annos). Näiden henkilöiden turvallisuusprofiili oli yhteneväinen koko populaation turvallisuusprofiilin kanssa, ja ripuli oli yleisin ilmoitettu haittavaikutus.
Jos yliannostusta tapahtuu, potilasta on hoidettava oireenmukaisesti ja ryhdyttävä tarvittaessa elintoimintoja tukeviin toimenpiteisiin.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Ummetuslääkkeet, muut ummetuslääkkeet, ATC‑koodi: A06AX04
Vaikutusmekanismi
Linaklotidi on guanylaattisyklaasi‑C (GC‑C) ‑reseptorin agonisti, jolla on sisäelinten kipua lievittäviä ja sekretorisia vaikutuksia.
Linaklotidi on 14 aminohapon synteettinen peptidi, joka on rakenteellisesti sukua endogeeniselle guanyliinipeptidien perheelle. Sekä linaklotidi että sen aktiivinen metaboliitti sitoutuvat GC‑C‑reseptoriin suolen epiteelin luminaalisella pinnalla. GC‑C‑reseptoriin kohdistuvan vaikutuksensa kautta linaklotidin on osoitettu vähentävän sisäelinkipua sekä lisäävän ruoansulatuskanavan läpikulkua eläinmalleissa ja paksusuolen läpikulkua ihmisissä. GC‑C:n aktivoituminen johtaa syklisen guanosiinimonofosfaatin (cGMP) pitoisuuksien suurenemiseen sekä solunulkoisesti että ‑sisäisesti. Solunulkoinen cGMP vähentää kipusyiden aktiivisuutta, mikä vähentää sisäelinkipua eläinmalleissa. Solunsisäinen cGMP aiheuttaa kloridin ja bikarbonaatin erittymistä suolen onteloon kystisen fibroosin transmembraanisen konduktanssinsäätäjän (cystic fibrosis transmembrane conductance regulator, CFTR) aktivoitumisen kautta. Tämä johtaa suolistonesteen määrän suurenemiseen ja nopeutuneeseen läpikulkuun.
Farmakodynaamiset vaikutukset
Vaihtovuoroisessa ruokayhteisvaikutustutkimuksessa 18 terveelle vapaaehtoiselle henkilölle annettiin Constellaa 290 mikrogrammaa 7 päivän ajan sekä paastotilassa että aterian yhteydessä. Kun Constellaa otettiin heti runsasrasvaisen aamiaisen jälkeen, tämä aiheutti tiheämpää ulostamista ja löysempiä ulosteita sekä enemmän ruoansulatuselimistöön liittyviä haittatapahtumia kuin otettaessa lääkettä paastotilassa.
Kliininen teho ja turvallisuus
Linaklotidin teho osoitettiin kahdessa satunnaistetussa, kaksoissokkoutetussa lumelääkekontrolloidussa faasin 3 kliinisessä tutkimuksessa, johon osallistui IBS‑C‑potilaita. Toisessa kliinisessä tutkimuksessa (tutkimus 1) 804:ää potilasta hoidettiin 290 mikrogramman Constella‑annoksella tai lumelääkkeellä kerran vuorokaudessa 26 viikon ajan. Toisessa kliinisessä tutkimuksessa (tutkimus 2) 800:aa potilasta hoidettiin 12 viikon ajan, ja sen jälkeen heidät uudelleensatunnaistettiin vielä 4 viikon hoitojaksolle. Hoitoa edeltävän kahden viikon lähtötasojakson aikana potilaiden keskimääräiset vatsakipupisteet olivat 5,6 (asteikolla 0-10), vatsakivuttomia päiviä oli 2,2 %, keskimääräiset vatsan turpoamispisteet olivat 6,6 (asteikolla 0-10) ja spontaaneja ulostamisia (SBM) oli keskimäärin 1,8/viikko.
Potilasryhmän ominaisuudet faasin 3 kliinisissä tutkimuksissa olivat seuraavat: keski‑ikä 43,9 vuotta (vaihteluväli 18-87 vuotta ja 5,3 % vähintään 65‑vuotiaita), 90,1 % naisia. Kaikki potilaat täyttivät IBS‑C‑sairauden Rooma II ‑kriteerit, ja heidän edellytettiin ilmoittavan keskimääräiset vatsakipupisteet, jotka olivat ≥ 3 (0–10 pisteen numeerisella asteikolla) (kriteerit, jotka vastaavat potilaita, joilla on keskivaikea tai vaikea IBS), < 3 täydellistä spontaania ulostamista sekä ≤ 5 SBM‑tapahtumaa viikkoa kohden 2 viikon lähtötasojakson aikana.
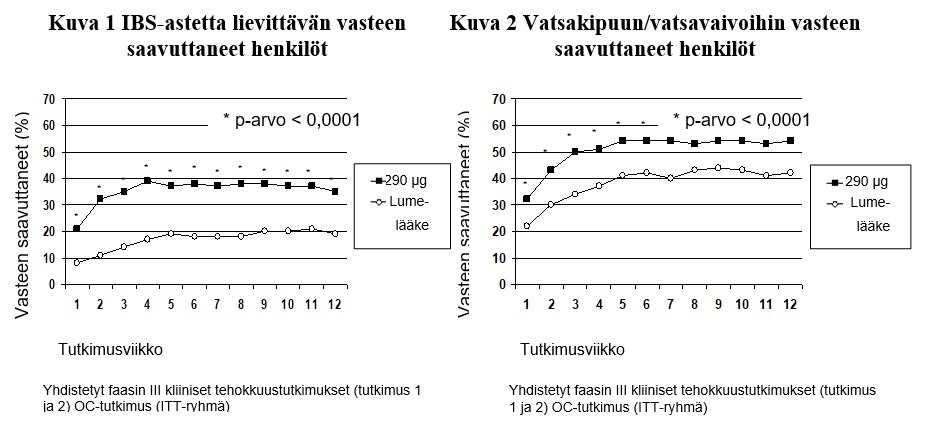
Molempien kliinisten tutkimusten yhteiset ensisijaiset päätetapahtumat olivat IBS‑astetta lievittävän vasteen saavuttaneiden henkilöiden määrä 12 viikon kohdalla ja vatsakipua/vatsavaivoja koskevan vasteen saavuttaneiden henkilöiden määrä 12 viikon kohdalla. IBS‑astetta lievittävän vasteen saavuttanut henkilö oli potilas, jonka vaiva oli lievittynyt merkittävästi tai kokonaan vähintään 50 %:n ajan hoitojaksosta; vatsakipuun/vatsavaivoihin vasteen saavuttanut oli potilas, jonka vaiva oli vähintään 30 % parempi vähintään 50 %:n ajan hoitojaksosta.
Tutkimuksen 1 tiedot 12 viikon ajalta osoittavat, että linaklotidilla hoidetuista potilaista 39 % saavutti IBS‑astetta lievittävän vasteen, kun lumelääkkeellä hoidettujen potilaiden vastaava määrä oli 17 % (p < 0,0001), ja 54 % linaklotidilla hoidetuista potilaista saavutti vasteen vatsakipuun/vatsavaivoihin, kun lumelääkkeellä hoidettujen potilaiden vastaava määrä oli 39 % (p < 0,0001). Tutkimuksessa 2 havaittiin, että linaklotidilla hoidetuista potilaista 37 % saavutti IBS‑astetta lievittävän vasteen, kun lumelääkkeellä hoidettujen potilaiden vastaava arvo oli 19 % (p < 0,0001), ja 55 % linaklotidilla hoidetuista potilaista saavutti vasteen vatsakipuun/vatsavaivoihin, kun lumelääkkeellä hoidettujen potilaiden vastaava arvo oli 42 % (p = 0,0002).
Tutkimuksen 1 tiedot 26 viikon ajalta osoittavat, että 37 % linoklotidilla hoidetuista potilaista saavutti IBS‑astetta lievittävän vasteen ja 54 % potilaista vasteen vatsakipuun/vatsavaivoihin, kun lumelääkkeellä hoidettujen potilaiden vastaavat arvot olivat 17 % (p < 0,0001) ja 36 % (p < 0,0001).
Molemmissa tutkimuksissa paranemista havaittiin viikolla 1, ja paraneminen jatkui koko hoitojaksojen ajan (kuvat 1 ja 2). Linaklotidin ei ole osoitettu aiheuttavan rebound-ilmiötä, kun lääkitys on lopetettu kolmen kuukauden yhtäjaksoisen hoidon jälkeen.
IBS‑C:n muut merkit ja oireet, esimerkiksi vatsan turpoaminen, täydellisen spontaanin ulostuksen (CSBM) esiintymistiheys, ulosteen ponnistaminen (straining) ja ulosteiden kiinteys, paranivat linaklotidilla hoidetuilla potilailla lumelääkehoitoon verrattuna (p < 0,0001) seuraavan taulukon mukaisesti. Nämä vaikutukset ilmaantuivat 1 viikossa, ja ne jatkuivat koko hoitojaksojen ajan.
Linaklotidin vaikutus IBS‑C‑oireisiin 12 ensimmäisen hoitoviikon aikana yhdistetyissä faasin 3 kliinisissä tehokkuustutkimuksissa (tutkimukset 1 ja 2).
Tärkeimmät toissijaiset tehokkuusparametrit | Lumelääke (N = 797) | Linaklotidi (N = 805) |
| ||||
Lähtötaso Keskiarvo | 12 viikkoa Keskiarvo | Muutos lähtötasosta Keskiarvo | Lähtötaso Keskiarvo | 12 viikkoa Keskiarvo | Muutos lähtötasosta Keskiarvo | Keskimääräinen LS‑ero | |
Vatsan turpoaminen (11 pisteen num. asteikko) | 6,5 | 5,4 | ‑1,0 | 6,7 | 4,6 | ‑1,9 | ‑0,9* |
CSBM/viikko | 0,2 | 1,0 | 0,7 | 0,2 | 2,5 | 2,2 | 1,6* |
Ulosteiden kiinteys (BSFS‑pisteet) | 2,3 | 3,0 | 0,6 | 2,3 | 4,4 | 2,0 | 1,4* |
Ulosteen ponnistaminen (5‑pisteinen järjestyslukuasteikko) | 3,5 | 2,8 | ‑0,6 | 3,6 | 2,2 | ‑1,3 | ‑0,6* |
*p < 0,0001, linaklotidi vs. lumelääke. LS: Pienin neliösumma CSBM: Täydellinen spontaani ulostaminen | |||||||
Linaklotidihoito aiheutti myös merkitsevää paranemista validoidussa tautikohtaisessa elämänlaatumittauksessa (IBS‑QoL; p < 0,0001) sekä EuroQoL‑mittauksessa (p = 0,001). Linaklotidilla hoidetuista potilaista 54 % saavutti kliinisesti merkitsevän vasteen kokonais‑IBS‑QoL (elämänlaatu) ‑pisteissä (> 14 pisteen ero) lumelääkkeellä hoidettujen potilaiden 39 %:iin verrattuna.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Constellan käytöstä toiminnallisen ummetuksen hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Linaklotidia havaitaan plasmassa yleensä vain vähän terapeuttisten suun kautta otettujen annosten jälkeen. Siksi tavallisia farmakokineettisiä parametrejä ei voida laskea.
Yhden enintään 966 mikrogramman linoklotidiannoksen ja usean enintään 290 mikrogramman linoklotidiannoksen jälkeen plasmassa ei ollut havaittavia määriä kanta-ainetta tai sen aktiivista metaboliittia (destyrosiinia). Kun päivänä 8 annettiin 2 897 mikrogrammaa linaklotidia (mitä ennen oli annettu 7 päivän ajan 290 mikrogrammaa vuorokaudessa), linaklotidia havaittiin vain 2 henkilöllä 18:sta pitoisuuksina, jotka juuri ylittivät kvantifioinnin alarajan 0,2 ng/ml (pitoisuudet olivat 0,212-0,735 ng/ml). Kahdessa keskeisessä faasin 3 avaintutkimuksessa, joissa potilaat saivat 290 mikrogrammaa linaklotidia kerran vuorokaudessa, linaklotidia havaittiin vain 2 potilaalla 162:sta noin 2 tunnin kuluttua ensimmäisen linaklotidiannoksen ottamisen jälkeen (pitoisuudet olivat 0,241-0,239 ng/ml) eikä yhdelläkään 162 potilaasta 4 viikon hoidon jälkeen. Aktiivista metaboliittia ei havaittu yhdelläkään 162 potilaasta missään aikapisteessä.
Jakautuminen
Koska linaklotidia harvoin kyetään havaitsemaan plasmassa terapeuttisten annosten jälkeen, tavanomaisia jakautumistutkimuksia ei ole tehty. Oletetaan, että linaklotidin jakautuminen on merkityksetöntä tai se ei jakaudu systeemisesti.
Biotransformaatio
Linaklotidi metabolisoituu paikallisesti maha‑suolikanavassa aktiiviseksi ensisijaiseksi metaboliitiksi, destyrosiiniksi. Sekä linaklotidi että aktiivinen metaboliitti destyrosiini hajotetaan ja pilkotaan maha‑suolikanavassa entsymaattisesti pienemmiksi peptideiksi ja luonnossa esiintyviksi aminohapoiksi.
Linaklotidin ja sen aktiivisen ensisijaisen metaboliitin MM‑419447:n mahdollista estävää vaikutusta ihmisen effluksikuljettajaproteiineihin BCRP, MRP2, MRP3 ja MRP4 sekä ihmisen sisäänkuljettajaproteiineihin OATP1B1, OATP1B3, OATP2B1, PEPT1 ja OCTN1 tutkittiin in vitro. Tämän tutkimuksen tulokset osoittivat, että kumpikaan peptidi ei ole yleisten effluksi‑ ja sisäänkuljettajaproteiinien inhibiittori kliinisesti merkityksellisinä pitoisuuksina.
Linaklotidin ja sen metaboliittien estävää vaikutusta yleisiin suolistoentsyymeihin (CYP2C9 ja CYP3A4) ja maksaentsyymeihin (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ja CYP3A4) sekä indusoivaa vaikutusta maksaentsyymeihin (CYP1A2, CYP2B6 ja CYP3A4/5) tutkittiin in vitro. Näiden tutkimusten tulokset osoittivat, että linaklotidi ja destyrosiinimetaboliitti eivät ole sytokromi P450 ‑entsyymijärjestelmän inhibiittoreita tai induktoreja.
Eliminaatio
Linaklotidin 7 päivän kuurin (annos 290 mikrogrammaa/vrk) jälkeen päivänä 8 suun kautta otetun yhden 2 897 mikrogramman annoksen jälkeen 18 terveellä vapaaehtoisella henkilöllä noin 3-5 % annoksesta kertyi ulosteisiin, käytännössä kaikki siitä aktiivisena destyrosiinimetaboliittina.
Ikä ja sukupuoli
Koska linaklotidia havaitaan harvoin plasmassa, ei ole tehty kliinisiä tutkimuksia, joissa määritettäisiin iän ja sukupuolen vaikutus linaklotidin kliiniseen farmakokinetiikkaan. Sukupuolella ei odoteta olevan mitään vaikutusta annostukseen. Ikää koskevat tiedot ovat kohdissa Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.
Munuaisten vajaatoiminta
Constellaa ei ole tutkittu potilailla, jotka sairastavat munuaisten vajaatoimintaa. Linaklotidia havaitaan harvoin plasmassa. Siksi munuaisten vajaatoiminnan ei odoteta vaikuttavan kantayhdisteen tai sen metaboliitin poistumaan.
Maksan vajaatoiminta
Constellaa ei ole tutkittu potilailla, jotka sairastavat maksan vajaatoimintaa. Linaklotidia havaitaan harvoin plasmassa, eivätkä maksan sytokromi P450 ‑entsyymit metaboloi linaklotidia. Sen vuoksi maksan vajaatoiminnan ei odoteta vaikuttavan lääkkeen kanta‑aineen tai sen metaboliitin aineenvaihduntaan tai poistumaan.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, karsinogeenisuutta sekä lisääntymis‑ ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Mikrokiteinen selluloosa
Hypromelloosi 4–6 mPa s – substituutiotyyppi 2910
Kalsiumklorididihydraatti
Leusiini
Kapselin kuori
Titaanidioksidi (E171)
Gelatiini
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Polyeteeniglykoli
Kapselin muste
Sellakka
Propeeniglykoli
Väkevä ammoniakki
Kaliumhydroksidi
Titaanidioksidi (E171)
Musta rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Avaamaton 28 tai 90 kapselin purkki ja monipakkaus, jossa on 112 kapselia (neljä 28 kapselin pakkausta): 3 vuotta.
Avaamaton 10 kapselin purkki: 2 vuotta.
Avattu purkki: 18 viikkoa.
Säilytys
Säilytä alle 30 ºC. Pidä purkki tiiviisti suljettuna. Herkkä kosteudelle.
Purkissa on yksi tai useampi suljettu säiliö, joka sisältää silikageeliä kapselien pitämiseksi kuivina. Pidä nämä säiliöt purkissa.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
CONSTELLA kapseli, kova
290 mikrog (L:ei) 28 kpl (73,46 €), 90 kpl (203,49 €)
PF-selosteen tieto
Valkoisesta suuritiheyksisestä polyeteenistä (HDPE) valmistettu purkki, jossa on turvasinetti ja turvasuljin sekä yksi tai useampia kuiva‑ainesäiliöitä, joissa on silikageeliä.
Pakkauskoot: 10, 28 tai 90 kapselia sekä monipakkaus, jossa 112 kapselia (neljä 28 kapselin pakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen ja oranssi läpinäkymätön kapseli (18 mm x 6,35 mm), jossa harmaalla musteella merkintä 290.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
CONSTELLA kapseli, kova
290 mikrog 28 kpl, 90 kpl
- Peruskorvaus (40 %).
ATC-koodi
A06AX04
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi