
MEKINIST tabletti, kalvopäällysteinen 0,5 mg, 2 mg
Vaikuttavat aineet ja niiden määrät
Mekinist 0,5 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää trametinibidimetyylisulfoksidia määrän, joka vastaa 0,5 mg trametinibiä.
Mekinist 2 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää trametinibidimetyylisulfoksidia määrän, joka vastaa 2 mg trametinibiä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
Melanooma
Trametinibi on tarkoitettu joko ainoana lääkkeenä tai yhdessä dabrafenibin kanssa BRAF V600 -mutaatiopositiivisen melanooman hoitoon aikuisille ja vähintään 12‑vuotiaille nuorille, joiden tauti on metastasoitunut tai jos kasvain ei ole kirurgisesti poistettavissa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Trametinibimonoterapialla ei ole todettu kliinistä tehoa niiden potilaiden hoidossa, joiden tauti on edennyt aikaisemman BRAF‑estäjähoidon aikana (ks. kohta Farmakodynamiikka).
Melanooman liitännäishoito
Trametinibi on tarkoitettu yhdessä dabrafenibin kanssa kirurgisesti kokonaan poistetun asteen III BRAF V600 -mutaatiopositiivisen melanooman liitännäishoitoon aikuisille ja vähintään 12‑vuotiaille nuorille.
Ei‑pienisoluinen keuhkosyöpä
Trametinibi on tarkoitettu yhdessä dabrafenibin kanssa edenneen BRAF V600 -mutaatiopositiivisen ei‑pienisoluisen keuhkosyövän hoitoon aikuisille.
Erilaistunut kilpirauhassyöpä (DTC)
Trametinibi on tarkoitettu yhdessä dabrafenibin kanssa aikuispotilaille, joilla on paikallisesti edennyt tai metastasoitunut, erilaistunut, BRAF V600E -mutaatiopositiivinen kilpirauhassyöpä ja joiden sairaus on radiojodihoidolle refraktaari tai kun radiojodihoito (RAI) ei sovellu, kun sairaus on edennyt aiemman systeemisen hoidon aikana tai sen jälkeen (biomarkkereihin perustuva potilasvalinta, ks. kohta Annostus ja antotapa).
Ehto
Hoidon tulee tapahtua solunsalpaajahoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Trametinibihoidon aloitus ja toteutus pitää tapahtua solunsalpaajahoitoon perehtyneen lääkärin valvonnassa.
Potilasvalinta
Kasvaimen BRAF V600 ‑mutaatio on vahvistettava CE-merkityllä, kyseiseen käyttötarkoitukseen tarkoitetulla in vitro -diagnostisella (IVD) lääkinnällisellä laitteella ennen trametinibihoidon aloittamista. Jos CE-merkittyä IVD-laitetta ei ole saatavilla, on käytettävä tähän käyttötarkoitukseen validoitua vaihtoehtoista testiä.
Annostus
Aikuisten suositeltu trametinibiannos on 2 mg kerran vuorokaudessa painosta riippumatta.
Melanoomaa sairastavien nuorten (12 – < 18-vuotiaat) suositeltu trametinibiannos perustuu painoon (ks. myös taulukko 1):
- 1 mg trametinibia kerran vuorokaudessa, jos paino on 26–37 kg
- 1,5 mg trametinibia kerran vuorokaudessa, jos paino on 38–50 kg
- 2 mg trametinibia kerran vuorokaudessa, jos paino on ≥ 51 kg.
Alle 26 kg painavien potilaiden suositeltua kalvopäällysteisten trametinibitablettien annosta ei ole määritetty.
Tiedot suositellusta dabrafenibiannoksesta yhdistelmähoidossa trametinibin kanssa, ks. dabrafenibin valmisteyhteenveto.
Hoidon kesto
Trametinibihoitoa jatketaan, kunnes potilas ei enää hyödy hoidosta tai hoito aiheuttaa ei‑hyväksyttäviä haittavaikutuksia (ks. taulukko 2). Melanooman liitännäishoidossa hoidon kesto on 12 kuukautta, jollei tauti puhkea uudelleen tai jollei ilmaannu ei‑hyväksyttäviä haittavaikutuksia.
Annoksen unohtuminen
Jos trametinibiannos unohtuu, voidaan unohtunut annos ottaa vain, jos seuraavaan aikataulun mukaiseen annokseen on yli 12 tuntia.
Jos dabrafenibiannos unohtuu, kun trametinibia käytetään yhdessä dabrafenibin kanssa, voidaan dabrafenibiannos ottaa vain, jos seuraavaan aikataulun mukaiseen annokseen on yli 6 tuntia.
Annoksen sovittaminen
Haittavaikutukset saattavat vaatia annoksen pienentämistä, hoidon keskeyttämistä tai hoidon lopettamista (ks. taulukot 1 ja 2).
Annoksen sovittamista ei suositella, jos potilaalle ilmaantuu haittavaikutuksena ihon okasolusyöpä tai uusi primaarimelanooma (tarkemmat tiedot, ks. dabrafenibin valmisteyhteenveto).
Taulukko 1 Suositukset annostasojen pienentämiseksi
Annostaso | Aikuiset | Nuoret | ||
Paino 26–37 kg | Paino 38–50 kg | Paino ≥ 51 kg | ||
Suositeltu aloitusannos | 2 mg kerran vuorokaudessa | 1 mg kerran vuorokaudessa | 1,5 mg kerran vuorokaudessa | 2 mg kerran vuorokaudessa |
1. pienennetty annostaso | 1,5 mg kerran vuorokaudessa | 0,5 mg kerran vuorokaudessa | 1 mg kerran vuorokaudessa | 1,5 mg kerran vuorokaudessa |
2. pienennetty annostaso | 1 mg kerran vuorokaudessa | NA | 0,5 mg kerran vuorokaudessa | 1 mg kerran vuorokaudessa |
NA = ei sovellu Trametinibiannoksen pienentämistä alle 1 mg:aan kerran vuorokaudessa ei suositella aikuisilla ja ≥ 51 kg painavilla nuorilla. | ||||
Taulukko 2 Annoksen muuttaminen minkä tahansa haittavaikutuksen vaikeusasteeseen perustuen (kuumetta lukuun ottamatta)
Vaikeusaste (CTCAE)* | Suositeltavia muutoksia trametinibiannoksissa Käytettäessä ainoana lääkkeenä tai yhdessä dabrafenibin kanssa |
1. tai 2. aste (siedettävissä) | Jatka hoitoa ja seuraa potilaan vointia. |
2. aste (kestämätön) tai 3. aste | Keskeytä hoito kunnes vaikeusaste on laskenut tasolle 0–1. Jatka hoitoa yhtä annostasoa pienemmällä annoksella. |
4. aste | Lopeta hoito pysyvästi tai keskeytä hoito, kunnes vaikeusaste on laskenut tasolle 0–1. Jatka hoitoa yhtä annostasoa pienemmällä annoksella. |
* Kliinisten haittavaikutusten vaikeusaste on määritetty CTCAE‑kriteerien (Common Terminology Criteria for Adverse Events) mukaan. | |
Kun haittavaikutukset on saatu tehokkaasti hallintaan, voidaan harkita annoksen nostamista uudelleen samojen annostasojen kautta kuin annosta pienennettäessä. Trametinibiannostus ei saa ylittää taulukossa 1 esitettyä suositeltua aloitusannosta.
Kuume
Hoito on keskeytettävä, jos potilaan ruumiinlämpö on ≥ 38 °C (trametinibin monoterapia ja molemmat trametinibi ja dabrafenibi yhdistelmähoidon yhteydessä). Kuumeilun toistuessa, hoito voidaan keskeyttää myös ensimmäisten kuumeen oireiden ilmaannuttua. Hoito kuumelääkkeillä, kuten ibuprofeenilla tai parasetamolilla on aloitettava. Oraalisten kortikosteroidien käyttöä on harkittava tapauksissa, joissa kuumelääkkeet ovat riittämättömiä. Potilaan infektio‑oireet ja ‑löydökset on arvioitava ja tarvittaessa hoidettava paikallisten hoitokäytäntöjen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Trametinibin monoterapia tai trametinibin ja dabrafenibin yhdistelmähoito on aloitettava uudelleen kun potilas on ollut oireeton vähintään 24 tunnin ajan joko (1) samalla annoksella, tai (2) yhtä pienemmällä annoksella, jos kuume oli toistuvaa ja/tai siihen liittyi muita vaikeita oireita kuten nestehukka, verenpaineen lasku tai munuaisten vajaatoiminta.
Jos trametinibin ja dabrafenibin yhdistelmän käytön aikana esiintyy hoitoon liittyviä haittoja, molempien annosta on pienennettävä samanaikaisesti tai hoidot on keskeytettävä tai lopetettava. Poikkeustapaukset, joissa vain toisen valmisteen annoksen muuttaminen on tarpeen, on esitetty jäljempänä seuraavien haittojen yhteydessä: uveiitti, muut RAS‑mutaatiopositiiviset syövät kuin ihosyöpä (liittyy ensisijaisesti dabrafenibiin), vasemman kammion ejektiofraktion pieneneminen, verkkokalvon laskimotukos, verkkokalvon pigmenttiepiteelin irtauma ja interstitiaalinen keuhkosairaus/pneumoniitti (liittyy ensisijaisesti trametinibiin).
Annosmuutoksia koskevat poikkeukset (kun vain toisen valmisteen annosta pienennetään) valikoitujen haittavaikutusten osalta
Uveiitti
Annosta ei tarvitse muuttaa uveiitin takia, jos silmätulehdus saadaan pidettyä hallinnassa tehokkailla paikallishoidoilla. Jos uveiitti ei reagoi silmän paikalliseen hoitoon, dabrafenibihoito on tauotettava, kunnes silmätulehdus on parantunut, ja aloitettava uudelleen yhtä annostasoa pienemmällä annoksella. Trametinibin annosta ei tarvitse muuttaa yhdistelmähoidossa dabrafenibin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muut RAS‑mutaatiopositiiviset syövät kuin ihosyöpä
Hyötyjä ja riskejä on punnittava ennen dabrafenibihoidon jatkamista, jos potilaalla on muu RAS‑mutaatiopositiivinen syöpä kuin ihosyöpä. Trametinibin annosta ei ole tarpeen muuttaa yhdistelmähoidossa dabrafenibin kanssa.
Vasemman kammion ejektiofraktion (LVEF) pieneneminen / vasemman kammion toimintahäiriö
Trametinibihoito on keskeytettävä, jos potilaalla todetaan oireeton > 10 %:n absoluuttinen vasemman kammion ejektiofraktion lasku lähtötasoon verrattuna ja ejektiofraktio alittaa hoitolaitoskohtaisen viitealueen alarajan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Dabrafenibin annosta ei tarvitse muuttaa yhdistelmähoidossa trametinibin kanssa. Jos vasemman kammion ejektiofraktio korjaantuu, trametinibihoito voidaan aloittaa uudelleen mutta annosta on pienennettävä yhden annostason verran ja potilaan vointia on seurattava tarkoin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Trametinibihoito on lopetettava pysyvästi, jos vasemman kammion toimintahäiriön vaikeusaste on 3 tai 4 tai jos vasemman kammion ejektiofraktio on pienentynyt kliinisesti merkittävästi eikä korjaannu 4 viikon kuluessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verkkokalvon laskimotukos ja verkkokalvon pigmenttiepiteelin irtauma
Jos potilas ilmoittaa uusista näköhäiriöistä, kuten keskeisen näön heikkenemisestä, näön hämärtymisestä tai näön menetyksestä, milloin tahansa trametinibihoidon aikana, on suositeltavaa tehdä oftalmologinen tutkimus viipymättä. Jos verkkokalvon laskimotukos diagnosoidaan, ainoana lääkkeenä tai yhdessä dabrafenibin kanssa käytettävä trametinibi on lopetettava pysyvästi. Dabrafenibin annosta ei tarvitse muuttaa yhdistelmähoidossa trametinibin kanssa. Jos potilaalla todetaan verkkokalvon pigmenttiepiteelin irtauma, trametinibiannosta muutetaan alla taulukossa 3 esitetyn ohjelman mukaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 3 Suositukset trametinibiannoksen muuttamisesta verkkokalvon pigmenttiepiteelin irtauman vuoksi
Verkkokalvon pigmenttiepiteelin 1. asteen irtauma | Jatka hoitoa ja tarkasta verkkokalvo kuukausittain, kunnes irtauma korjaantuu. Jos irtauma pahenee, noudata alla olevia ohjeita, ja keskeytä trametinibihoito enintään 3 viikon ajaksi. |
Verkkokalvon pigmenttiepiteelin 2.–3. asteen irtauma | Keskeytä trametinibihoito enintään 3 viikon ajaksi |
Verkkokalvon pigmenttiepiteelin 2.–3. asteen irtauma, joka lievittyy 0–1. asteen tasolle kolmessa viikossa | Aloita trametinibi uudelleen pienemmällä annoksella (annosta pienennetään 0,5 mg) tai lopeta trametinibi, jos trametinibiannos on 1 mg/vrk. |
Verkkokalvon pigmenttiepiteelin 2.–3. asteen irtauma, joka ei lievity vähintään 1. asteen tasolle kolmessa viikossa | Lopeta trametinibihoito pysyvästi. |
Interstitiaalinen keuhkosairaus/Pneumoniitti
Jos potilaalla epäillään interstitiaalista keuhkosairautta tai pneumoniittia ja myös jos potilaalla on uusia tai eteneviä keuhko‑oireita ja ‑löydöksiä, kuten yskää, hengenahdistusta, hypoksiaa, nestettä keuhkopussissa tai infiltraatteja, trametinibihoito on keskeytettävä, kunnes kliiniset tutkimukset on saatu päätökseen. Trametinibi on lopettava pysyvästi, jos potilaalla diagnosoidaan hoitoon liittyvä interstitiaalinen keuhkosairaus tai pneumoniitti. Dabrafenibin annosta ei tarvitse muuttaa yhdistelmähoidossa trametinibin kanssa interstitiaalisen keuhkosairauden tai pneumoniitin takia.
Erityisryhmät
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa lievän tai keskivaikean munuaisten vajaatoiminnan vuoksi (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden trametinibihoidosta ei ole tutkimustietoa, ja siksi ei voida arvioida, tarvitseeko aloitusannosta sovittaa. Trametinibin käytössä on noudatettava varovaisuutta vaikeassa munuaisten vajaatoiminnassa, kun valmistetta käytetään ainoana lääkkeenä tai yhdessä dabrafenibin kanssa.
Maksan vajaatoiminta
Annostuksen muuttaminen ei ole tarpeen lievän maksan vajaatoiminnan vuoksi. Kliinisen farmakologian tutkimus on osoittanut, että keskivaikealla tai vaikealla maksan vajaatoiminnalla on rajallinen vaikutus trametinibialtistukseen (ks. kohta Farmakokinetiikka). Trametinibin käytössä on noudatettava varovaisuutta keskivaikeassa tai vaikeassa maksan vajaatoiminnassa, kun valmistetta käytetään ainoana lääkkeenä tai yhdessä dabrafenibin kanssa.
Etninen tausta
Trametinibin turvallisuudesta ja tehosta muiden kuin kaukasialaista syntyperää olevien potilaiden hoidossa on vain rajoitetusti tietoa. Populaatiofarmakokineettisessä analyysissä ei havaittu merkittäviä eroja trametinibin farmakokinetiikassa aasialaisten ja kaukasialaista syntyperää olevien potilaiden välillä. Trametinibin annosta ei tarvitse muuttaa aasialaisille potilaille.
Iäkkäät
Aloitusannosta ei tarvitse sovittaa yli 65‑vuotiaita potilaita hoidettaessa. Tiheämpi annoksen sovittaminen saattaa olla tarpeen (ks. taulukot 1 ja 2 edellä) yli 65‑vuotiaita potilaita hoidettaessa (ks. kohta Haittavaikutukset).
Pediatriset potilaat
Kalvopäällysteisten trametinibitablettien turvallisuutta ja tehoa lasten ja nuorten (alle 18 vuoden ikäisten) hoidossa ei ole varmistettu lukuun ottamatta vähintään 12 vuoden ikäisten melanoomaa sairastavien nuorten hoitoa. Kliinisiä tietoja ei ole saatavilla. Eläimillä, jotka eivät ole saavuttaneet sukukypsyyttä, tehdyissä tutkimuksissa on havaittu trametinibin aiheuttamia haittavaikutuksia, joita ei havaittu täysikasvuisilla eläimillä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Antotapa
Trametinibi otetaan suun kautta täyden vesilasillisen kanssa. Tabletteja ei saa pureskella eikä murskata, ja ne otetaan tyhjään mahaan, vähintään tunti ennen ateriaa tai kaksi tuntia aterian jälkeen.
Trametinibiannos pitää ottaa joka päivä samaan aikaan. Kun trametinibia ja dabrafenibia käytetään yhdessä, kerran vuorokaudessa otettava trametinibiannos on otettava samaan aikaan joka päivä joko dabrafenibin aamuannoksen tai ilta‑annoksen yhteydessä.
Jos potilas oksentaa, kun hän on ottanut trametinibiannoksen, hänen ei pidä ottaa annosta uudelleen, ja seuraava annos on otettava normaaliin aikaan.
Tiedot dabrafenibin antotavasta yhdistelmähoidossa trametinibin kanssa, ks. dabrafenibin valmisteyhteenveto.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Kun trametinibia annetaan yhdessä dabrafenibin kanssa, dabrafenibin valmisteyhteenvetoon on perehdyttävä ennen hoidon aloittamista. Lisätiedot dabrafenibihoitoon liittyvistä varoituksista ja varotoimista, ks. dabrafenibin valmisteyhteenveto.
BRAF V600 ‑testaus
Trametinibin tehoa ja turvallisuutta ei ole arvioitu niiden potilaiden hoidossa, joiden melanooma on BRAF V600 ‑mutaation suhteen negatiivinen.
Trametinibihoito verrattuna BRAF‑estäjähoitoon
Trametinibia yksinään käytettynä ei ole verrattu BRAF:n estäjään kliinisessä tutkimuksessa BRAF V600 ‑mutaatiopositiivisen, leikkaukseen soveltumattoman tai metastasoituneen melanooman hoidossa. Tutkimusten välisissä vertailuissa trametinibi näyttäisi olevan yhtä tehokas kuin BRAF:n estäjät kokonais‑ ja etenemisvapaata elinaikaa koskevien tulosten perusteella. Kokonaisvasteosuudet olivat trametinibia saaneilla potilailla kuitenkin pienempiä kuin BRAF:n estäjiä saaneilla potilailla.
Trametinibin ja dabrafenibin yhdistelmähoito melanoomapotilailla, joiden tauti on edennyt BRAF‑estäjähoidon aikana
Tietoja on rajallisesti potilaista, jotka käyttävät trametinibin ja dabrafenibin yhdistelmää ja joiden tauti on edennyt aiemman BRAF‑estäjähoidon aikana. Nämä tiedot osoittavat yhdistelmähoidon tehon olevan heikompi tässä potilasryhmässä (ks. kohta Farmakodynamiikka). Tämän vuoksi on harkittava muita hoitovaihtoehtoja ennen yhdistelmähoidon käyttämistä aiemmin BRAF‑estäjähoitoa saaneessa populaatiossa. Hoitojen eri järjestyksiä BRAF‑estäjähoidon aikana tapahtuneen etenemisen jälkeen ei ole tutkittu.
Uudet maligniteetit
Uusia maligniteetteja (ihomaligniteetteja ja muita maligniteetteja) saattaa esiintyä, kun trametinibia käytetään yhdessä dabrafenibin kanssa.
Ihomaligniteetit
Ihon okasolusyöpä
Ihon okasolusyöpää (mukaan lukien keratoakantoomaa) on ilmoitettu potilailla, jotka ovat saaneet trametinibia yhdessä dabrafenibin kanssa. Ihon okasolusyöpä voidaan poistaa leikkauksella eikä hoidon muuttamista vaadita. Ks. dabrafenibin valmisteyhteenveto (kohta Varoitukset ja käyttöön liittyvät varotoimet).
Uusi primaarimelanooma
Uusia primaarimelanoomia ilmoitettiin potilailla, jotka käyttivät trametinibia yhdessä dabrafenibin kanssa. Uusi primaarimelanooma voidaan poistaa leikkauksella eikä hoidon muuttamista vaadita. Ks. dabrafenibin valmisteyhteenveto (kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muut syöpäsairaudet kuin ihosyöpä
Vaikutusmekanisminsa perusteella dabrafenibi saattaa suurentaa muiden syöpäsairauksien kuin ihosyövän riskiä, kun potilaalla on RAS‑mutaatio. Trametinibin ja dabrafenibin samanaikainen käyttö, ks. dabrafenibin valmisteyhteenveto (kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos potilaalla on RAS‑mutaatiopositiivinen maligniteetti, trametinibin annosta ei tarvitse muuttaa yhdistelmähoidossa dabrafenibin kanssa.
Verenvuoto
Trametinibia ainoana lääkkeenä tai yhdessä dabrafenibin kanssa saaneilla potilailla on esiintynyt verenvuototapahtumia, mukaan lukien merkittäviä verenvuototapahtumia ja kuolemaan johtaneita verenvuotoja (ks. kohta Haittavaikutukset). Näiden tapahtumien mahdollisuutta ei ole varmistettu potilailla, joilla on alhainen trombosyyttiarvo (< 75 000), sillä tällaiset potilaat suljettiin pois kliinisistä tutkimuksista. Samanaikainen antitromboottinen lääkitys tai antikoagulanttihoito saattaa suurentaa verenvuotoriskiä. Jos verenvuotoa esiintyy, potilasta on hoidettava kliinisen tilanteen mukaisesti.
Vasemman kammion ejektiofraktion pieneneminen/vasemman kammion toimintahäiriö
Trametinibin on raportoitu pienentävän vasemman kammion ejektiofraktiota, kun valmistetta käytetään ainoana lääkkeenä tai yhdessä dabrafenibin kanssa (ks. kohta Haittavaikutukset). Kliinisissä tutkimuksissa aika vasemman kammion toimintahäiriön ja sydämen vajaatoiminnan ensimmäiseen ilmaantumiseen ja ejektiofraktion ensimmäiseen pienenemiseen oli 2–5 kuukautta (mediaani).
Trametinibia on käyttävä varoen, jos potilaan vasemman kammion toiminta on heikentynyt. Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli vasemman kammion toimintahäiriö, NYHA (New York Heart Association) ‑luokan II, III tai IV sydämen vajaatoiminta, äkillinen sepelvaltimo‑oireyhtymä kuuden edellisen kuukauden aikana, kliinisesti merkittäviä hallitsemattomia rytmihäiriöitä ja hallitsematon hypertensio. Siksi lääkkeen käytön turvallisuutta tämän potilasryhmän hoidossa ei tunneta. Kaikkien potilaiden vasemman kammion ejektiofraktio on tarkistettava ennen trametinibihoidon aloittamista, kuukauden kuluttua hoidon aloittamisesta ja sen jälkeen noin 3 kuukauden välein hoidon aikana (ks. annoksen muuttamista koskevat ohjeet kohdasta Annostus ja antotapa).
Potilailla, jotka käyttävät trametinibia yhdessä dabrafenibin kanssa, on raportoitu yksittäisiä myokardiitista johtuvia akuutteja vakavia vasemman kammion toimintahäiriöitä. Potilaat toipuivat täysin, kun hoito lopetettiin. Lääkäreiden pitää ottaa huomioon myokardiitin mahdollisuus potilailla, joille ilmaantuu uusia tai pahenevia sydämeen liittyviä löydöksiä tai oireita.
Kuume
Kuumetta on ilmoitettu kliinisissä tutkimuksissa, kun trametinibia on käytetty ainoana lääkkeenä tai yhdessä dabrafenibin kanssa (ks. kohta Haittavaikutukset). Kuumeen ilmaantuvuus ja vaikeusaste ovat suurempia yhdistelmähoidossa (ks. dabrafenibin valmisteyhteenvedon kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos potilas käyttää trametinibia yhdessä dabrafenibin kanssa, kuumeeseen saattaa liittyä vaikeaa vilutusta, nestehukkaa ja hypotensiota. Tämä voi johtaa joissakin tapauksissa akuuttiin munuaisten vajaatoimintaan.
Hoito (trametinibin monoterapia ja molemmat trametinibi ja dabrafenibi yhdistelmähoidon yhteydessä) on keskeytettävä, jos potilaan ruumiinlämpö on ≥ 38 °C (ks. kohta Farmakodynamiikka). Toistuvassa kuumeilussa hoito voidaan keskeyttää myös ensimmäisten kuumeen oireiden ilmaannuttua. Hoito kuumelääkkeillä, kuten ibuprofeenilla tai parasetamolilla on aloitettava. Oraalisten kortikosteroidien käyttöä on harkittava tapauksissa, joissa kuumelääkkeet ovat riittämättömiä. Potilaan infektio‑oireet ja ‑löydökset on arvioitava. Hoito voidaan aloittaa uudelleen kuumeen laskettua. Jos kuumeeseen liittyy muita vaikeita oireita tai löydöksiä, hoitoa on jatkettava kuumeen laskettua pienemmällä annoksella ja kun se on kliinisesti tarkoituksenmukaista (ks. kohta Annostus ja antotapa).
Hypertensio
Verenpaineen nousua on raportoitu pelkän trametinibin ja trametinibin ja dabrafenibin yhdistelmän käytön aikana sekä potilailla, joilla on aiemmin ollut kohonnut verenpaine, että potilailla, joiden verenpaine on ollut normaali (ks. kohta Haittavaikutukset). Verenpaine on mitattava hoitoa aloitettaessa ja sitä on seurattava trametinibihoidon aikana, ja tarvittaessa hypertensio on pidettävä hallinnassa tavanomaisella hypertension hoidolla.
Interstitiaalinen keuhkosairaus/Pneumoniitti
Vaiheen III tutkimuksessa 2,4 %:lle (5/211) pelkkää trametinibihoitoa saaneista potilaista kehittyi interstitiaalinen keuhkosairaus tai pneumoniitti, ja kaikki viisi potilasta tarvitsivat sairaalahoitoa. Mediaaniaika interstitiaalisen keuhkosairauden tai pneumoniitin ensimmäiseen ilmaantumiseen oli 160 päivää (vaihteluväli: 60–172 päivää). MEK115306‑tutkimuksessa pneumoniitti tai interstitiaalinen keuhkosairaus kehittyi < 1 %:lle (2/209) trametinibin ja dabrafenibin yhdistelmää käyttäneistä potilaista ja MEK116513‑tutkimuksessa 1 %:lle (4/350) potilaista (ks. kohta Haittavaikutukset).
Jos potilaalla epäillään interstitiaalista keuhkosairautta tai pneumoniittia ja myös jos potilaalla on uusia tai eteneviä keuhko‑oireita ja ‑löydöksiä, kuten yskää, hengenahdistusta, hypoksiaa, nestettä keuhkopussissa tai infiltraatteja, trametinibihoito on keskeytettävä, kunnes kliiniset tutkimukset on saatu päätökseen. Trametinibi on lopettava pysyvästi, jos potilaalla diagnosoidaan hoitoon liittyvä interstitiaalinen keuhkosairaus tai pneumoniitti (ks. kohta Annostus ja antotapa). Jos trametinibia käytetään yhdessä dabrafenibin kanssa, dabrafenibihoitoa voidaan jatkaa samalla annoksella.
Näön heikkeneminen
Näköhäiriöitä aiheuttavia sairauksia, kuten verkkokalvon pigmenttiepiteelin irtaumia ja verkkokalvon laskimotukoksia saattaa esiintyä pelkän trametinibin ja trametinibin ja dabrafenibin yhdistelmän käytön yhteydessä. Oireita, kuten näön hämärtymistä, näöntarkkuuden heikkenemistä ja muita näköoireita on raportoitu trametinibin kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset). Kliinisissä tutkimuksissa on ilmoitettu myös uveiittia ja iridosykliittia, kun potilaille on annettu trametinibin ja dabrafenibin yhdistelmää.
Trametinibiä ei suositella potilaille, joilla on ollut verkkokalvon laskimotukos. Trametinibin turvallisuutta ei ole vahvistettu tapauksissa, joissa potilaalla on verkkokalvon laskimotukokselle altistavia tekijöitä, kuten huonossa hoitotasapainossa oleva glaukooma tai kohonnut silmänpaine, huonossa hoitotasapainossa oleva hypertensio, huonossa hoitotasapainossa oleva diabetes mellitus tai aiemmin todettu hyperviskositeettioireyhtymä tai veren liiallinen hyytymistaipumus.
Jos potilas ilmoittaa uusista näköhäiriöistä, kuten keskeisen näön heikkenemisestä, näön hämärtymisestä tai näön menetyksestä, milloin tahansa trametinibihoidon aikana, oftalmologinen tutkimus on suositeltavaa tehdä viipymättä. Jos potilaalla todetaan verkkokalvon pigmenttiepiteelin irtauma, annosta muutetaan taulukossa 3 esitetyn ohjelman mukaisesti (ks. kohta Annostus ja antotapa). Jos potilaalla todetaan uveiitti, ks. dabrafenibin valmisteyhteenvedon kohta Varoitukset ja käyttöön liittyvät varotoimet. Jos verkkokalvon laskimotukos diagnosoidaan, trametinibihoito on lopetettava pysyvästi. Dabrafenibin annosta ei tarvitse muuttaa yhdistelmähoidossa trametinibin kanssa, jos potilaalla on todettu verkkokalvon laskimotukos tai verkkokalvon pigmenttiepiteelin irtauma. Trametinibin annosta ei tarvitse muuttaa yhdistelmähoidossa dabrafenibin kanssa, jos potilaalla on todettu uveiitti.
Ihottuma
Ihottumaa on havaittu noin 60 %:lla potilaista trametinibimonoterapiatutkimuksissa ja noin 27 %:lla potilaista, jotka käyttivät trametinibin ja dabrafenibin yhdistelmähoitoa (ks. kohta Haittavaikutukset). Suurimmassa osassa näistä tapauksista ihottuman vaikeusaste oli 1 tai 2, eikä se vaatinut hoidon keskeyttämistä eikä annoksen pienentämistä.
Rabdomyolyysi
Rabdomyolyysiä on ilmoitettu potilailla, jotka ovat käyttäneet trametinibia ainoana lääkkeenä tai yhdessä dabrafenibin kanssa (ks. kohta Haittavaikutukset). Joissakin tapauksissa potilaat pystyivät jatkamaan trametinibihoitoa. Vaikeammat tapaukset vaativat sairaalahoitoa tai trametinibin tai trametinibin ja dabrafenibin yhdistelmän käytön keskeyttämistä tai pysyvää lopettamista. Rabdomyolyysin oireet ja löydökset vaativat asianmukaista kliinistä selvittelyä ja tarvittaessa hoitoa.
Munuaisten vajaatoiminta
Kliinisissä tutkimuksissa munuaisten vajaatoimintaa on todettu potilailla, jotka ovat käyttäneet trametinibia yhdessä dabrafenibin kanssa. Ks. dabrafenibin valmisteyhteenveto (kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haimatulehdus
Kliinisissä tutkimuksissa haimatulehdusta on ilmoitettu potilailla, jotka ovat käyttäneet trametinibia yhdessä dabrafenibin kanssa. Ks. dabrafenibin valmisteyhteenveto (kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksaan liittyvät tapahtumat
Trametinibin kliinisissä tutkimuksissa on raportoitu maksaan kohdistuvia haittavaikutuksia, kun valmistetta on käytetty ainoana lääkkeenä ja yhdessä dabrafenibin kanssa (ks. kohta Haittavaikutukset). Trametinibia ainoana lääkkeenä tai yhdessä dabrafenibin kanssa saavien potilaiden maksan toimintaa suositellaan seurattavan neljän viikon välein kuuden kuukauden ajan trametinibihoidon aloittamisen jälkeen. Maksa‑arvojen seurantaa voidaan jatkaa myös tämän jälkeen kliinisen tarpeen mukaan.
Maksan vajaatoiminta
Trametinibi eliminoituu ensisijaisesti metaboloitumalla ja erittymällä sappeen, ja siksi trametinibin käytössä on noudatettava varovaisuutta, jos potilaalla on keskivaikea tai vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Syvä laskimotukos/keuhkoembolia
Keuhkoembolioita tai syviä laskimotukoksia saattaa esiintyä, kun trametinibia käytetään ainoana lääkkeenä tai yhdessä dabrafenibin kanssa. Jos potilaalla ilmenee keuhkoembolian tai syvän laskimotukoksen oireita, kuten hengenahdistusta, rintakipua tai käsivarren tai jalan turvotusta, hänen on hakeuduttava välittömästi lääkärin hoitoon. Trametinibi‑ ja dabrafenibihoito on lopetettava pysyvästi, jos potilaalla on henkeä uhkaava keuhkoembolia.
Vaikeat ihoon kohdistuvat haittavaikutukset
Dabrafenibin ja trametinibin yhdistelmähoidon aikana on ilmoitettu vaikeita ihoon kohdistuneita haittavaikutuksia, mukaan lukien Stevens–Johnsonin oireyhtymää ja DRESS-reaktioita (lääkereaktio, johon liittyy eosinofiliaa ja systeemisiä oireita), jotka voivat olla henkeä uhkaavia tai johtaa kuolemaan. Ennen hoidon aloitusta potilaille on kerrottava ihoreaktioiden oireista ja löydöksistä, ja heitä on seurattava niiden varalta tarkoin. Jos vaikeisiin ihoon kohdistuviin haittavaikutuksiin viittaavia oireita ja löydöksiä esiintyy, dabrafenibin ja trametinibin käyttö on lopetettava.
Ruoansulatuselimistö
Koliittia ja ruoansulatuskanavan perforaatioita (myös kuolemaan johtaneita tapauksia) on raportoitu trametinibia yksinään ja yhdistelmänä dabrafenibin kanssa käyttäneillä potilailla (ks. kohta Haittavaikutukset). Trametinibin käytössä ainoana lääkkeenä tai yhdessä dabrafenibin kanssa on noudatettava varovaisuutta hoidettaessa potilaita, joilla on ruoansulatuskanavan perforaation riskitekijöitä, kuten aikaisempi divertikuliitti, ruoansulatuskanavan metastaasit, tai sellaisten lääkevalmisteiden samanaikainen käyttö, joihin tiedetään liittyvän ruoansulatuskanavan perforaation riski.
Sarkoidoosi
Trametinibin ja dabrafenibin yhdistelmällä hoidetuilta potilailta on ilmoitettu sarkoidoositapauksia. Haittavaikutukset ovat kohdistuneet pääasiassa ihoon, keuhkoihin, silmiin ja imusolmukkeisiin. Suurimmassa osassa tapauksia trametinibi- ja dabrafenibihoitoa jatkettiin. Sarkoidoosidiagnoosin yhteydessä on harkittava asianmukaista hoitoa. On tärkeää, ettei sarkoidoosia tulkita virheellisesti sairauden etenemiseksi.
Hemofagosyyttinen lymfohistiosytoosi
Myyntiluvan myöntämisen jälkeen trametinibia ja dabrafenibia yhdistelmähoitona saavilla potilailla on havaittu hemofagosyyttista lymfohistiosytoosia (HLH). Käytettäessä trametinibia yhdessä dabrafenibin kanssa on noudatettava varovaisuutta. Jos potilaalla on vahvistettu HLH, on lopetettava trametinibin ja dabrafenibin yhdistelmähoidon antaminen ja aloitettava HLH:n hoito.
Tuumorilyysioireyhtymä
Trametinibin ja dabrafenibin yhdistelmähoidon käyttöön on liittynyt tuumorilyysioireyhtymää, joka voi johtaa kuolemaan (ks. kohta Haittavaikutukset). Tuumorilyysioireyhtymän riskitekijöitä ovat suuri kasvaintaakka, olemassa oleva krooninen munuaisten vajaatoiminta, oliguria, nestehukka, hypotensio ja hapan virtsa. Potilaita, joilla on tuumorilyysioireyhtymän riskitekijöitä, on seurattava huolellisesti ja profylaktista nesteytystä on harkittava. Tuumorilyysioireyhtymä on hoidettava viipymättä kliinisen tarpeen mukaan.
Sädehoidon toksisuuden vahvistuminen
Potilailla, jotka saivat sädehoitoa ennen trametinibin ja dabrafenibin yhdistelmähoitoa, sen aikana tai sen jälkeen on raportoitu sädereaktion uusiutumista (radiation recall) ja herkistymistä sädehoidolle. Useimmat tapauksista liittyivät ihoon, mutta osa tapauksista kohdistui myös muualle, kuten selkäytimeen (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset). Trametinibin ja dabrafenibin yhdistelmähoidon käytössä on noudatettava varovaisuutta, kun sitä annetaan samanaikaisesti tai peräkkäin sädehoidon kanssa.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkkeiden vaikutus trametinibiin
Trametinibin metaboloituminen tapahtuu pääasiassa hydrolyyttisten entsyymien (esim. karboksyyliesteraasien) välittämän deasetylaation kautta, joten muut lääkeaineet eivät todennäköisesti vaikuta sen farmakokinetiikkaan metabolisten yhteisvaikutusten kautta (ks. kohta Farmakokinetiikka). Näiden hydrolyyttisten entsyymien välittämiä lääkeaineiden yhteisvaikutuksia ei voida sulkea pois, ja ne voivat vaikuttaa trametinibialtistukseen.
Trametinibi on P‑gp‑kuljetusproteiinin substraatti in vitro. Koska maksan P‑gp:n voimakkaan eston aiheuttamaa trametinibitason nousua ei voida sulkea pois, on varovaisuutta noudatettava annettaessa trametinibia yhdessä sellaisten lääkeavalmisteiden kanssa, jotka ovat voimakkaita P‑gp‑estäjiä (esim. verapamiili, siklosporiini, ritonaviiri, kinidiini, itrakonatsoli).
Trametinibin vaikutus muihin lääkkeisiin
In vitro ja in vivo ‑tutkimusten perusteella trametinibi ei todennäköisesti vaikuta merkittävästi muiden lääkevalmisteiden farmakokinetiikkaan CYP‑entsyymien tai kuljettajaproteiinien välityksellä (ks. kohta Farmakokinetiikka). Trametinibi saattaa estää tilapäisesti BCRP:n substraatteja (esim. pitavastatiinia) suolistossa. Tätä vaikutusta voidaan vähentää porrastamalla näiden lääkeaineiden ja trametinibin antaminen (lääkkeiden välillä on oltava 2 tuntia).
Kliinisten tietojen perusteella hormonaalisten ehkäisyvalmisteiden tehon ei odoteta heikkenevän, kun niitä annetaan samanaikaisesti trametinibimonoterapian kanssa (ks. kohta Farmakokinetiikka).
Käyttö yhdessä dabrafenibin kanssa
Trametinibin käyttö yhdessä dabrafenibin kanssa, ks. dabrafenibin valmisteyhteenvedon yhteisvaikutuksia koskevat kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset.
Sädehoito
Trametinibin ja dabrafenibin yhdistelmähoitoa saaneilla potilailla on raportoitu sädehoidon toksisuuden vahvistumista (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Ruoan vaikutus trametinibiin
Potilaiden on otettava pelkkä trametinibi tai trametinibi yhdessä dabrafenibin kanssa vähintään tunti ennen ateriaa tai kaksi tuntia aterian jälkeen, sillä ruoka vaikuttaa trametinibin imeytymiseen (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset/Raskauden ehkäisy naisilla
Naisia, jotka voivat tulla raskaaksi, on kehotettava käyttämään tehokkaita ehkäisymenetelmiä trametinibihoidon aikana ja 16 viikon ajan hoidon päättymisen jälkeen.
Käyttö yhdessä dabrafenibin kanssa saattaa heikentää hormonaalisten ehkäisyvalmisteiden tehoa, joten vaihtoehtoista ehkäisymenetelmää (kuten estemenetelmää) on käytettävä, kun trametinibia käytetään yhdessä dabrafenibin kanssa. Lisätiedot, ks. dabrafenibin valmisteyhteenveto.
Raskaus
Trametinibin käytöstä raskauden aikana ei ole tehty asianmukaisia ja hyvin kontrolloituja tutkimuksia. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Trametinibia ei pidä antaa raskaana oleville naisille. Jos trametinibia käytetään raskauden aikana tai jos potilas tulee raskaaksi trametinibihoidon aikana, hänelle on kerrottava mahdollisesta sikiöön kohdistuvasta vaarasta.
Imetys
Ei tiedetä, erittyykö trametinibi ihmisillä äidinmaitoon. Monet lääkeaineet erittyvät ihmisillä äidinmaitoon, joten imetettävään lapseen kohdistuvia riskejä ei voida sulkea pois. Trametinibia ei pidä antaa imettäville naisille. On päätettävä lopetetaanko imetys vai lopetetaanko trametinibihoito ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Tietoja pelkän trametinibin tai trametinibin ja dabrafenibin yhdistelmän vaikutuksista ihmisen hedelmällisyyteen ei ole. Eläimillä ei ole tehty hedelmällisyystutkimuksia, mutta naaraiden lisääntymiselimiin kohdistuvia haittavaikutuksia on havaittu (ks. kohta Prekliiniset tiedot turvallisuudesta). Trametinibi saattaa heikentää ihmisen hedelmällisyyttä.
Miehet, jotka käyttävät trametinibia yhdessä dabrafenibin kanssa
Dabrafenibia saaneilla eläimillä on havaittu spermatogeneesiin kohdistuvia vaikutuksia. Trametinibia yhdessä dabrafenibin kanssa käyttäville miespotilaille on kerrottava mahdollisesta spermatogeneesin heikentymisestä, joka voi olla korjautumatonta. Lisätiedot, ks. dabrafenibin valmisteyhteenveto.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Trametinibilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaan kliininen tila ja haittavaikutusprofiili on otettava huomioon, kun arvioidaan potilaan kykyä selviytyä harkintakykyä ja motorisia tai kognitiivisia taitoja vaativista tehtävistä. Potilaille on kerrottava että väsymys, huimaus tai silmäoireet voivat vaikuttaa näihin toimintoihin.
Haittavaikutukset
Yhteenveto turvallisuustiedoista
Trametinibimonoterapian turvallisuutta on arvioitu 329 potilaan yhdistettyjen turvallisuustietojen perusteella. BRAF V600 ‑mutatoitunutta leikkaukseen soveltumatonta tai metastasoitunutta melanoomaa sairastavat potilaat saivat trametinibia 2 mg kerran vuorokaudessa tutkimuksissa MEK114267, MEK113583 ja MEK111054. Näistä potilaista 211 sai trametinibia BRAF V600 ‑mutatoituneen melanooman hoitoon vaiheen III satunnaistetussa avoimessa MEK114267 (METRIC) ‑tutkimuksessa (ks. kohta Farmakodynamiikka). Trametinibin yleisimpiä haittavaikutuksia (esiintymistiheys ≥ 20 %) olivat ihottuma, ripuli, väsymys, perifeerinen edeema, pahoinvointi ja aknetyyppinen ihottuma.
Trametinibin ja dabrafenibin yhdistelmän turvallisuutta on arvioitu 1 188 aikuispotilaan yhdistettyjen turvallisuustietojen perusteella. Potilaat saivat 2 mg trametinibia kerran vuorokaudessa ja 150 mg dabrafenibia kahdesti vuorokaudessa BRAF V600 ‑mutatoituneen leikkaukseen soveltumattoman tai metastasoituneen melanooman, leikkauksella kokonaan poistetun asteen III BRAF V600 ‑mutatoituneen melanooman (liitännäishoito), edenneen ei‑pienisoluisen keuhkosyövän, tai edenneen erilaistuneen kilpirauhassyövän hoitoon. Keskimääräinen hoidon kesto oli 16 kuukautta (ks. kohta Farmakodynamiikka).
Yleisimpiä haittavaikutuksia (esiintymistiheys ≥ 20 %) trametinibin ja dabrafenibin yhdistelmähoidossa olivat: kuume, väsymys, pahoinvointi, ihottuma, vilunväristykset, ripuli, päänsärky, nivelkipu, oksentelu, yskä, verenvuoto ja perifeerinen turvotus.
Haittavaikutusluettelo
Kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä valvonnassa havaitut trametinibiin liittyvät haittavaikutukset on esitetty jäljempänä trametinibimonoterapian (taulukko 4) sekä trametinibin ja dabrafenibin yhdistelmähoidon (taulukko 5) osalta.
Haittavaikutukset luetellaan alla MedDRA‑elinjärjestelmän mukaan.
Yleisyysluokituksessa on noudatettu seuraavaa käytäntöä:
Hyvin yleinen ≥ 1/10
Yleinen ≥ 1/100, < 1/10
Melko harvinainen ≥ 1/1 000, < 1/100
Harvinainen ≥ 1/10 000, < 1/1 000
Hyvin harvinainen < 1/10 000
Tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin)
Jako yleisyysluokkiin perustuu kliinisistä tutkimuksista saatuihin absoluuttisiin esiintymistiheyksiin. Kunkin yleisyysluokan haittavaikutukset on esitetty vakavuusjärjestyksessä vakavimmasta alkaen.
Taulukko 4 Trametinibia ainoana lääkkeenä saaneilla potilailla raportoidut haittavaikutukset
| Elinjärjestelmä | Yleisyys (kaikki vaikeusasteet) | Haittavaikutukset |
| Infektiot | Yleinen | Karvatupentulehdus |
| Kynsivallintulehdus | ||
| Selluliitti | ||
| Märkärakkulainen ihottuma | ||
| Veri ja imukudos | Yleinen | Anemia |
| Immuunijärjestelmä | Yleinen | Yliherkkyysa |
| Aineenvaihdunta ja ravitsemus | Yleinen | Dehydraatio |
| Hermosto | Yleinen | Perifeerinen neuropatia (mukaan lukien sensorinen ja motorinen neuropatia) |
| Silmät | Yleinen | Näön hämärtyminen |
| Turvotus silmäkuopan ympärillä | ||
| Näön heikkeneminen | ||
| Melko harvinainen | Korioretinopatia | |
| Näköhermon nystyn turvotus | ||
| Verkkokalvon irtauma | ||
| Verkkokalvon laskimotukos | ||
| Sydän | Yleinen | Vasemman kammion toimintahäiriö |
| Ejektiofraktion pieneneminen | ||
| Bradykardia | ||
| Melko harvinainen | Sydämen vajaatoiminta | |
| Tuntematon | Eteis-kammiokatkosb | |
| Verisuonisto | Hyvin yleinen | Hypertensio |
| Verenvuotoc | ||
| Yleinen | Lymfedeema | |
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Yskä |
| Hengenahdistus | ||
| Yleinen | Pneumoniitti | |
| Melko harvinainen | Interstitiaalinen keuhkosairaus | |
| Ruoansulatuselimistö | Hyvin yleinen | Ripuli |
| Pahoinvointi | ||
| Oksentelu | ||
| Ummetus | ||
| Vatsakipu | ||
| Suun kuivuminen | ||
| Yleinen | Suutulehdus | |
| Melko harvinainen | Ruoansulatuskanavan perforaatio | |
| Koliitti | ||
| Iho ja ihonalainen kudos | Hyvin yleinen | Ihottuma |
| Aknetyyppinen ihottuma | ||
| Kuiva iho | ||
| Kutina | ||
| Hiustenlähtö | ||
| Yleinen | Punoitus | |
| Kämmenten ja jalkapohjien erytrodysestesia | ||
| Ihon haavaumat | ||
| Ihon halkeilu | ||
| Luusto, lihakset ja sidekudos | Melko harvinainen | Rabdomyolyysi |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Väsymys |
| Perifeerinen edeema | ||
| Kuume | ||
| Yleinen | Kasvojen turvotus | |
| Limakalvotulehdus | ||
| Voimattomuus | ||
| Tutkimukset | Hyvin yleinen | Kohonnut aspartaattiaminotransferaasiarvo |
| Yleinen | Kohonnut alaniiniaminotransferaasiarvo | |
| Kohonnut veren alkalinen fosfataasiarvo | ||
| Kohonnut veren kreatiinikinaasiarvo | ||
a Mahdollisia oireita ovat kuume, ihottuma, kohonneet maksan transaminaasiarvot ja näköhäiriöt. b Mukaan lukien täydellinen eteis-kammiokatkos. c Tapahtumia ovat mm.: nenäverenvuoto, veriulosteet, verenvuoto ikenistä, verivirtsaisuus ja peräsuolen, peräpukamien, mahalaukun, emättimen, sidekalvoalainen ja kallonsisäinen verenvuoto ja verenvuoto toimenpiteen jälkeen. | ||
Taulukko 5 Trametinibia ja dabrafenibia yhdistelmähoitona saaneilla potilailla raportoidut haittavaikutukset
| Elinjärjestelmä | Yleisyys (kaikki vaikeusasteet) | Haittavaikutukset |
| Infektiot | Hyvin yleinen | Nenänielutulehdus |
| Virtsatieinfektio | ||
| Yleinen | Selluliitti | |
| Karvatupen tulehdus | ||
| Kynnenvierustulehdus | ||
| Märkärakkulainen ihottuma | ||
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Yleinen | Ihon okasolusyöpäa |
| Papilloomab | ||
| Seborrooinen keratoosi | ||
| Melko harvinainen | Uusi primaarimelanoomac | |
| Ihopolyypit | ||
| Veri ja imukudos | Hyvin yleinen | Neutropenia |
| Anemia | ||
| Yleinen | Trombosytopenia | |
| Leukopenia | ||
| Immuunijärjestelmä | Melko harvinainen | Yliherkkyysd |
| Sarkoidoosi | ||
| Harvinainen | Hemofagosyyttinen lymfohistiosytoosi | |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Ruokahalun heikentyminen |
| Yleinen | Nestehukka | |
| Hyponatremia | ||
| Hypofosfatemia | ||
| Hyperglykemia | ||
| Tuntematon | Tuumorilyysioireyhtymä | |
| Hermosto | Hyvin yleinen | Päänsärky |
| Huimaus | ||
| Yleinen | Perifeerinen neuropatia (mukaan lukien sensorinen ja motorinen neuropatia) | |
| Silmät | Yleinen | Näön hämärtyminen |
| Näön heikkeneminen | ||
| Uveiitti | ||
| Melko harvinainen | Korioretinopatia | |
| Verkkokalvon irtauma | ||
| Turvotus silmäkuopan ympärillä | ||
| Sydän | Yleinen | Ejektiofraktion pieneneminen |
| Bradykardia | ||
| Melko harvinainen | Sydämen vajaatoiminta | |
| Vasemman kammion toimintahäiriö | ||
| Eteis-kammiokatkose | ||
| Tuntematon | Myokardiitti | |
| Verisuonisto | Hyvin yleinen | Hypertensio |
| Verenvuotof | ||
| Yleinen | Hypotensio | |
| Lymfedeema | ||
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Yskä |
| Yleinen | Pneumoniitti | |
| Hengenahdistus | ||
| Harvinainen | Interstitiaalinen keuhkosairaus | |
| Ruoansulatuselimistö | Hyvin yleinen | Vatsakipug |
| Ummetus | ||
| Ripuli | ||
| Pahoinvointi | ||
| Oksentelu | ||
| Yleinen | Suun kuivuus | |
| Suutulehdus | ||
| Melko harvinainen | Ruoansulatuskanavan perforaatio | |
| Haimatulehdus | ||
| Koliitti | ||
| Iho ja ihonalainen kudos | Hyvin yleinen | Ihon kuivuus |
| Kutina | ||
| Ihottuma | ||
| Punoitus | ||
| Aknetyyppinen ihottuma | ||
| Yleinen | Yleistynyt eksfoliatiivinen dermatiittih | |
| Aktiininen keratoosi | ||
| Yöhikoilu | ||
| Hyperkeratoosi | ||
| Hiustenlähtö | ||
| Kämmenten ja jalkapohjien erytrodysestesia | ||
| Ihomuutokset | ||
| Voimakas hikoilu | ||
| Pannikuliitti | ||
| Ihon fissuurat | ||
| Valoherkkyys | ||
| Melko harvinainen | Akuutti kuumeinen neutrofiilinen dermatoosi | |
| Tuntematon | Stevens–Johnsonin oireyhtymä | |
| DRESS-reaktio (lääkereaktio, johon liittyy eosinofiliaa ja systeemisiä oireita) | ||
| Tatuointeihin liittyvät ihoreaktiot | ||
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Nivelkipu |
| Lihaskipu | ||
| Raajakipu | ||
| Lihasspasmiti | ||
| Melko harvinainen | Rabdomyolyysi | |
| Munuaiset ja virtsatiet | Yleinen | Munuaisten vajaatoiminta, akuutti munuaisvaurio |
| Harvinainen | Munuaistulehdus | |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Väsymys |
| Vilunväristykset | ||
| Voimattomuus | ||
| Perifeerinen edeema | ||
| Kuume | ||
| Painonnousu (poikkeava painonnousu)j | ||
| Influenssan kaltainen sairaus | ||
| Yleinen | Limakalvotulehdus | |
| Kasvojen turvotus | ||
| Tutkimukset | Hyvin yleinen | Kohonnut ALAT‑arvo |
| Kohonnut ASAT‑arvo | ||
| Yleinen | Kohonnut AFOS‑arvo | |
| Kohonnut GGT‑arvo | ||
| Kohonnut veren kreatiinikinaasiarvo | ||
| Vammat, myrkytykset ja hoitokomplikaatiot | Yleinen | Sädehoidon toksisuuden vahvistuminen |
a Ihon okasolusyöpä: Okasolusyöpä, ihon okasolusyöpä, okasolusyöpä in situ (Bowenin tauti) ja keratoakantooma b Papillooma, ihon papillooma c Pahanlaatuinen melanooma ja pinnallisesti leviävä melanooma (aste määrittelemätön) d Sisältää lääkeyliherkkyyden e Mukaan lukien täydellinen eteis-kammiokatkos f Eri alueiden verenvuodot, mukaan lukien kallonsisäinen verenvuoto ja kuolemaan johtava verenvuoto g Sisältää ylävatsakivun ja alavatsakivun h Sisältää ihon hilseilyn i Lihasspasmit, lihasten ja luuston jäykkyys j Painonnousua (poikkeava painonnousu) on raportoitu ainoastaan pediatrisilla potilailla. | ||
Tärkeimpien haittavaikutusten kuvaus
Uudet maligniteetit
Uusia maligniteetteja (ihomaligniteetteja ja muita maligniteetteja) saattaa esiintyä, kun trametinibia käytetään yhdessä dabrafenibin kanssa. Ks. dabrafenibin valmisteyhteenveto.
Verenvuoto
Trametinibia ainoana lääkkeenä tai yhdessä dabrafenibin kanssa saaneilla potilailla esiintyi verenvuototapahtumia, mukaan lukien merkittäviä verenvuototapahtumia ja kuolemaan johtaneita verenvuotoja. Valtaosa verenvuototapahtumista oli lieviä. Yhdistetyssä turvallisuuspopulaatiossa kuolemaan johtaneita kallonsisäisiä verenvuotoja esiintyi trametinibin ja dabrafenibin yhdistelmähoidossa < 1 %:lla potilaista. Ensimmäisten verenvuototapahtumien ilmaantumiseen kuluneen ajan mediaani trametinibin ja dabrafenibin yhdistelmähoidossa oli 94 vuorokautta vaiheen III melanoomatutkimuksissa ja 75 vuorokautta ei‑pienisoluisen keuhkosyövän tutkimuksessa potilailla, jotka olivat aiemmin saaneet syöpähoitoa.
Samanaikainen antitromboottinen lääkitys tai antikoagulanttihoito saattaa suurentaa verenvuotoriskiä. Mahdollinen verenvuoto hoidetaan kliinisen tilanteen mukaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vasemman kammion ejektiofraktion pieneneminen / vasemman kammion toimintahäiriö
Trametinibin on raportoitu pienentävän vasemman kammion ejektiofraktiota, kun sitä käytetään ainoana lääkkeenä tai yhdessä dabrafenibin kanssa. Kliinisissä tutkimuksissa aika vasemman kammion toimintahäiriön ja sydämen vajaatoiminnan ensimmäiseen ilmaantumiseen ja ejektiofraktion ensimmäiseen pienenemiseen oli 2–5 kuukautta (mediaani). Yhdistetyssä turvallisuuspopulaatiossa trametinibin ja dabrafenibin yhdistelmähoidossa vasemman kammion ejektiofraktion pienenemistä on ilmoitettu 8 %:lla potilaista. Useimmat tapaukset olivat oireettomia ja korjautuvia. Kliinisiin trametinibitutkimuksiin ei otettu potilaita, joiden vasemman kammion ejektiofraktio alitti hoitolaitoskohtaisen viitearvojen alarajan. Trametinibia on käytettävä varoen, jos potilaalla on jokin sairaus, joka voi heikentää vasemman kammion toimintaa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kuume
Kliinisissä tutkimuksissa on ilmoitettu kuumetta, kun trametinibia on käytetty ainoana lääkkeenä tai yhdessä dabrafenibin kanssa. Kuumeen ilmaantuvuus ja vaikeusaste ovat kuitenkin suurempia yhdistelmähoidossa. Ks. dabrafenibin valmisteyhteenvedon kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.
Maksaan liittyvät tapahtumat
Kliinisissä tutkimuksissa on ilmoitettu maksaan kohdistuvia haittavaikutuksia, kun trametinibia on käytetty ainoana lääkkeenä tai yhdessä dabrafenibin kanssa. Yleisimpiä maksaan kohdistuneita haittavaikutuksia olivat ALAT‑ ja ASAT‑arvojen kohoaminen, ja suurimmassa osassa tapauksista vaikeusasteluokka oli 1 tai 2. Pelkkää trametinibia saaneilla potilailla yli 90 % näistä maksatapahtumista ilmaantui kuuden ensimmäisen hoitokuukauden aikana. Maksatapahtumat todettiin kliinisissä tutkimuksissa, joissa arvoja seurattiin neljän viikon välein. Trametinibia ainoana lääkkeenä tai yhdessä dabrafenibin kanssa saavien potilaiden maksatoimintaa on suositeltavaa seurata neljän viikon välein kuuden kuukauden ajan. Maksa‑arvojen seurantaa voidaan jatkaa myös tämän jälkeen, jos se on kliinisesti aiheellista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hypertensio
Verenpaineen nousua on raportoitu pelkän trametinibin ja trametinibin ja dabrafenibin yhdistelmän käytön aikana sekä potilailla, joilla on aiemmin ollut hypertensio, että potilailla, joiden verenpaine ei ole ollut koholla. Verenpaine on mitattava hoitoa aloitettaessa ja sitä on seurattava hoidon aikana, ja tarvittaessa hypertensio on pidettävä hallinnassa tavanomaisella hoidolla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Interstitiaalinen keuhkosairaus/Pneumoniitti
Trametinibia tai trametinibin ja dabrafenibin yhdistelmää saaville potilaille saattaa kehittyä interstitiaalinen keuhkosairaus tai pneumoniitti. Jos potilaalla epäillään interstitiaalista keuhkosairautta tai pneumoniittia ja myös jos potilaalla on uusia tai eteneviä keuhko‑oireita ja ‑löydöksiä, kuten yskää, hengenahdistusta, hypoksiaa, nestettä keuhkopussissa tai infiltraatteja, trametinibihoito on keskeytettävä, kunnes kliiniset tutkimukset on saatu päätökseen. Trametinibi on lopettava pysyvästi, jos potilaalla diagnosoidaan hoitoon liittyvä interstitiaalinen keuhkosairaus tai pneumoniitti (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Näön heikkeneminen
Näköhäiriöitä aiheuttavia sairauksia, kuten verkkokalvon pigmenttiepiteelin irtaumia ja verkkokalvon laskimotukoksia, on havaittu trametinibihoidon yhteydessä. Trametinibin kliinisissä tutkimuksissa on raportoitu oireita, kuten näön hämärtymistä, näöntarkkuuden heikkenemistä ja muita näköhäiriöitä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Ihottuma
Ihottumaa on havaittu noin 60 %:lla potilaista, kun trametinibia on käytetty ainoana lääkkeenä, ja noin 27 %:lla potilaista yhdistetyssä turvallisuuspopulaatiossa trametinibin ja dabrafenibin yhdistelmää koskevissa tutkimuksissa. Suurimmassa osassa näistä tapauksista vaikeusaste oli 1 tai 2, eikä se vaatinut hoidon keskeyttämistä eikä annoksen pienentämistä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Rabdomyolyysi
Rabdomyolyysiä on raportoitu pelkkää trametinibia tai trametinibin ja dabrafenibin yhdistelmää käyttävillä potilailla. Rabdomyolyysin oireet ja löydökset vaativat asianmukaista kliinistä selvittelyä ja tarvittaessa hoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haimatulehdus
Haimatulehdusta on ilmoitettu dabrafenibin ja trametinibin yhdistelmähoidossa. Ks. dabrafenibin valmisteyhteenveto.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa on ilmoitettu dabrafenibin ja trametinibin yhdistelmähoidossa. Ks. dabrafenibin valmisteyhteenveto.
Pediatriset potilaat
Kalvopäällysteisten trametinibitablettien turvallisuutta ei ole arvioitu prospektiivisissa kliinisissä tutkimuksissa melanoomaa sairastavilla nuorilla potilailla. Kun julkaistussa, Italiassa toteutetussa retrospektiivisessä tutkimuksessa arvioitiin 6:ta edennyttä BRAF V600 ‑mutaatiopositiivista melanoomaa sairastavaa pediatrista potilasta, jotka saivat trametinibia yhdessä dabrafenibin kanssa (ks. kohta Farmakodynamiikka), yleisimmin raportoidut haittatapahtumat olivat kuume ja kohonnut kreatiinikinaasiarvo. Kahdella nuorella potilaalla todettiin oireeton > 10 %:n pienenemä vasemman kammion ejektiofraktiossa. Pienenemä korjautui, kun trametinibihoito keskeytettiin tilapäisesti. Molemmat potilaat aloittivat trametinibihoidon uudelleen pienennetyllä annoksella, eikä sydäntoksisuutta enää havaittu.
Muissa käyttöaiheissa tehdyissä kliinisissä tutkimuksissa trametinibin ja dabrafenibin yhdistelmän turvallisuusprofiili pediatrisilla potilailla oli laajalti yhdenmukainen aikuispotilailla aiemmin todetun turvallisuusprofiilin kanssa. Painonnousua (poikkeava painonnousu) on raportoitu ainoastaan pediatrisilla potilailla. Sitä raportoitiin haittavaikutuksena 15 %:lla nuorista potilaista; asteen 3 haittavaikutus raportoitiin 4 %:lla potilaista.
Muut erityisryhmät
Iäkkäät
Vaiheen III trametinibitutkimuksessa, jossa potilailla oli leikkaukseen soveltumaton tai metastasoitunut melanooma (n = 211), 49 potilasta (23 %) oli ≥ 65‑vuotiaita ja 9 potilasta (4 %) ≥ 75‑vuotiaita. Haittavaikutuksia ja vakavia haittavaikutuksia esiintyi yhtä suurella osalla < 65‑vuotiaista ja ≥ 65‑vuotiaista potilaista. Lääkevalmisteen pysyvään lopettamiseen, annoksen pienentämiseen ja hoidon keskeyttämiseen johtaneet haittavaikutukset olivat todennäköisempiä ≥ 65‑vuotiailla kuin < 65‑vuotiailla potilailla.
Trametinibin ja dabrafenibin yhdistelmää saaneiden potilaiden yhdistetyssä turvallisuuspopulaatiossa (n = 1 188) 27 % potilaista oli ≥ 65‑vuotiaita ja 6 % potilaista ≥ 75‑vuotiaita. Haittavaikutuksen kokeneiden potilaiden osuus oli kaikissa tutkimuksissa samaa luokkaa < 65 vuoden ikäisillä ja ≥ 65 vuoden ikäisillä. Vakavia haittavaikutuksia ja lääkevalmisteen käytön pysyvään lopettamiseen, annoksen pienentämiseen ja hoidon keskeyttämiseen johtavia haittavaikutuksia esiintyi todennäköisemmin ≥ 65‑vuotiailla potilailla kuin < 65‑vuotiailla.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa lievän tai keskivaikean munuaisten vajaatoiminnan vuoksi (ks. kohta Farmakokinetiikka). Trametinibin käytössä on noudatettava varovaisuutta vaikeassa munuaisten vajaatoiminnassa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Annostuksen muuttaminen ei ole tarpeen lievän maksan vajaatoiminnan vuoksi (ks. kohta Farmakokinetiikka). Trametinibin käytössä on noudatettava varovaisuutta keskivaikeassa tai vaikeassa maksan vajaatoiminnassa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Trametinibin ja dabrafenibin yhdistelmähoito potilailla, joilla on aivometastaaseja
Trametinibin ja dabrafenibin yhdistelmän tehoa ja turvallisuutta on arvioitu avoimessa vaiheen II monikohorttitutkimuksessa potilailla, joilla on BRAF V600 ‑mutatoitunut, aivometastaaseja aiheuttanut melanooma. Näillä potilailla havaittu turvallisuusprofiili vaikuttaa yhdenmukaiselta tämän yhdistelmän yhdistetyn turvallisuusprofiilin kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä trametinibimonoterapiatutkimuksissa raportoitiin yksi tahaton yliannostustapaus, jossa kerta‑annos oli 4 mg. Haittatapahtumia ei raportoitu tämän trametinibin yliannostuksen jälkeen. Trametinibin ja dabrafenibin yhdistelmää koskevissa kliinisissä tutkimuksissa 11 potilaalla ilmoitettiin trametinibin yliannostus (4 mg). Vakavia haittatapahtumia ei ilmoitettu. Yliannostukseen ei ole spesifistä hoitoa. Yliannostapauksissa on annettava asianmukaista tukihoitoa, ja potilaan tilaa on seurattava tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, proteiinikinaasin estäjät, mitogeenin aktivoiman proteiinikinaasin (MEK) estäjät, ATC-koodi: L01EE01
Vaikutusmekanismi
Trametinibi on reversiibeli, erittäin selektiivinen, mitogeenin aktivoiman solunulkoisen signaalin säätelykinaasi 1:n (MEK1:n) ja MEK2:n aktivaation ja kinaasiaktiivisuuden allosteerinen estäjä. MEK‑proteiinit ovat ERK‑kinaasin signalointireitin osia. Melanoomassa ja muissa syövissä tämän reitin aktivoivat usein BRAF:n mutatoituneet muodot, mikä puolestaan aktivoi MEK:n. Trametinibi estää BRAF:n aiheuttaman MEK:n aktivoitumisen ja estää MEK:n kinaasiaktiivisuutta. Trametinibi estää BRAF V600 ‑mutatoituneiden melanoomasolulinjojen kasvua, ja sillä on osoitettu olevan syövän kasvua estäviä vaikutuksia BRAF V600 ‑mutatoituneen melanooman eläinmalleissa.
Käyttö yhdessä dabrafenibin kanssa
Dabrafenibi on RAF‑kinaasien estäjä. BRAF:n onkogeeniset mutaatiot johtavat RAS/RAF/MEK/ERK‑reitin konstitutiiviseen aktivaatioon. Trametinibi ja dabrafenibi estävät siis kahta tämän reitin kinaasia (MEK ja RAF), ja tästä syystä yhdistelmä estää reittiä samanaikaisesti. Trametinibin ja dabrafenibin yhdistelmän on osoitettu rajoittavan kasvua BRAF V600 ‑mutaatiopositiivisissa melanoomasolulinjoissa in vitro ja viivästyttävän resistenssin kehittymistä in vivo BRAF V600 ‑mutaatiopositiivisissa melanoomaksenografteissa.
BRAF‑mutaatiostatuksen määrittäminen
Ennen trametinibin tai trametinibin ja dabrafenibin yhdistelmän käytön aloittamista kasvaimen BRAF V600 ‑mutaatio on vahvistettava validoidulla testillä.
Kliinisissä melanoomatutkimuksissa BRAF V600 ‑mutaatio määritettiin keskitetysti BRAF‑mutaatioanalyysillä tuoreimmasta käytettävissä olevasta kasvainnäytteestä. Primaarikasvain tai etäpesäkekohdan kasvain testattiin validoidulla Response Genetics ‑yhtiön kehittämällä polymeraasiketjureaktiomenetelmällä (PCR). Testi on kehitetty erityisesti erottamaan V600E‑ ja V600K‑mutaatiot toisistaan. Tutkimukseen valittiin vain potilaita, joilla oli BRAF V600E‑ tai V600K ‑mutaatiopositiivinen kasvain.
Kaikki potilasnäytteet testattiin myöhemmin uudelleen käyttäen bioMerieux’n (bMx) validoitua THxID BRAF ‑testiä, jolla on CE‑merkintä. Tämä bHx:n THxID BRAF ‑testi on alleelispesifinen PCR, ja se tehdään DNA:sta, joka on eristetty formaliinilla kiinnitetystä parafiiniin valetusta (FFPE) kasvainkudoksesta. Testi on kehitetty tunnistamaan BRAF V600E ja V600K ‑mutaatiot erittäin suurella herkkyydellä (mikä tarkoittaa, että testi tunnstaa mikäli analysoitavassa näytteessä on vähintään 5 % V600E‑ ja V600K‑sekvenssiä sekoittuneena normaaliin DNA:han, kun käytetään FFPE‑kudoksesta eristettyä DNA:ta). Prekliinisissä ja kliinisissä tutkimuksissa, joissa on käytetty retrospektiivisiä kaksisuuntaisia Sanger‑sekvensointimenetelmiä, on osoitettu, että testi tunnistaa pienemmällä herkkyydellä myös harvinaisemmat BRAF V600D‑ ja V600E/K601E‑mutaatiot. Prekliinisten ja kliinisten tutkimusten näytteet (n = 876), jotka oli THxID BRAF ‑testillä todettu mutaatiopositiivisiksi, sekvensoitiin myöhemmin vertailumenetelmällä, ja testin spesifisyys oli 94 %.
Farmakodynaamiset vaikutukset
Trametinibi laski fosforyloituneen ERK:n tasoja BRAF‑mutatoituneissa melanoomasolulinjoissa ja melanooman ksenograftimalleissa.
Potilailla, joilla oli BRAF‑ ja NRAS‑mutaatiopositiivinen melanooma, trametinibi aiheutti annoksesta riippuvia muutoksia kasvainmerkkiaineissa, joita olivat fosforyloituneen ERK:n määrän väheneminen, Ki67:n (soluproliferaatiomerkkiaineen) määrän väheneminen ja p27:n (apoptoosimerkkiaineen) lisääntyminen. Kun trametinibia annettiin toistuvina annoksina 2 mg kerran vuorokaudessa, trametinibin pitoisuuden keskiarvo oli prekliinisen tavoitepitoisuuden yläpuolella koko 24 tunnin annosvälin ajan, joten sillä oli pitkäkestoinen MEK‑reittiä estävä vaikutus.
Kliininen teho ja turvallisuus
Leikkaukseen soveltumaton tai metastasoitunut melanooma
Kliinisissä tutkimuksissa oli mukana vain ihomelanoomaa sairastavia potilaita. Valmisteen tehoa ei ole tutkittu silmän uveaalimelanoomassa eikä limakalvon melanoomien hoidossa.
- Trametinibi yhdessä dabrafenibin kanssa
Potilaat, jotka eivät olleet aiemmin saaneet hoitoa
Trametinibin (2 mg kerran vuorokaudessa) ja dabrafenibin (150 mg kahdesti vuorokaudessa) yhdistelmän suositusannosten tehoa ja turvallisuutta tutkittiin kahdessa vaiheen III tutkimuksessa ja yhdessä vaiheen I/II lisätutkimuksessa aikuispotilailla, joilla oli leikkaukseen soveltumaton tai metastasoitunut BRAF V600 ‑mutaatiopositiivinen melanooma.
MEK115306 (COMBI‑d):
MEK115306 oli vaiheen III satunnaistettu, kaksoissokkoutettu tutkimus, jossa verrattiin dabrafenibin ja trametinibin yhdistelmää dabrafenibiin ja lumeeseen ensilinjan hoidossa tutkittavilla, joilla oli leikkaukseen soveltumaton (aste IIIC) tai metastasoitunut (aste IV) BRAF V600E/K ‑mutaatiopositiivinen ihomelanooma. Tutkimuksen ensisijainen päätetapahtuma oli etenemisvapaa elinaika ja tärkeä toissijainen päätetapahtuma oli kokonaiselinaika. Tutkittavat stratifioitiin laktaattidehydrogenaasiarvon (LDH) perusteella (> viitealueen yläraja [ULN] vs ≤ ULN) ja BRAF‑mutaation perusteella (V600E vs V600K).
Yhteensä 423 tutkittavaa satunnaistettiin suhteessa 1:1 saamaan joko yhdistelmähoitoa (n = 211) tai dabrafenibia (n = 212). Valtaosa tutkittavista oli kaukasialaista syntyperää (> 99 %) ja miehiä (53 %). Iän mediaani oli 56 vuotta (28 % oli ≥ 65‑vuotiaita). Valtaosalla tutkittavista oli asteen IVM1c tauti (67 %). Lähtötilanteessa valtaosalla tutkittavista LDH‑arvo oli ≤ ULN (65 %), Eastern Cooperative Oncology Group (ECOG) ‑toimintakykyluokka 0 (72 %) ja sisäelimet olivat affisioituneet (73 %). Valtaosalla tutkittavista oli BRAF V600E ‑mutaatio (85 %). Tutkimukseen ei otettu tutkittavia, joilla oli aivometastaaseja.
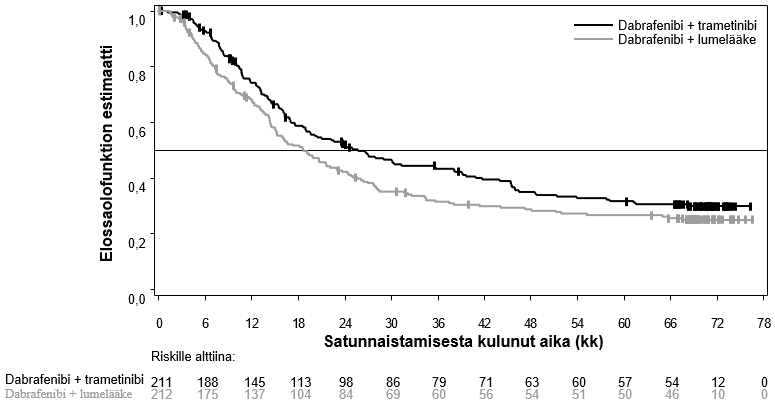
Kokonaiselinajan (OS) mediaani ja arvioidut 1 v, 2 v, 3 v, 4 v ja 5 v elossaolo-osuudet esitetään taulukossa 6. Viiden vuoden kohdalla tehdyssä OS-analyysissä kokonaiselinajan mediaani oli yhdistelmäryhmässä noin 7 kk pidempi kuin dabrafenibimonoterapiaryhmässä (25,8 kk vs 18,7 kk), ja 5 v elossaolo-osuus oli yhdistelmäryhmässä 32 %, kun taas dabrafenibimonoterapiaryhmän luku oli 27 % (taulukko 6, kuva 1). Kokonaiselinajan Kaplan–Meier-kuvaaja näyttää tasaantuvan 3 vuoden ja 5 vuoden välillä (ks. kuva 1). Potilailla, joiden lähtötilanteen laktaattidehydrogenaasiarvo oli normaali, 5 vuoden kokonaiselossaolo-osuus oli yhdistelmäryhmässä 40 % (95 % lv: 31,2–48,4) ja dabrafenibimonoterapiaryhmässä 33 % (95 % lv: 25,0–41,0). Potilailla, joiden lähtötilanteen laktaattidehydrogenaasiarvo oli koholla, luku puolestaan oli yhdistelmäryhmässä 16 % (95 % lv: 8,4–26,0) ja dabrafenibimonoterapiaryhmässä 14 % (95 % lv: 6,8–23,1).
Taulukko 6 Kokonaiselinajan tulokset, tutkimus MEK115306 (COMBI‑d)
OS-analyysi (tiedonkeruun katkaisu: 12.1.2015) | 5 vuoden OS-analyysi (tiedonkeruun katkaisu: 20.12.2018) | |||
| Dabrafenibi + trametinibi (n = 211) | Dabrafenibi + lumelääke (n = 212) | Dabrafenibi + trametinibi (n = 211) | Dabrafenibi + lumelääke (n = 212) | |
| Potilasmäärä | ||||
| Kuolemantapaukset (tapahtumat), n (%) | 99 (47) | 123 (58) | 135 (64) | 151 (71) |
| OS-estimaatit (kk) | ||||
| Mediaani (95 % lv) | 25,1 (19,2–NR) | 18,7 (15,2–23,7) | 25,8 (19,2–38,2) | 18,7 (15,2–23,1) |
| Riskitiheyssuhde (95 % lv) | 0,71 (0,55–0,92) | 0,80 (0,63–1,01) | ||
| p-arvo | 0,011 | NA | ||
| Kokonaiselinajan estimaatti, % (95 % lv) | Dabrafenibi + trametinibi (n = 211) | Dabrafenibi + lumelääke (n = 212) | ||
| 1 vuoden kohdalla | 74 (66,8–79,0) | 68 (60,8–73,5) | ||
| 2 vuoden kohdalla | 52 (44,7–58,6) | 42 (35,4–48,9) | ||
| 3 vuoden kohdalla | 43 (36,2–50,1) | 31 (25,1–37,9) | ||
| 4 vuoden kohdalla | 35 (28,2–41,8) | 29 (22,7–35,2) | ||
| 5 vuoden kohdalla | 32 (25,1–38,3) | 27 (20,7–33,0) | ||
| NR = ei saavutettu, NA = ei sovellu | ||||
Kuva 1 Kaplan–Meier‑kuvaajat kokonaiselinajasta MEK115306‑tutkimuksessa (ITT‑populaatio)
Ensisijaisen päätetapahtuman (etenemisvapaa elinaika) paremmuus yhdistelmäryhmässä verrattuna dabrafenibimonoterapiaan säilyi 5 vuoden ajan. Myös kokonaisvasteprosentissa havaittiin kohenemista ja vasteen kesto oli pidempi yhdistelmäryhmässä verrattuna dabrafenibimonoterapiaan (taulukko 7).
Taulukko 7 MEK115306‑tutkimuksen tehoa osoittavat tulokset (COMBI‑d)
| Ensisijainen analyysi (tiedonkeruun katkaisu: 26.8.2013) | Päivitetty analyysi (tiedonkeruun katkaisu: 12.1.2015) | 5 vuoden analyysi (tiedonkeruun katkaisu: 10.12.2018) | ||||
| Päätetapahtuma | Dabrafenibi + trametinibi (n = 211) | Dabrafenibi + lumelääke (n = 212) | Dabrafenibi + trametinibi (n = 211) | Dabrafenibi + lumelääke (n = 212) | Dabrafenibi + trametinibi (n = 211) | Dabrafenibi + lumelääke (n = 212) |
| Etenemisvapaa elinaika (PFS)a | ||||||
| Etenevä tauti tai kuolema, n (%) | 102 (48) | 109 (51) | 139 (66) | 162 (76) | 160 (76) | 166 (78) |
| PFS‑mediaani (kk) (95 % lv) | 9,3 (7,7–11,1) | 8,8 (5,9–10,9) | 11,0 (8,0–13,9) | 8,8 (5,9–9,3) | 10,2 (8,1–12,8) | 8,8 (5,9–9,3) |
Riskitiheyssuhde (95 % lv) | 0,75 (0,57–0,99) | 0,67 (0,53–0,84) | 0,73 (0,59–0,91) | |||
| p‑arvo | 0,035 | < 0,001f | NA | |||
Kokonaisvasteprosenttib % (95 % lv) | 67 (59,9–73,0) | 51 (44,5–58,4) | 69 (61,8–74,8) | 53 (46,3–60,2) | 69 (62,5–75,4) | 54 (46,8–60,6) |
Kokonaisvasteprosenttien ero (95 % lv) | 15e (5,9–24,5) | 15e (6,0–24,5) | NA | |||
| p‑arvo | 0,0015 | 0,0014f | NA | |||
Vasteen kestoc (kk) Mediaani (95 % lv) | 9,2d (7,4–NR) | 10,2d (7,5–NR) | 12,9 (9,4–19,5) | 10,6 (9,1–13,8) | 12,9 (9,3–18,4) | 10,2 (8,3–13,8) |
a – Etenemisvapaa elossaolo (tutkijan arvioima) b – Kokonaisvasteprosentti = täydellinen vaste + osittainen vaste c – Vasteen kesto d – Ilmoitusajankohtana valtaosa (≥ 59 %) tutkijan arvioimista vasteista jatkui yhä e – Kokonaisvasteprosenttien ero laskettiin pyöristämättömien kokonaisvasteprosenttitulosten perusteella f – Päivitetty analyysi ei ollut ennalta suunniteltu, eikä p-arvoa mukautettu useampaan testaamiseen NR = ei saavutettu NA = ei sovellu | ||||||
MEK116513 (COMBI‑v):
MEK116513 oli kaksiryhmäinen, satunnaistettu, avoin vaiheen III tutkimus, jossa verrattiin dabrafenibin ja trametinibin yhdistelmää pelkkään vemurafenibiin BRAF V600 ‑mutaatiopositiivisessa leikkaukseen soveltumattomassa tai metastasoituneessa melanoomassa. Tutkimuksen ensisijainen päätetapahtuma oli kokonaiselinaika ja tärkeä toissijainen päätetapahtuma etenemisvapaa elinaika. Tutkittavat stratifioitiin laktaattidehydrogenaasiarvon (LDH) perusteella (> viitealueen yläraja [ULN] vs ≤ ULN) ja BRAF‑mutaation perusteella (V600E vs V600K).
Yhteensä 704 tutkittavaa satunnaistettiin suhteessa 1:1 saamaan joko yhdistelmähoitoa tai vemurafenibia. Valtaosa tutkittavista oli kaukasialaista syntyperää (> 96 %) ja miehiä (55 %). Iän mediaani oli 55 vuotta (24 % oli ≥ 65‑vuotiaita). Valtaosalla tutkittavista oli asteen IVM1c tauti (yhteensä 61 %). Lähtötilanteessa valtaosalla tutkittavista LDH‑arvo oli ≤ ULN (67 %), ECOG‑toimintakykyluokka 0 (70 %) ja sisäelimet olivat affisioituneet (78 %). Kaiken kaikkiaan 54 %:lla tutkittavista oli lähtötilanteessa < 3 tautialuetta. Valtaosalla tutkittavista oli BRAF V600E ‑mutaatiopositiivinen melanooma (89 %). Tutkimukseen ei otettu tutkittavia, joilla oli aivometastaaseja.
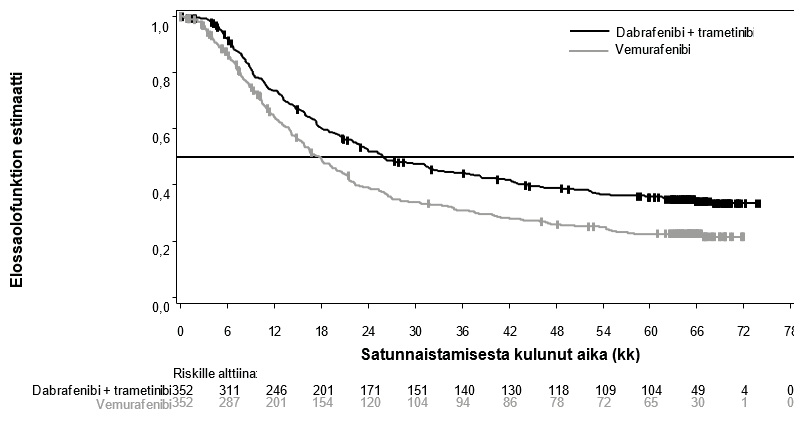
Kokonaiselinajan (OS) mediaani ja arvioidut 1 v, 2 v, 3 v, 4 v ja 5 v elossaolo-osuudet esitetään taulukossa 8. Viiden vuoden kohdalla tehdyssä OS-analyysissä kokonaiselinajan mediaani oli yhdistelmäryhmässä noin 8 kk pidempi kuin vemurafenibimonoterapiaryhmässä (26,0 kk vs 17,8 kk), ja 5 v elossaolo-osuus oli yhdistelmäryhmässä 36 %, kun taas vemurafenibimonoterapiaryhmän luku oli 23 % (taulukko 8, kuva 2). Kokonaiselinajan Kaplan–Meier-kuvaaja näyttää tasaantuvan 3 vuoden ja 5 vuoden välillä (ks. kuva 2). Potilailla, joiden lähtötilanteen laktaattidehydrogenaasiarvo oli normaali, 5 vuoden kokonaiselossaolo-osuus oli yhdistelmäryhmässä 46 % (95 % lv: 38,8–52,0) ja vemurafenibimonoterapiaryhmässä 28 % (95 % lv: 22,5–34,6). Potilailla, joiden lähtötilanteen laktaattidehydrogenaasiarvo oli koholla, luku puolestaan oli yhdistelmäryhmässä 16 % (95 % lv: 9,3–23,3) ja vemurafenibimonoterapiaryhmässä 10 % (95 % lv: 5,1–17,4).
Taulukko 8 Kokonaiselinajan tulokset, tutkimus MEK116513 (COMBI‑v)
OS-analyysi (tiedonkeruun katkaisu: 13.3.2015) | 5 vuoden OS-analyysi (tiedonkeruun katkaisu: 8.10.2018) | |||
Dabrafenibi + trametinibi (n = 352) | Vemurafenibi (n = 352) | Dabrafenibi + trametinibi (n = 352) | Vemurafenibi (n = 352) | |
| Potilasmäärä | ||||
| Kuolemantapaukset (tapahtumat), n (%) | 155 (44) | 194 (55) | 216 (61) | 246 (70) |
| OS-estimaatit (kk) | ||||
| Mediaani (95 % lv) | 25,6 (22,6–NR) | 18,0 (15,6–20,7) | 26,0 (22,1–33,8) | 17,8 (15,6–20,7) |
| Korjattu riskitiheyssuhde (95 % lv) | 0,66 (0,53–0,81) | 0,70 (0,58–0,84) | ||
| p-arvo | < 0,001 | NA | ||
| Kokonaiselinajan estimaatti, % (95 % lv) | Dabrafenibi + trametinibi (n = 352) | Vemurafenibi (n = 352) | ||
| 1 vuoden kohdalla | 72 (67–77) | 65 (59–70) | ||
| 2 vuoden kohdalla | 53 (47,1–57,8) | 39 (33,8–44,5) | ||
| 3 vuoden kohdalla | 44 (38,8–49,4) | 31 (25,9–36,2) | ||
| 4 vuoden kohdalla | 39 (33,4–44,0) | 26 (21,3–31,0) | ||
| 5 vuoden kohdalla | 36 (30,5–40,9) | 23 (18,1–27,4) | ||
| NR = ei saavutettu, NA = ei sovellu | ||||
Kuva 2 Kaplan–Meier‑kuvaajat MEK116513‑tutkimuksen päivitetystä kokonaiselinajan analyysistä
Toissijaisen päätetapahtuman (etenemisvapaa elinaika) paremmuus säilyi 5 vuoden ajan yhdistelmäryhmässä verrattuna vemurafenibimonoterapiaan. Kohenemista havaittiin myös kokonaisvasteprosentissa, ja vasteen kesto oli pidempi yhdistelmäryhmässä verrattuna vemurafenibimonoterapiaan (taulukko 9).
Taulukko 9 MEK116513‑tutkimuksen tehoa osoittavat tulokset (COMBI‑v)
| Ensisijainen analyysi (tiedonkeruun katkaisu: 17.4.2014) | 5 vuoden analyysi (tiedonkeruun katkaisu: 8.10.2018) | |||
| Päätetapahtuma | Dabrafenibi + trametinibi (n = 352) | Vemurafenibi (n = 352) | Dabrafenibi + trametinibi (n = 352) | Vemurafenibi (n = 352) |
| Etenemisvapaa elinaika (PFS)a | ||||
Etenevä tauti tai kuolema, n (%) | 166 (47) | 217 (62) | 257 (73) | 259 (74) |
PFS‑mediaani (kk) (95 % lv) | 11,4 (9,9–14,9) | 7,3 (5,8–7,8) | 12,1 (9,7–14,7) | 7,3 (6,0–8,1) |
Riskitiheyssuhde (95 % lv) | 0,56 (0,46–0,69) | 0,62 (0,52–0,74) | ||
| p‑arvo | < 0,001 | NA | ||
Kokonaisvasteprosenttib % (95 % lv) | 64 (59,1–69,4) | 51 (46,1–56,8) | 67 (62,2–72,2) | 53 (47,2–57,9) |
Kokonaisvasteprosenttien ero (95 % lv) | 13 (5,7–20,2) | NA | ||
| p‑arvo | 0,0005 | NA | ||
Vasteen kestoc (kk) Mediaani (95 % lv) | 13,8d (11,0–NR) | 7,5d (7,3–9,3) | 13,8 (11,3–18,6) | 8,5 (7,4–9,3) |
a – Etenemisvapaa elossaolo (tutkijan arvioima) b – Kokonaisvasteprosentti = täydellinen vaste + osittainen vaste c – Vasteen kesto d – Ilmoitusajankohtana valtaosa (dabrafenibi + trametinibiryhmässä 59 % ja vemurafenibiryhmässä 42 %) tutkijan arvioimista hoitovasteista jatkui yhä NR = ei saavutettu NA = ei sovellu | ||||
Aikaisempi BRAF‑estäjähoito
Tietoja on rajallisesti potilaista, jotka käyttävät trametinibin ja dabrafenibin yhdistelmää ja joiden tauti on edennyt aiemman BRAF‑estäjähoidon aikana.
BRF113220‑tutkimuksen B‑osaan kuului kohortti, jonka 26 potilaalla tauti oli edennyt BRAF‑estäjähoidon aikana. Trametinibin (2 mg kerran vuorokaudessa) ja dabrafenibin (150 mg kahdesti vuorokaudessa) yhdistelmällä oli rajallisesti kliinistä aktiivisuutta potilailla, joiden tauti oli edennyt BRAF‑estäjähoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tutkijan arvioima vahvistettu vasteprosentti oli 15 % (95 % lv: 4,4–34,9) ja etenemisvapaan elinajan mediaani 3,6 kuukautta (95 % lv: 1,9–5,2). Samankaltaisia tuloksia todettiin 45 potilaalla, jotka siirtyivät pelkästä dabrafenibista trametinibin (2 mg kerran vuorokaudessa) ja dabrafenibin (150 mg kahdesti vuorokaudessa) yhdistelmään tutkimuksen C‑osassa. Näillä potilailla vahvistettu vasteprosentti oli 13 % (95 % lv: 5,0–27,0) ja etenemisvapaan elinajan mediaani 3,6 kuukautta (95 % lv: 2–4).
Potilaat, joilla on aivometastaaseja
Trametinibin ja dabrafenibin yhdistelmän tehoa ja turvallisuutta on arvioitu ei‑satunnaistetussa avoimessa vaiheen II monikeskustutkimuksessa (COMBI-MB-tutkimus) potilailla, joilla on BRAF‑mutaatiopositiivinen, aivometastaaseja aiheuttanut melanooma. Yhteensä 125 potilasta otettiin mukaan neljään kohorttiin:
- Kohortti A: potilaat, joilla oli BRAF V600E -mutatoitunut melanooma, oireettomia aivometastaaseja, ei aiempaa paikallista aivoihin kohdistuvaa hoitoa ja ECOG-toimintakykyluokka 0 tai 1.
- Kohortti B: potilaat, joilla oli BRAF V600E -mutatoitunut melanooma, oireettomia aivometastaaseja, saaneet aiempaa paikallista aivoihin kohdistuvaa hoitoa ja ECOG-toimintakykyluokka 0 tai 1.
- Kohortti C: potilaat, joilla oli BRAF V600D/K/R -mutatoitunut melanooma, oireettomia aivometastaaseja, joko olivat tai eivät olleet saaneet aiempaa paikallista aivoihin kohdistuvaa hoitoa ja ECOG-toimintakykyluokka 0 tai 1.
- Kohortti D: potilaat, joilla oli BRAF V600D/E/K/R -mutatoitunut melanooma, oireisia aivometastaaseja, joko olivat tai eivät olleet saaneet aiempaa paikallista aivoihin kohdistuvaa hoitoa ja ECOG-toimintakykyluokka 0 tai 1 tai 2.
Tutkimuksen ensisijainen päätetapahtuma oli intrakraniaalinen vaste kohortissa A. Sen määritelmä oli prosenttiosuus potilaista, joilla oli vahvistettu tutkijan arvioima intrakraniaalinen vaste ‘modified Response Evaluation Criteria In Solid Tumors’ (RECIST) version 1.1 ‑kriteerejä käyttäen. Tutkijan arvioima intrakraniaalinen vaste kohorteissa B, C ja D olivat tutkimuksen toissijaisia päätetapahtumia. Leveän 95 % luottamusvälin kuvastaman pienen otoskoon vuoksi kohorttien B, C ja D tuloksia on tulkittava varovaisesti. Tehoa osoittavat tulokset on esitetty yhteenvetona taulukossa 10.
Taulukko 10 Tutkijan arvioon perustuvat tiedot tehosta COMBI-MB-tutkimuksessa
| Kaikkien hoidettujen potilaiden ryhmä | ||||
| Päätetapahtumat/ arvio | Kohortti A n = 76 | Kohortti B n = 16 | Kohortti C n = 16 | Kohortti D n = 17 |
| Intrakraniaalisen vasteen osuus, % (95 % lv) | ||||
59 % (47,3–70,4) | 56 % (29,9–80,2) | 44 % (19,8–70,1) | 59 % (32,9–81,6) | |
| Intrakraniaalisen vasteen kesto, mediaani (kk) (95 % lv) | ||||
6,5 (4,9–8,6) | 7,3 (3,6–12,6) | 8,3 (1,3–15,0) | 4,5 (2,8–5,9) | |
| Kokonaisvasteprosentti, % (95 % lv) | ||||
59 % (47,3–70,4) | 56 % (29,9–80,2) | 44 % (19,8–70,1) | 65 % (38,3–85,8) | |
| Etenemisvapaa elinaika, mediaani (kk) (95 % lv) | ||||
5,7 (5,3–7,3) | 7,2 (4,7–14,6) | 3,7 (1,7–6,5) | 5,5 (3,7–11,6) | |
| Kokonaiselinaika, mediaani (kk) (95 % lv) | ||||
10,8 (8,7–17,9) | 24,3 (7,9–NR) | 10,1 (4,6–17,6) | 11,5 (6,8–22,4) | |
lv = luottamusväli NR = ei saavutettu | ||||
- Trametinibi ainoana lääkkeenä
Potilaat, jotka eivät olleet aiemmin saaneet hoitoa
Trametinibin tehoa ja turvallisuutta arvioitiin vaiheen III satunnaistetussa, avoimessa tutkimuksessa (MEK114267 [METRIC]) potilailla, joilla oli BRAF‑mutatoitunut leikkaukseen soveltumaton tai metastasoitunut melanooma (V600E ja V600K). Kaikkien potilaiden BRAF V600 ‑mutaatiostatus määritettiin.
Potilaat (n = 322), jotka eivät olleet aiemmin saaneet hoitoa tai olivat voineet saada yhtä solunsalpaajahoitoa metastasoituneeseen tautiin [lähtöryhmien analyysiin (Intent to Treat, ITT) perustuva potilasjoukko], satunnaistettiin suhteessa 2:1 trametinibia 2 mg kerran vuorokaudessa tai solunsalpaajahoitoa (dakarbatsiinia 1 000 mg/m2 kolmen viikon välein tai paklitakselia 175 mg/m2 kolmen viikon välein) saavaan ryhmään. Kaikkien potilaiden hoito jatkui taudin etenemiseen, kuolemaan tai tutkimuksen keskeyttämiseen asti.
Tutkimuksen ensisijainen päätetapahtuma oli arvioida trametinibin tehoa solunsalpaajahoitoon verrattuna etenemisvapaan elinajan (PFS) perusteella potilailla, joilla oli pitkälle edennyt / metastasoitunut BRAF V600E/K ‑mutaatiopositiivinen melanooma ja joilla ei ollut aikaisemmin todettu aivometastaaseja (n = 273), ja tämän katsotaan olevan ensisijaista tehoa mittaava potilasjoukko. Toissijaiset päätetapahtumat olivat etenemisvapaa elinaika (PFS) ITT‑potilasjoukossa ja kokonaiselinaika (OS), kokonaisvasteprosentti (ORR) ja vasteen kesto (DoR) ensisijaisessa tehoa mittaavassa potilasjoukossa ja ITT‑potilasjoukossa. Solunsalpaajahoitohaaran potilaat saivat siirtyä trametinibihoitohaaraan, kun taudin eteneminen oli varmistettu riippumattomasti. Trametinibihoitoon siirtyi yhteensä 51 (47 %) niistä solunsalpaajahoitohaaran potilaista, joiden taudin eteneminen oli varmistettu.
Potilaiden ominaisuudet lähtötilanteessa olivat tasapainossa hoitoryhmien välillä ensisijaista tehoa mittaavassa potilasjoukossa ja ITT‑potilasjoukossa. ITT‑potilasjoukossa 54 % potilaista oli miehiä ja kaikki olivat kaukasialaista syntyperää. Mediaani‑ikä oli 54 vuotta (22 % oli ≥ 65‑vuotiaita), kaikkien potilaiden ECOG‑toimintakykyluokka oli 0 tai 1 ja 3 %:lla potilaista oli aikaisemmin todettu aivometastaaseja. Useimmilla ITT‑potilasjoukon potilailla (87 %) oli BRAF V600E ‑mutaatio, ja 12 %:lla potilaista oli BRAF V600K ‑mutaatio. Useimmat potilaat (66 %) eivät olleet aikaisemmin saaneet solunsalpaajahoitoa pitkälle edenneeseen tai metastasoituneeseen tautiin.
Tehoa osoittavat tulokset olivat ensisijaista tehoa mittaavassa potilasjoukossa yhdenmukaiset ITT‑potilasjoukon tulosten kanssa. Siksi vain ITT‑potilasjoukon tehoa osoittavat tiedot on esitetty taulukossa 11. Kaplan-Meier‑kuvaajat tutkijan arvioon perustuvasta kokonaiselinajasta (OS) (post hoc ‑analyysi 20.5.2013) on esitetty kuvassa 3.
Taulukko 11 Tutkijan arvioimat tehoa osoittavat tulokset (ITT‑populaatio)
| Päätetapahtuma | Trametinibi | Solunsalpaajahoitoa |
| Etenemisvapaa elinaika (PFS) | (n = 214) | (n = 108) |
PFS‑mediaani (kk) (95 % lv) | 4,8 (4,3–4,9) | 1,5 (1,4–2,7) |
Riskitiheyssuhde (HR) (95 % lv) P‑arvo | 0,45 (0,33–0,63) < 0,0001 | |
| Kokonaisvaste (%) | 22 | 8 |
ITT (intent to treat) = lähtöryhmien analyysiin perustuva potilasjoukko; PFS (progression‑free survival) = etenemisvapaa elinaika; lv = luottamusväli. a Solunsalpaajahoitoon kuului potilaita, jotka saivat dakarbatsiinia (DTIC) 1 000 mg/m2 kolmen viikon välein tai paklitakselia 175 mg/m2 kolmen viikon välein. | ||
PFS‑tulos oli johdonmukainen V600K‑mutaatiopositiivista melanoomaa sairastavien potilaiden alaryhmässä (HR = 0,50; [95 % lv: 0,18–1,35], p = 0,0788).
Lisäanalyysi kokonaiselinajasta (OS) tehtiin tiedonkeräyksen katkaisukohdan 20.5.2013 perusteella, ks. taulukko 12.
Lokakuussa 2011 toiseen hoitohaaraan oli siirtynyt 47 % tutkittavista, ja toukokuussa 2013 toiseen hoitohaaraan oli siirtynyt 65 % tutkittavista.
Taulukko 12 Elossaolotiedot primaarista ja post‑hoc‑analyysistä
| Tiedonkeräyksen katkaisukohdat | Hoito | Kuolemantapauksia (%) | Kokonaiselinajan mediaani, kk (95 % lv) | Riskitiheyssuhde (95 % lv) | 12 kuukauden elossaolo‑osuus (%) (95 % lv) | |
| 26.10.2011 | Solunsalpaajahoito (n = 108) | 29 (27) | NR | 0,54 (0,32–0,92) | NR | |
| Trametinibi (n = 214) | 35 (16) | NR | NR | |||
| 20.5.2013 | Solunsalpaajahoito (n = 108) | 67 (62) | 11,3 (7,2–14,8) | 0,78 (0,57–1,06) | 50 % (39 %–59 %) | |
| Trametinibi (n = 214) | 137 (64) | 15,6 (14,0–17,4) | 61 % (54 %–67 %) | |||
| NR = ei saavutettu | ||||||
Kuva 3 Kaplan-Meier‑kuvaajat tutkijan arvioimasta kokonaiselinajasta (OS) – ad hoc ‑analyysi 20.5.2013
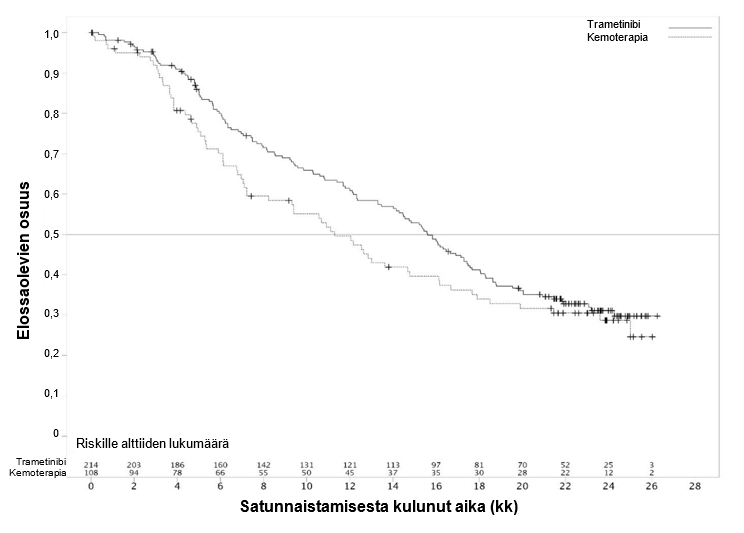
Aikaisempi BRAF‑estäjähoito
Vaiheen II yhden hoitohaaran tutkimuksen tavoitteena oli arvioida trametinibin objektiivista hoitovastetta, turvallisuutta ja farmakokinetiikkaa, kun sitä annettiin 2 mg kerran vuorokaudessa potilaille, joilla oli BRAF V600E‑, V600K‑ tai V600D ‑mutaatiopositiivinen metastasoitunut melanooma (MEK113583). Tutkimukseen otettiin mukaan kaksi erillistä kohorttia: kohortti A: potilaat, jotka olivat aikaisemmin saaneet BRAF:n estäjähoitoa aiemman hoidon jälkeen tai ilman aiempaa hoitoa; kohortti B: potilaat, jotka eivät olleet aikaisemmin saaneet BRAF:n estäjähoitoa mutta olivat aikaisemmin saaneet vähintään yhtä solunsalpaajahoitoa tai immunoterapiaa.
Tämän tutkimuksen kohortissa A trametinibilla ei ollut kliinistä tehoa potilaisiin, joiden tauti oli edennyt aikaisemman BRAF‑estäjähoidon aikana.
Asteen III melanooman liitännäishoito
BRF115532 (COMBI‑AD)
Trametinibin ja dabrafenibin yhdistelmähoidon tehoa ja turvallisuutta tutkittiin vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa monikeskustutkimuksessa, johon osallistui asteen III (aste IIIA [imusolmukemetastaasi > 1 mm], IIIB tai IIIC) BRAF V600 E/K -mutaatiopositiivista ihomelanoomaa sairastavia potilaita, joiden kasvain oli kokonaan poistettu leikkauksella.
Potilaat satunnaistettiin 1:1 suhteessa saamaan joko yhdistelmähoitoa (150 mg dabrafenibia kahdesti vuorokaudessa ja 2 mg trametinibia kerran vuorkaudessa) tai kahta lumelääkettä 12 kuukauden ajan. Tutkimukseen otettiin potilaita, joilta melanooma oli kokonaan poistettu leikkauksella ja joille oli tehty imusolmukkeidenpoistoleikkaus satunnaistamista edeltävien 12 viikon aikana. Mitään aikaisempaa systeemistä syöpähoitoa, mukaanlukien sädehoito, ei sallittu. Potilaat, joilla oli aikaisemmin ollut pahanlaatuinen kasvain, olivat soveltuvia tutkimukseen, jos tautivapaan ajan pituus oli vähintään 5 vuotta. Potilaat, joilla oli todettu pahanlaatuinen kasvain ja siinä aktivoiva RAS‑mutaatio, eivät olleet soveltuvia tutkimukseen. Potilaat stratifioitiin BRAF mutaation tyypin (V600E vs. V600K) ja leikkausta edeltäneen taudin asteen (asteen III alaryhmät, osoittaen eriasteisia imusolmukemetastasointeja sekä primaarikasvaimen kokoa ja ulseraatiota) perusteella. Taudin aste määriteltiin American Joint Committee on Cancer (AJCC) 7th edition Melanoma Staging System ‑kriteerien avulla. Ensisijainen päätetapahtuma oli tutkijan arvioima uusiutumisvapaa elinaika (RFS), joka määriteltiin satunnaistamisesta taudin uusiutumiseen tai mistä tahansa syystä johtuneeseen kuolemaan kuluneeksi ajaksi. Kasvain arvioitiin radiologisesti 3 kuukauden välein ensimmäisten kahden vuoden aikana, ja 6 kuukauden välein sen jälkeen, kunnes havaittiin ensimmäisen kerran taudin uusiutuminen. Toissijaisia päätetapahtumia olivat kokonaiselinaika (OS; tärkeä toissijainen päätetapahtuma), taudin uusiutumattomuus (freedom from recurrence = FFR) ja kaukoetäpesäkkeistä vapaa elinaika (DMFS).
870 potilasta satunnaistettiin yhdistelmähoitoryhmään (n = 438) ja lumelääkeryhmään (n = 432). Suurin osa potilaista oli kaukasialaista alkuperää (99 %) ja miehiä (55 %), ja potilaiden mediaani‑ikä oli 51 vuotta (18 % oli ≥ 65‑vuotiaita). Tutkimus sisälsi potilaita kaikista asteen III alaryhmistä (taudin aste ennen kasvaimen poistamista leikkauksella); 18 %:lla potilaista oli ainoastaan mikroskooppisesti havaittava imusolmukemetastasointi eikä primaarikasvaimeen liittynyt ulseraatiota. Suurimmalla osalla potilaista oli BRAF V600E -mutaatio (91 %).
Seuranta‑ajan mediaani primaarianalyysin ajankohtana oli 2,83 vuotta dabrafenibin ja trametinibin yhdistelmähoidon ryhmässä ja 2,75 vuotta lumelääkeryhmässä.
Uusiutumisvapaan elinajan primaarianalyysin tulokset esitetään taulukossa 13. Tutkimus osoitti, että hoitoryhmien välillä oli tilastollisesti merkitsevä ero tutkijan arvioimassa uusiutumisvapaassa elinajassa, joka oli tutkimuksen ensisijainen vastemuuttuja. Uusiutumisvapaan elinajan mediaani oli lumelääkeryhmässä 16,6 kuukautta, ja yhdistelmähoitoryhmässä sitä ei ole vielä saavutettu (HR = 0,47; 95 % lv: 0,39–0.58; p = 1,53×10‑14). Hyöty uusiutumisvapaassa elinajassa osoitettiin johdonmukaisesti eri potilasryhmissä mukaanlukien ikä, sukupuoli ja rotu. Tulokset olivat johdonmukaiset myös, kun huomioitiin taudin asteeseen ja BRAF V600 -mutaation tyyppiin liittyvät stratifikaatioperusteet.
Taulukko 13 Tutkijan arvioimat uusiutumisvapaan elinajan tulokset BRF115532 (COMBI-AD primaarianalyysi) ‑tutkimuksessa
| Dabrafenibi + Trametinibi | Lumelääke | |
| RFS parametri | n = 438 | n = 432 |
Tapahtumien lukumäärä, n (%) Taudin uusiutuminen Uusiutuminen johon liittyy kaukoetäpesäke Kuolema | 166 (38 %) 163 (37 %) 103 (24 %) 3 (< 1 %) | 248 (57 %) 247 (57 %) 133 (31 %) 1 (< 1 %) |
Mediaani (kk) (95 % lv) | Ei arvioitavissa (44,5–Ei arvioitavissa) | 16,6 (12,7–22,1) |
Riskitiheyssuhde[1] (95 % lv) p‑arvo[2] | 0,47 (0,39–0,58) 1,53 × 10-14 | |
| Osuus 1 vuoden kohdalla (95 % lv) | 0,88 (0,85–0,91) | 0,56 (0,51–0,61) |
| Osuus 2 vuoden kohdalla (95 % lv) | 0,67 (0,63–0,72) | 0,44 (0,40–0,49) |
| Osuus 3 vuoden kohdalla (95 % lv) | 0,58 (0,54–0,64) | 0,39 (0,35–0,44) |
[1] Riskitiheyssuhde saatiin stratifioidusta Pike ‑mallista. [2] P‑arvo saatiin kaksipuolisesta stratifioidusta log-rank ‑testistä (stratifiointitekijöitä olivat taudin aste IIIA vs. IIIB vs. IIIC – ja BRAF V600 -mutaation tyyppi – V600E vs. V600K) | ||
Kun verrattiin päivitettyjä 29 kuukautta pidempään kestäneen seurannan tietoja ja primaarianalyysin (seurannan kesto vähintään 59 kuukautta) tietoja, havaittiin, että RFS‑hyöty säilyi ja arvioitu HR oli 0,51 (95 % lv: 0,42–0,61) (Kuva 4). 5 vuoden RFS-lukema oli 52 % (95 % lv: 48, 58) yhdistelmähoitoryhmässä ja 36 % (95 % lv: 32, 41) lumelääkeryhmässä.
Kuva 4 Kaplan-Meier‑kuvaajat uusiutumisvapaasta elinajasta BRF115532‑tutkimuksessa (ITT‑populaatio, päivitetyt tulokset)
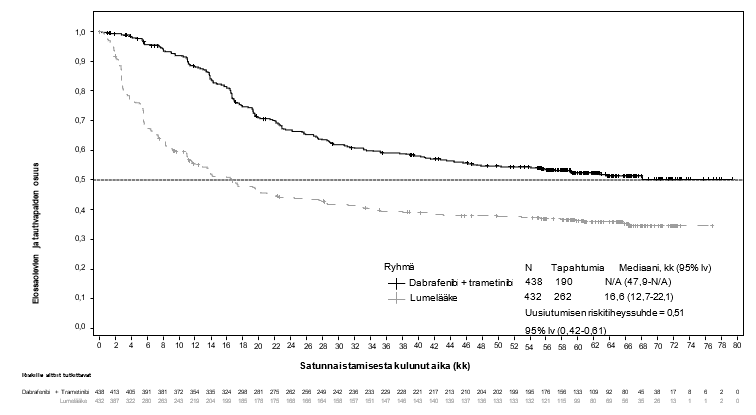
Lopullista kokonaiselinaikaa analysoitaessa seuranta-ajan mediaani oli yhdistelmähoitoryhmässä 8,3 vuotta ja lumelääkeryhmässä 6,9 vuotta. Kokonaiselinajassa havaittu ero ei ollut tilastollisesti merkitsevä (HR: 0,80; 95 % lv: 0,62–1,01). Yhdistelmähoitoryhmässä oli 125 tapahtumaa (29 %) ja lumelääkeryhmässä 136 tapahtumaa (31 %). Arvioitu kokonaiselossaolo-osuus 5 vuoden kohdalla oli yhdistelmähoitoryhmässä 79 % ja lumelääkeryhmässä 70 %, ja 10 vuoden kohdalla yhdistelmähoitoryhmässä 66 % ja lumelääkeryhmässä 63 %
Ei‑pienisoluinen keuhkosyöpä
Tutkimus BRF113928
Trametinibin ja dabrafenibin yhdistelmähoidon tehoa ja turvallisuutta tutkittiin vaiheen II avoimessa kolmen kohortin ei‑satunnaistetussa monikeskustutkimuksessa. Tutkimukseen otettiin potilaita, joilla oli IV asteen BRAF V600E -mutatoitunut ei‑pienisoluinen keuhkosyöpä. Ensisijainen päätetapahtuma oli tutkijan arvioima kokonaisvasteprosentti (ORR) RECIST 1.1 ‑kriteerejä käyttäen. Toissijaisia päätetapahtumia olivat vasteen kesto (DoR), etenemisvapaa elinaika (PFS), kokonaiselinaika (OS), turvallisuus ja populaatiofarmakokinetiikka. Myös riippumaton komitea (Independent Review Committee [IRC]) arvioi kokonaiselinaikaa, vasteen kestoa ja etenemisvapaata elinaikaa herkkyysanalyysina.
Kohortit otettiin tutkimukseen peräkkäisessä järjestyksessä:
- Kohortti A: Monoterapia (150 mg dabrafenibia kahdesti vuorokaudessa), 84 potilasta. 78 potilasta oli aiemmin saanut systeemistä hoitoa metastasoituneeseen sairauteensa.
- Kohortti B: Yhdistelmähoito (150 mg dabrafenibia kahdesti vuorokaudessa ja 2 mg trametinibia kerran vuorokaudessa), 59 potilasta. 57 potilasta oli saanut 1‑3 linjassa aikaisempaa systeemistä hoitoa metastasoituneeseen sairauteensa. 2 potilasta ei ollut aikaisemmin saanut systeemistä hoitoa ja nämä potilaat sisällytettiin kohorttin C otettujen potilaiden kanssa samaan analyysiin.
- Kohortti C: Yhdistelmähoito (150 mg dabrafenibia kahdesti vuorokaudessa ja 2 mg trametinibia kerran vuorokaudessa), 34 potilasta. Kaikki potilaat saivat tutkimuslääkevalmistetta ensilinjan hoitona metastasoituneen sairauden hoitoon.
Yhdistelmähoitokohortteihin B ja C otetuista 93 potilaasta enemmistö oli kaukasialaista syntyperää (> 90 %), ja potilaissa oli enemmän naisia (54 %) kuin miehiä (46 %). Toisen tai myöhäisemmän linjan hoitoa saaneiden potilaiden mediaani‑ikä oli 64 vuotta ja ensilinjan hoitoa saaneiden potilaiden 68 vuotta. Suurimmalla osalla (94 %) yhdistelmähoitokohorttien potilaista ECOG‑toimintakykyluokka oli 0 tai 1. 26 (28 %) potilasta ei ollut koskaan tupakoinut. Suurimmalla osalla potilaista oli ei‑epiteliaalinen histologia. Aikaisempaa hoitoa saaneessa populaatiossa 38 potilasta (67 %) oli saanut yhdessä linjassa systeemistä syöpähoitoa metastasoituneeseen sairauteen.
Primäärianalyysin ajankohtana ensisijainen päätetapahtuma, tutkijan arvioima kokonaisvasteprosentti (ORR), oli ensilinjan populaatiossa 61,1 % (95 % lv: 43,5 %–76,9 %) ja aikaisempaa hoitoa saaneessa populaatiossa 66,7 % (95 % lv: 52,9 %–78,6 %). Nämä tulokset olivat tilastollisesti merkittäviä ja niiden perusteella hylättiin oletus (null hypothesis), että dabrafenibin ja trametibinin yhdistelmähoidossa kokonaisvasteprosentti (ORR) olisi 30 % tai vähemmän. Riippumattoman komitean (IRC) arvio kokonaisvasteprosenttituloksista (ORR) oli yhtenevä tutkija‑arvion kanssa. Lopullinen tehoanalyysi, joka tehtiin 5 vuotta sen jälkeen kun viimeinen potilas oli saanut ensimmäisen annoksensa, on esitetty taulukossa 14.
Taulukko 14 Yhteenveto tehosta yhdistelmähoitokohorteissa tutkijan arvioon ja riippumattomaan radiologiseen arvioon perustuen
| Päätetapahtuma | Analyysi | Yhdistelmähoito 1. linja n = 361 | Yhdistelmähoito 2. tai myöhäisempi linja n = 571 |
Kokonaisvaste, varmistettu n (%) (95 % lv) | Tutkija IRC | 23 (63,9 %) (46,2–79,2) 23 (63,9 %) (46,2–79,2) | 39 (68,4 %) (54,8–80,1) 36 (63,2 %) (49,3–75,6) |
DoR mediaani kk (95 % lv) | Tutkija IRC | 10,2 (8,3–15,2) 15,2 (7,8–23,5) | 9,8 (6,9–18,3) 12,6 (5,8–26,2) |
PFS mediaani kk (95 % lv) | Tutkija IRC | 10,8 (7,0-14,5) 14,6 (7,0-22,1) | 10,2 (6,9–16,7) 8,6 (5,2–16,8) |
OS mediaani kk (95 % lv) | - | 17,3 (12,3–40,2) | 18,2 (14,3–28,6) |
| 1 Tietojen katkaisupiste: 7. tammikuuta 2021 | |||
Erilaistunut kilpirauhassyöpä (DTC)
Tutkimus CDRB436J12301 (J12301)
Trametinibin ja dabrafenibin yhdistelmähoidon (D+T) tehoa ja turvallisuutta arvioitiin vaiheen III satunnaistetussa (2:1), kaksoissokkoutetussa, lume-kontrolloidussa monikeskustutkimuksessa (J12301) aikuispotilailla, joilla oli BRAF V600E -mutaatiopositiivinen, paikallisesti edennyt tai metastasoitunut, radiojodihoidolle refraktaari erilaistunut kilpirauhassyöpä, joka oli edennyt enintään kahden aiemman VEGFR-täsmähoidon jälkeen (mukaan lukien, mutta ei rajoittuen, lenvatinibi tai sorafenibi). BRAF V600E -mutaatiostatus vahvistettiin keskitetysti Cobas® 4800 -testin avulla.
153 potilasta satunnaistettiin saamaan trametinibia 2 mg suun kautta kerran vuorokaudessa yhdessä dabrafenibin 150 mg suun kautta kahdesti vuorokaudessa (n = 101) tai lumelääkkeen (n = 52) kanssa. Potilaat, joilla oli RET-fuusiopositiivinen kasvain, suljettiin pois tutkimuksesta. Satunnaistaminen stratifioitiin aiemman lenvatinibihoidon (kyllä vs ei) ja aiempien VEGFR-täsmähoitojen lukumäärän (1 vs 2) perusteella. Lumelääkkeelle satunnaistetut, soveltumiskriteerit täyttävät potilaat saattoivat siirtyä D+T-hoitoon, kun sokkoutettu riippumaton radiologinen arviointikomitea (blinded independent radiology review committee, BIRC) oli vahvistanut taudin etenemisen. Potilaat jatkoivat sokkoutettua tutkimushoitoa niin kauan kuin he saivat siitä kliinistä hyötyä tai kunnes ilmeni toksisuutta, jota ei voitu hyväksyä. Ensisijainen päätetapahtuma oli etenemisvapaa elinaika (PFS), jonka arvioi BIRC. Keskeiset toissijaiset päätetapahtumat olivat kokonaisvasteprosentti (ORR) BIRC:n arvioimana ja kokonaiselinaika (OS).
Mediaani-ikä oli 64 vuotta (vaihteluväli 36–82 vuotta), ja 12 potilasta (8 %) oli ≥75-vuotiaita. Suurin osa potilaista oli aasialaisia (86 %) ja 52 % oli naisia. Kaikilla potilailla oli vahvistettu papillaarinen kilpirauhaskarsinoomadiagnoosi. Tutkimukseen ei otettu mukaan follikulaarista kilpirauhaskarsinoomaa sairastavia potilaita. Aiempi lenvatinibihoito oli raportoitu 31 %:lla potilaista, ja suurin osa (78 %) oli saanut yhden aiemman VEGFR-täsmähoidon. Lähtötilanteen ECOG‑toimintakykyluokka oli 0 (50 %), 1 (47 %) tai 2 (3 %). Hoidon mediaanikesto oli 14,5 kuukautta D+T-ryhmässä ja 5,5 kuukautta lumelääkeryhmässä.
Ensisijaisen analyysin tulokset (tiedonkeruun katkaisu 22. tammikuuta 2025 ja mediaaniseuranta 17 kuukautta) on esitetty taulukossa 15 ja kuvassa 5. Tutkimus osoitti tilastollisesti merkitsevän paranemisen sekä etenemisvapaassa elinajassa (PFS) että kokonaisvasteprosentissa (ORR).
Taulukko 15Tehoa mittaavat tulokset erilaistuneessa kilpirauhassyövässä (tutkimus J12301, ensisijainen analyysi, ITT‑populaatio)
Dabrafenibi + trametinibi n = 101 | Lumelääke n = 52 | |
| Etenemisvapaa elinaika* (PFS) BIRC:n arvioimana | ||
| Tapahtumia (%) | 54 (53,5 %) | 43 (82,7 %) |
| PFS Mediaani kuukausina (95 % lv)a | 12,8 (10,2–21,2) | 3,7 (2,3–7,5) |
| Riskitiheyssuhde (95 % lv)b | 0,38 (0,25–0,57) | |
| p-arvo (yksisuuntainen)c | <0,001 | |
| Vahvistettu paras kokonaisvaste BIRC:n arvioimana | ||
| Vahvistettu ORR, N (%) | 58 (57,4 %) | 2 (3,8 %) |
| (95 % lv)d | (47,2–67,2) | (0,5–13,2) |
| p-arvo (yksisuuntainen)e | <0,001 | |
| Kokonaiselinaika (OS)f | ||
| Kuolemia, N (%) | 27 (26,7 %) | 19 (36,5 %) |
| Riskitiheyssuhde (95 % lv)b | 0,66 (0,36–1,19) | |
* Ensisijainen PFS‑analyysi sisälsi sensuroinnin uuden syöpälääkehoidon aloittamisen kohdalla. PFS‑tulokset olivat yhdenmukaiset riippumatta siitä, sisällytettiinkö sensurointi uuden syöpälääkehoidon vuoksi vai ei. lv: luottamusväli, ORR= kokonaisvasteprosentti (CR+PR); PR ja CR vahvistetaan uusinta-arvioinneilla, jotka tehdään aikaisintaan 4 viikkoa sen jälkeen, kun vasteen kriteerit täyttyvät ensimmäisen kerran. a Kaplan–Meier‑estimaattiin perustuen. b Perustuu Coxin regressiomalliin, joka on stratifioitu aiemman lenvatinibihoidon (kyllä/ei) sekä aiempien VEGFR‑täsmähoitojen lukumäärän (1 vs 2) mukaan. c Perustuu log‑rank‑testiin, joka on stratifioitu aiemman lenvatinibihoidon (kyllä/ei) sekä aiempien VEGFR‑täsmähoitojen lukumäärän (1 vs 2) mukaan. d Clopper–Pearsonin eksakti binomijakaumaan perustuva 95 %:n luottamusväli. e Perustuu Cochran–Mantel–Haenszel‑testiin, joka on stratifioitu aiemman lenvatinibihoidon (kyllä/ei) sekä aiempien VEGFR‑täsmähoitojen lukumäärän (1 vs 2) mukaan. f Kokonaiselossaoloajan (OS) paraneminen ei ollut tilastollisesti merkitsevää (ensisijaisen analyysin ajankohtana 30 potilasta [57,7 %] oli jo siirtynyt lumelääkeryhmästä D+T‑ryhmään). | ||
Kuva 5Etenemisvapaan elinajan Kaplan–Meier kuvaajat (tutkimus J12301, ensisijainen analyysi)
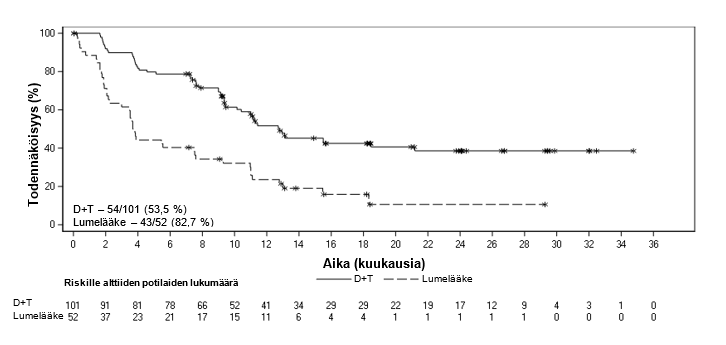
Muut tutkimukset – kuumeen hallinnan analyysi
Tutkimukset CPDR001F2301 (COMBI‑i) ja CDRB436F2410 (COMBI‑Aplus)
Kohonnutta ruumiin lämpöä havaittiin dabrafenibi-trametinibiyhdistelmähoitoa saaneilla potilailla. Ensimmäisissä yhdistelmähoidon rekisteröintitutkimusten asetelmissa, joissa kasvain oli leikkaukseen soveltumaton tai melanooma oli metastoitunut (COMBI-d ja COMBI-v; n=559 yhteensä) ja melanooman liitännäishoito (COMBI-AD, n=435) suositeltiin vain dabrafenibi hoidon keskeyttämistä ruumiin lämpötilan noustessa (kuume ≥ 38,5 °C). Kahden myöhemmän tutkimuksen asetelmissa, joissa kasvain oli leikkaukseen soveltumaton tai melanooma oli metastoitunut (COMBI-i control arm, n=264) ja melanooman liitännäishoito (COMBI-Aplus, n=552), molempien lääkevalmisteiden hoidon kesteyttämistä suositeltiin kun potilaan ruumiinlämpö oli ≥ 38 °C (COMBI-Aplus) tai ensimmäisten kuumeoireiden ilmetessä (COMBI-i; COMBI-Aplus uusiutunut lämmön nousu). Tutkimuksissa COMBI-i ja COMBI-Aplus ilmaantui vähemmän asteen 3/4 kuumetta, komplisoitunutta kuumetta, vakavia kuumeen aiheuttamia sairaalahoitoa vaatineita erityisen mielenkiinnon kohteena olevia haittatapahtumia (AESI), kuumeen kestoa (AESI) ja kuumeesta johtuvia pysyviä molempien lääkevalmisteiden hoidon lopettamispäätöksiä (AESI) (viimeksi mainittu vain liitännäishoidon yhteydessä) verrattuna tutkimuksiin COMBI-d, COMBI-v ja COMBI-AD. Ensisijainen päätetapahtuma, kuumeiden yhdistetty määrä 8,0 %, saavutettiin COMBI-Aplus-tutkimuksessa (lv 95 %: 5,9-10,6) 3/4 asteen kuume, kuumeen aiheuttama sairaalahoitoon joutuminen tai kuumeesta johtuvaa pysyvä hoidon keskeyttäminen verrattuna aiemman kontrollitutkimuksen (COMBI-AD) 20,0 % (lv 95 %: 16,3-24,1).
Pediatriset potilaat
Melanoomaa sairastavia nuoria potilaita koskevia kliinisistä tutkimuksista saatuja tietoja on erittäin niukasti, joten tehon määrittämiseen on käytetty ekstrapolointia aikuisia koskevista tiedoista. Julkaistussa, Italiassa toteutetussa retrospektiivisessä tutkimuksessa arvioitiin 6:ta edennyttä BRAF V600 ‑mutaatiopositiivista melanoomaa sairastavaa pediatrista potilasta, jotka saivat trametinibia yhdessä dabrafenibin kanssa. Iän mediaani diagnoosihetkellä oli 15 vuotta, ja aloitusannosten vaihteluvälit olivat 125–150 mg dabrafenibia kahdesti vuorokaudessa ja 1,5–2 mg trametinibia kerran vuorokaudessa. Taudin uusiutumista ei todettu 4:llä dabrafenibin ja trametinibin yhdistelmää liitännäishoitona saaneella potilaalla, kun seurannan mediaanikesto oli 22 kuukautta. Metastasoituneeseen tautiin hoitoa saaneista 1:llä raportoitiin täydellinen vaste ja 1:llä etenevä tauti.
Farmakokinetiikka
Imeytyminen
Suun kautta otettu trametinibi imeytyy ja saavuttaa huippupitoisuuden 1,5 tunnin (mediaani) kuluttua annoksesta. Tablettina annetun 2 mg:n kerta‑annoksen absoluuttinen hyötyosuus on 72 % (keskiarvo) laskimoon annettuun mikroannokseen verrattuna. Toistuvan annostelun jälkeen lääkeainealtistus (Cmax ja AUC) suureni suhteessa annokseen. Kun trametinibia annettiin 2 mg kerran vuorokaudessa, vakaan tilan aikaisten Cmax‑arvojen geometrinen keskiarvo oli 22,2 ng/ml, AUC(0‑τ) oli 370 ng*h/ml ja annosta edeltävä pitoisuus 12,1 ng/ml ja huippupitoisuuden ja jäännöspitoisuuden suhde (peak:trough) oli pieni (1,8). Tutkittavien väliset vaihtelut vakaan tilan aikana olivat vähäisiä (< 28 %).
Trametinibi kumuloituu toistuvan päivittäisen annostelun aikana, ja kumuloitumissuhde on 6,0 (keskiarvo), kun annostus on 2 mg kerran vuorokaudessa. Vakaa tila saavutettiin 15. päivään mennessä.
Kun trametinibi annettiin kerta‑annoksena rasvaisen ja kaloripitoisen aterian kanssa, Cmax‑arvo pieneni 70 % ja AUC‑arvo 10 % verrattuna arvoihin, jotka saatiin tyhjään mahaan annetun annoksen jälkeen (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Jakautuminen
Trametinibi sitoutuu ihmisen plasman proteiineihin 97,4‑prosenttisesti. Trametinibin jakautumistilavuus on noin 1 200 litraa määritettynä 5 µg:n laskimoon annetun mikroannoksen jälkeen.
Biotransformaatio
In vitro‑ ja in vivo ‑tutkimukset osoittivat, että trametinibi metaboloituu pääasiassa deasetylaation kautta joko pelkästään tai yhdessä mono‑oksygenaation kanssa. Deasetyloitunut metaboliitti metaboloitui edelleen glukuronidaation kautta. CYP3A4‑oksidaatiota pidetään vähäisempänä metaboloitumisreittinä. Deasetylaatio tapahtuu karboksyyliesteraasien 1b, 1c ja 2 välityksellä ja siihen osallistuu mahdollisesti myös muita hydrolyyttisia entsyymejä.
Trametinibin kerta‑annoksen ja toistuvien annosten jälkeen kanta-aine trametinibi on pääasiallinen plasmassa tavattava aineosa.
Eliminaatio
Terminaalisen puoliintumisajan keskiarvo on 127 tuntia (5,3 vuorokautta) kerta‑annoksen jälkeen. Laskimoon annetun trametinibin plasmapuhdistuma on 3,21 litraa tunnissa.
Kun radioaktiivisesti merkittyä trametinibiliuosta annettiin kerta‑annoksena, koko erittynyt lääkemäärä 10 vuorokauden keräämisajan jälkeen oli pieni (< 50 %), mikä johtuu lääkeaineen pitkästä eliminoitumisen puoliintumisajasta. Lääkkeeseen liittyvä aines erittyi lähinnä ulosteeseen (osuus kerätystä radioaktiivisuudesta oli > 80 %) ja vähäisemmässä määrin virtsaan (≤ 19 %). Alle 0,1 % erittyneestä annoksesta erittyi kanta-aineena virtsaan.
Erityisryhmät
Maksan vajaatoiminta
Populaatiofarmakokineettinen analyysi ja kliinisen farmakologian tutkimuksesta saadut tiedot potilailla, joilla on normaali maksan toiminta tai lievästi, keskivaikeasti tai vaikeasti koholla olevat bilirubiini‑ ja/tai ASAT‑arvot (NCI‑ [National Cancer Institute] ‑luokituksen mukaan), osoittavat ettei maksan toiminta vaikuta merkittävästi suun kautta annetun trametinibin puhdistumaan.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminta ei todennäköisesti vaikuta kliinisesti merkittävästi trametinibin farmakokinetiikkaan, koska trametinibi erittyy vain vähäisessä määrin munuaisten kautta. Trametinibin farmakokinetiikkaa tarkasteltiin populaatiofarmakokineettisen analyysin avulla kliinisiin trametinibitutkimuksiin osallistuneilla 223 potilaalla, joilla oli lievä munuaisten vajaatoiminta, ja 35 potilaalta, joilla oli keskivaikea munuaisten vajaatoiminta. Lievällä ja keskivaikealla munuaisten vajaatoiminnalla ei ollut vaikutusta trametinibialtistukseen (< 6 % kummassakin ryhmässä). Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista ei ole tutkimustietoa (ks. kohta Annostus ja antotapa).
Iäkkäät
Populaatiofarmakokineettisen analyysin perusteella (ikäjakauma 19–92 vuotta) iällä ei ollut merkittävää kliinistä vaikutusta trametinibin farmakokinetiikkaan. Turvallisuustiedot ≥ 75‑vuotiaista potilaista ovat vähäisiä (ks. kohta Haittavaikutukset).
Etninen tausta
Populaatiofarmakokineettisessä analyysissä ei havaittu merkittäviä eroja trametinibin farmakokinetiikassa aasialaisten ja valkoihoisten potilaiden välillä. Tietoja ei ole riittävästi, jotta muiden etnisten taustojen mahdollista vaikutusta trametinibin farmakokinetiikkaan voitaisiin arvioida.
Pediatriset potilaat
Nuorten potilaiden farmakokineettiset trametinibialtistukset suositelluilla painon mukaisilla annoksilla asettuivat aikuisilla havaittujen altistusten vaihteluvälille.
Kehon paino ja sukupuoli
Populaatiofarmakokineettisen analyysin perusteella sukupuoli ja paino vaikuttavat suun kautta annetun trametinibin puhdistumaan. Vaikka pienempikokoisten naispotilaiden lääkeainealtistuksen ennustetaan olevan suurempi kuin painavampien miespotilaiden, erot eivät todennäköisesti ole kliinisesti merkityksellisiä, eikä annoksen muuttaminen ole tarpeen.
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Trametinibin vaikutukset lääkeaineita metaboloiviin entsyymeihin ja kuljetusproteiineihin: In vitro ja in vivo ‑tutkimusten tulokset viittaavat siihen, ettei trametinibi todennäköisesti vaikuta muiden lääkevalmisteiden farmakokinetiikkaan. In vitro ‑tutkimusten perusteella trametinibi ei ole CYP1A2‑, CYP2A6‑, CYP2B6‑, CYP2D6- eikä CYP3A4‑entsyymien estäjä. Trametinibin todettiin estävän CYP2C8‑, CYP2C9‑ ja CYP2C19-entsyymejä ja indusoivan CYP3A4‑entsyymiä sekä estävän kuljetusproteiineja OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, P‑gp ja BCRP in vitro. Annos ja systeeminen altistus ovat kuitenkin pieniä verrattuna esto- tai induktiovaikutuksen aiheuttaneisiin arvoihin in vitro, joten trametinibin ei katsota estävän tai indusoivan näitä entsyymejä tai kuljetusproteiineja in vivo, vaikka BCRP:n substraattien ohimenevä estyminen suolistossa on mahdollinen (ks. kohta Yhteisvaikutukset).
Muiden lääkevalmisteiden vaikutukset trametinibiin: in vitro ja in vivo ‑tutkimusten perusteella muut lääkevalmisteet eivät todennäköisesti vaikuta trametinibin farmakokinetiikkaan. Trametinibi ei ole CYP‑entsyymien eikä BCRP‑, OATP1B1‑, OATP1B3‑, OATP2B1‑, OCT1‑, MRP2‑ tai MATE1‑kuljetusproteiinien substraatti. Trametinibi on BSEP:n ja P‑gp‑kuljetusproteiinin substraatti in vitro. Vaikka BSEP:n esto ei todennäköisesti vaikuta trametinibialtistukseen, maksan P‑gp:n voimakkaan eston aiheuttamaa trametinibitason nousua ei voida sulkea pois (ks. kohta Yhteisvaikutukset).
Trametinibin vaikutus muihin lääkevalmisteisiin: toistuvien trametinibiannosten vaikutusta noretisteronia ja etinyyliestradiolia sisältävien yhdistelmäehkäisytablettien vakaan tilan farmakokinetiikkaan arvioitiin kliinisessä tutkimuksessa 19 naispotilaalla, joilla oli kiinteitä kasvaimia. Noretisteronialtistus suureni 20 % ja etinyyliestradiolialtistus oli samaa luokkaa, kun näitä lääkeaineita annettiin yhtä aikaa trametinibin kanssa. Näiden tulosten pohjalta hormonaalisten ehkäisyvalmisteiden tehon ei oleteta heikkenevän, kun niitä käytetään samanaikaisesti trametinibimonoterapian kanssa.
Prekliiniset tiedot turvallisuudesta
Trametinibilla ei ole tehty karsinogeenisuustutkimuksia. Trametinibi ei ollut genotoksinen bakteereilla tehdyissä takaisinmutaatiokokeissa, nisäkässoluissa tehdyissä kromosomipoikkeavuuskokeissa eikä rottien luuytimen mikrotumatestissä.
Trametinibi saattaa heikentää naisten hedelmällisyyttä, sillä toistuvilla annoksilla tehdyissä eläinkokeissa havaittiin kystisten follikkeleiden lisääntymistä ja keltarauhasen pienenemistä naarasrotilla, kun altistukset olivat pienempiä kuin ihmisen kliininen altistus AUC‑arvon perusteella.
Lisäksi nuorilla, ei‑sukukypsillä, trametinibia saaneilla rotilla havaittiin munasarjojen painon laskua, vähäistä viivästymistä naaraiden sukukypsyyden tunnusmerkeissä (viivästynyt vaginan avautuminen ja lisääntynyt rintatiehyiden kasvusolukeskusten määrä) sekä vähäistä kohdun pintaepiteelin hypertrofiaa. Nämä vaikutukset korjaantuivat lääkkeettömän jakson jälkeen ja johtuivat farmakologiasta. Rotilla ja koirilla tehdyissä toksisuustutkimuksissa, joiden kesto oli enintään 13 viikkoa, hoidon ei kuitenkaan havaittu vaikuttavan urosten lisääntymiskudoksiin.
Rotilla ja kaniineilla tehdyissä alkion ja sikiön kehitystä koskeneissa toksisuustutkimuksissa trametinibilla oli emoon ja sikiönkehitykseen kohdistuvia haittavaikutuksia. Rotilla havaittiin sikiöiden painon laskua ja implantaation jälkeisten keskenmenojen lisääntymistä, kun altistukset olivat pienempiä tai vain vähän suurempia kuin ihmisen kliiniset altistukset AUC‑arvon perusteella. Alkion ja sikiön kehitystä koskeneessa toksisuustutkimuksessa kaniineilla havaittiin sikiöiden painon laskua, keskenmenojen lisääntymistä, epätäydellisen luutumisen ilmaantuvuuden suurenemista ja luuston epämuodostumia AUC‑arvon perusteella ihmisen subkliinisillä altistustasoilla.
Toistuvilla annoksilla tehdyissä tutkimuksissa trametinibialtistuksen jälkeen vaikutuksia havaittiin pääasiassa ihossa, ruoansulatuselimistössä, hematologisessa järjestelmässä, luustossa ja maksassa. Suurin osa löydöksistä korjaantuu lääkkeettömän jakson jälkeen. Maksasolunekroosia ja kohonneita aminotransferaasiarvoja havaittiin rotilla, jotka olivat saaneet 8 viikon ajan ≥ 0,062 mg/kg/vrk trametinibia (noin 0,8‑kertainen altistus verrattuna ihmisen kliiniseen altistukseen AUC‑arvon perusteella).
Hiirillä esiintyi sydämen sykkeen hidastumista, sydämen painon laskua ja vasemman kammion toiminnan heikkenemistä mutta ei sydämen histopatologisia muutoksia 3 viikon kuluttua, kun trametinibia annettiin ≥ 0,25 mg/kg/vrk (noin 3‑kertainen altistus verrattuna ihmisen kliiniseen altistukseen AUC‑arvon perusteella) enintään 3 viikon ajan. Täysikasvuisilla rotilla seerumin fosforipitoisuuden suurenemiseen liittyi useiden elinten mineralisaatiota, ja se oli läheisessä yhteydessä sydämen, maksan ja munuaisten nekroosiin ja keuhkoverenvuotoon altistustasoilla, jotka vastasivat ihmisen kliinistä altistusta. Rotilla havaittiin kasvulevyn hypertrofiaa ja luun aineenvaihdunnan kiihtymistä. Kasvulevyn hypertrofialla ei kuitenkaan odoteta olevan kliinistä merkitystä aikuispotilaiden hoidossa ihmisellä. Rotilla ja koirilla, joille annettiin trametinibia ihmisen kliinistä altistusta vastaavina tai sen alittavina annoksina, havaittiin luuytimen nekroosia, imukudoksen atrofiaa kateenkorvassa ja suolistoonliittyvässä imukudoksessa sekä imukudoksen nekroosia imusolmukkeissa, pernassa ja kateenkorvassa. Nämä ilmiöt voivat heikentää immuunijärjestelmän toimintaa. Nuorilla, ei‑sukukypsillä rotilla havaittiin sydämen painon nousua, mutta ei histopatologisia muutoksia annoksella 0,35 mg/kg/vrk (noin kaksinkertainen altistus verrattuna aikuisen kliiniseen altistukseen AUC‑arvon perusteella).
Trametinibi oli fototoksinen in vitro hiiren 3T3-fibroblastien NRU‑testissä (Neutral Red Uptake), kun pitoisuudet olivat huomattavasti korkeammat kuin ihmisen kliinisessä altistuksessa (IC50 pitoisuuden ollessa 2,92 mikrog/ml, ≥ 130 kertaa korkeampi kuin ihmisen kliininen altistus laskettuna Cmax‑arvosta), Trametinibia saavilla potilailla fototoksisuuden riski näyttäisi näin ollen olevan matala.
Käyttö yhdessä dabrafenibin kanssa
Kun koirille annettiin tutkimuksessa trametinibin ja dabrafenibin yhdistelmää 4 viikon ajan, niillä havaittiin maha‑suolikanavan haittojen ja kateenkorvan imukudoksen solukkuuden vähenemisen merkkejä, kun altistukset olivat pienempiä kuin pelkkää trametinibia saaneilla koirilla. Muutoin havaittiin samankaltaisia haittoja kuin vertailukelpoisissa monoterapiatutkimuksissa.
Farmaseuttiset tiedot
Apuaineet
Mekinist 0,5 mg kalvopäällysteiset tabletit
Tabletin ydin
Mannitoli (E421)
Mikrokiteinen selluloosa (E460)
Hypromelloosi (E464)
Kroskarmelloosinatrium (E468)
Magnesiumstearaatti (E470b)
Natriumlauryylisulfaatti
Kolloidinen piidioksidi (E551)
Tabletin kalvopäällyste
Hypromelloosi (E464)
Titaanidioksidi (E171)
Polyetyleeniglykoli
Keltainen rautaoksidi (E172)
Mekinist 2 mg kalvopäällysteiset tabletit
Tabletin ydin
Mannitoli (E421)
Mikrokiteinen selluloosa (E460)
Hypromelloosi (E464)
Kroskarmelloosinatrium (E468)
Magnesiumstearaatti (E470b)
Natriumlauryylisulfaatti
Kolloidinen piidioksidi (E551)
Tabletin kalvopäällyste
Hypromelloosi (E464)
Titaanidioksidi (E171)
Polyetyleeniglykoli
Polysorbaatti 80 (E433)
Punainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Avaamaton purkki
3 vuotta
Avattu purkki
Säilytä avaamisen jälkeen alle 30 °C:n lämpötilassa ja käytä 30 vuorokauden kuluessa.
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Säilytä alkuperäispakkauksessa. Herkkä valolle. Herkkä kosteudelle.
Pidä purkki tiiviisti suljettuna.
Avatun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
MEKINIST tabletti, kalvopäällysteinen
0,5 mg (L:ei) 30 kpl (1171,34 €)
2 mg (L:ei) 30 kpl (4354,46 €)
PF-selosteen tieto
Polyetyleenistä (HDPE) valmistettu purkki, jossa on polypropyleeninen turvakorkki. Purkissa on kuivatusainesäiliö.
Pakkauskoot: Yksi tablettipurkki sisältää 7 tai 30 tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Mekinist 0,5 mg kalvopäällysteiset tabletit
Keltaisia, soikeita, kaksoiskuperia kalvopäällysteisiä tabletteja, joiden koko on noin 5,0 x 9,0 mm ja joissa on toisella puolella yrityksen logo ja toisella puolella ”TT”.
Mekinist 2 mg kalvopäällysteiset tabletit
Vaaleanpunaisia, pyöreitä, kaksoiskuperia kalvopäällysteisiä tabletteja, joiden halkaisija on noin 7,6 mm ja joissa on toisella puolella yrityksen logo ja toisella puolella ”LL”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
MEKINIST tabletti, kalvopäällysteinen
0,5 mg 30 kpl
2 mg 30 kpl
- Ylempi erityiskorvaus (100 %). Dabrafenibi ja trametinibi: Melanooman, gliooman ja keuhkosyövän hoito erityisin edellytyksin (1509).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Dabrafenibi ja trametinibi: Dabrafenibin ja trametinibin yhdistelmähoito aikuisille, kun kyseessä BRAF V600 - mutaatiopositiivisen melanooman hoito erityisin edellytyksin (3024).
ATC-koodi
L01EE01
Valmisteyhteenvedon muuttamispäivämäärä
19.05.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com