
OPZELURA emulsiovoide 15 mg/g
Vaikuttavat aineet ja niiden määrät
Yksi gramma emulsiovoidetta sisältää 15 mg ruksolitinibia (fosfaattina).
Apuaineet, joiden vaikutus tunnetaan
Propyleeniglykoli (E 1520) 150 mg/g
Setyylialkoholi 30 mg/g
Stearyylialkoholi 17,5 mg/g
Metyyliparahydroksibentsoaatti (E 218) 1 mg/g
Propyyliparahydroksibentsoaatti 0,5 mg/g
Butyylihydroksitolueeni (antioksidanttina valkovaseliinissa) (E 321)
Polysorbaatti 20 (E 432)
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Emulsiovoide
Kliiniset tiedot
Käyttöaiheet
Opzelura on tarkoitettu aikuisten ja vähintään 12 vuoden ikäisten nuorten ei-segmentaalisen, kasvo-oireisen vitiligon hoitoon.
Ehto
Ei-segmentaalisen vitiligon diagnosointiin ja hoitoon perehtyneen lääkärin on aloitettava hoito ja valvottava sitä.
Annostus ja antotapa
Ei-segmentaalisen vitiligon diagnosointiin ja hoitoon perehtyneen lääkärin on aloitettava Opzelura-hoito ja valvottava sitä.
Annostus
Aikuiset
Suositeltu annos on ohut kerros emulsiovoidetta levitettynä kaksi kertaa vuorokaudessa depigmentoituneille ihoalueille, enintään 10 prosentille kehon pinta-alasta (BSA). Ruksolitinibiemulsiovoiteen levityskertojen välin on oltava vähintään 8 tuntia. Pinta-alana 10 prosenttia kehon pinta-alasta vastaa 10 kertaa yhden käden kämmenen (5 sormea mukaan lukien) kokoista aluetta. Ruksolitinibiemulsiovoidetta on käytettävä mahdollisimman pienelle tarvittavalle ihoalueelle.
Sallittu käyttömäärä kuukaudessa on enintään kaksi 100 gramman tuubia.
Tyydyttävän repigmentaatiotuloksen saavuttaminen saattaa edellyttää yli 24 viikkoa kestävää hoitoa. Jos alle 25 % hoidettavista alueista on repigmentoitunut viikolla 52, hoidon lopettamista on harkittava.
Kun tyydyttävä repigmentaatiotulos on saavutettu, hoito voidaan näillä alueilla lopettaa. Jos depigmentaatio uusiutuu hoidon lopettamisen jälkeen, hoito voidaan aloittaa ongelma-alueille uudelleen.
Hoitoa ei tarvitse lopettaa vähitellen.
Erityisryhmät
Maksan vajaatoiminta
Ruksolitinibiemulsiovoiteen käytöstä maksan vajaatoimintaa sairastavilla potilailla ei ole tehty tutkimuksia. Koska systeeminen altistus on vähäistä, annosta ei kuitenkaan tarvitse muuttaa potilailla, joilla on maksan vajaatoiminta.
Munuaisten vajaatoiminta
Ruksolitinibiemulsiovoiteen käytöstä munuaisten vajaatoimintaa sairastavilla potilailla ei ole tehty tutkimuksia. Koska systeeminen altistus on vähäistä, annosta ei kuitenkaan tarvitse muuttaa potilailla, joilla on munuaisten vajaatoiminta. Varotoimenpiteenä ruksolitinibiemulsiovoidetta ei saa käyttää potilailla, joilla on loppuvaiheen munuaissairaus, koska turvallisuutta koskevia tietoja ei ole.
Iäkkäät
Kliinisiin tutkimuksiin Opzelura-valmisteen käytöstä vitiligon hoidossa on otettu jonkin verran vähintään 65‑vuotiaita potilaita ja selvitetty, poikkeaako heidän vasteensa nuoremmista tutkittavista (ks. kohta Farmakodynamiikka). Annosta ei tarvitse muuttaa vähintään 65 vuotiaiden potilaiden osalta.
Pediatriset potilaat
Nuorilla (12–17‑vuotiailla) annostus on sama kuin aikuisilla.
Ruksolitinibiemulsiovoiteen turvallisuutta ja tehoa alle 12 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Emulsiovoide on tarkoitettu vain iholle.
Hoidetun ihon pesemistä on vältettävä vähintään 2 tunnin ajan ruksolitinibiemulsiovoiteen levittämisestä.
Emulsiovoidetta ei saa levittää huulille, jotta sitä ei niellä.
Potilaita on neuvottava pesemään kätensä emulsiovoiteen levittämisen jälkeen, ellei kyse ole käsien hoidosta. Jos joku muu levittää emulsiovoiteen potilaan iholle, hänen on pestävä kätensä emulsiovoiteen levittämisen jälkeen.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Raskaus ja imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Emulsiovoidetta ei ole tarkoitettu käytettäväksi silmiin, suuhun eikä emättimen sisään (ks. kohta Annostus ja antotapa). Jos valmistetta joutuu vahingossa silmiin tai limakalvoille, emulsiovoide on pyyhittävä pois huolellisesti ja/tai huuhdeltava vedellä.
Ei-melanoomatyyppinen ihosyöpä
Paikallista ruksolitinibihoitoa saaneilla potilailla on raportoitu ei-melanoomatyyppisiä ihosyöpiä (NMSC), pääasiassa tyvisolukarsinoomia. Useimmilla näistä potilaista oli riskitekijöitä, kuten aiempi valohoito tai aiempi NMSC. Syy-yhteyttä paikallisesti käytettävään ruksolitinibiin ei ole varmistettu. Iho on suositeltavaa tutkia säännöllisesti kaikilla potilailla, etenkin potilailla, joilla on ihosyövän riskitekijöitä.
Vyöruusu (Herpes zoster)
Paikallista ruksolitinibihoitoa saaneilla potilailla on raportoitu vyöruusun uudelleenaktivoitumista. Jos potilaalle kehittyy vyöruusu, on harkittava ruksolitinibiemulsiovoidehoidon keskeyttämistä, kunnes oireet poistuvat.
Apuaineet, joiden vaikutus tunnetaan
Propyleeniglykoli
Tämä lääkevalmiste sisältää 150 mg propyleeniglykolia (E 1520) per gramma, ja se saattaa aiheuttaa ihoärsytystä.
Setyylialkoholi ja stearyylialkoholi
Tämä lääkevalmiste sisältää setyylialkoholia ja stearyylialkoholia, jotka saattavat aiheuttaa paikallisia ihoreaktioita (esim. kosketusihottumaa).
Parahydroksibentsoaatit
Tämä lääkevalmiste sisältää metyyliparahydroksibentsoaattia (E 218) ja propyyliparahydroksibentsoaattia, jotka saattavat aiheuttaa allergisia reaktioita (mahdollisesti viivästyneitä).
Butyylihydroksitolueeni
Tämä lääkevalmiste sisältää butyylihydroksitolueenia (E 321), joka saattaa aiheuttaa paikallisia ihoreaktioita (esim. kosketusihottumaa) tai silmä- ja limakalvoärsytystä.
Polysorbaatti 20
Tämä lääkevalmiste sisältää polysorbaatti 20:tä (E 432), joka saattaa aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Paikallisesti käytettävällä ruksolitinibinilla ei ole tehty yhteisvaikutustutkimuksia.
Ruksolitinibin yhteisvaikutuspotentiaalia pidetään alhaisena, koska paikallisesta käytöstä aiheutuva systeeminen altistus on vähäistä.
In vitro ‑tietojen perusteella ruksolitinibi poistuu elimistöstä pääasiassa sytokromi P450 3A4 (CYP3A4) -välitteisen metabolian kautta. Suun kautta otetun ruksolitinibin yhteisvaikutuspotentiaalia on arvioitu erillisissä kliinis-farmakologisissa tutkimuksissa, joissa valmisteen kanssa annettiin voimakkaita tai kohtalaisen voimakkaita CYP3A4:n estäjiä tai voimakasta indusoria. AUC-arvo plasmassa suunnilleen kaksinkertaistuu, jos valmisteen kanssa annetaan voimakasta CYP3A4:n estäjää, kun taas kohtalaisen voimakkaan CYP3A4:n estäjän kanssa annettuna arvo suurenee vain vähän.
Ruksolitinibiemulsiovoiteen käyttöä muiden vitiligon hoitoon paikallisesti käytettävien lääkevalmisteiden kanssa ei ole arvioitu, eikä useiden valmisteiden käyttöä samalla ihoalueella suositella. Ruksolitinibiemulsiovoiteen tehoa ja turvallisuutta kapeakaistaisen UVB-valohoidon (NB‑UVB) kanssa ei ole arvioitu, eikä suositusta voida antaa.
Muiden sairauksien hoitoon tarkoitettuja, paikallisesti käytettäviä lääkevalmisteita saa levittää samalle ihoalueelle vähintään 2 tunnin kuluttua ruksolitinibiemulsiovoiteen levittämisestä. Tämä koskee myös aurinkovoiteiden ja perusvoiteiden käyttöä.
Raskaus ja imetys
Ehkäisy naisilla, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja 4 viikkoa hoidon päättymisen jälkeen.
Raskaus
Ruksolitinibin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Tietoja paikallisesti käytettävän ruksolitinibin systeemisestä imeytymisestä raskauden aikana ei ole. Systeemisen altistuksen kasvuun saattavat vaikuttaa myös yksilölliset tekijät (esim. ihoesteen vaurioituminen, liiallinen käyttö).
Eläimillä tehdyissä tutkimuksissa on havaittu, että suun kautta otettu ruksolitinibi on alkio- ja sikiötoksinen. Teratogeenisuutta ei ole havaittu rotilla eikä kaniineilla (ks kohta Prekliiniset tiedot turvallisuudesta). Opzelura-valmisteen käyttö raskauden aikana on vasta-aiheista (ks. kohta Vasta-aiheet).
Imetys
Ei tiedetä, erittyykö ruksolitinibi ihmisillä äidinmaitoon, vaikuttaako se imetettävään vauvaan tai vaikuttaako se rintamaidon erittymiseen Opzelura-valmisteen paikallisen käytön jälkeen. Kun ruksolitinibia annettiin imettäville rotille suun kautta, ruksolitinibia ja/tai sen metaboliitteja esiintyi maidossa 13‑kertainen pitoisuus emon plasmassa havaittuun pitoisuuteen nähden. Nuorilla rotilla tehdyissä tutkimuksissa ruksolitinibin antaminen suun kautta vaikutti kasvuun ja luiden mittoihin (ks. kohta Prekliiniset tiedot turvallisuudesta). Opzelura-valmisteen käyttö imetyksen aikana on vasta-aiheista (ks. kohta Vasta-aiheet), ja hoito on lopetettava noin 4 viikkoa ennen imetyksen aloittamista.
Hedelmällisyys
Ruksolitinibin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Eläinkokeissa oraalisella ruksolitinibilla ei havaittu olevan hedelmällisyyteen kohdistuvia vaikutuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ruksolitinibiemulsiovoiteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisin haittavaikutus on käyttökohdan akne (5,8 %).
Haittavaikutustaulukko
Haittavaikutukset esitetään seuraavassa niiden esiintymistiheyden mukaan yleisimmästä alkaen seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1: Haittavaikutukset
Elinjärjestelmä | Esiintymistiheys | Haittavaikutus |
Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Käyttökohdan akne |
Infektiot | Melko harvinainen | Vyöruusu (Herpes zoster) |
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostus iholle käytön jälkeen on epätodennäköistä. Jos emulsiovoidetta on levitetty liikaa, ylimääräisen valmisteen voi pyyhkiä pois.
Jos valmistetta joutuu vahingossa silmiin, suun limakalvoille tai emättimen sisään, emulsiovoide on pyyhittävä pois huolellisesti ja/tai huuhdeltava vedellä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: muut ihotautien lääkkeet, ihottumalääkkeet, lukuun ottamatta kortikosteroideja, ATC-koodi: D11AH09
Vaikutusmekanismi
Ruksolitinibi on januskinaasin estäjä eli JAK-estäjä, joka vaikuttaa selektiivisesti JAK1- ja JAK2-isoformeihin. Solunsisäiseen JAK-signalointiin liittyy STAT-transkriptiotekijöiden (signaalinvälittäjiä ja transkriptioaktivaattoreita) rekrytointi sytokiinireseptoreihin ja sitä seuraava geenien ilmentymisen modulointi. Sytotoksisia T-lymfosyyttejä tuottavan autoimmuuni-IFNγ:n arvellaan aiheuttavan suoraan ihmisen vitiligoon liittyvän melanosyyttien tuhoutumisen. Sytotoksisten lymfosyyttien rekrytointi vaurioituneeseen ihoon tapahtuu IFNγ-riippuvaisten kemokiinien, kuten CXCL10:n välityksellä. IFNγ:n myöhempi signalointi riippuu JAK1/2:sta, ja ruksolitinibihoito pienentää vitiligopotilaiden CXCL10-tasoja.
Kliininen teho ja turvallisuus
Teho
Kahteen tutkimusasetelmaltaan identtiseen kaksoissokkoutettuun, satunnaistettuun, vehikkelikontrolloituun tutkimukseen (TRuE‑V1 ja TRuE‑V2) otettiin yhteensä 674 potilasta, joiden kasvoissa oli vitiligoa ja joiden koko kehon (kasvot ja muut alueet) pinta-alasta (BSA) vitiligo peitti enintään 10 %. Sairauden laajuuden vaihteluväli oli tutkimuksen alussa 3,2–10,1 % BSA:sta ja potilaiden ikä oli vähintään 12 vuotta (10,7 % potilaista oli 12–17‑vuotiaita ja 6,7 % oli 65-vuotiaita tai vanhempia). Potilaista 53,1 % oli naisia. Potilaista 81,9 % oli valkoihoisia, 4,7 % mustia ja 4,2 % aasialaisia. Useimpien potilaiden Fitzpatrickin luokituksen mukainen ihotyyppi oli III, IV, V tai VI (67,5 %).
Potilaat satunnaistettiin molemmissa tutkimuksissa suhteessa 2:1 käyttämään hoitona joko ruksolitinibiemulsiovoidetta tai vehikkeliä kahdesti vuorokaudessa 24 viikon ajan kohdealueille, joiden BSA-osuus oli enintään 10 %; tämän jälkeen kaikki potilaat käyttivät ruksolitinibiemulsiovoidetta vielä 28 viikon ajan kahdesti vuorokaudessa. Tehon ensisijainen päätetapahtuma oli niiden potilaiden osuus, joilla saavutettiin kasvojen vitiligoalueen 75 %:n repigmentaatio (F‑VASI75) viikkoon 24 mennessä. Keskeiset toissijaiset päätetapahtumat olivat niiden potilaiden osuudet, joilla saavutettiin F‑VASI-asteikolla arvioituna kasvojen vitiligoalueen 90 %:n repigmentaatio (F‑VASI90), 50 %:n parannus koko kehon vitiligoalueen pisteytysindeksillä arvioituna (T‑VASI50) ja vitiligon havaittavuutta mittaavalla asteikolla (VNS) pistemäärä 4 tai 5 (vitiligo ”selvästi vähemmän havaittavissa” tai ”ei enää havaittavissa”).
Hoidettujen vitiligoleesioiden repigmentaatio ja ruksolitinibiemulsiovoiteen paremmuus vehikkeliemulsiovoiteeseen verrattuna todettiin molemmissa tutkimuksissa, sillä viikkoon 24 mennessä todettiin tilastollisesti merkitsevät erot F‑VASI75/90- ja T‑VASI50‑vasteissa sekä VNS-pistemäärässä 4 tai 5 (taulukko 2).
Hoitovaikutuksen ero vehikkeliin verrattuna on nähtävissä numeerisesti arvioituna jo viikolla 12. Potilailla, jotka käyttivät ruksolitinibiemulsiovoidetta jatkuvasti kahdesti vuorokaudessa lähtötilanteesta lähtien, todettiin VASI- ja VNS-pistemäärillä arvioituna repigmentaation jatkuvan viikolle 52 saakka. Kuvassa 1 on esitetty TRuE‑V1- ja TRuE‑V2-tutkimusten yhdistetyn datan perusteella niiden potilaiden osuus, joilla saavutettiin F‑VASI75-tulos 52 viikon hoitojakson aikana.
Viikolla 52 havaittiin samankaltainen hoitovaste potilailla, jotka olivat siirtyneet vehikkelistä ruksolitinibiiin (kuva 1).
Taulukko 2 Niiden vitiligopotilaiden osuus, joilla saavutettiin ensisijainen päätetapahtuma ja keskeiset toissijaiset päätetapahtumat viikkoon 24 mennessä (hoitoaikeen mukainen ryhmä)a
TRuE-V1 | TRuE-V2 | |||
Opzelura | Vehikkeli | Opzelura | Vehikkeli | |
(N = 221) | (N = 109) | (N = 222) | (N = 109) | |
F-VASI75 (%) | 29,8 | 7,4 | 30,9 | 11,4 |
Vasteosuuksien ero (95 %:n lv) | 22,3b | – | 19,5c | – |
F-VASI90 (%) | 15,3 | 2,2 | 16,3 | 1,3 |
Vasteosuuksien ero (95 %:n lv) | 13,2d | – | 15,0e | – |
T-VASI50 (%) | 20,6 | 5,1 | 23,9 | 6,8 |
Vasteosuuksien ero (95 %:n lv) | 15,5d | – | 17,1c | – |
VNS 4 tai 5 (%) | 24,5 | 3,3 | 20,5 | 4,9 |
Vasteosuuksien ero (95 %:n lv) | 21,2c | – | 15,5d | – |
a Ensisijaiseen päätetapahtumaan ja toissijaisiin päätetapahtumiin on tehty oikaisut moni-imputointimenetelmällä.
b p-arvo < 0,0001
c p-arvo < 0,001
d p-arvo < 0,005
e p-arvo < 0,01
Kuva 1 Niiden potilaiden osuus, joilla saavutettiin F-VASI75-tulos 52 viikon hoitojakson aikana (hoitoaikeen mukainen ryhmä) – tutkimusten TRuE‑V1 ja TRuE‑V2 koontitiedot
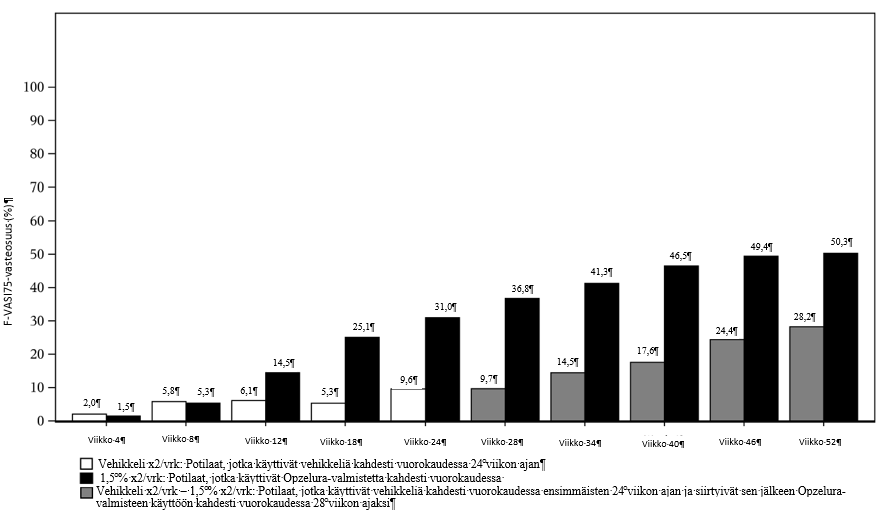
Viikolla 52 F‑VASI90-vasteosuus oli 30,3 %, T‑VASI50-vasteosuus 51,1 % ja VNS-vasteosuus 36,3 % yhdistetystä ITT-ryhmästä.
Vasteen kesto
Kahdesti vuorokaudessa annettavan ruksolitinibiemulsiovoidehoidon keskeyttämistä ja jatkamista tutkittiin vaiheen 3 kaksoissokkoutetussa, vehikkelikontrolloidussa, satunnaistetussa tutkimuksessa. Tutkimukseen osallistui 458 soveltuvaa vitiligo-potilasta, jotka olivat suorittaneet jommankumman aiemmista ruksolitinibitutkimuksista (TruE-v1 ja TruE-v2; viikko 52). Potilaat sijoitettiin joko kohorttiin A tai B, ja seurantaa jatkettiin enintään 104 viikon ajan.
Kohortissa A oli 116 potilasta, joilla oli saavutettu aiemmassa tutkimuksessa ≥ F-VASI90 viikon 52 kohdalla. Nämä potilaat satunnaistettiin uudelleen saamaan joko ruksolitinibia tai vehikkeliä (ts. hoidon keskeyttäminen) uusiutumisen tutkimiseksi (< F‑VASI75). Uusiutumista esiintyi 15 %:lla ruksolitinibiryhmän potilaista ja 29 %:lla vehikkeliryhmän potilaista. Jälkimmäisessä ryhmässä suurin osa uusiutumisista (9/16) esiintyi ensimmäisten 4 kuukauden aikana ruksolitinibiemulsiovoiteen lopettamisesta. Vehikkeliryhmän 16 potilaasta, joilla uusiutumista esiintyi ja joilla hoito aloitettiin uudelleen, 12 potilaalla (75 %) saavutettiin uudelleen F-VASI75 12 viikon kuluessa (mediaani) ja 11 potilaalla (69 %) saavutettiin uudelleen F-VASI90 15 viikon kuluessa (mediaani).
Kohortissa B oli 342 potilasta, joilla oli saavutettu aiemmassa tutkimuksessa < F-VASI90 viikon 52 kohdalla. Nämä potilaat jatkoivat avoimeen ruksolitinibitutkimukseen; niistä potilaista, jotka oli aluksi satunnaistettu saamaan ruksolitinibiemulsiovoidehoitoa kahdesti vuorokaudessa, 66 %:lla saavutettiin viikon 104 kohdalla F-VASI75 ja 34 %:lla F-VASI90.
Turvallisuus
Pitkäkestoisessa, enintään 104 viikkoon ulottuvassa jatkotutkimuksessa kumulatiiviset turvallisuustulokset olivat yhdenmukaisia tutkimuksissa raportoidun, enintään 52 viikkoon ulottuvan profiilin kanssa.
Pediatriset potilaat
Avaintutkimuksiin otettiin yhteensä 72 nuorta (ikä 12 - < 18 vuotta; n = 55 ruksolitinibiemulsiovoide, n = 17 vehikkeli). Nuorilla saavutettiin ensisijaisen päätetapahtuman ja keskeisten toissijaisten päätetapahtumien osalta ruksolitinibihoidolla vastaavat vasteosuudet 24 viikon kohdalla kuin 18–65‑vuotiailla aikuisilla.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Opzelura-valmisteen käytöstä vitiligon hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Ruksolitinibiemulsiovoiteen farmakokinetiikkaa tutkittiin 429 vitiligoa sairastavalla tutkittavalla, joiden ikä oli vähintään 12 vuotta (12,6 % oli 12–17‑vuotiaita) ja keskimääräinen ±STD BSA-osuus oli 7,31 ± 2,02 % (vaihteluväli 3,2–10,0 %). Tutkittavat levittivät ruksolitinibiemulsiovoidetta noin 1,58 mg/cm2 (annoksen vaihtelualue oli noin 0,18–8,4 grammaa ruksolitinibiemulsiovoidetta käyttökertaa kohden) samoille ihoalueille kaksi kertaa vuorokaudessa 24 viikon ajan.
Keskimääräinen ± STD vakaan tilan pienin pitoisuus plasmassa oli 56,9 ± 62,6 nM ja arvioitu AUC0–12h 683 ± 751 h*nM, mikä vastaa noin 25 prosenttia havaitusta vakaan tilan keskimääräisestä AUC0–12h-arvosta (2716 h*nM), joka saavutettiin, kun terveet osallistujat ottivat lääkeainetta 15 mg suun kautta kahdesti vuorokaudessa. Ruksolitinibiemulsiovoiteen keskimääräinen paikallinen hyötyosuus (geometrinen keskiarvo) vitiligopotilailla oli kahden vaiheen 3 tutkimuksen yhdistettyjen tietojen perusteella 9,72 % (5,78 %).
Jakautuminen
In vitro ‑tutkimuksen perusteella ruksolitinibi sitoutuu 97-prosenttisesti ihmisen plasman proteiineihin, pääasiassa albumiiniin.
Biotransformaatio
Ruksolitinibi metaboloituu CYP3A4‑välitteisesti ja vähäisemmässä määrin CYP2C9‑välitteisesti.
Eliminaatio
Suun kautta otetun ruksolitinibin eliminaation puoliintumisajan keskiarvo on noin 3 tuntia. Opzelura-valmisteen paikallista käyttöä seurannutta ruksolitinibin keskimääräistä näennäistä terminaalista puoliintumisaikaa arvioitiin 9 aikuisella ja nuorella potilaalla, joilla oli atooppista ihottumaa ≥ 25 % kehon pinta-alasta (BSA). Kyseinen aika on noin 116 tuntia, mikä kuvastaa pikemminkin lääkkeen hidasta imeytymistä kuin eliminaationopeutta.
Erityisryhmät
Munuaisten vajaatoiminta
Ruksolitinibin ja sen metaboliittien farmakologiseen aktiivisuuteen mukautettu arvioitu AUC-arvo suurenee noin kaksinkertaiseksi potilailla, joilla on loppuvaiheen munuaistauti (ESRD). Varotoimenpiteenä Opzelura-valmistetta ei saa käyttää potilailla, joilla on ESRD, koska turvallisuutta koskevia tietoja ei ole.
Maksan vajaatoiminta
Vaikka AUC-arvo suureni, kun ruksolitinibia annettiin suun kautta maksan vajaatoimintaa sairastaville potilaille, maksan vajaatoiminnan vaikeusasteen ja AUC-arvon suurenemisen välillä ei ollut selvää suhdetta. Annosta ei tarvitse muuttaa potilailla, joilla on maksan vajaatoiminta.
Prekliiniset tiedot turvallisuudesta
Suun kautta otettua ruksolitinibia on arvioitu farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta ja lisääntymistoksisuutta sekä karsinogeenisuutta koskevissa tutkimuksissa. Lisäksi on tutkittu iholle käytön vaikutuksia minisioilla ja hiirillä. Ruksolitinibin farmakologisen vaikutuksen kohde-elimiä tutkimuksissa, joissa ruksolitinibia otettiin toistuvasti suun kautta, olivat luuydin, ääreisveri ja imukudokset. Koirilla havaittiin yleisesti immunosuppressioon liittyviä infektioita. Kroonista toksisuutta koskevissa tutkimuksissa todettiin, että sitoutumattomaan AUC-arvoon perustuvat turvarajat, joilla ei aiheutunut haittavaikutuksia, olivat noin 6- ja 200‑kertaiset uros- ja naarasrotilla ja 10‑kertaiset koirilla verrattuna havaittuun systeemiseen altistukseen vitiligopotilailla, jotka käyttivät 1,5‑prosenttista ruksolitinibiemulsiovoidetta kahdesti vuorokaudessa. Koirilla tehdyssä telemetriatutkimuksessa todettiin haitallista verenpaineen laskua ja sykkeen nousua, ja rotilla tehdyssä hengitystutkimuksessa todettiin haitallista minuuttitilavuuden laskua. Koira- ja rottatutkimuksissa sitoutumattomaan Cmax-arvoon perustuvat turvarajat, joilla ei aiheutunut haittavaikutuksia, olivat koirilla noin 300‑kertaiset ja rotilla noin 100‑kertaiset verrattuna havaittuun systeemiseen altistukseen vitiligopotilailla, jotka käyttivät 1,5-prosenttista ruksolitinibiemulsiovoidetta kahdesti vuorokaudessa. Rotilla tehdyssä ruksolitinibin neurofarmakologisten vaikutusten arvioinnissa ei havaittu haittavaikutuksia.
Toistuvalla annoksella tehdyssä 3 kuukautta kestäneessä tutkimuksessa havaittiin lymfosyyttimäärien laskua hiirillä. Sitoutumattomaan AUC-arvoon perustuvat turvarajat, joilla ei aiheutunut haittavaikutuksia, olivat noin 10‑kertaiset uroshiirillä ja 24‑kertaiset naarashiirillä verrattuna havaittuun systeemiseen altistukseen vitiligopotilailla, jotka käyttivät 1,5-prosenttista ruksolitinibiemulsiovoidetta kahdesti vuorokaudessa. Minisioilla havaittiin myös ei-haitallista perifeeristen lymfosyyttiarvojen laskua 9 kuukautta kestäneessä ihotoksisuustutkimuksessa. Sitoutumattomaan AUC-arvoon perustuvat turvarajat, joilla ei aiheutunut haittavaikutuksia, olivat noin 3‑kertaiset minisioilla verrattuna havaittuun systeemiseen altistukseen vitiligopotilailla, jotka käyttivät 1,5‑prosenttista ruksolitinibiemulsiovoidetta kahdesti vuorokaudessa. Tätä vaikutusta ei havaittu minisioilla 3 kuukautta kestäneessä ihotoksisuustutkimuksessa. Tanskalaisilla maatiaisminisioilla ei havaittu merkkejä systeemisestä toksisuudesta, kun 1,5‑prosenttista ruksolitinibiemulsiovoidetta käytettiin paikallisesti kahdesti vuorokaudessa enintään 9 kuukauden ajan.
Nuorilla rotilla tehdyissä tutkimuksissa ruksolitinibin antaminen suun kautta vaikutti kasvuun ja luiden mittoihin. Luiden kasvun hidastumista havaittiin annoksella ≥ 5 mg/kg/vrk, kun hoito aloitettiin päivänä 7 syntymän jälkeen (verrattavissa vastasyntyneeseen ihmiseen) ja annoksella ≥ 15 mg/kg/vrk, kun hoito aloitettiin päivänä 14 tai 21 syntymän jälkeen (verrattavissa 1–3-vuotiaaseen pikkulapseen). Annoksilla ≥ 30 mg/kg/vrk rotilla havaittiin murtumia ja ennenaikaisia kuolemia, kun hoito aloitettiin päivänä 7 syntymän jälkeen. Sitoutumattoman AUC-arvon perusteella NOAEL-tason (haitaton vaikutustaso) altistuminen nuorilla rotilla, joilla hoito oli aloitettu jo päivänä 7 syntymän jälkeen, oli noin 20-kertainen vitiligoa sairastaviin aikuispotilaisiin nähden. Luiden kasvun hidastumista tapahtui vitiligoa sairastavien aikuispotilaiden altistumistasoon nähden 22‑kertaisella ja murtumia 150‑kertaisella altistumisella. Vaikutukset olivat yleisesti ottaen vaikea-asteisempia uroksilla ja silloin, kun antaminen aloitettiin nopeammin syntymän jälkeen. Luiden kehittymiseen kohdistuvia vaikutuksia lukuun ottamatta ruksolitinibin vaikutukset nuorilla rotilla olivat samankaltaisia kuin aikuisilla rotilla. Nuoret rotat ovat aikuisia rottia herkempiä ruksolitinibin toksisuudelle.
Alkio- ja sikiötoksisuustutkimuksissa ruksolitinibin antaminen rotille ja kaniineille tiineyden aikana alensi sikiöiden painoa ja lisäsi implantaation jälkeisiä keskenmenoja emolle toksisilla annoksilla. Rotilla ja kaniineilla ei havaittu merkkejä teratogeenisesta vaikutuksesta. Sitoutumattomaan AUC-arvoon perustuvat turvarajat, joilla rotilla ei aiheutunut kehitystoksisia haittavaikutuksia, olivat noin 25‑kertaiset verrattuna havaittuun systeemiseen altistukseen vitiligopotilailla, jotka käyttivät 1,5-prosenttista ruksolitinibiemulsiovoidetta kahdesti vuorokaudessa. Uros- tai naarasrotilla ei havaittu hedelmällisyyteen kohdistuvia vaikutuksia, kun ruksolitinibia annettiin suun kautta. Pre- ja postnataalisessa kehitystutkimuksessa havaittiin hieman tavallista pidempi tiineyden kesto, pienempi implantaatiokohtien lukumäärä ja pienempi syntyneiden poikasten määrä. Poikasten keskimääräinen syntymäpaino oli tavanomaista pienempi ja keskimääräinen painonnousu oli lyhyen ajan tavanomaista pienempi. Imettävillä rotilla ruksolitinibia ja/tai sen metaboliitteja erittyi maitoon 13‑kertainen pitoisuus emon plasmassa havaittuun pitoisuuteen nähden. Ruksolitinibi ei ollut mutageeninen eikä klastogeeninen. Ruksolitinibilla ei todettu karsinogeenista potentiaalia paikallisesti käytettynä hiirillä eikä suun kautta annettuna Sprague–Dawley-rotilla eikä Tg.rasH2‑hiirillä.
Farmaseuttiset tiedot
Apuaineet
Butyylihydroksitolueeni (antioksidanttina valkovaseliinissa) (E 321)
Setyylialkoholi
Dimetikoni (E 900)
Dinatriumedetaatti (E 385)
Itse-emulgoituva glyseryylistearaatti
Makrogoli
Keskipitkäketjuiset triglyseridit
Metyyliparahydroksibentsoaatti (E 218)
Parafiini (E 905), kevyt nestemäinen
Valkovaseliini (E 905)
Fenoksietanoli
Fosforihappo (E 338)
Polysorbaatti 20 (E 432)
Propyleeniglykoli (E 1520)
Propyyliparahydroksibentsoaatti
Puhdistettu vesi
Stearyylialkoholi
Ksantaanikumi (E 415)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
21 kuukautta
Ensimmäisen avaamisen jälkeen: 6 kuukautta
Säilytys
Säilytä alle 30 °C.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
OPZELURA emulsiovoide
15 mg/g (L:ei) 100 g (963,75 €)
PF-selosteen tieto
Laminaattituubi, jossa pientiheyspolyeteeni ja korkeatiheyspolyeteeni sisäkerros ja polypropyleeninen korkki, tai sisäpuolelta lakkapinnoitettu alumiinituubi, jossa on puhkaistava polypropeenikorkki (100 g tuubi) tai korkeatiheyspolyeteeni (5 g tuubi).
5 g ja 100 g:n tuubit. Yksi tuubi koteloa kohden.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen emulsiovoide.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
OPZELURA emulsiovoide
15 mg/g 100 g
- Ei korvausta.
ATC-koodi
D11AH09
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2026
Yhteystiedot
Paasheuvelweg 25
1105 BP Amsterdam
Netherlands
09 7479 0132
eumedinfo@incyte.com