
FILSPARI tabletti, kalvopäällysteinen 200 mg, 400 mg
Huomioitavaa

Tähän lääkkeeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Filspari 200 mg tabletti, kalvopäällysteinen
Yksi tabletti sisältää 200 mg sparsentaania.
Apuaine, jonka vaikutus tunnetaan
Yksi tabletti sisältää 42 mg laktoosia.
Filspari 400 mg tabletti, kalvopäällysteinen
Yksi tabletti sisältää 400 mg sparsentaania.
Apuaine, jonka vaikutus tunnetaan
Yksi tabletti sisältää 84 mg laktoosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen
Kliiniset tiedot
Käyttöaiheet
Filspari on tarkoitettu primaarista immunoglobuliini A- nefropatiaa (IgA‑nefropatiaa) sairastavien aikuisten hoitoon, joilla virtsaan erittyvän proteiinin määrä on ≥ 1,0 g/vrk (tai virtsan proteiini-kreatiniinisuhde on ≥ 0,75 g/g, ks. kohta Farmakodynamiikka).
Annostus ja antotapa
Annostus
Sparsentaanihoito aloitetaan annoksella 200 mg kerran vuorokaudessa 14 vuorokauden ajan, minkä jälkeen annos suurennetaan ylläpitoannokseen 400 mg kerran vuorokaudessa siedettävyydestä riippuen.
Annoksen sovittamiseen aloitusannoksesta 200 mg kerran vuorokaudessa ylläpitoannokseen 400 mg kerran vuorokaudessa on saatavilla 200 mg:n ja 400 mg:n kalvopäällysteisiä tabletteja ylläpitoannoksen saavuttamiseksi.
Jos potilailla on siedettävyysongelmia (systolinen verenpaine ≤ 100 mmHg, diastolinen verenpaine ≤ 60 mmHg, edeema pahenee tai hyperkalemia), on suositeltavaa muokata muita samanaikaisia lääkehoitoja ja sen jälkeen pienentää sparsentaaniannosta väliaikaisesti tai lopettaa sparsentaanihoito (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Jos sparsentaanihoito aloitetaan uudelleen hoidon keskeyttämisen jälkeen, voidaan harkita alkuperäisen antoaikataulun toistamista. Jos ilmenee pitkäkestoista hypotensiota tai muutoksia maksan toiminnassa, hoidon keskeyttämistä voidaan harkita joko sparsentaaniannoksen asteittaisen pienentämisen kautta tai ilman sitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annoksen unohtuminen
Jos annos unohtuu, unohtunut annos jätetään väliin ja seuraava annos otetaan tavanomaiseen aikaan. Kaksinkertaisia tai ylimääräisiä annoksia ei pidä ottaa.
Erityisryhmät
Iäkkäät
Iäkkäiden potilaiden annoksen muuttamista ei suositella (ks. kohta Farmakokinetiikka). Iäkkäillä potilailla sparsentaanihoito tulee aloittaa annoksella 200 mg kerran vuorokaudessa 14 vuorokauden ajan. Annoksen suurentaminen ylläpitoannokseen 400 mg kerran vuorokaudessa tulee tehdä varoen, siedettävyyden mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Farmakokineettisten tietojen perusteella lievää tai keskivaikeaa maksan vajaatoimintaa (Child‑Pugh-luokka A tai Child‑Pugh-luokka B, ks. kohta Farmakokinetiikka) sairastavien potilaiden sparsentaaniannosta ei tarvitse muuttaa.
Kliinistä kokemusta on vain vähän potilaista, joilla on keskivaikea maksan vajaatoiminta. Siksi sparsentaania on käytettävä varoen näille potilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sparsentaania ei ole tutkittu vaikeaa maksan vajaatoimintaa (Child‑Pugh-luokka C) sairastavilla potilailla, eikä sen käyttämistä näille potilaille siksi suositella.
Kliinistä kokemusta on vain vähän potilaista, joiden aspartaattiaminotransferaasi (ASAT) / alaniiniaminotransferaasi (ALAT) -arvot ovat yli kaksinkertaiset normaalialueen ylärajaan (ULN) nähden. Siksi sparsentaania ei pidä aloittaa potilaille, joiden ASAT/ALAT > 2 × ULN (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä (vaiheen 2 krooninen munuaistauti [CKD]; arvioitu glomerulusten suodatusnopeus [eGFR] 60–89 ml/min/1,73 m2) tai keskivaikea (vaiheen 3a ja 3b CKD; eGFR 30–59 ml/min/1,73 m2) munuaistauti. Farmakokineettisten tietojen perusteella annosmuutoksia ei voida suositella potilaille, joilla on vaikea munuaistauti (vaiheen 4 CKD; eGFR < 30 ml/min/1,73 m2) (ks. kohta Farmakokinetiikka). Vaikeaa munuaistautia sairastavista potilaista on vain vähän tietoa, joten sparsentaania ei suositella näille potilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sparsentaania ei ole tutkittu potilailla, jotka ovat saaneet munuaissiirteen, joten sparsentaania tulee käyttää tällaisille potilaille varoen.
Sparsentaania ei ole tutkittu potilailla, jotka saavat dialyysihoitoa. Tällaisille potilaille ei suositella sparsentaanihoitoa.
Pediatriset potilaat
Filsparin turvallisuutta ja tehoa alle 18 vuoden ikäisten, IgA‑nefropatiaa sairastavien lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Tablettien karvaan maun vuoksi ne on suositeltavaa niellä kokonaisina veden kera. Sparsentaani voidaan ottaa ruoan kanssa tai ilman sitä.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Raskaus (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys).
- Angiotensiinireseptorin salpaajien (ARB), endoteliinireseptorin salpaajien (ERA) tai reniininestäjien samanaikainen käyttö (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Naiset, jotka voivat tulla raskaaksi
Sparsentaanihoidon saa aloittaa naisille, jotka voivat tulla raskaaksi, vasta kun on varmistettu, ettei nainen ole raskaana ja että hän käyttää tehokasta ehkäisyä (ks. kohdat Vasta-aiheet ja Raskaus ja imetys).
Hypotensio
Reniini-angiotensiini-aldosteronijärjestelmän (RAAS) estäjien, kuten sparsentaanin, käyttöön on liittynyt hypotensiota. Sparsentaanihoidon aikana voi esiintyä hypotensiota, ja sitä on raportoitu enemmän iäkkäillä potilailla (ks. kohta Haittavaikutukset).
Jos potilaalla on hypotension riski, tulee harkita muiden verenpainetta alentavien lääkevalmisteiden käytön lopettamista tai annoksen muuttamista ja asianmukaisesta nesteytyksestä huolehtimista. Jos hypotensio kehittyy muiden verenpainetta alentavien lääkevalmisteen käytön lopettamisesta tai annoksen pienentämisestä huolimatta, tulee harkita sparsentaaniannoksen pienentämistä tai hoidon keskeyttämistä. Ohimenevä hypotensiivinen vaste ei ole vasta-aihe sparsentaanin käytön jatkamiselle. Hoitoa voidaan jatkaa verenpaineen vakautumisen jälkeen.
Jos hypotensio jatkuu verenpainetta alentavien lääkevalmisteiden käytön lopettamisesta tai annoksen pienentämisestä huolimatta, sparsentaaniannos tulee pienentää alkuperäiseen aloitusannokseen, kunnes verenpaine vakautuu. Sparsentaanihoidon keskeyttämistä on harkittava, jos hypotension oireet jatkuvat vielä 2 viikkoa annoksen pienentämisen jälkeen. Sparsentaania on käytettävä varoen potilaille, joiden systolinen verenpaine on ≤ 100 mmHg ( ks. kohta Annostus ja antotapa). Sparsentaaniannosta ei tule suurentaa potilailla, joiden systolinen verenpaine on ≤ 100 mmHg (ks. kohta Annostus ja antotapa).
Munuaistoiminnan häiriöt
RAAS-estäjien, kuten sparsentaanin, käyttöön on liittynyt ohimenevää seerumin kreatiniiniarvon nousua. Seerumin kreatiniiniarvon ohimenevää nousua voi esiintyä etenkin sparsentaanihoidon alussa (ks. kohta Haittavaikutukset). Riskipotilaiden seerumin kreatiniiniarvoa ja seerumin kaliumarvoa on seurattava määräajoin. Sparsentaania on käytettävä varoen potilaille, joilla on molemminpuolinen munuaisvaltimon ahtauma.
Potilaista, joiden eGFR on < 30 ml/min/1,73 m2, on vain vähän tietoa, joten sparsentaania ei suositella näille potilaille (ks. kohta Annostus ja antotapa).
Nesteen kertyminen elimistöön
Nesteen kertymistä elimistöön on liittynyt tyypin A endoteliinireseptoria (ETAR) salpaavien lääkevalmisteiden, kuten sparsentaanin, käyttöön. Nesteen kertymistä elimistöön voi esiintyä sparsentaanihoidon aikana (ks. kohta Haittavaikutukset). Jos sparsentaanihoidon aikana ilmenee nesteen kertymistä, diureettihoitoa tai jo käytössä olevien diureettien annoksen suurentamista suositellaan ennen sparsentaaniannoksen muuttamista. Diureettihoitoa voidaan harkita potilaille, joilla on merkkejä nesteen kertymisestä ennen sparsentaanihoidon aloittamista.
Sparsentaania ei ole tutkittu sydämen vajaatoimintaa sairastavilla potilailla. Siksi sparsentaania on käytettävä varoen sydämen vajaatoimintaa sairastaville potilaille.
Maksan toiminta
Sparsentaanihoidon yhteydessä on todettu ALAT- tai ASAT-arvojen nousua vähintään tasolle 3 × ULN (ks. kohta Haittavaikutukset). Sparsentaanihoitoa saavilla potilailla ei ole todettu samanaikaista bilirubiiniarvon nousua tasolle > 2 × ULN eikä maksan vajaatoimintatapauksia. Näin ollen mahdollisesti vakavan maksatoksisuuden riskin pienentämiseksi seerumin aminotransferaasiarvot ja kokonaisbilirubiini on tarkistettava ennen hoidon aloittamista, ja arvoja on seurattava kolmen kuukauden välein.
Potilaita on seurattava maksavaurion merkkien varalta. Jos potilaalla ilmenee pitkäkestoista, selittämätöntä, kliinisesti merkittävää ALAT- ja/tai ASAT-arvojen nousua, tai jos arvojen nousuun liittyy bilirubiiniarvon nousu tasolle > 2 × ULN, tai jos ALAT- ja/tai ASAT-arvojen nousun yhteydessä esiintyy maksavaurion merkkejä tai oireita (esim. keltaisuutta), sparsentaanihoito on lopetettava.
Sparsentaanihoidon aloittamista uudelleen voidaan harkita vasta, kun maksaentsyymi- ja bilirubiiniarvot ovat palanneet hoitoa edeltävälle tasolle ja vain, jos potilaalla ei ole maksatoksisuuteen viittaavia kliinisiä oireita. Sparsentaanihoidon aloittamista tulee välttää, jos potilaan aminotransferaasiarvot ovat koholla (> 2 × ULN) ennen lääkkeen käytön aloittamista (ks. kohta Annostus ja antotapa).
Kliinistä kokemusta on vain vähän potilaista, joilla on keskivaikea maksan vajaatoiminta. Siksi sparsentaania on käytettävä varoen näille potilaille (ks. kohta Annostus ja antotapa).
Reniini-angiotensiini-aldosteronijärjestelmän (RAAS) tuplasalpaus
On osoitettu, että angiotensiinikonvertaasin (ACE:n) estäjien, angiotensiini II -reseptorin salpaajien tai aliskireenin samanaikainen käyttö suurentaa hypotension, hyperkalemian ja munuaistoiminnan heikkenemisen (mukaan lukien munuaisten akuutin vajaatoiminnan) riskiä. Sen vuoksi RAAS:n tuplasalpausta ACE:n estäjiä, angiotensiini II -reseptorin salpaajia (sparsentaanin osittainen vaikutusmekanismi) tai reniinin estäjiä yhdistelemällä ei suositella (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Jos tuplasalpausta pidetään täysin välttämättömänä, se tulee tehdä ainoastaan erikoislääkärin valvonnassa ja sen yhteydessä tulee seurata tiheästi munuaisten toimintaa, elektrolyyttejä ja verenpainetta.
Hyperkalemia
Hoitoa ei pidä aloittaa potilaille, joiden seerumin kaliumpitoisuus on > 5,5 mmol/l. Kuten muidenkin reniini-angiotensiini-aldosteronijärjestelmään vaikuttavien lääkevalmisteiden kohdalla, hyperkalemiaa voi esiintyä sparsentaanihoidon aikana erityisesti munuaisten vajaatoiminnan ja/tai sydämen vajaatoiminnan yhteydessä. Seerumin kaliumin tarkkaa seurantaa suositellaan riskipotilailla. Jos potilailla ilmenee kliinisesti merkittävää hyperkalemiaa, suositellaan samanaikaisten lääkevalmisteiden säätämistä tai tilapäistä annoksen pienentämistä tai lopettamista. Jos seerumin kaliumpitoisuus on > 5,5 mmol/l, on harkittava lääkityksen lopettamista.
Laktoosi
Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi‑galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Angiotensiinireseptorin salpaajien (ARB), endoteliinireseptorin salpaajien (ERA) ja reniininestäjien samanaikainen käyttö
Sparsentaanin samanaikainen käyttö endoteliinireseptorin salpaajien, kuten bosentaanin, ambrisentaanin, masitentaanin, sitaksentaanin, kanssa, angiotensiinireseptorin salpaajien, kuten irbesartaanin, losartaanin, valsartaanin, kandesartaanin, telmisartaanin, kanssa tai reniininestäjien, kuten aliskireenin, kanssa on vasta-aiheista (ks. kohta Vasta-aiheet).
Sparsentaanin samanaikainen käyttö ACE:n estäjien ja mineralokortikoidireseptorin salpaajien kanssa
Sparsentaanin samanaikaiseen käyttöön mineralokortikoidireseptorin (aldosteronireseptorin) salpaajien, kuten spironolaktonin ja finerenonin, kanssa odotetaan liittyvän hyperkalemian riskin suurenemista.
Sparsentaanin ja ACE:n estäjien, kuten enalapriilin tai lisinopriilin, yhdistelmän käytöstä ei ole tietoja. Kliinisistä tutkimuksista saadut tiedot ovat osoittaneet, että reniini-angiotensiini-aldosteronijärjestelmän (RAAS) tuplasalpaukseen ACE:n estäjien, angiotensiini II -reseptorien salpaajien tai aliskireenin yhdistelmäkäytöllä liittyy suurempi haittavaikutusten, kuten hypotension, hyperkalemian ja munuaistoiminnan heikkenemisen (mukaan lukien munuaisten akuutin vajaatoiminnan) esiintymistiheys kuin käytettäessä yksittäistä RAA-järjestelmään vaikuttavaa lääkeainetta (ks. kohta Farmakodynamiikka).
Sparsentaanin käyttö ACE:n estäjien, kuten enalapriilin tai lisinopriilin, kanssa vaatii varovaisuutta, ja potilaan verenpainetta, kaliumarvoja ja munuaisten toimintaa on seurattava (katso kohta Varoitukset ja käyttöön liittyvät varotoimet).
Samanaikainen käyttö kaliumlisien ja kaliumia säästävien diureettien kanssa
Tyypin 1 angiotensiini II -reseptoria (AT1R) salpaavia lääkevalmisteita saavilla potilailla voi esiintyä hyperkalemiaa (ks. kohta Haittavaikutukset). Kaliumlisien, kaliumia säästävien diureettien, kuten spironolaktonin, eplerenonin, triamtereenin tai amiloridin, kanssa tai kaliumia sisältävien ruokasuolavalmisteiden samanaikainen käyttö voi suurentaa hyperkalemian riskiä, joten sitä ei suositella.
Muiden lääkevalmisteiden vaikutukset sparsentaaniin
Sparsentaani metaboloituu pääasiassa sytokromi P450 (CYP)3A:n välityksellä.
Voimakkaat ja kohtalaisen voimakkaat CYP3A:n estäjät
Sparsentaanin ja itrakonatsolin (voimakas CYP3A:n estäjä) samanaikainen käyttö suurensi sparsentaanin suurinta pitoisuutta plasmassa (Cmax) 1,3-kertaisesti ja pitoisuus-aikakuvaajan pinta-alaa nollasta äärettömään (AUC0‑inf) 2,7-kertaisesti. Voimakkaiden CYP3A:n estäjien, kuten bosepreviirin, telapreviirin, klaritromysiinin, indinaviirin, lopinaviiri/ritonaviirin, itrakonatsolin, nefatsodonin, ritonaviirin, greipin ja greippimehun, samanaikaista käyttöä ei suositella. Jos voimakkaan CYP3A:n estäjän käyttöä ei voida välttää, on harkittava sparsentaanihoidon keskeyttämistä. Sparsentaanihoitoa voidaan jatkaa voimakkaan CYP3A:n estäjän käytön lopettamisen jälkeen.
Sparsentaanin ja siklosporiinin (kohtalaisen voimakas CYP3A:n estäjä) samanaikainen käyttö suurensi sparsentaanin Cmax-arvoa 1,4-kertaisesti ja AUC0‑inf-arvoa 1,7-kertaisesti. Kohtalaisen voimakkaiden CYP3A:n estäjien, kuten konivaptaanin, flukonatsolin ja nelfinaviirin, samanaikainen käyttö vaatii varovaisuutta. Kun käytetään kohtalaisen voimakasta CYP3A:n estäjää, potilaita on seurattava hypotonian, hyperkalemian ja turvotuksen varalta ja/tai munuaisten toiminnan osalta.
CYP3A:n indusorit
Sparsentaani on CYP3A:n substraatti. Samanaikainen käyttö kohtalaisen voimakkaan CYP3A:n indusorin (efavirentsin) kanssa alensi sparsentaanin AUC0‑inf-arvoa 62 % ja Cmax-arvoa 38 %. Sparsentaanin terapeuttisen tehon mahdollisen heikkenemisen vuoksi voimakkaiden CYP3A:n indusorien (esimerkiksi rifampisiinin, karbamatsepiinin, fenytoiinin ja mäkikuisman), samanaikaista käyttöä ei suositella. Samanaikainen käyttö kohtalaisen voimakkaan CYP3A:n indusorin (esim. efavirentsi, deksametasoni ja fenobarbitaali) kanssa pitää toteuttaa varoen. Kun kortikosteroidihoito on tarpeen, suositellaan lääkeaineita, joiden ei tiedetä indusoivan CYP3A:ta in vivo tai, joiden kyky indusoida sitä on minimaalinen (esim. budesonidi, prednisoni, prednisoloni, metyyliprednisoloni).
Mahahappoa vähentävät aineet
Populaatiofarmakokineettisen analyysin perusteella happoa vähentävän aineen samanaikaisella käytöllä sparsentaanihoidon aikana ei pitäisi olla tilastollisesti merkittävää vaikutusta sparsentaanin farmakokineettiseen vaihteluun. Mahalaukun pH-arvoa muuttavia aineita, kuten antasideja, protonipumpun estäjiä ja histamiinin H2-reseptorin antagonisteja, saa käyttää samanaikaisesti sparsentaanin kanssa.
Sparsentaanin vaikutus muihin lääkevalmisteisiin
CYP-entsyymit
In vivo sparsentaani sekä esti että indusoi CYP3A4:ää ja indusoi CYP2B6:ta, CYP2C9:ää ja CYP2C19:ää.
Sparsentaani on sekä CYP3A4:n kohtalaisen voimakas estäjä että indusori. In vivo, 800 mg:n sparsentaanikerta-annoksen samanaikainen antaminen CYP3A4:n substraatin (midatsolaamin) kanssa suurensi midatsolaamin Cmax-arvoa 1,4-kertaisesti ja AUC0-inf-arvoa 1,6-kertaisesti. Useiden 800 mg:n sparsentaaniannosten samanaikainen antaminen CYP3A4:n substraatin (midatsolaamin) kanssa ei vaikuttanut midatsolaamin systeemiseen altistukseen. Varovaisuutta on noudatettava, kun sparsentaanihoito aloitetaan CYP3A4:n kautta metaboloituvien lääkevalmisteiden (esim. alfentaniilin, konivaptaanin, indinaviirin, simvastatiinin) kanssa. Jos samanaikainen käyttö on välttämätöntä, erityisesti CYP3A4:n substraattien kanssa, joilla on kapea terapeuttinen indeksi (esim. syklosporiini, fentanyyli ja takrolimuusi), potilaita on seurattava haittavaikutusten varalta ja näiden substraattien annoksen sovittaminen saattaa olla tarpeen.
Sparsentaani on heikko CYP2B6:n indusori. In vivo, useiden 800 mg:n sparsentaaniannosten samanaikainen antaminen CYP2B6:n substraatin (bupropionin) kanssa pienensi bupropionin Cmax‑arvoa 32 % ja AUC0-inf-arvoa 33 %. Pääasiassa CYP2B6:n kautta metaboloituvien lääkkeiden annostusta ei tarvitse muuttaa. Varovaisuutta on kuitenkin noudatettava, kun sparsentaania annetaan samanaikaisesti CYP2B6:n substraattien (esim. efavirentsin) kanssa, joiden terapeuttinen indeksi on kapea, koska se voi alentaa niiden pitoisuutta plasmassa.
Sparsentaani on heikko CYP2C9:n indusori. In vivo, useiden 800 mg:n sparsentaaniannosten samanaikainen antaminen CYP2C9:n substraatin (tolbutamidin) kanssa pienensi tolbutamidin Cmax‑arvoa 9 % ja AUC0-inf-arvoa 25 %. Pääasiassa CYP2C9:n kautta metaboloituvien lääkkeiden annostusta ei tarvitse muuttaa. Varovaisuutta on kuitenkin noudatettava, kun sparsentaania annetaan samanaikaisesti CYP2C9:n substraattien (esim. kumariinin, varfariinin, fenytoiinin) kanssa, joiden terapeuttinen indeksi on kapea, koska se voi alentaa niiden pitoisuutta plasmassa.
Sparsentaani on kohtalaisen voimakas CYP2C19:n indusori. In vivo, useiden 800 mg:n sparsentaaniannosten samanaikainen antaminen pienensi CYP2C19:n substraatin (omepratsolin) Cmax-arvoa 49 % ja AUC0‑inf-arvoa 60 %, mikä viittaa siihen, että sparsentaani on CYP2C19:n kohtalaisen voimakas indusori. CYP2C19:n substraatteja on käytettävä varoen, koska sparsentaani voi alentaa niiden pitoisuutta plasmassa, mikä voi johtaa subterapeuttisiin pitoisuuksiin. Jos sparsentaanin samanaikainen käyttö on välttämätöntä, erityisesti lääkkeiden kanssa, joiden terapeuttinen indeksi on kapea (esim. S‑mefenytoiini, diatsepaami), näiden substraattien annoksen sovittaminen saattaa olla tarpeen.
Kuljettajaproteiinit
Sparsentaani on heikko P-gp:n estäjä. In vivo, useiden 800 mg:n sparsentaaniannosten samanaikainen antaminen suurensi P-gp:n substraatin (digoksiinin) Cmax-arvoa 1,6-kertaisesti ja AUC0‑inf‑arvoa 1,2-kertaisesti. Sparsentaani voi suurentaa P-gp:n substraattien pitoisuutta plasmassa. Potilaita on seurattava haittavaikutusten varalta kun sparsentaania käytetään samanaikaisesti lääkkeiden kanssa, joilla on kapea terapeuttinen indeksi (esim. dabigatraani) kanssa, ja näiden substraattien annoksen sovittaminen saattaa olla tarpeen.
Useiden 800 mg:n sparsentaaniannosten samanaikainen antaminen suurensi rosuvastatiinin (BCRP:n substraatti) Cmax-arvoa 1,6-kertaisesti ja pienensi AUC0‑inf-arvoa 5 %, mikä viittaa siihen, että sparsentaani ei vaikuta BCRP-substraattien biologiseen hyötyosuuteen. BCRP:n säätelemien kuljetusprosessien kompleksisuus lukuisissa eri kudoksissa on mahdollisesti johtanut AUC-arvossa havaittuun neutraaliin nettovaikutukseen.
Sparsentaanin antaminen ei vaikuttanut seerumin kreatiniinipitoisuuteen (OAT2:n, OCT2:n MATE1:n ja MATE2K:n substraatti), 6-beeta-hydroksikortisolipitoisuuteen (OAT3:n substraatti) tai seerumin sappihappopitoisuuteen (BSEP:n substraatti).
Samanaikainen 800 mg:n sparsentaaniannoksen antaminen pienensi pitavastatiinin (OATP1B1:n ja OATP1B3:n substraatti) Cmax-arvoa 19 % ja AUC0‑inf-arvoa 30 %, mikä viittaa siihen, että sparsentaani ei ole OATP1B1:n ja OATP1B3:n estäjä.
Annostusta ei tarvitse muuttaa, kun sparsentaania annetaan samanaikaisesti BCRP:n, OAT2:n, OCT2:n, MATE1:n, MATE2K:n, OAT3:n, BSEP:n, OATP1B1:n ja OATP1B3:n substraatin kanssa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Sparsentaanihoidon saa aloittaa naisille, jotka voivat tulla raskaaksi, vasta kun on varmistettu, ettei nainen ole raskaana. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja edelleen 1 kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja sparsentaanin käytöstä raskaana oleville naisille.
Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Filspari on vasta-aiheista raskauden aikana (ks. kohta Vasta-aiheet).
Imetys
Fysikaalis-kemialliset tiedot viittaavat siihen, että sparsentaani erittyy ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Sparsentaania ei pidä käyttää rintaruokinnan aikana.
Hedelmällisyys
Ei ole olemassa tietoja sparsentaanin vaikutuksesta ihmisen hedelmällisyyteen. Eläinkokeissa ei havaittu urosten tai naaraiden hedelmällisyyden heikentymistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Filsparilla voi olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Tutkimuksia sparsentaanin vaikutuksesta ajokykyyn ja koneidenkäyttökykyyn ei ole tehty. On kuitenkin otettava huomioon, että sparsentaanihoidon aikana voi esiintyä heitehuimausta (ks. kohta Haittavaikutukset). Potilaita, joilla esiintyy heitehuimausta, on ohjeistettava olemaan ajamatta ja käyttämättä koneita, kunnes oireet ovat laantuneet.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoituja lääkkeen haittavaikutuksia olivat hypotensio (10,8 %), hyperkalemia (9,6 %), heitehuimaus (7,8 %) ja perifeerinen edeema (5,4 %). Yleisimmin raportoitu vakava haittavaikutus oli akuutti munuaisvaurio (0,9 %).
Haittavaikutustaulukko
Aktiivikontrolloiduissa vaiheen 2 ja 3 kliinisissä tutkimuksissa raportoidut haittavaikutukset sparsentaania saaneilla potilailla, jotka sairastivat kroonista munuaistautia, mukaan lukien IgA-nefropatia ja fokaalinen segmentaalinen glomeruloskleroosi (FSGS) (N=446), on lueteltu alla olevassa taulukossa MedDRA-järjestelmän mukaisen elin- ja yleisyysluokituksen perusteella seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000).
Taulukko 1: Kliinisissä tutkimuksissa todetut haittavaikutukset
| Elinjärjestelmäluokka | Hyvin yleinen | Yleinen | Melko harvinainen |
| Veri ja imukudos | - | Anemia | |
| Aineenvaihdunta ja ravitsemus | Hyperkalemia | - | |
| Hermosto | Heitehuimaus Päänsärky | - | |
| Verisuonisto | Hypotensio | Ortostaattinen hypotensio | - |
| Munuaiset ja virtsatiet | Munuaisten vajaatoiminta Akuutti munuaisvaurio | - | |
| Yleisoireet ja antopaikassa todettavat haitat | Perifeerinen edeema Väsymys | - | |
| Tutkimukset | Veren kreatiniiniarvon suureneminen Transaminaasiarvojen suureneminena | - |
a Transaminaasiarvojen suureneminen sisältää seuraavat suositellut termit: alaniiniaminotransferaasiarvon suureneminen, aspartaattiaminotransferaasiarvon suureneminen, gammaglutamyylitransferaasiarvon suureneminen ja maksaentsyymiarvojen suureneminen..
Valikoitujen haittavaikutusten kuvaus
Hemoglobiiniarvon pieneneminen
PROTECT-tutkimuksessa anemiaa tai hemoglobiiniarvon pienenemistä raportoitiin haittavaikutuksena kahdella (1 %) sparsentaania saaneella tutkittavalla ja neljällä (2 %) irbesartaania saaneella tutkittavalla. Yleisesti ottaen hemoglobiiniarvoa ≤ 90 g/l milloin tahansa hoidon jälkeen raportoitiin sparsentaaniryhmässä seitsemällä tutkittavalla (3 %) ja irbesartaaniryhmässä neljällä tutkittavalla (2 %). Tämän laskun uskotaan johtuvan osittain hemodiluutiosta. Yksikään potilas ei lopettanut hoitoa anemian takia.
Maksaan liittyvät haittavaikutukset
PROTECT-tutkimuksessa yhteensä kuudella (3 %) sparsentaania saaneella tutkittavalla ja neljällä (2 %) irbesartaania saaneella tutkittavalla maksan transaminaasiarvot suurenivat yli kolminkertaisiksi normaalialueen ylärajaan verrattuna, ilman kokonaisbilirubiinin suurenemista, kun sparsentaaniryhmän tutkittavat olivat saaneet tutkimuslääkettä 168 päivän ajan ja irbesartaaniryhmän tutkittavat 407 päivän ajan. Mitkään tapahtumista eivät olleet vakavia, ne olivat oireettomia, suurin osa voimakkuudeltaan lieviä tai keskivaikeita ja kaikki ennalleen palautuvia. Lisäksi transaminaasiarvojen suurenemiselle tunnistettiin myös muita mahdollisia aiheuttavia tai vaikuttavia tekijöitä. Maksavaurion kliinisiä oireita ei havaittu. Sparsentaaniryhmässä tutkimuslääke lopetettiin kolmelta tutkittavalta haittavaikutusten uusiuduttua hoidon uudelleen aloittamisen jälkeen, kun taas kahdella tutkittavalla sparsentaanihoito aloitettiin uudelleen ilman toistuvia maksaentsyymien kohoamisia.
Akuutti munuaisvaurio (AKI)
PROTECT-tutkimuksessa raportoitiin haittatapahtumana akuutti munuaisvaurio neljällä (2 %) sparsentaania saaneella tutkittavalla ja kolmella (1 %) irbesartaania saaneella tutkittavalla. Neljällä (2 %) sparsentaania saaneella tutkittavalla raportoitiin vakava akuutti munuaisvaurio, jotka olivat kaikki ennalleen palautuvia. Yksikään vakavista akuuteista munuaisvaurioista ei vaatinut dialyysihoitoa. Sparsentaaniryhmässä tutkimuslääkkeen antaminen lopetettiin kolmella tutkittavalla.
Hyperkalemia
PROTECT-tutkimuksessa raportoitiin haittatapahtumana hyperkalemiaa 20:lla (10 %) sparsentaania saaneella tutkittavalla ja 16:lla (8 %) irbesartaania saaneella tutkittavalla. Yksikään sparsentaania saaneiden tutkittavien tapahtumista ei ollut vakava, suurin osa oli voimakkuudeltaan lieviä tai keskivaikeita, ja kaikki olivat ennalleen palautuvia. Hyperkalemiasta johtuvia keskeytyksiä ei tehty. Hyperkalemian riski on suurentunut potilailla, joiden eGFR on alhainen.
Hypotensio
Sparsentaanihoidon yhteydessä on raportoitu hypotensiota. PROTECT-tutkimuksessa raportoitiin 12 %:lla sparsentaania saavista tutkittavista systolisen verenpaineen arvoa < 100 mmHg ja 10 %:lla systolisen verenpaineen laskua yli 30 mmHg, kun irbesartaania saavilla tutkittavilla vastaavat arvot olivat 11 % ja 10 %. Sparsentaania saaneista tutkittavista vain 15 (7,4 %) oli ≥ 65-vuotiaita. Hypotensiota raportoitiin 20:llä (11 %) < 65-vuotiaalla tutkittavalla ja 6:llä (40 %) 65–74-vuotiaalla tutkittavalla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‐haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Sparsentaania on annettu terveille henkilöille enintään 1 600 mg:n vuorokausiannoksina ilman näyttöä annosta rajoittavasta toksisuudesta. Yliannostuksen (mahdollisesti hypotension merkkien ja oireiden) ilmetessä potilasta on seurattava tiiviisti ja hänelle on annettava asiaankuuluvaa oireenmukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Reniini-angiotensiinijärjestelmään vaikuttavat lääkeaineet, ATC-koodi: C09XX01
Vaikutusmekanismi
Sparsentaani on sekä endoteliini- että angiotensiinireseptorien salpaaja.
Se on yksittäinen molekyyli, joka toimii kaksoisvaikutteisena suuren affiniteetin ETAR- ja AT1R-salpaajana. Endoteliini 1 (ETAR:n kautta) ja angiotensiini II (AT1R:n kautta) toimivat IgA‑nefropatian etenemiseen johtavien prosessien välittäjinä hemodynaamisten vaikutusten ja mesangiaalisten solujen lisääntymisen, proinflammatoristen ja profibroottisten välittäjäaineiden lisääntyneen ekspression ja aktiivisuuden, podosyyttien vaurioitumisen sekä oksidatiivisen stressin kautta. Sparsentaani estää sekä ETAR:n että AT1R:n aktivaatiota ja siten vähentää proteinuriaa ja hidastaa munuaistaudin etenemistä.
Farmakodynaamiset vaikutukset
Satunnaistetussa, aktiivisella vertailuvalmisteella ja plasebolla kontrolloidussa tutkimuksessa terveillä tutkittavilla sparsentaani aiheutti vähäistä QTcF-ajan pidentymistä, 8,8 millisekuntia (90 %:n CI: 5,9 - 11,8) annoksella 800 mg ja 8,1 millisekuntia ( 5,2 - 11,0) annoksella 1 600 mg. Toisessa tutkimuksessa terveillä tutkittavilla sparsentaanialtistus, joka ylitti ihmisen enimmäisannossuosituksen yli 2-kertaisesti, muutos oli 8,3 millisekuntia (6,69 - 9,90). Siten on epätodennäköistä, että sparsentaanilla olisi kliinisesti olennaista vaikutusta QT-ajan pitenemiseen.
Kliininen teho ja turvallisuus
Sparsentaanin, joka ei ole immunosuppressiivinen lääke, tehoa ja turvallisuutta on arvioitu IgA‑nefropatiaa sairastavilla potilailla tehdyssä PROTECT-tutkimuksessa.
PROTECT on satunnaistettu, kaksoissokkoutettu (110 viikkoa), aktiivikontrolloitu, maailmanlaajuinen vaiheen 3 monikeskustutkimus IgA‑nefropatiaa sairastavilla potilailla. Tutkimukseen otettiin ≥ 18‑vuotiaita potilaita, mukaan lukien 15 (7,4 %) ≥ 65‑vuotiasta sparsentaanihoitoa saanutta potilasta, joiden eGFR oli ≥ 30 ml/min/1,73 m2 ja virtsaan erittyneen proteiinin kokonaismäärä ≥ 1,0 g/vrk. Ennen tutkimukseenottoa potilaat olivat saaneet suurinta siedettyä ACE:n estäjän ja/tai angiotensiinireseptorin salpaajan annosta vähintään 3 kuukauden ajan. ACE:n estäjän ja/tai angiotensiinireseptorin salpaajan käyttö lopetettiin ennen sparsentaanihoidon aloittamista. Tutkimuksesta suljettiin pois potilaat, joiden kaliumarvo oli lähtötilanteessa yli 5,5 mmol/l.
Yhteensä 404 potilasta satunnaistettiin saamaan sparsentaania (n = 202) tai irbesartaania (n = 202). Hoito aloitettiin sparsentaaniannoksella 200 mg kerran vuorokaudessa tai irbesartaaniannoksella 150 mg kerran vuorokaudessa. Kun hoito oli kestänyt 14 vuorokautta, annosta titrattiin siedettävyyden mukaan suositusannokseen 400 mg sparsentaania kerran vuorokaudessa tai 300 mg irbesartaania kerran vuorokaudessa. Annoksen siedettävyys määriteltiin systoliseksi verenpaineeksi > 100 mmHg ja diastoliseksi verenpaineeksi > 60 mmHg 2 viikkoa kestäneen hoidon jälkeen sekä haittatapahtumien (esim. turvotuksen pahenemisen) ja poikkeavien laboratoriolöydösten (esim. seerumin kalium > 5,5 mEq/l [5,5 mmol/l]) puuttumiseksi. RAAS- tai endoteliinijärjestelmän estäjien käyttö oli kiellettyä tutkimuksen aikana. Muiden luokkien verenpainelääkkeet olivat sallittuja siinä määrin kuin niiden käyttö oli tarpeellista tavoiteverenpaineen saavuttamiseksi. Immunosuppressiivisten aineiden käyttö oli sallittua tutkimuksen aikana tutkijan harkinnan mukaan.
Lähtötilanteen eGFR- ja proteinuriaominaisuudet olivat vertailukelpoisia hoitoryhmien välillä. Kokonaispopulaatiossa eGFR:n keskiarvo (keskihajonta) oli 57 (24) ml/min/1,73 m2 ja virtsan proteiini-kreatiniinisuhteen (UP/C) mediaani oli 1,24 g/g (kvartiilien välinen alue: 0,83 - 1,77). Keski-ikä oli 46 vuotta (vaihteluväli 18–76 vuotta); 70 % oli miehiä, 67 % valkoihoisia, 28 % aasialaisia, 1 % tummaihoisia tai afroamerikkalaisia ja 3 % muiden rotujen edustajia.
Proteinurian ensisijainen analyysi tehtiin 36 viikon kuluttua noin 280 tutkittavan satunnaistamisesta sen määrittämiseksi, onko tehon ensisijainen päätetapahtuma eli UP/C-suhteen muutos lähtötilanteesta viikolla 36 tilastollisesti merkitsevä. Tutkimuksen ensisijainen päätetapahtuma eli UP/C-suhteen muutos lähtötilanteesta viikolla 36 saavutettiin. Geometrinen keskiarvo UP/C viikolla 36 oli 0,62 g/g sparsentaanihaarassa ja 1,07 g/g irbesartaanihaarassa. UP/C-suhteen pienimmän neliösumman geometrisen keskiarvon prosentuaalinen muutos lähtötilanteesta viikolla 36 oli -49,8 % (95 %:n luottamusväli [CI]: -54,98; -43,95) sparsentaaniryhmässä vs. -15,1 % (95 %:n CI: ‑23,72; ‑5,39) irbesartaaniryhmässä (p < 0,0001). Lopullisessa analyysissa sparsentaani osoitti nopean ja yli 2 vuotta kestävän antiproteinurisen hoitotehon. UP/C:n geometrinen keskiarvo viikolla 110 oli 0,64 g/g sparsentaanihaarassa ja 1,09 g/g irbesartaanihaarassa. Se siis pieneni sparsentaaniryhmässä keskimäärin 42,8 % (95 % CI: ‑49.75, ‑34.97) lähtötilanteesta, kun irbesartaaniryhmässä vain 4,4 % (95 % CI: ‑15.84, 8.70). Sparsentaaniryhmässä todettiin johdonmukaisesti parannusta proteinurian vähenemisessä jo 4 viikon kohdalla, ja parannukset säilyivät viikolle 110 asti (kuva 1).
Kuva 1: Käyntikohtainen prosentuaalinen muutos lähtötilanteen virtsan proteiini/kreatiniini -suhteesta (PROTECT)
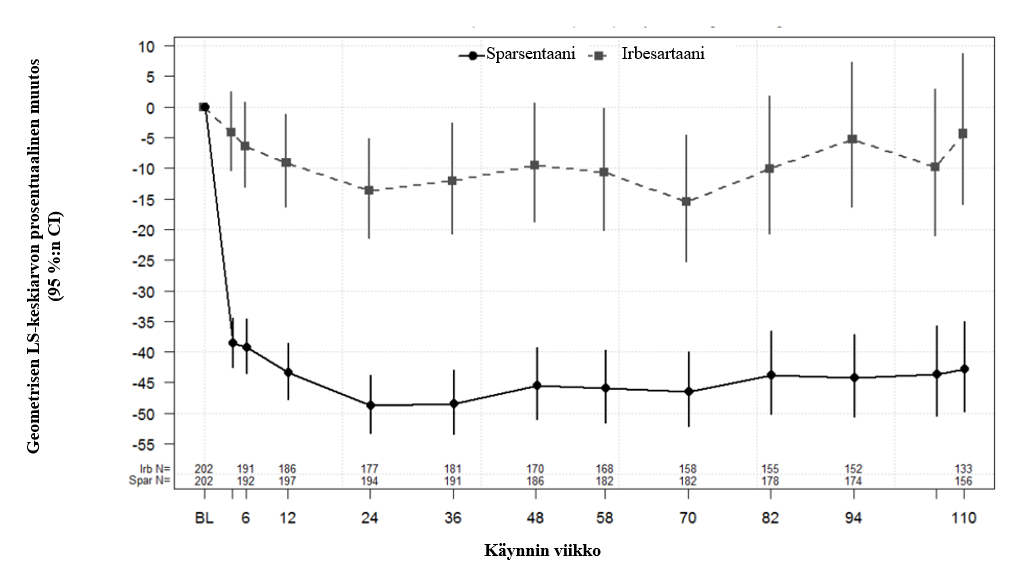
Huomautukset: UP/C:n pienimmän neliösumman mukautettu geometrinen keskiarvo lähtötilanteeseen nähden perustui longitudinaaliseen toistomittausmalliin, joka stratifioitiin seulontajakson eGFR-arvon ja proteinurian mukaan ja raportoitiin prosentuaalisena muutoksena 95 %:n luottamusvälillä. Analyysi sisältää kaksoissokkoutetun jakson UP/C-tiedot kaikista potilaista, jotka satunnaistettiin ja jotka saivat vähintään 1 annoksen tutkimuslääkettä. Lähtötilanne määriteltiin viimeiseksi vahvistetuksi havainnoksi ennen lääkkeenannon aloittamista, aloitushetki mukaan lukien.
Lyhenteet: CI = luottamusväli; eGFR = arvioitu glomerulusten suodatusnopeus; LS = pienin neliösumma; UP/C = virtsan proteiini-kreatiniinisuhde.
Arvioitu glomerulusten suodatusnopeus (eGFR)
Vahvistavassa analyysissä sparsentaanin parannus irbesartaaniin verrattuna oli kahden vuoden eGFR:n pitkäaikaisessa käyrässä (eli kuudesta viikosta eteenpäin) 1,1 ml/min/1,73 m2 vuodessa (95 %:n CI: 0,07, 2,12; p = 0,037). Vastaava kahden vuoden eGFR-käyrä parani kokonaisuudessaan (eli lähtötasosta eteenpäin) 1,0 ml/min/1,73 m2 vuodessa (95 %:n CI: -0,03, 1,94; p = 0,058). EGFR:n absoluuttinen muutos lähtötilanteesta kahden vuoden kuluttua oli ‑5,8 ml/min/1,73 m2 (95 %:n CI: ‑7,38, ‑4,24) sparsentaanilla ja ‑9,5 ml/min/1,73 m2 (95 %:n CI: ‑11,17, ‑7,89) irbesartaanilla.
Sparsentaani- ja irbesartaaniryhmien välisten samanaikaisten tapahtumien epätasapainon selvittämiseksi suoritettiin post hoc -analyysi, joka sisälsi kaikki havaitut tiedot hoidon lopettamisen tai immunosuppressiivisen rescue-hoidon aloittamisen jälkeen. Kahden vuoden eGFR:n pitkäaikaisessa käyrässä (kuudesta viikosta eteenpäin) sparsentaanin parannus irbesartaaniin verrattuna oli 1,3 ml/min/1,73 m2 vuodessa (95 % CI: 0,33, 2,31; p = 0,0087), ja vastaava kahden vuoden eGFR-käyrä parani kokonaisuudessaan (lähtötasosta alkaen) 1,2 ml/min/1,73 m2 vuodessa (95 % CI: 0,21, 2,13; p = 0,0168). Absoluuttinen muutos eGFR:ssä 2 vuoden kuluttua lähtötasosta oli ‑6,1 ml/min/1,73 m2 (95 % CI: ‑7,65, ‑4,54) sparsentaanilla ja ‑9,9 ml/min/1,73 m2 (95 % CI: ‑11,54, ‑8,31) irbesartaanilla.
Lisätietoa
ACE:n estäjän ja angiotensiini II -reseptorin salpaajan käyttämistä yhdistelmänä on tutkittu kahdessa suuressa satunnaistetussa ja kontrolloidussa tutkimuksessa, jotka olivat ONTARGET (Ongoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial) ja VA NEPHRON-D (The Veterans Affairs Nephropathy in Diabetes). ONTARGET-tutkimuksessa tutkittavilla oli aiempi sydän- ja verisuonisairaus tai aivoverisuonisairaus tai tyypin 2 diabetes mellitus, johon liittyi näyttöä pääte-elimen vauriosta. VA NEPHRON-D -tutkimuksessa tutkittavilla oli tyypin 2 diabetes mellitus ja diabeettinen nefropatia. Kumpikaan tutkimus ei osoittanut merkitseviä suotuisia vaikutuksia munuaisia ja/tai sydäntä ja verisuonia koskevissa tuloksissa tai kuolevuudessa. Sen sijaan hyperkalemian, akuutin munuaisvaurion ja/tai hypotension riskin havaittiin suurentuneen monoterapiaan verrattuna. Samankaltaisten farmakodynaamisten ominaisuuksien vuoksi nämä tulokset ovat relevantteja myös muiden ACE:n estäjien ja angiotensiini II -reseptorin salpaajien osalta. Sen vuoksi potilailla, joilla on diabeettinen nefropatia, ei pidä käyttää yhtä aikaa ACE:n estäjiä ja angiotensiini II -reseptorin salpaajia. ALTITUDE-tutkimuksessa (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) selvitettiin, millaista hyötyä olisi aliskireenin lisäämisestä standardihoitoon ACE:n estäjällä tai angiotensiini II -reseptorin salpaajalla potilailla, joilla oli tyypin 2 diabetes mellitus sekä joko krooninen munuaistauti tai sydän- ja verisuonitauti tai molemmat. Tutkimus lopetettiin ennenaikaisesti haitallisten tulosten suurentuneen riskin vuoksi. Kardiovaskulaariset kuolemat ja aivohalvaukset olivat molemmat numeerisesti yleisempiä aliskireeniryhmässä kuin plaseboryhmässä, ja aliskireeniryhmässä raportoitiin kiinnostuksen kohteena olevia haittatapahtumia ja vakavia haittatapahtumia (hyperkalemiaa, hypotensiota ja munuaisten vajaatoimintaa) useammin kuin plaseboryhmässä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Filspari-valmisteen käytöstä immunoglobuliini A -nefropatian hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Sparsentaanin suun kautta annetun 400 mg:n kerta-annoksen jälkeen mediaaniaika huippupitoisuuden saavuttamiseen plasmassa on noin 3 tuntia.
Sparsentaanin suun kautta annetun 400 mg:n kerta-annoksen jälkeen geometrinen Cmax-keskiarvo on 6,97 μg/ml ja AUC-keskiarvo 83 μg × h/ml. Vakaan tilan pitoisuudet plasmassa saavutetaan 7 vuorokauden kuluessa, eikä altistuksen kertymistä tapahdu suositellulla annostuksella.
Kun sparsentaania annetaan 400 mg vuorokaudessa suun kautta, vakaan tilan geometrinen Cmax‑keskiarvo on 6,47 μg/ml ja AUC-keskiarvo 63,6 μg × h/ml.
Ruoan vaikutus
Enintään 400 mg:n annoksia käytettäessä runsaasti rasvaa sisältävällä aterialla ei ollut kliinisesti merkityksellistä vaikutusta sparsentaanialtistukseen. Sparsentaani voidaan ottaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella näennäinen vakaan tilan jakautumistilavuus on 61,4 l.
Sparsentaani sitoutuu suuressa määrin (> 99 %) ihmisen plasman proteiineihin, voimakkaimmin albumiiniin ja kohtalaisessa määrin happamaan α1‑glykoproteiiniin.
Biotransformaatio
Sparsentaani metaboloituu pääosin CYP3A4-välitteisesti mutta siihen osallistuvat vähäisessä määrin myös CYP2C8, 2C9 ja 3A5. Kanta-aine on tärkein ihmisen plasmassa oleva entiteetti, ja sitä oli noin 90 % verenkierrossa olevasta kokonaisradioaktiivisuudesta. Eräs vähäisempi hydroksyloitunut metaboliitti oli plasmassa ainoa metaboliitti, jonka osuus oli > 1 % kokonaisradioaktiivisuudesta (noin 3 %). Sparsentaanin tärkein metaboliareitti oli oksidaatio ja dealkylaatio, ja ihmisen ulosteesta, plasmasta ja virtsasta tunnistettiin 9 metaboliittia.
Eliminaatio
Sparsentaanin puhdistuma riippuu ajasta. Populaatiofarmakokineettisen analyysin perusteella näennäinen puhdistuma on 3,88 l/h, ja se suurenee vakaassa tilassa tasolle 5,11 l/h.
Sparsentaanin puoliintumisaika vakaassa tilassa on arviolta noin 9,6 tuntia.
Yhden 400 mg:n radiomerkityn sparsentaaniannoksen jälkeen 82 % annoksen radioaktiivisuudesta mitattiin 10 vuorokauden keräysjakson aikana seuraavasti: 80 % ulosteesta, josta 9 % erittyi muuttumattomassa muodossa, ja 2 % virtsasta, johon vain vähäinen määrä erittyi muuttumattomassa muodossa.
Lineaarisuus/ei-lineaarisuus
Sparsentaanin Cmax- ja AUC-arvot suurenevat vähemmän kuin suhteessa annokseen 200–1 600 mg:n kerta-annosten antamisen jälkeen. Sparsentaanin farmakokinetiikan todettiin olevan riippuvainen ajasta, eikä vakaassa tilassa tapahtunut Cmax:n kertymistä mutta AUC-arvo laski annoksilla 400–800 mg vuorokaudessa.
Erityisryhmät
Iäkkäät
Populaatiofarmakokineettisessä analyysissa iän ei havaittu vaikuttavan merkitsevästi plasman sparsentaanialtistukseen. Iäkkäiden potilaiden annostusta ei tarvitse muuttaa (ks. kohta Annostus ja antotapa). Sparsentaania ei ole tutkittu yli 75-vuotiailla potilailla.
Maksan vajaatoiminta
Nimenomaan maksan vajaatoimintaa koskeneessa tutkimuksessa systeeminen altistus yhden 400 mg:n sparsentaaniannoksen jälkeen oli samaa luokkaa potilailla, joilla oli lähtötilanteessa lievä tai keskivaikea maksan vajaatoiminta (Child‑Pugh-luokka A tai Child‑Pugh-luokka B), ja potilailla, joiden maksa toimi normaalisti. Lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Sparsentaania tulee käyttää varoen potilailla, joilla on keskivaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Vaikeaa maksan vajaatoimintaa sairastavista potilaista ei ole saatavilla tietoja, eikä sparsentaanin käyttöä näille potilaille (Child‑Pugh-luokka C) siksi suositella (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Populaatiofarmakokineettinen analyysi tehtiin kroonista munuaistautia sairastavilla potilailla, joilla oli lievä (kreatiniinipuhdistuma 60–89 ml/min), keskivaikea (kreatiniinipuhdistuma 30–59 ml/min) tai vaikea (kreatiniinipuhdistuma 15–29 ml/min) munuaistauti. Sen perusteella munuaisten vajaatoiminnalla ei ole kliinisesti merkityksellistä vaikutusta farmakokinetiikkaan verrattuna henkilöihin, joiden munuaiset toimivat normaalisti (kreatiniinipuhdistuma ≥ 90 ml/min). Loppuvaiheen munuaistautia (kreatiniinipuhdistuma < 15 ml/min) sairastavista potilaista ei ole saatavilla tietoja.
Saatavilla olevien vähäisten tietojen perusteella annosmuutoksia ei voida suositella potilaille, joilla on vaikea munuaistauti (eGFR < 30 ml/min/1,73 m2, ks. kohta Annostus ja antotapa). Sparsentaania ei ole tutkittu potilailla, joilla on vaikea munuaistauti tai jotka saavat dialyysihoitoa, joten sparsentaania ei suositella näille potilaille. Sparsentaania ei ole tutkittu potilailla, jotka ovat saaneet munuaissiirteen, joten tässä potilasryhmässä sparsentaania tulee käyttää varoen (ks. kohta Annostus ja antotapa).
Muut erityisryhmät
Populaatiofarmakokineettisten analyysien mukaan iällä, sukupuolella tai rodulla ei ole kliinisesti merkityksellistä vaikutusta sparsentaanin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, karsinogeenisuutta sekä lisääntymistoksisuutta ja poikasten kehitystä koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Seuraavia haittavaikutuksia ei ole todettu kliinisissä tutkimuksissa, mutta niitä on todettu koe-eläimillä, jotka ovat saaneet hoitoannoksia vastaavia määriä lääkeainetta. Siksi haitoilla voi olla kliinistä merkitystä.
Rotilla ja kaniineilla tehdyissä alkion- ja sikiönkehitystutkimuksissa molemmilla lajeilla havaittiin kehitystoksisuutta. Rotilla annoksesta riippuvia teratogeenisia vaikutuksia, kuten kraniofasiaalisia epämuodostumia, luuston poikkeavuuksia, alkio- ja sikiökuolleisuuden lisääntymistä ja sikiöiden painon pienenemistä todettiin kaikilla testatuilla sparsentaaniannoksilla, joissa altistukset olivat 8‑kertaisia ihmisten altistukseen annoksella 800 mg/vrk ja 13‑kertaisia ihmisten altistukseen annoksella 400 mg/vrk . Kaniineilla ei esiintynyt sikiöiden epämuodostumia eikä alkioiden ja sikiöiden elinkelpoisuuteen tai sikiönkasvuun kohdistuneita vaikutuksia, mutta luuston poikkeavuuksia (ylimääräisiä servikaalisia kylkiluita) esiintyi altistuksilla, jotka olivat noin 0,10–0,2‑kertaisia ihmisten annoksilla 800 mg/vrk ja 400 mg/vrk saavutettuihin AUC-arvoihin nähden.
Rotilla tehdyssä pre- ja postnataalista kehitystä koskeneessa tutkimuksessa emoon kohdistuvaa toksisuutta ja myös kuolemia havaittiin noin 8‑kertaisilla ja 13‑kertaisilla altistuksilla. Emoon kohdistuvaa toksisuutta havaittiin noin 2‑kertaisilla ja 3‑kertaisilla altistuksilla ihmisten annoksilla 800 mg/vrk ja 400 mg/vrk saavutettuihin AUC-arvoihin nähden. Poikaskuolemien lisääntymistä ja kasvun heikentymistä esiintyi noin 8‑kertaisilla ja 13‑kertaisilla altistuksilla ja kasvun heikentymistä noin 2‑kertaisilla ja 3‑kertaisilla altistuksilla ihmisten annoksilla 800 mg/vrk ja 400 mg/vrk saavutettuihin AUC-arvoihin nähden.
Tutkimukset nuorilla eläimillä
Nuorilla rotilla tehdyissä tutkimuksissa osoitettiin, ettei yleisiä toksikologisia haittavaikutuksia esiintynyt enintään 10 mg/kg/vrk annoksin eikä uroksilla tai naarailla esiintynyt lisääntymistoksisuutta enintään 60 mg/kg/vrk annoksin, kun lääkkeenanto aloitettiin postnataalisena päivänä (PND) 14 (vastaa 1‑vuotiaita lapsia). Vaskulaarista toksisuutta esiintyi annoksilla ≥ 3 mg/kg/vrk, kun lääkkeenanto aloitettiin PND-päivänä 7 (vastaa vastasyntyneitä vauvoja).
Ympäristöön kohdistuvien riskien arviointi
Sparsentaania koskevien tutkimusten päätelmät osoittavat, että sparsentaania ei pidetä hitaasti hajoavana, biokertyvänä ja myrkyllisenä eikä erittäin hitaasti hajoavana ja erittäin voimakkaasti biokertyvänä. Sparsentaanin lääkemääräyksen mukaisen käytön ei odoteta aiheuttavan riskiä jätevedenpuhdistamolle, pinta- ja pohjavedelle, sedimentille eikä maaperälle (ks. kohta Käyttö- ja käsittelyohjeet).
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa, laktoosi, natriumtärkkelysglykolaatti (tyyppi A), kolloidinen vedetön piidioksidi, magnesiumstearaatti
Kalvopäällyste
Poly(vinyylialkoholi), makrogoli, talkki, titaanidioksidi (E171)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Tämä lääkevalmiste ei edellytä erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
FILSPARI tabletti, kalvopäällysteinen
200 mg (L:kyllä) 30 kpl (3941,26 €)
400 mg (L:kyllä) 30 kpl (3941,26 €)
PF-selosteen tieto
Korkeatiheyksisestä polyeteenistä (HDPE) valmistettu pullo, jossa lapsiturvallinen polypropeenikorkki.
Pakkauskoko 30 kalvopäällysteistä tablettia tai monipakkaus, jossa on 90 kalvopäällysteistä tablettia (kolme 30 tabletin pakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Filspari 200 mg tabletti, kalvopäällysteinen
Valkoinen tai luonnonvalkoinen, soikea kalvopäällysteinen tabletti, jonka toiselle puolelle on painettu merkintä ”105” ja toisella puolella ei ole mitään merkintöjä. Tabletin mitat ovat noin 13 mm × 7 mm.
Filspari 400 mg tabletti, kalvopäällysteinen
Valkoinen tai luonnonvalkoinen, soikea kalvopäällysteinen tabletti, jonka toiselle puolelle on painettu merkintä ”021” ja toisella puolella ei ole mitään merkintöjä. Tabletin mitat ovat noin 18 mm × 8 mm.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
FILSPARI tabletti, kalvopäällysteinen
200 mg 30 kpl
400 mg 30 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Sparsentaani: Aikuisten primaarisen immunoglobuliini A-nefropatian (IgA-nefropatia) hoito erityisin edellytyksin (3114).
ATC-koodi
C09XX01
Valmisteyhteenvedon muuttamispäivämäärä
01.11.2025
Yhteystiedot
Gustav III:s Boulevard 46
SE-169 73 Solna
Sverige
+46 8 558 066 00
www.viforpharma.se
Info.nordic@viforpharma.com