
QALSODY injektioneste, liuos 100 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi 15 ml:n injektiopullo sisältää 100 mg toferseenia (tofersen).
Yksi millilitra sisältää 6,7 mg toferseenia.
Apuaine, jonka vaikutus tunnetaan
Yksi 15 ml:n injektiopullo sisältää 52 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos
Kliiniset tiedot
Käyttöaiheet
Qalsody on tarkoitettu aikuisille superoksididismutaasi 1 (SOD1) ‑geenin mutaatioon liittyvän amyotrofisen lateraaliskleroosin (ALS-taudin) hoitoon.
Ehto
Valmisteen käyttöaiheissa mainittujen sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito.
Annostus ja antotapa
Toferseenihoidon saa aloittaa vain lääkäri, jolla on kokemusta ALS-taudin hoidosta.
Qalsody-valmistetta saavat antaa vain terveydenhuollon ammattilaiset, joilla on kokemusta lannepistojen tekemisestä, tai valmiste on annettava tällaisten henkilöiden valvonnassa.
Annostus
Suositeltu annos on 100 mg toferseenia hoitokertaa kohti.
Toferseenihoito pitää aloittaa kolmella latausannoksella, jotka annetaan 14 vuorokauden välein.
Sen jälkeen annetaan ylläpitoannos 28 vuorokauden välein.
Väliin jääneet tai viivästyneet annokset
Jos toinen latausannos viivästyy tai jää väliin, toferseeni pitää antaa mahdollisimman pian ja kolmas latausannos annetaan 14 vuorokautta myöhemmin.
Jos kolmas latausannos viivästyy tai jää väliin, toferseeni pitää antaa mahdollisimman pian ja ensimmäinen ylläpitoannos annetaan 28 vuorokautta myöhemmin.
Jos ylläpitoannos viivästyy tai jää väliin, toferseeni pitää antaa mahdollisimman pian. Seuraavat ylläpitoannokset pitää antaa 28 vuorokauden välein edellisestä annoksesta lukien.
Hoidon kesto
Hoidon jatkamisen tarvetta pitää arvioida säännöllisesti ja harkita yksilöllisesti potilaan kliinisen kuvan ja hoitovasteen mukaan.
Erityisryhmät
Iäkkäät potilaat
Toferseenin käytöstä iäkkäille potilaille on vähän kokemusta. Saatavissa olevien kliinisten tietojen perusteella toferseenin teho ja turvallisuus ovat kuitenkin oletettavasti samankaltaiset kuin muilla tutkituilla ikäryhmillä.
Toferseenin käytön yhteydessä ei ole näyttöä ikään perustuvista erityistä annostusta edellyttävistä seikoista.
Munuaisten vajaatoiminta
Toferseenia ei ole tutkittu munuaisten vajaatoimintaa sairastavilla potilailla.
Maksan vajaatoiminta
Toferseenia ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla.
Pediatriset potilaat
Qalsody-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Qalsody on tarkoitettu annettavaksi lannepiston avulla selkäydinnesteeseen.
- Intratekaalinen yhteys selkäydinnesteeseen on suositeltavaa varmistaa ennen muovikannen poistamista injektiopullosta ja toferseenin vetämistä injektiopullosta.
-
Muovikansi poistetaan injektiopullosta juuri ennen antoa, ja ruiskuun kiinnitetään ei-spinaalinen neula toferseenin vetämiseksi injektiopullosta ruiskuun. Ruiskun neula työnnetään injektiopulloon päällyssinetin keskikohdan läpi, ja tarvittava 15 ml:n annos (vastaa 100 milligrammaa) vedetään injektiopullosta ruiskuun.
- Qalsody-valmistetta ei saa laimentaa.
- Ulkoisia suodattimia, mukaan lukien bakteeri- tai hiukkassuodattimia, ei tarvita.
- Lannepistoneulalla on suositeltavaa poistaa noin 10 ml aivo-selkäydinnestettä ennen toferseenin antamista.
- Toferseeni annetaan lannepistoneulalla selkäydinnesteeseen 1–3 minuutin kestoisena bolusinjektiona.
Ohjeet toimenpiteen valmisteluun:
- Sedaatiota voidaan harkita, jos potilaan kliininen tila sitä edellyttää.
- Toferseenin antoa selkäydinnesteeseen kuvantamisohjauksessa voidaan harkita, jos potilaan kliininen tila sitä edellyttää.
- Ennen injektiopullon kannen poistamista alumiinisen päällyssinetin päältä on varmistettava, että potilas on valmiina. Avaamaton injektiopullo voidaan palauttaa jääkaappiin. Katso sallittu kokonaisaika kohdasta Kestoaika.
- Toimenpiteestä aiheutuvien vakavien komplikaatioiden välttämiseksi potilaat on tutkittava ennen injektion antamista selkäydinnesteeseen ja sen jälkeen lannepistoon liittyvien mahdollisten vakavien jälkitilojen varalta.
Injektion jälkeen suositellaan tavanomaista lannepiston jälkeistä hoitoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Lannepistotoimenpide
Lannepistoon liittyy haittavaikutusten (esim. päänsärky, selkäkipu, lannepiston jälkeinen oireyhtymä, infektio) ilmaantumisen riski.
Myeliitti ja/tai radikuliitti
Toferseenihoitoa saaneilla potilailla on raportoitu vakavaa myeliittiä ja radikuliittia. Jos näihin haittavaikutuksiin sopivia oireita kehittyy, hoitokäytännön mukaiset diagnostiset tutkimukset ja hoito pitää aloittaa.
Kohonnut kallonsisäinen paine ja/tai papilledeema
Toferseenihoitoa saaneilla potilailla on raportoitu vakavaa kohonnutta kallonsisäistä painetta ja/tai papilledeemaa. Jos näihin haittavaikutuksiin sopivia oireita kehittyy, hoitokäytännön mukaiset diagnostiset tutkimukset ja hoito pitää aloittaa.
Trombosytopenia ja hyytymishäiriöt
Trombosytopeniaa ja hyytymishäiriöitä, mukaan lukien akuuttia vaikeaa trombosytopeniaa, on havaittu antisense-oligonukleotidien ihon alle tai laskimoon annon jälkeen. Verihiutaleita ja veren hyytymistä koskevia laboratoriotutkimuksia suositellaan ennen toferseenin antamista, jos ne ovat kliinisesti aiheellisia.
Munuaistoksisuus
Munuaistoksisuutta on havaittu antisense-oligonukleotidien ihon alle tai laskimoon annon jälkeen. Virtsan proteiinien tutkimista (mielellään aamuvirtsanäytteestä) suositellaan, jos se on kliinisesti tarpeen. Pitkään koholla olevien virtsan proteiiniarvojen yhteydessä pitää harkita lisätutkimuksia.
Apuaineet
Natrium
Tämä lääkevalmiste sisältää 52 mg natriumia per 15 ml, mikä vastaa 3 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Kalium
Tämä lääkevalmiste sisältää kaliumia alle 1 mmol (39 mg) per 15 ml:n annos, eli sen voidaan sanoa olevan ”kaliumiton”.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Muiden selkäydinnesteeseen annettavien lääkevalmisteiden samanaikaista antamista toferseenin kanssa ei ole tutkittu eikä näiden yhdistelmien turvallisuutta tunneta.
Toferseeni ei ole CYP450-välitteisen oksidatiivisen aineenvaihdunnan indusoija eikä estäjä, joten se ei oletettavasti häiritse muiden näihin metaboliareitteihin liittyvien lääkevalmisteiden toimintaa.
Raskaus ja imetys
Raskaus
Toferseenin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Tutkimuksissa eläimillä, joilla toferseeni ei ole farmakologisesti aktiivinen, ei ole havaittu suoria eikä epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Toferseenin käyttöä ei suositella raskauden aikana eikä niiden naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Toferseenin käytöstä ihmisille imetyksen aikana ei ole olemassa tietoja. Olemassa olevat farmakodynaamiset tiedot koe-eläimistä ovat osoittaneet toferseenin erittyvän maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois.
On päätettävä, lopetetaanko imetys vai pidättäydytäänkö toferseenihoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Tietoja mahdollisista vaikutuksista ihmisen hedelmällisyyteen ei ole saatavilla. Eläimillä tehdyt toksisuustutkimukset ovat osoittaneet, että toferseenilla ei ilmeisesti ole haitallisia vaikutuksia urosten tai naaraiden hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Toferseenilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaita, joille kehittyy näköhäiriöitä toferseenihoidon aikana, pitää neuvoa välttämään ajamista ja koneiden käyttämistä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Toferseenihoitoa saaneilla tutkittavilla havaittuja vakavia haittavaikutuksia olivat myeliitti (4,1 %), suurentunut kallonsisäinen paine ja/tai papilledeema (2,7 %), radikuliitti (1,4 %) ja aseptinen aivokalvotulehdus (1,4 %). Yleisimmät haittavaikutukset 100 mg:n toferseeniannoksia saaneilla tutkittavilla (n = 147) olivat kipu (68,7 %), nivelkipu (36,7 %), väsymys (30,6 %), veren valkosolujen lisääntynyt määrä aivo-selkäydinnesteessä (27,9 %), proteiinien lisääntynyt määrä aivo-selkäydinnesteessä (26,5 %), lihaskipu (22,4 %) ja kuume (20,4 %).
Haittavaikutustaulukko
Haittavaikutukset on lueteltu elinjärjestelmäluokan ja esiintyvyyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1: Haittavaikutukset Qalsody-hoitoa tutkimuksissa 101 ja 102 saaneilla tutkittavilla
Elinjärjestelmäluokka | Haittavaikutus | Esiintyvyys |
|---|---|---|
Hermosto | Veren valkosolujen lisääntynyt määrä aivo-selkäydinnesteessä* | Hyvin yleinen |
Proteiinien lisääntyminen aivo-selkäydinnesteessä | Hyvin yleinen | |
Papilledeema‡ | Yleinen | |
Neuralgia | Yleinen | |
Aseptinen aivokalvotulehdus†† | Yleinen | |
Radikuliitti† | Yleinen | |
Myeliitti§ | Yleinen | |
Luusto, lihakset ja sidekudos | Nivelkipu | Hyvin yleinen |
Lihaskipu | Hyvin yleinen | |
Muskuloskeletaalinen jäykkyys | Yleinen | |
Yleisoireet ja antopaikassa todettavat haitat | Kipu‡‡ | Hyvin yleinen |
Väsymys | Hyvin yleinen | |
Kuume | Hyvin yleinen |
* Veren valkosolujen lisääntynyt määrä aivo-selkäydinnesteessä sisältää termit veren valkosolujen lisääntynyt määrä aivo-selkäydinnesteessä ja pleosytoosi.
† Radikuliitti sisältää termit radikulopatia ja lumbaalinen radikulopatia.
‡ Papilledeema sisältää termit papilledeema ja suurentunut kallonsisäinen paine. Ks. lisätietoja kohdasta Valikoitujen haittavaikutusten kuvaus.
§ Myeliitti sisältää termit myeliitti, selkäytimen poikittaistulehdus ja neurosarkoidoosi. Ks. lisätietoja kohdasta Valikoitujen haittavaikutusten kuvaus.
†† Aseptinen aivokalvotulehdus sisältää termit kemiallinen aivokalvotulehdus ja aseptinen aivokalvotulehdus. Ks. lisätietoja kohdasta Valikoitujen haittavaikutusten kuvaus.
‡‡ Kipu sisältää termit kipu, selkäkipu ja raajakipu.
Valikoitujen haittavaikutusten kuvaus
Lannepistotoimenpide
Toferseenin antoon lannepiston avulla liittyviä haittavaikutuksia on havaittu. Lannepistoon yleisesti liittyviä haittavaikutuksia ovat päänsärky, selkäkipu, lannepiston jälkeinen oireyhtymä ja infektio. Näiden tapahtumien ilmaantuvuus ja vaikeusaste olivat yhdenmukaiset niiden tapahtumien kanssa, joita lannepiston yhteydessä oletettavasti ilmenee.
Myeliitti ja/tai radikuliitti
Kliinisissä tutkimuksissa kuudella 100 mg toferseenia saaneella tutkittavalla raportoitiin vakavana reaktiona myeliitti (4,1 %). Ennen myeliitin ilmenemistä oli annettu 5–58 toferseeniannosta. Neljällä tutkittavalla oli oireita, ja kaksi tutkittavaa oli oireettomia. Kaikilla kuudella tutkittavalla todettiin magneettikuvauksessa (MRI) poikkeavuuksia, jotka liittyivät tapahtumaan. Kaksi tutkittavaa lopetti hoidon, ja oireet hävisivät. Neljän muun osallistujan kohdalla tapahtuma ei johtanut hoidon lopettamiseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.).
Kahdella 100 mg toferseenia saaneella tutkittavalla raportoitiin vakavana reaktiona radikuliitti (1,4 %). Ennen radikuliitin ilmenemistä oli annettu 1–24 toferseeniannosta. Molemmista reaktioista aiheutui oireita. Toisella tutkittavalla todettiin magneettikuvauksessa poikkeavuuksia, jotka liittyivät tapahtumaan, ja toisen tutkittavan magneettikuvat olivat normaalit. Kumpikaan tutkittavista ei lopettanut hoitoa, ja reaktio hävisi toisella tutkittavalla ilman jälkiseurauksia, ja toiselle tutkittavalle jäi jälkiseurauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kallonsisäisen paineen nousu ja/tai papilledeema
Neljällä 100 mg toferseenia saaneella tutkittavalla raportoitiin vakavina reaktioina kallonsisäisen paineen nousua ja/tai papilledeema (2,7 %). Ennen kallonsisäisen paineen nousua ja/tai papilledeeman ilmenemistä oli annettu 7–18 toferseeniannosta. Kaikissa neljässä tapauksessa kallonsisäisen paineen noususta ja/tai papilledeemasta aiheutui oireita. Neljällä tutkittavalla ei todettu magneettikuvauksessa oleellisia tapahtumaan liittyviä löydöksiä. Yksi reaktio johti lopulta toferseenihoidon pysyvään lopettamiseen, ja yksi reaktio johti toferseenihoidon keskeyttämiseen. Kaikki reaktiot olivat hoidettavissa tavanomaisella hoidolla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Aseptinen tai kemiallinen aivokalvotulehdus
Kahdella 100 mg toferseenia saaneella tutkittavalla raportoitiin vakavana reaktiona aseptinen tai kemiallinen aivokalvotulehdus (1,4 %). Ennen aseptisen tai kemiallisen aivokalvotulehduksen ilmenemistä oli annettu 5–7 toferseeniannosta. Kummassakin tapauksessa aseptisesta tai kemiallisesta aivokalvotulehduksesta aiheutui oireita. Toisella tutkittavalla ei todettu magneettikuvauksessa oleellisia tapahtumaan liittyviä löydöksiä. Toinen osallistujista lopetti toferseenihoidon, mutta toinen ei.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa ei raportoitu toferseeniin liittyviä yliannostuksia.
Yliannostuksen yhteydessä potilaalle pitää antaa tukihoitoa, johon sisältyy mm. terveydenhuollon ammattilaisen konsultointi ja potilaan kliinisen tilan tarkka seuranta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut hermostoon vaikuttavat lääkeaineet, ATC-koodi: N07XX22
SOD1-ALS on pääasiassa autosomaalisesti vallitseva sairaus, jota sairastaa noin 2 % ALS-potilaista. SOD1-geenin mutaatiot johtavat SOD1-proteiinin toksisen muodon kertymiseen. ALS-tautiin liittyviä yksilöllisiä SOD1-mutaatioita on tunnistettu yli 200, ja sairauden keston mediaani on noin 2,3 vuotta.
Vaikutusmekanismi
Ihmisen SOD1-geeni koodaa runsaasti esiintyvää dimeeristä entsyymiä, kupari-sinkkisuperoksididismutaasia (Cu/ZnSOD eli SOD1), joka katalysoi superoksidin (O2-) transmutaatiota hapeksi (O2) ja vetyperoksidiksi (H2O2). SOD1-ALS-potilailla SOD1-geenin mutaatiot johtavat SOD1-proteiinin toksisen muodon kertymiseen, mistä aiheutuu aksonivaurio ja hermostorappeuma.
Toferseeni on antisense-oligonukleotidi (ASO), joka on komplementaarinen osalle ihmisen SOD1:n mRNA:n 3′-translatoitumatonta aluetta (3′UTR) ja joka sitoutuu mRNA:han muodostamalla Watson-Crick-emäspareja (hybridisaatio). Toferseenin ja sitä vastaavan mRNA:n tällainen hybridisaatio johtaa SOD1:n mRNA:n ribonukleaasi H (Rnase-H) ‑välitteiseen hajoamiseen, mikä vähentää SOD1-proteiinin synteesiä.
Farmakodynaamiset vaikutukset
Aivo-selkäydinnesteen SOD1-proteiinin määrä
Aivo-selkäydinnesteen SOD1:n määrää mitattiin tutkimuksen 101 osassa C (VALOR) ja tutkimuksessa 102 kohteeseen liittymisen epäsuorana mittarina.
Tutkimuksen 101 osassa C viikolla 28 aivo-selkäydinnesteen SOD1-proteiinin määrä väheni ITT-potilasjoukossa toferseenilla hoidetussa ryhmässä 35 % (geometrisen keskiarvon suhde lähtötilanteeseen) verrattuna 2 %:n vähenemään lähtötilanteesta vastaavilla lumelääkettä saaneilla tutkittavilla (toferseenin ja lumelääkkeen geometristen keskiarvojen suhteiden ero: 34 % (95 %:n luottamusväli: 23 % ˗ 43 %). Aivo-selkäydinnesteen SOD1-proteiinin määrä väheni noin päivään 56 saakka, minkä jälkeen vähenemä säilyi ajan mittaan.
Plasman neurofilamentin kevytketjun (NfL) pitoisuus biomarkkerina
Neurofilamentin kevytketjun (NfL) pitoisuus plasmassa mitattiin tutkimuksen 101 osassa C (VALOR) ja tutkimuksessa 102 aksonivaurion ja hermostorappeuman markkerina.
Tutkimuksen 101 osassa C viikolla 28 plasman keskimääräinen NfL-pitoisuus pieneni toferseenilla hoidetussa ryhmässä (ITT) 55 % (geometrisen keskiarvon suhde lähtötilanteeseen) ja suureni lumelääkettä saaneilla tutkittavilla 12 % (toferseenin ja lumelääkkeen geometristen keskiarvojen suhteiden ero: 60 % (95 %:n luottamusväli: 51 % ˗ 67 %). Plasman NfL-pitoisuudet pienenivät noin päivään 113 saakka, minkä jälkeen vähenemä säilyi ajan mittaan. Aivo-selkäydinnesteessä NfL:n vähenemä oli yhdenmukainen plasmassa todettuun vähenemään verrattuna.
Kuva 1: Tutkimuksen 101 osa C ja tutkimus 102: ITT-potilasjoukon plasman NfL-pitoisuuden korjattu geometrisen keskiarvon suhde lähtötilanteeseen tutkimusviikon mukaan
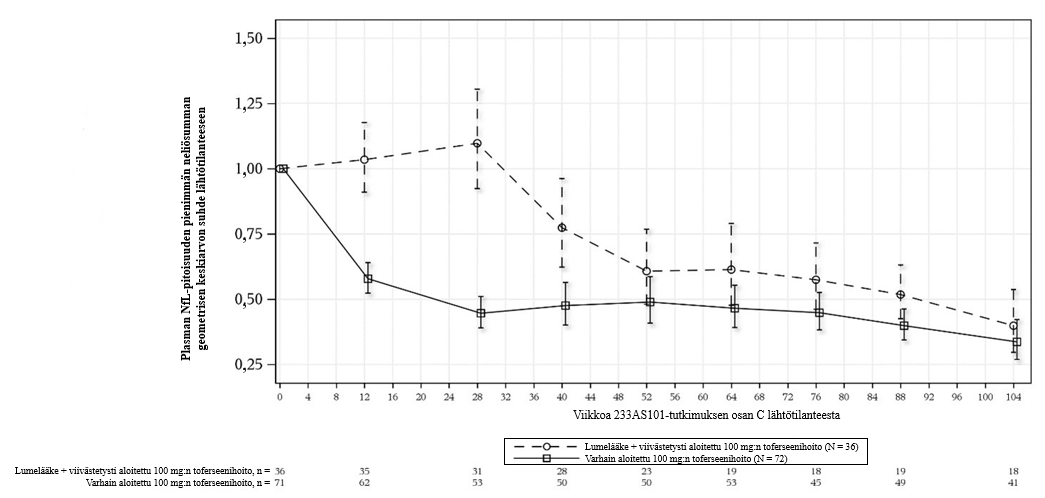
Lyhenteet: NfL (neurofilament light chain) = neurofilamentin kevytketju.
Huomautus 1: Lähtötilanteeksi määriteltiin päivän 1 arvo ennen kliinisen tutkimuslääkkeen saamista. Jos päivän 1 arvo puuttuu, lähtötilanteen arvona käytettiin luotettavaa arvoa (non-missing value) (mukaan lukien seulontakäynti), joka oli lähimpänä ennen ensimmäistä annosta.
Huomautus 2: Määritysrajan alittavat arvot on määritetty laskelmissa puoleen määrityksen alarajasta (4,9 pg/ml). Puuttuville tiedoille käytettiin moni-imputointia.
Huomautus 3: ITT-analyysi perustuu ANCOVA (kovarianssianalyysi) ‑malliin, joka käyttää luonnollisella logaritmilla muunnettuja tietoja. Malli sisältää kovariaatit vastaaville lähtötilanteen arvoille eli log-arvolle, sairauden kestolle lähtötilanteessa oireiden alkamisesta ja rilutsolin tai edaravonin käytölle.
Huomautus 4: Alaosassa oleva taulukko esittää niiden tutkittavien lukumäärän, joista oli luotettavia tietoja (non-missing data) kullakin käynnillä.
Sydämen elektrofysiologia
EKG-rekisteröinnit ja ‑arvot tutkimuksen 101 osassa C olivat 100 mg toferseenia saaneessa ryhmässä (n = 41) samankaltaiset kuin lumelääkeryhmässä (n = 34). EKG-rekisteröinneissä havaittujen poikkeavuuksien ilmaantuvuus oli toferseeniryhmässä suurempi kuin lumelääkeryhmässä: 8 tutkittavalla toferseeniryhmässä (11,3 %) verrattuna 2 tutkittavaan lumelääkeryhmässä (5,6 %) QTcF-ajan (Friderician kaava) suurin pidentymä lähtötilanteesta oli > 30–60 ms. Tämän epätasapainon kliinistä merkitystä ei tiedetä. QTcF-ajan pidentymä lähtötilanteesta ei ollut yhdelläkään toferseeni- tai lumelääkeryhmän tutkittavalla yli 60 ms, eikä pisin lähtötilanteen jälkeinen QTcF-aika ollut yhdelläkään tutkittavalla yli 480 ms.
Immunogeenisyys
Lääkevasta-aineita (ADA) havaittiin hyvin yleisesti. Lääkevasta-aineiden vaikutuksesta tehoon tai turvallisuuteen ei ollut havaintoja.
Kliininen teho ja turvallisuus
Toferseenin tehoa arvioitiin 28 viikkoa kestäneessä satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa kliinisessä tutkimuksessa (tutkimus 101, osa C) 23–78-vuotiailla tutkittavilla, joilla oli ALS-tautiin liittyvää heikkoutta ja keskuslaboratorion varmistama SOD1-mutaatio. Satakahdeksan (108) tutkittavaa satunnaistettiin suhteessa 2:1 saamaan 24 viikon ajan hoitona joko 100 mg:n toferseenia tai lumelääkettä (3 latausannosta, joiden jälkeen 5 ylläpitoannosta). Neljääkymmentäkahta (42) yksilöllistä SOD1-mutaatiota arvioitiin, niistä yleisimmät olivat p.Ile114Thr (n = 20), p.Ala5Val (n = 17), p.Gly94Cys (n = 6) ja p.His47Arg (n = 5). Rilutsolin ja/tai edaravonin samanaikainen käyttö sallittiin tutkittaville, jotka olivat saaneet niitä vakaalla annostuksella vähintään 30 päivän (rilutsoli) tai 60 päivän (edaravoni) ajan ennen tutkimuksen lähtötilannetta.
Sairauden lähtötilanteen ominaisuudet koko ITT-potilasjoukossa olivat yleisesti samankaltaiset toferseenihoitoa saaneilla (n = 72) ja lumelääkehoitoa saaneilla (n = 36) tutkittavilla. Lähtötilanteen ALSFRS-R (ALS Functional Rating Scale–Revised) ‑kokonaispisteet olivat toferseeniryhmässä 36,9 (keskihajonta: 5,9) ja lumelääkeryhmässä 37,3 (keskihajonta: 5,81). Toferseeniryhmässä oireiden alkamisesta kuluneen ajan mediaani oli lyhyempi (11,4 kuukautta; vaihteluväli: 1,7–145,7) kuin lumelääkeryhmässä (14,6 kuukautta; vaihteluväli: 2,4–103,2) ja plasman NfL-pitoisuuden mediaani oli lähtötilanteessa suurempi (78,5 pg/ml; vaihteluväli 5–329) kuin lumelääkeryhmässä (64,6 pg/ml; vaihteluväli: 8–370).
Ensisijainen tehon päätetapahtuma oli ALSFRS‑R-kokonaispisteiden muutos lähtötilanteesta viikkoon 28. Tulokset olivat toferseenin kannalta numeerisesti suotuisat, mutta eivät olleet tilastollisesti merkitseviä (ITT-potilasjoukko: toferseenin ja lumelääkkeen korjattu keskimääräinen ero [95 %:n luottamusväli]: 1,4 [‑1,3–4,1]). 28 viikon aikana toferseenin ja lumelääkkeen välillä havaitut erot olivat numeerisesti suurempia potilailla, joiden lähtötilanteen NfL-pitoisuus oli mediaania suurempi (keskimääräinen ero [95 %:n luottamusväli]: 3,9 [‑1,0–8,9]), verrattuna potilaisiin, joiden lähtötilanteen NfL-pitoisuus oli mediaania pienempi (0,6 [‑1,3–4,2]). Myöskään toissijaiset kliiniset tulokset eivät olleet tilastollisesti merkitseviä.
Jotta pitkäkestoinen seuranta olisi mahdollista, tutkimuksen 101 osaan C sen päättymiseen saakka osallistuneilla oli mahdollisuus osallistua avoimeen jatkotutkimukseen (tutkimus 102), jossa kaikki tutkittavat saivat 100 mg:n toferseeniannoksia. Tutkimukseen 102 tuli mukaan yhteensä 95 (88 %) tutkittavaa: varhaisvaiheessa aloitettua hoitoa saaneessa ryhmässä n = 63 (ES [early-start] ‑ryhmä: tutkimuksen 101 osassa C toferseenia saamaan satunnaistetut) ja lumelääke- / viivästyneesti aloitettua hoitoa saaneessa ryhmässä n = 32 (DS [delayed-start] ‑ryhmä: tutkimuksen 101 osassa C lumelääkettä saamaan satunnaistetut). Lopullisen integroidun analyysin ajankohtana 33,3 % (12/36) tutkittavista lumelääke- / viivästyneesti aloitettua hoitoa saaneessa ryhmässä ja 47,2 % (34/72) tutkittavista varhaisvaiheessa aloitettua hoitoa saaneessa ryhmässä oli mukana tutkimuksessa 102 sen päättymiseen saakka, jolloin näiden kahden tutkimuksen seurantamahdollisuuden mediaani oli 4,9 vuotta (vaihteluväli: 3,6–5,4 vuotta). Toferseenin varhaisempaan aloittamiseen (ES-ryhmä) liittyi ALSFRS-R-pisteiden, hitaan vitaalikapasiteetin ennustetun prosenttiarvon ja kädessä pidettävällä laitteella saatujen megapisteiden pienenemistä verrattuna lumelääke- / viivästyneesti aloitettua toferseenihoitoa saaneeseen ryhmään (DS-ryhmä), mutta erot eivät olleet tilastollisesti merkitseviä.
Varhaisempaan toferseenihoidon aloittamiseen (ES-ryhmä) liittyi kuoleman tai pysyvän ventilaation riskin selvä pieneneminen (riskitiheyksien suhde [95 %:n luottamusväli]: 0,64 [0,28–1.46]) sekä kuoleman riski (riskitiheyksien suhde [95 %:n luottamusväli]: 0,52 [0,20–1,36]). Tapahtumaan kuluneen ajan mediaani ei ollut arvioitavissa havaittujen tapahtumien vähäisen lukumäärän vuoksi. Tutkimuksen 101 osan C ja tutkimuksen 102 integroidussa analyysissä tutkittavilla, joilla sairaus todennäköisemmin eteni nopeammin (suurempien lähtötilanteen plasman NfL-pitoisuuksien perusteella), kuolemaan tai pysyvään ventilaatioon kuluneen ajan mediaani ES-ryhmässä oli 253,6 viikkoa verrattuna lumelääke- / viivästyneesti aloitettua hoitoa saaneen ryhmän 76,0 viikkoon.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset toferseenin käytöstä ALS-taudin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Tämän lääkevalmisteen myyntilupa on myönnetty poikkeuksellisin perustein. Se tarkoittaa, että lääkevalmisteesta ei ole ollut mahdollista saada täydellisiä tietoja sairauden harvinaisuuden vuoksi. Euroopan lääkevirasto arvioi vuosittain mahdolliset uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Injektiona selkäydinnesteeseen annetun toferseenin kerta-annoksen ja usean annoksen farmakokinetiikka määritettiin niiden aikuisten ALS-tautia sairastavien tutkittavien plasmasta ja aivo-selkäydinnesteestä, joilla oli SOD1-mutaatio, sekä kliiniseen tutkimukseen osallistuneiden (n = 3) kuolleiden tutkittavien ruumiinavauksissa kerätystä kudoksesta.
Imeytyminen
Suurin pitoisuus aivo-selkäydinnesteessä havaittiin kolmannen annoksen yhteydessä, joka oli latausjakson viimeinen annos. Latausvaiheen jälkeisessä kuukausittaisessa annossa kertymistä tapahtui vähän tai ei lainkaan; kumuloitumissuhde vaikuttaa olevan alle 2-kertainen. Toferseeni siirtyy nopeasti aivo-selkäydinnesteestä systeemiseen verenkiertoon. Plasman huippupitoisuus (Tmax) saavutettiin 2–6 tuntia (mediaani) intratekaalisen annon jälkeen. Toistuvat kuukausittaiset ylläpitoannokset eivät altistuksen mittarien (Cmax ja AUC) perusteella aiheuttaneet aineen kertymistä plasmassa.
Jakautuminen
Selkäydinnesteeseen annettu toferseeni jakautui laajalti keskushermostossa ja saavutti hoitopitoisuuden kohteena olevissa selkäydinkudoksissa. Annoksia 100 mg käytettäessä (tutkimuksen 101 osan C tiedot) plasman AUC-arvon mediaani ensimmäisen annoksen jälkeen oli 13 973,1 ng/ml*h. Plasman huippupitoisuuden (Cmax) mediaani oli 824,3 ng/ml, joka saavutettiin 4–6 tuntia annoksen jälkeen. Plasman jakautumistilavuuden mediaaniksi tutkimuksissa 101 ja 102 arvioitiin 48,7 l (variaatiokerroin 124 %) ja 100 mg:n annoksen ryhmässä se oli 43,0 l (variaatiokerroin 123 %). Farmakokineettinen analyysi osoittaa, että selkäydinnesteeseen annettu toferseeni jakautuu laajalti keskushermoston kudoksiin ja siirtyy nopeasti aivo-selkäydinnesteestä systeemiseen verenkiertoon.
Plasman proteiineihin sitoutuminen
Toferseeni sitoutuu voimakkaasti ihmisen plasman proteiineihin (≥ 98 %) kliinisesti oleellisilla tai sitä suuremmilla pitoisuuksilla plasmassa (0,1 ja 3 mikrog/ml), mikä rajoittaa glomerulussuodatusta ja vähentää vaikuttavan aineen erittymistä virtsaan. Lääkkeiden välisten yhteisvaikutusten todennäköisyys kilpailevan plasman proteiineihin sitoutumisen vuoksi on hyvin pieni.
Biotransformaatio
Toferseeni metaboloituu eksonukleaasi (3'‑ ja 5’) -välitteisen hydrolyysin kautta, eikä se ole CYP450-entsyymien substraatti, estäjä eikä indusori.
Eliminaatio
Ensisijaisen eliminaatioreitin oletetaan olevan muuttumattoman toferseenin ja sen metaboliittien erittyminen virtsaan. Puoliintumisaikaa keskushermostokudoksessa ei voida mitata ihmisillä, mutta jaavanmakakien keskushermostokudoksesta mitattu keskimääräinen terminaalisen eliminaation puoliintumisaika oli 31–40 vuorokautta. Plasmapuhdistuman mediaani tutkimusten 101 ja 102 tutkittavilla oli arvion mukaan 8,20 l/h (variaatiokerroin 47,3 %), ja 100 mg:n annoksella se oli 6,02 l/h (variaatiokerroin 48,2 %).
Lineaarisuus/ei-lineaarisuus
Selkäydinnesteeseen annetun toferseenin farmakokinetiikka aivo-selkäydinnesteessä lisääntyy vähemmän kuin suhteessa annokseen, kun annos on 20–100 mg.
Selkäydinnesteeseen annetun toferseenin farmakokinetiikka plasmassa lisääntyy enemmän kuin suhteessa annokseen, kun annos on 20–100 mg.
Immunogeenisyys
Lääkevasta-aineiden esiintyminen vaikutti vähentävän plasmapuhdistumaa 37,9 %.
Ominaisuudet erityisissä potilasryhmissä
Iäkkäät potilaat
Toferseenia kliinisissä tutkimuksissa saaneista 166 potilaasta yhteensä 22 potilasta oli vähintään 65-vuotiaita ja näistä kaksi oli vähintään 75-vuotiaita. Kaiken kaikkiaan kliinisessä farmakokinetiikassa ei havaittu eroja näiden potilaiden välillä, mutta tiedot ovat suppeat.
Munuaisten vajaatoiminta
Toferseenin farmakokinetiikkaa ei ole tutkittu munuaisten vajaatoimintaa sairastavilla potilailla.
Maksan vajaatoiminta
Toferseenin farmakokinetiikkaa ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla.
Prekliiniset tiedot turvallisuudesta
Karsinogeneesi
Toferseenilla ei ole tehty karsinogeenisuustutkimuksia.
Mutageneesi
Toferseenilla ei ole prekliinisten genotoksisuustutkimusten perusteella (Amesin bakteerimutageenisuustesti in vitro, kromosomipoikkeavuustesti in vitro ja hiirten mikrotumatesti in vivo) ilmennyt näyttöä mutageenisuudesta.
Lisääntymistoksisuus
Lisääntymistä koskevissa toksikologisissa tutkimuksissa toferseenia annettiin hiirille ja kaniineille ihon alle. Hiirillä tehdyssä hedelmällisyyttä ja alkio- ja sikiökehitystä selvittäneessä tutkimuksessa uroshiirillä esiintyi suuren annoksen ryhmässä (30 mg/kg eli > 50 kertaa ihmisen altistus [AUC] 100 mg:n toferseeniannoksen jälkeen) minimaalista tai lievää siementiehyiden rappeutumista, siementiehyiden laajentumista, spermatidien retentiota, epiteelisolujen apoptoosia, solujäämien lisääntymistä kiveksissä ja hypospermiaa lisäkiveksissä. Toferseeniin liittyviä haittavaikutuksia paritteluun tai hedelmällisyys- tai spermaparametreihin ei kuitenkaan havaittu. Naarashiirillä ei esiintynyt toferseeniin liittyvää kuolleisuutta tai ennenaikaisia synnytyksiä eikä vaikutuksia paritteluun tai hedelmällisyyteen. Hiirillä ja kaniineilla ei todettu toferseeniin liittyviä haittavaikutuksia alkio- ja sikiökehitykseen (altistuksilla, jotka olivat yli 40-kertaisia verrattuna ihmisen altistukseen suurimmalla ihmisille suositellulla annoksella). Hiirten perinataalisessa/postnataalisessa lisääntymistutkimuksessa suurimmasta arvioidusta annoksesta (30 mg/kg) ei aiheutunut haittavaikutuksia F0-naaraille eikä F1-poikasten kasvuun ja kehitykseen. Toferseenia havaittiin hiiren maitonäytteissä kaikilla toferseenia saaneilla eläimillä. Toferseeni ei ole hiirillä ja kaniineilla farmakologisesti aktiivinen, mikä vähentää näiden tutkimusten validiteettia, sillä SOD1:n vaimennussäätelyyn liittyviä haitallisia vaikutuksia ei voida arvioida näillä lajeilla.
Sekä urosten että naaraiden lisääntymiskudoksia arvioitiin mikroskooppisesti 13 viikkoa ja 39 viikkoa kestäneissä toksikologiatutkimuksissa ihmisapinoihin kuulumattomilla kädellisillä, joilla toferseeni on farmakologisesti aktiivinen, eikä vaikutuksia lisääntymiskudoksiin havaittu.
Toksikologia
Toistuvaa altistusta koskeneessa toksikologisessa tutkimuksessa (9 kuukautta) aikuiset jaavanmakakit sietivät selkäydinnesteeseen annetun toferseenin yleisesti ottaen hyvin. Poikkeuksena oli yksi naaras suuren annoksen ryhmässä (35 mg, joka vastaa ihmisellä 350 mg:n injektiota selkäydinnesteeseen), jolla esiintyi lihaskramppeja, pään/niskan dorsifleksiota ja opistotonuksen kaltaista selän taipumista kaarelle selkäydinnesteeseen antamisen jälkeen. Aivosähkökäyrä (EEG) osoitti, ettei kyse ollut kouristuskohtauksesta. Toistuvaa, kroonista altistusta koskevissa toksikologisissa tutkimuksissa haitaton vaikutustaso (NOAEL-arvo) oli hiirille ihon alle annettaessa 150 mg/kg sekä ihmisapinoihin kuulumattomilla kädellisillä tehdyssä 9 kuukauden pituisessa tutkimuksessa selkäydinnesteeseen annettaessa 12 mg. Herkimmällä lajilla eli ihmisapinoihin kuulumattomilla kädellisillä 12 mg:n annos vastaa ihmisellä 120 mg:n annosta (HED, Human Equivalent Dose) (apinoiden ja ihmisten aivo-selkäydinnesteen tilavuuden perusteella). Apinoille selkäydinnesteeseen annettujen annosten turvamarginaali (1,2-kertainen) suhteessa ihmisille selkäydinnesteeseen annettuihin annoksiin perustuu ihmiselle muunnettuun vastaavaan annokseen (HED), jossa otetaan huomioon aivo-selkäydinnesteen tilavuusero (joka on ihmisten ja apinoiden välillä noin 10-kertainen). Näin ollen annostasoilla, jotka vastasivat 120 mg:n annosta ihmisille, ei havaittu toksisuuteen liittyviä vaikutuksia.
Farmaseuttiset tiedot
Apuaineet
Dinatriumfosfaatti
Kaliumkloridi
Kalsiumklorididihydraatti
Magnesiumkloridiheksahydraatti
Natriumkloridi
Natriumdivetyfosfaattidihydraatti
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
60 kuukautta
Tilapäiset säilytysolosuhteet
Qalsody-injektiopulloa voidaan säilyttää alkuperäisessä pahvikotelossa huoneenlämmössä (säilytä alle 30 °C) enintään 14 vuorokautta.
Avaamattomat Qalsody-injektiopullot voidaan tarvittaessa ottaa pois jääkaapista ja palauttaa sinne. Avaamattomat injektiopullot voidaan ottaa pois alkuperäisestä pahvikotelosta huoneenlämpöön enintään 6 tunnin ajaksi vuorokautta kohden enintään 6 päivänä.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Lääkevalmisteen avaamattomien injektiopullojen tilapäinen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
QALSODY injektioneste, liuos
100 mg (L:ei) 1 kpl (15 ml (6,7 mg/ml)) (23003,18 €)
PF-selosteen tieto
20 ml:n kirkas tyypin I lasinen injektiopullo, jossa on klooributyylikumitulppa ja alumiininen päällyssinetti sekä irti napsautettava (flip-off) muovikansi.
Qalsody on saatavana yhden injektiopullon pakkauksina.
Valmisteen kuvaus:
Kirkas ja väritön tai hieman kellertävä liuos, jonka pH on 6,7–7,7.
Käyttö- ja käsittelyohjeet
Toferseenin valmistelussa ja selkäydinnesteeseen antamisessa on käytettävä aseptista tekniikkaa.
Vain kertakäyttöön.
Injektiopullon valmisteluohjeet:
- Jääkaapissa säilytetyn injektiopullon pitää antaa lämmetä huoneenlämpöiseksi (25 °C) ennen antoa ulkoisella lämmönlähteellä lämmittämättä.
- Injektiopulloa ei saa ravistaa.
- Qalsody ei sisällä säilytysaineita. Liuos on annettava heti ruiskuun vetämisen jälkeen (4 tunnin kuluessa jääkaapista poistamisen jälkeen) huoneenlämmössä. Muussa tapauksessa se pitää hävittää.
- Liuos pitää tarkistaa silmämääräisesti ennen kuin se vedetään injektiopullosta. Liuoksessa ei saa näkyä hiukkasia. Vain kirkkaan ja värittömän tai hieman kellertävän liuoksen saa antaa. Jos liuos ei ole tällaista, injektiopulloa ei saa käyttää.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
QALSODY injektioneste, liuos
100 mg 1 kpl
- Ei korvausta.
ATC-koodi
N07XX22
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2026
Yhteystiedot
Bertel Jungin aukio 5 C
02600 Espoo
0207 401 200
www.biogen.fi
www.MS-nyt.fi