
BREYANZI infuusioneste, dispersio 1,1-70 x 10exp6 solua/ml / 1,1-70 x 10exp6 solua/ml
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Breyanzi (lisokabtageenimaraleuseeli) on autologinen T‑soluhoito, jonka sisältämät solut on geneettisesti muunneltu ja kohdennettu CD19‑antigeenin tunnistamiseen. Soluhoito koostuu puhdistetuista CD8‑positiivisista ja CD4‑positiivisista T‑soluista tietyssä suhteessa. Solut on transduktoitu erillisinä CD8‑positiivisina ja CD4‑positiivisina solukomponentteina ex vivo käyttämällä monistumiskyvytöntä lentivirusvektoria, joka ilmentää CD19‑antigeenia tunnistavaa kimeeristä antigeenireseptoria (CAR). Kimeerinen antigeenireseptori koostuu yksiketjuista vaihtelevaa fragmenttia (scFv) sitovasta domeenista, joka on peräisin hiiren monoklonaalisesta CD19‑spesifisestä vasta-aineesta (mAb; FMC63), kostimulatorisen 4‑1BB-endodomeenin osasta, CD3‑zeeta-(ζ)-ketjun signalointidomeenista ja toimimattomasta typistetystä epidermaalisen kasvutekijän reseptorista (EGFRt).
Breyanzi sisältää CAR‑positiivisia elinkykyisiä T‑soluja ja koostuu CD8‑positiivisista ja CD4‑positiivisista solukomponenteista tietyssä suhteessa:
CD8‑positiivinen solukomponentti
Jokainen injektiopullo sisältää lisokabtageenimaraleuseelia, jossa on tietty eräkohtainen pitoisuus autologisia, geneettisen muuntelun vuoksi kimeeristä anti-CD19‑antigeenireseptoria ilmentäviä T‑soluja (CAR‑positiivisia elinkykyisiä T‑soluja). Lääkevalmiste on pakattu yhteen tai useampaan injektiopulloon, jotka yhteensä sisältävät soludispersiona 5,1– 322 × 106 CAR‑positiivista elinkykyistä T‑solua (1,1–70 × 106 CAR‑positiivista elinkykyistä T‑solua/ml) suspendoituina pakastusliuokseen.
Yksi injektiopullo sisältää 4,6 ml CD8‑positiivista solukomponenttia.
CD4‑positiivinen solukomponentti
Jokainen injektiopullo sisältää lisokabtageenimaraleuseelia, jossa on tietty eräkohtainen pitoisuus autologisia, geneettisen muuntelun vuoksi kimeeristä anti-CD19‑antigeenireseptoria ilmentäviä T‑soluja (CAR‑positiivisia elinkykyisiä T‑soluja). Lääkevalmiste on pakattu yhteen tai useampaan injektiopulloon, jotka yhteensä sisältävät soludispersiona 5,1–322 × 106 CAR‑positiivista elinkykyistä T‑solua (1,1–70 × 106 CAR‑positiivista elinkykyistä T‑solua/ml) suspendoituina pakastusliuokseen.
Yksi injektiopullo sisältää 4,6 ml CD4‑positiivista solukomponenttia.
Yhteen Breyanzi-annokseen voidaan tarvita useampi kuin yksi injektiopullo CD8‑positiivista solukomponenttia ja/tai CD4‑positiivista solukomponenttia. Kummankin solukomponentin annettava kokonaistilavuus ja tarvittavien injektiopullojen määrä kummankin solukomponentin osalta voivat vaihdella.
Lääkevalmisteen kummankin solukomponentin määrälliset tiedot, mukaan lukien annettavien injektiopullojen määrä (katso kohta 6), ovat luovutussertifikaatissa (RfIC), joka sijaitsee kuljetuksessa käytettävän kylmäkuljetuspakkauksen kannen sisäpuolella. Kunkin komponentin luovutussertifikaatti (RfIC) sisältää annettavan kokonaistilavuuden, tarvittavien injektiopullojen määrän ja kustakin injektiopullosta annosteltavan tilavuuden. Määrät perustuvat pakastettujen CAR‑positiivisten elinkykyisten T‑solujen pitoisuuteen.
Apuaineet, joiden vaikutus tunnetaan:
Tämä lääkevalmiste sisältää 12,5 mg natriumia, 6,5 mg kaliumia ja 0,35 ml (7,5 % v/v) dimetyylisulfoksidia per injektiopullo (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusioneste, dispersio (infuusioneste).
Kliiniset tiedot
Käyttöaiheet
Breyanzi on tarkoitettu diffuusin suurisoluisen B‑solulymfooman (DLBCL), korkean maligniteettiasteen B‑solulymfooman (HGBCL), primaarisen välikarsinan suurisoluisen B‑solulymfooman (PMBCL) ja gradus 3B follikulaarisen lymfooman (FL3B) hoitoon aikuispotilailla, joiden sairaus on uusiutunut 12 kuukauden kuluessa ensilinjan kemoimmunoterapian päättymisestä tai ei reagoi ensilinjan kemoimmunoterapiaan.
Breyanzi on tarkoitettu uusiutuneen tai hoitoon reagoimattoman diffuusin suurisoluisen B‑solulymfooman (DLBCL), primaarisen välikarsinan suurisoluisen B‑solulymfooman (PMBCL) ja gradus 3B follikulaarisen lymfooman (FL3B) hoitoon aikuispotilailla kahden tai useamman systeemistä hoitoa sisältäneen hoitolinjan jälkeen.
Breyanzi on tarkoitettu uusiutuneen tai hoitoon reagoimattoman follikulaarisen lymfooman (FL) hoitoon aikuispotilailla kahden tai useamman systeemistä hoitoa sisältäneen hoitolinjan jälkeen.
Breyanzi on tarkoitettu uusiutuneen tai hoitoon reagoimattoman manttelisolulymfooman (MCL) hoitoon aikuispotilailla vähintään kahden systeemistä hoitoa sisältäneen hoitolinjan jälkeen, mukaan lukien Brutonin tyrosiinikinaasin (BTK) estäjä.
Ehto
CAR-T-soluhoito annetaan kvalifioidussa hoitokeskuksessa hematologisten syöpien hoitoon perehtyneen ja CAR-T-soluhoitojen antamiseen koulutetun lääkärin ohjauksessa ja valvonnassa.
Annostus ja antotapa
Breyanzi on annettava pätevässä hoitokeskuksessa.
Hoito on aloitettava hematologisten syöpien hoitoon perehtyneen ja Breyanzi-valmisteen antamiseen ja valmisteella hoitamiseen koulutetun terveydenhoitoalan ammattilaisen ohjauksessa ja valvonnassa.
Ennen Breyanzi-infuusion antamista on jokaista potilasta kohden oltava käytettävissä ainakin yksi annos tosilitsumabia ja ensiapuvälineet sytokiinioireyhtymän (cytokine release syndrome, CRS) varalta. Hoitokeskuksella on oltava käytettävissään tosilitsumabin lisäannos 8 tunnin sisällä edellisen annoksen antamisesta. Jos tosilitsumabia ei poikkeustapauksessa ole saatavilla Euroopan lääkeviraston saatavuushäiriöluettelossa ilmoitetun saatavuushäiriön takia, jokin muu sopiva vaihtoehto sytokiinioireyhtymän hoitamiseksi on oltava saatavilla tosilitsumabin sijaan ennen infuusion antamista.
Annostus
Breyanzi on tarkoitettu autologiseen käyttöön (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoito koostuu yhdestä infuusioannoksesta. Yksi annos on CAR‑positiivisia, elinkykyisiä T‑soluja sisältävä infuusiona annettava dispersio, joka on yhdessä tai useammassa injektiopullossa.
Tavoiteannos on 100 × 106 CAR‑positiivista, elinkykyistä T‑solua (CD4‑positiivisten ja CD8‑positiivisten solukomponenttien osuuksien tavoitesuhde on 1:1), ja vaihtelualue on 44–120 × 106 CAR‑positiivista elinkykyistä T‑solua. Katso annosta koskevat lisätiedot valmisteen mukana toimitetusta luovutussertifikaatista (RfIC).
Breyanzi-valmisteen saatavuus on varmistettava ennen lymfosyyttejä vähentävän kemoterapian aloittamista.
Potilaiden tila on arvioitava kliinisesti ennen lymfosyyttejä vähentävän kemoterapian ja Breyanzi-hoidon aloittamista, jotta voidaan varmistaa, ettei hoidon siirtämiselle ole syytä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Esihoito (lymfosyyttejä vähentävä kemoterapia)
Lymfosyyttejä vähentävää (nk. lymfodepletiivistä) kemoterapiaa, jonka muodostavat laskimoon infusoitavat syklofosfamidi 300 mg/m2/vrk ja fludarabiini 30 mg/m2/vrk, annetaan kolmen vuorokauden ajan. Katso syklofosfamidin ja fludarabiinin valmisteyhteenvedoista lisää tietoa annosmuutoksista potilaille, joilla on munuaisten vajaatoiminta.
Breyanzi on annettava 2–7 vuorokautta lymfosyyttejä vähentävän kemoterapian päättymisen jälkeen.
Jos lymfosyyttejä vähentävän kemoterapian päättymisen ja Breyanzi-infuusion välillä on yli kaksi viikkoa, potilaalle on annettava uudestaan lymfosyyttejä vähentävää kemoterapiaa ennen infuusion antamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Esilääkitys
On suositeltavaa, että esilääkitys parasetamolilla ja difenhydramiinilla (25–50 mg, laskimoon tai suun kautta) tai jollakin toisella H1‑antihistamiinilla annetaan 30–60 minuuttia ennen Breyanzi-valmisteen infusointia. Näin vähennetään infuusioreaktion todennäköisyyttä.
Systeemisten kortikosteroidien profylaktista käyttöä tulisi välttää, sillä ne saattavat häiritä Breyanzi-valmisteen aktiivisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infuusion jälkeinen seuranta
- Potilaiden vointi on tarkistettava 2–3 kertaa infuusiota seuraavan viikon aikana sytokiinioireyhtymän, neurologisten tapahtumien ja muiden haittatapahtumien merkkien ja oireiden varalta. Lääkärin on harkittava sairaalahoitoa ensimmäisten sytokiinioireyhtymän merkkien ja oireiden tai neurologisten tapahtumien ilmaantuessa.
- Ensimmäisen viikon jälkeen seurantaa on jatkettava lääkärin harkinnan mukaisella tiheydellä yhteensä vähintään kahden viikon ajan infuusion jälkeen.
- Potilaita on ohjeistettava pysymään pätevän hoitokeskuksen läheisyydessä vähintään kaksi viikkoa infuusion jälkeen.
Erityisryhmät
Potilaat, joilla on ihmisen immuunikatoviruksen (HIV), hepatiitti B -viruksen (HBV) tai hepatiitti C -viruksen (HCV) aiheuttama infektio
Kliinistä kokemusta ei ole potilaista, joilla on aktiivinen HIV‑, HBV‑ tai HCV‑infektio.
Ennen kuin soluja kerätään valmistusta varten, on tehtävä hoitosuositusten mukaiset seulontatestit HIV:n, aktiivisen HBV:n ja aktiivisen HCV:n varalta. Valmistamiseen ei hyväksytä leukafereesimateriaalia potilailta, joilla on aktiivinen HIV‑ tai aktiivinen HCV‑infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Heikentynyt munuaisten toiminta
Kliinistä kokemusta ei ole potilaista, joilla on heikentynyt munuaisten toiminta (kreatiniinipuhdistuma ≤ 30 ml/min).
Iäkkäät potilaat
Annoksen muuttaminen yli 65‑vuotiaille potilaille ei ole tarpeen.
Pediatriset potilaat
Breyanzi-valmisteen turvallisuutta ja tehoa lasten ja alle 18-vuoden ikäisten nuorten hoidossa ei ole varmistettu.
Antotapa
Breyanzi on tarkoitettu vain laskimonsisäiseen käyttöön.
Breyanzi-valmisteen valmistelu
Ennen valmisteen sulattamista on varmistettava, että potilaan henkilöllisyys vastaa kuljetuslaatikossa, ulkokotelossa ja luovutussertifikaatissa (RfIC) ilmoitettuja yksilöllisiä potilastunnisteita. Myös annettavien injektiopullojen kokonaismäärä on tarkistettava luovutussertifikaatissa (RfIC) olevista potilaskohtaisen etiketin tiedoista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). On otettava välittömästi yhteyttä yritykseen, jos etikettien ja potilastunnisteiden välillä on eroja.
Valmisteen antaminen
- Leukosyyttejä poistavaa suodatinta EI SAA käyttää.
- Varmista, että saatavilla on ennen infuusion antamista ja toipumisjakson ajan ensiapuvälineet ja tosilitsumabia tai muuta sopivaa vaihtoehtoa, jos tosilitsumabia ei poikkeustapauksessa ole saatavilla Euroopan lääkeviraston saatavuushäiriöluettelossa ilmoitetun saatavuushäiriön takia.
- Varmista, että potilaan henkilöllisyys vastaa luovutussertifikaatin (RfIC) mukana toimitetun ruiskun etiketin potilastunnisteita.
- Kun Breyanzi-valmisteen komponentit on vedetty ruiskuihin, anna valmiste mahdollisimman pian. Kokonaisaika siitä, kun valmiste poistetaan pakkassäilytyksestä, valmisteen antamiseen ei saa ylittää kahta tuntia.
Yksityiskohtaiset ohjeet, jotka koskevat Breyanzi-valmisteen valmistelua ja antamista sekä vahinkoaltistumisen tapahduttua tai Breyanzi-valmistetta hävitettäessä tehtäviä toimenpiteitä, on esitetty kohdassa Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys kohdassa Apuaineet mainituille apuaineille.
Lymfosyyttejä vähentävän kemoterapian vasta-aiheet on otettava huomioon.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Jäljitettävyysvaatimuksia, jotka koskevat solupohjaisia pitkälle kehitetyissä hoidoissa käytettäviä lääkkeitä, on noudatettava. Jäljitettävyyden varmistamiseksi valmisteen nimeä, eränumeroa ja hoidetun potilaan nimeä on säilytettävä 30 vuoden ajan valmisteen viimeisen käyttöpäivän jälkeen.
Autologinen käyttö
Breyanzi on tarkoitettu ainoastaan autologiseen käyttöön, eikä sitä saa missään tapauksessa antaa muille potilaille. Breyanzi-valmistetta ei saa antaa, jos valmisteen merkinnöissä ja luovutussertifikaatissa (RfIC) olevat tiedot eivät vastaa potilaan henkilötietoja.
Hoidon lykkäämisen syyt
Breyanzi-hoitoon liittyvien riskien vuoksi infuusion antoa on siirrettävä myöhäisempään ajankohtaan, jos potilaalla on jokin seuraavista tiloista:
- vakava, selvittämätön haittatapahtuma (etenkin keuhkoihin tai sydämeen liittyvä tapahtuma tai matala verenpaine), mukaan lukien aiemmista solunsalpaajahoidoista johtuvat haittatapahtumat
- aktiivinen hallitsematon infektio tai tulehdustila
- aktiivinen käänteishyljintäsairaus (GVHD).
Jos Breyanzi-infuusiota on lykättävä, ks. kohta Annostus ja antotapa.
Veren, elinten, kudosten ja solujen luovutus
Breyanzi-hoitoa saaneet potilaat eivät saa luovuttaa verta eivätkä elimiä, kudoksia tai soluja transplantaatiota varten.
Keskushermoston lymfooma
Breyanzi-valmisteen käytöstä ei ole kokemusta potilailla, joilla on primaarinen keskushermoston lymfooma. Breyanzi-valmisteen käytöstä potilailla, joilla on sekundaarinen keskushermoston lymfooma, on vain vähän kliinistä kokemusta (ks. kohta Farmakodynamiikka).
Aiempi CD19‑antigeeniin kohdistuva hoito
Breyanzi-valmisteen käytöstä on vain vähän kliinistä kokemuksia potilailla, jotka ovat aiemmin saaneet CD19‑antigeeniin kohdistuvaa hoitoa (ks. kohta Farmakodynamiikka). Breyanzi-hoitoa saaneista CD19‑negatiivisista potilaista on saatavilla vain vähän kliinistä tietoa. Potilaat, joilla on immunohistokemiallisesti CD19‑negatiivinen status, saattavat silti ilmentää CD19:ää. CD19‑negatiivisten potilaiden Breyanzi‑hoitoon liittyvät mahdolliset riskit ja hyödyt on punnittava.
Sytokiinioireyhtymä (CRS, cytokine release syndrome)
Breyanzi-infuusion jälkeen voi esiintyä sytokiinioireyhtymää, mukaan lukien hengenvaarallisia tai kuolemaan johtavia reaktioita. Potilailla, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon, mediaaniaika sytokiinioireyhtymän alkamiselle oli 4 päivää (vaihteluväli: 1–63 päivää; yläraja yhdellä potilaalla ilmoitetun kuumeettoman sytokiinioireyhtymän alkamisen mukaan). Potilailla, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon, mediaaniaika sytokiinioireyhtymän alkamiselle oli 4 päivää (vaihteluväli: 1–14 päivää). Potilailla, jotka olivat saaneet Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon, mediaaniaika sytokiinioireyhtymän alkamiselle oli 6 päivää (vaihteluväli: 1–17 päivää). Potilailla, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon, mediaaniaika sytokiinioireyhtymän alkamiselle oli 4 päivää (vaihteluväli: 1–10 päivää). Alle puolella kaikista Breyanzi-valmisteella hoidetuista potilaista ilmeni jonkinasteista sytokiinioireyhtymää (ks. kohta Haittavaikutukset).
Kliinisissä tutkimuksissa suuri kasvaintaakka ennen Breyanzi-infuusiota yhdistettiin suurempaan sytokiinioireyhtymän ilmenemiseen.
Breyanzi-infuusion jälkeistä sytokiinioireyhtymää hoidettiin tosilitsumabilla ja/tai kortikosteroideilla (ks. kohta Haittavaikutukset).
Sytokiinioireyhtymän seuranta ja hoito
Sytokiinioireyhtymä tunnistetaan kliinisen kuvan perusteella. Potilaat on arvioitava ja heitä on hoidettava muiden kuumeen, hypoksian ja matalan verenpaineen aiheuttajien varalta.
Ennen Breyanzi-infuusion antamista hoitokeskuksessa on oltava saatavilla ainakin yksi annos tosilitsumabia potilasta kohden. Hoitokeskuksella on oltava käytettävissään tosilitsumabin lisäannos 8 tunnin sisällä edellisen annoksen antamisesta. Jos tosilitsumabia ei poikkeustapauksessa ole saatavilla Euroopan lääkeviraston saatavuushäiriöluettelossa ilmoitetun saatavuushäiriön takia, hoitokeskuksella on oltava saatavilla jokin muu sopiva vaihtoehto sytokiinioireyhtymän hoitamiseksi tosilitsumabin sijaan. Potilaiden vointi on tarkistettava 2–3 kertaa Breyanzi-infuusiota seuraavan viikon aikana pätevässä hoitokeskuksessa sytokiinioireyhtymän merkkien ja oireiden varalta. Ensimmäisen viikon jälkeen seurantaa on jatkettava lääkärin harkinnan mukaisella tiheydellä vähintään kahden viikon ajan infuusion jälkeen. Potilaille ja heitä hoitaville henkilöille on kerrottava, että sytokiinioireyhtymä saattaa ilmetä viiveellä, ja kehotettava hakeutumaan välittömästi lääkäriin, jos potilailla ilmenee sytokiinioireyhtymän merkkejä tai oireita.
Jo ensimmäisten sytokiinioireyhtymän merkkien ilmetessä on aloitettava tukihoito sekä tosilitsumabin tai tosilitsumabin ja kortikosteroidien käyttö taulukon 1 mukaisesti. Breyanzi jatkaa monistumista tosilitsumabin ja kortikosteroidien antamisen jälkeen (ks. kohta Farmakokinetiikka).
Sydämen ja elinten toimintaa on seurattava tarkoin potilailla, joilla on sytokiinioireyhtymä, kunnes oireet häviävät. Kun kyseessä on vaikea tai hengenvaarallinen sytokiinioireyhtymä, tehohoitotason tarkkailua ja tukihoitoja on harkittava.
Hemofagosyyttisen lymfohistiosytoosin / makrofagiaktivaatio-oireyhtymän (HLH/MAS) tutkimista on harkittava potilailla, joilla on vaikea tai hoitoihin reagoimaton sytokiinioireyhtymä. Hemofagosyyttistä lymfohistiosytoosia / makrofagiaktivaatio-oireyhtymää on hoidettava hoitolaitoksen hoito-ohjeistuksen mukaisesti.
Jos epäillään sytokiinioireyhtymän kanssa samanaikaista neurologista haittatapahtumaa, sitä on hoidettava seuraavalla tavalla:
- Kortikosteroideilla taulukossa 1 esitetyn sytokiinioireyhtymän vaikeusasteen mukaisesti ja taulukossa 2 esitetyn neurologisen haittatapahtuman vaikeusasteen mukaisesti siten, että hoitosuosituksista valitaan aggressiivisemmat hoidot
- Tosilitsumabilla taulukossa 1 esitetyn sytokiinioireyhtymän vaikeusasteen mukaisesti
- Epilepsialääkkeillä taulukossa 2 esitetyn neurologisen haittatapahtuman vaikeusasteen mukaisesti.
Taulukko 1: Sytokiinioireyhtymän vaikeusasteen määrittäminen ja hoito-ohjeet
| Sytokiinioireyhtymän vaikeusastea | Tosilitsumabi | Kortikosteroiditb |
Aste 1 Kuume | Jos oireilu alkaa vähintään 72 tunnin kuluttua infuusiosta, hoidetaan oireenmukaisesti. Jos oireilu alkaa korkeintaan 72 tunnin kuluttua infuusiosta, on harkittava 8 mg/kg ‑tosilitsumabi-annoksen antamista 1 tunnin aikana laskimoon (annos korkeintaan 800 mg). | Jos oireilu alkaa vähintään 72 tunnin kuluttua infuusiosta, hoidetaan oireenmukaisesti. Jos oireilu alkaa korkeintaan 72 tunnin kuluttua infuusiosta, on harkittava 10 mg:n deksametasoniannoksen antamista laskimoon 24 tunnin välein. |
Aste 2 Oireet edellyttävät kohtalaisia hoitotoimenpiteitä, joilla saadaan vaste. Kuume, hapentarve alle 40 % (sisäänhengitysilman happipitoisuus, FiO2); tai matala verenpaine, joka reagoi nestehoitoon tai hoitoon yhdellä pieniannoksisella vasopressorilla; tai vaikeusasteen 2 elintoksisuus. | Annetaan tosilitsumabia 8 mg/kg laskimoon 1 tunnin aikana (annos korkeintaan 800 mg). | Jos oireilu alkaa vähintään 72 tunnin kuluttua infuusiosta, harkitaan 10 mg:n deksametasoniannoksen antamista laskimoon 12–24 tunnin välein. Jos oireilu alkaa korkeintaan 72 tunnin kuluttua infuusiosta, annetaan deksametasonia 10 mg laskimoon 12–24 tunnin välein. |
Jos paranemista ei havaita 24 tunnin kuluessa tai jos sytokiinioireyhtymä etenee nopeasti, toistetaan tosilitsumabin anto ja korotetaan deksametasonin annosta ja antokertoja (10–20 mg laskimoon 6–12 tunnin välein). Jos paranemista ei havaita tai jos sytokiinioireyhtymä jatkaa nopeaa etenemistä, nostetaan deksametasonin annos enimmäistasolle ja vaihdetaan tarvittaessa suuriannoksiseen metyyliprednisoloniin, jota annetaan 2 mg/kg. Kahden tosilitsumabiannoksen jälkeen harkitaan vaihtoehtoisen immunosuppressiivisen lääkkeen antamista. Tosilitsumabia saa antaa korkeintaan 3 annosta 24 tunnin kuluessa tai yhteensä enintään 4 annosta. | ||
Aste 3 Oireet edellyttävät aggressiivisia hoitotoimenpiteitä, joilla saadaan vaste. Kuume, hapentarve vähintään 40 % (sisäänhengitysilman happipitoisuus, FiO2) tai matala verenpaine, joka edellyttää hoitoa suuriannoksisella vasopressorilla tai useilla vasopressoreilla, tai vaikeusasteen 3 elintoksisuus tai vaikeusasteen 4 transaminaasipitoisuuksien suureneminen. | Asteen 2 ohjeiden mukaisesti. | Annetaan 10 mg deksametasonia laskimoon 12 tunnin välein. |
| Jos paranemista ei havaita 24 tunnin kuluessa tai jos sytokiinioireyhtymä etenee nopeasti, tosilitsumabin ja kortikosteroidien annosta nostetaan vaikeusasteen 2 ohjeiden mukaisesti. | ||
Aste 4 Hengenvaaralliset oireet. Edellyttää hengityslaitteen käyttöä tai jatkuvaa venovenoosista hemodialyysiä (CVVHD); tai vaikeusasteen 4 elintoksisuus (transaminaasipitoisuuksien suurenemista lukuun ottamatta). | Asteen 2 ohjeiden mukaisesti. | Annetaan 20 mg deksametasonia laskimoon 6 tunnin välein. |
| Jos paranemista ei havaita 24 tunnin kuluessa tai jos sytokiinioireyhtymä etenee nopeasti, siirrytään antamaan tosilitsumabia ja kortikosteroideja vaikeusasteen 2 ohjeiden mukaisesti. | ||
a Lee et al 2014.
b Jos kortikosteroidihoito aloitetaan, sitä jatketaan ainakin 3 annoksen ajan tai kunnes oireet häviävät kokonaan, jolloin kortikosteroidihoiton asteittaista lopettamista harkitaan.
Neurologiset haittavaikutukset
Neurologisia haittavaikutuksia, kuten immuuniefektorisoluihin liittyvää neurotoksisuusoireyhtymää (ICANS), jotka voivat olla kuolemaan johtavia tai hengenvaarallisia, ilmeni Breyanzi-hoidon jälkeen samanaikaisesti sytokiinioireyhtymän kanssa, sytokiinioireyhtymän mentyä ohi tai ilman sytokiinioireyhtymää. Potilailla, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon, mediaaniaika ensimmäisen tapahtuman alkamiselle oli 8 päivää (vaihteluväli: 1–63 päivää), potilailla, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon, mediaaniaika ensimmäisen tapahtuman alkamiselle oli 9 päivää (vaihteluväli: 1–66 päivää), potilailla, jotka olivat saaneet Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon, mediaaniaika ensimmäisen tapahtuman alkamiselle oli 8 päivää (vaihteluväli: 4–16 päivää), ja potilailla, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon, mediaaniaika ensimmäisen tapahtuman alkamiselle oli 8 päivää (vaihteluväli: 1–25 päivää). Yleisimpiin neurologisiin oireisiin kuuluivat enkefalopatia, vapina, afasia, delirium, huimaus ja päänsärky (ks. kohta Haittavaikutukset).
Neurologisten haittatapahtumien seuranta ja hoito
Potilaiden vointi on tarkistettava 2–3 kertaa infuusiota seuraavan viikon aikana pätevässä hoitokeskuksessa neurologisten haittatapahtumien merkkien ja oireiden varalta. Ensimmäisen viikon jälkeen seurantaa on jatkettava lääkärin harkinnan mukaisella tiheydellä yhteensä vähintään kahden viikon ajan infuusion jälkeen. Potilaille ja heitä hoitaville henkilöille on kerrottava, että neurologisia haittatapahtumia saattaa ilmetä viiveellä, ja kehotettava hakeutumaan välittömästi lääkäriin, jos potilailla ilmenee neurologisten haittatapahtumien merkkejä tai oireita.
Jos neurologista haittatapahtumaa epäillään, se on hoidettava taulukon 2 suositusten mukaisesti. Muut neurologisten oireiden syyt, mukaan lukien verisuonistosta johtuvat tapahtumat, on suljettava pois. Tehohoitoa täydentävää hoitoa on annettava vakavia tai hengenvaarallisia neurologisia haittatapahtumia hoidettaessa.
Jos epäillään neurologisen toksisuusreaktion kanssa samanaikaista sytokiinioireyhtymää, sitä on hoidettava seuraavalla tavalla:
- Kortikosteroideilla taulukossa 1 esitetyn sytokiinioireyhtymän vaikeusasteen mukaisesti ja taulukossa 2 esitetyn neurologisen haittatapahtuman vaikeusasteen mukaisesti siten, että hoitosuosituksista valitaan aggressiivisemmat hoidot
- Tosilitsumabilla taulukossa 1 esitetyn sytokiinioireyhtymän vaikeusasteen mukaisesti
- Epilepsialääkkeillä taulukossa 2 esitetyn neurologisen haittatapahtuman vaikeusasteen mukaisesti.
Taulukko 2: Neurologisen toksisuuden (ml. ICANS) vaikeusasteen määrittäminen ja hoito-ohjeet
| Neurologisen toksisuuden vaikeusaste (ml. esiintyvät oireet)a | Kortikosteroidit ja epilepsialääkkeet |
Aste 1* Lievä tai oireeton tai ICE-pisteytys 7–9b tai alentunut tajunnantasoc: herää spontaanisti | Aloitetaan väsyttämättömien epilepsialääkkeiden (kuten levetirasetaamin) käyttö kohtausten ennaltaehkäisemiseksi. Jos infuusiosta on kulunut vähintään 72 tuntia, potilasta tarkkaillaan. Jos infuusiosta on kulunut alle 72 tuntia, annetaan 10 mg deksametasonia laskimoon 12–24 tunnin välein 2–3 vuorokauden ajan. |
Aste 2* Kohtalainen tai ICE-pisteytys 3–6b tai alentunut tajunnantasoc: herää ääneen | Aloitetaan väsyttämättömien epilepsialääkkeiden (kuten levetirasetaamin) käyttö kohtausten ennaltaehkäisemiseksi. Annetaan 10 mg deksametasonia laskimoon 12 tunnin välein 2–3 vuorokauden ajan tai pidempään, jos oireet jatkuvat. Harkittava steroidien lopettamista asteittain, jos steroidialtistusta on kestänyt pidempään kuin 3 vuorokautta. Jos paranemista ei tapahdu 24 tunnin kuluessa tai jos neurologinen haittatapahtuma pahenee, lisätään deksametasonin annosta ja/tai antokertoja. Enimmäismääränä on 20 mg laskimoon 6 tunnin välein. Jos paranemista ei tapahdu 24 tunnin kuluessa, oireet pahenevat nopeasti tai jos ilmenee henkeä uhkaavia komplikaatioita, siirrytään antamaan metyyliprednisolonia (2 mg:n/kg latausannos, jota seuraa 2 mg/kg jaettuna neljään annokseen vuorokaudessa; lopetus asteittain 7 vuorokauden kuluessa). |
Aste 3* Vakava tai kliinisesti merkittävä, mutta ei välittömästi hengenvaarallinen; sairaalahoito tai pitkittyminen; invalidisoiva tai ICE-pisteytys 0–2b jos ICE-pisteytys on 0, mutta potilas on herätettävissä (esim. hereillä laaja-alainen afasia) ja kykenee suorittamaan arvioinnin tai alentunut tajunnantasoc: herää vain tuntoärsykkeeseen tai kouristuksiac, joko:
tai kohonnut ICPc: paikallisalkuinen/paikallinen edeema neurokuvantamisessa. | Aloitetaan väsyttämättömien epilepsialääkkeiden (kuten levetirasetaamin) käyttö kohtausten ennaltaehkäisemiseksi. Annetaan 10–20 mg deksametasonia laskimoon 8–12 tunnin välein. Kortikosteroideja ei suositella asteen 3 päänsärkyjen hoitoon, jos muita oireita ei ilmene. Jos paranemista ei tapahdu 24 tunnin kuluessa tai jos neurologinen haittatapahtuma pahenee, siirrytään antamaan metyyliprednisolonia (annos ja antokerrat asteen 2 ohjeiden mukaisesti). Jos epäillään aivoedeemaa, on harkittava hyperventilaatiota ja hyperosmolaarista hoitoa. Annetaan korkea-annoksista metyyliprednisolonia (1–2 g, toistetaan tarvittaessa 24 tunnin välein; lopetetaan asteittain kliinisen tarpeen mukaisesti) ja syklofosfamidia 1,5 g/m2. |
Aste 4* Hengenvaarallinen tai ICE-pisteytysb 0 tai alentunut tajunnantasoc, joko:
tai kouristuskohtauksetc, joko:
tai motoriset löydöksetc:
tai kohonnut ICP/aivoedeemac, jossa merkkejä/oireita, kuten:
| Aloitetaan väsyttämättömien epilepsialääkkeiden (kuten levetirasetaamin) käyttö kohtausten ennaltaehkäisemiseksi. Annetaan 20 mg deksametasonia laskimoon 6 tunnin välein. Jos paranemista ei tapahdu 24 tunnin kuluessa tai jos neurologinen haittatapahtuma pahenee, siirrytään antamaan metyyliprednisolonia (annos ja antokerrat asteen 2 ohjeiden mukaisesti). Jos epäillään aivoedeemaa, on harkittava hyperventilaatiota ja hyperosmolaarista hoitoa. Annetaan korkea-annoksista metyyliprednisolonia (1–2 g, toistetaan tarvittaessa 24 tunnin välein; lopetetaan asteittain kliinisen tarpeen mukaisesti) ja syklofosfamidia 1,5 g/m2. |
EEG = elektroenkefalogrammi; ICE = immuunijärjestelmän efektorisoluihin liittyvä enkefalopatia; ICP = kallonsisäinen paine.
* Asteen määritys NCI CTCAE:n tai ASTCT/ICANS:n mukaan
a Hoito määritetään vaikeimman tapahtuman mukaan, joka ei johdu mistään muusta syystä.
b Jos potilas on herätettävissä ja pystyy tekemään ICE-arvioinnin, arvioi: orientaatio (tietää vuoden, kuukauden, paikkakunnan, sairaalan = 4 pistettä); nimeäminen (nimeää 3 esinettä; osoittaa esimerkiksi kelloa, kynää, nappia = 3 pistettä); kehotusten noudattaminen (esim. ”näytä minulle 2 sormea” tai ”sulje silmäsi ja työnnä kieli ulos” = 1 piste); kirjoittaminen (kykenee kirjoittamaan normaalin virkkeen = 1 piste); ja tarkkaavaisuus (taaksepäin laskeminen sadasta kymmenen välein = 1 piste). Jos potilas ei ole herätettävissä eikä pysty suorittamaan ICE-arviointia (asteen 4 ICANS), pisteytys = 0 pistettä.
c Ei katsota johtuvan mistään muusta syystä.
Infektiot ja kuumeinen neutropenia
Breyanzi-valmistetta ei saa antaa potilaille, joilla on kliinisesti merkittäviä aktiivisia infektioita tai tulehdussairauksia. Potilailla on havaittu vakavia, myös hengenvaarallisia tai kuolemaan johtaneita, infektioita tällä lääkevalmisteella annetun hoidon jälkeen (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava infektioiden merkkien ja oireiden varalta ennen lääkevalmisteen antoa ja sen jälkeen, ja infektiot on hoidettava asianmukaisesti. Profylaktisia mikrobilääkkeitä annetaan hoitolaitoksen tavanomaisten ohjeiden mukaisesti.
Kuumeista neutropeniaa on havaittu potilailla Breyanzi-infuusion jälkeen (ks. kohta Haittavaikutukset), ja sitä saattaa ilmetä samanaikaisesti sytokiinioireyhtymän kanssa. Jos potilaalla havaitaan kuumeista neutropeniaa, infektio on arvioitava ja hoidettava laajakirjoisilla antibiooteilla, nesteytyksellä ja muilla tukihoidoilla kliinisen kuvan edellyttämällä tavalla.
Breyanzi-hoitoa saaneilla potilailla voi olla tavanomaista suurempi vaikea-asteisen tai kuolemaan johtavan COVID‑19-infektion riski. Potilaille pitää kertoa, että sen ehkäisytoimenpiteet ovat tärkeitä.
Virusten uudelleenaktivoituminen
Immunosuppressiopotilailla voi ilmetä virusten (kuten hepatiitti B -viruksen [HBV] ja ihmisen herpesvirus 6:n [HHV‑6] sekä John Cunningham -viruksen [JC]) uudelleenaktivoitumista.
Virusten uudelleenaktivoitumisen aiheuttamat oireet voivat vaikeuttaa ja viivästyttää CAR‑T‑soluihin liittyvien haittatapahtumien diagnosointia ja asianmukaista hoitoa. Asianmukainen diagnostinen arviointi on tehtävä, jotta tällaiset oireet voidaan erottaa CAR‑T‑soluihin liittyvistä haittatapahtumista.
Hepatiitti B -virus voi aktivoitua uudelleen potilailla, joita hoidetaan B‑soluja vastaan kohdistetuilla lääkkeillä, ja aktivoituminen voi joissakin tapauksissa johtaa fulminanttiin hepatiittiin, maksan vajaatoimintaan ja kuolemaan. Potilaille, joilla on aiemmin ollut hepatiitti B -virus, suositellaan ennaltaehkäisevää antiviraalista estohoitoa, jotta voidaan ehkäistä hepatiitti B -viruksen uudelleen aktivoituminen Breyanzi-hoidon aikana ja sen jälkeen (ks. kohta Farmakodynamiikka).
JC-viruksen uudelleenaktivoitumista, joka johtaa progressiiviseen multifokaaliseen leukoenkefalopatiaan (PML), on raportoitu potilailla, joita on hoidettu Breyanzi-valmisteella ja jotka ovat saaneet aiempaa hoitoa myös muilla immunosuppressiivisilla lääkkeillä. Kuolemaan johtaneita tapauksia on raportoitu.
Serologiset tutkimukset
HBV‑, HCV‑ ja HIV‑seulontakokeet on tehtävä ennen solujen keräämistä valmisteen valmistusta varten (ks. kohta Annostus ja antotapa).
Pitkittyneet sytopeniat
Potilailla saattaa ilmetä pitkittyneitä sytopenioita useiden viikkojen ajan lymfosyyttejä vähentävän kemoterapian ja Breyanzi-infuusion jälkeen (ks. kohta Haittavaikutukset). Verenkuvaa on seurattava ennen Breyanzi-valmisteen antoa ja sen jälkeen. Pitkittyneitä sytopenioita on hoidettava hoitosuositusten mukaisesti.
Hypogammaglobulinemia
Breyanzi-hoitoa saaneilla potilailla voi ilmetä hypogammaglobulinemiaan johtavaa B‑soluaplasiaa. Breyanzi-valmistetta saaneilla potilailla on havaittu hyvin yleisesti hypogammaglobulinemiaa (ks. kohta Haittavaikutukset). Immunoglobuliinipitoisuuksia on tarkkailtava hoidon jälkeen ja hoidettava hoitosuositusten mukaisesti, mukaan lukien infektioita ehkäisevien varotoimien, antibioottiprofylaksian ja/tai immunoglobuliinikorvaushoidon käyttö.
Sekundaariset maligniteetit mukaan luettuina T-soluperäiset sekundaariset syövät
Breyanzi-hoitoa saaneille potilaille saattaa kehittyä sekundaarisia maligniteetteja. T-soluperäisiä syöpiä on raportoitu sen jälkeen, kun hematologisia syöpiä on hoidettu BCMA- tai CD19‑kohdennetulla CAR-T‑soluhoidolla, kuten Breyanzi-valmisteella. T-soluperäisiä syöpiä, myös CAR-positiivisia syöpiä, on raportoitu viikkoja ja jopa useita vuosia BCMA- tai CD19-kohdennetun CAR-T-soluhoidon jälkeen. Kuolemaan johtaneita tapauksia on esiintynyt. Potilaita on seurattava elinikäisesti sekundaaristen syöpien varalta. Jos potilaalla ilmenee T‑soluperäinen sekundaarinen syöpä, on otettava yhteyttä yritykseen, jolta saa ohjeet kasvainnäytteiden keräämiseen testausta varten.
Tuumorilyysioireyhtymä
CAR‑T‑soluhoitoa saaneilla potilailla saattaa ilmetä tuumorilyysioireyhtymää. Tuumorilyysioireyhtymän riskin minimoimiseksi potilaille, joiden virtsahappopitoisuudet ovat koholla tai joiden kasvaintaakka on suuri, on annettava allopurinolia tai muuta estohoitoa ennen Breyanzi-infuusiota. Potilasta on tarkkailtava tuumorilyysioireyhtymän merkkien ja oireiden varalta, ja niitä on hoidettava hoitosuositusten mukaisesti.
Yliherkkyysreaktiot
Allergisia reaktioita saattaa ilmetä Breyanzi-infuusion yhteydessä. Vakavia yliherkkyysreaktioita, kuten anafylaksiaa, saattaa aiheutua dimetyylisulfoksidista.
Taudinaiheuttajien siirtyminen
Vaikka testeillä tarkistetaan, että Breyanzi on steriili eikä sisällä mykoplasmaa, tartunnanaiheuttajien siirtymisen vaara on olemassa. Tämän vuoksi Breyanzi-valmistetta antavien terveydenhuollon ammattilaisten on tarkkailtava potilaita hoidon jälkeen infektio-oireiden varalta ja tarvittaessa annettava asianmukainen hoito.
Interferenssi virustestauksessa
Koska Breyanzi-valmisteen valmistuksessa käytettävässä lentivirusvektorissa on pieni määrä lyhyitä, HI‑viruksen kanssa identtisiä geneettisen tiedon jaksoja, jotkin HIV‑nukleiinihappotestit (NAT‑testit) saattavat antaa väärän positiivisen tuloksen.
Aiempi kantasolusiirto (käänteishyljintäsairaus, GVHD)
Hoidon antamista allogeenisen kantasolusiirron saaneille potilaille, joilla on aktiivinen akuutti tai krooninen käänteishyljintäsairaus, ei suositella, koska Breyanzi saattaa pahentaa käänteishyljintää.
Pitkäaikainen seuranta
Potilaiden odotetaan tulevan mukaan rekisteritutkimukseen, jotta heitä voidaan seurata ja jotta voidaan selvittää paremmin Breyanzi-valmisteen turvallisuutta ja tehoa pitkällä aikavälillä.
Apuaineet
Tämä lääkevalmiste sisältää 12,5 mg natriumia per injektiopullo, joka vastaa 0,6 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Tämä lääkevalmiste sisältää 0,2 mmol (tai 6,5 mg) kaliumia per injektiopullo. Potilaiden, joilla on munuaisten vajaatoimintaa tai ruokavalion kaliumrajoitus, on otettava tämä huomioon.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ihmisillä ei ole tehty.
Epidermaaliseen kasvutekijäreseptoriin kohdistuvat monoklonaaliset vasta-aineet (anti-EGFR-vasta-aineet)
Monoklonaalisten anti-EGFR-vasta-aineiden käyttö Breyanzi-hoidon jälkeen voi vaikuttaa CAR‑T‑solujen pitkäaikaiseen pysyvyyteen. Tietoa on kuitenkin vain rajallisesti saatavilla monoklonaalisten anti-EGFR-vasta-aineiden kliinisestä käytöstä Breyanzi-hoitoa saaneilla potilailla.
Eläviä taudinaiheuttajia sisältävät rokotteet
Eläviä viruksia sisältävien rokotteiden käytön turvallisuutta Breyanzi-hoidon aikana tai sen jälkeen ei ole tutkittu. Varotoimenpiteenä suositellaan, että eläviä taudinaiheuttajia sisältäviä rokotteita ei anneta vähintään 6 viikkoon ennen lymfosyyttejä vähentävän kemoterapian aloittamista, Breyanzi-hoidon aikana eikä ennen kuin immuniteetti on elpynyt hoidon jälkeen.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy miehillä ja naisilla
Naisille, jotka voivat tulla raskaaksi, täytyy tehdä raskaustesti mahdollisen raskauden selvittämiseksi ennen Breyanzi-hoidon aloittamista.
Tarkista fludarabiinin ja syklofosfamidinin valmisteyhteenvedoista tiedot tehokkaan ehkäisyn tarpeesta potilailla, jotka saavat lymfosyyttejä vähentävää kemoterapiaa.
Altistusta koskevia tietoja ei ole riittävästi, jotta voitaisiin antaa suosituksia ehkäisyn kestosta Breyanzi-hoidon jälkeen.
Raskaus
Ei ole olemassa tietoja lisokabtageenimaraleuseelin käytöstä raskaana oleville naisille. Ei ole tutkittu lisääntymis- ja kehitystoksisuutta koskevilla eläinkokeilla, voiko valmisteen antaminen raskaana olevalle naiselle vahingoittaa sikiötä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Ei tiedetä, voiko lisokabtageenimaraleuseeli siirtyä sikiöön. Toimintamekanismin perusteella voidaan olettaa, että jos transduktoituja soluja kulkeutuu istukan läpi, ne saattavat aiheuttaa sikiötoksisuutta, mukaan lukien B‑solulinjan lymfosytopeniaa. Siksi Breyanzi-valmistetta ei suositella raskaana oleville naisille eikä naisille, jotka voivat tulla raskaaksi eivätkä käytä ehkäisyä. Raskaana oleville naisille on kerrottava mahdollisista sikiöön kohdistuvista riskeistä. Raskaudesta Breyanzi-hoidon antamisen jälkeen on keskusteltava hoitavan lääkärin kanssa.
Breyanzi-hoitoa saaneen äidin vastasyntyneen lapsen immunoglobuliinitasojen ja B‑solujen määrittämistä on harkittava.
Imetys
Ei tiedetä, erittyykö lisokabtageenimaraleuseeli ihmisen rintamaitoon tai siirtyykö se imetettävään lapseen. Imettävälle naiselle on kerrottava imetettävään lapseen mahdollisesti kohdistuvasta riskistä.
Hedelmällisyys
Lisokabtageenimaraleuseelin vaikutuksesta hedelmällisyyteen ei ole saatavilla tietoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Breyanzi-valmisteella voi olla huomattava vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Breyanzi-valmisteen käyttöön liittyvien mahdollisten neurologisten vaikutusten vuoksi, mukaan lukien psyykkisen tilan muutokset tai kouristuskohtaukset, Breyanzi-valmistetta saavan potilaan on vältettävä ajamista tai raskaiden tai mahdollisesti vaarallisten koneiden käyttöä vähintään 4 viikon ajan Breyanzi-infuusion jälkeen tai pidempään lääkärin harkinnan mukaan.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
LBCL
Potilaat, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluiseen B‑solulymfoomaan (LBCL)
Tässä kohdassa kuvattavia haittavaikutuksia havaittiin populaatiossa, jossa 177 potilasta sai Breyanzi-hoitoa infuusiona kolmessa yhdistetyssä tutkimuksessa (TRANSFORM [BCM‑003], PILOT [017006] ja TRANSCEND WORLD [JCAR017‑BCM‑001, kohortti 2]).
Yleisimmät haittavaikutukset (kaikki vaikeusasteet) olivat neutropenia (71 %), anemia (45 %), sytokiinioireyhtymä (45 %) ja trombosytopenia (43 %).
Yleisimpiä vakavia haittavaikutuksia olivat sytokiinioireyhtymä (12 %), neutropenia (3 %), bakteeri-infektiot (3 %), tarkemmin määrittelemättömän patogeenin aiheuttama infektio (3 %), trombosytopenia (2 %), kuumeinen neutropenia (2 %), kuume (2 %), afasia (2 %), päänsärky (2 %), sekavuustila (2 %), keuhkoembolia (2 %), anemia (1 %), maha-suolikanavan yläosan verenvuoto (1 %) ja vapina (1 %).
Yleisimpiä vähintään vaikeusasteen 3 haittavaikutuksia olivat neutropenia (68 %), trombosytopenia (33 %), anemia (31 %), lymfopenia (17 %), leukopenia (17 %), kuumeinen neutropenia (5 %) ja bakteeri-infektiot (5 %).
Potilaat, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluiseen B‑solulymfoomaan (LBCL)
Tässä kohdassa kuvattavia haittavaikutuksia havaittiin populaatiossa, jossa 384 potilasta sai Breyanzi-hoitoa infuusiona neljässä yhdistetyssä tutkimuksessa (TRANSCEND [017001], TRANSCEND WORLD [JCAR017‑BCM‑001, kohortit 1, 3 ja 7], PLATFORM [JCAR017‑BCM‑002] ja OUTREACH [017007].
Yleisimmät haittavaikutukset (kaikki vaikeusasteet) olivat neutropenia (68 %), anemia (45 %), sytokiinioireyhtymä (38 %), uupumus (37 %) ja trombosytopenia (36 %).
Yleisimpiä vakavia haittavaikutuksia olivat sytokiinioireyhtymä (18 %), tarkemmin määrittelemättömän patogeenin aiheuttama infektio (6 %), kuume (4 %), enkefalopatia (4 %), kuumeinen neutropenia (4 %), neutropenia (3 %), trombosytopenia (3 %), afasia (3 %), bakteeri-infektiot (3 %), vapina (3 %), sekavuustila (3 %), anemia (2 %) ja matala verenpaine (2 %).
Yleisimpiä vähintään vaikeusasteen 3 haittavaikutuksia olivat neutropenia (64 %), anemia (34 %), trombosytopenia (29 %), leukopenia (25 %), lymfopenia (9 %), tarkemmin määrittelemättömän patogeenin aiheuttama infektio (8 %) ja kuumeinen neutropenia (8 %).
FL
Tässä kohdassa kuvatut haittavaikutukset havaittiin 130 potilaalla, jotka saivat Breyanzi-infuusion TRANSCEND‑FL-tutkimuksessa (FOL-001).
Yleisimmät haittavaikutukset (kaikki vaikeusasteet) olivat neutropenia (68 %), sytokiinioireyhtymä (58 %), anemia (40 %), päänsärky (29 %), trombosytopenia (29 %) ja ummetus (21 %).
Yleisimpiä vakavia haittavaikutuksia olivat sytokiinioireyhtymä (9 %), afasia (4 %), kuumeinen neutropenia (3 %), kuume (2 %) ja vapina (2 %).
Yleisimpiä vähintään vaikeusasteen 3 haittavaikutuksia olivat neutropenia (61 %), leukopenia (12 %), lymfopenia (12 %), trombosytopenia (12 %) ja anemia (10 %).
MCL
Tässä kohdassa kuvatut haittavaikutukset havaittiin 88 potilaalla, jotka saivat Breyanzi-infuusion TRANSCEND-tutkimuksen MCL-kohortissa (017001).
Yleisimmät haittavaikutukset (kaikki vaikeusasteet) olivat sytokiinioireyhtymä (61 %), neutropenia (59 %), anemia (44 %), uupumus (35 %), trombosytopenia (30 %) ja päänsärky (23 %).
Yleisimpiä vakavia haittavaikutuksia olivat sytokiinioireyhtymä (24 %), sekavuustila (6 %), kuume (3 %), psyykkisen tilan muutokset (2 %), enkefalopatia (2 %), ylähengitystieinfektio (2 %) ja pleuraeffuusio (2 %).
Yleisimpiä vähintään vaikeusasteen 3 haittavaikutuksia olivat neutropenia (56 %), anemia (38 %), trombosytopenia (25 %), hypofosfatemia (9 %) ja leukopenia (7 %).
Taulukkomuotoinen luettelo haittavaikutuksista
Haittavaikutusten esiintymistiheydet perustuvat seitsemän tutkimuksen (TRANSCEND [017001], mukaan lukien LBCL- ja MCL-kohortit, TRANSCEND WORLD [JCAR017‑BCM‑001, kohortit 1, 2, 3 ja 7], PLATFORM [JCAR017‑BCM‑002], OUTREACH [017007], TRANSFORM [BCM‑003], PILOT [017006] ja TRANSCEND‑FL [JCAR017-FOL-001]) yhdistettyihin tietoihin myyntiin tulon jälkeisistä ilmoituksista sekä 779 aikuispotilaasta, jotka olivat saaneet annoksen lisokabtageenimaraleuseelia. Kliinisissä tutkimuksissa havaittujen haittavaikutusten esiintymistiheydet perustuvat mistä tahansa syystä aiheutuneiden haittatapahtumien esiintymistiheyksiin, jolloin osaan tietyn haittavaikutuksen tapahtumista voi olla muitakin syitä.
Ilmoitetut haittavaikutukset on esitetty alla. Haittavaikutukset on esitetty MedDRA-elinjärjestelmäluokan ja esiintyvyyden mukaisesti jaoteltuina. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3: Breyanzi-hoidon yhteydessä todetut lääkkeen haittavaikutukset
| Elinjärjestelmäluokka | Esiintymistiheys | Haittavaikutus |
| Infektiota | Hyvin yleinen | Tarkemmin määrittelemättömän patogeenin aiheuttamat infektiot |
| Yleinen | Bakteeri-infektiot Virusinfektiot Sieni-infektiot | |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Melko harvinainen | T-soluperäinen sekundaarinen syöpä |
| Veri ja imukudos | Hyvin yleinen | Neutropenia Anemia Trombosytopenia Leukopenia Lymfopenia |
| Yleinen | Kuumeinen neutropenia Hypofibrinogenemiaw | |
| Melko harvinainen | Pansytopenia | |
| Immuunijärjestelmä | Hyvin yleinen | Sytokiinioireyhtymä |
| Yleinen | Hypogammaglobulinemiav | |
| Melko harvinainen | Hemofagosyyttinen lymfohistiosytoosi | |
| Aineenvaihdunta ja ravitsemus | Yleinen | Hypofosfatemia |
| Melko harvinainen | Tuumorilyysioireyhtymä | |
| Psyykkiset häiriöt | Hyvin yleinen | Unettomuus |
| Yleinen | Deliriumb Ahdistuneisuus | |
| Hermosto | Hyvin yleinen | Päänsärkyc Enkefalopatiad Huimause Vapinaf |
| Yleinen | Afasiag Perifeerinen neuropatiah Näköhäiriöi Ataksiaj Makuhäiriök Pikkuaivo-oireyhtymäl | |
| Melko harvinainen | Aivoverenkiertohäiriöm Kouristuksetn Pareesio Aivoedeema | |
| Tuntematon | Immuuniefektorisoluihin liittyvä neurotoksisuusoireyhtymä* | |
| Sydän | Hyvin yleinen | Takykardia |
| Yleinen | Rytmihäiriöp | |
| Melko harvinainen | Kardiomyopatia | |
| Verisuonisto | Hyvin yleinen | Matala verenpaine |
| Yleinen | Korkea verenpaine Tromboosiq | |
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Yskä |
| Yleinen | Dyspnear Pleuraeffuusio Hypoksia | |
| Melko harvinainen | Keuhkoedeema | |
| Ruoansulatuselimistö | Hyvin yleinen | Pahoinvointi Ripuli Ummetus Vatsakipu Oksentelu |
| Yleinen | Maha-suolikanavan verenvuotos | |
| Iho ja ihonalainen kudos | Hyvin yleinen | Ihottuma |
| Munuaiset ja virtsatiet | Yleinen | Akuutti munuaisvauriot |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Uupumus Kuume Edeemau |
| Yleinen | Vilunväristykset | |
| Vammat ja myrkytykset | Yleinen | Infuusioon liittyvät reaktiot |
* Haittavaikutustietoa ei kerätty kliinisissä tutkimuksissa systemaattisesti.
a Infektiot on ryhmitelty MedDRA:n ylätason ryhmätermien mukaan.
b Deliriumiin kuuluvat kiihtyneisyys, delirium, harhaluulot, desorientaatio, aistiharhat, näköharhat, ärtyneisyys ja levottomuus
c Päänsärkyyn kuuluvat päänsärky, migreeni, silmämigreeni ja sivuontelopäänsärky.
d Enkefalopatiaan kuuluvat amnesia, kognitiiviset häiriöt, sekavuustilat, depersonalisaatio-/derealisaatiohäiriöt, alentunut tajunnantaso, huomiokyvyn häiriöt, enkefalopatia, tunneilmaisun huomattava latistuminen, letargia, leukoenkefalopatia, tajuttomuus, muistin heikentyminen, henkisen suorituskyvyn heikkeneminen, psyykkisen tilan muutokset, harhaluuloisuus, uneliaisuus ja tokkuraisuus.
e Huimaukseen kuuluvat huimaus, asentohuimaus, pyörrytys ja pyörtyminen.
f Vapinaan kuuluvat essentiaalinen vapina, kohdennusvapina, lepovapina ja vapina.
g Afasiaan kuuluvat afasia, puheen sekavuus, dysartria, ääntöhäiriö, hidas puhe ja puhehäiriö.
h Perifeeriseen neuropatiaan kuuluvat demyelinoiva polyneuropatia, hyperestesia, hypoestesia, hyporeflexia, proprioseptiikan menetys, perifeerinen neuropatia, parestesia, perifeerinen motorinen neuropatia, perifeerinen sensorinen neuropatia ja tuntopuutokset ja rannekanavaoireyhtymä.
i Näköhäiriöön kuuluvat sokeus, toisen silmän sokeus, katsehalvaus, mydriaasi, silmävärve, näön sumeneminen, näkökentän puutokset ja näön heikkeneminen.
j Ataksiaan kuuluvat ataksia ja kävelyn häiriö.
k Makuhäiröön kuuluvat dysgeusia ja makuhäiriö.
l Pikkuaivo-oireyhtymään kuuluvat tasapainohäiriö, dysdiadokokineesi, dyskinesia, dysmetria ja heikentynyt käden ja silmän koordinaatio.
m Aivoverenkiertohäiriöön kuuluvat aivoinfarkti, aivolaskimosinustromboosi, embolia ja aivoinfarkti, kallonsisäinen verenvuoto ja ohimenevä aivoverenkiertohäiriö.
n Kouristuksiin kuuluvat kouristukset ja status epileptikus.
o Pareesiin kuuluvat kasvohermohalvaus, kasvojen halvaus ja äänihuulihalvaus.
o Rytmihäiriöön kuuluvat rytmihäiriö, eteisvärinä, täydellinen eteis-kammiokatkos, toisen asteen eteis-kammiokatkos, supraventrikulaarinen takykardia, lisälyönnit, kammiolisälyönnit ja kammiotakykardia.
q Tromboosiin kuuluvat syvä laskimotromboosi, embolia, keuhkoemblia, tromboosi, onttolaskimotromboosi, laskimotromboosi ja raajan laskimotukos.
qrDyspneaan kuuluvat akuutti hengitysvajaus, dyspnea, rasitukseen liittyvä hengenahdistus ja hengitysvajaus.
s Maha-suolikanavan verenvuotoon kuuluvat mahaverenvuoto, mahahaavaverenvuoto, maha-suolikanavan verenvuoto, ulosteen verisyys, maha-suolikanavan alaosan verenvuoto, meleena, verenvuoto peräsuolesta ja maha-suolikanavan yläosan verenvuoto.
t Akuuttiin munuaisvaurioon kuuluvat akuutti munuaisvaurio, veren kreatiniiniarvon nousu, glomerulusten suodatusnopeuden lasku, munuaisten vajaatoiminta, heikentynyt munuaisten toiminta ja munuaisvaurio.
tuEdeemaan kuuluvat kasvojen edeema, yleistynyt edeema, paikallinen edeema, edeema, sukuelinten edeema, perifeerinen edeema, perifeerinen turvotus, kivespussin edeema, turvotus, kasvojen turvotus.
v Hypogammaglobulinemiaan kuuluvat pienentynyt veren immunoglobuliini A:n arvo, pienentynyt veren immunoglobuliini G:n arvo, pienentynyt veren immunoglobuliini M:n arvo, hypogammaglobulinemia ja vähentyneet immunoglobuliinit.
w Hypofibrinogenemiaan kuuluvat pienentynyt veren fibrinogeenipitoisuus ja hypofibrinogenemia
Valikoitujen haittavaikutusten kuvaus
Sytokiinioireyhtymä
Sytokiinioireyhtymää ilmeni 45 prosentilla potilaista, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon. Näistä potilaista 1 prosentilla ilmeni vaikeusasteen 3 sytokiinioireyhtymää. Mediaaniaika sytokiinioireyhtymän alkamiselle oli 4 päivää (vaihteluväli: 1–63 päivää, yläraja yhdellä potilaalla ilmoitetun kuumeettoman sytokiinioireyhtymän alkamisen mukaan), ja sytokiinioireyhtymän mediaanikesto oli 4 päivää (vaihteluväli: 1–16 päivää).
Yleisimpiä sytokiinioireyhtymän ilmentymiä olivat kuume (44 %), matala verenpaine (12 %), vilunväristykset (5 %), hypoksia (5 %), takykardia (4 %), päänsärky (3 %) ja uupumus (2 %).
Suurisoluista B‑solulymfoomaa (LBCL) koskeneissa kliinisissä tutkimuksissa 42 potilaalle 177:stä (24 %) annettiin tosilitsumabia ja/tai kortikosteroidia sytokiinioireyhtymän hoitoon Breyanzi-infuusion jälkeen. 18 potilasta (10 %) sai vain tosilitsumabia, 24 potilasta (14 %) sai tosilitsumabia ja kortikosteroideja. Yhdenkään potilaan sytokiinioireyhtymää ei hoidettu ainoastaan kortikosteroideilla.
Sytokiinioireyhtymää ilmeni 38 prosentilla potilaista, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon. Näistä potilaista 2 prosentilla ilmeni vaikeusasteen 3 tai 4 (vaikeaa tai henkeä uhkaavaa) sytokiinoireyhtymää. Niistä potilaista, jotka kuolivat saatuaan Breyanzi-valmistetta, neljällä oli kuoleman aikaan sytokiinioireyhtymä. Mediaaniaika sytokiinioireyhtymän alkamiselle oli 4 päivää (vaihteluväli: 1–14 päivää), ja sytokiinioireyhtymän mediaanikesto oli 5 päivää (vaihteluväli: 1–17 päivää).
Yleisimpiä sytokiinioireyhtymän ilmentymiä olivat kuume (38 %), matala verenpaine (18 %), takykardia (13 %), vilunväristykset (9 %) ja hypoksia (8 %).
Suurisoluista B‑solulymfoomaa (LBCL) koskeneissa kliinisissä tutkimuksissa 74 potilaalle 384:stä (19 %) annettiin tosilitsumabia ja/tai kortikosteroidia sytokiinioireyhtymän hoitoon Breyanzi-infuusion jälkeen. 37 potilasta (10 %) sai vain tosilitsumabia, 29 potilasta (8 %) sai tosilitsumabia ja kortikosteroideja ja 8 potilasta (2 %) sai ainoastaan kortikosteroideja.
Sytokiinioireyhtymää ilmeni 58 prosentilla potilaista, jotka olivat saaneet Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon. Näistä potilaista 0,8 prosentilla ilmeni vaikeusasteen 3 sytokiinioireyhtymää. Mediaaniaika sytokiinioireyhtymän alkamiselle oli 6 päivää (vaihteluväli: 1–17 päivää), ja sytokiinioireyhtymän mediaanikesto oli 3 päivää (vaihteluväli: 1–10 päivää).
Yleisimpiä sytokiinioireyhtymän ilmentymiä olivat kuume (57 %), matala verenpaine (14 %), vilunväristykset (4 %), hypoksia (2 %) ja takykardia (0,8 %).
Follikulaarisen lymfooman (FL) kliinisessä tutkimuksessa 33 potilaalle 130:stä (25 %) annettiin tosilitsumabia ja/tai kortikosteroidia sytokiinioireyhtymän hoitoon Breyanzi-infuusion jälkeen. 18 potilasta (14 %) sai vain tosilitsumabia, 15 potilasta (12 %) sai tosilitsumabia ja kortikosteroideja. Yhdenkään potilaan sytokiinioireyhtymää ei hoidettu ainoastaan kortikosteroideilla. Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Sytokiinioireyhtymää ilmeni 61 prosentilla potilaista, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon. Näistä potilaista 1 prosentilla ilmeni vaikeusasteen 3 tai 4 sytokiinioireyhtymää. Mediaaniaika sytokiinioireyhtymän alkamiselle oli 4 päivää (vaihteluväli: 1–10 päivää), ja sytokiinioireyhtymän mediaanikesto oli 4 päivää (vaihteluväli: 1–14 päivää).
Yleisimpiä sytokiinioireyhtymän ilmentymiä olivat kuume (60 %), matala verenpaine (22 %), hypoksia (11 %), takykardia (10 %), vilunväristykset (8 %), päänsärky (8 %), pahoinvointi (3 %) ja hengenahdistus (2 %).
TRANSCEND-tutkimuksen manttelisolulymfooman (MCL) kohortissa 24 potilaalle 88:sta (27 %) annettiin tosilitsumabia ja/tai kortikosteroidia sytokiinioireyhtymän hoitoon Breyanzi-infuusion jälkeen. 15 potilasta (17 %) sai vain tosilitsumabia ja 8 potilasta (9 %) sai tosilitsumabia ja kortikosteroideja. Yhden potilaan (1 %) sytokiinioireyhtymää hoidettiin ainoastaan kortikosteroideilla.
Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Neurologiset haittavaikutukset
Potilailla, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon, CAR‑T‑soluihin liittyviä neurologisia haittatapahtumia ilmeni tutkijan arvion mukaan 18 prosentilla Breyanzi-hoitoa saaneista potilaista, mukaan lukien vaikeusasteen 3 haittatapahtumat 5 prosentilla potilaista. Mediaaniaika ensimmäisen tapahtuman alkamiselle oli 8 päivää (vaihteluväli: 1–63 päivää), ja 84 prosenttia kaikista neurologisista haittatapahtumista ilmeni 2 viikon kuluessa Breyanzi-infuusiosta. Neurologisten haittatapahtumien mediaanikesto oli 6 päivää (vaihteluväli: 1–89 päivää).
Yleisimpiä neurologisia haittatapahtumia olivat enkefalopatia (10 %), vapina (8 %), afasia (5 %), huimaus (2 %) ja päänsärky (1 %).
Potilailla, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon, CAR‑T‑soluihin liittyviä neurologisia haittatapahtumia ilmeni tutkijan arvion mukaan 26 prosentilla Breyanzi-hoitoa saaneista potilaista, mukaan lukien vaikeusasteen 3 ja 4 haittatapahtumat 10 prosentilla potilaista. Mediaaniaika ensimmäisen tapahtuman alkamiselle oli 9 päivää (vaihteluväli: 1–66 päivää), ja 83 prosenttia kaikista neurologisista haittatapahtumista ilmeni 2 viikon kuluessa Breyanzi-infuusiosta. Neurologisten haittatapahtumien mediaanikesto oli 10 päivää (vaihteluväli: 1–84 päivää).
Yleisimpiä neurologisia haittatapahtumia olivat enkefalopatia (18 %), vapina (9 %), afasia (8 %), delirium (7 %), päänsärky (4 %), ataksia (3 %) ja huimaus (3 %). Breyanzi-hoitoa saaneilla potilailla ilmeni myös kouristuksia (2 %:lla) ja aivoedeemaa (0,3 %:lla).
Potilailla, jotka olivat saaneet Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon, CAR‑T‑soluihin liittyviä neurologisia haittatapahtumia ilmeni tutkijan arvion mukaan 16 prosentilla Breyanzi-hoitoa saaneista potilaista, mukaan lukien vaikeusasteen 3 haittatapahtumat 3 prosentilla potilaista. Mediaaniaika ensimmäisen tapahtuman alkamiselle oli 8 päivää (vaihteluväli: 4–16 päivää), ja 95 prosenttia kaikista neurologisista haittatapahtumista ilmeni 2 viikon kuluessa Breyanzi-infuusiosta. Neurologisten haittatapahtumien mediaanikesto oli 3 päivää (vaihteluväli: 1–17 päivää).
Yleisimpiä neurologisia haittatapahtumia olivat vapina (8 %), afasia (8 %), enkefalopatia (5 %), delirium (4 %) ja päänsärky (2 %). Ks. neurologisten haittatapahtumien tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Potilailla, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon, CAR‑T‑soluihin liittyviä neurologisia haittatapahtumia ilmeni tutkijan arvion mukaan 31 prosentilla Breyanzi-hoitoa saaneista potilaista, mukaan lukien vaikeusasteen 3 ja 4 haittatapahtumat 9 prosentilla potilaista. Mediaaniaika ensimmäisen tapahtuman alkamiselle oli 8 päivää (vaihteluväli: 1–25 päivää), ja 100 prosenttia kaikista neurologisista haittatapahtumista ilmeni 8 viikon kuluessa Breyanzi-infuusiosta. Neurologisten haittatapahtumien mediaanikesto oli 5 päivää (vaihteluväli: 1–45 päivää).
Yleisimpiä neurologisia haittatapahtumia olivat enkefalopatia (26 %), vapina (7 %), delirium (6 %), afasia (6 %), päänsärky (5 %) ja huimaus (3 %). Breyanzi-hoitoa saaneilla potilailla on ilmennyt myös kouristuksia (1 %:lla).
Ks. neurologisten haittatapahtumien tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Kuolemaan johtaneita ICANS-oireyhtymätapauksia on ilmoitettu myyntiin tulon jälkeen.
Kuumeinen neutropenia ja infektiot
Kuumeista neutropeniaa on havaittu Breyanzi-hoidon jälkeen 7 %:lla ja 9 %:lla niistä potilaista, jotka olivat saaneet Breyanzi-valmistetta yhden aiemman hoitolinjan ja kahden tai useamman ensisijaisen hoitolinjan jälkeen suurisoluiseen B‑solulymfoomaan (LBCL), 5 %:lla niistä potilaista, jotka olivat saaneet Breyanzi-valmistetta follikulaariseen lymfoomaan (FL), ja 6 %:lla niistä potilaista, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfoomaan (MCL).
Infektioita (kaikki vaikeusasteet) ilmeni 25 %:lla niistä potilaista, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon. Vähintään vaikeusasteen 3 infektioita ilmeni 10 %:lla potilaista. Vähintään vaikeusasteen 3 infektioita, joiden aiheuttaja oli tarkemmin määrittelemätön patogeeni, ilmeni 3 %:lla potilaista, bakteeri-infektioita 5 %:lla potilaista, virusinfektioita 2 %:lla potilaista ja sieni-infektioita ei ilmennyt yhdelläkään potilaalla.
Potilailla, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon, infektioita (kaikki vaikeusasteet) ilmeni 38 %:lla potilaista. Vähintään vaikeusasteen 3 infektioita ilmeni 12 %:lla potilaista. Vähintään vaikeusasteen 3 infektioita, joiden aiheuttaja oli tarkemmin määrittelemätön patogeeni, ilmeni 8 %:lla potilaista, bakteeri-infektioita ilmeni 4 %:lla ja virus- ja sieni-infektioita ilmeni 1 %:lla potilaista.
Infektioita (kaikki vaikeusasteet) ilmeni 20 %:lla niistä potilaista, jotka olivat saaneet Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon. Vaikeusasteen 3 infektioita ilmeni 5 %:lla potilaista. Vähintään vaikeusasteen 3 infektioita, joiden aiheuttaja oli tarkemmin määrittelemätön patogeeni, ilmeni 4 %:lla potilaista, bakteeri-infektioita 2 %:lla potilaista, virusinfektioita 1 %:lla potilaista ja sieni-infektioita ei ilmennyt yhdelläkään potilaalla.
Infektioita (kaikki vaikeusasteet) ilmeni 35 %:lla niistä potilaista, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon. Vähintään vaikeusasteen 3 infektioita ilmeni 15 %:lla potilaista. Vähintään vaikeusasteen 3 infektioita, joiden aiheuttaja oli tarkemmin määrittelemätön patogeeni, ilmeni 6 %:lla potilaista, bakteeri-infektioita 5 %:lla potilaista, virusinfektioita 5 %:lla potilaista ja sieni-infektioita 1 %:lla potilaista.
Opportunistisia infektioita (kaikki vaikeusasteet) havaittiin 2 %:lla niistä 117:sta Breyanzi-hoitoa saaneista potilaista, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon. Vähintään vaikeusasteen 3 opportunistisia infektioita ilmeni 0,6 %:lla näistä potilaista. Opportunistisia infektioita (kaikki vaikeusasteet) havaittiin 3 %:lla niistä 384:stä Breyanzi-hoitoa saaneesta potilaasta, jotka olivat saaneet kahta tai useampaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon. Vähintään vaikeusasteen 3 opportunistisia infektioita ilmeni 1 %:lla näistä potilaista. Opportunistisia infektioita (kaikki vaikeusasteet) havaittiin 0,8 %:lla 130:stä Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon saaneista potilaista. Vähintään vaikeusasteen 3 opportunistisia infektioita ei ilmennyt yhdelläkään potilaalla. Opportunistisia infektioita (kaikki vaikeusasteet) havaittiin 1 %:lla 88:sta Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon saaneista potilaista. Kaikissa tapauksissa opportunistisen infektion vaikeusaste oli vähintään 3.
Niillä 177:llä Breyanzi-hoitoa saaneella potilaalla, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman hoitoon, ilmoitettiin kaksi kuolemaan johtanutta infektiotapausta. Yhdistetyissä LBCL-tutkimuksissa 384:llä Breyanzi-hoitoa saaneella potilaalla, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman (LBCL) hoitoon, ilmoitettiin yhteensä neljä kuolemaan johtanutta infektiota. Näistä yksi tapaus ilmoitettiin kuolemaan johtaneena opportunistisena infektiona. Kuolemaan johtaneita infektioita ei ilmoitettu 130 potilaalla, jotka olivat saaneet Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon. Niillä 88:lla potilaalla, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon, ilmoitettiin kaksi kuolemaan johtanutta infektiota.
Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Pitkittyneet sytopeniat
35 %:lla potilaista, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluiseen B‑solulymfoomaan, ilmeni 35 päivää Breyanzi-hoidon jälkeen vähintään vaikeusasteen 3 sytopenioita, kuten trombosytopeniaa (28 %), neutropeniaa (26 %) ja anemiaa (9 %).
TRANSFORM-, PILOT- ja TRANSCEND WORLD (kohortti 2) -tutkimuksissa hoidetuista yhteensä 177 potilaasta niillä potilailla, joiden sytopenian seurannan laboratoriotulokset olivat saatavilla ja joilla oli laboratoriotutkimusten mukaan päivänä 35 ja päivänä 29 vaikeusasteen 3–4 trombosytopenia (n = 50), vaikeusasteen 3–4 neutropenia (n = 46) tai vaikeusasteen 3–4 anemia (n = 15), haittavaikutuksen väistymisen mediaaniaika (minimi, maksimi) oli päivinä mitattuna seuraava: trombosytopenia 32 päivää (4, 309), neutropenia 32 päivää (8, 339) ja anemia 22 päivää (4, 64). Haittavaikutuksen väistymisellä tarkoitetaan sitä, että sytopenia lieveni vaikeusasteelle 2 tai tätä lievemmäksi.
38 %:lla potilaista, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman hoitoon, ilmeni Breyanzi-infuusion jälkeisenä päivänä 29 vähintään vaikeusasteen 3 sytopenioita, kuten trombosytopeniaa (31 %:lla), neutropeniaa (21 %:lla) ja anemiaa (7 %:lla).
TRANSCEND-, TRANSCEND WORLD- (kohortit 1, 3 ja 7), PLATFORM- ja OUTREACH-tutkimuksissa hoidetuista yhteensä 384 potilaasta niillä potilailla, joiden sytopenian seurannan laboratoriotulokset olivat saatavilla ja joilla oli laboratoriotutkimusten mukaan päivänä 29 vaikeusasteen 3–4 trombosytopenia (n = 117), vaikeusasteen 3–4 neutropenia (n = 80) tai vaikeusasteen 3–4 anemia (n = 27), haittavaikutuksen väistymisen mediaaniaika (minimi, maksimi) oli päivinä mitattuna seuraava: trombosytopenia 30 päivää (2, 329); neutropenia 29 päivää (3, 337) ja anemia 15 päivää (3, 78). Haittavaikutuksen väistymisellä tarkoitetaan sitä, että sytopenia lieveni vaikeusasteelle 2 tai tätä lievemmäksi.
22 %:lla potilaista, jotka olivat saaneet Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon, ilmeni 29 päivää Breyanzi-hoidon jälkeen vähintään vaikeusasteen 3 sytopenioita, kuten trombosytopeniaa (15 %), neutropeniaa (15 %) ja anemiaa (5 %). Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
TRANSCEND‑FL-tutkimuksessa hoidetuista yhteensä 130 potilaasta niillä potilailla, joiden sytopenian seurannan laboratoriotulokset olivat saatavilla ja joilla oli laboratoriotutkimusten mukaan päivänä 29 vaikeusasteen 3–4 trombosytopenia (n = 19), vaikeusasteen 3–4 neutropenia (n = 20) tai vaikeusasteen 3–4 anemia (n = 6), haittavaikutuksen väistymisen mediaaniaika (minimi, maksimi) oli päivinä mitattuna seuraava: trombosytopenia 36 päivää (16, 694), neutropenia 30 päivää (5, 110) ja anemia 36 päivää (8, 64). Haittavaikutuksen väistymisellä tarkoitetaan sitä, että sytopenia lieveni vaikeusasteelle 2 tai tätä lievemmäksi.
40 %:lla potilaista, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon, ilmeni 29 päivää Breyanzi-hoidon jälkeen vähintään vaikeusasteen 3 sytopenioita, kuten trombosytopeniaa (32 %), neutropeniaa (24 %) ja anemiaa (5 %).
TRANSCEND‑tutkimuksen MCL-kohortissa hoidetuista yhteensä 88 potilaasta niillä potilailla, joiden sytopenian seurannan laboratoriotulokset olivat saatavilla ja joilla oli laboratoriotutkimusten mukaan päivänä 29 vaikeusasteen 3–4 trombosytopenia (n = 28), vaikeusasteen 3–4 neutropenia (n = 21) tai vaikeusasteen 3–4 anemia (n = 4), haittavaikutuksen väistymisen mediaaniaika (minimi, maksimi) oli päivinä mitattuna seuraava: trombosytopenia 30 päivää (5, 302), neutropenia 30 päivää (8, 275) ja anemia 18 päivää (9, 32). Haittavaikutuksen väistymisellä tarkoitetaan sitä, että sytopenia lieveni vaikeusasteelle 2 tai tätä lievemmäksi.
Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Hypogammaglobulinemia
Yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman hoitoon saaneilla potilailla hypogammaglobulinemiaa ilmeni 7 %:lla potilaista. Kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman hoitoon saaneilla potilailla hypogammaglobulinemiaa ilmeni 11 %:lla potilaista. Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon saaneilla potilailla hypogammaglobulinemiaa ilmeni 2 %:lla potilaista. Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon saaneilla potilailla hypogammaglobulinemiaa ilmeni 7 %:lla potilaista. Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Immunogeenisuus
Breyanzi voi aiheuttaa vasta-aineiden muodostumista tätä lääkevalmistetta kohtaan. Breyanzi-valmisteen humoraalista immunogeenisuutta on mitattu määrittämällä anti‑CAR-vasta-aineet ennen valmisteen antoa ja annon jälkeen. Potilailla, jotka olivat saaneet yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman hoitoon (TRANSFORM- ja PILOT-tutkimukset ja TRANSCEND WORLD ‑tutkimuksen kohortti 2), Breyanzi-hoitoa edeltäviä lääkevasta-aineita (anti-therapeutic antibodies; ATA) havaittiin 0,6 %:lla (1/172) potilaista ja hoidon aiheuttamia vasta-aineita havaittiin 19 %:lla (32/172) potilaista. Yhdistetyissä tutkimuksissa potilailla, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman hoitoon (TRANSEND-tutkimus ja TRANSCEND WORLD ‑tutkimus, kohortit 1 ja 3), 9 %:lla (29/309) potilaista havaittiin lääkevasta-aineita ennen Breyanzi-valmisteen antoa ja Breyanzi-hoidon aiheuttamia tai lisäämiä vasta-aineita havaittiin 16 %:lla (48/304) potilaista. Potilailla, jotka olivat saaneet Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon (TRANSCEND‑FL), Breyanzi-hoitoa edeltäviä lääkevasta-aineita (anti-therapeutic antibodies; ATA) havaittiin 1,6 %:lla (2/124) potilaista ja hoidon aiheuttamia tai vahvistamia vasta-aineita havaittiin 26,8 %:lla (33/123) potilaista. Potilailla, jotka olivat saaneet Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon (TRANSCEND‑MCL-kohortti), Breyanzi-hoitoa edeltäviä lääkevasta-aineita (anti-therapeutic antibodies; ATA) havaittiin 13 %:lla (11/88) potilaista ja hoidon aiheuttamia tai vahvistamia vasta-aineita havaittiin 20 %:lla (17/86) potilaista. Koska potilaita, joilla oli lääkevasta-aineita, oli niin vähän tutkimustasolla, lääkevasta-aineiden vaikutuksesta tehoon, turvallisuuteen ja farmakokinetiikkaan ei voida tehdä johtopäätöksiä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Breyanzi-valmisteen yliannostuksesta ei ole kliinisistä tutkimuksista saatua tietoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut antineoplastiset lääkeaineet, ATC-koodi: L01XL08
Vaikutusmekanismi
Breyanzi on CD19‑antigeeniin kohdistuva immunologinen soluhoito, joka sisältää geenimuunneltuja autologisia T‑soluja. Solut annetaan hoidon yhteydessä ennalta määriteltynä koostumuksena CD8‑positiivisten ja CD4‑positiivisten T‑solujen annosvaihtelun vähentämiseksi. Kimeerinen antigeenireseptori (CAR) koostuu hiiren yksiketjuisesta vaihtelevasta vasta-ainefragmentista (FMC63:n monoklonaalinen vasta-aine; scFv), joka on yhdistetty IgG4:n sarana-alueeseen, CD28‑transmembraanidomeeniin, 4‑1BB (CD137) -kostimulaattoridomeeniin ja CD3‑zeeta-aktivointidomeeniin. CD3‑zeeta-signalointi on tärkeää T‑soluaktivaatiossa ja syöpäsoluun kohdentuvan aktiivisuuden kannalta, kun taas 4‑1BB (CD137) -signalointi edistää Breyanzi-valmisteessa solujen lisääntymistä ja elinkyvyn jatkuvuutta (ks. kohta Farmakokinetiikka).
Kun kimeerinen antigeenireseptori (CAR) sitoutuu B‑solujen ja kasvainsolujen pinnalla esiintyvään CD19‑antigeeniin, se saa CAR‑T‑solut aktivoitumaan, proliferoitumaan, erittämään proinflammatorisia sytokiinejä ja tappamaan kohdesoluja sytotoksisesti.
Kliininen teho ja turvallisuus
TRANSFORM
Breyanzi-valmisteen tehoa ja turvallisuutta verrattiin standardihoitoon (standard of care, SOC) satunnaistetussa, avoimessa, rinnakkaisryhmillä tehdyssä vaiheen 3 monikeskustutkimuksessa TRANSFORM (BCM‑003). Tutkimukseen osallistui aikuispotilaita, joilla oli suurisoluinen non‑Hodgkin-tyyppinen B‑solulymfooma, joka oli primaaristi refraktaarinen ensimmäiselle hoitolinjalle tai uusiutunut 12 kuukauden kuluessa ensimmäisen hoitolinjan jälkeen, ja jotka olivat soveluvia hematopoieettiseen kantasolusiirtoon (HSCT). Tutkimuksessa käytetty standardihoito koostui salvage-tyyppisestä immunokemoterapiasta, jonka jälkeen annettiin suuriannoksista kemoterapiaa (HDCT) ja tehtiin autologinen hematopoieettinen kantasolusiirto. Tutkimuksessa mukana olleilla potilailla oli WHO:n vuoden 2016 luokituksen mukainen muutoin määrittämätön (NOS) diffuusi suurisoluinen B‑solulymfooma (DLBCL) (de novo tai muuntunut indolentista non-Hodginin lymfoomasta); korkean maligniteettiasteen B‑solulymfooma (HGBCL), jossa oli histologisesti määritetty DLBC sekä MYC‑ ja BCL2‑ ja/tai BCL6‑geenien uudelleenryhmittyminen (rearrangement) (double hit / triple hit ‑lymfooma [DHL/THL]); primaarinen välikarsinan suurisoluinen B‑solulymfooma (PMBCL); T‑solu-/histiosyyttirikas B‑solulymfooma (THRBCL) tai gradus 3B follikulaarinen lymfooma (FL3B). Tutkimukseen osallistuvien potilaiden Eastern Cooperative Oncology Group (ECOG) ‑suorituskykyluokka oli ≤ 1. Potilaita, joilla oli sekundaarinen keskushermostolymfooma, voitiin ottaa mukaan BCM‑003-tutkimukseen, jos yksittäisen potilaan hyöty-riskisuhde oli tutkijan arvion mukaan positiivinen.
Tutkimukseen ottamisen ja tutkimuksen ulkopuolelle jättämisen kriteerit valittiin niin, että mukaan otettujen potilaiden elintoiminnot ja veriarvot vastasivat hematopoieettisen kantasolusiirtoon vaadittavia arvoja. Tutkimuksen ulkopuolelle jätettiin potilaat, joiden kreatiniinipuhdistuma oli alle 45 ml/min, alaniiniaminotransferaasiarvo (ALAT) oli > 5 kertaa normaalin yläraja (ULN) tai vasemman kammion ejektiofraktio (LVEF) < 40 % ja absoluuttinen neutrofiilien määrä (ANC) oli < 1,0 × 109 solua/l ja trombosyyttien määrä < 50 × 109 solua/l, kun tauti ei ollut levinnyt luuytimeen.
Potilaat satunnaistettiin (1:1) saamaan joko Breyanzi-hoitoa tai standardihoitoa. Satunnaistaminen ositettiin ensilinjan hoidon vasteen ja sekundaarisen ikävakioidun indeksin (secondary age adjusted international prognostic index, sAAIPI-luokitus) (0–1 vs 2–3) perusteella. Breyanzi-haaran potilaille annettiin lymfosyyttejä vähentävää kemoterapiaa, johon kuului fludarabiinia 30 mg/m2/vrk and syklofosfamidia 300 mg/m2/vkr samanaikaisesti kolmen päivän ajan, minkä jälkeen potilaille annettiin Breyanzi-infuusio 2–7 päivää lymfosyyttejä vähentävän kemoterapian päättymisen jälkeen.
Siltahoitona annettava kemoterapia oli sallittu tutkimuksen Breyanzi-haarassa afereesin ja lymfosyyttejä vähentävän kemoterapian aloituksen välillä. Siltahoitona annettiiin yksi sykli immunokemoterapiaa (eli rituksimabia, deksametasonia, sytarabiinia ja sisplatiinia [R‑DHAP], rituksimabia, ifosfamidia, karboplatiini ja etoposidia [R‑ICE] tai rituksimabia, gemsitabiinia, deksametasonia ja sisplatiinia [R‑GDP]). Kaikki standardihoitoa saavien haaraan satunnaistetut potilaat saivat kolme sykliä salvage-tyyppistä immunokemoterapiaa (kuten R‑DHAP, R‑ICE tai R‑GDP). Potilaille, jotka olivat kolmen syklin jälkeen saavuttaneet vasteen (täydellinen vaste [CR] tai osittainen vaste [PR]), annettiin suuriannoksista kemoterapiaa (HDCT) ja tehtiin autologinen kantasolusiirto. Standardihoitoa saaneille potilaille voitiin antaa Breyanzi-hoitoa, jos he eivät saavuttaneet täydellistä tai osittaista vastetta saatuaan kolme sykliä salvage-tyyppistä immunokemoterapiaa, jos heidän sairautensa eteni milloin tahansa tutkimuksen aikana tai jos potilaan oli aloitettava uusi hoito tehoon liittyvien huolenaiheiden takia.
Breyanzi-haaraan satunnaistetusta 92 potilaasta 58 potilasta (63 %) sai taudin kontrolloimiseen tarkoittettua syöpähoitoa (siltahoito), 89 potilasta (97 %) sai Breyanzi-hoitoa ja 1 potilas (1 %) sai valmistetta, joka ei ollut vaatimusten mukainen (non‑conforming product). Potilaista kaksi ei saanut Breyanzi-hoitoa. Näistä kahdesta (2 %) potilaasta yksi (1 %) ei saanut Breyanzi-hoitoa valmistusvirheen takia ja yksi (1 %) potilas perui suostumuksensa ennen hoitoa. Breyanzi-valmisteen mediaaniannos oli 99,9 × 106 CAR‑positiivista elinkykyistä T‑solua (vaihteluväli: 97–103 × 106 CAR‑positiivista elinkykyistä T‑solua).
Standardihoitohaaraan satunnaistetusta 92 potilaasta 91 potilasta (99 %) aloitti hoidon. Yksi potilas (1 %) perui suostumuksensa ennen hoidon aloittamista. 43 potilaalle (47 %) annettiin immunokemoterapiaa ja suuriannoksista kemoterapiaa ja tehtiin kantasolusiirto. 58 potilasta (63 %) sai Breyanzi-hoitoa sen jälkeen, kun standardihoito ei ollut toiminut.
Tehon analyysit perustuvat hoitoaikeen mukaiseen (ITT) analyysijoukkoon (n = 184), joka määriteltiin kaikiksi potilaiksi, jotka oli satunnaistettu jompaankumpaan hoitohaaraan.
Mediaaniaika leukafereesistä valmisteen saatavuuteen oli 26 päivää (vaihteluväli: 19–84 päivää), ja mediaaniaika leukafereesistä infuusioon oli 36 päivää (vaihteluväli: 25–91 päivää).
Taulukossa 4 on yhteenveto lähtötilanteen potilas- ja sairaustiedoista TRANSFORM-tutkimuksessa.
Taulukko 4: Lähtötilanteen demografiset ja sairautta koskevat tiedot TRANSFORM-tutkimuksessa (hoitoaikeen [intention-to-treat, ITT] mukainen analyysijoukko)
| Ominaisuus | Breyanzi (N = 92) | Standardihoito (N = 92) |
Iän mediaani, vuosina (vaihteluväli) ≥ 65 – < 75 vuotta, n (%) ≥ 75 vuotta, n (%) | 60,0 (20, 74) 36 (39,1) 0 | 58,0 (26, 75) 23 (25,0) 2 (2,2) |
Sukupuoli, n (%) Mies Nainen |
44 (47,8) 48 (52,2) |
61 (66,3) 31 (33,7) |
ECOG-toimintakykyluokka (seulontavaiheessa) ECOG 0, n (%) ECOG 1, n (%) |
48 (52,2) 44 (47,8) |
57 (62,0) 35 (38,0) |
Taudin histologinen alatyyppi, n (%) Diffuusi suurisoluinen B‑solulymfooma (DLBCL), muutoin määrittämätön (NOS) DLBCL, joka on muuntunut indolentista lymfoomasta Korkean maligniteettiasteen B‑solulymfooma (HGBCL) Primaarinen välikarsinan B‑solulymfooma (PMBCL) Gradus 3B follikulaarinen lymfooma (FL3B) T‑solu-/histiosyyttirikas B‑solulymfooma (THRBCL) |
53 (57,6)
7 (7,6) 22 (23,9) 8 (8,7) 1 (1,1) 1 (1,1) |
50 (54,3)
8 (8,7) 21 (22,8) 9 (9,8) 0 4 (4,3) |
| Kemoterapiaan reagoimatona, n (%) | 26 (28,3) | 18 (19,6) |
| Refraktaarinenb, n (%) | 67 (72,8) | 70 (76,1) |
| Uusiutunutc, n (%) | 25 (27,2) | 22 (23,9) |
| Taudin leviäminen keskushermostoon vahvistettu, n (%) | 1 (1,1) | 3 (3,3) |
| Ei saavuttanut koskaan täydellistä vastetta aiemmissa hoidoissa, n (%) | 62 (67,4) | 64 (69,6) |
a Kemoterapiaan reagoimattoman taudin määritelmänä oli, että potilaalla oli vakaa tauti (SD) tai etenevä tauti (PD) viimeiseen kemoterapiaa sisältävään hoito-ohjelmaan asti.
b Taudin tila oli refraktaarinen, jos potilaan tauti määritettiin vakaaksi taudiksi (SD) tai eteneväksi taudiksi (PD) tai jos potilas saavutti osittaisen vasteen (PR) tai jos potilas saavutti täydellisen vasteen (CR), minkä jälkeen tauti uusiutui kolmen kuukauden kuluessa.
c Taudin tila oli uusiutunut, jos potilas saavutti täydellisen vasteen (CR), minkä jälkeen tauti uusiutui 3–12 kuukauden kuluessa.
Tutkimus osoitti Breyanzi-haaraan satunnaistetuilla potilailla standardihoitohaaraan satunnaistettuihin potilaisiin verrattuna tilastollisesti merkitsevää paranemista niin ensisijaisen päätetapahtuman kuin toissijaisten päätetapahtumienkin suhteen. Ensisijainen päätetapahtuma oli tapahtumavapaa elinaika (event free survival, EFS) ja tärkeimpiä toissijaisia päätetapahtumia olivat täydellisen vasteen saavuttaneiden osuus (complete response, CR) ja etenemisvapaa elinaika (progression-free survival, PFS). Teho perustui riippumattoman arviointiryhmän (IRC) vuoden 2014 Lugano-kriteerien mukaan arvioituun tapahtumavapaaseen elinaikaan. Tapahtumavapaa elinaika määriteltiin ajaksi satunnaistamisesta johonkin seuraavista tapahtumista (mikä tahansa tapahtui ensin): mistä tahansa syystä aiheutunut kuolema, taudin eteneminen, täydellisen tai osittaisen vasteen saavuttamatta jättäminen 9 viikkoa satunnaistamisen jälkeen (kolmen salvage-tyyppisen immunokemoterapiasyklin ja viisi viikkoa Breyanzi-infuusion jälkeen) tai uuden antineoplastisen hoidon aloittaminen tilanteessa, jossa on tehosta aiheutuvia huolenaiheita. Ennalta määritetyn välianalyysin ajankohtana, jolloin oli saavutettu 80 prosenttia kokonaistavoitteena olevista tapahtumista ja tutkimuksen aikaisen (on-study) seuranta-ajan mediaani oli 6,2 kuukautta (vaihteluväli 0,9–20 kuukautta), Breyanzi-hoitoa saaneilla potilailla oli tilastollisesti merkitsevää paranemista etenemisvapaassa elinajassa verrattuna standardihoitoa saaneisiin potilaisiin (HR = 0,349 [95 %:n CI: 0,229; 0,530], yksitahoinen p‑arvo < 0,0001). P‑arvoa verrattiin allokoituun α 0,012:een ennalta määritettyä välianalyysia varten.
Breyanzi-hoito osoittautui paremmaksi kuin standardihoito diffuusissa suurisoluisessa B‑solulymfoomassa (DLBCL) (n = 60, HR: 0,357 [95 %:n CI: 0,204; 0,625]) ja korkean maligniteettiasteen B‑solulymfoomassa (HGBCL) (n = 22, HR: 0,413 [95 %:n CI: 0,189; 0,904]).
Lopullisen analyysin tulokset (esitetty taulukossa 5 ja kuvassa 1), kun tutkimuksen aikaisen (on-study) seuranta-ajan mediaani oli 33,86 kuukautta (vaihteluväli 0,9–53,0 kuukautta), olivat yhdenmukaiset sekä väli- että primaarianalyysin kanssa.
Taulukko 5: TRANSFORM-tutkimus: kokonaisvasteen saavuttaneiden osuus, tapahtumavapaa elinaika ja etenemisvapaa elinaika sekä kokonaiselinaika potilailla, joilla on refraktaarinen tai uusiutunut suurisoluinen B‑solulymfooma (hoitoaikeen [intention-to-treat, ITT] mukainen analyysijoukko)
| Tulosa | Breyanzi-haara (N = 92) | Standardihoitohaara (N = 92) |
| Tapahtumavapaa elinaika, (kuukausina) | ||
| Tapahtumien määrä n, (%) | 48 (52,2) | 73 (79,3) |
| Mediaani [95 %:n CI]b | 29,5 (9,5; NR) | 2,4 (2,2; 4,9) |
| Riskitiheyssuhde [95 %:n CI]c | 0,375 [0,259; 0,542] | |
| Täydellisen vasteen saavuttaneiden osuus | ||
| n (%) | 68 (73,9) | 40 (43,5) |
| Kaksitahoinen [95 %:n CI] | [63,7; 82,5] | [33,2; 54,2] |
| Etenemisvapaa elinaika, (kuukausina) | ||
| Tapahtumien määrä n, (%) | 41 (44,6) | 54 (58,7) |
| Mediaani [95 %:n CI]b | NR (12,6; NR) | 6,2 (4,3; 8,6) |
| Riskitiheyssuhde [95 %:n CI]c | 0,422 [0,279; 0,639] | |
| Kokonaiselinaika (OS), (kuukausina) | ||
| Tapahtumien määrä n, (%) | 34 (37,0) | 42 (45,7) |
| Mediaani [95 %:n CI]b | NR (42,8; NR) | NR (18,2; NR) |
| Riskitiheyssuhde [95 %:n CI]c | 0,757 [0,481; 1,191] | |
NR = ei saavutettu (not reached). CI = luottamusväli (confidence interval).
a Luganon kriteerien (2014) perusteella riippumattoman arviointiryhmän (IRC) arvion mukaan.
b Kaplan–Meier-arvio.
c Perustuu stratifioituun Coxin suhteellisen vaaran malliin.
d Greenwoodin kaava
Breyanzi-haaran 92 potilaasta 80 potilasta sai vasteen (68 täydellisen vasteen,12 osittaisen vasteen), jolloin kokonaisvasteen saavuttaneiden osuus oli 87 %.
Kuva 1: Kaplan–Meier-kuvaaja tapahtumavapaasta eliniästä riippumattoman arviointiryhmän (IRC) arvioimana (hoitoaikeen [intention-to-treat, ITT] mukainen analyysijoukko)
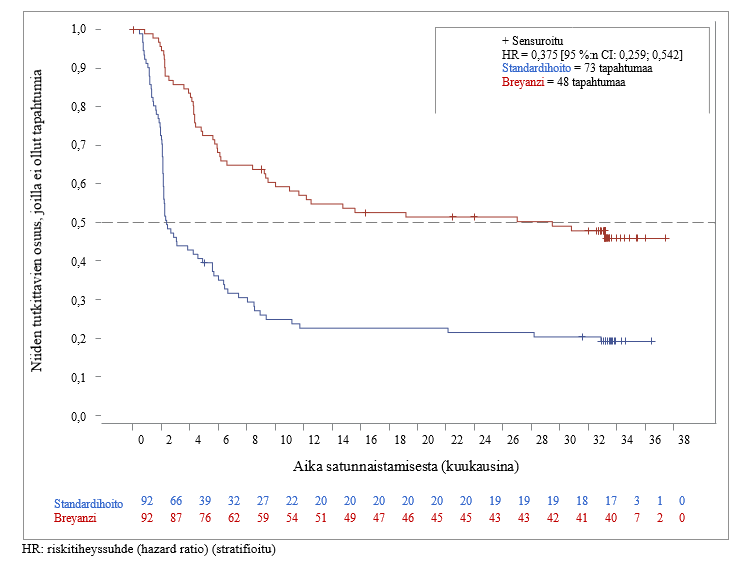
TRANSCEND‑LBCL-kohortti
Breyanzi-valmisteen tehoa ja turvallisuutta arvioitiin avoimessa yksihaaraisessa TRANSCEND (017001) -monikeskustutkimuksessa, jossa potilailla oli uusiutunut tai refraktaarinen aggressiivinen B‑soluinen non‑Hodgkin-lymfooma (NHL). Soveltuvat potilaat olivat ≥ 18‑vuotiaita, ja heillä oli WHO:n vuoden 2008 luokituksen mukainen uusiutunut/refraktaarinen muutoin määrittämätön (NOS, not otherwise specified) diffuusi suurisoluinen B‑solulymfooma (DLBCL), mukaan lukien diffuusi suurisoluinen B‑solulymfooma, joka on kehittynyt indolentista lymfoomasta (muuntunut esim. follikulaarisesta lymfoomasta, marginaalivyöhykkeen lymfoomasta, kroonisesta lymfaattisesta leukemiasta / pienilymfosyyttisesta lymfoomasta tai Waldenströmin makroglobulinemiasta), korkean maligniteettiasteen B‑solulymfooma (HGBCL), primaarinen välikarsinan B‑solulymfooma (PMBCL) ja gradus 3B follikulaarinen lymfooma (FL3B), ja he olivat saaneet vähintään kaksi aiempaa hoitolinjaa tai autologisen hematopoieettisen kantasolusiirron. Tutkimukseen ei otettu potilaita, joilla oli muita diffuusin suurisoluisen B‑solulymfooman alatyyppejä, ja riski-hyötyprofiilia tässä ryhmässä ei ole varmistettu. Tutkimukseen otettujen potilaiden ECOG-suorituskykyluokka oli ≤ 2, heille oli tehty autologinen ja/tai allogeeninen hematopoieettinen kantasolusiirto (HSCT) tai heillä oli sekundaarinen keskushermostolymfooma. Potilaat, jotka olivat aiemmin saaneet CD19‑antigeeniin kohdistuvaa hoitoa, soveltuivat tutkimukseen, jos CD19‑positiivisuus voitiin vahvistaa kasvainnäytteestä CD19‑antigeeniin kohdistuneen hoidon jälkeen. Tutkimuksen ulkopuolelle jätettiin potilaat, joiden kreatiniinipuhdistuma oli alle 30 ml/min, alaniiniaminotransferaasiarvo > 5 kertaa normaalin yläraja tai vasemman kammion ejektiofraktio < 40 prosenttia.
Verisolujen määrälle ei ollut vähimmäisvaatimuksia: potilaat soveltuivat tutkimukseen, jos heillä oli tutkijan arvion mukaan lymfosyyttejä poistavaan solunsalpaajahoitoon riittävä luuytimen toiminta. Taulukossa 6 on esitetty tutkittavien lähtötilanteen demografiset tekijät ja sairauteen liittyvät ominaisuudet.
Hoitoon kuului lymfosyyttejä poistava solunsalpaajahoito, fludarabiinia 30 mg/m2/vrk ja syklofosfamidinia 300 mg/m2/vrk kolmen päivän ajan, minkä jälkeen tutkittaville annettiin Breyanzi-valmistetta 2–7 päivää myöhemmin.
Syöpähoito taudin kontrolloimiseksi sallittiin afereesin ja lymfosyyttejä poistavan hoidon välillä (siltahoito). 137 potilasta (60 %) 229:stä Breyanzi-hoitoa saaneesta potilaasta sai syöpähoitoa taudin hallintaan. Siltahoidon tyyppi ja kesto päätettiin tutkijan harkinnan mukaan.
Mediaaniaika leukafereesistä valmisteen saatavuuteen oli 24 päivää (vaihteluväli: 17–51 päivää). Lisäksi mediaaniaika leukafereesistä infuusioon oli 38,5 päivää (vaihteluväli: 27–156 päivää).
Niistä 298 potilaasta, joille tehtiin leukafereesi ja joille valmistettiin Breyanzi-valmistetta annosvälillä 44–120 × 106 CAR‑positiivista elinkykyistä T‑solua, 229 potilasta (77 %) sai Breyanzi-hoitoa ja 69 potilasta (23 %) ei saanut. Näistä 69 potilaasta 27 tapauksessa (39 %) oli kyse valmistusvirheestä, mukaan lukien 2 potilasta, jotka eivät saaneet Breyanzi-hoitoa, ja 25 potilasta, jotka saivat hoitoa tutkimusvalmisteella, joka ei täyttänyt vapauttamiseen liittyviä vaatimuksia. Loppuja 42 potilasta (61 %) ei hoidettu Breyanzi-valmisteella: yleisin syy oli tutkittavan kuolema (n = 29) tai sairauden komplikaatiot (n = 6). Potilailla, joiden annos oli vaihteluvälillä 44–120 × 106 CAR‑positiivista elinkykyistä T‑solua, Breyanzi-valmisteen mediaaniannos oli 87 × 106 CAR‑positiivista elinkykyistä T‑solua.
Tehon suhteen voitiin arvioida 216 potilasta (tehoaineisto). Potilaista 13:a ei voitu arvioida tehon suhteen, mukaan lukien 10 potilasta, joilla ei ollut lähtötilanteessa PET‑positiivista tautia tai joilla ei voitu vahvistaa olevan PET-positiivista tautia IRC:n arvioimana sen jälkeen, kun potilaille oli annettu syöpähoitoa taudin kontrolloimiseksi, kolmea potilasta muusta syystä.
Taulukossa 6 on yhteenveto lähtötilanteen potilas- ja sairaustiedoista TRANSCEND-tutkimuksessa.
Taulukko 6: Lähtötilanteen demografiset ja sairautta koskevat tiedot TRANSCEND-tutkimuksessa
| Ominaisuus | Kaikki potilaat, joille tehtiin leukafereesi (N = 298) | Kaikki potilaat, jotka saivat Breyanzi-hoitoa (N = 229) |
Iän mediaani, vuosina (vaihteluväli) ≥ 65 vuotta, n (%) ≥ 75 vuotta, n (%) | 62,0 (18; 82) 116 (38,9) 25 (8,4) | 62,0 (18, 82) 89 (38,9) 19 (8,3) |
Sukupuoli, n (%) Mies Nainen |
197 (66,1) 101 (33,9) |
153 (66,8) 76 (33,2) |
Aiempi kantasolusiirto, n (%) Autologinen kantasolusiirto Allogeeninen kantasolusiirto | 106 (35,6) 100 (33,6) 11 (3,7) | 87 (38,0) 84 (36,7) 8 (3,5) |
ECOG-toimintakykyluokka (seulontavaiheessa) ECOG 0–1, n (%) ECOG 2, n (%) |
290 (97,3) 8 (2,7) |
225 (98,3) 4 (1,7) |
Taudin histologinen alatyyppi, n (%) Diffuusi suurisoluinen B‑solulymfooma (DLBCL), muutoin määrittämätön (NOS) DLBCL, joka on muuntunut indolentista lymfoomasta Korkean maligniteettiasteen B‑solulymfoomaa (HGBCL) Primaarinen välikarsinan B‑solulymfooma (PMBCL) Gradus 3B follikulaarinen lymfooma (FL3B) |
142 (47,7) 87 (29,2) 48 (16,1) 15 (5,0) 6 (2,0) |
117 (51,1) 60 (26.2) 33 (14,4) 15 (6,6) 4 (1,7) |
| Aiempien hoitojen lukumäärän mediaani (vaihteluväli) | 3 (1–12) | 3 (1–8) |
| Solunsalpaajahoitoon reagoimatonb, n (%) | 212 (71,1) | 160 (69,9) |
| Refraktaarinenc, n (%) | 246 (82,6) | 186 (81,2) |
| Uusiutunutd, n (%) | 52 (17,4) | 43 (18,8) |
| Sekundaarinen keskushermoston lymfooma Breyanzi-infuusion aikaan, n (%) | 7 (2,3) | 6 (2,6) |
| Ei saavuttanut koskaan täydellistä vastetta aiemmissa hoidoissa, n (%) | 141 (47,3) | 103 (45,0) |
a Histologisesti määritetty DLBCL, jossa MYC‑ ja BCL2‑ ja/tai BCL6‑uudelleenryhmittyminen mutaatioiden kautta.
b Solunsalpaajahoitoon reagoimaton tauti määritettiin vakaaksi tai eteneväksi taudiksi viimeisen solunsalpaajahoitoa sisältäneen hoito-ohjelman jälkeen tai uusiutuneeksi taudiksi < 12 kuukautta autologisen kantasolusiirron jälkeen.
c Taudin tila oli refraktaarinen, jos potilas ei edellisessä hoidossa saavuttanut täydellistä vastetta.
d Taudin tila oli uusiutunut, jos potilas oli saavuttanut edellisessä hoidossa täydellisen vasteen.
Tehokkuus arvioitiin primaarisen päätetapahtuman, kokonaisvasteen saavuttaneiden osuuden (overall response rate, ORR), ja sekundaaristen päätetapahtumien, kuten täydellisen vasteen saavuttaneiden osuuden (complete response, CR) ja IRC:n arvioiman vasteen keston (duration of response, DOR), perusteella (taulukko 7 ja kuva 2). Tutkimuksen aikaisen (on-study) seuranta-ajan mediaani oli 20,5 kuukautta (vaihteluväli: 0,2–60,9 kuukautta).
Taulukko 7: TRANSCEND-tutkimus: Vasteen saavuttaneiden osuus ja vasteen kesto (IRC:n arvioimana)
Kaikki potilaat, joille tehtiin leukafereesi (N = 298) | Tehoaineisto (N = 216) | |
Kokonaisvasteen saavuttaneiden osuusa, n (%) [95 %:n CI] | 179 (60,1) [54,3; 65,7] | 157 (72,7) [66,2; 78,5] |
Täydellinen vaste, n (%) [95 %:n CI] | 128 (43,0) [37,3; 48,8] | 115 (53,2) [46,4; 60,0] |
Osittainen vaste, n (%) [95 %:n CI] | 51 (17,1) [13,0; 21,9] | 42 (19,4) [14,4; 25,4] |
Vasteen kesto (DOR)a,b (kuukausina) Mediaani [95 %:n CI]c Vaihteluväli | n = 179 16,8 [8.0; NR] 0,0; 34,3+ | n = 157 20,5 [8,2; NR] 0,0; 34,3+ |
Vasteen kesto, jos paras vaste on täydellinen vastea,b (kuukausina) Mediaani [95 %:n CI]c Vaihteluväli | n = 128 26,1 [23,1; NR] 0,0; 34,3+ | n = 115 26,1 [23,1; NR] 0,0; 34,3+ |
CI = luottamusväli (confidence interval), CR = täydellinen vaste (complete response), IRC = riippumaton arviointiryhmä (Independent Review Committee), KM = Kaplan–Meier, NR = ei saavutettu (not reached)
a Luganon 2014 kriteerien perusteella riippumattoman arviointiryhmän (IRC) arvion mukaan.
b Syöpähoidon aloittamisen jälkeiset kuolemat katsotaan tapahtumiksi.
c Kaksisuuntainen 95 %:n luottamusväli saatiin käyttämällä Kaplan–Meierin menetelmää.
+ Jatkuu edelleen.
Mediaaniaika vasteeseen (CR tai osittainen vaste [PR]) oli 1,0 kuukautta (vaihteluväli: 0,7–8,9 kuukautta). Mediaaniaika täydelliseen vasteeseen (CR) oli 1,0 kuukautta (vaihteluväli: 0,8–12,5 kuukautta). Vasteen kesto oli pidempi potilailla, jotka saavuttivat täydellisen hoitovasteen, kuin potilailla, joiden paras vaste oli osittainen vaste.
TRANSCEND-tutkimuksessa oli kuusi potilasta, jotka saivat hoitoa sekundaariseen keskushermoston lymfoomaan ja joilla hoidon teho oli arvioitavissa. Näistä kuudesta potilaasta kolme saavutti täydellisen vasteen; kaksi kolmesta saavutti kestävän 23 kuukauden remission, joka jatkui tutkimuksen päättyessä. Sekundaarista keskushermoston lymfoomaa sairastavien potilaiden turvallisuusprofiili oli yhdenmukainen koko tutkimuspopulaation turvallisuusprofiilin kanssa.
Tehoaineistossa primaarisen välikarsinan B‑solulymfooman kokonaisvasteosuus (ORR) oli 79 prosenttia (11/14 potilasta) ja gradus 3B follikulaarisen lymfooman kokonaisvasteosuus oli 100 prosenttia (4/4 potilasta). Primaarisen välikarsinan B‑solulymfooman CR‑vasteosuus oli 50 prosenttia ja gradus 3B follikulaarisen lymfooman CR‑vasteosuus 100 prosenttia. Näiden alatyyppien turvallisuusprofiilit olivat yhdenmukaiset.
Tehoaineistossa diffuusin suurisoluisen B‑solylymfooman (DLBCLt) kokonaisvasteosuus aiemmasta indolentista lymfoomasta muuntuneelle diffuusille suurisoluiselle B‑solulymfoomalle olivat seuraavat: 86 prosenttia (38/44 potilasta) follikulaarisesta lymfoomasta muuntuneelle alatyypille, 43 prosenttia (3/7 potilasta) marginaalivyöhykkeen lymfoomasta muuntuneelle alatyypille (MZL), 50 prosenttia (2/4 potilasta) kroonisesta lymfaattisesta leukemiasta / pienilymfosyyttisesta lymfoomasta (CLL/SLL) muuntuneelle alatyypille ja 50 prosenttia (1/2 potilasta) Waldenströmin makroglobulinemiasta (WM) muuntuneelle alatyypille. CR‑vasteosuudet olivat 61,4 prosenttia follikulaarisesta lymfoomasta muuntuneelle alatyypille (tFL), 29 prosenttia marginaalivyöhykkeen lymfoomasta muuntuneelle alatyypille (tMZL), 25 prosenttia kroonisesta lymfaattisesta leukemiasta / pienilymfosyyttisesta lymfoomasta muuntuneelle alatyypille (tCLL/SLL, Richterin syndrooma) ja 0 prosenttia Waldenströmin makroglobulinemiasta muuntuneelle alatyypille (WM). Näiden alatyyppien turvallisuusprofiilit olivat yhdenmukaiset. Pitkäaikaista remissiota (vasteen kesto ≥ 12 kuukautta) havaittiin potilailla, joilla oli follikulaarisesta lymfoomasta (tFL) tai marginaalivyöhykkeen lymfoomasta muuntunut alatyyppi (tMZL). Potilaista, joilla oli kroonisesta lymfaattisesta leukemiasta / pienilymfosyyttisesta lymfoomasta muuntunut alatyyppi (tCLL/SLL, 4 potilasta) tai Waldenströmin makroglobulinemiasta muuntunut alatyyppi (tWM, 2 potilasta) ja joilla pisin havaittu vasteen kesto oli 2 kuukautta (tCLL/SLL-potilaat) ja 5,3 kuukautta (tWM‑potilaat), on vain hyvin vähän tietoa. Näiden alatyyppien turvallisuusprofiilit olivat yhdenmukaiset.
Breyanzi-valmisteen kliinisessä TRANSCEND-tutkimuksessa 89 potilasta (39 prosenttia) 229 potilaasta oli vähintään 65‑vuotiaita ja 19 potilasta (8 prosenttia) oli vähintään 75‑vuotiaita. Breyanzi-valmisteen havaittu turvallisuus ja teho olivat samankaltaiset näillä potilailla ja nuoremmilla potilailla.
11 potilasta oli saanut aiempaa CD19‑antigeeniin kohdistuvaa hoitoa, ja näiden potilaiden tehoa ja turvallisuutta koskevat tulokset olivat samankaltaiset kuin muussa populaatiossa. Kaikilla potilailla oli todettu CD19‑antigeenin ilmentyminen ennen Breyanzi-infuusiota.
Breyanzi-valmisteen käytöstä potilaille, joiden ECOG-toimintakykyluokka oli 2 ennen afereesia (4 potilasta), ja potilaille, joille oli aiemmin tehty allogeeninen hematopoieettinen kantasolusiirto (8 potilasta), on vain vähän kokemusta.
229:sta Breyanzi-hoitoa saaneesta potilaasta suurin osa (n = 209) sai Breyanzi-valmistetta solukomponenttien CD4 ja CD8 suositussuhteessa (suhteen vaihteluväli: 0,8–1,2). Breyanzi-valmisteen käytöstä tämän CD4:CD8‑suhteen ulkopuolella (n = 19 yli 1,2, n = 1 alle 0,8) on vain vähän kokemusta, mikä rajoittaa tietojen tulkintaa tässä alaryhmässä.
115:sta täydellisen vasteen (CR) saavuttaneesta potilaasta 82 (71 prosenttia) saavutti remission, joka kesti vähintään 6 kuukautta, ja 74 potilasta (64 prosenttia) saavutti remission, joka kesti vähintään 12 kuukautta.
Kuva 2: Vasteen kesto IRC:n arvioimana potilailla, jotka saavuttivat vasteen (TRANSCEND-tutkimuksen tehoaineisto)
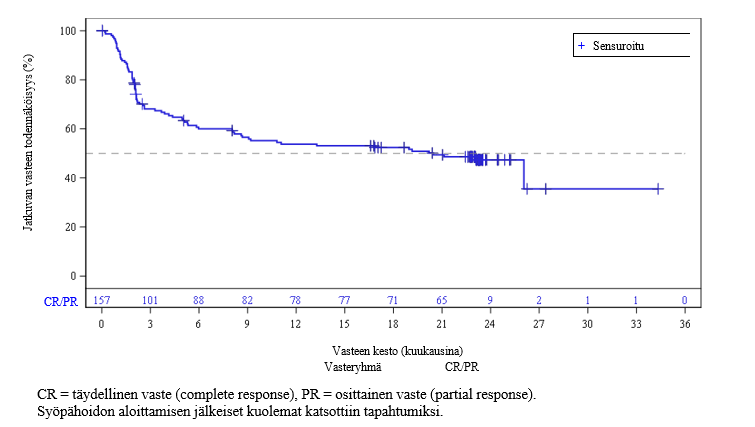
Breyanzi-hoitoa annettiiin 11 potilaalle, joilla oli aiemmin ollut B‑hepatiitti tai C‑hepatiitti, eikä hepatiitti aktivoitunut uudelleen. Potilaat saivat samalla hoitosuositusten mukaista antiviraalista estohoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
TRANSCEND WORLD
TRANSCEND WORLD on edelleen käynnissä oleva yksihaarainen vaiheen 2 monikeskustutkimus. Tutkimuksessa kohortin 1 tarkoitus on tuottaa kliinistä kokemusta Breyanzi-hoidon käytöstä Euroopassa sellaisten aikuispotilaiden hoidossa, joilla on kolmannen hoitolinjan jälkeinen suurisoluinen B‑solulymfooma, joka määritellään WHO:n vuoden 2016 luokituksen mukaan joksikin seuraavista: uusiutunut/refraktaarinen diffuusi suurisoluinen B‑solulymfooma (DLBCL, muutoin määrittämätön (NOS, not otherwise specified) [uusi], muuntunut FL), korkean maligniteettiasteen B‑solulymfooma (HGBCL), jossa histologisesti määritetty DLBC ja MYC‑ ja BCL2‑ ja/tai BCL6‑uudelleenryhmittyminen mutaatioiden kautta, ja gradus 3B follikulaarinen lymfooma. Potilaita, jotka olivat saaneet aiempaa CD19‑antigeeniin kohdistuvaa hoitoa, ei otettu mukaan tutkimukseen. Seuraavassa taulukossa 8 on esitetty lähtötilanteen demografiset tekijät ja sairauteen liittyvät ominaisuudet.
Taulukko 8: Lähtötilanteen demografiset ja sairautta koskevat tiedot TRANSCEND WORLD ‑tutkimuksessa (kohortti 1)
| Ominaisuus | Kaikki potilaat, joille tehtiin leukafereesi (N = 45) | Kaikki potilaat, jotka saivat Breyanzi-hoitoa (N = 36) |
Iän mediaani, vuosina (vaihteluväli) ≥ 65 vuotta, n (%) ≥ 75 vuotta, n (%) | 64,0 (26; 73) 19 (42,2) 0 | 61,5 (26,0; 72,0) 14 (38,9) 0 |
Sukupuoli, n (%) Mies Nainen |
30 (66,7) 15 (33,3) |
25 (69,4) 11 (30,6) |
Aiempi kantasolusiirto, n (%) Autologinen kantasolusiirto Allogeeninen kantasolusiirto | 14 (31,1) 14 (31,1) 0 | 12 (33,3) 12 (33,3) 0 |
ECOG-toimintakykyluokka (seulontavaiheessa) ECOG 0, n (%) ECOG 1, n (%) ECOG 2, n (%) |
26 (57,8) 18 (40,0) 1 (2,2) |
19 (52,8) 16 (44,4) 1 (2,8) |
Taudin histologinen alatyyppi, n (%) Diffuusi suurisoluinen B‑solulymfooma (DLBCL), muutoin määrittämätön (NOS) Korkean maligniteettiasteen B‑solulymfoomaa (HGBCL) Primaarinen välikarsinan B‑solulymfooma (PMBCL) Gradus 3B follikulaarinen lymfooma (FL3B) |
36 (80,0) 7 (15,6) 0 2 (4,4) |
31 (86,1) 4 (11,1) 0 1 (2,8) |
| Solunsalpaajahoitoon reagoimatonb, n (%) | 37 (82,2) | 29 (80,6) |
| Refraktaarinenc, n (%) | 36 (80,0) | 28 (77,8) |
| Uusiutunutd, n (%) | 9 (20,0) | 8 (22,2) |
a Histologisesti määritetty DLBCL, jossa MYC‑ ja BCL2‑ ja/tai BCL6‑uudelleenryhmittyminen mutaatioiden kautta.
b Solunsalpaajahoitoon reagoimaton tauti määritettiin vakaaksi tai eteneväksi taudiksi viimeisen solunsalpaajahoitoa sisältäneen hoito-ohjelman jälkeen tai uusiutuneeksi taudiksi < 12 kuukautta autologisen kantasolusiirron jälkeen.
c Taudin tila oli refraktaarinen, jos potilas ei edellisessä hoidossa saavuttanut täydellistä vastetta.
d Taudin tila oli uusiutunut, jos potilas oli saavuttanut edellisessä hoidossa täydellisen vasteen.
Lopullisen analyysin hetkellä 45 potilaalle kohortissa 1 oli tehty leukafereesi ja 36 potilasta oli saanut Breyanzi-hoitoa; seuranta-ajan mediaani oli 15,8 kuukautta. Mediaaniaika leukafereesistä valmisteen saatavuuteen oli 29 päivää (vaihteluväli: 24–38 päivää). Breyanzi-hoitoa saaneiden ryhmässä kokonaisvaste (ORR) oli 61,1 prosenttia (95 %:n luottamusväli: 43,5‑76,9), ja täydellisen vasteen (CR) saavuttaneiden osuus oli 33,3 prosenttia (95:%:n luottamusväli: 18,6‑51,0). Tautitaakka ja lähtötilanteen demografiset tekijät viittasivat edenneen, aggressiivisen taudin ominaisuuksiin. Breyanzi-valmisteen turvallisuusprofiili oli yhdenmukainen yhdistetyssä turvallisuuspopulaatiossa. Ks. lisokabtageenimaraleuseeliin liittyvät haittavaikutukset kohdasta Haittavaikutukset.
TRANSCEND‑FL
Breyanzi-valmisteen tehoa ja turvallisuutta arvioitiin vaiheen 2 avoimessa yksiryhmäisessä monikeskustutkimuksessa (TRANSCEND‑FL) aikuispotilailla, joilla oli uusiutunut tai hoitoon reagoimaton follikulaarinen lymfooma vaikeusasteeltaan 1, 2 ja 3A kahden tai useamman systeemistä hoitoa sisältäneen hoitolinjan jälkeen. Tutkimukseen otettiin potilaita, joiden ECOG-suorituskyky oli ≤ 1. Tutkimuksen ulkopuolelle jätettiin potilaat, joiden kreatiniinipuhdistuma oli alle 30 ml/min, alaniiniaminotransferaasiarvo > 5 kertaa normaalin yläraja tai vasemman kammion ejektiofraktio (LVEF) ≤ 40 prosenttia. Verisolujen määrälle ei ollut esimääritettyä rajaa: potilaat soveltuivat tutkimukseen, jos heillä oli tutkijan arvion mukaan lymfosyyttejä poistavaan solunsalpaajahoitoon riittävä luuytimen toiminta.
Hoitoon kuului lymfosyyttejä poistava solunsalpaajahoito, fludarabiinia 30 mg/m2/vrk ja syklofosfamidia 300 mg/m2/vrk kolmen päivän ajan, minkä jälkeen tutkittaville annettiin Breyanzi-valmistetta 2–7 päivää myöhemmin. Breyanzi-valmisteen mediaaniannos oli 100 × 106 CAR‑positiivista elinkykyistä T‑solua (vaihteluväli: 93,4–109,2 × 106 CAR‑positiivista elinkykyistä T‑solua).
Syöpähoito taudin kontrolloimiseksi sallittiin afereesin ja lymfosyyttejä poistavan hoidon välillä (siltahoito). 44 potilasta (41 %) 107:stä Breyanzi-hoitoa saaneesta potilaasta sai syöpähoitoa taudin hallintaan tutkijan harkinnan mukaan.
107 potilasta (93,8 %) 114 potilaasta, joille tehtiin leukafereesi, sai Breyanzi-valmistetta, ja neljä (3,5 %) potilasta valmistetta, joka ei ollut vaatimusten mukainen. Kolme (2,7 %) potilasta ei saanut Breyanzi-valmistetta seuraavista syistä: yksi (0,9 %) potilas haittatapahtuman vuoksi, yksi (0,9 %) potilas ei täyttänyt tutkimuksen kriteereitä ja yksi (0,9 %) potilas muusta syystä.
Tehon suhteen voitiin arvioida 103 potilasta (tehoaineisto). Potilaista neljää ei voitu arvioida tehon suhteen, koska potilailla ei ollut lähtötilanteessa PET-positiivista tautia tai joilla ei voitu vahvistaa olevan PET-positiivista tautia IRC:n arvioimana sen jälkeen, kun potilaille oli annettu syöpähoitoa taudin kontrolloimiseksi.
Mediaaniaika leukafereesistä valmisteen saatavuuteen oli 29 päivää (vaihteluväli: 20–55 päivää), ja mediaaniaika leukafereesistä infuusioon oli 50 päivää (vaihteluväli: 31–313 päivää).
Taulukko 9: Lähtötilanteen demografiset ja sairautta koskevat tiedot TRANSCEND‑FL-tutkimuksessa
| Ominaisuus | Kaikki potilaat, joille tehtiin leukafereesi (N = 114) | Kaikki potilaat, jotka saivat Breyanzi-hoitoa (N = 107) |
Iän mediaani, vuosina (vaihteluväli) ≥ 65 – < 75 vuotta, n (%) ≥ 75 vuotta, n (%) | 62,0 (23; 80) 36 (31,6) 10 (8,8) | 62,0 (23; 80) 32 (29,9) 10 (9,3) |
| Miessukupuoli, n (%) | 72 (63,2) | 66 (61,7) |
Aiempi kantasolusiirto, n (%) Autologinen kantasolusiirto |
34 (29,8) |
33 (30,8) |
| Korkeat FLIPI-pisteet (3–5), n (%) | 66 (57,9) | 61 (57,0) |
| Vaikeusasteen III–IV sairaus seulontavaiheessa, n (%) | 102 (89,4) | 95 (88,7) |
ECOG-toimintakykyluokka (seulontavaiheessa) ECOG 0, n (%) ECOG 1, n (%) |
68 (59,6) 46 (40,4) |
65 (60,7) 42 (39,3) |
| Kahteen hoitoon reagoimaton, n (%) | 74 (64,9) | 69 (64,5) |
Eteneminen 24 kuukauden sisällä ensilinjan hoidosta anti-CD20:llä ja alkylaattorilla, n (%) Kyllä Ei Ei arvioitavissa |
63 (55,3) 50 (43,9) 1 (0,9) |
58 (54,2) 48 (44,9) 1 (0,9) |
| Aiempien systeemisten hoitojen lukumäärän mediaani (vaihteluväli) | 3 (2,10) | 3 (2, 10) |
Teho perustui kokonaisvasteen saavuttaneiden osuuteen (ORR), joka on määritetty niiden potilaiden prosenttiosuutena, joilla oli IRC:n mukaan paras yleinen vaste (BOR, best overall response) täydellisestä vasteesta (CR) tai osittaisesta vasteesta (PR) Breyanzi-infuusion jälkeen (taulukko 10). Tutkimuksen aikaisen (on-study) seuranta-ajan mediaani oli 30,0 kuukautta (vaihteluväli: 0,3–39,6 kuukautta).
Mediaaniaika ensimmäiseen vasteeseen (CR tai PR) ja mediaaniaika ensimmäiseen täydelliseen vasteeseen oli 0,95 kuukautta (vaihteluväli: 0,6–3,3 kuukautta).
Taulukko 10: TRANSCEND‑FL-tutkimus: Vasteen saavuttaneiden osuus ja vasteen kesto (IRC:n arvioimana)
Kaikki potilaat, joille tehtiin leukafereesi (N = 114) | Tehoaineisto (N = 103) | |
Kokonaisvasteen saavuttaneiden osuusa, n (%) [95 %:n CI]b | 106 (93,0) [86,6; 96,9] | 100 (97,1) [91,7; 99,4] |
Täydellinen vaste, n (%) [95 %:n CI]b | 103 (90,4) [83,4; 95,1] | 97 (94,2) [87,8; 97,8] |
Osittainen vaste, n (%) [95 %:n CI]b | 3 (2,6) [0,5; 7,5] | 3 (2,9) [0,6; 8,3] |
Vasteen kesto (DOR) (kuukausina) Mediaani [95 %:n CI]c Vaihteluväli Jatkuvan remission asted, % [95 %:n CI] 18. kuukautena |
NR (30,85; NR) 1,9; 35,0+
76,1 (66,7; 83,2) |
NR (30,85; NR) 1,9; 35,0+
75,7 (66,0; 83,0) |
CI = luottamusväli; CR = täydellinen vaste; NR = ei saavutettu;
+ tarkoittaa sensuroitua arvoa
a Luganon kriteerien (2014) perusteella riippumattoman arviointiryhmän (IRC) arvion mukaan.
b Kaksipuolinen 95 %:n luottamusväli tarkan Clopper-Pearsonin menetelmän perusteella.
c Mediaani, Q1 ja Q3 on arvioitu KM-tulorajaestimaateista
d Perustuu vasteen keston KM-estimaatteihin
Kuva 3: Vasteen kesto IRC:n arvion mukaan, TRANSCEND‑FL-tutkimuksen tehoaineisto
TRANSCEND‑MCL-kohortti
Breyanzi-valmisteen tehoa ja turvallisuutta arvioitiin avoimessa, yksihaaraisessa monikeskustutkimuksessa (TRANSCEND‑MCL-kohortti) aikuispotilailla, joilla oli uusiutunut tai hoitoon reagoimaton manttelisolulymfooma (MCL) ja jotka olivat saaneet vähintään kahta aiempaa hoitolinjaa, mukaan lukien Brutonin tyrosiinikinaasin estäjä, alkyloiva aine ja anti-CD20-vasta-aine. Tutkimukseen otettujen potilaiden ECOG-suorituskykyluokka oli ≤ 2, heille oli tehty autologinen ja/tai allogeeninen HSCT tai heillä oli sekundaarinen keskushermostolymfooma. Tutkimuksen ulkopuolelle jätettiin potilaat, joiden kreatiniinipuhdistuma oli ≤ 30 ml/min, alaniiniaminotransferaasiarvo > 5 kertaa normaalin yläraja tai vasemman kammion ejektiofraktio (LVEF) < 40 prosenttia. Verisolujen määrälle ei ollut esimääritettyä rajaa: potilaat soveltuivat tutkimukseen, jos heillä oli tutkijan arvion mukaan lymfosyyttejä poistavaan solunsalpaajahoitoon riittävä luuytimen toiminta.
Hoitoon kuului lymfosyyttejä poistava solunsalpaajahoito, fludarabiinia 30 mg/m2/vrk ja syklofosfamidia 300 mg/m2/vrk kolmen päivän ajan, minkä jälkeen tutkittaville annettiin Breyanzi-valmistetta 2–7 päivää myöhemmin.
Syöpähoito taudin kontrolloimiseksi sallittiin afereesin ja lymfosyyttejä poistavan hoidon välillä (siltahoito). 58 potilasta (65,9 %) 88:sta Breyanzi-hoitoa saaneesta potilaasta sai taudin kontrolloimiseen tarkoitettua syöpähoitoa tutkijan harkinnan perusteella.
Niistä 104 potilaasta, joille tehtiin leukafereesi, 88 (84,6 %) sai Breyanzi-valmistetta; Breyanzi-valmisteen mediaaniannos oli 99,5 × 106 CAR‑positiivista elinkykyistä T‑solua (vaihteluväli: 46–103 × 106 CAR‑positiivista elinkykyistä T‑solua). Neljä (3,8 %) potilasta sai valmistetta, joka ei ollut vaatimusten mukainen (non‑conforming product). Kaksitoista (11,5 %) potilasta ei saanut Breyanzi-valmistetta seuraavista syistä: kahdeksan (7,6 %) potilasta kuoleman vuoksi, yksi (0,9 %) potilas ei enää täyttänyt tutkimuksen kriteereitä ja kolme (2,8 %) potilasta muusta syystä.
88:sta Breyanzi-valmistetta saaneesta potilaasta tehon suhteen voitiin arvioida 81 potilasta, jotka olivat saaneet vähintään kahta aiempaa systeemistä hoitolinjaa, mukaan lukien Brutonin tyrosiinikinaasin estäjä, ja nämä potilaat sisällytettiin tehoaineistoon. Viittä potilasta ei voitu arvioida tehon suhteen, koska potilailla ei ollut IRC:n arvion mukaan lähtötilanteessa PET-positiivista sairautta tai vahvistetusti PET-positiivista sairautta sen jälkeen, kun he olivat saaneet syöpähoitoa sairauden kontrolloimiseksi; yksi potilas ei ollut saanut vähintään kahta aiempaa systeemistä hoitolinjaa ja Brutonin tyrosiinikinaasin estäjää, ja yksi potilas ei ollut saanut aiemmin Brutonin tyrosiinikinaasin estäjää.
Mediaaniaika leukafereesistä valmisteen saatavuuteen oli 24,5 päivää (vaihteluväli: 17–80 päivää). Lisäksi mediaaniaika leukafereesistä infuusioon oli 39 päivää (vaihteluväli: 28–489 päivää).
Taulukko 11: Lähtötilanteen demografiset ja sairautta koskevat tiedot TRANSCEND‑MCL-kohortissa
| Ominaisuus | Kaikki potilaat, joille tehtiin leukafereesi (N = 104) | Kaikki potilaat, jotka saivat Breyanzi-hoitoa (N = 88) |
Iän mediaani, vuosina (vaihteluväli) ≥ 65 vuotta, n (%) ≥ 75 vuotta, n (%) | 68,0 (36, 86) 71 (68,3) 22 (21,2) | 68,5 (36, 86) 64 (72,7) 18 (20,5) |
Sukupuoli, n (%) Mies Nainen |
81 (77,9) 23 (22,1) |
67 (76,1) 21 (23,9) |
Aiempi kantasolusiirto, n (%) Autologinen kantasolusiirto Allogeeninen kantasolusiirto |
33 (31,7) 8 (7,7) |
26 (29,5) 6 (6,8) |
ECOG-toimintakykyluokka (seulontavaiheessa) ECOG 0, n (%) ECOG 1, n (%) ECOG 2, n (%) |
56 (53,8) 47 (45,2) 1 (1,0) |
48 (54,5) 40 (45,5) 0 |
Suuren riskin ominaisuudet, n (%) Ki67:n proliferaatioaste ≥ 30 % TP53-mutaatio Blastoidinen morfologia Monimuotoinen karyotyyppi |
82 (78,8) 25 (24,0) 30 (28,8) 30 (28,8) |
66 (75,0) 20 (22,7) 27 (30,7) 26 (29,5) |
| Sekundaarinen keskushermoston lymfooma Breyanzi-infuusion aikaan, n (%) | 7 (6,7) | 7 (8,0) |
| Aiempien systeemisten hoitojen lukumäärän mediaani (vaihteluväli) | 3 (1, 11) | 3 (1, 11) |
Refraktaarinen tai uusiutunut edellisessä hoidossa, n (%) Refraktaarinena Uusiutunutb |
70 (67,3) 34 (32,7) |
58 (65,9) 30 (34,1) |
a Taudin tila oli refraktaarinen, jos potilas ei edellisessä hoidossa saavuttanut täydellistä vastetta.
b Taudin tila oli uusiutunut, jos potilas oli saavuttanut edellisessä hoidossa täydellisen vasteen.
Teho perustui kokonaisvasteen saavuttaneiden osuuteen (ORR), joka on määritelty niiden potilaiden prosenttiosuutena, joilla oli IRC:n mukaan paras yleinen vaste (BOR, best overall response) täydellisestä vasteesta (CR) tai osittaisesta vasteesta (PR) Breyanzi-infuusion jälkeen (taulukko 12). Tutkimuksen aikaisen (on-study) seuranta-ajan mediaani oli 19,5 kuukautta (vaihteluväli: 0,4–72 kuukautta).
Tehoaineistoon kuuluneilla 81 potilaalla mediaaniaika ensimmäiseen vasteeseen (täydellinen tai osittainen vaste) oli 0,95 kuukautta (vaihteluväli: 0,7–3,0 kuukautta) ja mediaaniaika ensimmäiseen täydelliseen vasteeseen 0,95 kuukautta (0,7–4,9 kuukautta). Vasteen kesto oli pidempi potilailla, joiden paras yleinen vaste oli täydellinen hoitovaste, kuin potilailla, joiden paras vaste oli osittainen vaste.
Taulukko 12: TRANSCEND‑MCL-kohortti: Vasteen saavuttaneiden osuus ja vasteen kesto (IRC:n arvioimana)
Kaikki potilaat, joille tehtiin leukafereesi (N = 104) | Tehoaineisto (N = 81) | |
Kokonaisvasteen saavuttaneiden osuusa, n (%) [95 %:n CI] | 73 (70,2) [60,4; 78,8] | 67 (82,7) [72,7; 90,2] |
Täydellinen vaste, n (%) [95 %:n CI]b | 64 (61,5) [51,5; 70,9] | 58 (71,6) [60,5; 81,1] |
Osittainen vaste, n (%) [95 %:n CI]b | 9 (8,7) [4,0; 15,8] | 9 (11,1) [5,2; 20,0] |
Vasteen saaneiden lukumäärä Vasteen kesto (DOR) (kuukausina) Mediaani [95 %:n CI]c Vaihteluväli Jatkuvan remission asted, % [95 %:n CI] 24. kuukautena |
15,2 [7,0; 24,0] 0,0+; 24,0
44,8 (32,9; 55,9) |
11,5 [6,2; 24,0] 0,0+; 24,0
41,2 (29,2; 52,9) |
Vasteen keston (DOR) seuranta-ajan mediaani (kuukausina) Mediaani [95 %:n CI] Vaihteluväli |
23,0 [22,8; 23,1] 0,0+; 24,0 |
22,9 [22,8; 23,0] 0,0+; 24,0 |
CI = luottamusväli; CR = täydellinen vaste; NR = ei saavutettu;
+ tarkoittaa sensuroitua arvoa
a Luganon kriteerien (2014) perusteella riippumattoman arviointiryhmän (IRC) arvion mukaan.
b Kaksipuolinen 95 %:n luottamusväli tarkan Clopper-Pearsonin menetelmän perusteella.
c Mediaani, Q1 ja Q3 on arvioitu KM-tulorajaestimaateista.
d Perustuu vasteen keston KM-estimaatteihin.
Kuva 4 Vasteen kesto IRC:n arvion mukaan, TRANSCEND‑MCL-kohortin tehoaineisto
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Breyanzi-valmisteen käytöstä kypsien B‑solukasvainten hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Breyanzi-infuusion jälkeen ilmeni alkuvaiheen ekspansio ja sen jälkeen kaksivaiheinen väheneminen.
LBCL
TRANSCEND‑LBCL-kohortissa potilailla, jotka olivat saaneet yhtä tai useampaa aiempaa hoitolinjaa suurisoluisen B‑solulymfooman hoitoon, maksimiekspansion mediaaniaika ääreisveressä oli 11 päivää ensimmäisen infuusion jälkeen. Breyanzi säilyi ääreisveressä enintään 2 vuotta.
Yhtä aiempaa hoitolinjaa suurisoluisen B‑solulymfooman hoitoon saaneilla potilailla (TRANSFORM-tutkimus) mediaani‑Cmax oli vasteen saavuttaneilla potilailla (N = 76) 33 285 kopiota/mikrog ja 95 618 kopioita/mikrog niillä potilailla, jotka eivät saavuttaneet vastetta (N = 7). Mediaani‑AUC0‑28d oli vasteen saavuttaneilla potilailla 268 887 vrk*kopiota/mikrog ja 733 406 vrk*kopiota/mikrog niillä potilailla, jotka eivät saavuttaneet vastetta.
TRANSCEND-tutkimuksessa vasteen saavuttaneilla potilailla (N = 150) oli 2,85 kertaa suurempi mediaani‑Cmax kuin vastetta saavuttamattomilla potilailla (N = 45) (33 766,0 vs. 11 846,0 kopiota/mikrog). Vasteen saavuttaneilla potilailla oli 2,22 kertaa suurempi mediaani‑AUC0‑28d kuin vastetta saavuttamattomilla potilailla (257 769,0 vs. 116 237,3 vrk*kopiota/mikrog).
TRANSCEND-tutkimuksessa alle 65‑vuotiailla potilailla (N = 145) oli 2,93 kertaa suurempi mediaani‑Cmax ja 2,35 kertaa suurempi AUC0‑28d verrattuna 65‑vuotiaisiin tai tätä vanhempiin potilaisiin (N = 102, mukaan lukien 77 potilasta ikäryhmässä 65–74 vuotta, 24 potilasta ikäryhmässä 75–84 vuotta ja yksi yli 85‑vuotias potilas). Sukupuolella ja kehonpainolla ei ollut selvää yhteyttä Cmax- ja AUC0‑28d-arvoihin.
FL
TRANSCEND‑FL-tutkimuksessa potilailla, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa follikulaarisen lymfooman (FL) hoitoon, maksimiekspansion mediaaniaika ääreisveressä oli 10 päivää ensimmäisen infuusion jälkeen. Breyanzi säilyi ääreisveressä enintään 3 vuotta.
Breyanzi-valmistetta follikulaarisen lymfooman (FL) hoitoon saaneista potilaista (TRANSCEND‑FL-tutkimus) mediaani‑Cmax oli vasteen saavuttaneilla potilailla (N = 100) 31 336 kopiota/mikrog ja 15 568 kopioita/mikrog niillä potilailla, jotka eivät saavuttaneet vastetta (N = 2). Mediaani‑AUC0‑28d oli vasteen saavuttaneilla potilailla (N = 96) 245 730 vrk*kopiota/mikrog ja 161 935 vrk*kopiota/mikrog niillä potilailla, jotka eivät saavuttaneet vastetta (N = 2).
MCL
TRANSCEND‑MCL-kohortissa potilailla, jotka olivat saaneet kahta tai useampaa aiempaa hoitolinjaa manttelisolulymfooman (MCL) hoitoon, maksimiekspansion mediaaniaika ääreisveressä oli 10 päivää ensimmäisen infuusion jälkeen. Breyanzi säilyi ääreisveressä enintään 2 vuotta.
Breyanzi-valmistetta manttelisolulymfooman (MCL) hoitoon saaneista potilaista (TRANSCEND‑MCL-kohortti) mediaani‑Cmax oli vasteen saavuttaneilla potilailla (N = 67) 31 631 kopiota/mikrog ja 12 444 kopiota/mikrog niillä potilailla, jotka eivät saavuttaneet vastetta (N = 8). Mediaani‑AUC0–28d oli vasteen saavuttaneilla potilailla (N = 67) 309 578 vrk*kopiota/mikrog ja 142 462 vrk*kopiota/mikrog niillä potilailla, jotka eivät saavuttaneet vastetta (N = 8).
Prekliiniset tiedot turvallisuudesta
Genotoksisuutta tai karsinogeenisuutta koskevia tutkimuksia ei ole tehty.
In vitro -solunjakautumiskokeissa, joissa luovuttajina oli terveitä henkilöitä ja potilaita, ei havaittu todisteita muuntumisesta ja/tai immortalisaatiosta. Breyanzi‑T‑soluissa ei myöskään havaittu suosivaa integraatiota lähelle huolta aiheuttavia geenejä.
Valmisteen luonne huomioon ottaen ei ole tehty tutkimuksia, joilla arvioitaisiin sen vaikutuksia hedelmällisyyteen, lisääntymiseen ja kehitykseen.
Farmaseuttiset tiedot
Apuaineet
Cryostor CS10
Natriumkloridi
Natriumglukonaatti
Natriumasetaattitrihydraatti
Kaliumkloridi
Magnesiumkloridi
Ihmisen albumiini
N‑asetyyli-DL‑tryptofaani
Kapryylihappo
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo säilytettynä nestetypen höyryfaasissa
13 kuukautta.
Sulattamisen jälkeen
Valmiste on annettava heti sulattamisen jälkeen. Käytön aikana valmiste säilyy korkeintaan 2 tuntia huoneenlämmössä (15 °C – 25 °C).
Ei saa pakastaa uudelleen.
Säilytys
Breyanzi-valmistetta on säilytettävä ja kuljetettava pakastettuna nestetypen höyryfaasissa (≤ ‑130 °C), ja valmisteen on pysyttävä jäätyneenä, kunnes potilas on valmis hoitoon. Näin varmistetaan, että potilaalle on käytettävissä elinkykyisiä soluja. Sulatettua lääkevalmistetta ei saa pakastaa uudelleen.
Sulatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BREYANZI infuusioneste, dispersio
1,1-70 x 10exp6 solua/ml / 1,1-70 x 10exp6 solua/ml (L:ei) 1 annos (371325,60 €)
PF-selosteen tieto
Breyanzi toimitetaan kryosäilytysinjektiopulloissa, jotka on valmistettu syklisestä olefiinikopolymeeristä. Yksi 5 ml:n injektiopullo sisältää 4,6 ml soludispersiota.
CAR‑positiiviset elinkykyiset T‑solut (CD8‑positiivinen solukomponentti tai CD4‑positiivinen solukomponentti) on pakattu yksittäisiin koteloihin, jotka sisältävät korkeintaan 4 injektiopulloa kutakin komponenttia, riippuen kryosäilytetyn lääkevalmisteen CAR‑positiivisten elinkykyisten T‑solujen pitoisuudesta.
CD8‑positiivisen ja CD4‑positiivisen solukomponentin kotelot toimitetaan yhdessä ulkopakkauksessa.
Valmisteen kuvaus:
Hieman samea tai samea dispersio, jonka väri voi vaihdella värittömästä keltaiseen tai ruskehtavan keltaiseen.
Käyttö- ja käsittelyohjeet
Ennen lääkkeen käsittelyä tai antamista tehtävät varotoimet
- Breyanzi on kuljetettava hoitokeskuksen sisällä suljetuissa, hajoamattomissa ja vuotamattomissa pakkauksissa.
- Tämä lääkevalmiste sisältää ihmisen verisoluja. Breyanzi-valmistetta käsittelevien terveydenhuollon ammattilaisten on noudatettava asianmukaisia varotoimia (käytettävä suojakäsineitä, suojavaatetusta ja silmiensuojaimia), jotta vältetään infektiotautien mahdollinen tarttuminen.
Valmistelu ennen antamista
Ennen injektiopullojen/lääkepullojen sulattamista
- Varmista, että potilaan henkilöllisyys vastaa kuljetuslaatikon potilastunnisteita.
- Breyanzi koostuu CAR‑positiivisista elinkykyisistä T‑soluista, jotka on muunneltu erillisiksi CD8‑positiivisiksi ja CD4‑positiivisiksi solukomponenteiksi; kummallekin solukomponentille on erillinen luovutussertifikaatti (RfIC, Release for Infusion Certificate). Lue luovutussertifikaatista (kiinnitetty kuljetuslaatikon sisään) tarvittavien ruiskujen lukumäärä sekä CD8‑positiivisten ja CD4‑positiivisten solukomponenttien annettava tilavuus (ruiskujen etiketit toimitetaan luovutussertifikaatin mukana).
-
Varmista infuusioaika etukäteen ja ajoita Breyanzi-valmisteen sulattamisen aloitus siten, että valmiste on valmis infusoitavaksi, kun potilaskin on valmis.
Huomio: Kun CAR‑positiivisia elinkykyisiä T‑soluja sisältävät injektiopullot (CD8‑positiiviset ja CD4‑positiiviset solukomponentit) on poistettu pakkassäilytyksestä, sulattaminen on tehtävä loppuun ja solut annettava kahden tunnin kuluessa.
Injektiopullojen sulattaminen
- Varmista, että potilaan henkilöllisyys vastaa ulkopakkauksen ja luovutussertifikaatin (RfIC) potilastunnisteita.
- Poista CD8‑positiivisen solukomponentin pakkaus ja CD4‑positiivisen solukomponentin pakkaus ulkopakkauksesta.
- Avaa jokainen sisäpakkaus ja tarkista injektiopullot silmämääräisesti vaurioiden varalta. Jos injektiopullot ovat vahingoittuneet, ota yhteyttä yritykseen.
- Poista injektiopullot varovasti koteloista ja aseta ne sulamaan huoneenlämpöön suoja-alustalle. Sulata kaikki injektiopullot samanaikaisesti. Varmista, että CD8‑positiiviset ja CD4‑positiiviset solukomponentit pysyvät erillään.
Annoksen valmistelu
-
Yhteen annokseen saatetaan tarvita useampi kuin yksi injektiopullo CD8‑positiivista ja CD4‑positiivista solukomponenttia kunkin komponentin CAR‑positiivisten elinkykyisten T‑solujen pitoisuuden mukaan. Kaikille vastaanotetuille CD8‑positiivisten ja CD4‑positiivisten solukomponenttien injektiopulloille on valmisteltava oma ruisku.
Huomio:Ruiskuun vedettävät ja infusoitavat tilavuudet voivat olla erilaiset eri komponenteille. - Yksi 5 ml:n injektiopullo sisältää yhteensä 4,6 ml ruiskuun vedettävää CD8‑positiivista tai CD4‑positiivista T‑solun solukomponenttia. Kunkin komponentin luovutussertifikaatti (RfIC) kertoo kuhunkin ruiskuun vedettävän soludispersion tilavuuden (ml). Käytä pienintä mahdollista Luer-lock-kärjellä varustettua ruiskua (1 ml – 5 ml) ja vedä kustakin injektiopullosta määritelty tilavuus ruiskuun. Alle 3 ml:n tilavuuksiin ei saa käyttää 5 ml:n ruiskua.
- Valmistele ensin CD8‑postiivisen solukomponentin ruisku(t). Varmista, että CD8‑positiivisen solukomponentin ruiskun etiketin potilastunnisteet vastaavat CD8‑positiivisen solukomponentin injektiopullon etikettiä. Liitä CD8‑positiivisen solukomponentin ruiskun etiketti ruiskuun/ruiskuihin ennen kuin vedät tarvittavan tilavuuden ruiskuun/ruiskuihin.
-
Tee sama CD4‑positiiviselle solukomponentille.
Huomio: On tärkeää varmistaa, että kunkin solukomponentin ruiskuun vedetty tilavuus vastaa vastaavassa luovutussertifikaatissa (RfIC) määritettyä tilavuutta.
Vaadittavan soludispersion tilavuuden vetäminen kustakin injektiopullosta erilliseen ruiskuun on tehtävä seuraavien ohjeiden mukaisesti:
1. Pidä sulanutta injektiopulloa / sulaneita injektiopulloja pystyasennossa ja käännä injektiopullo/injektiopullot varovaisesti ylösalaisin, jotta soluvalmiste sekoittuu. Jos valmisteessa näkyy paakkuja, jatka injektiopullon/injektiopullojen kääntämistä, kunnes paakut ovat hajonneet ja soluaines näyttää tasaiselta suspensiolta.
2. Tarkasta sulanut injektiopullo / sulaneet injektiopullot silmämääräisesti vaurioiden tai vuotojen varalta. Älä käytä injektiopulloa, jos se on vaurioitunut tai siinä on paakkuja, jotka eivät hajoa: ota yhteyttä yritykseen. Injektiopullojen sisältämän nesteen pitäisi olla hieman sameaa tai sameaa, väritöntä tai keltaista tai rusehtavan keltaista.
3. Poista polyalumiinikansi (jos sellainen on) injektiopullon pohjasta ja puhdista injektiopullon kalvo alkoholia sisältävällä puhdistuslapulla. Anna kuivua ennen kuin jatkat.
HUOM.: Polyalumiinikannen puuttuminen ei vaikuta injektiopullon steriiliyteen.
4. Pidä injektiopullo/injektiopullot pystyasennossa, leikkaa letkun sinetti injektiopullon yläosasta heti suodattimen yläpuolelta, jotta injektiopullon ilmaventtiili aukeaa.
HUOM.: Varmista, että valitset oikean letkun, jossa on suodatin. Leikkaa VAIN letku, jossa on suodatin.

5. Pidä 20 G:n, 1–1 ½ tuuman neulaa siten, että neulan kärjen aukko on poispäin annosteluaukon kalvosta.
- Vie neula kalvoon 45–60°:n kulmassa siten, että se lävistää annosteluaukon kalvon.
- Lisää neulan kulmaa asteittain samalla kun neula menee sisälle injektiopulloon.
6. Vedä (luovutussertifikaatin mukainen) tavoitetilavuus hitaasti ruiskuun VETÄMÄTTÄ ruiskuun ilmaa.
7. Tarkista ruisku huolellisesti sakan varalta ennen kuin jatkat. Jos sakkaa näkyy, ota yhteyttä yritykseen.
8. Varmista, että CD8‑/CD4‑positiivisen solukomponentin tilavuus vastaa komponentille luovutussertifikaatissa (RfIC) ilmoitettua tilavuutta.
Kun tilavuus on varmistettu, aseta injektiopullo ja ruisku vaakatasoon ja poista ruisku/neula injektiopullosta.
Irrota neula varovasti ruiskusta ja laita ruiskuun korkki.

9. Pidä injektiopullo edelleen vaakatasossa ja palauta se koteloon vuotojen estämiseksi.
10. Hävitä käyttämätön Breyanzi-valmiste.
Valmisteen antaminen
Ks. lisätietoja valmisteen annosta kohdasta Annostus ja antotapa.
- Huuhtele koko infuusioletku 0,9‑prosenttisella (9 mg/ml) natriumkloridi-injektionesteellä (i.v.) ennen jokaista CD8‑positiivisten ja CD4‑positiivisten solukomponenttien antoa sekä niiden jälkeen.
- Anna CD8‑positiivinen solukomponentti ensin. CD8‑positiivinen solukomponentti annetaan ensin kokonaan laskimoon infuusionopeudella 0,5 ml minuutissa lähimmän portin tai Y‑haaran (rinnakkaisletkun) kautta.
- Jos CD8‑positiivisen solukomponentin koko soluannokseen tarvitaan useampi kuin yksi ruisku, anna jokaisen ruiskun sisältämä valmiste peräkkäin siten, että ruiskujen sisällön antaminen tapahtuu yhtäjaksoisesti (ellei annoksen antamisen siirtämiseen ole kliinistä syytä, kuten infuusion aiheuttama reaktio). Kun CD8‑positiivinen solukomponentti on annettu, huuhtele letku 0,9‑prosenttisella (9 mg/ml) natriumkloridi-injektionesteellä.
- Anna CD4‑positiivinen solukomponentti heti, kun CD8‑positiivisen solukomponentin anto on valmis. Noudata samoja vaiheita ja infuusionopeuksia kuin CD8‑positiivisen solukomponentin annossa. Huuhtele letku CD4‑positiivisen solukomponentin annon jälkeen 0,9‑prosenttisella (9 mg/ml) natriumkloridi-injektionesteellä. Käytä riittävästi huuhdetta, jotta letku ja i.v.‑katetri puhdistuvat kokonaan. Infuusioaika vaihtelee ja on yleensä alle 15 minuuttia kumpaakin komponenttia kohden.
Toimenpiteet vahinkoaltistumisen tapahtuessa
- Vahinkoaltistumisen tapahduttua on noudatettava ihmisperäisen materiaalin käsittelyä koskevia paikallisia ohjeita. Työtasot ja materiaalit, jotka ovat mahdollisesti joutuneet kosketuksiin Breyanzi-valmisteen kanssa, on dekontaminoitava asianmukaisella desinfiointiaineella.
Lääkevalmisteen hävittämiseen liittyvät varotoimet
- Käyttämätön lääkevalmiste ja kaikki materiaalit, jotka ovat olleet kosketuksissa Breyanzi-valmisteen kanssa (kiinteä ja nestemäinen jäte) on käsiteltävä ja hävitettävä mahdollisesti tartuntavaarallisena jätteenä ihmisperäisen materiaalin käsittelyä koskevien paikallisten ohjeiden mukaisesti.
Korvattavuus
BREYANZI infuusioneste, dispersio
1,1-70 x 10exp6 solua/ml / 1,1-70 x 10exp6 solua/ml 1 annos
- Ei korvausta.
ATC-koodi
L01XL08
Valmisteyhteenvedon muuttamispäivämäärä
21.11.2025
Yhteystiedot
09 2512 1244
www.bms.com/fi
medinfo.finland@bms.com