
LAZCLUZE tabletti, kalvopäällysteinen 80 mg, 240 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Lazcluze 80 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 80 mg latsertinibia (mesylaattimonohydraattina).
Lazcluze 240 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 240 mg latsertinibia (mesylaattimonohydraattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen.
Kliiniset tiedot
Käyttöaiheet
Lazcluze on tarkoitettu yhdistelmänä amivantamabin kanssa edenneen ei‑pienisoluisen keuhkosyövän (NSCLC) ensilinjan hoitoon aikuispotilaille, joilla on epidermaalisen kasvutekijän reseptorin (EGFR) eksonin 19 deleetioita tai eksonin 21 L858R-substituutiomutaatioita.
Ehto
Ainoastaan syöpälääkkeiden antoon perehtyneen lääkärin tulee aloittaa hoito.
Annostus ja antotapa
Lazcluze-hoidon saa aloittaa vain syöpälääkkeiden käyttöön perehtynyt lääkäri.
Ennen Lazcluze-hoidon aloittamista on tarkistettava EGFR-mutaatiopositiivisuus kasvainkudos- tai plasmanäytteestä käyttämällä validoitua testimenetelmää. Jos plasmanäytteessä ei havaita mutaatiota, testi on tehtävä kasvainkudosnäytteestä, jos saatavissa on riittävän suuri ja laadukas näyte, koska plasmatestin negatiivinen tulos saattaa olla virheellinen.
Annostus
Suositeltu Lazcluze-annos on 240 mg kerran vuorokaudessa yhdessä amivantamabin kanssa.
On suositeltavaa antaa Lazcluze-valmistetta mihin tahansa aikaan vuorokaudesta ennen amivantamabia, kun valmisteita annetaan samana päivänä. Katso amivantamabin suositeltua annostusta koskevat tiedot amivantamabin valmisteyhteenvedon kohdasta Annostus ja antotapa.
Laskimotromboemboliset (VTE) tapahtumat amivantamabin samanaikaisen käytön yhteydessä
Potilaille, jotka saavat Lazcluze-valmistetta yhdistelmänä amivantamabin kanssa, pitää hoitoa aloitettaessa antaa estolääkityksenä antikoagulantteja laskimotromboembolisten tapahtumien ennaltaehkäisyyn. Kliinisten hoitosuositusten mukaisesti potilaille pitää antaa estolääkityksenä joko suun kautta otettavaa suoraa antikoagulanttia tai pienimolekyylistä hepariinia. K‑vitamiiniantagonistien käyttöä ei suositella.
Iho- ja kynsireaktiot
Iho- ja kynsireaktioiden riskin ja vaikeusasteen vähentämiseksi Lazcluze-valmisteen ja amivantamabin yhdistelmää saavilla potilailla suositellaan estohoitoa suun kautta otettavilla ja paikallisesti käytettävillä antibiooteilla. Kasvoihin ja koko kehoon (päänahkaa lukuun ottamatta) suositellaan käyttämään ei-komedogeenista kosteusvoidetta (mieluiten keramidipohjainen tai muu ihoa pitkäkestoisesti kosteuttava koostumus, joka ei sisällä kuivattavia ainesosia), ja käsien ja jalkaterien pesemiseen suositellaan myös klooriheksidiiniliuosta. Potilaita on neuvottava rajoittamaan altistumista auringolle Lazcluze-yhdistelmähoidon aikana ja 2 kuukauden ajan sen jälkeen. Lisätietoja iho- ja kynsireaktioiden estohoidosta, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Hoidon kesto
Hoitoa on jatkettava, kunnes sairaus etenee tai ilmenee toksisuutta, joka ei ole hyväksyttävissä.
Unohtunut annos
Jos suunniteltu Lazcluze-annos jää ottamatta, se voidaan ottaa 12 tunnin kuluessa. Jos suunnitellusta annoksen ottamisajankohdasta on kulunut yli 12 tuntia, unohtunutta annosta ei saa ottaa, vaan seuraava annos on otettava tavanomaisen annostusaikataulun mukaisesti.
Annosmuutokset
Suositukset annoksen pienentämisestä haittavaikutusten vuoksi esitetään taulukossa 1.
Taulukko 1: Suositukset Lazcluze-annoksen pienentämisestä haittavaikutusten vuoksi | |
Annoksen pienennys | Suositeltu annos |
Aloitusannos | 240 mg kerran vuorokaudessa |
Annoksen 1. pienennys | 160 mg kerran vuorokaudessa |
Annoksen 2. pienennys | 80 mg kerran vuorokaudessa |
Annoksen 3. pienennys | Lopeta Lazcluze-hoito |
Annosmuutokset tiettyjen haittavaikutusten vuoksi esitetään taulukossa 2.
Katso amivantamabin annosmuutoksia koskevat tiedot amivantamabin valmisteyhteenvedon kohdasta Annostus ja antotapa.
Taulukko 2: Suositukset Lazcluze- ja amivantamabiannosten muuttamisesta haittavaikutusten vuoksi* | ||
Haittavaikutus | Vaikeusaste | Annosmuutos |
Interstitiaalinen keuhkosairaus (ILD) / pneumoniitti | Mikä tahansa vaikeusaste |
|
Laskimotromboemboliset tapahtumat (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Tapahtumat, joihin liittyy kliinistä epävakautta (esim. hengitysvajaus tai sydämen toimintahäiriö) |
|
Toistuva laskimotromboembolinen tapahtuma huolimatta antikoagulanttihoidosta terapeuttisella annoksella |
| |
Iho- ja kynsireaktiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | 1. aste |
|
2. aste |
| |
3. aste |
| |
4. aste (mukaan lukien vaikeat ihotaudit, joihin liittyy vesikelloja, rakkuloita tai ihon kesimistä, kuten toksinen epidermaalinen nekrolyysi) |
| |
Maksatoksisuus | 3.–4. aste |
|
Parestesia | 3.–4. aste |
|
Ripuli | 3. aste |
|
4. aste |
| |
Suutulehdus | 3.–4. aste |
|
Muut haittavaikutukset | 3.–4. aste |
|
* Katso amivantamabin suositeltua annostusta koskevat tiedot amivantamabin valmisteyhteenvedon kohdasta Annostus ja antotapa. | ||
Erityiset potilasryhmät
Iäkkäät
Annosta ei tarvitse muuttaa (ks. kohdat Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Populaatiofarmakokineettisen analyysin perusteella lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Tiedot vaikeaa munuaisten vajaatoimintaa sairastavista potilaista ovat suppeat. Latsertinibin farmakokinetiikkaa ei tunneta potilailla, joilla on loppuvaiheen munuaissairaus. Varovaisuutta on noudatettava, jos potilaalla on loppuvaiheen munuaissairaus (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Latsertinibin farmakokinetiikkaa ei tunneta potilailla, joilla on vaikea maksan vajaatoiminta. Varovaisuutta on noudatettava, jos potilaalla on vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää latsertinibia pediatrisille potilaille ei‑pienisoluisen keuhkosyövän hoitoon.
Antotapa
Lazcluze otetaan suun kautta. Tabletit niellään kokonaisina aterian yhteydessä tai tyhjään mahaan. Tabletteja ei pidä murskata, halkaista eikä pureskella.
Jos milloin tahansa Lazcluze-valmisteen ottamisen jälkeen ilmenee oksentelua, seuraava annos pitää ottaa seuraavana päivänä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle (vaikuttaville aineille) tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Interstitiaalinen keuhkosairaus / pneumoniitti
Latsertinibia ja amivantamabia saaneilla potilailla on raportoitu interstitiaalista keuhkosairautta (ILD) tai interstitiaalisen keuhkosairauden kaltaisia haittavaikutuksia (kuten pneumoniittia), mukaan lukien kuolemaan johtaneita tapahtumia (ks. kohta Haittavaikutukset). Potilaat, joilla oli anamneesissa interstitiaalinen keuhkosairaus, lääkkeen aiheuttama interstitiaalinen keuhkosairaus, steroidihoitoa vaatinut sädepneumoniitti tai näyttöä kliinisesti aktiivisesta interstitiaalisesta keuhkosairaudesta, suljettiin pois kliinisestä pivotaalitutkimuksesta.
Potilasta on seurattava interstitiaaliseen keuhkosairauteen / pneumoniittiin viittaavien oireiden (esim. hengenahdistuksen, yskän, kuumeen) varalta. Jos oireita kehittyy, Lazcluze-hoito on keskeytettävä näiden oireiden tutkimisen ajaksi. Epäilty interstitiaalinen keuhkosairaus tai interstitiaalisen keuhkosairauden kaltaiset haittavaikutukset on arvioitava, ja asianmukainen hoito on aloitettava tarpeen mukaan. Potilaan Lazcluze-hoito pitää lopettaa pysyvästi, jos interstitiaalinen keuhkosairaus tai interstitiaalisen keuhkosairauden kaltaiset haittavaikutukset varmistuvat (ks. kohta Annostus ja antotapa).
Laskimotromboemboliset tapahtumat (VTE)
Potilailla, jotka saivat Lazcluze-valmistetta yhdistelmänä amivantamabin kanssa, raportoitiin laskimotromboembolisia tapahtumia (myös kuolemaan johtaneita), joita olivat mm. syvä laskimotukos ja keuhkoembolia (ks. kohta Haittavaikutukset). Kliinisten hoitosuositusten mukaisesti potilaille pitää antaa estolääkityksenä joko suun kautta otettavaa suoraa antikoagulanttia tai pienimolekyylistä hepariinia. K‑vitamiiniantagonistien käyttöä ei suositella.
Laskimotromboembolisten tapahtumien oireita ja löydöksiä on seurattava. Potilaille, joilla ilmenee laskimotromboembolisia tapahtumia, on annettava hyytymisenestohoitoa kliinisen tarpeen mukaan. Jos laskimotromboembolisiin tapahtumiin liittyy kliinistä epävakautta, hoito pitää keskeyttää, kunnes potilaan tila on kliinisesti vakaa. Sen jälkeen kummankin lääkevalmisteen käyttöä voidaan jatkaa aiemmalla annoksella.
Jos laskimotromboembolisia tapahtumia ilmenee uudelleen asianmukaisesta hyytymisenestohoidosta huolimatta, amivantamabihoito on lopetettava. Lazcluze-hoitoa voidaan jatkaa aiemmalla annoksella (ks. kohta Annostus ja antotapa).
Iho- ja kynsireaktiot
Latsertinibin ja amivantamabin yhdistelmähoitoa saaneilla potilailla esiintyi ihottumaa (mukaan lukien aknetyyppistä ihottumaa), kutinaa ja ihon kuivumista (ks. kohta Haittavaikutukset). Potilaita on neuvottava rajoittamaan altistumista auringolle Lazcluze-yhdistelmähoidon aikana ja 2 kuukauden ajan sen jälkeen. On suositeltavaa käyttää suojavaatetusta ja laajakirjoista UVA/UVB-aurinkovoidetta. Ihottuman ehkäisemiseksi suositellaan estohoitoa. Se käsittää hoidon alussa estohoidon jollakin suun kautta otettavalla antibiootilla (esim. 100 mg doksisykliiniä tai minosykliiniä kaksi kertaa vuorokaudessa) päivästä 1 alkaen 12 ensimmäisen hoitoviikon ajan sekä päänahan paikallishoidon antibioottivoiteella (esim. 1 % klindamysiini) suun kautta otettavan antibioottihoidon päättymisen jälkeen seuraavien 9 kuukauden ajan. Kasvoihin ja koko kehoon (päänahkaa lukuun ottamatta) suositellaan ei-komedogeenista kosteusvoidetta (mieluiten keramidipohjainen tai muu ihoa pitkäkestoisesti kosteuttava koostumus, joka ei sisällä kuivattavia ainesosia), ja käsien ja jalkaterien pesemiseen suositellaan klooriheksidiiniliuosta päivästä 1 alkaen. Näitä toimia suositellaan jatkamaan koko hoidon ajan.
Hoidon alussa on suositeltavaa olla valmiina lääkemääräykset muita paikallisesti käytettäviä ja/tai suun kautta otettavia antibiootteja sekä paikallisesti käytettäviä kortikosteroideja varten, jotta estohoidosta huolimatta mahdollisesti kehittyvä ihottuma voidaan hoitaa viiveettä. Jos ilmenee iho- tai kynsireaktioita, on annettava tukihoitoa, paikallisesti käytettäviä kortikosteroideja ja paikallisesti käytettäviä ja/tai suun kautta otettavia antibiootteja. Asteen 3 tai huonosti siedettyjen asteen 2 tapahtumien yhteydessä on annettava lisäksi systeemisiä antibiootteja ja suun kautta otettavia steroideja sekä harkittava dermatologin konsultointia. Lazcluze-valmisteen annosta on pienennettävä, hoito on keskeytettävä tai lopetettava pysyvästi vaikeusasteen perusteella (ks. kohta Annostus ja antotapa).
Silmiin liittyvät häiriöt
Silmiin liittyviä häiriöitä, mukaan lukien sarveiskalvotulehdusta, esiintyi latsertinibin ja amivantamabin yhdistelmähoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Potilaat, joilla on pahenevia silmäoireita, on ohjattava viipymättä silmälääkärin vastaanotolle, ja piilolinssien käyttö on lopetettava, kunnes oireet on arvioitu.
Apuaineet
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti, eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Voimakkaat CYP3A4:n indusorit voivat pienentää latsertinibipitoisuutta plasmassa. Latsertinibi voi suurentaa CYP3A4:n ja BCRP:n substraattien pitoisuuksia plasmassa.
Lääkeaineet, jotka voivat muuttaa latsertinibipitoisuutta plasmassa
CYP3A4:n indusorit
Useiden rifampisiiniannosten (rifampisiini on voimakas CYP3A4:n indusori) antaminen samanaikaisesti latsertinibin kanssa pienensi latsertinibin Cmax‑arvoa 72 % ja AUC‑arvoa 83 % terveillä tutkittavilla. Lazcluze-valmisteen ja voimakkaiden CYP3A4:n indusorien (esim. karbamatsepiini, fenytoiini, rifampisiini, mäkikuisma) samanaikaista antoa on vältettävä. Lazcluze-valmisteen ja kohtalaisten CYP3A4:n indusorien samanaikainen käyttö voi myös pienentää latsertinibin pitoisuutta plasmassa, joten kohtalaisten CYP3A4:n indusorien (esim. bosentaani, efavirentsi, modafiniili) käytössä on noudatettava varovaisuutta.
CYP3A4:n estäjät
Useiden itrakonatsoliannosten (itrakonatsoli on voimakas CYP3A4:n estäjä) antaminen samanaikaisesti latsertinibin kanssa suurensi latsertinibin Cmax‑arvoa 1,19‑kertaisesti ja AUC‑arvoa 1,46‑kertaisesti terveillä tutkittavilla. Aloitusannosta ei tarvitse muuttaa, kun Lazcluze-valmistetta käytetään samanaikaisesti CYP3A4:n estäjien kanssa.
Mahahappoja vähentävät lääkeaineet
Latsertinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja, kun sitä annettiin samanaikaisesti mahahappoja vähentävien lääkeaineiden (protonipumpun estäjien ja H2‑reseptorin salpaajien) kanssa. Annosta ei tarvitse muuttaa, kun Lazcluze-valmistetta käytetään yhdessä mahahappoja vähentävien lääkeaineiden kanssa.
Lääkeaineet, joiden pitoisuus plasmassa voi muuttua Lazcluze-valmisteen vaikutuksesta
CYP3A4:n substraatit
Useiden 160 mg:n Lazcluze-annosten antaminen kerran vuorokaudessa samanaikaisesti midatsolaamin (CYP3A4:n substraatti) kanssa suurensi midatsolaamin Cmax‑arvoa 1,39‑kertaisesti ja AUC‑arvoa 1,47‑kertaisesti. Lääkevalmisteita, joilla on kapea terapeuttinen indeksi ja jotka ovat CYP3A4:n substraatteja (esim. siklosporiini, everolimuusi, pimotsidi, kinidiini, sirolimuusi, takrolimuusi), on käytettävä varoen, koska latsertinibi voi suurentaa näiden lääkevalmisteiden pitoisuutta plasmassa.
BCRP:n substraatit
Useiden 160 mg:n Lazcluze-annosten antaminen kerran vuorokaudessa samanaikaisesti rosuvastatiinin (BCRP:n substraatti) kanssa suurensi rosuvastatiinin Cmax‑arvoa 2,24‑kertaisesti ja AUC‑arvoa 2,02‑kertaisesti. Lääkevalmisteita, joilla on kapea terapeuttinen indeksi ja jotka ovat BCRP:n substraatteja (esim. sunitinibi), on käytettävä varoen, koska latsertinibi voi suurentaa näiden lääkevalmisteiden pitoisuutta plasmassa.
CYP1A2:n substraatit
CYP1A2:n induktiota ei voida sulkea pois. Sen vuoksi varovaisuutta suositellaan käytettäessä samanaikaisesti CYP1A2:n substraatteja (esim. titsanidiinia).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehille ja naisille
Naisia, jotka voivat tulla raskaaksi, on kehotettava käyttämään tehokasta ehkäisyä hoidon aikana ja 3 viikon ajan hoidon päättymisen jälkeen.
Miespotilaita, joiden naispuolinen kumppani voi tulla raskaaksi, on kehotettava käyttämään tehokasta ehkäisyä (esim. kondomia) ja olemaan luovuttamatta tai ottamatta talteen siemennestettä hoidon aikana ja 3 viikon ajan viimeisen latsertinibiannoksen jälkeen.
Raskaus
Latsertinibin käytöstä raskaana oleville naisille ei ole tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (alkion tai sikiön eloonjäämisen vähenemistä ja sikiön alentunutta painoa) (ks. kohta Prekliiniset tiedot turvallisuudesta). Latsertinibin vaikutusmekanismin ja koe-eläimistä saatujen tietojen perusteella latsertinibi saattaa raskauden aikana käytettynä vahingoittaa sikiötä. Latsertinibia ei pidä käyttää raskauden aikana, ellei hoidosta naiselle koituvan hyödyn katsota olevan suurempi kuin sikiölle mahdollisesti aiheutuvat riskit. Jos potilas tulee raskaaksi tämän lääkevalmisteen käytön aikana, potilaalle on kerrottava sikiölle mahdollisesti aiheutuvista riskeistä.
Imetys
Ei tiedetä, erittyvätkö latsertinibi tai sen metaboliitit ihmisillä äidinmaitoon tai vaikuttavatko ne maidontuotantoon. Koska imetettävään lapseen kohdistuvia riskejä ei voida sulkea pois, naispotilaita on kehotettava olemaan imettämättä hoidon aikana ja 3 viikon ajan viimeisen latsertinibiannoksen jälkeen.
Hedelmällisyys
Lazcluze-valmisteen vaikutuksista ihmisen hedelmällisyyteen ei ole tietoja. Eläimillä tehdyt tutkimukset ovat osoittaneet, että latsertinibi vaikuttaa sukupuolielimiin naarailla (vähentämällä kiima-aikoja ja keltarauhasten määrää) ja uroksilla (aiheuttamalla kivesten degeneratiivisia muutoksia) ja että se voi heikentää naaraiden ja urosten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lazcluze-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Jos potilaalla ilmenee hoitoon liittyviä oireita (kuten uupumusta), jotka vaikuttavat hänen keskittymis- ja reaktiokykyynsä, on suositeltavaa, ettei hän aja autoa tai käytä koneita, ennen kuin vaikutus loppuu.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Yleisimpiä haittavaikutuksia kaikki vaikeusasteet mukaan lukien olivat ihottuma (89 %), kynsitoksisuus (71 %), infuusioon liittyvä reaktio (amivantamabi) (63 %), hypoalbuminemia (amivantamabi) (48 %), maksatoksisuus (47 %), turvotus (amivantamabi) (47 %), suutulehdus (43 %), laskimotromboembolia (37 %), parestesia (34 %), uupumus (32 %), ummetus (29 %), ripuli (29 %), ihon kuivuminen (26 %), ruokahalun heikentyminen (24 %), kutina (24 %), hypokalsemia (21 %), muut silmiin liittyvät häiriöt (21 %) ja pahoinvointi (21 %).
Yleisimpiä vakavia haittavaikutuksia olivat laskimotromboembolia (11 %), keuhkokuume (4,0 %), ihottuma (3,1 %), interstitiaalinen keuhkosairaus / pneumoniitti (2,9 %), COVID‑19-tauti (2,4 %), maksatoksisuus (2,4 %), pleuraeffuusio (2,1 %), infuusioon liittyvä reaktio (amivantamabi) (2,1 %), hengitysvajaus (1,4 %), uupumus (1,2 %), turvotus (amivantamabi) (1,2 %), hypoalbuminemia (amivantamabi) (1,2 %) ja hyponatremia (1,2 %).
Yleisimpiä kumman tahansa hoidon lopettamiseen johtaneita haittavaikutuksia Lazcluze-valmisteen ja amivantamabin yhdistelmää saaneilla potilailla olivat ihottuma (6 %), infuusioon liittyvä reaktio (amivantamabi) (4,5 %), kynsitoksisuus (3,6 %), interstitiaalinen keuhkosairaus / pneumoniitti (2,9 %), laskimotromboembolia (2,9 %), keuhkokuume (1,9 %) ja turvotus (amivantamabi) (1,7 %).
Haittavaikutustaulukko
Taulukossa 3 esitetään yhteenveto latsertinibin ja amivantamabin yhdistelmähoitoa saaneilla potilailla esiintyneistä haittavaikutuksista.
Tiedot kuvaavat 421 potilaan altistusta latsertinibille MARIPOSA-tutkimuksessa, jossa potilaat saivat latsertinibin ja amivantamabin yhdistelmähoitoa. Latsertinibialtistuksen mediaani oli 18,5 kuukautta (vaihteluväli: 0,2–31,4 kuukautta).
Kliinisissä tutkimuksissa havaitut haittavaikutukset on lueteltu jäljempänä esiintyvyysluokittain seuraavan esitystavan mukaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin esiintyvyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 3: Haittavaikutukset latsertinibin ja amivantamabin yhdistelmähoitoa saaneilla potilailla | |||
Elinjärjestelmäluokka Haittavaikutus | Esiintyvyysluokka | Mikä tahansa aste (%) | 3.–4. aste (%) |
| Aineenvaihdunta ja ravitsemus | |||
| Hypoalbuminemiaa, b | Hyvin yleinen | 48 | 5 |
| Ruokahalun heikentyminen | 24 | 1,0 | |
| Hypokalsemia | 21 | 2,1 | |
| Hypokalemia | 14 | 3,1 | |
| Hypomagnesemia | Yleinen | 5 | 0 |
| Hermosto | |||
| Parestesiaa | Hyvin yleinen | 34 | 1,7 |
| Huimausa | 13 | 0 | |
| Silmät | |||
| Muut silmiin liittyvät häiriöta | Hyvin yleinen | 21 | 0,5 |
| Näön heikkeneminena | Yleinen | 4,5 | 0 |
| Sarveiskalvotulehdus | 2,6 | 0,5 | |
| Silmäripsien kasvua | 1,9 | 0 | |
| Verisuonisto | |||
| Laskimotromboemboliaa | Hyvin yleinen | 37 | 11 |
| Hengityselimet, rintakehä ja välikarsina | |||
| Interstitiaalinen keuhkosairaus / pneumoniittia | Yleinen | 3,1 | 1,2 |
| Ruoansulatuselimistö | |||
| Suutulehdusa | Hyvin yleinen | 43 | 2,4 |
| Ripuli | 29 | 2,1 | |
| Ummetus | 29 | 0 | |
| Pahoinvointi | 21 | 1,2 | |
| Oksentelu | 12 | 0,5 | |
| Vatsakipua | 11 | 0 | |
| Peräpukamat | Yleinen | 10 | 0,2 |
| Maksa ja sappi | |||
| Maksatoksisuusa | Hyvin yleinen | 47 | 9 |
| Iho ja ihonalainen kudos | |||
| Ihottumaa | Hyvin yleinen | 89 | 27 |
| Kynsitoksisuusa | 71 | 11 | |
| Ihon kuivuminena | 26 | 1,0 | |
| Kutina | 24 | 0,5 | |
| Käsi-jalkaoireyhtymä | Yleinen | 6 | 0,2 |
| Nokkosihottuma | 1,2 | 0 | |
| Luusto, lihakset ja sidekudos | |||
| Lihaskrampit | Hyvin yleinen | 17 | 0,5 |
| Lihassärky | 13 | 0,7 | |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Turvotusa, b | Hyvin yleinen | 47 | 2,9 |
| Uupumusa | 32 | 3,8 | |
| Kuume | 12 | 0 | |
| Vammat, myrkytykset ja hoitokomplikaatiot | |||
| Infuusioon liittyvä reaktiob | Hyvin yleinen | 63 | 6 |
a yhdistelmätermejä b koskee vain amivantamabia | |||
Valikoitujen haittavaikutusten kuvaus
Laskimotromboembolia
Laskimotromboembolisia tapahtumia, mukaan lukien syvä laskimotukos (14,5 %) ja keuhkoembolia (17,3 %), raportoitiin 37 %:lla latsertinibin ja amivantamabin yhdistelmähoitoa saaneista potilaista. Useimmat tapauksista olivat asteen 1 tai 2 tapahtumia. Asteen 3–4 tapahtumia ilmeni 11 %:lla potilaista, ja 0,5 % latsertinibin ja amivantamabin yhdistelmähoitoa saaneista potilaista kuoli. Tietoja estolääkityksenä käytettävistä antikoagulanteista ja laskimotromboembolisten tapahtumien hoidosta, ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Latsertinibin ja amivantamabin yhdistelmähoitoa saaneilla potilailla laskimotromboembolisen tapahtuman ensimmäiseen ilmenemiseen kuluneen ajan mediaani oli 84 vuorokautta. Laskimotromboemboliset tapahtumat johtivat kumman tahansa hoidon lopettamiseen 2,9 %:lla potilaista.
Interstitiaalinen keuhkosairaus (ILD) / pneumoniitti
Interstitiaalista keuhkosairautta tai interstitiaalisen keuhkosairauden kaltaisia haittavaikutuksia (kuten pneumoniittia) on raportoitu käytettäessä latsertinibia yhdessä amivantamabin kanssa sekä muiden EGFR:n estäjien kanssa. Interstitiaalista keuhkosairautta tai pneumoniittia raportoitiin 3,1 %:lla latsertinibin ja amivantamabin yhdistelmähoitoa saaneista potilaista, ja 0,2 % tapauksista johti kuolemaan. Potilaat, joilla oli anamneesissa interstitiaalinen keuhkosairaus, lääkkeen aiheuttama interstitiaalinen keuhkosairaus, steroidihoitoa vaatinut sädepneumoniitti tai näyttöä kliinisesti aktiivisesta interstitiaalisesta keuhkosairaudesta, suljettiin pois kliinisestä tutkimuksesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iho- ja kynsireaktiot
On ilmennyt ihottumaa (mukaan aknetyyppinen ihottuma), kutinaa ja ihon kuivumista. Ihottumaa ilmeni 89 %:lla latsertinibin ja amivantamabin yhdistelmähoitoa saaneista potilaista. Useimmat tapauksista olivat asteen 1 tai 2 tapahtumia. Asteen 3 tapahtumia ilmeni 27 %:lla potilaista. Ihottumaa, joka johti kumman tahansa hoidon lopettamiseen, ilmeni 6 %:lla potilaista. Yleensä ihottuma kehittyi neljän ensimmäisen hoitoviikon aikana, ja ihottuman ilmenemiseen kuluneen ajan mediaani oli 14 vuorokautta. Latsertinibin ja amivantamabin yhdistelmähoitoa saaneilla potilailla ilmeni kynsitoksisuutta. Useimmat tapauksista olivat asteen 1 tai 2 tapahtumia. Asteen 3 kynsitoksisuutta ilmeni 11 %:lla potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilailla, jotka saivat hoitona Lazcluze-valmisteen ja amivantamabin yhdistelmää, tehtiin vaiheen 2 tutkimus estohoidon käytön arvioimiseksi. Estohoito käsitti suun kautta otettavan antibiootin, päänahkaan paikallisesti käytettävän antibiootin, kosteusvoiteen kasvoihin ja koko kehoon (päänahkaa lukuun ottamatta) ja antiseptisen aineen käsiin ja jalkateriin (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Asteen ≥ 2 dermatologisten haittavaikutusten ilmaantuvuuden osoitettiin vähentyneen ensimmäisten 12 hoitoviikon aikana verrattuna tavanomaiseen kliinisessä hoitokäytännössä käytettävään dermatologiseen hoitoon (38,6 % vs. 76,5 %, p < 0,0001). Lisäksi päänahkaan liittyvät asteen ≥ 2 haittavaikutukset vähenivät ensimmäisten 12 hoitoviikon aikana (8,6 % vs. 29,4 %), ja myös annosta pienennettiin (7,1 % vs. 19,1 %), hoito keskeytettiin (15,7 % vs. 33,8 %) ja hoito lopetettiin (1,4 % vs. 4,4 %) dermatologisten haittavaikutusten vuoksi harvemmin.
Silmiin liittyvät häiriöt
Silmiin liittyviä häiriöitä, mukaan lukien sarveiskalvotulehdus (2,6 %), ilmeni latsertinibin ja amivantamabin yhdistelmähoitoa saaneilla potilailla. Muita raportoituja haittavaikutuksia olivat silmäripsien kasvu, näön heikkeneminen ja muut silmiin liittyvät häiriöt. Useimmat tapauksista olivat asteen 1–2 tapahtumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksatoksisuus
Maksatoksisuuteen liittyviä reaktioita ilmeni 47 %:lla latsertinibin ja amivantamabin yhdistelmähoitoa saaneista potilaista. Useimmat tapauksista olivat asteen 1–2 tapahtumia, ja asteen 3–4 maksatoksisuutta ilmeni 9 %:lla potilaista. Useimmat tapaukset liittyivät kohonneisiin seerumin transaminaasipitoisuuksiin (36 %:lla alaniiniaminotransferaasipitoisuus suureni ja 29 %:lla aspartaattiaminotransferaasipitoisuus suureni). Valtaosa potilaista, joilla oli kohonneita transaminaasipitoisuuksia, pystyi jatkamaan tutkimushoitoa ilman muutoksia tutkimushoitoon, mutta pieni joukko hoidettiin keskeyttämällä hoito tai pienentämällä annosta. Latsertinibin ja amivantamabin yhdistelmällä tehdyissä kliinisissä tutkimuksissa ei todettu maksan vajaatoimintaa eikä kuolemaan johtanutta maksatoksisuutta.
Latsertinibimonoterapiassa on todettu yksittäisinä tapauksina raportoituja suurentuneita alkalisen fosfataasin pitoisuuksia ja pitkään koholla olleita bilirubiinipitoisuuksia.
Parestesia
Parestesiaa ilmeni 34 %:lla latsertinibin ja amivantamabin yhdistelmähoitoa saaneista potilaista. Useimmat tapauksista olivat asteen 1–2 tapahtumia, ja asteen 3 parestesiaa ilmeni 1,7 %:lla potilaista. Valtaosalla potilaista parestesia hävisi, kun hoito keskeytettiin tai annosta pienennettiin.
Suutulehdus
Suutulehdusta ilmeni 43 %:lla latsertinibin ja amivantamabin yhdistelmähoitoa saaneista potilaista. Useimmat tapauksista olivat asteen 1–2 tapahtumia, ja asteen 3 suutulehdusta ilmeni 2,4 %:lla potilaista.
Ripuli
Ripulia ilmeni 29 %:lla latsertinibin ja amivantamabin yhdistelmähoitoa saaneista potilaista. Useimmat tapauksista olivat asteen 1–2 tapahtumia, ja asteen 3 ripulia ilmeni 2,1 %:lla potilaista.
Erityisryhmät
Iäkkäät
Latsertinibin käytöstä 75-vuotiaille ja sitä vanhemmille potilaille on vain vähän kliinistä tietoa (ks. kohta Farmakodynamiikka). Asteen 3 tai sitä vaikeampiasteisia haittatapahtumia raportoitiin enemmän iäkkäämmillä potilailla (≥ 65-vuotiailla) kuin < 65-vuotiailla potilailla (81 % vs. 70 %). Lääkityksen keskeyttäminen ja annoksen pienentäminen olivat yhtä yleisiä, mutta kumman tahansa hoidon lopettamiseen johtaneet haittatapahtumat olivat ≥ 65-vuotiailla yleisempiä kuin < 65-vuotiailla potilailla (47 % vs. 25 %).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Lazcluze-valmisteen yliannostukseen ei ole tunnettua spesifistä vastalääkettä. Yliannostuksen ilmetessä lopeta Lazcluze-hoito ja aloita yleiset tukitoimet. Potilaita pitää seurata tarkoin mahdollisten haittavaikutusten oireiden ja löydösten havaitsemiseksi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, proteiinikinaasin estäjät, ATC-koodi: L01EB09.
Vaikutusmekanismi
Latsertinibi on irreversiibeli epidermaalisen kasvutekijäreseptorin tyrosiinikinaasin estäjä. Se estää selektiivisesti sekä primaarisia aktivoivia EGFR-geenin mutaatioita (eksonin 19 deleetioita ja eksonin 21 L858R-substituutiomutaatioita) että resistenssin aiheuttavaa EGFR-geenin T790M-mutaatiota, mutta aktiivisuus villityypin EGFR-geenejä vastaan on vähäisempää.
Farmakodynaamiset vaikutukset
Turvallisuutta koskevat altistus-vasteanalyysit osoittivat, että parestesian ja suutulehduksen esiintyvyys vaikuttaa lisääntyvän latsertinibialtistuksen kasvaessa.
Sydämen elektrofysiologia
Latsertinibin QTc-aikaa pidentävää vaikutusta arvioitiin altistus-vasteanalyysilla käyttämällä kliinisiä tietoja vaiheen I/II tutkimuksesta, jossa 243 ei‑pienisoluista keuhkosyöpää sairastavaa potilasta sai 20, 40, 80, 120, 160, 240 tai 320 mg latsertinibia kerran vuorokaudessa. Altistus-vasteanalyysissa ei ilmennyt kliinisesti merkittävää yhteyttä plasman latsertinibipitoisuuden ja QTc-ajan muutoksen välillä.
Kliininen teho ja turvallisuus
MARIPOSA on satunnaistettu, avoin, aktiivikontrolloitu, vaiheen 3 monikeskustutkimus, jossa arvioidaan Lazcluze-valmisteen ja amivantamabin yhdistelmähoidon tehoa ja turvallisuutta verrattuna osimertinibimonoterapiaan, kun niitä käytetään ensilinjan hoitona potilaille, joilla on EGFR-mutaatiopositiivinen, paikallisesti edennyt tai metastasoitunut ei‑pienisoluinen keuhkosyöpä, joka ei ole hoidettavissa kuratiivisella hoidolla. Edellytyksenä oli, että potilasnäytteistä oli paikallisessa testauksessa tunnistettu jompikumpi kahdesta yleisestä EGFR-mutaatiosta (eksonin 19 deleetio tai eksonin 21 L858R-substituutiomutaatio). Kaikkien potilaiden kasvainkudosnäytteet (94 %) ja/tai plasmanäytteet (6 %) testattiin paikallisesti EGFR-geenin mutaatiostatuksen (eksonin 19 deleetio ja/tai eksonin 21 L858R-substituutiomutaatio) määrittämiseksi käyttämällä polymeraasiketjureaktiota (PCR, polymerase chain reaction) 65 %:lla ja uuden sukupolven sekvensointia (NGS, next generation sequencing) 35 %:lla potilaista.
Yhteensä 1 074 potilasta satunnaistettiin (2:2:1) saamaan Lazcluze-valmisteen ja amivantamabin yhdistelmähoitoa, osimertinibimonoterapiaa tai Lazcluze-monoterapiaa, kunnes sairaus etenee tai ilmenee toksisuutta, joka ei ole hyväksyttävissä. Lazcluze-valmistetta annettiin 240 mg suun kautta kerran vuorokaudessa. Amivantamabia annettiin laskimoon 1050 mg (alle 80 kg painaville potilaille) tai 1400 mg (vähintään 80 kg painaville potilaille) kerran viikossa 4 viikon ajan ja sen jälkeen joka toinen viikko viikosta 5 alkaen. Osimertinibia annettiin 80 mg suun kautta kerran vuorokaudessa. Satunnaistaminen ositettiin EGFR-mutaatiotyypin (eksonin 19 deleetio tai eksonin 21 L858R-substituutiomutaatio), etnisen taustan (aasialainen tai muu kuin aasialainen) ja aiempien aivometastaasien (kyllä tai ei) mukaan.
Lähtötilanteen demografiset tiedot ja sairauden ominaisuudet olivat samankaltaiset eri hoitohaaroissa. Iän mediaani oli 63 vuotta (vaihteluväli: 25–88 vuotta), 45 % potilaista oli ≥ 65-vuotiaita ja 11 % ≥ 75-vuotiaita, 62 % oli naisia ja 59 % oli aasialaisia ja 38 % valkoihoisia. Lähtötilanteen ECOG (Eastern Cooperative Oncology Group) ‑toimintakykyluokka oli 0 (34 %) tai 1 (66 %), 69 % potilaista ei ollut koskaan tupakoinut, 41 %:lla oli aiemmin ollut aivometastaaseja ja 90 %:lla todettiin alkuperäisen diagnosoinnin yhteydessä levinneisyysasteen IV syöpä. EGFR-mutaatioista 60 % oli eksonin 19 deleetioita ja 40 % eksonin 21 L858R-substituutiomutaatioita.
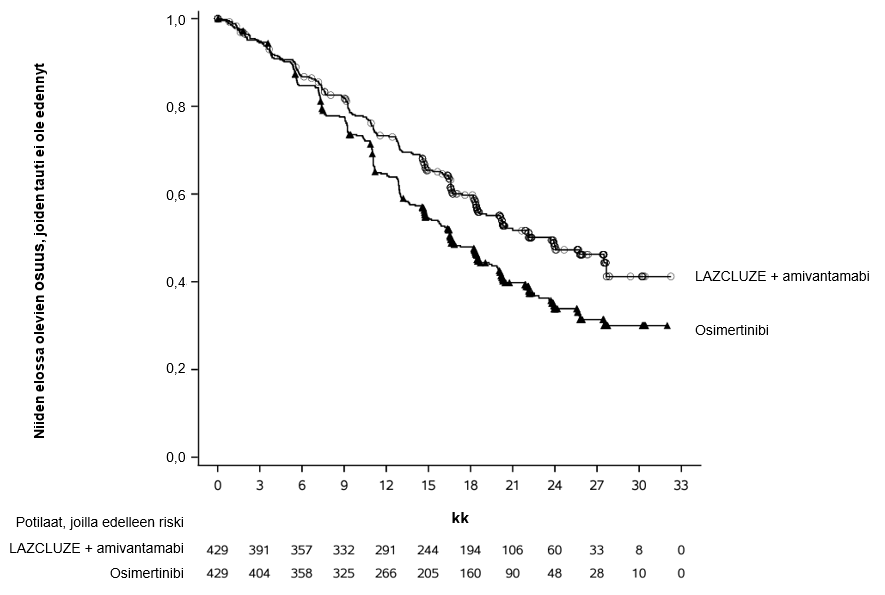
Sokkoutettu riippumaton keskitetty arviointi (BICR) osoitti, että Lazcluze-valmisteen ja amivantamabin yhdistelmähoidolla saavutettiin tilastollisesti merkitsevä etenemättömyysajan (PFS) pidentyminen.
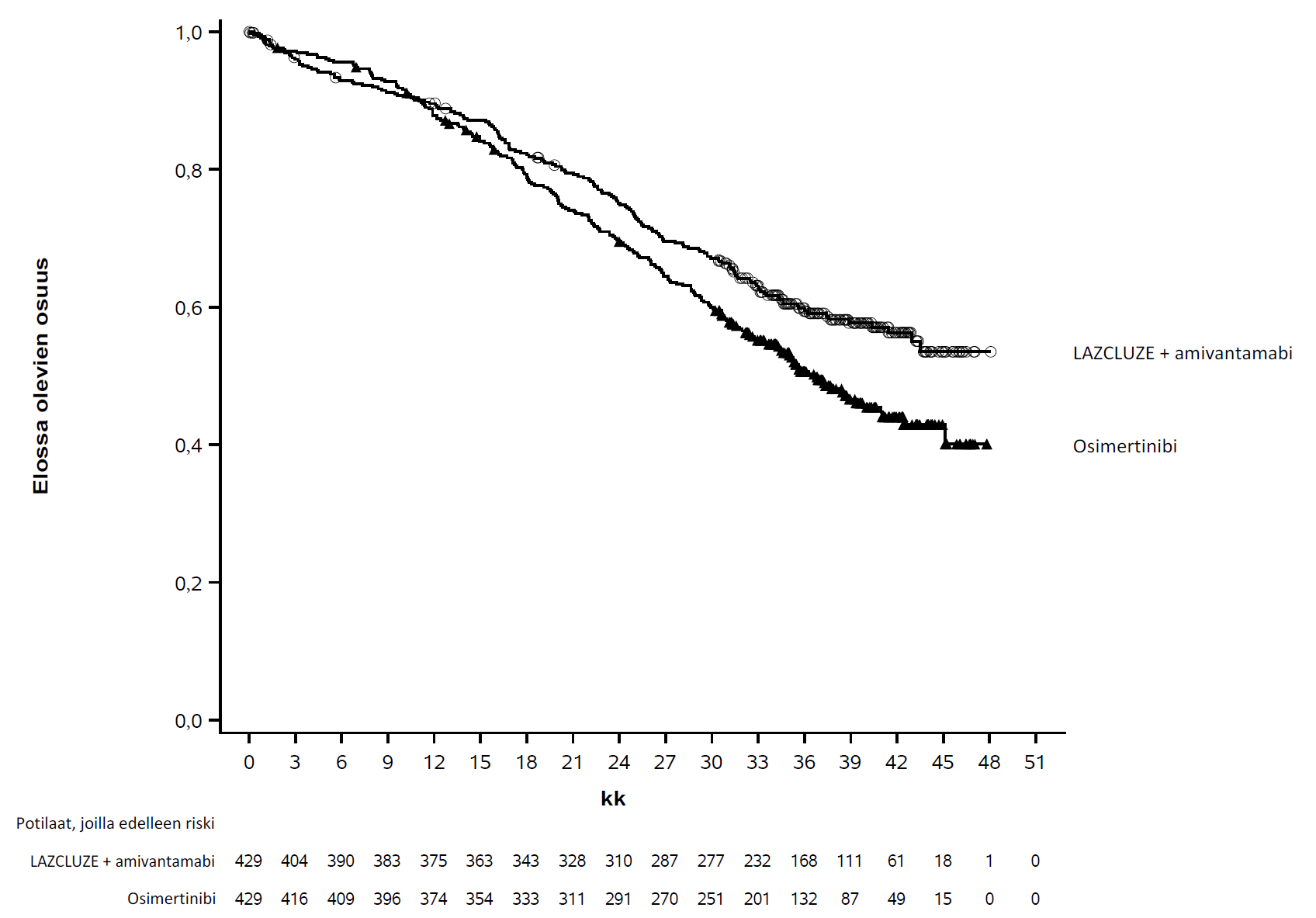
Kokonaiselinajan loppuanalyysissa todettiin kokonaiselinajan tilastollisesti merkitsevä piteneminen Lazcluze-valmisteen ja amivantamabin yhdistelmän käytössä osimertinibiin verrattuna (ks. taulukko 4 ja kuva 2).
Taulukossa 4, kuvassa 1 ja kuvassa 2 esitetään yhteenveto Lazcluze-valmisteen ja amivantamabin yhdistelmähoidon tehosta.
Taulukko 4: Tehoa koskevat tulokset MARIPOSA-tutkimuksesta | ||
Lazcluze + amivantamabi (N = 429) | Osimertinibi (N = 429) | |
Etenemättömyysaika (PFS)a | ||
Tapahtumien lukumäärä | 192 (45 %) | 252 (59 %) |
Mediaani, kk (95 %:n luottamusväli) | 23,7 (19,1–27,7) | 16,6 (14,8–18,5) |
Riskitiheyksien suhde (95 %:n luottamusväli); p‑arvo | 0,70 (0,58–0,85); p = 0,0002 | |
Kokonaiselinaika (OS) | ||
Tapahtumien lukumäärä | 173 (40 %) | 217 (51 %) |
Mediaani, kk (95 %:n luottamusväli) | NE (42,9–NE) | 36,7 (33,4–41,0) |
Riskitiheyksien suhde (95 %:n luottamusväli); p‑arvo | 0,75 (0,61–0,92); p = 0,0048 | |
Objektiivinen hoitovaste (ORR)a, b | ||
ORR, % (95 %:n luottamusväli) | 80 % (76–84 %) | 77 % (72–81 %) |
Vasteen kesto (DOR)a,b | ||
Mediaani, kk (95 %:n luottamusväli) | 25,8 (20,3–33,9) | 18,1 (14,8–20,1) |
PFS = Progression-free survival; OS = Overall survival; ORR = Objective response rate; DOR = Duration of response; NE = ei arvioitavissa (not estimable). Etenemättömyysaikaa koskevien tietojen tiedonkeruun päättymispäivä oli 11.8.2023 ja seurannan mediaani oli 22,0 kuukautta. Objektiivista hoitovastetta ja vasteen kestoa koskevien tietojen tiedonkeruun päättymispäivä oli 13.5.2024 ja seurannan mediaani oli 31,3 kuukautta. Kokonaiselinaikaa koskevien tietojen tiedonkeruun päättymispäivä oli 4.12.2024 ja seurannan mediaani oli 37,8 kuukautta. a Sokkoutettu riippumaton keskitetty arvio (BICR) RECIST v1.1 ‑kriteerien mukaan. b Perustuu varmistettuihin vasteen saaneisiin. | ||
Kuva 1: Kaplan–Meierin käyrä, joka kuvaa sokkoutetun riippumattoman keskitetyn arvion (BICR) mukaista etenemättömyysaikaa aiemmin hoitamattomilla ei‑pienisoluista keuhkosyöpää sairastavilla potilailla
Kuva 2: Kaplan–Meierin käyrä, joka kuvaa aiemmin hoitamattomien ei‑pienisoluista keuhkosyöpää sairastavien potilaiden kokonaiselinaikaa
MARIPOSA-tutkimuksessa ennalta määriteltyjä päätetapahtumia olivat kallonsisäinen objektiivinen hoitovaste (ORR) ja vasteen kesto (DOR) riippumattoman keskitetyn arvion (BICR) mukaan. Alaryhmässä, jossa potilailla oli lähtötilanteessa kallonsisäisiä leesioita, Lazcluze-valmisteen ja amivantamabin yhdistelmähoidolla saavutettiin vastaava kallonsisäinen objektiivinen hoitovaste kuin verrokkivalmisteella. Tutkimussuunnitelman mukaisesti kaikille potilaille MARIPOSA-tutkimuksessa tehtiin useita aivojen magneettikuvauksia kallonsisäisen vasteen ja vasteen keston arvioimiseksi. Tulosten yhteenveto esitetään taulukossa 5.
Taulukko 5: Kallonsisäinen objektiivinen hoitovaste ja vasteen kesto sokkoutetun riippumattoman keskitetyn arvion (BICR) mukaan tutkittavilla, joilla oli lähtötilanteessa kallonsisäisiä leesioita | |||
Lazcluze + amivantamabi (N = 180) | Osimertinibi (N = 186) | ||
Kallonsisäisen kasvaimen vasteen arviointi | |||
Kallonsisäinen objektiivinen hoitovaste (täydellinen vaste + osittainen vaste), % (95 %:n luottamusväli) | 78 % (71–84 %) | 77 % (71–83 %) | |
Täydellinen vaste | 64 % | 59 % | |
Kallonsisäisen vasteen kesto | |||
Mediaani, kk (95 %:n luottamusväli) | 35,0 (20,4–NE) | 25,1 (22,1–31,2) | |
NE = ei arvioitavissa. Kallonsisäistä objektiivista hoitovastetta ja vasteen kestoa koskevien tietojen tiedonkeruun päättymispäivä oli 4.12.2024 ja seurannan mediaani oli 37,8 kuukautta. | |||
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Lazcluze-valmisteen käytöstä ei‑pienisoluisen keuhkosyövän hoidossa kaikissa pediatrisissa potilasryhmissä.
Farmakokinetiikka
Kerta-annoksen ja toistuvien kerran vuorokaudessa annettavien annosten suun kautta annon jälkeen latsertinibin enimmäispitoisuus plasmassa (Cmax) ja plasman pitoisuus-aikakäyrän alle jäävä pinta-ala (AUC) suurenivat suunnilleen suhteessa annokseen 20–320 mg:n annosalueella.
Kerran vuorokaudessa tapahtuvalla annostuksella vakaan tilan altistus plasmassa saavutettiin päivään 15 mennessä, ja annoksella 240 mg kerran vuorokaudessa vakaan tilaan kertymän havaittiin olevan noin kaksinkertainen.
Latsertinibialtistus plasmassa oli vastaava annettaessa latsertinibia yhdessä amivantamabin kanssa kuin käytettäessä sitä monoterapiana.
Imeytyminen
Cmax-arvon saavuttamiseen kuluneen ajan mediaani oli kerta-annoksen jälkeen vastaava kuin vakaassa tilassa, ja se vaihteli kahdesta neljään tuntiin.
Kun latsertinibia annettiin 240 mg runsasrasvaisen aterian (noin 800 – 1 000 kcal, noin 50 % rasvaa) yhteydessä, latsertinibin Cmax- ja AUC-arvot olivat verrattavissa paastoarvoihin, mikä viittaa siihen, että latsertinibi voidaan ottaa joko aterian yhteydessä tai tyhjään mahaan.
Jakautuminen
Latsertinibi jakautui laajasti; sen keskimääräinen (CV %) näennäinen jakautumistilavuus oli 240 mg:n annoksella 4 264 l (43,2 %). Latsertinibin keskimääräinen (CV %) sitoutuminen plasman proteiineihin oli ihmisillä noin 99,2 % (0,13 %). Latsertinibin todettiin suun kautta annettuna sekä in vitro ‑inkubaation aikana sitoutuvan kovalenttisesti ihmisen veren ja plasman proteiineihin.
Metabolia
Latsertinibi metaboloituu pääasiassa glutationikonjugaation välityksellä (joko entsymaattisesti glutationi‑S-transferaasin kautta tai ei‑entsymaattisesti) sekä CYP3A4:n välityksellä. Yleisimpiä metaboliitteja ovat glutationin kataboliitit, joita pidetään kliinisesti inaktiivisina. Plasman latsertinibialtistukseen vaikutti GSTM1-geenin välityksellä tapahtuva metabolia, joka johti pienempään altistukseen (ero oli vähemmän kuin kaksinkertainen) potilailla, joilla oli muu kuin GSTM1 tyhjä ‑genotyyppi. Annosta ei tarvitse muuttaa GSTM1‑statuksen perusteella.
Eliminaatio
240 mg:n annoksella latsertinibin keskimääräinen (CV %) näennäinen puhdistuma oli 44,5 l/h (29,5 %) ja terminaalinen puoliintumisaika 64,7 tuntia (32,8 %).
Erittyminen
Suun kautta annetun radioaktiivisesti merkityn latsertinibin kerta-annoksen jälkeen noin 86 % annoksesta erittyi ulosteeseen (< 5% muuttumattomana) ja 4 % virtsaan (< 0,5% muuttumattomana).
Anto samanaikaisesti OCT1:n ja UGT1A1:n substraattien kanssa
Useiden Lazcluze-annosten antaminen samanaikaisesti metformiinin (OCT1:n substraatti) kanssa ei suurentanut metmorfiinin Cmax-arvoa eikä AUC-arvoa. Lazcluze ei estä OCT1:tä.
In vitro ‑tutkimusten perusteella Lazcluze saattaa estää UGT1A1:tä. Koska vaikutusta epäsuoran bilirubiinin pitoisuuteen ei kliinisessä tutkimuksessa ollut, kliinisesti merkittäviä yhteisvaikutuksia UGT1A1:n substraattien kanssa ei kuitenkaan ole odotettavissa.
Erityiset potilasryhmät
Iäkkäät
Populaatiofarmakokineettisen analyysin perusteella latsertinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä ikään perustuvia eroja.
Munuaisten vajaatoiminta
Populaatiofarmakokineettisen analyysin perusteella annosta ei tarvitse muuttaa potilailla, joilla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta ja joiden laskennallinen glomerulusten suodatusnopeus (eGFR) on 15–89 ml/min. Vaikeaa munuaisten vajaatoimintaa (eGFR 15–29 ml/min) sairastavista potilaista on vähän tietoja (n = 3), mutta ei ole näyttöä siitä, että näiden potilaiden annosta olisi tarpeen muuttaa. Tietoja ei ole saatavissa potilaista, joilla on loppuvaiheen munuaissairaus (eGFR < 15 ml/min).
Maksan vajaatoiminta
Kliinisen farmakologian tutkimuksessa tehtyjen havaintojen perusteella keskivaikea maksan vajaatoiminta (Child–Pughin luokka B) ei vaikuttanut kliinisesti merkittävästi latsertinibin kerta-annoksen farmakokinetiikkaan. Populaatiofarmakokineettisen analyysin perusteella annosta ei tarvitse muuttaa potilailla, joilla on lievä (kokonaisbilirubiini ≤ viitealueen yläraja [ULN] ja ASAT > ULN tai ULN < kokonaisbilirubiini ≤ 1,5 × ULN ja mikä tahansa ASAT) tai keskivaikea (1,5 × ULN < kokonaisbilirubiini ≤ 3 × ULN ja mikä tahansa ASAT) maksan vajaatoiminta. Tietoja ei ole saatavissa potilaista, joilla on vaikea maksan vajaatoiminta (kokonaisbilirubiini > 3 × ULN ja mikä tahansa ASAT).
Pediatriset potilaat
Latsertinibin farmakokinetiikkaa ei ole tutkittu pediatrisilla potilailla.
Muut potilasryhmät
Latsertinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja sukupuolen, painon, etnisen taustan, lähtötilanteen laboratorioarvojen (kreatiniinipuhdistuma, albumiini, alaniiniaminotransferaasi, alkalinen fosfataasi, aspartaattiaminotransferaasi), ECOG-toimintakykyluokan, EGFR-mutaatiotyypin, alkuperäisen diagnoosin mukaisen syövän vaiheen, aiempien hoitojen, aivometastaasien ja tupakointihistorian perusteella.
Prekliiniset tiedot turvallisuudesta
Tärkeimmät löydökset rotilla ja koirilla tehdyissä toistuvan latsertinibialtistuksen aiheuttamaa toksisuutta koskeneissa tutkimuksissa olivat lievä epiteelin atrofia aina degeneratiiviseen eroosioon asti, tulehdus ja nekroosi, jotka vaikuttivat silmiin (sarveiskalvon atrofia), ihoon (ohut ja karkea turkki, karvatupen rappeuma, alopesia, haavauma), maksaan (suurentuneet maksan entsyymipitoisuudet, Kupfferin solujen liikakasvu ja hepatosellulaarinen nekroosi), keuhkoihin (alveolaaristen makrofagien infiltraatio, keuhkotulehdus ja tyypin II alveolaaristen solujen hyperplasia), munuaisiin (tubulusdilataatio, papillaarinen nekroosi, suurentunut ureatyppipitoisuus, kreatiniinipitoisuus [vain naarailla], epäorgaanisen fosforin pitoisuus ja kaliumpitoisuus), maha-suolikanavaan (ruokatorven epiteelin atrofia, pohjukaissuolen ja tyhjäsuolen suolinukan madaltuminen/yhteensulautuminen, nestemäinen uloste) ja sukupuolielimiin (kivesten siementiehyiden degeneraatio, hypospermia, kiimakiertojen ja keltarauhasten väheneminen, kohdun ja emättimen atrofia). Näitä löydöksiä havaittiin eläimillä altistuksilla, jotka olivat 0,9–3,4-kertaisia suositellulla annoksella (240 mg) hoidettujen potilaiden arvioituun altistukseen nähden, ja ne hävisivät täysin tai osittain toipumisvaiheiden aikana. Sydän katsottiin kohde-elimeksi ainoastaan koiralla, ja siihen liittyviä tapahtumia ilmeni altistustasoilla, jotka olivat 7‑kertaisia verrattuna ihmiselle suositellusta annoksesta oletettavasti saataviin altistustasoihin.
Karsinogeenisuus ja mutageenisuus
Latsertinibin genotoksisuudesta ei havaittu näyttöä bakteerimutageenisuustestissä in vitro, kromosomipoikkeavuusmäärityksessä in vitro eikä mikrotumamäärityksissä rotilla in vivo. Latsertinibin mahdollisen karsinogeenisuuden arvioimiseksi ei ole tehty pitkäkestoisia eläinkokeita.
Lisääntymistoksikologia
Eläimillä tehtyjen tutkimusten perusteella latsertinibi voi heikentää urosten ja naaraiden hedelmällisyyttä. Rottien ja koirien kiveksissä oli degeneratiivisia muutoksia, jotka johtivat koirilla 1 kuukauden ajan kliinisesti merkityksellisinä altistustasoina tapahtuneen latsertinibialtistuksen jälkeen siittiösolujen määrän vähenemiseen siementiehyissä. Vähintään kuukauden ajan kliinisesti merkityksellisinä altistustasoina latsertinibille altistuneiden rottien munasarjoissa havaittiin keltarauhasten määrän vähenemistä. Uros- ja naarasrotilla tehdyssä hedelmällisyyttä ja varhaista alkiokehitystä koskeneessa tutkimuksessa latsertinibi sai aikaan kiimakiertojen määrän vähenemisen, kiinnittymisen jälkeisten alkionmenetysten määrän suurenemisen ja elävien poikasten määrän pienenemisen poikueissa annostasolla, joka oli vastaava tai pienempi kuin ihmisten kliininen altistus likimäärin käytettäessä suositeltua 240 mg:n annosta.
Rotilla ja kaniineilla tehdyissä alkion ja sikiön kehitystä koskeneissa tutkimuksissa havaittiin kehitystoksisuutta. Rotilla havaittiin sikiön painon laskua, joka liittyi emolle aiheutuvaan toksisuuteen, kun emon altistus oli noin neljä kertaa suurempi kuin ihmisten kliininen altistus 240 mg:n annoksella. Kaniineilla havaittiin sikiön kallon luiden yhteensulautumisen (poskikaaren ja yläleukaluun yhteensulautumisen) lisääntymistä, kun emon altistus oli selkeästi pienempi kuin ihmisten kliininen altistus 240 mg:n annoksella.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Piidioksidi, hydrofobinen, kolloidinen
Kroskarmelloosinatrium (E468)
Selluloosa, mikrokiteinen (E460[i])
Mannitoli (E421)
Magnesiumstearaatti (E572)
Kalvopäällyste
Lazcluze 80 mg kalvopäällysteiset tabletit
Makrogolipoly(vinyylialkoholi)oksaskopolymeeri (E1209)
Polyvinyylialkoholi (E1203)
Glyserolimonokaprylokapraatti, tyyppi I (E471)
Titaanidioksidi (E171)
Talkki (E553b)
Keltainen rautaoksidi (E172)
Lazcluze 240 mg kalvopäällysteiset tabletit
Makrogolipoly(vinyylialkoholi)oksaskopolymeeri (E1209)
Polyvinyylialkoholi (E1203)
Glyserolimonokaprylokapraatti, tyyppi I (E471)
Titaanidioksidi (E171)
Talkki (E553b)
Punainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
2 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LAZCLUZE tabletti, kalvopäällysteinen
80 mg (L:ei) 56 fol (5231,35 €)
240 mg (L:ei) 28 fol (7756,73 €)
PF-selosteen tieto
Lazcluze 80 mg kalvopäällysteiset tabletit
Läpipainopakkaus
Polyvinyylikloridista ja polyklooritrifluorieteenistä (PVC‑PCTFE) valmistettu kalvo ja läpi painettava alumiinifolio.
- Yksi kotelo sisältää 56 kalvopäällysteistä tablettia (kaksi taskupakkausta, joissa on kummassakin 28 tablettia).
Purkki
Valkoinen, läpinäkymätön suurtiheyspolyeteenistä (HDPE) valmistettu purkki, jossa on polypropeeninen turvasuljin. Yksi purkki sisältää 60 tai 90 tablettia. Kussakin kotelossa on yksi purkki.
Lazcluze 240 mg kalvopäällysteiset tabletit
Läpipainopakkaus
Polyvinyylikloridista ja polyklooritrifluorieteenistä (PVC‑PCTFE) valmistettu kalvo ja läpi painettava alumiinifolio.
- Yksi kotelo sisältää 14 kalvopäällysteistä tablettia (yksi taskupakkaus, jossa on 14 tablettia).
- Yksi kotelo sisältää 28 kalvopäällysteistä tablettia (kaksi taskupakkausta, joissa on kummassakin 14 tablettia).
Purkki
Valkoinen, läpinäkymätön suurtiheyspolyeteenistä (HDPE) valmistettu purkki, jossa on polypropeeninen turvasuljin. Yksi purkki sisältää 30 kalvopäällysteistä tablettia. Kussakin kotelossa on yksi purkki.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Lazcluze 80 mg kalvopäällysteiset tabletit
Keltainen, 14 mm pitkä, soikea tabletti, jonka toiselle puolelle on kaiverrettu LZ ja vastakkaiselle puolelle 80.
Lazcluze 240 mg kalvopäällysteiset tabletit
Punertavanvioletti, 20 mm pitkä, soikea tabletti, jonka toiselle puolelle on kaiverrettu LZ ja vastakkaiselle puolelle 240.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LAZCLUZE tabletti, kalvopäällysteinen
80 mg 56 fol
240 mg 28 fol
- Ei korvausta.
ATC-koodi
L01EB09
Valmisteyhteenvedon muuttamispäivämäärä
12.02.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com