
NILEMDO tabletti, kalvopäällysteinen 180 mg
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 180 mg bempedoiinihappoa.
Apuaine(et), joiden vaikutus tunnetaan:
Yksi 180 mg kalvopäällysteinen tabletti sisältää 28,5 mg laktoosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen.
Kliiniset tiedot
Käyttöaiheet
Hyperkolesterolemia ja sekamuotoinen dyslipidemia
Nilemdo on tarkoitettu aikuisille, joilla on primäärinen (heterotsygoottinen familiaalinen tai ei-familiaalinen) hyperkolesterolemia tai sekamuotoinen dyslipidemia, ruokavalion ohella:
- yhdessä statiinin kanssa tai statiinin ja muun lipidejä alentavan lääkityksen kanssa potilaille, joiden LDL (pienitiheyksinen lipoproteiini) -kolesterolipitoisuus ei pienene tavoitetasolle suurimmalla siedetyllä statiiniannoksella (ks. kohdat Annostus ja antotapa, Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet) tai
- yksinään tai muun lipidejä alentavan lääkityksen kanssa potilaille, jotka eivät siedä statiinia tai joille statiini on vasta-aiheinen
Sydän- ja verisuonitauti
Nilemdo on tarkoitettu LDL-kolesterolipitoisuuden pienentämiseen kardiovaskulaarisen riskin vähentämiseksi aikuisille, joilla on vakiintunut ateroskleroottinen sydän- ja verisuonitauti tai suuri riski sairastua siihen, muiden riskitekijöiden korjaamisen ohella:
- potilaille, jotka saavat suurinta siedettyä statiiniannosta etsetimibin kanssa tai ilman etsetimibiä, tai
- yksinään tai etsetimibin kanssa potilaille, jotka eivät siedä statiinia tai joille statiini on vasta-aiheinen
Tutkimustulokset valmisteen vaikutuksista LDL-kolesteroliin ja sydän- ja verisuonitapahtumiin sekä valmisteen vaikutuksista eri populaatioissa, ks. kohta Farmakodynamiikka.
Annostus ja antotapa
Annostus
Nilemdon suositeltu annos on yksi kalvopäällysteinen 180 mg:n tabletti kerran päivässä.
Samanaikainen simvastatiinihoito
Kun Nilemdoa käytetään samanaikaisesti simvastatiinin kanssa, simvastatiinin enimmäisannos on 20 mg vuorokaudessa (tai 40 mg vuorokaudessa potilailla, joilla on vaikea hyperkolesterolemia ja suuri kardiovaskulaaristen komplikaatioiden riski tai jotka eivät ole saavuttaneet hoitotavoitetta pienemmällä annoksella ja joilla hyötyjen odotetaan olevan mahdollisia riskejä suuremmat) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Erityispotilasryhmät
Vanhukset
Annosta ei ole tarpeen muuttaa vanhuksilla (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoimintaa sairastavat potilaat
Annosta ei ole tarpeen muuttaa potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta. Potilaista, joilla on vaikea munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus [eGFR] < 30 ml/min/1,73 m2), ja dialyysipotilaista, joilla on loppuvaiheen munuaissairaus, on vain vähän tietoa (ks. kohta Farmakokinetiikka). Näillä potilailla haittavaikutusten lisäseuranta voi olla tarpeen Nilemdon käytön aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoimintaa sairastavat potilaat
Annosta ei ole tarpeen muuttaa potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta (Child-Pughin luokitus A tai B). Tietoja ei ole saatavissa potilaista, joilla on vaikea maksan vajaatoiminta (Child-Pughin luokitus C). Potilaille, joilla on vaikea maksan vajaatoiminta, on harkittava säännöllisiä maksantoimintakokeita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Nilemdon turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Kalvopäällysteiset tabletit otetaan suun kautta aterian yhteydessä tai aterioiden välillä. Tabletit on nieltävä kokonaisina.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Raskaus (ks. kohta Raskaus ja imetys).
- Samanaikainen käyttö simvastatiinin kanssa, kun simvastatiiniannos on > 40 mg vuorokaudessa (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Statiinien samanaikaisen käytön aiheuttama myopatian riski
Bempedoiinihappo suurentaa plasman statiinipitoisuutta (ks. kohta Yhteisvaikutukset). Potilaita, jotka saavat Nilemdoa statiinilääkitystä täydentävänä hoitona, on tarkkailtava suuriin statiiniannoksiin liittyvien haittavaikutusten varalta. Statiinit saattavat toisinaan aiheuttaa myopatiaa. Harvinaisissa tapauksissa myopatia voi ilmetä rabdomyolyysinä. Siihen saattaa liittyä myoglobinurian aiheuttama munuaisten äkillinen vajaatoiminta, ja se voi johtaa kuolemaan. Kaikille potilaille, jotka saavat Nilemdoa statiinin lisäksi, on kerrottava suurentuneesta myopatian riskistä, ja heitä on pyydettävä ilmoittamaan heti selittämättömästä lihaskivusta, -arkuudesta tai -heikkoudesta. Jos tällaisia oireita ilmenee, kun potilas saa Nilemdo-hoitoa ja statiinia, on harkittava saman tai vaihtoehtoisen statiinin pienempää enimmäisannosta tai Nilemdo-hoidon lopettamista ja vaihtoehtoisen lipidilääkityksen aloittamista. Lipidipitoisuuksia ja haittavaikutuksia on seurattava huolellisesti. Jos myopatia vahvistetaan sillä, että kreatiinikinaasin (CPK) pitoisuus on > 10 × normaaliarvon yläraja, Nilemdon ja potilaan samanaikaisesti ottaman statiinin käyttö on lopetettava välittömästi.
Myosiittia, jossa kreatiinikinaasin pitoisuus on > 10 × normaaliarvon yläraja, on ilmoitettu harvoin bempedoiinihapon ja peruslääkityksenä käytetyn 40 mg:n simvastatiiniannoksen yhteydessä. Nilemdon kanssa ei saa käyttää yli 40 mg:n simvastatiiniannoksia (ks. kohdat Annostus ja antotapa ja Vasta-aiheet).
Samanaikainen fibraattien käyttö
Fibraattien käyttö samanaikaisesti bempedoiinihapon kanssa aiheutti triglyseridiarvojen nousua ja HDL-kolesteroliarvon laskua joillakin potilailla kliinisissä tutkimuksissa ja markkinoille tulon jälkeen tehdyissä ilmoituksissa. HDL-kolesteroli- ja triglyseridiarvoja on seurattava (ks. kohta Yhteisvaikutukset).
Suurentunut seerumin virtsahappopitoisuus
Bempedoiinihappo saattaa suurentaa seerumin virtsahappopitoisuutta, sillä se estää OAT2-kuljettajaproteiinia munuaistiehyissä ja saattaa aiheuttaa tai pahentaa hyperurikemiaa ja laukaista kihdin potilailla, joilla on ollut aiemmin kihti tai joilla on taipumus kihtiin (ks. kohta Haittavaikutukset). Nilemdo-hoito on lopetettava, jos potilaalla ilmenee hyperurikemiaa ja kihdin oireita.
Kohonneet maksaentsyymiarvot
Kliinisissä tutkimuksissa bempedoiinihapon käytön yhteydessä on ilmoitettu maksaentsyymien alaniiniaminotransferaasin (ALAT) ja aspartaattiaminotransferaasin (ASAT) arvojen suurenemista tasolle > 3 × normaaliarvon yläraja. Tällaiset arvojen suurenemiset ovat olleet oireettomia eikä niihin ole liittynyt bilirubiiniarvon suurenemista tasolle ≥ 2 × normaaliarvon yläraja tai kolestaasia. Arvot ovat palautuneet lähtötasolle hoitoa jatkettaessa tai hoidon lopettamisen jälkeen. Maksan toimintakokeet on tehtävä ennen hoidon aloittamista. Nilemdo-hoito on lopetettava, jos transaminaasiarvot pysyvät tasolla > 3 × normaaliarvon yläraja (ks. kohta Haittavaikutukset).
Munuaisten vajaatoiminta
Bempedoiinihapon käytöstä potilailla, joilla on vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min/1,73 m2), ja dialyysipotilailla, joilla on loppuvaiheen munuaissairaus, on vain vähän kokemusta (ks. kohta Farmakokinetiikka). Näillä potilailla haittavaikutusten lisäseuranta voi olla tarpeen Nilemdon käytön aikana.
Maksan vajaatoiminta
Tutkimuksia ei ole tehty potilailla, joilla on vaikea maksan vajaatoiminta (Child-Pughin luokitus C) (ks. kohta Farmakokinetiikka). Säännöllisiä maksan toimintakokeita on harkittava, jos potilaalla on vaikea maksan vajaatoiminta.
Ehkäisy naisilla, jotka voivat tulla raskaaksi
Naisille, jotka voivat tulla raskaaksi, on annettava neuvontaa tehokkaista ehkäisymenetelmistä, ja tehokkaan ehkäisyn käyttö on aloitettava ennen hoidon aloittamista.
Potilaille, jotka käyttävät suun kautta otettavia estrogeenipohjaisia ehkäisytabletteja, on kerrottava ehkäisytehon mahdollisesta heikkenemisestä, jos ripulia ja/tai oksentelua esiintyy. Potilaita on kehotettava ottamaan välittömästi yhteys lääkäriin ja lopettamaan hoito, jos he suunnittelevat raskautta tai tulevat raskaaksi (ks. kohta Raskaus ja imetys).
Apuaineet
Nilemdo sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per yksi 180 mg:n vahvuinen kalvopäällysteinen tabletti (päiväannos) eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus bempedoiinihappoon
Kuljettajaproteiinivälitteiset yhteisvaikutukset
In vitro -yhteisvaikutustutkimukset viittaavat siihen, että bempedoiinihappo sekä sen aktiivinen aineenvaihduntatuote ja glukuronidimuoto eivät ole yleisesti kuvattujen lääkeaineiden kuljettajaproteiinien substraatteja, lukuun ottamatta bempedoiinihappoglukuronidia, joka on OAT3:n substraatti.
Probenesidi
Probenesidi on glukuronidikonjugaation estäjä. Sitä koskevan tutkimuksen tarkoituksena oli arvioida näiden estäjien mahdollista vaikutusta bempedoiinihapon farmakokinetiikkaan. 180 mg:n bempedoiinihappoannos probenesidin vakaassa tilassa suurensi bempedoiinihapon AUC-arvon 1,7-kertaiseksi ja bempedoiinihapon aktiivisen metaboliitin (ESP15228) AUC-arvon 1,9-kertaiseksi. Nämä AUC-arvojen suurenemiset eivät ole kliinisesti merkittäviä, eivätkä ne vaikuta annossuosituksiin.
Bempedoiinihapon vaikutus muihin lääkevalmisteisiin
Statiinit
Kliinisissä tutkimuksissa arvioitiin 180 mg:n bempedoiinihappoannoksen ja 40 mg:n simvastatiiniannoksen, 80 mg:n atorvastatiiniannoksen, 80 mg:n pravastatiiniannoksen ja 40 mg:n rosuvastatiiniannoksen farmakokineettisiä yhteisvaikutuksia. Simvastatiinihappoaltistus suureni kaksinkertaiseksi, kun simvastatiinin 40 mg:n kerta-annos otettiin bempedoiinihapon (180 mg, vakaa tila) kanssa. Kerta-annoksina annetun atorvastatiinin, pravastatiinin ja rosuvastatiinin ja/tai niiden päämetaboliittien AUC-arvot suurenivat 1,4–1,5-kertaisiksi, kun samanaikaisesti annettiin 180 mg bempedoiinihappoa. Tätä suurempaakin arvojen kohoamista on havaittu, kun näiden statiinien yhteydessä annettiin bempedoiinihapon hoitoannosta suurempi annos 240 mg (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kuljettajaproteiinivälitteiset yhteisvaikutukset
Bempedoiinihappo ja sen glukuronidi estävät heikosti OATP1B1:tä ja OATP1B3:a kliinisesti merkittävinä pitoisuuksina. Bempedoiinihapon samanaikainen käyttö sellaisten lääkevalmisteiden, jotka ovat OATP1B1:n tai OATP1B3:n substraatteja (esim. bosentaani, fimasartaani, asunapreviiri, glekapreviiri, gratsopreviiri ja voksilapreviiri, ja statiinit, kuten atorvastatiini, pravastatiini, fluvastatiini, pitavastatiini, rosuvastatiini ja simvastatiini [ks. kohta Varoitukset ja käyttöön liittyvät varotoimet]), kanssa voi suurentaa kyseisten lääkevalmisteiden pitoisuuksia plasmassa.
Bempedoiinihappo estää OAT2:ta in vitro, ja tämä mekanismi saattaa aiheuttaa vähäistä seerumin kreatiniini- ja virtsahappopitoisuuksien suurenemista (ks. kohta Haittavaikutukset). Lisäksi bempedoiinihapon OAT2:ta estävä vaikutus saattaa suurentaa sellaisten lääkevalmisteiden pitoisuuksia plasmassa, jotka ovat OAT2:n substraatteja. Bempedoiinihappo saattaa myös estää heikosti OAT3:a kliinisesti merkittävinä pitoisuuksina.
Etsetimibi
Etsetimibin kokonaispitoisuuden (etsetimibi ja sen glukuronidimuoto) ja etsetimibiglukuronidin AUC-arvo nousi noin 1,6-kertaiseksi ja seerumin enimmäispitoisuus (Cmax) noin 1,8-kertaiseksi, kun kerta-annos etsetimibiä otettiin yhdessä vakaan tilan bempedoiinihappoannoksen kanssa. Tämän suurenemisen aiheuttaa todennäköisesti se, että bempedoiinihappo estää OATP1B1:tä, mikä vähentää ottoa maksaan ja vähentää etsetimibiglukuronidin eliminaatiota. Etsetimibin AUC- ja Cmax-arvot suurenivat alle 20 prosenttia. Nämä arvojen suurenemiset eivät ole kliinisesti merkittäviä eivätkä vaikuta annossuosituksiin.
Fibraatit
Fibraattien käyttö samanaikaisesti bempedoiinihapon kanssa aiheutti triglyseridiarvojen nousua ja HDL-kolesteroliarvon laskua joillakin potilailla kliinisissä tutkimuksissa ja markkinoille tulon jälkeen tehdyissä ilmoituksissa. Sekä triglyseridiarvon nousun että HDL-kolesteroliarvon laskun korjautumista havaittiin, kun joko bempedoiinihappo- tai fibraattihoito keskeytettiin.
Triglyseridi- ja HDL-kolesteroliarvoja on seurattava neljän viikon jälkeen hoidon aloittamisesta ja säännöllisesti sen jälkeen, kun bempedoiinihappoa käytetään samanaikaisesti fibraatin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Mikäli havaitaan kliinisesti relevanttia triglyseridiarvojen nousua tai HDL-kolesteroliarvon laskua, bempedoiinihappo- tai fibraattihoito on keskeytettävä kliinisen harkinnan mukaan. Triglyseridi- ja HDL-kolesteroliarvoja on seurattava, kunnes ne palaavat lähtötilanteen arvoihin.
Anemian ja hyperurikemian esiintyvyyden on havaittu olevan suurempi potilailla, jotka käyttävät samanaikaisesti bempedoiinihappoa ja fibraatteja (ks. kohta Haittavaikutukset).
Muut tutkitut yhteisvaikutukset
Bempedoiinihapolla ei ollut vaikutusta metformiinin farmakokinetiikkaan tai farmakodynamiikkaan tai noretisteronia/etinyyliestradiolia sisältävien suun kautta otettavien ehkäisytablettien farmakokinetiikkaan.
Raskaus ja imetys
Raskaus
Nilemdon käyttö raskauden aikana on vasta-aiheista (ks. kohta Vasta-aiheet).
Bempedoiinihapon käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Bempedoiinihappoa koskevissa eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Koska bempedoiinihappo vähentää kolesterolisynteesiä ja mahdollisesti muiden sikiön normaaliin kehitykseen tarvittavien kolesterolijohdannaisten synteesiä, Nilemdo voi aiheuttaa haittaa sikiölle, jos sitä käytetään raskauden aikana. Nilemdon käyttö on lopetettava ennen hedelmöittymistä tai heti, kun raskautta suunnitellaan tai raskaus todetaan (ks. kohta Vasta-aiheet).
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Bempedoiinihappo ja sen aktiivinen metaboliitti erittyvät ihmisillä äidinmaitoon hyvin pieninä määrinä (keskimääräinen lapsen annososuus on noin 0,5 % bempedoiinihapolla) ja siksi Nilemdon hoitoannoksilla imetettävään vauvaan/lapseen kohdistuvia vaikutuksia ei odoteta (ks. kohta Farmakokinetiikka).
Nilemdon käyttöä imetyksen aikana voidaan harkita, ottaen huomioon imetyksen hyödyt lapselle ja Nilemdo-hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Nilemdon vaikutuksesta ihmisen hedelmällisyyteen ei ole saatavissa tietoja. Eläinkokeiden perusteella Nilemdon ei odoteta vaikuttavan lisääntymiseen tai hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Nilemdolla ei ole haitallista vaikutusta tai on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Bempedoiinihappoa koskevissa päätutkimuksissa yleisimmät ilmoitetut haittavaikutukset olivat hyperurikemia (3,8 %), raajojen kipu (3,1 %), anemia (2,5 %) ja kihti (1,4 %). Bempedoiinihappoa saaneet potilaat lopettivat tutkimuksen lumelääkettä saavia potilaita useammin lihaskouristusten (0,7 % bempedoiinihappoa saaneista potilaista ja 0,3 % lumelääkettä saaneista potilaista), ripulin (0,5 % vs. < 0,1 %), raajojen kivun (0,4 % vs. 0 %) ja pahoinvoinnin (0,3 % vs. 0,2 %) vuoksi, joskaan bempedoiinihapon ja lumelääkkeen erot eivät olleet merkittäviä.
Taulukko haittavaikutuksista
Bempedoiinihapon haittavaikutukset, jotka perustuvat primääristä hyperlipidemiaa koskevissa vaiheen 3 tutkimuksissa todettuihin ilmaantuvuuksiin ja CLEAR Outcomes -tutkimuksessa todettuihin, altistuksen suhteen mukautettuihin ilmaantuvuuksiin, on esitetty elinluokan ja yleisyyden mukaan taulukossa 1.
Haittavaikutusten yleisyys määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1: Haittavaikutukset
| Elinluokka | Haittavaikutukset | Yleisyysluokitus |
| Veri ja imukudos | Anemiaa | Yleinen |
| Hemoglobiiniarvon pieneneminen | Melko harvinainen | |
| Aineenvaihdunta ja ravitsemus | Kihti | Yleinen |
| Hyperurikemiaa,b | Yleinen | |
| Painon laskuc | Melko harvinainen | |
| Maksa ja sappi | Aspartaattiaminotransferaasiarvon suureneminen | Yleinen |
| Alaniiniaminotransferaasin suureneminen | Melko harvinainen | |
| Maksantoimintakokeiden suurentuneet arvot | Melko harvinainen | |
| Luusto, lihakset ja sidekudos | Raajojen kipu | Yleinen |
| Munuaiset ja virtsatiet | Glomerulusten suodatusnopeuden pieneneminen | Yleinen |
| Veren kreatiniiniarvon suureneminen | Melko harvinainen | |
| Veren ureapitoisuuden suureneminen | Melko harvinainen |
- Ks. kohta Yhteisvaikutukset
- Hyperurikemia sisältää liiallisen virtsahapon määrän veressä ja veren virtsahappopitoisuuden nousun
- (CLEAR Outcomes -tutkimus) Painon laskua todettiin vain potilailla, joiden painoindeksi (BMI) oli lähtötilanteessa ≥ 30 kg/m2, ja kuukauden 36 kohdalla paino oli laskenut keskimäärin -2,28 kg. Potilailla, joiden painoindeksi oli lähtötilanteessa 25 – < 30 kg/m2, paino laski keskimäärin < 0,5 kg. Potilailla, joiden painoindeksi oli lähtötilanteessa < 25 kg/m2, bempedoiinihappoon ei liittynyt keskimääräistä painon muutosta
Tiettyjen haittavaikutusten kuvaus
Maksaentsyymiarvojen suureneminen
Bempedoiinihapon käytön yhteydessä on ilmoitettu seerumin transaminaasiarvojen (ASAT ja/tai ALAT) suurenemista. Primääristä hyperlipidemiaa koskevissa vaiheen 3 tutkimuksissa maksan transaminaasiarvot suurenivat tasolle ≥ 3 × normaaliarvon yläraja 0,7 prosentilla bempedoiinihappohoitoa saaneista potilasta ja 0,3 prosentilla lumelääkettä saaneista potilaista. Myös CLEAR Outcomes -tutkimuksessa maksan transaminaasiarvojen suurenemista tasolle ≥ 3 × normaaliarvon yläraja esiintyi useammin bempedoiinihappohoitoa saaneilla potilailla (1,6 %) kuin lumelääkettä saaneilla potilailla (1,0 %). Näihin transaminaasiarvojen suurenemisiin ei liittynyt muita maksan toimintahäiriön merkkejä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Suurentunut seerumin virtsahappopitoisuus
Bempedoiinihappoa koskevissa kliinisissä tutkimuksissa havaittiin seerumin virtsahappopitoisuuden suurenemista. Se saattaa liittyä OAT2:n estämiseen munuaistiehyissä (ks. kohta Yhteisvaikutukset). Primääristä hyperlipidemiaa koskevissa vaiheen 3 tutkimuksissa virtsahappopitoisuuden keskimääräinen suureneminen 47,6 mikromoolia/l (0,8 mg/dl) lähtötasosta havaittiin bempedoiinihapon käytön yhteydessä viikolla 12. Seerumin virtsahappopitoisuuden suureneminen ilmeni yleensä neljän ensimmäisen hoitoviikon aikana ja palautui lähtötasolle hoidon lopettamisen jälkeen. Primääristä hyperlipidemiaa koskevissa vaiheen 3 tutkimuksissa kihtiä ilmoitettiin 1,4 prosentilla bempedoiinihappoa saaneista potilaista ja 0,4 prosentilla lumelääkettä saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). CLEAR Outcomes -tutkimuksessa bempedoiinihappohoitoa saaneiden potilaiden virtsahappopitoisuus oli suurentunut keskimäärin 47,6 mikromoolia/l (0,8 mg/dl) lähtötasosta kuukauden 3 kohdalla, ja kihtiä myös ilmoitettiin useammin bempedoiinihappohoitoa saaneilla potilailla (3,1 %) kuin lumelääkettä saaneilla potilailla (2,1 %). Kummassakin hoitoryhmässä potilailla, jotka ilmoittivat kihdistä, oli muita todennäköisemmin ollut aiemmin kihti, ja/tai virtsahapon lähtöpitoisuus oli lähtötilanteessa normaaliarvon ylärajan yläpuolella. Hyperurikemian esiintyvyyden havaittiin olevan suurempi potilailla, jotka saivat samanaikaisesti bempedoiinihappoa ja fibraattia. CLEAR Outcomes -tutkimuksessa hyperurikemiaa raportoitiin useammin bempedoiinihappoa saavilla potilailla, jotka käyttivät fibraattia lähtötilanteessa (19,5 %), verrattuna potilaisiin, jotka eivät käyttäneet fibraattia (10,4 %), ks. kohta Yhteisvaikutukset. Kihdin esiintyvyys ei lisääntynyt bempedoiinihappohoitoa saaneilla potilailla, jotka käyttivät fibraattia lähtötilanteessa (1,1 %), verrattuna potilaisiin, jotka eivät käyttäneet fibraattia (3,2 %).
Vaikutukset seerumin kreatiniinipitoisuuteen ja veren ureatyppeen
Bempedoiinihapon on osoitettu suurentavan seerumin kreatiniinipitoisuutta ja veren ureatyppeä (BUN). Primääristä hyperlipidemiaa koskevissa vaiheen 3 tutkimuksissa seerumin kreatiniinipitoisuuden keskimääräinen suureneminen 4,4 mikromoolia/l (0,05 mg/dl) lähtötasosta ja BUN-arvon keskimääräinen suureneminen 0,61 mikromoolia/l (1,7 mg/dl) lähtötasosta havaittiin bempedoiinihapon käytön yhteydessä viikolla 12. Seerumin kreatiniinipitoisuuden ja BUN-arvon suureneminen ilmeni yleensä neljän ensimmäisen hoitoviikon aikana, pysyi vakaana ja palautui lähtötasolle hoidon lopettamisen jälkeen. CLEAR Outcomes -tutkimuksessa bempedoiinihapon käytön yhteydessä havaittu seerumin kreatiniinipitoisuuden keskimääräinen suureneminen (5,8 mikromoolia/l (0,066 mg/dl)) ja BUN-arvon keskimääräinen suureneminen (0,82 mmol/l (2,3 mg/dl)) olivat samaa luokkaa.
Havaittu seerumin kreatiniinipitoisuuden suureneminen saattaa liittyä siihen, että bempedoiinihappo estää kreatiniinin OAT2:sta riippuvaista eritystä munuaistiehyissä (ks. kohta Yhteisvaikutukset). Kyseessä on lääkkeen ja endogeenisen substraatin välinen yhteisvaikutus, eikä se vaikuta viittaavan munuaisten toiminnan heikkenemiseen. Tämä vaikutus on huomioitava, kun tulkitaan muutoksia arvioidussa kreatiniinipuhdistumassa Nilemdo-hoitoa saavilla potilailla ja erityisesti potilailla, joilla on sairauksia tai jotka käyttävät arvioidun kreatiniinipuhdistuman seurantaa edellyttäviä lääkevalmisteita.
Hemoglobiiniarvojen pieneneminen
Bempedoiinihappoa koskevissa kliinisissä tutkimuksissa havaittiin hemoglobiiniarvojen pienenemistä. Primääristä hyperlipidemiaa koskevissa vaiheen 3 tutkimuksissa hemoglobiinipitoisuus pieneni lähtötasosta ≥ 20 g/l ja alle normaaliarvon alarajan (LLN) 4,6 prosentilla potilaista bempedoiinihapporyhmässä ja 1,9 prosentilla potilaista lumelääkeryhmässä. Hemoglobiinipitoisuuden pienenemistä yli 50 g/l ja alle normaaliarvon alarajan ilmoitettiin yhtä paljon bempedoiinihapporyhmässä (0,2 %) kuin lumelääkeryhmässä (0,2 %). Hemoglobiinipitoisuuden pieneneminen ilmeni yleensä neljän ensimmäisen hoitoviikon aikana ja palautui lähtötasolle hoidon lopettamisen jälkeen. Niistä potilaista, joiden hemoglobiiniarvo oli normaali lähtötilanteessa, hemoglobiiniarvo pieneni alle normaalin alarajan hoidon aikana 1,4 prosentilla bempedoiinihapporyhmässä ja 0,4 prosentilla lumelääkeryhmässä. Primääristä hyperlipidemiaa koskevissa vaiheen 3 tutkimuksissa anemiaa ilmoitettiin 2,5 prosentilla bempedoiinihappoa saaneista potilaista ja 1,6 prosentilla lumelääkettä saaneista potilaista. CLEAR Outcomes -tutkimuksessa havaittiin samankaltaista hemoglobiiniarvojen pienenemistä, ja anemiaa myös ilmoitettiin useammin bempedoiinihappohoitoa saaneilla potilailla (4,7 %) kuin lumelääkettä saaneilla potilailla (3,9 %). Anemian esiintyvyyden havaittiin olevan suurempi potilailla, jotka saivat samanaikaisesti bempedoiinihappoa ja fibraattia. CLEAR Outcomes -tutkimuksessa anemiaa raportoitiin useammin bempedoiinihappohoitoa saaneilla potilailla, jotka käyttivät fibraattia lähtötilanteessa (9,6 %), verrattuna potilaisiin, jotka eivät käyttäneet fibraattia (4,5 %).
Vanhukset
Primääristä hyperlipidemiaa koskevissa vaiheen 3 tutkimuksissa 3 621 potilasta sai bempedoiinihappohoitoa, ja tästä ryhmästä 2 098 (58 %) potilasta oli > 65-vuotiaita. CLEAR Outcomes -tutkimuksessa 4 141 bempedoiinihappohoitoa saanutta potilasta (59 %) oli ≥ 65-vuotiaita ja 1 066 bempedoiinihappohoitoa saanutta potilasta (15 %) oli ≥ 75-vuotiaita. Yleisessä turvallisuudessa ei havaittu eroja vanhusten ja nuorempien potilasryhmien välillä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa annetut annokset ovat olleet enintään 240 mg vuorokaudessa (1,3 kertaa hyväksytty suositeltu annos), eikä viitteitä annosta rajoittavasta toksisuudesta ole ilmennyt.
Haittatapahtumia ei havaittu eläinkokeissa, joissa eläimet altistuivat jopa 14 kertaa suuremmille annoksille kuin potilaat, jotka saavat 180 mg bempedoiinihappoa kerran vuorokaudessa.
Nilemdo-yliannostukselle ei ole olemassa tiettyä hoitoa. Yliannostustapauksissa potilasta hoidetaan oireenmukaisesti, ja tukitoimia aloitetaan tarvittaessa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: lipidejä muuntavat lääkeaineet, muut lipidejä muuntavat lääkeaineet, ATC-koodi: C10AX15
Vaikutusmekanismi
Bempedoiinihappo on adenosiinitrifosfaatti-sitraattilyaasin (ATP-sitraattilyaasin) estäjä, joka pienentää LDL-kolesterolin pitoisuutta estämällä kolesterolisynteesiä maksassa. ATP-sitraattilyaasi (ACL) on entsyymi, joka sijaitsee 3-hydroksi-3-metyyliglutaryylikoentsyymi-A-reduktaasista eli HMG-CoA-reduktaasista ylävirtaan kolesterolin biosynteesireitillä. Bempedoiinihappo edellyttää, että hyvin pitkäketjuinen asyyli-CoA-syntetaasi 1 (ACSVL1) aktivoi koentsyymi-A:n ETC-1002-CoA:ksi. ACSVL1 ilmentyy pääasiassa maksassa eikä luustolihaksissa. ACL:n esto ETC-1002-CoA:lla vähentää kolesterolisynteesiä maksassa ja pienentää veren LDL-kolesterolipitoisuutta LDL-reseptorien voimistussäätelyllä. Lisäksi ETC-1002-CoA:n aiheuttama ACL:n estäminen johtaa samanaikaiseen maksan rasvahappojen biosynteesin suppressioon.
Farmakodynaamiset vaikutukset
Bempedoiinihappo yksinään ja muiden lipidipitoisuutta muuttavien lääkkeiden kanssa käytettynä pienentää LDL-kolesterolin, non-HDL-kolesterolin, apolipoproteiini B:n (apo B), kokonaiskolesterolin ja C-reaktiivisen proteiinin (CRP) pitoisuutta potilailla, joilla on hyperkolesterolemia tai sekamuotoinen dyslipidemia.
Koska diabetespotilailla on suurentunut ateroskleroottisten sydän- ja verisuonisairauksien riski, bempedoiinihappoa koskeviin kliinisiin kokeisiin osallistui potilaita, joilla on diabetes mellitus. Diabetesta sairastavien potilaiden alaryhmässä havaittiin pienempiä hemoglobiinin A1c (HbA1c) -arvoja kuin lumelääkeryhmässä (keskimäärin 0,2 %). Potilailla, joilla ei ollut diabetesta, ei havaittu eroa HbA1c-arvoissa bempedoiinihappo- ja lumelääkeryhmän välillä, eikä hypoglykemian yleisyydessä ollut eroja.
Sydämen sähköfysiologia
Kun annos on 240 mg (1,3 kertaa hyväksytty suositeltu annos), bempedoiinihappo ei pidennä QT-aikaa kliinisesti merkittävästi.
Kliininen teho ja turvallisuus
Kliininen teho ja turvallisuus primäärisessä hyperkolesterolemiassa ja sekamuotoisessa dyslipidemiassa
Nilemdon tehoa tutkittiin neljässä primääristä hyperlipidemiaa koskevassa satunnaistetussa kaksoissokkoutetussa ja lumekontrolloidussa vaiheen 3 monikeskustutkimuksessa. Niihin osallistui 3 623 aikuispotilasta, joilla oli hyperkolesterolemia tai sekamuotoinen dyslipidemia. 2 425 potilasta satunnaistettiin saamaan bempedoiinihappoa. Kaikki potilaat saivat bempedoiinihappoa 180 mg tai lumelääkettä kerran vuorokaudessa suun kautta. Kahdessa tutkimuksessa potilaat saivat peruslääkityksenä lipidejä muuntavaa lääkitystä, johon kuului suurin siedetty statiiniannos sekä mahdollisesti myös muita lipidejä muuntavia lääkeaineita. Kaksi tutkimusta tehtiin potilailla, joilla oli dokumentoitu statiini-intoleranssi. Tehon ensisijainen päätetapahtuma oli kaikissa vaiheen 3 tutkimuksissa LDL-kolesterolipitoisuuden keskimääräinen pieneneminen prosentteina lähtötilanteesta viikolla 12 lumelääkkeeseen verrattuna.
Yhdistelmähoito statiinien kanssa
CLEAR Wisdom (tutkimus 1002-047) oli primääristä hyperlipidemiaa koskeva satunnaistettu, kaksoissokkoutettu ja lumekontrolloitu vaiheen 3 monikeskustutkimus. Tutkimus kesti 52 viikkoa, ja siihen osallistui potilaita, joilla oli hyperkolesterolemia tai sekamuotoinen dyslipidemia. Nilemdon tehoa arvioitiin viikolla 12. Tutkimukseen osallistuneet 779 potilasta satunnaistettiin suhteessa 2:1 saamaan bempedoiinihappoa (n = 522) tai lumelääkettä (n = 257) lipidejä alentavan lääkehoidon suurimman siedetyn annoksen lisäksi. Lipidejä alentavan lääkkeen suurimman siedetyn annoksen määritelmä oli statiinin suurin siedetty annos (mukaan lukien muut statiinihoito-ohjelmat kuin anto kerran vuorokaudessa, hyvin pienet annokset tai ei statiinia) annettuna yksinään tai yhdessä muiden lipidejä alentavien lääkkeiden kanssa. Tutkimuksesta suljettiin pois potilaat, jotka saivat simvastatiinia vähintään 40 mg vuorokaudessa.
Keski-ikä lähtötilanteessa oli 64 vuotta (vaihteluväli: 28–91 vuotta), ja 51 prosenttia potilaista oli ≥ 65-vuotiaita, 36 prosenttia oli naisia, 94 prosenttia oli valkoihoisia, 5 prosenttia oli tummaihoisia ja 1 prosentti oli aasialaisia. Lähtötason LDL-kolesteroli oli keskimäärin 3,1 mmol/l (120,4 mg/dl). Satunnaistamishetkellä statiinihoitoa sai 91 prosenttia potilaista ja 53 prosenttia potilaista sai suuriannoksista statiinihoitoa. Bempedoiinihappo pienensi LDL-kolesterolin pitoisuutta lähtötilanteesta viikolle 12 mennessä merkitsevästi enemmän kuin lumelääke (p < 0,001). Lisäksi bempedoiinihappo pienensi merkitsevästi non-HDL-kolesterolin, apolipoproteiini B:n ja kokonaiskolesterolin pitoisuutta.
CLEAR Harmony (tutkimus 1002-040) oli primääristä hyperlipidemiaa koskeva satunnaistettu, kaksoissokkoutettu ja lumekontrolloitu vaiheen 3 monikeskustutkimus. Tutkimus kesti 52 viikkoa, ja siinä arvioitiin bempedoiinihapon turvallisuutta ja tehoa potilailla, joilla oli hyperkolesterolemia tai sekamuotoinen dyslipidemia. Nilemdon tehoa arvioitiin viikolla 12. Tutkimukseen osallistuneet 2 230 potilasta satunnaistettiin suhteessa 2:1 saamaan joko bempedoiinihappoa (n = 1 488) tai lumelääkettä (n = 742) lipidejä alentavien lääkkeiden suurimman siedetyn annoksen lisäksi. Lipidilääkkeen suurimman siedetyn annoksen määritelmä oli statiinin suurin siedetty annos (mukaan lukien muut statiinihoito-ohjelmat kuin anto kerran vuorokaudessa ja hyvin pienet annokset) annettuna yksinään tai yhdessä muiden lipidilääkkeiden kanssa. Tutkimuksesta suljettiin pois potilaat, jotka saivat simvastatiinia vähintään 40 mg vuorokaudessa, ja potilaat, jotka saivat PCSK9:n estäjiä.
Keski-ikä lähtötilanteessa oli 66 vuotta (vaihteluväli: 24–88 vuotta), Potilaista 61 prosenttia oli ≥ 65-vuotiaita, 27 prosenttia oli naisia, 96 prosenttia oli valkoihoisia, 3 prosenttia oli tummaihoisia ja 1 prosentti oli aasialaisia. Keskimääräinen LDL-kolesteroli lähtötilanteessa oli 2,7 mmol/l (103,2 mg/dl). Satunnaistamishetkellä kaikki potilaat saivat statiinihoitoa ja 50 prosenttia potilaista sai suuriannoksista statiinihoitoa. Bempedoiinihappo pienensi LDL-kolesterolin pitoisuutta lähtötilanteesta viikolle 12 merkitsevästi enemmän kuin lumelääke (p < 0,001). Bempedoiinihapporyhmässä LDL-kolesteroli laski tasolle ˂ 1,81 mmol/l (˂ 70 mg/dl) merkitsevästi useammilla potilailla (32 %) kuin lumelääkeryhmässä (9 %, p < 0,001). Lisäksi bempedoiinihappo pienensi merkitsevästi non-HDL-kolesterolin, apolipoproteiini B:n ja kokonaiskolesterolin pitoisuutta (ks. taulukko 2).
Taulukko 2: Nilemdo-hoidon teho lumelääkkeeseen verrattuna potilailla, joilla on primaarinen hyperkolesterolemia tai sekamuotoinen dyslipidemia, keskimääräisenä prosentuaalisena muutoksena lähtötilanteesta viikolle 12.
| CLEAR Wisdom (tutkimus 1002-047) (N = 779) | CLEAR Harmony (tutkimus 1002-040) (N = 2 230) | |||
| Nilemdo n = 522 | Lumelääke n = 257 | Nilemdo n = 1 488 | Lumelääke n = 742 | |
| LDL-Ca, n | 498 | 253 | 1 488 | 742 |
| PN-keskiarvo | –15,1 | 2,4 | –16,5 | 1,6 |
| non-HDL-Ca, n | 498 | 253 | 1 488 | 742 |
| PN-keskiarvo | –10,8 | 2,3 | –11,9 | 1,5 |
| apo Ba, n | 479 | 245 | 1 485 | 736 |
| PN-keskiarvo | –9,3 | 3,7 | –8,6 | 3,3 |
| TCa, n | 499 | 253 | 1 488 | 742 |
| PN-keskiarvo | –9,9 | 1,3 | –10,3 | 0,8 |
apo B = apolipoproteiini B, HDL-C = HDL-kolesteroli, LDL-C = LDL-kolesteroli, PN-keskiarvo = pienimmän neliösumman keskiarvo, TC = kokonaiskolesteroli.
Peruslääkityksenä käytetty statiini (1002-047): atorvastatiini, simvastatiini, rosuvastatiini, pravastatiini, fluvastatiini, pitavastatiini ja lovastatiini.
Peruslääkityksenä käytetty statiini (1002-040): atorvastatiini, simvastatiini, pravastatiini.
a. Prosenttimääräinen muutos lähtötilanteesta analysoitiin kovarianssianalyysillä (ANCOVA), jossa selittäviä muuttujia olivat hoito- ja satunnaisositus ja kovariaatti oli lähtötason lipidiparametri.
Potilaat, joilla on statiini-intoleranssi
CLEAR Tranquility (tutkimus 1002-048) oli primääristä hyperlipidemiaa koskeva satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen 3 monikeskustutkimus. Tässä 12 viikkoa kestäneessä tutkimuksessa verrattiin lumelääkkeen ja Nilemdon tehoa LDL-kolesterolipitoisuuden pienentämisessä, kun lääke lisättiin etsetimibiin potilailla, joilla oli kohonnut LDL-kolesteroliarvo, joilla oli aiemmin esiintynyt statiini-intoleranssia ja jotka eivät sietäneet statiinin pienintä hyväksyttyä aloitusannosta suurempaa annosta. Tutkimukseen osallistui 269 potilasta, jotka satunnaistettiin suhteessa 2:1 saamaan bempedoiinihappoa (n = 181) tai lumelääkettä (n = 88) etsetimibin 10 mg:n päiväannoksen lisänä 12 viikon ajan.
Keski-ikä lähtötilanteessa oli 64 vuotta (vaihteluväli: 30–86 vuotta). Potilaista 55 prosenttia oli ≥ 65-vuotiaita, 61 prosenttia oli naisia, 89 prosenttia oli valkoihoisia, 8 prosenttia oli tummaihoisia, 2 prosenttia oli aasialaisia ja 1 prosentti oli muita. Keskimääräinen LDL-kolesterolipitoisuus lähtötilanteessa oli 3,3 mmol/l (127,6 mg/dl). Satunnaistamishetkellä 33 prosenttia bempedoiinihappoa saavista potilaista ja 28 prosenttia lumelääkettä saavista potilaista sai statiinihoitoa, jossa käytettiin enintään pienintä hyväksyttyä annosta. Bempedoiinihappo pienensi LDL-kolesterolipitoisuutta lähtötilanteesta viikolle 12 mennessä merkitsevästi enemmän kuin lumelääke (p < 0,001). Bempedoiinihappo pienensi myös merkitsevästi non-HDL-kolesterolin, apolipoproteiini B:n ja kokonaiskolesterolin pitoisuuksia (ks. taulukko 3).
CLEAR Serenity (tutkimus 1002-046) oli primääristä hyperlipidemiaa koskeva satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen 3 monikeskustutkimus, joka kesti 24 viikkoa. Tutkimuksessa verrattiin Nilemdon tehoa lumelääkkeeseen potilailla, joilla oli suurentuneen LDL-kolesterolin pitoisuuden lisäksi statiini-intoleranssi tai he eivät sietäneet kahta tai useampaa statiinia, joista yhtä pienimmällä mahdollisella annoksella. Potilaille, jotka sietivät annosta, joka oli pienempi kuin statiinin hyväksytty aloitusannos, annettiin kyseistä annosta tutkimuksen ajan. Bempedoiinihapon tehoa arvioitiin viikolla 12. Tutkimukseen osallistuneet 345 potilasta satunnaistettiin suhteessa 2:1 saamaan bempedoiinihappoa (n = 234) tai lumelääkettä (n = 111) 24 viikon ajan. Satunnaistamishetkellä 8 prosenttia bempedoiinihappoa saaneista potilaista ja 10 prosenttia lumelääkettä saaneista potilasta sai statiinihoitoa, jossa annos oli pienempi kuin pienin hyväksytty annos. 36 prosenttia bempedoiinihappoa saaneista potilaista ja 30 prosenttia lumelääkettä saaneista potilasta sai muuta lipidilääkitystä kuin statiinia.
Keski-ikä lähtötilanteessa oli 65 vuotta (vaihteluväli: 26–88 vuotta). Potilaista 58 prosenttia oli ≥ 65-vuotiaita, 56 prosenttia oli naisia, 89 prosenttia oli valkoihoisia, 8 prosenttia oli tummaihoisia, 2 prosenttia oli aasialaisia ja 1 prosentti oli muita. Keskimääräinen LDL-kolesterolipitoisuus lähtötilanteessa oli 4,1 mmol/l (157,6 mg/dl).
Bempedoiinihappo pienensi LDL-kolesterolin pitoisuutta lähtötilanteesta viikolle 12 mennessä merkitsevästi enemmän kuin lumelääke (p < 0,001). Lisäksi bempedoiinihappo pienensi merkitsevästi non-HDL-kolesterolin, apolipoproteiini B:n ja kokonaiskolesterolin pitoisuutta (katso taulukko 3).
Hoito ilman lipidilääkitystä
CLEAR Serenity -tutkimukseen (tutkimukseen 1002-046) osallistui 133 potilasta bempedoiinihapporyhmässä ja 67 potilasta lumelääkeryhmässä. He eivät saaneet peruslääkityksenä lipidilääkettä. Bempedoiinihappo pienensi LDL-kolesterolin pitoisuutta lähtötilanteesta viikolle 12 mennessä merkitsevästi enemmän kuin lumelääke tässä alaryhmässä. Ero LDL-kolesterolin pitoisuuden pienenemisessä lähtötilanteesta viikolle 12 oli bempedoiinihappo- ja lumelääkeryhmän välillä –22,1 % (CI: -26,8 %, -17,4 %; p < 0,001).
Taulukko 3: Nilemdo-hoidon vaikutukset lumelääkkeeseen verrattuna potilailla, joilla on statiini-intoleranssi, keskimääräisenä prosentuaalisena muutoksena lähtötilanteesta viikolle 12.
| CLEAR Tranquility (tutkimus 1002-048) (N = 269) | CLEAR Serenity (tutkimus 1002-046) (N = 345) | |||
| Nilemdo n = 181 | Lumelääke n = 88 | Nilemdo n = 234 | Lumelääke n = 111 | |
| LDL-Ca, n | 175 | 82 | 224 | 107 |
| PN-keskiarvo | –23,5 | 5,0 | –22,6 | –1,2 |
| non-HDL-Ca, n | 175 | 82 | 224 | 107 |
| PN-keskiarvo | –18,4 | 5,2 | –18,1 | –0,1 |
| apo Ba, n | 174 | 81 | 218 | 104 |
| PN-keskiarvo | –14,6 | 4,7 | –14,7 | 0,3 |
| TCa, n | 176 | 82 | 224 | 107 |
| PN-keskiarvo | –15,1 | 2,9 | –15,4 | –0,6 |
apo B = apolipoproteiini B, HDL-C = HDL-kolesteroli, LDL-C = LDL-kolesteroli, PN-keskiarvo = pienimmän neliösumman keskiarvo, TC = kokonaiskolesteroli.
Peruslääkityksenä käytetty statiini (1002-048): atorvastatiini, simvastatiini, rosuvastatiini, pravastatiini, lovastatiini.
Peruslääkityksenä käytetty statiini (1002-046): atorvastatiini, simvastatiini, pitavastatiini, rosuvastatiini, pravastatiini, lovastatiini.
a. Prosenttimääräinen muutos lähtötilanteesta analysoitiin kovarianssianalyysillä (ANCOVA), jossa selittäviä muuttujia olivat hoito- ja satunnaisositus ja kovariaatti oli lähtötason lipidiparametri.
Kaikissa neljässä tutkimuksessa suurimmat vaikutukset LDL-kolesterolipitoisuuden pienenemiseen havaittiin jo viikolla 4, ja teho säilyi koko tutkimuksen ajan. Nämä tulokset olivat johdonmukaisia kaikissa tutkimuksissa tarkastelluissa alaryhmissä, mukaan lukien ikä, sukupuoli, rotu, etnisyys, alue, diabeteshistoria, lähtötason LDL-kolesteroli, painoindeksi (BMI), heterotsygoottisen familiaalisen hyperkolesterolemian (HeFH) tila ja muu lääkitys.
Kliininen teho ja turvallisuus sydän- ja verisuonitapahtumien ehkäisyssä
CLEAR Outcomes (tutkimus 1002-043) oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu, tapahtumalähtöinen monikeskustutkimus, johon osallistuneilla 13 970 aikuispotilaalla oli vakiintunut ateroskleroottinen sydän- ja verisuonitauti (70 %) tai suuri riski sairastua ateroskleroottiseen sydän- ja verisuonitautiin (30 %). Potilailla, joilla oli vakiintunut sydän- ja verisuonitauti, oli dokumentoidusti anamneesissa sepelvaltimotauti, oireita aiheuttava ääreisvaltimotauti ja/tai aivoverisuonten ateroskleroosi. Potilailla, joilla ei ollut vakiintunutta sydän- ja verisuonitautia, katsottiin olevan suuri sydän- ja verisuonitaudin riski, koska he täyttivät vähintään yhden seuraavista kriteereistä: (1) diabetes mellitus (tyyppi 1 tai tyyppi 2) yli 65-vuotiailla naisilla tai yli 60-vuotiailla miehillä, tai (2) Reynoldsin riskipisteet > 30 % tai SCORE-riskipisteet > 7,5 % kymmenen vuoden aikana, tai (3) sepelvaltimon kalsiumarvo > 400 Agatston-yksikköä milloin tahansa aiemmin. Potilaat satunnaistettiin suhteessa 1:1 saamaan joko 180 mg Nilemdoa vuorokaudessa (n = 6 992) tai lumelääkettä (n = 6 978) joko yksin tai muiden lipidejä alentavien lääkkeiden, kuten hyvin pienten statiiniannosten, lisäksi. Kaikkiaan yli 95 %:a potilaista seurattiin tutkimuksen päättymiseen tai kuolemaan asti, ja seuranta epäonnistui alle 1 %:n kohdalla. Seurannan mediaanikesto oli 3,4 vuotta.
Keski-ikä lähtötilanteessa oli 65,5 vuotta, ja potilaista 48 % oli naisia ja 91 % oli valkoihoisia. Muita valittuja lähtötilanteen ominaisuuksia olivat hypertensio (85 %), diabetes mellitus (46 %), esidiabetes (42 %), senhetkinen tupakointi (22 %), eGFR < 60 ml/min/1,73 m2 (21 %) ja keskimääräinen painoindeksi 29,9 kg/m2. Keskimääräinen LDL-kolesterolipitoisuus lähtötilanteessa oli 3,6 mmol/l (139 mg/dl). Lähtötilanteessa 41 % potilaista sai vähintään yhtä lipidejä alentavaa hoitoa, kuten etsetimibiä (12 %) ja hyvin pieniä statiiniannoksia (23 %).
Nilemdo pienensi merkitsevästi ensisijaisen yhdistetyn päätetapahtuman eli merkittävien sydän- ja verisuonihaittatapahtumien (MACE-4) riskiä. Näitä olivat sydän- ja verisuoniperäinen kuolema, ei-fataali sydäninfarkti, ei-fataali aivohalvaus ja sepelvaltimoiden revaskularisaatio, ja niiden riski pieneni 13 %:lla verrattuna lumelääkkeeseen (riskisuhde: 0,87; 95 %:n CI: 0,79; 0,96; p = 0,0037). Myös tärkeimmän toissijaisen yhdistetyn MACE-3-päätetapahtuman (sydän- ja verisuoniperäinen kuolema, ei-fataali sydäninfarkti ja ei-fataali aivohalvaus) riski pieneni merkitsevästi 15 %:lla verrattuna lumelääkkeeseen (riskisuhde: 0,85; 95 %:n CI: 0,76; 0,96; p = 0,0058). Ensisijaisen yhdistetyn päätetapahtuman tulokset olivat yleisesti ottaen johdonmukaiset etukäteen määritellyissä alaryhmissä (mukaan lukien lähtötilanteen ikä, rotu, etninen tausta, sukupuoli, LDL-kolesteroliluokka, statiinien käyttö, etsetimibin käyttö ja diabetes). Nilemdon vaikutuksia ensisijaisen päätetapahtuman yksittäisiin osatekijöihin olivat mm. 27 %:n vähenemä ei-fataalin sydäninfarktin riskissä ja 19 %:n vähenemä sepelvaltimoiden revaskularisaation riskissä verrattuna lumelääkkeeseen. Ei-fataalit aivohalvaukset ja sydän- ja verisuoniperäisen kuoleman riski eivät vähentyneet tilastollisesti merkitsevästi verrattuna lumelääkkeeseen. Ensisijaisen ja tärkeimmän toissijaisen tehon päätetapahtuman tulokset esitetään taulukossa 4. Kaplan-Meierin käyrät ensisijaisen MACE-4-päätetapahtuman ja toissijaisen MACE-3-päätetapahtuman arvioidusta kumulatiivisesta ilmaantuvuudesta esitetään alla olevissa kuvissa 1 ja 2. Ensisijaisen MACE-4-päätetapahtuman kumulatiivinen ilmaantuvuus eriytyy kuukauteen 6 mennessä.
Lisäksi Nilemdon ja lumelääkkeen ero LDL-kolesterolin keskimääräisessä prosentuaalisessa muutoksessa lähtötilanteen ja kuukauden 6 välisenä aikana oli -20 % (95 %:n CI: -21 %, -19 %).
Taulukko 4:Nilemdon vaikutus merkittäviin sydän- ja verisuonitapahtumiin
| Päätetapahtuma | Nilemdo N = 6 992 | Lumelääke N = 6 978 | Nilemdo vs. lumelääke |
| n (%) | n (%) | Riskisuhdea (95 %:n CI) p-arvob | |
| Ensisijainen yhdistetty päätetapahtuma | |||
| Sydän- ja verisuoniperäinen kuolema, ei-fataali sydäninfarkti, ei-fataali aivohalvaus, sepelvaltimoiden revaskularisaatio (MACE-4) | 819 (11,7) | 927 (13,3) | 0,87 (0,79; 0,96) 0,0037 |
| Ensisijaisen päätetapahtuman osatekijät | |||
| Ei-fataali sydäninfarkti | 236 (3,4) | 317 (4,5) | 0,73 (0,62; 0,87) |
| Sepelvaltimoiden revaskularisaatio | 435 (6,2) | 529 (7,6) | 0,81 (0,72; 0,92) |
| Ei-fataali aivohalvaus | 119 (1,7) | 144 (2,1) | 0,82 (0,64; 1,05) |
| Sydän- ja verisuoniperäinen kuolema | 269 (3,8) | 257 (3,7) | 1,04 (0,88; 1,24) |
| Tärkeimmät toissijaiset päätetapahtumat | |||
| Sydän- ja verisuoniperäinen kuolema, ei-fataali sydäninfarkti, ei-fataali aivohalvaus (MACE-3) | 575 (8,2) | 663 (9,5) | 0,85 (0,76; 0,96) 0,0058 |
| Fataali ja ei-fataali sydäninfarkti | 261 (3,7) | 334 (4,8) | 0,77 (0,66; 0,91) 0,0016 |
| Sepelvaltimoiden revaskularisaatio | 435 (6,2) | 529 (7,6) | 0,81 (0,72; 0,92) 0,0013 |
| Fataali ja ei-fataali aivohalvaus | 135 (1,9) | 158 (2,3) | 0,85 (0,67; 1,07) NS |
CI = luottamusväli; MACE = merkittävä sydän- ja verisuonihaittatapahtuma; NS = ei merkitsevä
a. Riskisuhde ja vastaava 95 %:n CI perustuivat Coxin suhteellisen vaaran malliin, jossa hoito oli selittävä muuttuja.
b. p-arvo perustui log rank -testiin.
Huomautus: tässä taulukossa esitetään myös kunkin MACE-osatekijän ensimmäiseen esiintymiseen kulunut aika. Sama potilas voi olla mukana useammassa kuin yhdessä kategoriassa.
Kuva 1: Kaplan-Meierin käyrä MACE-4:n ensimmäiseen esiintymiseen kuluneesta ajasta
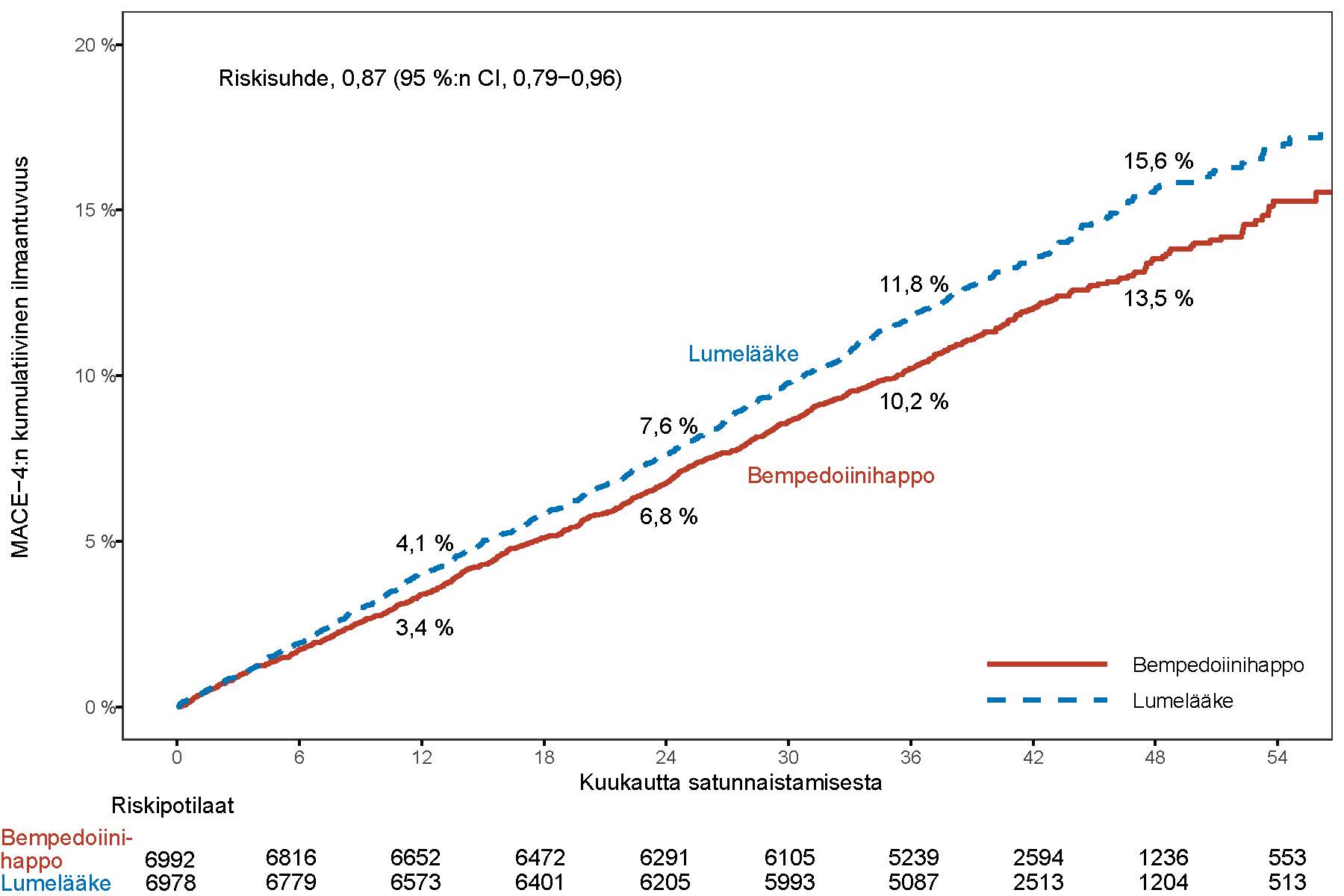
MACE = merkittävä sydän- ja verisuonihaittatapahtuma
Huomautus: MACE-4 määritellään sydän- ja verisuoniperäisestä kuolemasta, ei-fataalista sydäninfarktista, ei-fataalista aivohalvauksesta ja sepelvaltimoiden revaskularisaatiosta koostuvaksi yhdistetyksi päätetapahtumaksi.
Kuva 2: Kaplan-Meierin käyrä MACE-3:n ensimmäiseen esiintymiseen kuluneesta ajasta
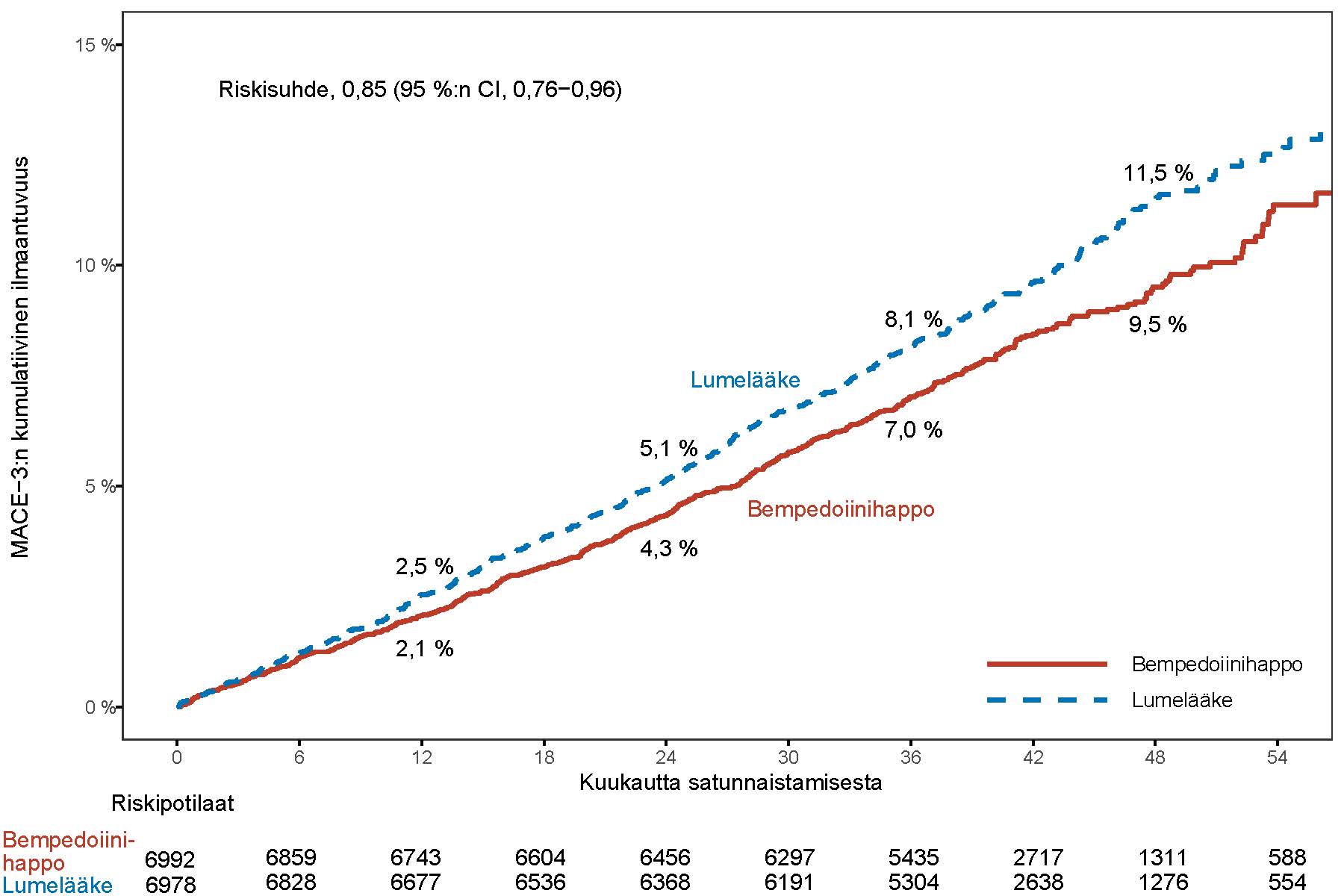
MACE = merkittävä sydän- ja verisuonihaittatapahtuma
Huomautus: MACE-3 määritellään sydän- ja verisuoniperäisestä kuolemasta, ei-fataalista sydäninfarktista ja ei-fataalista aivohalvauksesta koostuvaksi yhdistetyksi päätetapahtumaksi.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset bempedoiinihapon käytöstä kohonneen kolesterolin hoidossa pediatrisissa potilasryhmissä, joissa potilaiden ikä on 4–17 vuotta (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Farmakokineettiset tiedot osoittavat, että bempedoiinihapon imeytymisessä mediaaniaika enimmäispitoisuuden saavuttamiseen on 3,5 tuntia 180 mg:n Nilemdo-tabletteja käytettäessä. Bempedoiinihapon farmakokineettiset parametrit esitetään keskiarvoina (keskihajonta, SD), ellei toisin ole ilmoitettu. Bempedoiinihappoa voidaan pitää aihiolääkkeenä, jonka ACSVL1 aktivoi solunsisäisesti ETC-1002-CoA:ksi. Vakaan tilan Cmax oli 24,8 (6,9) mikrogrammaa/ml ja AUC-arvo 348 (120) mikrog.h/ml, kun hyperkolesterolemiaa sairastaville potilaille annettiin useita annoksia. Bempedoiinihapon vakaan tilan farmakokinetiikka oli yleisesti lineaarinen alueella 120–220 mg. Bempedoiinihapon farmakokinetiikassa ei tapahtunut aikasidonnaisia muutoksia, kun potilaalle annettiin toistuvasti suositeltu annos, ja bempedoiinihapon vakaa tila saavutettiin 7 vuorokaudessa. Bempedoiinihapon keskimääräinen kertymissuhde oli noin 2,3-kertainen.
Anto ruokailun yhteydessä ei vaikuttanut bempedoiinihapon oraaliseen biologiseen hyötyosuuteen, kun bempedoiinihappo annettiin 180 mg:n Nilemdo-tabletteina. Ruoka hidastaa bempedoiinihapon imeytymisnopeutta. Imeytymisvakio ruokailun yhteydessä on 0,32/h.
Jakautuminen
Bempedoiinihapon näennäinen jakautumistilavuus (V/F) oli 18 l. Bempedoiinihappo sitoutui plasman proteiineihin 99,3-prosenttisesti, sen glukuronidi 98,8-prosenttisesti ja sen aktiivinen metaboliitti ESP15228 99,2-prosenttisesti. Bempedoiinihappo ei jakaudu punasoluihin.
Kahdeksan terveen imettävän naisen imetystutkimuksessa arvioitiin bempedoiinihapon pitoisuuksia kypsässä äidinmaidossa. Naiset saivat 180 mg:n Nilemdo-tabletin suun kautta kerran vuorokaudessa kuutena peräkkäisenä päivänä. Geometrinen keskimääräinen bempedoiinin Cmax-arvon arvio äidinmaidossa oli 118 ng/ml (vaihteluväli 79,6–251 ng/ml) ja mediaani Tmax-aika noin 3 tuntia.
Bempedoiinihappoa havaittiin imettävien naisten äidinmaidossa, kun naiset saivat kuutena peräkkäisenä päivänä 180 mg:n annoksen bempedoiinihappoa. Bempedoiinihapon keskimääräinen päivittäinen annos vauvalle äidinmaidon kautta oli noin 0,03 mg/vrk (95 %:n CI: 0,02; 0,05) ja keskimääräinen laskettu päivittäinen vauvan oraalinen annos oli 0,012 mg/kg/vrk vauvan vakioidun maidonjuonnin 150 ml/kg/vrk perusteella. Keskimääräinen (SD) lapsen annososuus (RID) oli noin 0,5 (0,2) % äidin painoon suhtautetusta vastaavasta annoksesta. Aktiivisen metaboliitin, ESP15228:n, pitoisuudet äidinmaidossa olivat kvantitointirajan (20 ng/ml) alapuolella seitsemällä tutkitusta 8 tutkittavasta. Tietoa Nilemdon vaikutuksista imetettävään vauvaan tai äidin maidontuotantoon ei ole. Imetyksen kehitykselliset ja terveydelliset hyödyt on otettava huomioon samoin kuin äidin kliininen tarve Nilemdo-hoidolle ja Nilemdon tai äidin perussairauden imetettävälle vauvalle mahdollisesti aiheuttamat haittavaikutukset.
Biotransformaatio
Metabolisia yhteisvaikutuksia koskevat in vitro -tutkimukset viittaavat siihen, että bempedoiinihappo, sen aktiivinen metaboliitti ja glukuronidimuodot eivät metaboloidu sytokromi P450 -entsyymien kautta eivätkä estä tai indusoi niitä.
Bempedoiinihapon ensisijainen eliminaatioreitti on metaboloituminen asyyliglukuronidiksi. Lisäksi bempedoiinihappo muuntuu palautuvasti aktiiviseksi metaboliitiksi (ESP15228) ihmisen maksassa in vitro havaitun aldo-ketoreduktaasiaktiviteetin perusteella. Toistuvan annon jälkeen ESP15228:n metaboliitti-kanta-ainesuhteen keskimääräinen plasman AUC oli 18 %, ja arvo pysyi vakaana ajan kuluessa. Kumpikin yhdiste muuntuu epäaktiiviseksi glukuronidikonjugaatiksi in vitro UDP‑glukuronyylitransferaasi‑2B7:n (UGT2B7) vaikutuksesta. Bempedoiinihappoa, ESP15228:ta ja niiden konjugoituneita muotoja havaittiin plasmassa. Bempedoiinihapon osuus AUC0–48 h:sta oli suurin (46 %) ja toiseksi suurin oli sen glukuronidin osuus (30 %). ESP15228:n osuus plasma AUC0-48 h-arvosta oli 10 prosenttia ja sen glukuronidin osuus 11 prosenttia.
Hyperkolesterolemiapotilailla bempedoiinihapon ekvipotentin aktiivisen metaboliitin (ESP15228) vakaan tilan Cmax oli 3,0 (1,4) mikrogrammaa/ml ja AUC-arvo 54,1 (26,4) mikrog.h/ml. ESP15228:lla oli todennäköisesti vähäinen vaikutus bempedoiinihapon yleiseen kliiniseen aktiivisuuteen systeemisen altistumisen ja farmakokineettisten ominaisuuksien perusteella.
Eliminaatio
Bempedoiinihapon puhdistuma vakaassa tilassa (CL/F), joka määritettiin hyperkolesterolemiaa sairastavien potilaiden populaatiofarmakokineettisessä (PK) analyysissä, oli 12,1 ml/min kerran päivässä otettavan annoksen jälkeen. Muuttumattoman bempedoiinihapon munuaispuhdistuma oli alle 2 prosenttia kokonaispuhdistumasta. Bempedoiinihapon keskimääräinen (SD) puoliintumisaika ihmisillä oli 19 (10) tuntia vakaassa tilassa.
Kun potilaille annettiin suun kautta 240 mg:n kerta-annos bempedoiinihappoa (1,3 kertaa hyväksytty suositeltu annos), 62,1 prosenttia kokonaisannoksesta (bempedoiinihaposta ja sen metaboliiteista) havaittiin virtsassa, pääasiassa bempedoiinihapon asyyliglukuronidikonjugaattina, ja 25,4 % havaittiin ulosteessa. Yhteensä alle 5 prosenttia annetusta annoksesta erittyi muuttumattomana bempedoiinihappona ulosteeseen ja virtsaan.
Erityispotilasryhmät
Munuaisten vajaatoiminta
Bempedoiinihapon farmakokinetiikkaa arvioitiin kerta-annostutkimuksissa ja populaatiofarmakokineettisissä analyyseissa potilailla, joiden munuaisten vajaatoiminnan aste vaihteli. Potilailla, joilla oli lievä, keskivaikea tai vaikea munuaisten vajaatoiminta, bempedoiinihapon AUC-arvo oli 1,4–2,2-kertainen verrattuna tutkittaviin, joiden munuaiset toimivat normaalisti. Bempedoiinihapon AUC-arvo oli 1,47-kertainen (90 %:n luottamusväli: 1,01; 2,15) tutkittavilla, joilla oli loppuvaiheen munuaissairaus ja jotka saivat bempedoiinihappoa (kerta-annos 180 mg) tuntia ennen hemodialyysia ja 1,75-kertainen (90 %:n luottamusväli: 1,15; 2,68) tutkittavilla, joilla oli loppuvaiheen munuaissairaus ja jotka saivat bempedoiinihappoa 23 tunnin kuluttua hemodialyysista, verrattuna terveisiin tutkittaviin, joiden munuaiset toimivat normaalisti.
Munuaisten kautta tapahtuva erittyminen on vähäisempi muuttumattomassa muodossa olevan bempedoiinihapon eliminaatioreitti (ks. kohta Farmakokinetiikka, eliminaatio), ja AUC-altistusten geometrinen keskiarvo oli 392–480 mikrog.h/ml tutkittavilla, joiden munuaistoiminta vaihteli keskivaikeasta munuaisten vajaatoiminnasta hemodialyysia vaativaan loppuvaiheen munuaissairauteen kerta-annostutkimuksissa.
Maksan vajaatoiminta
Bempedoiinihapon ja sen metaboliitin (ESP15228) farmakokinetiikkaa tutkittiin potilailla, joilla oli normaali maksan toiminta tai lievä tai keskivaikea maksan vajaatoiminta (Child-Pughin luokitus A tai B), kerta-annoksen jälkeen (n = 8/ryhmä). Verrattuna potilaisiin, joiden maksa toimi normaalisti, bempedoiinihapon keskimääräinen Cmax-arvo oli 11 prosenttia pienempi ja AUC-arvo 22 prosenttia pienempi potilailla, joilla oli lievä maksan vajaatoiminta. Potilailla, joilla oli keskivaikea maksan vajaatoiminta, vastaavat arvot pienenivät 14 ja 16 prosenttia. Tämän ei odoteta heikentävän tehoa. Siksi annosta ei ole tarpeen muuttaa potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta.
Bempedoiinihappoa ei ole tutkittu potilailla, joilla on vaikea maksan vajaatoiminta (Child-Pughin luokitus C).
Muut erityispotilasryhmät
Ikä, sukupuoli tai rotu eivät vaikuttaneet bempedoiinihapon farmakokinetiikkaan. Paino oli tilastollisesti merkitsevä kovariaatti. Painon alimpaan kvartiiliin (< 73 kg) liittyi noin 30 prosenttia suurempi altistuminen. Altistumisen suurentuminen ei ollut kliinisesti merkittävää, eikä annoksen muuttamista painon perusteella suositella.
Prekliiniset tiedot turvallisuudesta
Tavanomaisissa genotoksisuustutkimuksissa ei ole havaittu mutageenisiä tai klastogeenisiä vaikutuksia. Bempedoiinihappo lisäsi jyrsijöiden elinikäisissä karsinogeenisuustutkimuksissa hepatosellulaaristen ja kilpirauhasen follikulaaristen kasvainten ilmaantuvuutta urosrotilla sekä hepatosellulaaristen kasvainten ilmaantuvuutta uroshiirillä. Koska nämä ovat yleisiä kasvaimia jyrsijöiden elinikäisissä biotesteissä ja kasvainten syntymekanismi johtuu jyrsijäspesifisen peroksisomiproliferaattoreilla aktivoituvan reseptori (PPAR) alfan aktivoitumisesta, näiden kasvainten ei katsota aiheuttavan riskiä ihmisille.
Maksan painon suurenemista ja hepatosellulaarista hypertrofiaa havaittiin ainoastaan rotilla, ja nämä vaikutukset palautuivat osittain kuukauden mittaisen palautumisjakson jälkeen. Tämän jakson aikana annostus oli ≥ 30 mg/kg/vrk tai neljä kertaa ihmisten altistus annoksella 180 mg. Palautuvia, ei-haitallisia laboratorioarvojen muutoksia, jotka viittasivat näihin maksaan kohdistuneisiin vaikutuksiin, punasolu- ja koagulaatioarvojen pienenemistä sekä ureatyppi- ja kreatiniiniarvojen suurenemista havaittiin rotilla ja apinoilla siedettyjä annoksia käytettäessä. Pitkäaikaistutkimuksissa annos, jolla ei todettu haittavaikutuksia (NOAEL), oli 10 mg/kg/vrk rotilla ja 60 mg/kg/vrk apinoilla, vastaten altistumista alle ihmisten altistumistason (annoksella 180 mg) ja 15 kertaa tätä altistumistasoa suurempi.
Bempedoiinihappo ei ollut teratogeeninen tai toksinen tiineenä olevien kanien alkioille tai sikiöille, kun annos oli enintään 80 mg/kg/vrk tai 12 kertaa suurempi kuin ihmisten systeeminen altistuminen (annoksella 180 mg). Elinkykyisten sikiöiden määrä väheni tiineillä rotilla, joille annettiin bempedoiinihappoa 10, 30 ja 60 mg/kg/vrk organogeneesin aikana, ja sikiön paino pieneni, kun annos oli ≥ 30 mg/kg/vrk tai neljä kertaa suurempi kuin ihmisten systeeminen altistuminen (180 mg). Sikiön luustolöydösten (lapaluun tai kylkiluiden taipumisten) määrä kasvoi kaikilla annoksilla, kun altistuminen oli vähäisempää kuin ihmisten systeeminen altistuminen (annoksella 180 mg). Syntymää edeltävässä ja syntymänjälkeisessä kehitystutkimuksessa tiineille rotille annettiin bempedoiinihappoa 5, 10, 20 ja 30 mg/kg/vrk koko tiineyden ja imetyksen ajan. Emoon kohdistui haittavaikutuksia, kun annos oli ≥ 20 mg/kg/vrk. Elävien ja eloonjääneiden poikasten määrä pieneni sekä poikasten kasvu, oppiminen ja muisti heikkenivät, kun annos oli ≥ 10 mg/kg/vrk, jolloin emon altistuminen oli 10 mg/kg/vrk eli pienempi kuin ihmisten altistumistaso annoksella 180 mg.
Nilemdon vaikutuksesta ihmisen hedelmällisyyteen ei ole saatavissa tietoja. Kun uros- ja naarasrotille annettiin bempedoiinihappoa ennen parittelua ja naaraille tiineyspäivään 7 asti, kiimakierrossa tapahtui muutoksia ja keltarauhasten ja kiinnittymisten määrä pieneni, kun annos oli ≥ 30 mg/kg/vrk. Vaikutuksia urosten tai naaraiden hedelmällisyyteen tai spermaparametreihin ei havaittu, kun annos oli 60 mg/kg/vrk (4 ja 9 kertaa suurempi kuin ihmisten systeeminen altistumistaso 180 mg).
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti
Mikrokiteinen selluloosa (E460)
Natriumtärkkelysglykolaatti (tyyppi A)
Hydroksipropyyliselluloosa (E463)
Magnesiumstearaatti (E470b)
Vedetön kolloidinen piidioksidi (E551)
Kalvopäällyste
Osittain hydrolysoitu polyvinyylialkoholi (E1203)
Talkki (E553b)
Titaanidioksidi (E171)
Makrogoli/PEG (E1521)
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
NILEMDO tabletti, kalvopäällysteinen
180 mg (L:kyllä) 28 fol (98,02 €)
PF-selosteen tieto
Polyvinyylikloridista (PVC) / alumiinista valmistetut läpipainopakkaukset.
Pakkauskoot: 10, 14, 28, 30, 84, 90, 98 tai 100 kalvopäällysteistä tablettia.
Rei’itetyt polyvinyylikloridista (PVC) / alumiinista valmistetut läpipainopakkaukset.
Pakkauskoot: 10 × 1, 50 × 1 tai 100 × 1 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen, soikea kalvopäällysteinen tabletti, koko noin 13,97 mm × 6,60 mm × 4,80 mm. Tabletin yhdellä puolella on kaiverrus ”180” ja toisella puolella ”ESP”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
NILEMDO tabletti, kalvopäällysteinen
180 mg 28 fol
- Ei korvausta.
ATC-koodi
C10AX15
Valmisteyhteenvedon muuttamispäivämäärä
15.01.2026
Yhteystiedot
 Organon Finland Oy
Organon Finland Oy Puolikkotie 8, 5. krs
02230 Espoo
Suomi
029 170 3520
dpoc.finland@organon.com