
LAZCLUZE filmdragerad tablett 80 mg, 240 mg
Observera
▼Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Lazcluze 80 mg filmdragerade tabletter
En filmdragerad tablett innehåller 80 mg lazertinib (som mesylatmonohydrat).
Lazcluze 240 mg filmdragerade tabletter
En filmdragerad tablett innehåller 240 mg lazertinib (som mesylatmonohydrat).
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Filmdragerad tablett.
Lazcluze 80 mg filmdragerade tabletter
Gul, 14 mm, oval tablett, präglad med ”LZ” på ena sidan och ”80” på andra sidan.
Lazcluze 240 mg filmdragerade tabletter
Rödlila, 20 mm, oval tablett, präglad med ”LZ” på ena sidan och ”240” på andra sidan.
Kliniska uppgifter
Terapeutiska indikationer
Lazcluze i kombination med amivantamab är avsett för första linjens behandling av vuxna patienter med avancerad icke-småcellig lungcancer (NSCLC) med EGFR exon 19-deletion eller substitutionsmutation L858R i exon 21.
Villkor
Ainoastaan syöpälääkkeiden antoon perehtyneen lääkärin tulee aloittaa hoito.
Dosering och administreringssätt
Behandling med Lazcluze ska sättas in av läkare med erfarenhet av att använda cancerläkemedel.
Innan behandling med Lazcluze inleds måste positiv EGFR-status i tumörvävnads- eller plasmaprover fastställas med en validerad testmetod. Om ingen mutation detekteras i ett plasmaprov ska tumörvävnad testas, om sådan finns i tillgänglig i tillräcklig mängd och kvalitet, på grund av risken för falskt negativa resultat med plasmatest.
Dosering
Den rekommenderade dosen av Lazcluze är 240 mg en gång dagligen i kombination med amivantamab.
Det rekommenderas att Lazcluze administreras när som helst före amivantamab när det ges samma dag. Se avsnitt Dosering och administreringssätt i produktresumén för amivantamab-läkemedlet för information om rekommenderad dosering av amivantamab.
Venösa tromboemboliska händelser (VTE) vid samtidig användning av amivantamab
Vid inledande behandling ska profylaktisk antikoagulantia administreras för att förhindra venösa tromboemboliska händelser (VTE) hos patienter som får Lazcluze i kombination med amivantamab. I enlighet med kliniska riktlinjer ska patienterna få profylaktisk dos av antingen ett direktverkande oralt antikoagulantium (DOAC) eller ett lågmolekylärt heparin (LMWH). Användning av vitamin K-antagonister rekommenderas inte.
Hud- och nagelreaktioner
Profylaktisk behandling med orala och topikala antibiotika rekommenderas för att minska risken för och svårighetsgraden av hud- och nagelreaktioner hos patienter som får Lazcluze i kombination med amivantamab. Icke-komedogen fuktkräm för huden (ceramidbaserad eller andra formuleringar som ger långvarig återfuktning av huden och utan torkmedel är att föredra) i ansiktet och på hela kroppen (utom hårbotten) och klorhexidinlösning för att tvätta händer och fötter rekommenderas också. Patienterna ska instrueras att begränsa sin exponering för sol under kombinationsbehandlingen och i 2 månader efter kombinationsbehandlingen med Lazcluze. För ytterligare information om profylax mot hud- och nagelreaktioner, se avsnitt Varningar och försiktighet.
Behandlingstid
Behandlingen ska fortsätta fram till sjukdomsprogression eller oacceptabel toxicitet.
Missad dos
Om en planerad dos av Lazcluze missas kan den administreras inom 12 timmar. Om det har gått mer än 12 timmar sedan dosen skulle ha getts, ska den missade dosen inte administreras och nästa dos ska administreras enligt det vanliga doseringsschemat.
Dosändringar
Rekommenderade dosminskningar vid biverkningar finns i tabell 1.
Tabell 1: Rekommenderade dosminskningar av Lazcluze vid biverkningar | |
Dosminskning | Rekommenderad dos |
Startdos | 240 mg en gång dagligen |
1:a dosminskning | 160 mg en gång dagligen |
2:a dosminskning | 80 mg en gång dagligen |
3:e dosminskning | Sätt ut Lazcluze |
Dosändringar vid specifika biverkningar finns i tabell 2.
Se avsnitt Dosering och administreringssätt i produktresumén för amivantamab för information om rekommenderade dosändringar av amivantamab.
Tabell 2: Rekommenderade dosändringar för Lazcluze och amivantamab vid biverkningar* | ||
Biverkning | Allvarlighetsgrad | Dosändring |
Interstitiell lungsjukdom (ILD)/pneumonit | Alla grader |
|
Venösa tromboemboliska händelser (VTE) (se avsnitt Varningar och försiktighet) | Händelser med klinisk instabilitet (t.ex. andningssvikt eller hjärtdysfunktion) |
|
Återkommande VTE‑händelse trots antikoagulation på terapeutisk nivå |
| |
Hud- och nagelreaktioner (se avsnitt Varningar och försiktighet) | Grad 1 |
|
Grad 2 |
| |
Grad 3 |
| |
Grad 4 (inklusive allvarliga bullösa, blåsbildande eller exfolierande hudreaktioner, t.ex. toxisk epidermal nekrolys) |
| |
Levertoxicitet | Grad 3–4 |
|
Parestesi | Grad 3–4 |
|
Diarré | Grad 3 |
|
Grad 4 |
| |
Stomatit | Grad 3–4 |
|
Övriga biverkningar | Grad 3–4 |
|
* Se avsnitt Dosering och administreringssätt i produktresumén för amivantamab för information om rekommenderad dosering av amivantamab. | ||
Särskilda populationer
Äldre
Inga dosjusteringar är nödvändiga (se avsnitt Biverkningar, Farmakodynamiska egenskaper och Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Baserat på farmakokinetiska (PK) populationsanalyser behövs ingen dosjustering hos patienter med lätt, måttligt eller allvarligt nedsatt njurfunktion. Data för patienter med allvarligt nedsatt njurfunktion är begränsade. Lazertinibs farmakokinetik hos patienter med terminal njursjukdom är okänd. Försiktighet ska iakttas hos patienter med terminal njursjukdom (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Ingen dosjustering behövs hos patienter med lätt eller måttligt nedsatt leverfunktion. Lazertinibs farmakokinetik hos patienter med allvarligt nedsatt leverfunktion är okänd. Försiktighet ska iakttas hos patienter med allvarligt nedsatt leverfunktion (se avsnitt Farmakokinetiska egenskaper).
Pediatrisk population
Det finns ingen relevant användning av lazertinib för en pediatrisk population vid behandling av icke-småcellig lungcancer.
Administreringssätt
Lazcluze ska sväljas. Tabletterna ska sväljas hela med eller utan mat. Tabletterna får inte krossas, delas eller tuggas.
Om kräkningar inträffar ska nästa dos tas nästa dag, oavsett när Lazcluze har intagits.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Interstitiell lungsjukdom/pneumonit
Interstitiell lungsjukdom (ILD) eller ILD-liknande biverkningar (t.ex. pneumonit), inklusive dödsfall, har rapporterats hos patienter som behandlats med lazertinib och amivantamab (se avsnitt Biverkningar). Patienter med ILD i anamnesen, läkemedelsinducerad ILD, strålningspneumonit som krävde steroidbehandling eller andra tecken på kliniskt aktiv ILD uteslöts från den pivotala kliniska studien.
Patienter ska övervakas avseende symtom som tyder på interstitiell lungsjukdom/pneumonit (t.ex. andnöd, hosta, feber). Om symtom uppträder ska behandlingen med Lazcluze avbrytas i väntan på utredning av dessa symtom. Misstänkt interstitiell lungsjukdom eller ILD-liknande biverkningar ska utvärderas och lämplig behandling ska påbörjas efter behov. Lazcluze ska sättas ut permanent hos patienter med bekräftad interstitiell lungsjukdom eller ILD-liknande biverkningar (se avsnitt Dosering och administreringssätt).
Venösa tromboemboliska händelser (VTE)
Hos patienter som fick Lazcluze i kombination med amivantamab rapporterades venösa tromboemboliska händelser (VTE), inklusive djup ventrombos (DVT) och lungemboli (PE), inklusive dödsfall (se avsnitt Biverkningar). I enlighet med kliniska riktlinjer ska patienterna få profylaktisk dos av antingen ett direktverkande oralt antikoagulantium (DOAC) eller lågmolekylärt heparin (LMWH). Användning av K-vitaminantagonister rekommenderas inte.
Tecken och symtom på VTE-händelser ska övervakas. Patienter med VTE-händelser ska behandlas med antikoagulantium enligt klinisk indikation. Vid VTE-händelser i samband med klinisk instabilitet ska behandlingen sättas ut tillfälligt tills patienten är kliniskt stabil. Därefter kan båda läkemedlen återinsättas med samma dos.
I händelse av återfall trots lämplig antikoagulation ska amivantamab sättas ut. Behandlingen med Lazcluze kan fortsätta med samma dos (se avsnitt Dosering och administreringssätt).
Hud- och nagelreaktioner
Utslag (inklusive akneliknande dermatit), klåda och torr hud har förekommit hos patienter behandlade med lazertinib i kombination med amivantamab (se avsnitt Biverkningar). Patienter ska instrueras att begränsa sin exponering för sol under och i 2 månader efter kombinationsbehandling med Lazcluze. Skyddande kläder och användning av solskyddsmedel som skyddar mot både UVA och UVB rekommenderas. En profylaktisk strategi för att förebygga utslag rekommenderas. Detta inkluderar profylaktisk behandling vid behandlingsstart med ett oralt antibiotikum (t.ex. doxycyklin eller minocyklin, 100 mg två gånger dagligen) med början på dag 1 under de första 12 behandlingsveckorna och efter avslutad oral antibiotikabehandling ska lokalverkande antibiotika i form av lotion i hårbotten (t.ex. klindamycin 1 %) användas under de följande 9 behandlingsmånaderna. En icke-komedogen fuktkräm som inte täpper porerna (ceramidbaserad eller andra formuleringar som ger långvarig återfuktning av huden och utan torkmedel är att föredra) för ansiktet och hela kroppen (utom hårbotten) samt klorhexidinlösning till att tvätta händer och fötter med rekommenderas med början dag 1 och fortsatt under hela behandlingstiden.
Det rekommenderas att recept på ytterligare topikala och/eller orala antibiotika samt topikala kortikosteroider finns tillhands vid tidpunkten för den första dosen. Detta för att minimera eventuella fördröjningar i den reaktiva behandlingen om utslag skulle förekomma trots förebyggande åtgärder. Om hud- eller nagelreaktioner uppträder ska stödjande behandling, topikala kortikosteroider och topikala och/eller orala antibiotika administreras. Vid förekomst av hudreaktioner av grad 3 eller reaktioner av grad 2 som tolereras dåligt ska även systemisk antibiotika och orala steroider administreras samt dermatologisk konsultation övervägas. Baserat på allvarlighetsgrad ska Lazcluze dosreduceras eller sättas ut, antingen tillfälligt eller permanent (se avsnitt Dosering och administreringssätt).
Ögon
Ögonsjukdomar, inklusive keratit, har förekommit hos patienter behandlade med lazertinib i kombination med amivantamab (se avsnitt Biverkningar). Patienter som uppvisar förvärrade ögonsymtom ska omedelbart remitteras till ögonläkare och sluta använda kontaktlinser tills symtomen har utvärderats.
Hjälpämnen
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per tablett, d.v.s. är näst intill ”natriumfritt”.
Interaktioner
Starka CYP3A4-inducerare kan minska plasmakoncentrationen av lazertinib. Lazertinib kan öka plasmakoncentrationerna av CYP3A4- och BCRP-substrat.
Läkemedel som kan förändra plasmakoncentrationen av lazertinib
CYP3A4-inducerare
Samtidig administrering av flera rifampicindoser (stark CYP3A4-inducerare) minskade lazertinibs Cmax med 72 % och AUC med 83 % hos friska försökspersoner. Samtidig administrering av Lazcluze och starka CYP3A4-inducerare (t.ex. karbamazepin, fenytoin, rifampicin, johannesört) ska undvikas. Samtidig administrering av Lazcluze och måttliga CYP3A4-inducerare kan också minska plasmakoncentrationen av lazertinib och därför ska måttliga CYP3A4-inducerare (t.ex. bosentan, efavirenz, modafinil) användas med försiktighet.
CYP3A4-hämmare
Samtidig administrering av flera itrakonazoldoser (stark CYP3A4-hämmare) ökade lazertinibs Cmax med faktor 1,19 och AUC med faktor 1,46 hos friska försökspersoner. Ingen initial dosjustering är nödvändig när Lazcluze administreras tillsammans med CYP3A4-hämmare.
Magsyra-reducerande medel
Inga kliniskt relevanta skillnader i farmakokinetiken för lazertinib observerades vid samtidig administrering med medel som minskar magsyran (protonpumpshämmare och H2‑receptorantagonister). Inga dosjusteringar krävs när Lazcluze används tillsammans med läkemedel som minskar magsyran.
Läkemedel vars plasmakoncentrationer kan påverkas av Lazcluze
CYP3A4-substrat
Upprepad samtidig administrering av Lazcluze 160 mg en gång dagligen ökade midazolams (CYP3A4-substrat) Cmax med faktor 1,39 och AUC med faktor 1,47. Läkemedel med smalt terapeutiskt index som är CYP3A4-substrat (t.ex. ciklosporin, everolimus, pimozid, kinidin, sirolimus, takrolimus) ska användas med försiktighet, eftersom lazertinib kan öka plasmakoncentrationerna av dessa läkemedel.
BCRP-substrat
Upprepad samtidig administrering av Lazcluze 160 mg en gång dagligen ökade rosuvastatins (BCRP-substrat) Cmax med faktor 2,24 och AUC med faktor 2,02. Läkemedel med smalt terapeutiskt index som är BCRP-substrat (t.ex. sunitinib) ska användas med försiktighet, eftersom lazertinib kan öka plasmakoncentrationerna av dessa läkemedel.
CYP1A2-substrat
Induktion av CYP1A2 kan inte uteslutas. Därför rekommenderas försiktighet vid samtidig administrering med CYP1A2-substrat (t.ex. tizanidin).
Fertilitet, graviditet och amning
Fertila kvinnor/preventivmetoder för män och kvinnor
Fertila kvinnor ska uppmanas att använda effektiv preventivmetod under behandlingen och upp till 3 veckor efter avslutad behandling.
Manliga patienter med kvinnlig fertil partner ska uppmanas att använda effektiv preventivmetod (t.ex. kondom) och inte donera eller lagra sperma under behandlingen och under 3 veckor efter den sista dosen av lazertinib.
Graviditet
Det finns inga data från användningen av lazertinib hos gravida kvinnor. Data från djurstudier har visat reproduktionstoxikologiska effekter (minskad embryo- och fosteröverlevnad och kroppsvikt hos fostret) (se avsnitt Prekliniska säkerhetsuppgifter). Baserat på verkningsmekanismen och data från djurstudier kan lazertinib orsaka fosterskador när det ges till en gravid kvinna. Lazertinib ska inte användas under graviditet, om inte nyttan med behandlingen av kvinnan anses uppväga potentiella risker för fostret. Om patienten blir gravid under behandlingen med detta läkemedel, ska hon informeras om den eventuella risken för fostret.
Amning
Det är okänt om lazertinib eller dess metaboliter utsöndras i bröstmjölk eller påverkar mjölkproduktionen. Eftersom risk för det ammade barnet inte kan uteslutas, ska kvinnliga patienter rådas att inte amma under behandlingen och under 3 veckor efter den sista dosen lazertinib.
Fertilitet
Det finns inga data om effekten av Lazcluze på fertiliteten hos människa. Djurstudier har visat att lazertinib har effekter på fortplantningsorganen hos honor (minskat antal brunstcykler och corpora lutea) och hanar (degenerativa förändringar i testiklarna) och kan försämra fertiliteten hos honor och hanar (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
Lazcluze har mindre effekt på förmågan att framföra fordon och använda maskiner. Om patienter upplever behandlingsrelaterade symtom som påverkar koncentrations- och reaktionsförmågan (t.ex. trötthet) bör de inte framföra fordon eller använda maskiner förrän symtomen försvunnit.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligaste biverkningarna i alla grader var hudutslag (89 %), nageltoxicitet (71 %), infusionsrelaterad reaktion (amivantamab) (63 %), hypoalbuminemi (amivantamab) (48 %), levertoxicitet (47 %), ödem (amivantamab) (47 %), stomatit (43 %), venös tromboembolism (37 %), parestesi (34 %), trötthet (32 %), förstoppning (29 %), diarré (29 %), torr hud (26 %), minskad aptit (24 %), klåda (24 %), hypokalcemi (21 %), andra ögonsjukdomar (21 %) och illamående (21 %).
De vanligaste allvarliga biverkningarna var venös tromboembolism (11 %), lunginflammation (4,0 %), hudutslag (3,1 %), interstitiell lungsjukdom/pneumonit (2,9 %), covid-19 (2,4 %), levertoxicitet (2,4 %), pleurautgjutning (2,1 %), infusionsrelaterad reaktion (amivantamab) (2,1 %), andningssvikt (1,4 %), trötthet (1,2 %), ödem (amivantamab) (1,2 %), hypoalbuminemi (amivantamab) (1,2 %) och hyponatremi (1,2 %).
De vanligaste biverkningarna som ledde till utsättning av Lazcluse och/eller amivantamab i samband med behandling med Lazcluze i kombination med amivantamab var hudutslag (6 %), infusionsrelaterad reaktion (amivantamab) (4,5 %), nageltoxicitet (3,6 %), interstitiell lungsjukdom/pneumonit (2,9 %), venös tromboembolism (2,9 %), lunginflammation (1,9 %) och ödem (amivantamab) (1,7 %).
Tabell över biverkningar
Tabell 3 sammanfattar de biverkningar som uppträdde hos patienter som fick lazertinib i kombination med amivantamab.
Dessa data återspeglar exponering för lazertinib hos 421 patienter som fick lazertinib i kombination med amivantamab i MARIPOSA-studien. Medianexponeringen för lazertinib var 18,5 månader (intervall: 0,2 till 31,4 månader).
Biverkningar observerade under kliniska studier listas nedan efter frekvenskategori. Frekvenskategorierna definieras enligt följande: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), sällsynta (≥ 1/10 000, < 1/1 000), mycket sällsynta (< 1/10 000) samt ingen känd frekvens (kan inte beräknas från tillgängliga data). Biverkningarna presenteras inom varje frekvensområde efter fallande allvarlighetsgrad.
| Tabell 3: Biverkningar hos patienter som fått lazertinib i kombination med amivantamab | |||
Organsystemklass Biverkning | Frekvenskategori | Alla grader (%) | Grad 3–4 (%) |
| Metabolism och nutrition | |||
| Hypoalbuminemia, b | Mycket vanliga | 48 | 5 |
| Minskad aptit | 24 | 1,0 | |
| Hypokalcemi | 21 | 2,1 | |
| Hypokalemi | 14 | 3,1 | |
| Hypomagnesemi | Vanliga | 5 | 0 |
| Centrala och perifera nervsystemet | |||
| Parestesia | Mycket vanliga | 34 | 1,7 |
| Yrsela | 13 | 0 | |
| Ögon | |||
| Andra ögonsjukdomara | Mycket vanliga | 21 | 0,5 |
| Synnedsättninga | Vanliga | 4,5 | 0 |
| Keratit | 2,6 | 0,5 | |
| Tillväxt av ögonfransara | 1,9 | 0 | |
| Blodkärl | |||
| Venös tromboembolisma | Mycket vanliga | 37 | 11 |
| Andningsvägar, bröstkorg och mediastinum | |||
| Interstitiell lungsjukdom/pneumonita | Vanliga | 3,1 | 1,2 |
| Magtarmkanalen | |||
| Stomatita | Mycket vanliga | 43 | 2,4 |
| Diarré | 29 | 2,1 | |
| Förstoppning | 29 | 0 | |
| Illamående | 21 | 1,2 | |
| Kräkningar | 12 | 0,5 | |
| Magsmärtora | 11 | 0 | |
| Hemorrojder | Vanliga | 10 | 0,2 |
| Lever och gallvägar | |||
| Levertoxiciteta | Mycket vanliga | 47 | 9 |
| Hud och subkutan vävnad | |||
| Utslaga | Mycket vanliga | 89 | 27 |
| Nageltoxiciteta | 71 | 11 | |
| Torr huda | 26 | 1,0 | |
| Klåda | 24 | 0,5 | |
| Hand-fotsyndrom (palmoplantar erytrodysestesi) | Vanliga | 6 | 0,2 |
| Urtikaria | 1,2 | 0 | |
| Muskuloskeletala systemet och bindväv | |||
| Muskelspasmer | Mycket vanliga | 17 | 0,5 |
| Myalgi | 13 | 0,7 | |
| Allmänna symtom och/eller symtom vid administreringsstället | |||
| Ödema, b | Mycket vanliga | 47 | 2,9 |
| Tröttheta | 32 | 3,8 | |
| Feber | 12 | 0 | |
| Skador, förgiftningar och behandlingskomplikationer | |||
| Infusionsrelaterad reaktionb | Mycket vanliga | 63 | 6 |
a grupperade termer b gäller endast för amivantamab. | |||
Beskrivning av utvalda biverkningar
Venös tromboembolism
Venösa tromboemboliska händelser (VTE), inklusive djup ventrombos (14,5 %) och lungemboli (PE) (17,3 %), rapporterades hos 37 % av de patienter som fick lazertinib i kombination med amivantamab. De flesta fallen var av grad 1 eller 2, medan händelser av grad 3–4 inträffade hos 11 % och dödsfall inträffade hos 0,5 % av patienterna som fick lazertinib i kombination med amivantamab. För information om profylaktiska antikoagulantia och hantering av VTE-händelser, se avsnitt Dosering och administreringssätt och Varningar och försiktighet.
Hos patienter som fick lazertinib i kombination med amivantamab var mediantiden till första VTE-händelsen 84 dagar. VTE-händelser ledde till att all behandling avbröts hos 2,9 % av patienterna.
Interstitiell lungsjukdom (ILD)/pneumonit
Interstitiell lungsjukdom eller ILD-liknande biverkningar (t.ex. pneumonit) har rapporterats vid användning av lazertinib i kombination med amivantamab, liksom vid användning av andra EGFR-hämmare. Interstitiell lungsjukdom eller pneumonit rapporterades hos 3,1 % av patienterna som behandlades med lazertinib i kombination med amivantamab, inklusive 0,2 % fall med dödlig utgång. Patienter med ILD i anamnesen, läkemedelsinducerad ILD, strålningspneumonit som krävt steroidbehandling eller med belägg för kliniskt aktiv ILD, uteslöts från den kliniska studien (se avsnitt Varningar och försiktighet).
Hud- och nagelreaktioner
Utslag (inklusive akneliknande dermatit), klåda och torr hud har förekommit. Utslag förekom hos 89 % av patienter som behandlats med lazertinib i kombination med amivantamab. De flesta fallen var av grad 1 eller 2; fall av grad 3 förekom hos 27 % av patienterna. Utslag som ledde till utsättning av all behandling förekom hos 6 % av patienterna. Utslagen uppträdde vanligtvis inom behandlingens första 4 veckor, med en mediantid till uppkomst på 14 dagar. Nageltoxicitet förekom hos patienter som behandlades med lazertinib i kombination med amivantamab. De flesta fallen var av grad 1 eller 2; nageltoxicitet av grad 3 förekom hos 11 % av patienterna (se avsnitt Varningar och försiktighet).
En fas 2-studie på patienter som behandlades med Lazcluze i kombination med amivantamab genomfördes för att utvärdera användningen av profylaktisk behandling med ett oralt antibiotikum, ett topikalt antibiotikum i hårbotten, en fuktighetskräm i ansiktet och på hela kroppen (utom hårbotten) samt ett antiseptiskt medel på händer och fötter (se avsnitt Dosering och administreringssätt och Varningar och försiktighet). En minskning av förekomsten av dermatologiska biverkningar av ≥ grad 2 under de första 12 veckorna av behandlingen påvisades, jämfört med den dermatologiska standardbehandling som används enligt klinisk praxis (38,6 % jämfört med 76,5 %, p < 0,0001). Dessutom sågs en minskning av biverkningar av ≥ grad 2 i hårbotten under de första 12 veckorna av behandlingen (8,6 % jämfört med 29,4 %) samt en lägre förekomst av dosreduktioner (7,1 % jämfört med 19,1 %), dosavbrott (15,7 % jämfört med 33,8 %) och behandlingsavbrott (1,4 % jämfört med 4,4 %) på grund av dermatologiska biverkningar.
Ögon
Ögonsjukdomar, inklusive keratit (2,6 %) förekom hos patienter som behandlats med lazertinib i kombination med amivantamab. Andra rapporterade biverkningar var tillväxt av ögonfransar, synnedsättning och andra ögonsjukdomar. De flesta händelser var av grad 1‑2 (se avsnitt Varningar och försiktighet).
Levertoxicitet
Levertoxiska reaktioner förekom hos 47 % av patienterna som behandlades med lazertinib i kombination med amivantamab. De flesta händelserna var av grad 1 eller 2, medan levertoxicitet av grad 3 eller 4 förekom hos 9 % av patienterna. De flesta händelserna var relaterade till förhöjningar av serumtransaminaser (36 % förhöjt alaninaminotransferas och 29 % förhöjt aspartataminotransferas). I studien kunde de flesta patienter med förhöjda transaminasvärden fortsätta behandlingen utan modifiering av behandlingen, men för ett litet antal gjordes dosavbrott eller dosreduktion. Det förekom inga fall av leversvikt eller dödlig levertoxicitet i kliniska studier med lazertinib i kombination med amivantamab.
Enstaka rapporter om förhöjt alkaliskt fosfatas och långvarigt förhöjt bilirubin har identifierats med lazertinib som monoterapi.
Parestesi
Parestesi förekom hos 34 % av patienterna som behandlades med lazertinib i kombination med amivantamab. De flesta händelserna var av grad 1 eller 2, medan parestesi av grad 3 förekom hos 1,7 % av patienterna. Hos de flesta patienter med parestesi försvann besvären vid dosavbrott eller dosreduktion.
Stomatit
Stomatit förekom hos 43 % av patienterna som behandlades med lazertinib i kombination med amivantamab. De flesta händelserna var av grad 1 eller 2, medan stomatit av grad 3 förekom hos 2,4 % av patienterna.
Diarré
Diarré förekom hos 29 % av patienterna som behandlades med lazertinib i kombination med amivantamab. De flesta händelserna var av grad 1 eller 2, medan diarré av grad 3 förekom hos 2,1 % av patienterna.
Särskilda populationer
Äldre
Det finns begränsade kliniska data för lazertinib hos patienter som är 75 år och äldre (se avsnitt Farmakodynamiska egenskaper). Äldre patienter (≥ 65 år) rapporterade fler biverkningar av grad 3 eller högre jämfört med patienter < 65 år (81 % jämfört med 70 %). Medan frekvensen av läkemedelsavbrott och dosreduktioner var likartad, var frekvensen av biverkningar som ledde till behandlingsavbrott högre hos patienter ≥ 65 år jämfört med patienter < 65 år (47 % jämfört med 25 %).
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Det finns ingen känd specifik antidot mot överdosering av Lazcluze. Vid överdosering ska Lazcluze sättas ut och allmänna stödåtgärder vidtas. Patienterna ska övervakas noggrant för tecken och symtom på biverkningar.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: antineoplastiska medel, proteinkinashämmare, ATC‑kod: L01EB09.
Verkningsmekanism
Lazertinib är en irreversibel EGFR-tyrosinkinashämmare (TKI). Den hämmar selektivt både primära aktiverande EGFR-mutationer (exon 19-deletion och substitutionsmutation L858R i exon 21) och EGFR T790M-resistensmutation, medan den har mindre aktivitet mot EGFR av vildtyp.
Farmakodynamisk effekt
Baserat på dos-responsanalyserna för säkerhet verkade parestesi och stomatit uppvisa en trend av ökande förekomst vid ökad exponering för lazertinib.
Hjärtelektrofysiologi
Lazertinibs potential gällande förlängning av QTc-intervallet utvärderades med en dos-responsanalys som genomfördes med kliniska data från 243 patienter med icke-småcellig lungcancer (NSCLC) som fick 20, 40, 80, 120, 160, 240 eller 320 mg lazertinib en gång dagligen i en fas 1/II-studie. Dos-responsanalysen visade inget kliniskt relevant samband mellan plasmakoncentrationen av lazertinib och förändringen av QTc-intervallet.
Klinisk effekt och säkerhet
MARIPOSA är en randomiserad, öppen multicenterstudie i fas 3 med aktiv kontroll som utvärderar effekt och säkerhet för Lazcluze i kombination med amivantamab jämfört med osimertinib som monoterapi vid första linjens behandling av patienter med EGFR-muterad lokalt avancerad eller metastaserad icke-småcellig lungcancer (NSCLC) som inte är mottaglig för kurativ behandling. Patientproverna måste ha en av de två vanligaste EGFR-mutationerna (exon 19-deletion eller substitutionsmutation L858R i exon 21), som identifierats med lokala tester. Prover på tumörvävnad (94 %) och/eller plasma (6 %) för alla patienter testades lokalt för att fastställa mutationsstatus i EGFR-genen (exon 19-deletion och/eller substitutionsmutation L858R i exon 21) med hjälp av Polymerase Chain Reaction (PCR) hos 65 % av patienterna och Next generation Sequencing (NGS) hos 35 % av patienterna.
Totalt 1 074 patienter randomiserades (2:2:1) till att få Lazcluze i kombination med amivantamab, osimertinib som monoterapi eller Lazcluze som monoterapi fram till sjukdomsprogression eller oacceptabel toxicitet. Lazcluze administrerades oralt i dosen 240 mg en gång dagligen. Amivantamab administrerades intravenöst i dosen 1 050 mg (hos patienter < 80 kg) eller 1 400 mg (hos patienter ≥ 80 kg) en gång i veckan i 4 veckor och därefter varannan vecka från vecka 5. Osimertinib administrerades oralt i en dos på 80 mg en gång dagligen. Randomiseringen stratifierades efter EGFR-mutationstyp (exon 19-deletion eller substitutionsmutation L858R i exon 21), etnicitet (asiatisk eller icke-asiatisk) och tidigare hjärnmetastaser (ja eller nej).
Demografi och sjukdomsegenskaper vid baseline var balanserade mellan behandlingsgrupperna. Medianåldern var 63 (intervall: 25–88) år med 45 % av patienterna var ≥ 65 år och 11 % ≥ 75 år; 62 % var kvinnor; 59 % var asiater och 38 % var vita. Vid baseline var funktionsstatus 0 (34 %) eller 1 (66 %) enligt Eastern Cooperative Oncology Group (ECOG); 69 % hade aldrig rökt; 41 % hade tidigare haft hjärnmetastaser och 90 % hade cancer i stadie IV vid första diagnos. När det gäller EGFR-mutationsstatus var 60 % exon 19-deletion och 40 % substitutionsmutation L858R i exon 21.
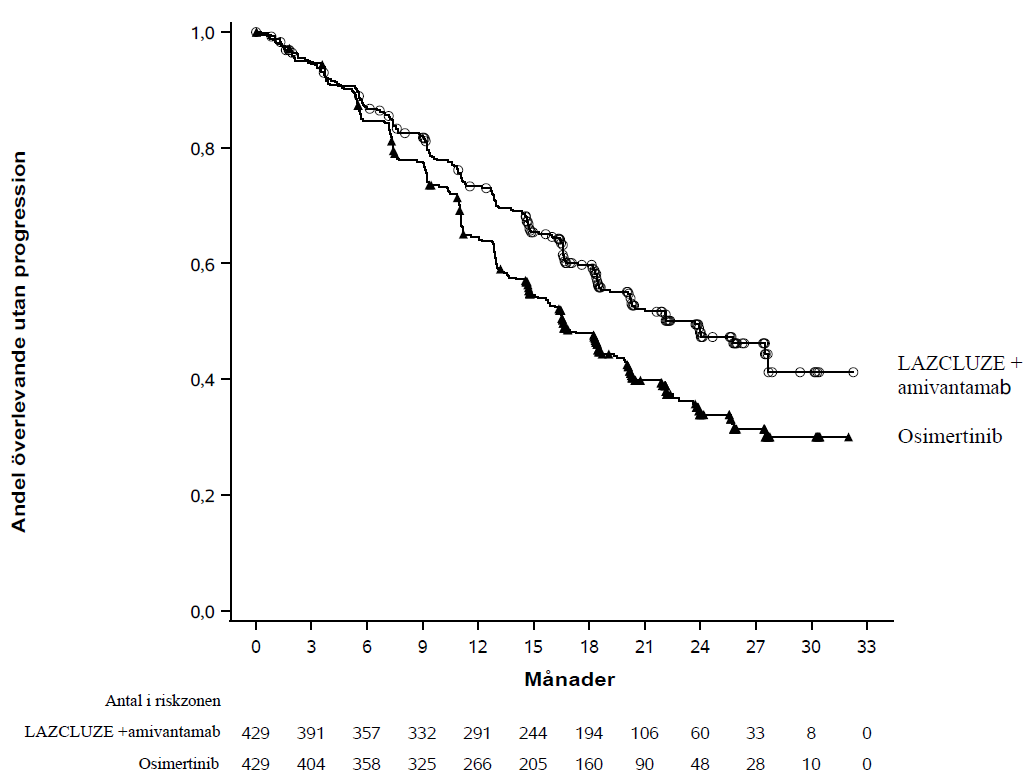
Lazcluze i kombination med amivantamab uppvisade en statistiskt signifikant förbättring av progressionsfri överlevnad (PFS) enligt blindad oberoende central granskning (BICR).
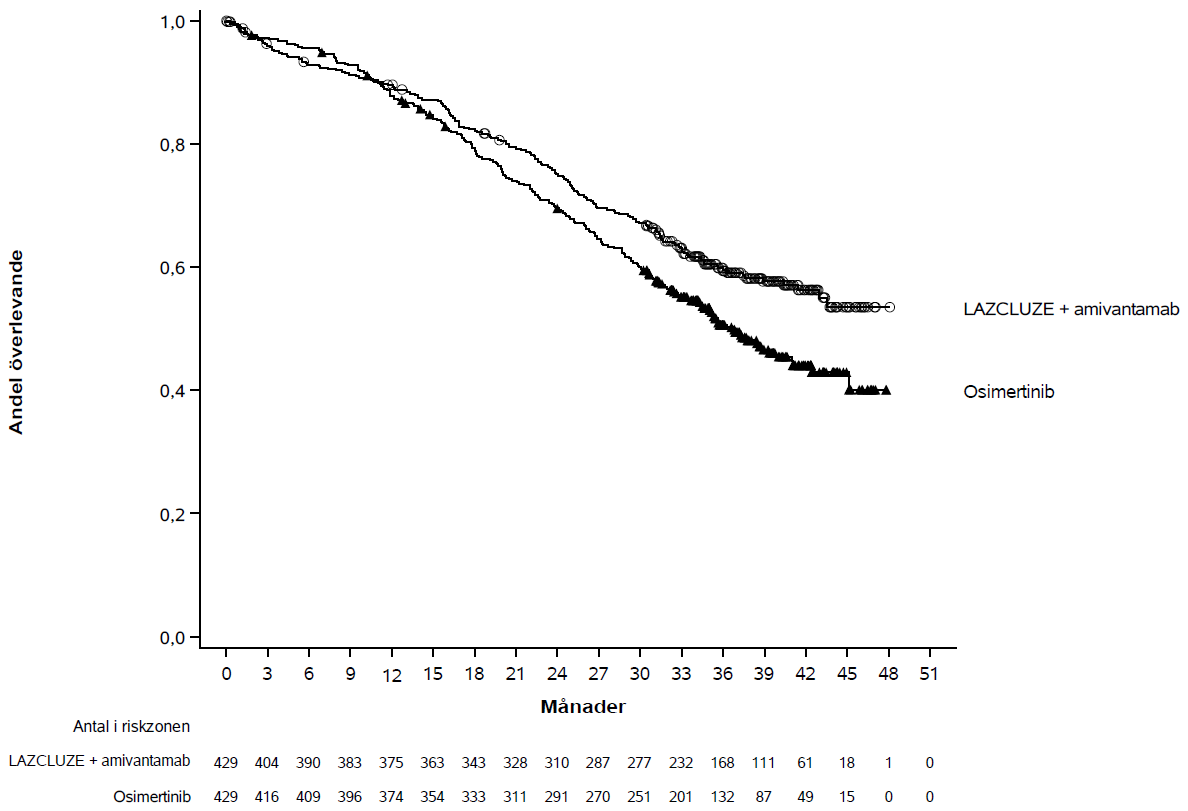
Den slutliga analysen av OS visade en statistiskt signifikant förbättring i OS för Lazcluze i kombination med amivantamab jämfört med osimertinib (se tabell 4 och figur 2).
I tabell 4, figur 1 och figur 2 sammanfattas effektresultaten för Lazcluze i kombination med amivantamab.
Tabell 4: Effektresultat i MARIPOSA | ||
Lazcluze + amivantamab (N=429) | Osimertinib (N=429) | |
Progressionsfri överlevnad (PFS)a | ||
Antal händelser | 192 (45 %) | 252 (59 %) |
Median, månader (95 % CI) | 23,7 (19,1; 27,7) | 16,6 (14,8; 18,5) |
HR (95 % CI); p-värde | 0,70 (0,58; 0,85); p=0,0002 | |
Total överlevnad (OS) | ||
Antal händelser | 173 (40 %) | 217 (51 %) |
Median, månader (95 % CI) | NE (42,9; NE) | 36,7 (33,4; 41,0) |
HR (95 % CI); p-värde | 0,75 (0,61; 0,92); p=0,0048 | |
Objektiv svarsfrekvens (ORR)a, b | ||
ORR % (95 % CI) | 80 % (76 %; 84 %) | 77 % (72 %; 81 %) |
Svarets varaktighet (DOR)a, b | ||
Median, månader (95 % CI) | 25,8 (20,3; 33,9) | 18,1 (14,8; 20,1) |
BICR = blindad oberoende central granskning; CI = konfidensintervall; NE = kan inte uppskattas. PFS-resultaten är från datagränsvärde 11 augusti 2023 med mediantid för uppföljning 22,0 månader. ORR- och DOR-resultat är från datagränsvärde 13 maj 2024 med mediantid för uppföljning 31,3 månader. OS-resultat är från datagränsvärde 4 december 2024 med en medianuppföljning på 37,8 månader. a BICR enligt RECIST v1.1. b Baserat på personer med bekräftad respons. | ||
Figur 1: Kaplan-Meier-kurva för PFS hos tidigare obehandlade patienter med NSCLC enligt BICR
Figur 2: Kaplan-Meier-kurva för OS hos tidigare obehandlade patienter med NSCLC
Intrakraniell ORR och DOR enligt BICR var förutbestämda effektmått i MARIPOSA. I undergruppen av patienter med intrakraniella lesioner vid baseline visade kombinationen av Lazcluze och amivantamab liknande intrakraniell ORR som kontrollgruppen. I enlighet med protokollet genomgick alla patienter i MARIPOSA på varandra följande magnetkameraundersökningar av hjärnan för att bedöma intrakraniellt svar och varaktighet. Resultaten sammanfattas i tabell 5.
Tabell 5: Intrakraniell ORR och DOR enligt BICR hos försökspersoner med intrakraniella lesioner vid baseline | ||
Lazcluze + amivantamab (N=180) | Osimertinib (N=186) | |
Intrakraniell tumörresponsbedömning | ||
Intrakraniell ORR (CR+PR), % (95 % CI) | 78 % (71 %; 84 %) | 77 % (71 %; 83 %) |
Komplett svar | 64 % | 59 % |
Intrakraniell DOR | ||
Median, månader (95 % CI) | 35,0 (20,4; NE) | 25,1 (22,1; 31,2) |
CI = konfidensintervall; NE = kan inte uppskattas Intrakraniell ORR och DOR-resultaten är från datagränsvärde 4 december 2024 med mediantid för uppföljning på 37,8 månader. | ||
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för Lazcluze för alla grupper av den pediatriska populationen vid NSCLC, icke-småcellig lungcancer.
Farmakokinetiska egenskaper
Efter oral administrering en eller flera gånger dagligen ökade den maximala plasmakoncentrationen av lazertinib (Cmax) och arean under plasmakoncentrationskurvan (AUC) ungefär dosproportionellt över dosintervallet 20 till 320 mg.
Plasmaexponering vid steady state uppnåddes dag 15 efter administrering en gång dagligen och ungefär tvåfaldig ackumulering observerades vid steady state med dosen 240 mg en gång dagligen.
Plasmaexponeringen för lazertinib var jämförbar när lazertinib administrerades antingen i kombination med amivantamab eller som monoterapi.
Absorption
Mediantiden för att uppnå Cmax vid engångsdos och steady state var jämförbar och varierade mellan 2 och 4 timmar.
Efter administrering av 240 mg lazertinib med en fettrik måltid (800~1 000 kcal, fetthalt cirka 50 %) var Cmax och AUC för lazertinib jämförbara med de under fastande förhållanden, vilket tyder på att lazertinib kan tas med eller utan föda.
Distribution
Lazertinib distribuerades i stor utsträckning, med en genomsnittlig (CV %) skenbar distributionsvolym på 4 264 (43,2 %) l vid dosen 240 mg. Lazertinibs genomsnittliga (CV %) plasmaproteinbindningsgrad var cirka 99,2 % (0,13 %) hos människor. Lazertinib uppvisade kovalent bindning till humana blod- och plasmaproteiner efter oral dosering och under in vitro-inkubation.
Metabolism
Lazertinib metaboliseras främst genom glutationkonjugering, antingen enzymatiskt via glutation-S-transferas (GST) eller icke-enzymatiskt, samt av CYP3A4. De mest förekommande metaboliterna är glutationskataboliter som anses vara kliniskt inaktiva. Plasmaexponeringen av lazertinib påverkades av GSTM1-medierad metabolism, vilket ledde till lägre exponering (mindre än tvåfaldig skillnad) hos patienter med icke-null GSTM1. Ingen dosjustering krävs baserat på GSTM1-status.
Eliminering
Medelvärdet (CV %) för skenbar clearance och terminal halveringstid för lazertinib vid dosen 240 mg var 44,5 (29,5 %) l/tim respektive 64,7 (32,8 %) timmar.
Exkretion
Efter en oral engångsdos av radioaktivt märkt lazertinib återfanns cirka 86 % av dosen i feces (< 5 % som oförändrad substans) och 4 % i urin (< 0,5 % som oförändrad substans).
Samtidig administrering med OCT1- och UGT1A1-substrat
Samtidig administrering av flera doser Lazcluze ökade inte metformins (OCT1-substrat) Cmax och AUC. Lazcluze hämmar inte OCT1.
Baserat på in vitro-studier kan Lazcluze hämma UGT1A1. På grund av avsaknad av effekt på indirekta bilirubinnivåer i kliniska studier förväntas dock ingen kliniskt relevant interaktion med UGT1A1-substrat.
Särskilda populationer
Äldre
Baserat på farmakokinetisk populationsanalys observerades inga kliniskt betydelsefulla åldersbaserade skillnader i farmakokinetiken för lazertinib.
Nedsatt njurfunktion
Baserat på farmakokinetisk populationsanalys krävs ingen dosjustering för patienter med lätt, måttligt eller allvarligt nedsatt njurfunktion med uppskattad glomerulär filtrationshastighet (eGFR) på 15 till 89 ml/min. Data för patienter med allvarligt nedsatt njurfunktion (eGFR på 15 till 29 ml/min) är begränsade (n=3), men det finns inga belägg för att dosjustering krävs för dessa patienter. Inga data finns för patienter med terminal njursjukdom (eGFR < 15 ml/min).
Nedsatt leverfunktion
Baserat på resultaten från den kliniska farmakologistudien hade måttligt nedsatt leverfunktion (Child-Pugh klass B) ingen kliniskt betydelsefull effekt på farmakokinetiken för lazertinib som engångsdos. Baserat på farmakokinetisk populationsanalys krävs ingen dosjustering för patienter med lätt (totalt bilirubin ≤ ULN och ASAT > ULN eller ULN < totalt bilirubin ≤ 1,5×ULN och något ASAT) eller måttligt (1,5×ULN < totalt bilirubin ≤ 3×ULN och något ASAT) nedsatt leverfunktion. Inga data finns för patienter med allvarligt nedsatt leverfunktion (totalt bilirubin > 3×ULN och något ASAT).
Pediatrisk population
Farmakokinetiken för lazertinib hos barn har inte undersökts.
Andra populationer
Inga kliniskt betydelsefulla skillnader i farmakokinetiken för lazertinib observerades baserat på kön, kroppsvikt, etnicitet, laboratorievärden vid baseline (kreatininclearance, albumin, alaninaminotransferas, alkaliskt fosfatas, aspartataminotransferas), funktionsstatus enligt ECOG, EGFR-mutationstyp, cancerstadium vid första diagnos, tidigare behandlingar, hjärnmetastaser och tidigare rökning.
Prekliniska säkerhetsuppgifter
De viktigaste fynden som observerades i toxicitetsstudier med upprepad dosering av lazertinib på råttor och hundar omfattade mild epitelatrofi till degenerativa erosioner, inflammation och nekros som påverkade ögat (hornhinneatrofi), huden (tunn och sträv hårväxt, hårsäcksdegeneration, alopeci, sår), lever (förhöjda leverenzymer, kupffercellhypertrofi och hepatocellulär nekros), lungor (alveolärt makrofaginfiltrat, lunginflammation och hyperplasi av alveolära celler av typ II) njure (tubulär dilatation, papillär nekros, högre ureakväve, kreatinin (endast kvinnor), oorganisk fosfor och kalium), magtarmkanal (esofagusepitelatrofi, villusblunting/fusion i duodenum och jejunum, flytande avföring), fortplantningssystem (degeneration av testikeltubuli, hypospermi, färre brunstcykler och corpora lutea, atrofi i livmoder och vagina). Dessa fynd observerades hos djur i exponeringsintervall på 0,9–3,4 gånger högre än den beräknade exponeringen hos patienter som administrerats med den rekommenderade dosen (240 mg) och försvann helt eller delvis under återhämtningsfaserna. Hjärtat ansågs vara ett målorgan enbart hos hundar och förekom vid exponeringsnivåer som var 7 gånger högre än de exponeringsnivåer som förväntas vid den rekommenderade dosen för människor.
Karcinogenicitet och mutagenicitet
Inga tecken på genotoxicitet för lazertinib observerades i tester av bakteriell mutagenicitet in vitro, kromosomavvikelsetester in vitro och mikronukleustester in vivo på råttor. Långtidsstudier på djur har inte utförts för att utvärdera den karcinogena potentialen av lazertinib.
Reproduktionstoxikologi
Baserat på djurstudier kan fertiliteten hos män och kvinnor försämras vid behandling med lazertinib. Degenerativa förändringar förekom i testiklarna hos råttor och hundar, vilket resulterade i minskad luminal spermieproduktion hos hundar efter exponering av lazertinib under 1 månad vid kliniskt relevanta exponeringsnivåer. Minskat antal corpora lutea noterades i äggstockarna hos råttor som exponerats för lazertinib i ≥ 1 månad vid kliniskt relevanta exponeringsnivåer. I en studie av fertilitet och tidig embryonal utveckling på han- och honråttor inducerade lazertinib en minskning av antalet brunstcykler, en ökning av förlust efter implantation och minskad levande kullstorlek vid eller under den dosnivå som motsvarar den kliniska exponeringen hos människa vid den rekommenderade dosen 240 mg.
Utvecklingstoxicitet observerades i studier av embryo- och fosterutveckling hos råttor och kaniner. Hos råttor observerades låg kroppsvikt hos foster i samband med toxicitet hos modern vid en exponering hos modern som var ungefär 4 gånger högre än den kliniska exponeringen hos människa vid 240 mg. Hos kaniner observerades en ökad förekomst av skallbensfusion hos foster (okbensbågen sammanvuxen med överkäksutskottet) då mödrar exponerats för doser väl under den kliniska exponeringen för människa vid 240 mg.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Tablettkärna
Kiseldioxid, kolloidal, vattenfri
Kroskarmellosnatrium (E 468)
Cellulosa, mikrokristallin (E 460i)
Mannitol (E 421)
Magnesiumstearat (E 572)
Filmdragering
Lazcluze 80 mg filmdragerade tabletter
Makrogol-poly(vinylalkohol)-ympsampolymer (E 1209)
Polyvinylalkohol (E 1203)
Glycerolmonokaprylokaprat typ I (E 471)
Titandioxid (E 171)
Talk (E 553b)
Gul järnoxid (E 172)
Lazcluze 240 mg filmdragerade tabletter
Makrogol-poly(vinylalkohol)-ympsampolymer (E 1209)
Polyvinylalkohol (E 1203)
Glycerolmonokaprylokaprat typ I (E 471)
Titandioxid (E 171)
Talk (E 553b)
Röd järnoxid (E 172)
Svart järnoxid (E 172)
Inkompatibiliteter
Ej relevant.
Hållbarhet
2 år
Särskilda förvaringsanvisningar
Inga särskilda förvaringsanvisningar.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
LAZCLUZE tabletti, kalvopäällysteinen
80 mg (L:ei) 56 fol (5231,35 €)
240 mg (L:ei) 28 fol (7756,73 €)
PF-selosteen tieto
Lazcluze 80 mg filmdragerade tabletter
Blisterförpackning
Polyvinylklorid-polyklorotrifluoroetylenfilm (PVC/PCTFE) med tryckfolie av aluminium.
- En kartong innehåller 56 filmdragerade tabletter (2 vikförpackningar med 28 tabletter vardera).
Burk
Vit ogenomskinlig burk av högdensitetspolyeten (HDPE) med barnskyddande förslutning av polypropen med 60 eller 90 tabletter. Varje kartong innehåller en burk.
Lazcluze 240 mg filmdragerade tabletter
Blisterförpackning
Polyvinylklorid-polyklorotrifluoroetylenfilm (PVC/PCTFE) med tryckfolie av aluminium.
- En kartong innehåller 14 filmdragerade tabletter (1 vikförpackning med 14 tabletter).
- En kartong innehåller 28 filmdragerade tabletter (2 vikförpackningar med 14 tabletter vardera).
Burk
Vit ogenomskinlig burk av högdensitetspolyeten (HDPE) med barnskyddande förslutning av polypropen med 30 filmdragerade tabletter. Varje kartong innehåller en burk.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Särskilda anvisningar för destruktion och övrig hantering
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
LAZCLUZE tabletti, kalvopäällysteinen
80 mg 56 fol
240 mg 28 fol
- Ei korvausta.
Atc-kod
L01EB09
Datum för översyn av produktresumén
12.02.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com