
HETRONIFLY koncentrat till infusionsvätska, lösning 10 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Yleinen
Observera

Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Serplulimab är en humaniserad antikropp (IgG4/kappa-isotyp med en stabiliserande sekvensförändring i hinge-regionen) som produceras i ovarieceller från kinesisk hamster med rekombinant DNA-teknologi.
Hjälpämne med känd effekt
Varje 10 ml injektionsflaska innehåller 0,98 mmol (22,5 mg) natrium och 2,0 mg polysorbat 80 (E 433).
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Varje ml koncentrat till infusionsvätska, lösning innehåller 10 mg serplulimab.
En injektionsflaska med 10 ml koncentrat innehåller 100 mg serplulimab.
Läkemedelsform
Koncentrat till infusionsvätska, lösning (sterilt koncentrat).
Kliniska uppgifter
Terapeutiska indikationer
Småcellig lungcancer (SCLC)
HETRONIFLY i kombination med karboplatin och etoposid är indicerat för första linjens behandling av vuxna patienter med utbredd småcellig lungcancer (ES‑SCLC).
Icke-småcellig lungcancer (NSCLC)
HETRONIFLY i kombination med karboplatin och pemetrexed är indicerat för första linjens behandling av vuxna patienter med NSCLC av icke-skivepiteltyp utan positiva EGFR-, ALK- eller ROS1-mutationer och som har:
-
lokalt avancerad NSCLC och som inte är kandidater för operation eller strålbehandling
-
metastaserad NSCLC.
HETRONIFLY i kombination med karboplatin och nab‑paklitaxel är indicerat för första linjens behandling av vuxna patienter med icke-resektabel, lokalt avancerad eller metastaserad icke-småcellig lungcancer av skivepiteltyp.
Esofaguscancer av skivepiteltyp (OSCC)
HETRONIFLY i kombination med fluoropyrimidin- och platinabaserad kemoterapi är indicerat för första linjens behandling av vuxna patienter med icke-resektabel, lokalt avancerad, recidiverande eller metastaserad esofaguscancer av skivepiteltyp vars tumörer uttrycker PD‑L1 med CPS ≥ 5.
Villkor
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Behandlingen måste initieras och övervakas av en läkare med erfarenhet av cancerbehandling.
PD‑L1-testning
Om det är specificerat i indikationen bör urvalet av patienter som ska behandlas med HETRONIFLY baserat på tumöruttrycket av PD‑L1, bekräftas med ett CE-märkt IVD-test med motsvarande avsett syfte. Om det CE-märkta IVD-testet inte är tillgängligt bör ett validerat test användas (se avsnitt Terapeutiska indikationer, Varningar och försiktighet och Farmakodynamiska egenskaper).
Dosering
Småcellig lungcancer (SCLC)
Den rekommenderade dosen under både induktions- och underhållsfaserna är serplulimab 4,5 mg/kg kroppsvikt var tredje vecka fram till sjukdomsprogression eller oacceptabel toxicitet. Under induktionsfasen (4 cykler) administreras karboplatin dag 1 och etoposid dag 1, 2 och 3 i varje 3‑veckorscykel.
Icke-småcellig lungcancer (NSCLC)
För behandling av icke-skivepiteltyp NSCLC är den rekommenderade dosen under både induktions- och underhållsfaserna serplulimab 4,5 mg/kg kroppsvikt var tredje vecka fram till sjukdomsprogression eller oacceptabel toxicitet. Under induktionsfasen (4 cykler) administreras karboplatin och pemetrexed dag 1 i varje 3‑veckorscykel. Under underhållsfasen fortsätter administreringen av pemetrexed enligt läkarens beslut.
För behandling av skivepiteltyp NSCLC är den rekommenderade dosen under både induktions- och underhållsfaserna serplulimab 4,5 mg/kg kroppsvikt var tredje vecka fram till sjukdomsprogression eller oacceptabel toxicitet. Under induktionsfasen (4‑6 cykler) administreras karboplatin dag 1 och nab‑paklitaxel dag 1, 8 och 15 i varje 3‑veckorscykel.
Esofaguscancer av skivepiteltyp (OSCC)
Den rekommenderade dosen under både induktions- och underhållsfaserna är serplulimab 3,0 mg/kg kroppsvikt varannan vecka fram till sjukdomsprogression eller oacceptabel toxicitet. Under induktionsfasen administreras cisplatin dag 1 i varje 2‑veckorscykel i upp till 8 cykler och 5‑fluorouracil dag 1 i varje 2-veckorscykel i upp till 12 cykler.
För kombinerad användning, se produktresumén för de samtidiga behandlingarna.
Senareläggning av dos eller utsättning av behandling (se även avsnitt Varningar och försiktighet)
Ökning eller minskning av HETRONIFLY-dosen rekommenderas inte. Dosuppehåll eller utsättning kan krävas baserat på individuell säkerhet och tolerabilitet. Dosuppehåll i upp till 12 veckor för tolerabilitet är acceptabelt (se avsnitt Varningar och försiktighet).
Rekommenderad hantering av immunmedierade biverkningar beskrivs i Tabell 1.
Tabell 1. Rekommenderade behandlingsändringar
Biverkningar | Allvarlighetsgrad | Behandlingsändring# |
Immunmedierad lungsjukdom | Grad 2 | Uppehåll tills biverkningarna upphör eller förbättras till grad 1 |
Grad 3 eller 4 eller återkommande grad 2 | Sätt ut permanent | |
Immunmedierad kolit | Grad 2 eller 3 | Uppehåll tills biverkningarna upphör eller förbättras till grad 1 |
Grad 4 eller återkommande grad 3 | Sätt ut permanent | |
Immunmedierad hepatit | Grad 2 med ASAT eller ALAT > 3 till 5 gånger ULN, eller totalt bilirubin > 1,5 till 3 gånger ULN | Uppehåll tills biverkningarna upphör eller förbättras till grad 1 |
Grad 3 eller 4 med ASAT eller ALAT > 5 gånger ULN, eller totalt bilirubin > 3 gånger ULN† | Sätt ut permanent | |
Immunmedierad njurinflammation och njursvikt | Grad 2 förhöjning av serumkreatinin | Uppehåll tills biverkningarna upphör eller förbättras till grad 1 |
Grad 3 eller 4 förhöjning av serumkreatinin | Sätt ut permanent | |
Immunmedierade endokrinopatier | Symptomatisk | Uppehåll tills symtomen försvinner och behandlingen med kortikosteroider är klar. Behandlingen ska fortsätta i närvaro av hormonersättningsterapi så länge som inga symtom finns |
Grad 4 hypotyreos | Sätt ut permanent | |
Immunmedierade hudbiverkningar | Grad 3 | Uppehåll tills biverkningarna upphör eller förbättras till grad 1 |
Grad 4 Stevens Johnsons syndrom (SJS) eller toxisk epidermal nekrolys (TEN) | Sätt ut permanent | |
Andra immunmedierade biverkningar | Grad 2 myasthenia gravis/myastent syndrom* | Uppehåll tills biverkningarna upphör eller förbättras till grad 1 |
Grad 3 eller 4 myasthenia gravis/myastent syndrom | Sätt ut permanent | |
Infusionsrelaterade reaktioner | Grad 2 | Minska infusionshastigheten till halv hastighet eller avbryt. Behandlingen kan återupptas när problemet är löst |
Grad 3 eller 4 | Sätt ut permanent |
Obs: Allvarlighetsgrad i enlighet med National Cancer Institute Common Terminology Criteria for Adverse Events Version 5.0 (NCI‑CTCAE v5.0).
#: Serplulimab måste sättas ut permanent för alla immunmedierade biverkningar av grad 3 som återkommer och för alla immunmedierade biverkningar av grad 4, förutom endokrinopatier som kontrolleras med ersättningshormoner (se avsnitt Varningar och försiktighet och Biverkningar).
†: ALAT: alaninaminotransferas; ASAT: aspartataminotransferas; ULN: övre normalgränsen (Upper Limit of Normal).
*: Säkerheten vid återbehandling med serplulimab hos patienter som upplevt immunmedierad myasthenia gravis/myastent syndrom eller myokardit är inte klarlagd.
Särskilda grupper
Äldre
Ingen dosjustering behövs för äldre patienter (≥ 65 år) (se avsnitt Farmakodynamiska egenskaper och avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Ingen dosjustering behövs för patienter med lätt nedsatt (CRCL=60‑89 ml/min) eller måttligt (CRCL=30‑59 ml/min) nedsatt njurfunktion. Det finns otillräckliga data och ingen dosrekommendation kan göras för patienter med svårt (CRCL=15‑29 ml/min) nedsatt njurfunktion (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Ingen dosjustering behövs för patienter med lindrig (totalt bilirubin ≤ ULN och ASAT > ULN eller totalt bilirubin > 1 till 1,5 × ULN och eventuell ASAT-stegring) leverfunktionsnedsättning. Det finns otillräckliga data för patienter med måttlig (totalt bilirubin > 1,5 till 3 × ULN och eventuell ASAT-stegring) leverfunktionsnedsättning och inga data finns tillgängliga för svår (totalt bilirubin > 3 × ULN och eventuell ASAT-stegring) leverfunktionsnedsättning. Ingen dosrekommendation kan göras för patienter med måttlig eller svår leverfunktionsnedsättning (se avsnitt Farmakokinetiska egenskaper).
Pediatrisk population
Det finns ingen relevant användning av serplulimab i den pediatriska populationen.
Administreringssätt
HETRONIFLY är avsett för intravenös användning.
Den initiala infusionshastigheten bör ställas in på 100 ml per timme. Om den första infusionen tolereras, kan alla efterföljande infusioner förkortas till 30 minuter (± 10 minuter).
Vid administrering i kombination med kemoterapi ska HETRONIFLY ges först följt av kemoterapi samma dag. Använd separata infusionspåsar för varje infusion.
HETRONIFLY får inte administreras som intravenös push- eller bolusinjektion.
Den totala dosen av HETRONIFLY som krävs ska spädas med natriumklorid 9 mg/ml (0,9 %) injektionsvätska, lösning (se avsnitt Särskilda anvisningar för destruktion och övrig hantering).
För instruktioner om spädning och hantering av läkemedlet före administrering, se avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarheten av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Bedömning av PD‑L1-status
Vid bedömning av tumörens PD‑L1-status är det viktigt att välja en väl validerad metod för att minska falska negativa eller falska positiva resultat.
Immunmedierade biverkningar
Immunmedierade biverkningar, inklusive allvarliga och dödliga fall, har inträffat hos patienter som fått serplulimab (se avsnitt Biverkningar). De flesta immunmedierade biverkningar som inträffade under behandlingen var reversibla och hanterades genom att göra uppehåll i behandlingen, administrera kortikosteroider och/eller stödjande behandling (se avsnitt Dosering och administreringssätt). Immunmedierade biverkningar har också inträffat upp till 3,6 månader efter den sista dosen. Immunmedieradee biverkningar som påverkar mer än ett kroppssystem kan inträffa samtidigt.
Vid misstänkta immunmedierade biverkningar, bör adekvat utvärdering säkerställas för att bekräfta etiologin eller utesluta andra orsaker. Beroende på biverkningens allvarlighetsgrad, bör uppehåll göras i behandlingen och kortikosteroid administreras. För de flesta immunmedierade biverkningar av grad 2 och vissa specifika immunmedierade biverkningar av grad 3 eller 4 ska administreringen avbrytas tills biverkningarna har upphört eller förbättrats till grad 1. Serplulimab måste sättas ut permanent för alla grad 4 och vissa specifika immunmedierade biverkningar av grad 3. För immunmedierade biverkningar av grad 3 och 4 och vissa specifika immunmedierade biverkningar av grad 2 (t.ex. immunmedierad pneumonit, immunmedierad myokardit), bör kortikosteroid (1‑2 mg/kg/dag prednison eller motsvarande) och andra symtomatiska behandlingar ges i enlighet med de kliniska symtomen tills de upphört eller förbättrats till grad 1. Vid förbättring till grad ≤ 1 bör nedtrappning av kortikosteroider påbörjas och fortsätta under minst 1 månad. Snabb nedtrappning kan leda till att biverkningen förvärras eller återkommer. Icke‑kortikosteroid immunsuppressiv behandling (t.ex. infliximab) bör läggas till vid försämring eller utebliven förbättring trots kortikosteroidbehandling.
Immunmedierad lungsjukdom
Immunmedierad pneumonit, inklusive dödliga fall, har rapporterats hos patienter som får HETRONIFLY (se avsnitt Biverkningar). Patienter bör övervakas med avseende på tecken och symtom på immunmedierad pneumonit såsom röntgenförändringar (t.ex. fokala GGO ”ground glass opacities”, fläckiga infiltrat), dyspné och hypoxi. Misstänkt immunmedierad pneumonit bör bekräftas med röntgenundersökning och andra orsaker ska uteslutas. För modifiering av behandlingen, se avsnitt Dosering och administreringssätt.
Immunmedierad kolit
Immunmedierad kolit, inklusive dödliga fall, har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Patienter bör övervakas avseende tecken och symtom på immunmedierad kolit, såsom buksmärtor, diarré, slem eller blod i avföring. Infektion och andra sjukdomsmedierade etiologier bör uteslutas. För modifiering av behandlingen, se avsnitt Dosering och administreringssätt. Den potentiella risken för gastrointestinal perforation bör beaktas och bekräftas med röntgenundersökning och/eller endoskopi vid behov.
Immunmedierad hepatit
Immunmedierad hepatit, inklusive dödliga fall, har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Patienter bör övervakas med jämna mellanrum (varje månad) med avseende på förändringar i leverfunktionen och kliniska tecken och symtom på immunmedierad hepatit såsom förhöjda nivåer av transaminas och totalt bilirubin. Infektion och sjukdomsrelaterade etiologier bör uteslutas. Frekvensen av leverfunktionstest bör ökas, om immunmedierad hepatit uppkommer. För modifiering av behandlingen, se avsnitt Dosering och administreringssätt.
Immunmedierad njurinflammation och njursvikt
Immunmedierad njurinflammation och njursvikt har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Patienter bör övervakas med jämna mellanrum (varje månad) med avseende på förändringar i njurfunktion och kliniska tecken och symtom på immunmedierad njurinflammation och njursvikt. Frekvensen av njurfunktionstest bör ökas om immunmedierad njurinflammation uppstår. De flesta patienter uppvisar asymtomatisk ökning av serumkreatinin. Sjukdomsrelaterade etiologier bör uteslutas. För modifiering av behandlingen, se avsnitt Dosering och administreringssätt.
Immunmedierade endokrinopatier
Sköldkörtelsjukdomar
Sköldkörtelrubbningar, inklusive hypertyreos, hypotyreos och tyreoidit har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Patienter bör övervakas med avseende på förändringar i sköldkörtelfunktionen och kliniska tecken och symtom på sköldkörtelrubbningar. För symtomatisk hypotyreos av grad 2 eller 3 ska uppehåll i behandlingen med serplulimab göras och sköldkörtelhormonersättning ska påbörjas vid behov. För symtomatisk hypertyreos av grad 2 eller 3 ska uppehåll i behandlingen med serplulimab göras och behandling med antityreoida läkemedel sättas in vid behov. Vid misstanke om akut inflammation i sköldkörteln ska uppehåll i behandlingen med serplulimab göras och hormonbehandling påbörjas. Behandlingen kan återupptas när symtom på hypotyreos eller hypertyreos är kontrollerade och sköldkörtelfunktionen förbättrats. Vid livshotande hypertyreos eller hypotyreos måste serplulimab sättas ut permanent. Sköldkörtelfunktionen bör övervakas kontinuerligt för att säkerställa lämplig hormonersättning (se avsnitt Dosering och administreringssätt).
Hypofyssjukdomar
Hypofysit har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Patienter bör övervakas med avseende på tecken och symtom på hypofysit, och andra orsaker bör uteslutas. För symtomatisk hypofysit av grad 2 eller 3 ska uppehåll i behandlingen med serplulimab göras och hormonersättning påbörjas vid behov. Vid misstanke om akut hypofysit ska kortikosteroider sättas in. Vid livshotande hypofysit av grad 4 måste serplulimab sättas ut permanent (se avsnitt Dosering och administreringssätt).
Binjureinsufficiens
Binjureinsufficiens har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Patienter bör övervakas med avseende på tecken och symtom, och andra orsaker bör uteslutas. Vid binjureinsufficiens av grad 2 ska uppehåll i behandlingen med serplulimab göras och hormonersättning ska påbörjas vid behov. Vid livshotande binjureinsufficiens av grad 3 eller 4 måste serplulimab sättas ut permanent. Binjurefunktion och hormonnivåer bör övervakas kontinuerligt för att säkerställa lämplig hormonersättning (se avsnitt Dosering och administreringssätt).
Hyperglykemi
Hyperglykemi eller typ 1-diabetes mellitus har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Patienter bör övervakas med avseende på blodsockernivåer och relaterade kliniska tecken och symtom. Insulinersättningsterapi bör initieras vid behov. För typ 1-diabetes mellitus med dålig blodsockerkontroll ska uppehåll i behandling med serplulimab göras och insulinersättningsbehandling påbörjas tills symtomen förbättrats. Vid livshotande typ 1-diabetes mellitus av grad 4 måste serplulimab sättas ut permanent. Blodsockernivåerna bör övervakas kontinuerligt för att säkerställa lämplig insulinersättning (se avsnitt Dosering och administreringssätt).
Immunmedierade hudbiverkningar
Immunmedierade hudreaktioner har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Vid utslag av grad 1 eller 2 kan behandling med serplulimab fortsätta och symtomatisk behandling eller behandling med lokala kortikosteroider kan ges. Vid utslag av grad 3 ska uppehåll i behandlingen med serplulimab göras och symtomatisk behandling eller lokal kortikosteroidbehandling bör ges. Vid utslag av grad 4, Stevens‑Johnsons syndrom (SJS), eller toxisk epidermal nekrolys (TEN) bör serplulimab sättas ut permanent (se avsnitt Dosering och administreringssätt).
Immunmedierad pankreatit
Immunmedierad pankreatit, inklusive ökning av serumamylas- och lipasnivåer samt dödliga fall, har rapporterats hos patienter som får serplulimab, (se avsnitt Biverkningar). Patienter bör övervakas med avseende på förändringar i serumlipas och amylas (i början av behandlingen, periodiskt under behandlingen och enligt indikation baserat på klinisk utvärdering), samt kliniska tecken och symtom på pankreatit. Uppehåll i behandlingen med serplulimab ska göras för ökning av serumamylas- eller lipasnivåer av grad 3 eller 4 och pankreatit av grad 2 eller 3. Vid pankreatit av grad 4 eller återkommande pankreatit av vilken grad som helst, bör serplulimab sättas ut permanent (se avsnitt Dosering och administreringssätt).
Immunmedierad myokardit
Immunmedierad myokardit, inklusive dödsfall, har rapporterats hos patienter som får serplulimab (se avsnitt Biverkningar). Patienter bör övervakas med avseende på kliniska tecken och symtom på myokardit. Misstänkt immunmedierad myokardit ska bekräftas med myokardenzymundersökningar och andra orsaker bör uteslutas. Vid myokardit av grad 2 ska uppehåll i behandlingen med serplulimab göras och kortikosteroidbehandling ska ges. Säkerheten att återuppta serplulimabbehandling hos patienter som tidigare fått immunmedierad myokardit är inte klarlagd. En multidisciplinär diskussion rekommenderas innan serplulimab återupptas hos patienter med tidigare myokardit av grad 2, och beslutet bör baseras på olika kliniska faktorer, inklusive graden av hjärtåterhämtning, onkologisk respons på behandlingen samt tillgång till alternativa onkologiska behandlingar och prognos. Vid myokardit av grad 3 eller 4 måste serplulimab sättas ut permanent och kortikosteroidbehandling bör inledas. När en diagnos av myokardit har fastställts bör uppehåll i behandlingen med serplulimab göras eller sättas ut permanent. Myokardenzymer och hjärtfunktion bör övervakas noggrant med avseende på myokardit av någon grad (se avsnitt Dosering och administreringssätt).
Immunmedierad uveit
Om uveit och andra immunmedierade biverkningar inträffar samtidigt, såsom Vogt‑Koyanagi‑Haradas syndrom bör systemiska kortikosteroider ges för att förhindra permanent blindhet.
Andra immunmedierade biverkningar
Med tanke på verkningsmekanismen för serplulimab kan andra potentiella immunmedierade biverkningar förekomma. Andra dödliga och livshotande immunmedierade biverkningar har observerats hos patienter som behandlats med serplulimab i kliniska prövningar för olika doser och tumörtyper: trombocytopeni, akut koronarsyndrom, hjärtinfarkt, immunmedierad encefalit, myasthenia gravis och myastent syndrom (se avsnitt Biverkningar). Patienter ska vara medvetna om symtomen på myasthenia gravis och myastent syndrom (t.ex. muskelsvaghet och snabb uttröttbarhet) och rådas att omedelbart söka läkare om symtom uppkommer.
Vid misstänkta immunmedierade biverkningar bör adekvat utvärdering göras för att bekräfta etiologin och utesluta andra orsaker. Baserat på svårighetsgraden av biverkningarna ska uppehåll i behandlingen med serplulimab göras vid immunmedierade biverkningar av grad 2 eller 3 som inträffar för första gången. Vid återkommande immunmedierade biverkningar av grad 3 (förutom endokrinopatier) och immunmedierade biverkningar av grad 4 måste behandlingen med serplulimab sättas ut permanent. Kortikosteroider kan initieras efter vad som är klinisk indicerat (se avsnitt Dosering och administreringssätt).
Infusionsrelaterade reaktioner
Infusionsrelaterade reaktioner har rapporterats hos patienter som får serplulimab. Patienter bör övervakas med avseende på kliniska tecken och symtom på infusionsrelaterade reaktioner. Patienter med infusionsrelaterade reaktioner av grad 1 kan fortsätta administreringen under noggrann övervakning. Hos patienter med infusionsrelaterade reaktioner av grad 2 bör infusionshastigheten minskas eller behandlingen avbrytas. Antipyretika och antihistaminer kan övervägas. Behandling med serplulimab kan återupptas under noggrann övervakning när infusionsrelaterade reaktioner av grad 2 är under kontroll. Vid infusionsrelaterade reaktioner av grad ≥ 3 ska infusionen avbrytas omedelbart, behandlingen sättas ut permanent och lämplig behandling ges (se avsnitt Dosering och administreringssätt).
Patienter som uteslutits från kliniska prövningar
Patienter med följande tillstånd exkluderades från kliniska prövningar: aktiv eller tidigare dokumenterad autoimmun sjukdom, patienter med aktiv tuberkulos eller hepatit B eller C eller HIV-infektion eller patienter som fått levande försvagat vaccin inom 28 dagar före administrering av serplulimab, patienter med en aktiv infektion som kräver systemisk infektionsbehandling inom 14 dagar före den första dosen, pneumonit eller interstitiell lungsjukdom i anamnesen, patienter med aktiva hjärnmetastaser, betydande kardiovaskulär sjukdom (t.ex. hjärtinfarkt inom det senaste halvåret) i anamnesen, överkänslighet mot en annan monoklonal antikropp i anamnesen och systemiska immunsuppressiva läkemedel inom 2 veckor före behandling med serplulimab.
Hjälpämnen med känd effekt
Detta läkemedel innehåller 0,98 mmol (eller 22,5 mg) natrium per 10 ml injektionsflaska, motsvarande 1,1 % av WHO:s högsta rekommenderat dagligt intag (2 g natrium för vuxna).
Detta läkemedel innehåller 2,0 mg polysorbat 80 (E 433) i varje 10 ml injektionsflaska. Polysorbater kan orsaka allergiska reaktioner.
För instruktioner om spädning och hantering av läkemedlet före administrering, se avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Patientkort
Förskrivaren måste diskutera riskerna med serplulimabbehandling med patienten. Patienten ska förses med ett patientkort vid varje ordination.
Interaktioner
Läkemedelsinteraktionsstudier har inte utförts. Eftersom monoklonala antikroppar inte metaboliseras av cytokrom P450 (CYP) enzymer eller andra läkemedelsmetaboliserande enzymer, förväntas inte hämning eller induktion av dessa enzymer orsakade av samtidigt administrerade läkemedel påverka farmakokinetiken för HETRONIFLY.
Användning av systemiska kortikosteroider eller immunsuppressiv behandling bör undvikas inför att behandling med serplulimab påbörjas på grund av deras potentiella påverkan på den farmakodynamiska aktiviteten och effekten. Systemiska kortikosteroider eller andra immunsuppressiva läkemedel kan dock användas för att behandla immunmedierade biverkningar efter att behandling med serplulimab påbörjats (se avsnitt Varningar och försiktighet).
Fertilitet, graviditet och amning
Fertila kvinnor/preventivmedel
Fertila kvinnor bör använda effektivt preventivmedel under behandlingen och i minst 6 månader efter den sista dosen av serplulimab.
Graviditet
Det finns inga data om användning av serplulimab hos gravida kvinnor. Djurstudier har visat att hämning av PD‑1-signalvägen orsakar embryofetal toxicitet (se Prekliniska säkerhetsuppgifter). Humant IgG är känt för att passera placentabarriären och serplulimab är en IgG4; därför har den potential att överföras från modern till fostret. Serplulimab rekommenderas inte under graviditet eller till fertila kvinnor som inte använder preventivmedel.
Amning
Det är okänt om serplulimab utsöndras i bröstmjölk. Det är känt att humant IgG utsöndras i bröstmjölk under de första dagarna efter födseln och minskar till låga koncentrationer kort därefter; Följaktligen kan en risk för spädbarn som ammas inte uteslutas under denna korta period. Efter denna period skulle serplulimab kunna användas under amning om det bedöms kliniskt nödvändigt.
Fertilitet
Studier för att utvärdera fertilitet har inte utförts. Effekten av serplulimab på manlig och kvinnlig fertilitet är därför okänd.
Effekter på förmågan att framföra fordon och använda maskiner
Serplulimab har mindre effekt på förmågan att framföra fordon och använda maskiner. På grund av potentiella biverkningar såsom trötthet (se avsnitt Biverkningar), bör patienter rådas att vara försiktiga när de kör bil eller använder maskiner tills de är säkra på att serplulimab inte påverkar dem negativt.
Biverkningar
Sammanfattning av säkerhetsprofilen
Säkerheten hos serplulimab i kombination med kemoterapi baseras på poolade data från 1 343 patienter. De vanligaste biverkningarna var anemi (80,2 %), neutropeni (74,5 %), leukopeni (71,6 %), trombocytopeni (53,8 %), illamående (44,4 %), minskad aptit (36,0 %), alopeci (33,6 %), hypoproteinemi, (31,3 %) asteni (29,9 %) och kräkningar (28,2 %).
De vanligaste biverkningarna av grad ≥ 3 var neutropeni (46,2 %, leukopeni (27,3 %), anemi (25,8 %), trombocytopeni (15,3 %), hyponatremi (6,9 %) och pneumoni (5,6 %).
De vanligaste allvarliga biverkningarna var trombocytopeni (8,4 %), pneumoni (6,6 %), neutropeni (6,0 %), leukopeni (5,9 %), anemi (4,5 %) och pneumonit (3,3 %).
De vanligaste immunmedierade biverkningarna var hypotyreos (11,5 %), immunmedierade reaktioner (7,6 %), hypertyreos (7,1 %), immunmedierad lungsjukdom (5,5 %), onormal leverfunktion (3,2 %), immunmedierad nefrit och njursvikt (3,1 %) och immunmedierad kolit (1,6 %).
Serplulimab avbröts på grund av biverkningar hos 8,1 % av patienterna. De vanligaste biverkningen som ledde till att behandlingen avbröts var pneumonit (1,6 %) och pneumoni (1,3 %).
Lista över biverkningar i tabellform
Biverkningar rapporterade i klinisk prövning och efter godkännandet för försäljning listas enligt organsystem och frekvens (se tabell 2). Om inget annat anges är frekvensen av biverkningar baserad på alla biverkningsfrekvenser identifierade i prövningar, där 1 343 patienter exponerades för serplulimab i kombination med kemoterapi. Se avsnitt Farmakodynamiska egenskaper för information om de viktigaste egenskaperna hos patienterna i de pivotala kliniska prövningarna.
Frekvenser definieras som: mycket vanliga (≥ 1/10); vanliga (≥ 1/100 till < 1/10); mindre vanliga (≥ 1/1 000 till < 1/100); sällsynta (≥ 1/10 000 till < 1/1 000); mycket sällsynta (< 1/10 000); ingen känd frekvens (kan inte beräknas utifrån tillgängliga data). Inom varje frekvensgrupp presenteras biverkningarna efter fallande allvarlighetsgrad.
Tabell 2. Biverkningar hos patienter som behandlats med serplulimab*
Serplulimab i kombination med kemoterapi | |
Infektioner och infestationer | |
Mycket vanliga | lunginflammationa |
Vanliga | urinvägsinfektionb, luftvägsinfektionc, hudinfektion |
Mindre vanliga | septisk chock |
Sällsynta | gastrointestinal infektion, herpetisk meningoencefalit |
Blodet och lymfsystemet | |
Mycket vanliga | neutropeni, leukopeni, anemi, trombocytopeni, lymfopeni |
Vanliga | onormalt koagulationsfunktionstestd, granulocytopeni, febril neutropeni |
Sällsynta | Lymfadenit |
Immunsystemet | |
Vanliga | infusionsrelaterad reaktione |
Mindre vanliga | anafylaktisk reaktion |
Endokrina systemet | |
Mycket vanliga | hypotyreosf, hypertyreosg, hyperglykemi eller typ 1-diabetes mellitush |
Vanliga | tyreoiditi |
Mindre vanliga | binjureinsufficiensj, annan sköldkörtelsjukdomk, hypofysit, onormalt tyreoideafunktionstestl, hypoparatyreoidism |
Sällsynta | hyperadrenokorticism |
Metabolism och nutrition | |
Mycket vanliga | hyperlipidemi, minskad aptit, hypoproteinemi, hyperurikemi, elektrolytobalansm, viktminskning |
Vanliga | hypoglykemi |
Mindre vanliga | onormalt lipoprotein |
Psykiatriska tillstånd | |
Mycket vanliga | Sömnlöshet |
Centrala och perifera nervsystemet | |
Vanliga | parestesi, huvudvärk, yrsel, perifer neuropatin |
Mindre vanliga | vertigo, immunmedierad encefalito, neurotoxicitet, cerebral infarkt, smakstörningar, nedsatt minnesfunktion |
Sällsynta | motorisk dysfunktion, myasthenia gravis, myastent syndrom |
Ögon | |
Mindre vanliga | suddig syn, keratit, konjunktivit |
Hjärtat | |
Mycket vanliga | arytmip |
Vanliga | sinustakykardi, ledningsdefekterq, sinusbradykardi, hjärtsviktr, förhöjt troponin, myokardskada |
Mindre vanliga | myokardischemi, perikardiell effusion, myokardit |
Sällsynta | kardiomyopati |
Blodkärl | |
Vanliga | hypertoni, vaskulit, hypotoni |
Mindre vanliga | ventrombos |
Andningsvägar, bröstkorg och mediastinum | |
Mycket vanliga | hosta, bröstsmärta |
Vanliga | pneumonits, dyspné, dysfoni, lungemboli |
Mindre vanliga | andningssvikt |
Magtarmkanalen | |
Mycket vanliga | illamående, förstoppning, diarré, kräkningar |
Vanliga | dysfagi, buksmärta, flatulens, gastrointestinala störningart, stomatit, dyspepsi, muntorrhet, gastrit |
Mindre vanliga | enteritu, immunmedierad pankreatit, gingival blödning, esofagit, magsår |
Lever och gallvägar | |
Mycket vanliga | förhöjt alaninaminotransferas, förhöjt aspartataminotransferas |
Vanliga | förhöjt gamma‑glutamyltransferas, hyperbilirubinemi, leverskadav |
Hud och subkutan vävnad | |
Mycket vanliga | utslagw, alopeci |
Vanliga | klåda, dermatitx |
Mindre vanliga | pigmentstörning, psoriasis, torr hud, hyperhidros |
Sällsynta | toxisk epidermal nekrolys |
Muskuloskeletala systemet och bindväv | |
Mycket vanliga | muskuloskeletal smärta |
Vanliga | artrit |
Sällsynta | myosity |
Njurar och urinvägar | |
Mycket vanliga | protein i urinen, förhöjt kreatinin i blodet |
Vanliga | förhöjt urea i blodet, hematuri, njurskadaz |
Mindre vanliga | dysuri, pollakiuri |
Allmänna symtom och/eller symtom vid administreringsstället | |
Mycket vanliga | pyrexi, asteni |
Vanliga | sjukdomskänsla, ödem, |
Mindre vanliga | frossa |
Undersökningar och provtagningar | |
Vanliga | förhöjt alkaliskt fosfatas i blodet, förhöjt myoglobin i blodet, förhöjt kreatinfosfokinas i blodet, förhöjt amylas |
Mindre vanliga | förhöjt lipas |
*Biverkningsfrekvenser som presenteras i Tabell 2 kanske inte enbart kan tillskrivas serplulimab utan kan innehålla bidrag från den underliggande sjukdomen eller från andra läkemedel som används i en kombination.
Följande termer representerar en grupp relaterade händelser som beskriver ett medicinskt tillstånd snarare än en enskild händelse:
a. Inkluderar pneumoni, lungabscess.
b. Inkluderar urinvägsinfektion, asymtomatisk bakteriuri, positiva vita blodkroppar i urin.
c. Inkluderar övre luftvägsinfektion, faryngotonsillit, tonsillit, influensaliknande sjukdom, nedre luftvägsinfektion.
d. Inkluderar förlängd aktiverad partiell tromboplastintid, aktiverad partiell tromboplastintid, förkortad aktiverad partiell tromboplastintid, minskat INR (international normalised ratio), förhöjd protrombinnivå, koagulopati, hyperkoagulering.
e. Inkluderar läkemedelsöverkänslighet, infusionsrelaterad reaktion.
f. Inkluderar hypotyreos, förhöjt sköldkörtelstimulerande hormon i blodet, minskat fritt tyroxin, minskat tyroxin, central hypotyreos, minskat trijodtyronin, minskat fritt trijodtyronin, immunmedierad hypotyreos.
g. Inkluderar hypertyreos, minskat sköldkörtelstimulerande hormon i blodet, förhöjt tyroxin, förhöjt trijodtyroxin, förhöjt fritt trijodtyroxin, förhöjt fritt tyroxin, immunmedierad hypertyreos.
h. Inkluderar hyperglykemi, typ 1-diabetes mellitus, ökad nivå av glukos i blodet, typ 2-diabetes mellitus, nedsatt fasteglukos, diabetisk ketoacidos, ökad nivå ketonkroppar i blodet, nedsatt glukostolerans, ketoacidos, glukosuri.
i. Inkluderar sköldkörtelsjukdom, tyreoidit.
j. Inkluderar binjureinsufficiens, minskad kortisolnivå.
k. Inkluderar euthyroid sick syndrome, onormalt ultraljud av tyreoidea.
l. Inkluderar positivt test för antikroppar riktade mot tyreoidea, ökat tyroglobulin.
m. Inkluderar hyponatremi, hypokalcemi, hypokalemi, hypomagnesemi, hypofosfatemi, hypokloremi, hyperfosfatemi, hyperkalemi, hypermagnesemi, hyperkalcemi.
n. Inkluderar perifer neuropati, perifer sensorimotorisk neuropati, immunmedierad neuropati.
o. Inkluderar immunmedierad encefalit, autoimmun encefalit.
p. Inkluderar supraventrikulära extrasystoler, supraventrikulär takykardi, arytmi, ventrikulära extrasystoler, supraventrikulär arytmi, förmaksflimmer, förmakstakykardi, bradyarytmi, tidigt repolarisationssyndrom, kammararytmi, palpitationer, onormalt elektrokardiogram.
q. Inkluderar atrioventrikulärt block av första graden, högersidigt grenblock, förlängd förmaksledningstid, vänstersidigt grenblock, defekt ledning intraventrikulärt.
r. Inkluderar hjärtsvikt, akut hjärtsvikt, vänsterkammarsvikt, hjärt-lungsvikt, N‑terminal prohormon B‑typ natriuretisk peptid.
s. Inkluderar immunmedierad lungsjukdom, pneumonit, interstitiell lungsjukdom.
t. Inkluderar förvärvad trakeoesofagal fistel, gastrointestinala blödningar, gastrointestinala störningar, intestinal obstruktion.
u. Inkluderar enterit, infektiös enterit, immunmedierad enterokolit, kolit, ulcerös kolit, enterokolit, duodenit.
v. Inkluderar onormal leverfunktion, läkemedelsinducerad leverskada, leverskada, immunmedierad hepatit, immunmedierad leverstörning**, leversvikt**.
w. Inkluderar utslag, makulopapulärt utslag, eksem, läkemedelsutslag, erytem, hudtoxicitet, palmar-plantar erytrodysestesisyndrom.
x. Inkluderar autoimmun dermatit, dermatit, allergisk dermatit, bullös dermatit, seborroisk dermatit.
y. Inkluderar myosit**, immunmedierad myosit.
z. Inkluderar akut njurskada, njursvikt, nedsatt njurfunktion, njurskada, kronisk njursjukdom, minskad njurclearance av kreatinin, immunmedierad nefrit.
**Händelse efter godkännande för försäljning.
Beskrivning av utvalda biverkningar
Serplulimab är associerat med immunmedierade biverkningar. Uppgifterna för följande immunmedierade biverkningar baseras på 2 086 patienter som fick serplulimab monoterapi (n=292) eller i kombination med andra läkemedel (n=1 794) över nio doser (0,3, 1, 3, 10 mg/kg varannan vecka, 4,5 mg/kg var tredje vecka, 200 mg varannan vecka, 300 mg var tredje vecka, 400 mg var fjärde vecka eller 600 mg var sjätte vecka) i tio kliniska prövningar. Riktlinjerna för hantering av dessa biverkningar beskrivs i avsnitt Dosering och administreringssätt och Varningar och försiktighet.
Immunmedierad lungsjukdom
Immunmedierad lungsjukdom förekom hos 4,9 % av patienterna, inklusive grad 3, 4 eller 5 hos 1,2 %, 0,2 % respektive 0,3 % av patienterna. Mediantiden till debut var 4,40 månader (intervall: 0,03‑34,53 månader). Medianvaraktigheten var 1,76 månader (intervall: 0,10‑13,34 månader). 2,5 % av patienterna fick högdosbehandling med kortikosteroider. Immunmedierad lungsjukdom ledde till att behandlingen avbröts hos 1,3 % av patienterna.
Immunmedierad kolit
Immunmedierad kolit förekom hos 2,0 % av patienterna, inklusive grad 3 hos 0,6 % av patienterna och grad 5 hos < 0,1 % av patienterna. Mediantiden till debut var 3,35 månader (intervall: 0,03‑30,55 månader). Medianvaraktigheten var 0,43 månader (intervall: 0,03‑8,94 månader). 0,7 % av patienterna fick högdosbehandling med kortikosteroider. Immunmedierad kolit ledde till att behandlingen avbröts hos 0,2 % av patienterna.
Immunmedierad hepatit
Hepatit förekom hos 0,8 % av patienterna, inklusive grad 3 hos 0,3 % av patienterna, grad 4 hos 0,1 % av patienterna och grad 5 hos 0,1 % av patienterna. Mediantiden till debut var 2,48 månader (intervall: 0,36‑26,78 månader). Medianvaraktigheten var 0,95 månader (intervall: 0,10‑8,48 månader). 0,4 % av patienterna fick högdosbehandling med kortikosteroider. Hepatit ledde till utsättning av behandlingen hos 0,3 % av patienterna. Onormal leverfunktion förekom hos 3,7 % av patienterna, inklusive grad 3 hos 0,8 % av patienterna och grad 4 hos 0,1 % av patienterna. Mediantiden till debut var 2,30 månader (intervall: 0,07‑45,31 månader). Medianvaraktigheten var 1,31 månader (intervall: 0,26‑17,54 månader). 0,5 % av patienterna fick högdosbehandling med kortikosteroider. Onormal leverfunktion ledde till att behandlingen avbröts hos 0,2 % av patienterna.
Immunmedierad njurinflammation och njursvikt
Immunmedierad njurinflammation och njursvikt förekom hos 3,0 % av patienterna, inklusive grad 3 hos 0,3 % av patienterna och grad 4 hos < 0,1% av patienterna. Mediantiden till debut var 2,83 månader (intervall: 0,23‑17,77 månader). Medianvaraktigheten var 1,48 månader (intervall: 0,13‑17,94 månader). 0,4 % av patienterna fick högdosbehandling med kortikosteroider. Immunmedierad njurinflammation och njursvikt ledde till att behandlingen avbröts hos 0,2% av patienterna.
Immunmedierade endokrinopatier
Hypotyreos
Hypotyreos förekom hos 11,7 % av patienterna, inklusive grad 3 hos 0,2 % av patienterna. Mediantiden till debut var 3,83 månader (intervall: 0,46‑34,10 månader). Medianvaraktigheten var 2,73 månader (intervall: 0,13‑29,08 månader). 6,7 % av patienterna fick sköldkörtelhormonersättningsterapi. < 0,1 % patienter avbröt behandlingen med serplulimab på grund av hypotyreos.
Hypertyreos
Hypertyreos förekom hos 6,7 % av patienterna och det fanns ingen grad ≥ 3 hypertyreos. Mediantiden till debut var 2,73 månader (intervall: 0,62‑31,18 månader). Medianvaraktigheten var 1,45 månader (intervall: 0,07‑17,77 månader). Inga patienter avbröt behandlingen med serplulimab på grund av hypertyreos.
Tyreoidit
Tyreoidit förekom hos 0,7 % av patienterna och det fanns ingen grad ≥ 3 tyreoidit. Mediantiden till debut var 6,64 månader (intervall: 0,99‑13,50 månader). Medianvaraktigheten var 1,30 månader (intervall: 0,56‑11,30 månader). 0,2 % av patienterna fick sköldkörtelhormonersättningsterapi. Inga patienter avbröt behandlingen med serplulimab på grund av tyreoidit.
Binjurebesvär
Binjurebesvär förekom hos 0,5 % av patienterna, inklusive grad 3 hos 0,1 % av patienterna. Mediantiden till debut var 6,24 månader (intervall: 3,55‑21,45 månader). Medianvaraktigheten var 4,60 månader. < 0,1 % av patienterna fick behandling med högdos kortikosteroid. Inga patienter avbröt behandlingen med serplulimab på grund av binjurebesvär.
Hypofyssjukdomar
Hypofyssjukdomar förekom hos 0,8 % av patienterna, inklusive grad 3 hos 0,1 % av patienterna. Mediantiden till debut var 6,72 månader (intervall: 1,41‑20,53 månader). Medianvaraktigheten var 3,25 månader. 0,2 % av patienterna fick högdosbehandling med kortikosteroider. Hypofyssjukdomar ledde till att behandlingen avbröts hos 0,1 % av patienterna.
Typ 1-diabetes mellitus/hyperglykemi
Typ 1-diabetes mellitus/hyperglykemi förekom hos 0,9 % av patienterna, inklusive grad 3 hos 0,4 % av patienterna och grad 4 hos 0,1% av patienterna. Mediantiden till debut var 4,34 månader (intervall: 0,69‑40,28 månader). Medianvaraktigheten var 3,48 månader (intervall: 0,53‑10,68). 0,5 % av patienterna fick insulinersättningsterapi. Typ 1-diabetes mellitus/hyperglykemi ledde till att behandlingen avbröts hos < 0,1% av patienterna.
Immunmedierade hudreaktioner
Immunmedierade hudreaktioner förekom hos 7,8 % av patienterna, inklusive grad 3 hos 0,8 % av patienterna, grad 4 hos < 0,1 % av patienterna och grad 5 hos < 0,1 % av patienterna. Mediantiden till debut var 2,96 månader (intervall: 0,03‑30,52 månader). Medianvaraktigheten var 1,56 månader (intervall: 0,07‑19,06 månader). 1,2 % av patienterna fick högdosbehandling med kortikosteroider. Immunmedierade hudbiverkningar ledde till att behandlingen avbröts hos 0,5 % av patienterna.
Immunmedierad pankreatit
Immunmedierad pankreatit förekom hos 1,0 % av patienterna, inklusive grad 3 hos 0,3 % av patienterna, grad 4 hos 0,1 % av patienterna och grad 5 hos < 0,1% av patienterna. Mediantiden till debut var 2,86 månader (intervall: 0,23‑13,67 månader). Medianvaraktigheten var 0,76 månader (intervall: 0,16‑10,12 månader). 0,1 % av patienterna fick högdosbehandling med kortikosteroider. Immunmedierad pankreatit ledde till att behandlingen avbröts hos 0,2 % av patienter.
Immunmedierad myokardit
Immunmedierad myokardit förekom hos 0,7 % av patienterna, inklusive grad 3 hos 0,1 % av patienterna, grad 4 hos < 0,1 % och grad 5 hos 0,2 % av patienterna. Mediantiden till debut var 1,71 månader (intervall: 0,26‑20,70 månader). Medianvaraktigheten var 0,79 månader (intervall: 0,30‑5,72 månader). 0,5 % av patienterna fick högdosbehandling med kortikosteroider. Immunmedierad myokardit ledde till att behandlingen avbröts hos 0,3 % av patienterna.
Immunmedierad uveit
Immunmedierad uveit inträffade hos < 0,1% av patienterna, vilken var av grad 1. Tiden till debut var 6,90 månader. Varaktigheten av immunmedierad uveit var 1,35 månader. Händelsen löste sig för patienten.
Andra immunmedierade biverkningar
Andra kliniskt signifikanta immunmedierade biverkningar rapporterade hos patienter som fick serplulimab var följande. Allvarliga eller dödliga fall har rapporterats för några av dessa biverkningar.
Blodet och lymfsystemet: anemi, leukopeni, trombocytopeni, neutropeni.
Centrala och perifera nervsystemet: immunmedierad encefalit, perifer neuropati, epilepsi, encefalopati, perifer sensomotorisk neuropati.
Ögon: suddig syn.
Hjärtat/blodkärl: akut kranskärlssyndrom, hjärtinfarkt, hjärtsvikt, kardiotoxicitet, förhöjt troponin onormalt test av hjärtfunktion.
Andningsvägar, bröstkorg och mediastinum: dyspné, kronisk obstruktiv lungsjukdom, andningssvikt.
Magtarmkanalen: munsår, kräkningar, proktit, övre gastrointestinal blödning.
Allmänna symtom och/eller symtom vid administreringsstället: asteni, trötthet, pyrexi.
Övrigt: panikångest, onormalt beteende, akut kolangit, sepsis, peritonit, förhöjt alkaliskt fosfatas i blodet, förhöjt kreatininfosfokinas i blodet, förhöjt laktatdehydrogenas i blodet, N‑terminal prohormon B-typ natriuretisk peptid, förhöjt kolesterol i blodet, obalans i elektrolyterna, kronisk njursjukdom, urinvägsinflammation.
Infusionsrelaterade reaktioner
Infusionrelaterade reaktioner inträffade hos 1,7 % av patienterna, inklusive grad 3 hos 0,1 % av patienterna och grad 4 hos 0,1 % av patienterna. Mediantiden till debut var 1,74 månader (intervall: 0,03‑34,04 månader). Medianvaraktigheten var 0,07 månader (intervall: 0,03‑6,70 månader). Inga patienter avbröt behandlingen med serplulimab på grund av infusionsrelaterade reaktioner.
Laboratorieavvikelser
Andelen patienter som upplevde en förändring från baslinjen till en laboratorieavvikelse av grad ≥ 3 var följande: 0,5 % för minskat trombocytantal, 0,3 % för minskat antal neutrofiler, 0,2 % för förhöjt kreatinfosfokinas i blodet, 0,1 % för minskat antal vita blodkroppar, 0,1 % för förhöjt troponin I.
Äldre
Inga övergripande skillnader i säkerhet rapporterades mellan äldre (≥ 65 år) och yngre patienter. Data för patienter ≥ 75 år är för begränsade för att dra slutsatser om denna population.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det möjliggör fortsatt övervakning av läkemedlets nytta/risk-förhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera alla misstänkta biverkningar
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Vid överdosering måste patienterna övervakas noga med avseende på tecken eller symtom på biverkningar och lämplig symtomatisk behandling inledas omedelbart.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Antineoplastiska medel, monoklonala antikroppar och antikroppsläkemedelskonjugat, PD-1/PD-L1 (Programmerad celldöd-1/dödsligand 1) hämmare
ATC-kod: L01FF12.
Verkningsmekanism
Serplulimab (HLX10) är en humaniserad monoklonal IgG4-antikropp, som binder till programmerad celldöd‑1 (PD‑1) receptorn och blockerar dess interaktion med ligand PD‑L1 och PD‑ L2. PD‑1-receptorn är en negativ regulator av T‑cellsaktivitet som har visat sig vara involverad i kontrollen av T‑cellsimmunsvar. Engagemang av PD‑1 med liganderna PD‑L1 och PD‑L2, som uttrycks i antigenpresenterande celler och kan uttryckas av tumörer eller andra celler i tumörmikromiljön, resulterar i hämning av T‑cellsproliferation och cytokinutsöndring. Serplulimab potentierar T‑cellssvar, inklusive antitumörsvar, genom blockad av PD‑1 bindning till PD‑L1 och PD‑L2-ligander.
PD‑1 receptorockupation av perifera T-celler och interleukin‑2 (IL‑2) frisättningsförmåga in vitro studerades i en fas 1-studie med 29 kinesiska patienter med avancerade solida tumörer som injicerades med enstaka och multipla doser (0,3 mg/kg, 1 mg/kg, 3 mg/kg, 10 mg/kg) av serplulimab. Resultatet visade att serplulimab stabilt kunde upprätthålla mättnadstillståndet av receptorockupation och bibehållen funktionell blockering vid dosering från 0,3 mg/kg till 10 mg/kg varannan vecka.
Klinisk effekt och säkerhet
Småcellig lungcancer (SCLC)
ASTRUM‑005: Randomiserad fas III‑prövning hos patienter med kemoterapinaiv utbredd SCLC, i kombination med karboplatin och etoposid
Effekten av serplulimab i kombination med kemoterapi (karboplatin plus etoposid) för första linjens behandling av ES‑SCLC utvärderades i ASTRUM‑005-studien (NCT04063163), en fas 3, randomiserad, dubbelblind, multiregional klinisk prövning. Det primära effektmåttet var total överlevnad (OS). Sekundära effektmått var progressionsfri överlevnad (PFS), objektiv svarsfrekvens (ORR) och svarstiden (DOR) och utvärderades av en oberoende radiologigranskningskommitté (IRRC) och utredare baserat på RECIST 1.1. Analys för det primära effektmåttet utfördes 25 och 33 månader efter starten av den kliniska prövningen. Studiens behandlingsregimer avblindades efter den primära analysen.
Studien inkluderade vuxna patienter (18 år eller äldre) med ES‑SCLC (enligt Veterans Administration Lung Study Group [VALG] utvärderingssystem) som inte hade behandlats med systemisk terapi och med ett ECOG-funktionsstatus på 0 eller 1. Patienter exkluderades om de hade aktiva eller obehandlade metastaser i det centrala nervsystemet; aktiv autoimmun sjukdom; eller om administrering av systemiska immunsuppressiva läkemedel skett inom 14 dagar före den första dosen.
Totalt 585 patienter inkluderades och randomiserades (2:1) för att få en av behandlingsregimerna som beskrivs i Tabell 3. Randomisering stratifierades av PD‑L1-uttrycksnivå (negativ: tumörandelspoäng [TPS] < 1%, positiv: TPS ≥ 1%, eller ej utvärderbar/ej tillgänglig, mätt med PD‑L1 IHC 22C3 pharmDxkit), hjärnmetastaser (ja mot nej) och ålder (≥ 65 år mot < 65 år).
Tabell 3. Intravenösa behandlingsregimer
Behandling | Induktion | Underhåll |
A | Serplulimab (4,5 mg/kg)a + karboplatin (AUC=5, upp till 750 mg)b + etoposid (100 mg/m2)b,c | Serplulimab (4,5 mg/kg)a |
B | Placebo + karboplatin (AUC=5, upp till 750 mg)b + etoposid (100 mg/m2)b,c | Placebo |
a. Serplulimab administrerades tills sjukdomsprogression eller oacceptabel toxicitet.
b. Karboplatin och etoposid administrerades tills fullbordandet av 4 cykler, eller progressiv sjukdom eller oacceptabel toxicitet, beroende på vilket som inträffade först.
c. Etoposid administrerades dag 1, 2 och 3 i varje cykel.
Basegenskaperna balanserades mellan behandlingsarmarna. Bland de inskrivna patienterna var 68,5% asiater (401 patienter) och 31,5% icke‑asiater (184 patienter), som alla var vita. Medianåldern var 62 år (intervall: 28‑83) med 39,3% av patienterna ≥ 65 år och 1,9% av patienterna ≥ 75 år. 82,2% av patienterna var män. Baslinje ECOG-funktionsstatus var 0 (17,6%) eller 1 (82,4%). 16,9% av patienterna var PD‑L1 positiv (TPS ≥ 1%). 13,3% av patienterna hade en historia av hjärnmetastaser.
Vid tidpunkten för den interimanalysens avslutande den 22 oktober 2021 när 66% av fördefinierade OS-händelser observerades (definierade cirka 226, faktiska 246 OS-händelser), hade patienterna en medianöverlevnadsuppföljningstid på 12,3 månader. OS-, PFS- och ORR-resultaten från interimsanalysen sammanfattas i tabell 4.
Tabell 4. Effektdata för den primära analysen (Sista datum för datainsamling: den 22 oktober 2021)
| Arm A | Arm B | |
Antal patienter | 389 | 196 | |
Primär slutpunkt | |||
OS | Antal patienter med händelser, n (%) | 146 (37,5%) | 100 (51,0%) |
Median OS (månader) | 15,4 | 10,9 | |
Riskförhållande (95% KI) | 0,63 (0,49‑0,82) | ||
p‑värde | < 0,001 | ||
Sekundära slutpunkter | |||
PFS | Median PFS (månader) | 5,7 | 4,3 |
Riskförhållande (95% KI) | 0,48 (0,38‑0,59) | ||
Bekräftad ORR | (%) | 67,4% | 58,7% |
Median DOR | Månader (95% CI) | 5,8 (5,2-7,5) | 4,1 (3,0-4,2) |
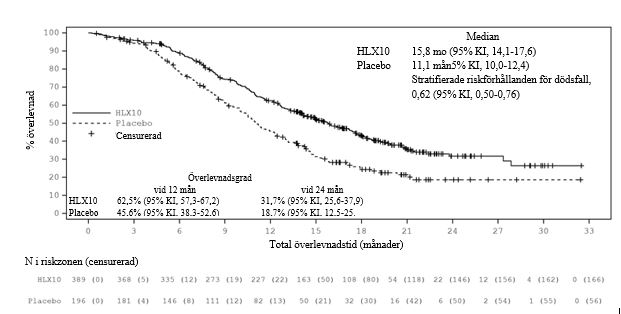
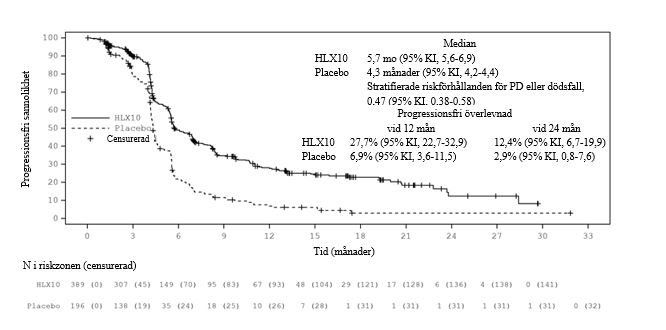
Uppdaterad analys efter avblindning med längre uppföljningsvaraktighet (median: 19,7 månader) genomfördes innan slutdatumet den 13 juni 2022 då 100% av fördefinierade OS-händelser observerades (definierade cirka 342, faktiska 363 OS-händelser). Median OS var 15,8 månader i serplulimabgruppen och 11,1 månader i placebogruppen. Den stratifierade HR (95% KI) var 0,62 (0,50, 0,76). Medianvärdet för PFS genom IRRC-bedömning per RECIST 1,1 var 5,7 månader respektive 4,3 månader med en stratifierad HR (95% KI) på 0,47 (0,38, 0,58). Effektresultaten av den slutliga analysen överensstämde med den primära analysen. Kaplan‑Meierkurvor för OS och PFS för slutlig analys presenteras i figurerna 1 och 2.
Figur 1. Kaplan‑Meier-kurva för OS i total population i den uppdaterade analysen (ITT) (Sista datum för datainsamling: den 13 juni 2022)
Figur 2. Kaplan‑Meier-kurva för PFS (RECIST 1.1) av IRRC i total population i den uppdaterade analysen (ITT) (sista datum för datainsamling: den 13 juni 2022)
Icke-småcellig lungcancer (NSCLC)
ASTRUM‑002: Randomiserad fas III-prövning hos kemoterapinaiva patienter med lokalt avancerad eller metastaserad NSCLCav icke‑skivepiteltyp i kombination med karboplatin och pemetrexed.
ASTRUM‑002 bestod av två stadier.Stadium I var en säkerhets-run-in med en arm utformad för att utvärdera säkerhet, tolerans och preliminär effekt av serplulimab i kombination med bevacizumab och kemoterapi (karboplatin + pemetrexed) som första linjens behandling för avancerad, NSCLC av icke‑skivepiteltyp. Stadium II var en randomiserad, dubbelblind, klinisk multicenterprövning i fas III med tre armar. Det primära effektmåttet var progressionsfri överlevnad (PFS) som utvärderades av en oberoende radiologigranskningskommitté (IRRC), det viktiga sekundära effektmåttet var total överlevnad (OS). Andra sekundära effektmått var PFS enligt utvärdering av prövare, objektiv svarsfrekvens (ORR) och svarstiden (DOR) enligt utvärdering av IRRC och prövare baserat på RECIST 1.1.
Prövningen inkluderade vuxna (≥ 18 år och ≤ 75 år) patienter med histologiskt eller cytologiskt bekräftad, icke-resektabel eller radiografiskt olämplig, NSCLC av icke‑skivepiteltyp i stadium IIIB, IIIC eller IV, utan EGFR-sensibiliserande mutationer eller ALK/ROS1-rearrangemang, och utan tidigare systemisk behandling för avancerad sjukdom. Patienterna måste ha ≥ 1 mätbar lesion enligt RECIST 1.1 utvärderad av IRRC och ECOG PS 0‑1.
Patienter med aktiva/misstänkta autoimmuna sjukdomar, aktiva CNS-metastaser och/eller malign meningit, eller tidigare behandling med immuncheckpointhämmare (t.ex. PD‑1-, PD‑L1-, CTLA‑4-antikroppar) exkluderades.
Sex patienter var inskrivna i stadium I av studien. Totalt 636 patienter var inskrivna i stadium II och randomiserades (1:1:1). I arm A fick patienterna serplulimab 4,5 mg/kg, bevacizumab 15 mg/kg, karboplatin (AUC = 5, upp till 800 mg, i upp till fyra cykler) och pemetrexed (500 mg/m²) var tredje vecka fram till sjukdomsprogression eller oacceptabel toxicitet. Patienter randomiserade till arm B eller arm C fick behandlingar enligt tabell 5. Randomiseringen stratifierades av PD‑L1-uttryck mätt med PD‑L1 IHC 22C3 pharmDx kit (negativ [CPS < 1] mot positiv [CPS ≥ 1] mot obestämbar), rökanamnes (ja mot nej) och hjärnmetastas (ja mot nej).
Tabell 5. Intravenösa behandlingsregimer
Behandlingsregim | Induktion | Underhåll |
B | Serplulimab (4,5 mg/kg)a + placebo (15 mg/kg)a + karboplatin (AUC = 5, upp till 800 mg)b + pemetrexed (500 mg/m2)a | Serplulimab (4,5 mg/kg)a + placebo (15 mg/kg)a + pemetrexed (500 mg/m2)a |
C | Placebo (4,5 mg/kg)a + placebo (15 mg/kg)a + karboplatin (AUC = 5, upp till 800 mg)b + pemetrexed (500 mg/m2)a | Placebo (4,5 mg/kg)a + placebo (15 mg/kg)a + pemetrexed (500 mg/m2)a |
a. Serplulimab och pemetrexed administrerades fram till sjukdomsprogression eller oacceptabel toxicitet.
b. Karboplatin administrerades tills fullbordandet av 4 cykler eller progressiv sjukdom eller oacceptabel toxicitet, beroende på vilket som inträffade först.
c. Byte var tillåtet från arm C för att få serplulimab 4,5 mg/kg var tredje vecka och behandling med bevacizumab 15 mg/kg var tredje vecka.
Baslinjeegenskaperna var balanserade mellan behandlingsarmarna. Bland de inskrivna patienterna var 100 % asiater (636 patienter). Medianåldern var 61 år (intervall: 27 till 75) och 73,1 % av patienterna var män och de flesta patienterna var aktuella eller tidigare rökare (66,8 %). Funktionsstatus enligt ECOG vid baslinjen var 0 (26,9 %) eller 1 (73,0 %). Trettionio procent hade tumör PD‑L1-uttryck TPS < 1 % [negativ], 31 % hade TPS 1‑49 %, 28 % hade TPS ≥ 50 %. Totalt 78,0 % av patienternas tumörer hade positivt PD‑L1-uttryck (CPS ≥ 1) och 18,7 % hade hjärnmetastas vid baslinjen. 79 patienter (37,6 %) i arm C fick behandling med serplulimab i kombination med bevacizumab efter bekräftad sjukdomsprogression.
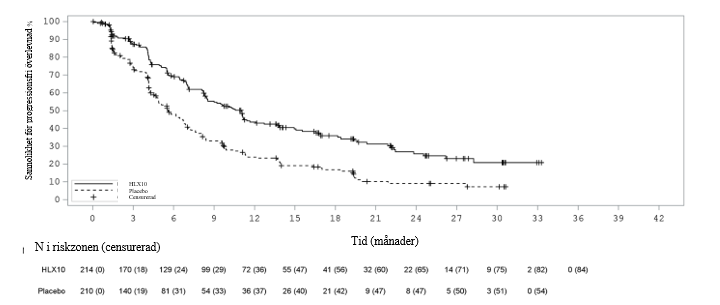
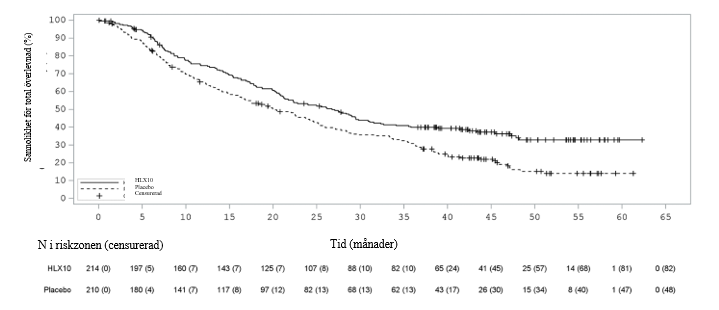
Medianöverlevnadsuppföljning var 23,1 månader vid den primära analysen (sista datum för datainsamling: 15 juni 2023) och 45,4 månader vid den uppdaterade analysen (sista datum för datainsamling: 7 augusti 2025). PFS-, ORR- och DOR-resultat från den primära analysen och OS-resultat från den uppdaterade analysen sammanfattas i tabell 6. Kaplan‑Meier-kurvor för PFS av primär analys och OS av uppdaterad analys presenteras i figur 3 och figur 4.
Tabell 6. Effektdata i ASTRUM-002
Arm B | Arm C | ||
Antal patienter | 214 | 210 | |
Primärt effektmått | |||
PFS1 | Antal patienter med händelser, n (%) | 130 (60,7 %) | 156 (74,3 %) |
Median PFS (månader, 95 % KI) | 11,0 (8,4, 12,7) | 5,6 (4,8, 6,8) | |
Riskkvot (95 % KI) | 0,55 (0,43‑0,69) | ||
p‑värde | < 0,0001 | ||
Sekundära effektmått | |||
OS2 | Antal patienter med händelser, n (%) | 132 (61,7 %) | 162 (77,1 %) |
Median OS (månader, 95 % KI) | 26,8 (21,2, 30,9) | 20,3 (16,2, 24,6) | |
Riskkvot (95 % KI) | 0,66 (0,52‑0,83) | ||
Bekräftad ORR1 | (%, 95 % KI) | 52,8 % (45,9 %‑59,7 %) | 27,6 % (21,7 %‑34,2 %) |
1PFS- och ORR-resultat är baserade på den förspecificerade interimsanalysen med sista datum för datainsamling den 15 juni 2023.
2OOS-result är baserade på den slutliga analysen med sista datum för datainsamling den 7 augusti 2025.
Figur 3 Kaplan‑Meier-kurva av PFS (RECIST 1.1) enligt IRRC hos den totala populationen vid den primära analysen (sista datum för datainsamling: 15 juni 2023)
Figur 4 Kaplan‑Meier-kurva av OSS hos den totala populationen vid den uppdaterade analysen (sista datum för datainsamling: 7 augusti 2025).
I tabell 7 sammanfattas effektresultaten av PFS för PD‑L1-undergrupper (TPS < 1 %, 1 % ≤ TPS < 50% och TPS ≥ 50 %) från den uppdaterade analysen.
Tabell 7. IRRC-utvärderad PFS enligt PD‑L1-uttryck (sista datum för datainsamling: 7 augusti 2025).
Arm B | Arm C | ||||
PD-L1-uttryck | Händelser/N (%) | Median (månader, 95 % KI) | Händelser/N (%) | Median (månader, 95 % KI) | Stratifierad riskkvot (95 % KI) |
TPS < 1 % | 59/84 (70,2 %) | 8,5 (5,6, 13,9) | 51/68 (75,0 %) | 6,8 (4,6, 9,8) | 0,83 (0,55, 1,26) |
1 % ≤ TPS < 50 % | 45/64 (70,3 %) | 10,3 (8,1, 15,5) | 62/73 (84,9 %) | 6,9 (5,1, 8,4) | 0,63 (0,43, 0,94) |
TPS ≥ 50 % | 37/62 (59,7 %) | 12,1 (9,5, 45,4) | 51/62 (82,3 %) | 4,4 (4,0, 5,8) | 0,36 (0,23, 0,57) |
ASTRUM‑004: Randomiserad fas III-prövning hos kemoterapinaiva patienter med lokalt avancerad eller metastaserad skivepiteltyp NSCLC i kombination med karboplatin och- nab-paklitaxel
Effekten av serplulimab i kombination med kemoterapi (karboplatin plus nab-paklitaxel) för första linjens behandling av skivepiteltyp NSCLC utvärderades i ASTRUM‑004-studien (NCT04033354), en randomiserad, dubbelblind, multiregional klinisk fas 3-prövning. Det primära effektmåttet var progressionsfri överlevnad (PFS) enligt bedömning av en oberoende radiologisk granskningskommitté (IRRC). De sekundära effektmåtten var total överlevnad (OS), PFS enligt utvärdering av prövare, objektiv svarsfrekvens (ORR) och svarstiden (DOR) av IRRC och av prövaren baserad på RECIST 1.1.
Prövningen inkluderade patienter (18 år eller äldre) med skivepiteltyp NSCLC i stadium IIIB/IIIC eller stadium IV (American Joint Committee on Cancer Edition 8) utan EGFR-sensibiliserande mutationer eller ALK/ROS1-rearrangemang, och ingen tidigare systemisk behandling för avancerad sjukdom. Patienterna måste ha ≥ 1 mätbar mållesion enligt RECIST 1.1 bedömd av IRRC och funktionsstatus enligt ECOG på 0‑1. Patienter exkluderades om de hade aktiva eller obehandlade metastaser i centrala nervsystemet, aktiv autoimmun sjukdom eller om administrering av systemiska immunsuppressiva läkemedel skett inom 14 dagar före den första dosen.
Totalt 537 patienter inkluderades och randomiserades (2:1) att få en av behandlingsregimerna som beskrivs i tabell 8. Randomisering stratifierades av PD‑L1-uttrycksnivå (negativ: tumörandelspoäng [TPS] ≥ 50 %, 50 % > TPS ≥ 1 %, TPS < 1 %, mätt med PD‑L1 IHC 22C3 pharmDx kit), asiatisk population (ja eller nej) och stadium på skivepiteltyp NSCLC (stadium IIIB/IIIC eller stadium IV).
Tabell 8. Intravenösa behandlingsregimer
Behandlings-regim | Induktion | Underhåll |
A | Serplulimab (4,5 mg/kg) a + Nab-paklitaxel (100 mg/m2)b,c + karboplatin (AUC = 5, upp till 750 mg eller AUC = 6, upp till 900 mg)b | Serplulimab (4,5 mg/kg)a |
B | Placebo + Nab-paklitaxel (100 mg/m2)b,c + karboplatin (AUC = 5, upp till 750 mg eller AUC = 6, upp till 900 mg)b | Placebod |
a. Serplulimab administrerades fram till sjukdomsprogression eller oacceptabel toxicitet eller upp till 2 år.
b. Nab-paklitaxel och karboplatin administrerades tills fullbordandet av 4‑6 cykler eller progressiv sjukdom eller oacceptabel toxicitet, beroende på vilket som inträffade först.
c. Nab-paklitaxel administrerades dag 1, 8 och 15 i varje cykel.
d. Byte var tillåtet från arm B för att få serplulimab 4,5 mg/kg som monoterapi var tredje vecka efter patientens första PD-bedömning baserad på RECIST 1.1.
Baslinjeegenskaperna var balanserade mellan behandlingsarmarna. Bland de inskrivna patienterna var 66,9 % asiater (359 patienter) och 33,1 % var icke-asiater (178 patienter), som alla var vita. Medianåldern var 63 år (intervall: 35‑86) och 42,3 % av patienterna var ≥ 65 år och 5,2 % av patienterna var ≥ 75 år. 90,9 % av patienterna var män. Funktionsstatus enligt ECOG vid baslinjen var 0 (16,9 %) eller 1 (83,1 %). 62,2 % av patienterna var PD‑L1-positiva (TPS ≥ 1 %). 71,7 % av patienterna var i stadium IV. 13,0 % av patienterna hade aldrig rökt och 87,0 % av patienterna var tidigare/aktuella rökare; 7,1 % med hjärnmetastaser.
Vid tidpunkten för cut‑off för interimsanalysen den 30 mars 2021 hade patienterna en medianöverlevnadsuppföljningstid på 8,9 månader. Uppdaterad analys med längre uppföljning (median: 31,1 månader) utfördes vid cut‑off den 31 januari 2023.
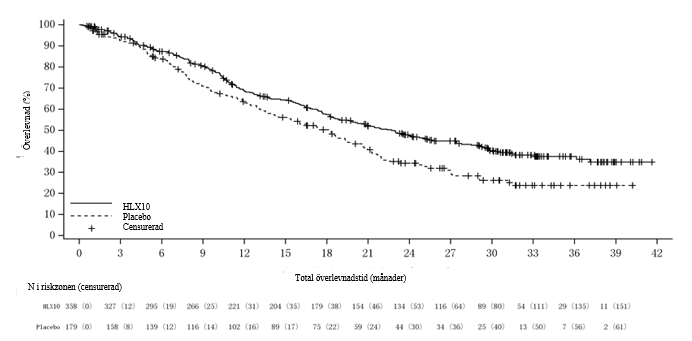
PFS- och ORR-resultat från interimsanalysen (primär analys) och OS-resultat från den slutliga analysen sammanfattas i tabell 9. Kaplan‑Meier-kurvor för PFS av primär analys och OS för slutlig analys presenteras i figur 5 och figur 6.
Tabell 9. Effektdatai ASTRUM-004
Arm A | Arm B | ||
Antal patienter | 358 | 179 | |
Primärt effektmått | |||
PFS1 | Antal patienter med händelser, n (%) | 146 (40,8 %) | 93 (52,0 %) |
Median PFS (månader, 95 % KI) | 8,3 (6,9, 10,4) | 5,7 (5,2, 6,8) | |
Riskkvot (95 % KI) | 0,55 (0,42, 0,73) | ||
P-värde | < 0,001 | ||
Sekundära effektmått | |||
OS2 | Antal patienter med händelser, n (%) | 196 (54,7 %) | 116 (64,8 %) |
Median OS (månader, 95 % KI) | 22,7 (18,6, 27,4) | 18,2 (14,1, 20,6) | |
Riskkvot (95 % KI) | 0,73 (0,58, 0,93) | ||
Bekräftad ORR1 | (%, 95 % KI) | 52,8% (47,5, 58,1) | 34,6% (27,7, 42,1) |
1PFS- och ORR-resultat är baserade på den prespecifierade primära analysen med data-cut-off den 30 mars 2021.
2OOS-resulat är baserade på den slutliga analysen med data-cut-off den 31 januari 2023.
Figur 5 Kaplan‑Meier-kurva av PFS (RECIST 1.1) enligt IRRC hos den totala populationen vid en primära analysen (datum för data-cut‑off: 30 mars 2021)
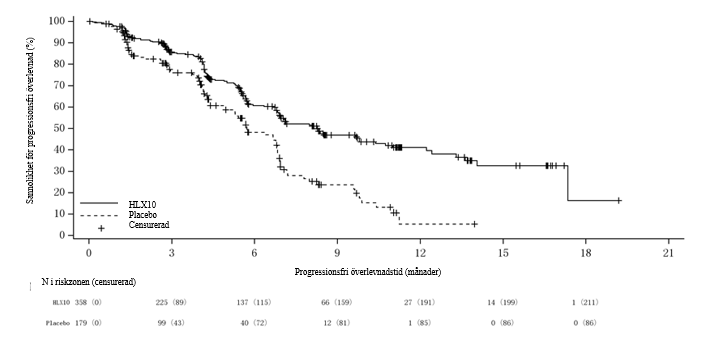
Figur 6 Kaplan‑Meier-kurva av OS hos den totala populationen vid den slutliga analysen (datum för data-cut‑off: 31 januari 2023)
Undergruppsanalyserna av PFS vid den primära analysen och OS vid den uppdaterade analysen utfördes per etnicitet. Hos icke-asiatiska patienter var median-PFS 4,21 månader (95 % KI: 4,11, 5,55) i serplulimabarmen mot 6,41 månader (95 % KI: 6,41, 6,83) i placeboarmen, med en stratifierad KI på 1,10 (95 % KI: 0,64, 1, 91). Median-OS var 16,30 månader (95 % KI: 10,64, 19,78) i serplulimabarmen mot 15,41 månader (95 % KI: 9,56, 23,29) i placeboarmen, med en stratifierad KI på 1,04 (95 % KI: 0,68, 1,60).
Hos asiatiska patienter var median-PFS 9,79 månader (95 % KI: 8,28, 13,63) i serplulimabarmen mot 5,72 månader (95 % KI: 5,22, 6,90) i placeboarmen, med en stratifierad KI på 0,44 (95 % KI: 0,32, 0,59). Median-OS var 27,40 månader (95 % KI: 21,82, 31,41) i serplulimabarmen mot 18,37 månader (95 % KI: 14,49, 21,26) i placeboarmen, med en stratifierad KI på 0,62 (95 % KI: 0,47, 0,82).
Esofaguscancer av skivepiteltyp (OSCC)
ASTRUM‑007: Randomiserad fas III-prövning av kombinationsbehandling hos patienter med esofaguscancer av skivepiteltyp
Effekten av serplulimab i kombination med kemoterapi undersöktes i ASTRUM‑007 (NCT03958890), en randomiserad, dubbelblind, placebokontrollerad multicenterstudie hos patienter med icke-resektabel, lokalt avancerad, återkommande eller metastaserad esofaguscancer av skivepiteltyp. De dubbla primära effektmåtten var progressionsfri överlevnad (PFS) utvärderad av en oberoende radiologigranskningskommitté (IRRC) baserat på RECIST v1.1 och total överlevnad (OS) hos ITT (intent-to-treat)-populationen. De sekundära effektmåtten inkluderade PFS utvärderad av prövare, objektiv svarsfrekvens (ORR) och svarstiden (DOR) utvärderad av IRRC och av prövaren. Studiebehandlingsregimerna var oblindade efter den primära analysen.
Prövningen inkluderade vuxna (≥ 18 år och ≤ 75 år) patienter med histologiskt diagnostiserad lokalt avancerad, återkommande eller fjärrmetastaserad esofageal (inklusive gastroesofageal) skivepitelcancer och ingen tidigare systemisk behandling för återkomst eller metastas. Patienter med återkommande OSCC som hade fått neoadjuvant/adjuvant behandling eller kurativ samtidig kemoterapi eller strålbehandling kunde inkluderas om den sista behandlingen gavs mer än 6 månader från återfall eller sjukdomsprogression. Patienterna måste ha ≥ 1 mätbar lesion enligt RECIST 1.1 utvärderad av IRRC, positivt PD‑L1-uttryck med CPS ≥ 1 baserat på PD‑L1 IHC 22C3 pharmDx Kit och ECOG PS 0 eller 1. Patienter med anamnes på gastrointestinal perforation och/eller fistlar inom 6 månader före den första dosen av studieläkemedel, aktiva autoimmuna sjukdomar, CNS-metastaser eller tidigare behandling med anti-PD‑1- eller anti-PD‑L1-antikroppar exkluderades.
Totalt 551 patienter var inskrivna och randomiserade (2:1) att få en av de behandlingsregimer som beskrivs i tabell 10. Randomisering stratifierades enligt grad av PD‑L1-uttryck (1 ≤ CPS < 10 mot CPS ≥ 10), ålder (≥ 65 år mot < 65 år) och tumörstatus (lokalt avancerad mot fjärrmetastas).
Tabell 10. Intravenösa behandlingsregimer
Behandlings-regim | Induktion | Underhåll |
A | Serplulimab (3,0 mg/kg)a + cisplatin (50 mg/m2)b + 5-fluorouracil (5-FU, 2 400 mg/m2)c | Serplulimab (3,0 mg/kg)a |
B | Placebo + cisplatin (50 mg/m2)b + 5-FU (2 400 mg/m2)c | Placebo |
a. Serplulimab administrerades fram till sjukdomsprogression, oacceptabel toxicitet eller upp till 2 år.
b. Cisplatin administrerades tills fullbordandet av 8 cykler eller progressiv sjukdom eller oacceptabel toxicitet, beroende på vilket som inträffade först.
c. 5‑FU administrerades som kontinuerligt intravenöst dropp under 44–48 timmar tills fullbordandet av 12 cykler eller progressiv sjukdom eller oacceptabel toxicitet, beroende på vilket som inträffade först.
Av de 551 inskrivna patienterna hade 343 (62,3 %) tumörer som uttryckte PD‑L1 med CPS ≥ 5. Bland dessa 343 patienter var alla asiater. Medianåldern var 64 år (intervall: 34‑75) och 48,4 % av patienterna var ≥ 65 år. 85,7 % av patienterna var män. Funktionsstatus enligt ECOG vid baslinjen var 0 (26,2 %) eller 1 (73,8 %). 70,3 % av patienterna var CPS ≥ 10. 14,0 % av patienterna hade lokalt avancerade sjukdomar. 34,7 % av patienterna hade tidigare fått cancerbehandling inklusive kirurgi, strålbehandling och systemisk cancerbehandling före inskrivning i studien.
Vid sista datum för datainsamling för interimsanalysen den 15 april 2022 hade patienterna en medianöverlevnadsuppföljningstid på 14,9 månader. För alla patienter inskrivna i studien var median-PFS vid IRRC-utvärdering enligt RECIST 1.1 5,8 (95 % KI: 5,7, 6,9) månader i serplulimabgruppen och 5,3 (95 % KI: 4,3, 5,6) månader i placebogruppen, med en stratifierad HR (95 % KI) på 0,60 (0,48, 0,75). Median-OS var 15,3 (95 % KI: 14,0, 18,6) månader i serplulimabgruppen och 11,8 (95 % KI: 9,7, 14,0) månader i placebogruppen. Stratifierad HR (95 % KI) var 0,68 (0,53, 0,87).
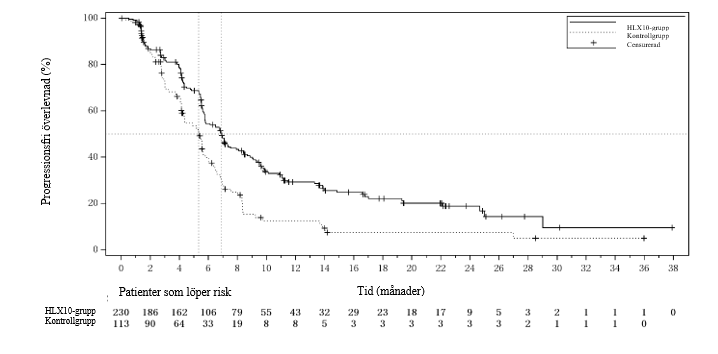
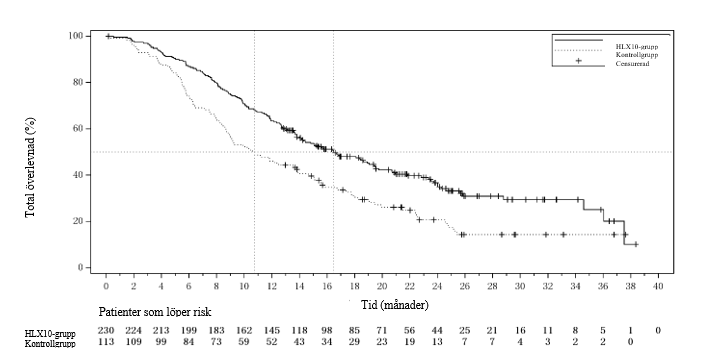
En uppdaterad analys efter avblindning med längre uppföljningsvaraktighet (median: 24,3 månader) utfördes med sista datum för datainsamling den 9 januari 2023 och visade effektresultat som överensstämde med interimsanalysen. Effektresultat från den uppdaterade analysen (sista datum för datainsamling den 9 januari 2023) för patienter med CPS ≥ 5 sammanfattas i tabell 11. Kaplan‑Meier-kurvor för PFS och OS av uppdaterad analys hos patienter med CPS ≥ 5 visas i figur 7 och figur 8.
Tabell 11. Effektdata vid den uppdaterade analysen för patienter med CPS ≥ 5 (sista datum för datainsamling: 9 januari 2023)
Arm A | Arm B | ||
Antal patienter | 230 | 113 | |
Dubbelt primärt effektmått | |||
PFS | Antal patienter med händelser, n (%) | 154 (67,0 %) | 84 (74,3 %) |
Median PFS (95 % KI) (månader) | 6,9 (5,7, 8,1) | 5,3 (4,1, 5,8) | |
Riskkvot (95 % KI) | 0,57 (0,43, 0,75) | ||
OS | Antal patienter med händelser, n (%) | 144 (62,6 %) | 89 (78,8 %) |
Median OS (95 % KI) (månader) | 16,5 (13,8, 19,5) | 10,7 (8,7, 13,9) | |
Riskkvot (95 % KI) | 0,60 (0,46, 0,79) | ||
Sekundära effektmått | |||
Bekräftad ORR | % (95 % KI) | 65,2 % (58,7 %, 71,4 %) | 39,8 % (30,7 %, 49,5 %) |
Figur 7 Kaplan‑Meier-kurva för PFS (RECIST 1.1) enligt IRRC hos patienter med CPS-poäng ≥ 5 vid den uppdaterade analysen (sista datum för datainsamling: 9 januari 2023)
Figur 8 Kaplan‑Meier-kurva för OS hos patienter med CPS ≥ 5 vid den primära analysen (sista datum för datainsamling: 9 januari 2023)
Immunogenicitet
Serplulimabs immunogenicitet utvärderades hos alla 1 795 utvärderbara patienter i 5 kliniska studier med doser från 0,3 mg/kg till 10 mg/kg varannan vecka (Q2W), inklusive godkända och föreslagna regimer som monoterapi eller i kombination med andra läkemedel mot cancer.
Hos alla utvärderbara patienter testade 82 (4,6 %) av patienterna positivt för anti-läkemedelsantikroppar (ADA) vid något besök och 70 (3,9 %) av patienterna hade behandlingsutlösta ADA (TE‑ADA)-svar, definierat som minst en post-baslinje ADA-positivt. Neutraliserande antikroppar (NAb) mot serpulimab detekterades hos 0,2 % (3/1 795) av patienterna.
Inga tecken på ADA-påverkan på farmakokinetik, effekt eller säkerhet observerades..
Äldre patienter
I ASTRUM‑005-, ASTRUM‑002-, ASTRUM-007- och ASTRUM‑004-studierna, av de 1 343 patienterna i serplulimabgruppen i den totala populationen, var 637 (47,4 %) ≥ 65 år. Inga övergripande skillnader i effekt observerades mellan äldre patienter och yngre patienter. Data för patienter ≥ 75 år är för begränsade för att några slutsatser om den här populationen.
Pediatrisk population
Europeiska läkemedelsmyndigheten har avstått från skyldigheten att lämna in resultat från studier med serplulimab i alla undergrupper av den pediatriska populationen för maligna tumörer (förutom hematopoetisk och lymfoid vävnad) (se avsnitt Dosering och administreringssätt för information om pediatrisk användning).
Farmakokinetiska egenskaper
Serplulimabs farmakokinetik har undersökts i en populationsfarmakokinetisk (popPK) analys som inkluderade 2 110 patienter med cancer (inklusive OSCC, SCLC, NSCLC) och andra solida cancertyper från 11 studier. Patienterna fick serplulimab intravenöst som monoterapi eller kombinationsterapi i dosintervallet 0,3 till 10 mg/kg Q2W, 4,5 mg/kg Q3W, 200 mg Q2W, 300 mg Q3W och 400 mg Q4W. PK beskrevs av en tvådelad modell med tidsberoende clearance (CL). Variation mellan individer (variationskoefficient, CV) intervall från 16,3 % till 54,3 %. Medelvärdet (CV) observerat genom koncentrationen vid stabilt tillstånd är mellan 44,2 µg/ml (34,7 %) och 60,7 µg/ml (30,3 %) för alla tumörtyper.
Absorption
Serplulimab administreras som intravenös infusion och är därför omedelbart och fullständigt biotillgängligt. Andra administreringsvägar har inte undersökts.
Distribution
Baserat på en popPK-analys är distributionsvolymen för serplulimab ungefär i intervallet 6,17 l till 6,46 l.
Metabolism
Den metaboliska vägen för serplulimab har inte karakteriserats. Serplulimab förväntas kataboliseras till små peptider och aminosyror genom allmänna proteinnedbrytningsprocesser.
Eliminering
Baserat på en popPK-analys är serplulimab-clearance (CL) efter den första dosen i intervallet 0,171 l/dag till 0,211 l/dag. Clearance minskar över tiden med maximalt 8,8 % (CV 34,1 %) med 221 dagar för att nå hälften av den maximala effekten. Halveringstiden vid jämvikt är i intervallet 25,0‑31,2 dagar.
Linjäritet/icke‑linjäritet
Serplulimab uppvisade linjär farmakokinetik över dosintervallet 0,3 till 10 mg/kg Q2W (inklusive platta doser på 200 mg Q2W, 300 mg Q3W och 400 mg Q4W) både efter enstaka och multipla doser.
Särskilda grupper
Inga särskilda studier har utförts i speciella populationer. En popPK-analys antydde ingen skillnad i totalt systemiskt clearance av serplulimab baserat på ålder (23‑83 år), ras (n=265 vita och n=1 845 asiater) och ECOG-prestanda‑statuspoäng (0 eller 1). Serplulimab-clearance ökade med ökad kroppsvikt.
Nedsatt njurfunktion
Ingen effekt av kreatinin eller kreatininclearance (CRCL) (Cockcroft‑Gault) hittades på serplulimab CL baserat på en popPK-analys hos patienter med mild (CRCL=60‑89 ml/min; n=917), måttlig (CRCL=30‑59 ml/min; n=216) och svår (CRCL=15‑29 ml/min; n=1) nedsatt njurfunktion och normal njurfunktion (CRCL≥ 90 ml/min, n=973). Det finns otillräckliga data från patienter med gravt nedsatt njurfunktion för dosrekommendationer (se avsnitt Dosering och administreringssätt).
Nedsatt leverfunktion
Ingen effekt av ALAT, ASAT eller totalt bilirubin hittades på serplulimab CL baserat på en popPK-analys hos patienter med mild (totalt bilirubin ≤ ULN och ASAT > ULN eller totalt bilirubin > 1 till 1,5 × ULN och eventuell ASAT; n=279) och måttlig (totalt bilirubin > 1,5 till 3 × ULN och eventuell AST n=4) nedsatt leverfunktion och normal (totalt bilirubin ≤ ULN och AST ≤ ULN; n=1 819) leverfunktion. Det finns otillräckliga data från patienter med måttligt nedsatt leverfunktion för dosrekommendationer. Serplulimab har inte studerats hos patienter med gravt (totalt bilirubin > 3 × ULN och eventuell ASAT) nedsatt leverfunktion (se avsnitt Dosering och administreringssätt).
Prekliniska säkerhetsuppgifter
Toxicitet vid upprepad dosering
I studier som undersökte toxicitet vid upprepad dosering på cynomolgusapor som doserats i upp till 31 veckor, observerades en hög incidens av farmakologirelaterad perivaskulär mononukleär cellinfiltration i hjärnans choroidplexus vid 100 mg/kg. Ingen observerad negativ effektnivå (NOAEL) i 31‑veckors toxicitetsstudie var 50 mg/kg/vecka, vilket gav exponering 36 gånger (beräknat med AUC0‑ t) exponeringen hos människor vid dosen 3 mg/kg varannan vecka.
Reproduktionstoxicitet
Reproduktionstoxicitetsstudier har inte genomförts.
PD‑1/PD‑L1-vägen tros vara inblandad i att bibehålla tolerans mot fostret under hela graviditeten. Blockad av PD‑L1-signalering har visats i murina graviditetsmodeller störa toleransen mot fostret och resultera i en ökning av fosterförlusten.
Två anti‑PD‑L1 monoklonala antikroppar utvärderades i cynomolgusapor med avseende på reproduktions- och utvecklingstoxicitet och visades orsaka för tidig förlossning, fosterförlust och för tidig neonatal död när de administrerades till gravida apor.
Därför inkluderar potentiella risker med att administrera serplulimab under graviditet ökad frekvens av abort eller dödfödsel. Baserat på dess verkningsmekanism kan fostrets exponering för serplulimab öka risken för att utveckla immunmedierade störningar eller förändra det normala immunsvaret och immunmedierade störningar som har rapporterats i PD‑1 knockoutmöss.
Genotoxicitet och carcinogenitet
Inga studier har utförts för att bedöma den genotoxiska eller cancerframkallande potentialen hos serplulimab.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Citronsyramonohydrat (för pH-justering), natriumcitrat (E331) (för pH-justering), natriumklorid, mannitol (E421), polysorbat 80 (E433), vatten för injektioner
Inkompatibiliteter
I avsaknad av kompatibilitetsstudier får detta läkemedel inte blandas med andra läkemedel, förutom de som nämns i avsnitt Särskilda anvisningar för destruktion och övrig hantering. HETRONIFLY ska inte infunderas samtidigt i samma intravenösa linje som andra läkemedel.
Hållbarhet
Oöppnad injektionsflaska
3 år.
Spädd lösning
Ur mikrobiologisk synvinkel bör produkten, när den är utspädd, användas omedelbart. Den utspädda lösningen får inte frysas. Om den inte används omedelbart, är användningstiden och förvaringsförhållanden före användning användarens ansvar och bör inte vara längre än 24 timmar vid 2 °C till 8 °C. Denna 24‑timmarsperiod kan innefatta upp till 6 timmar vid rumstemperatur (≤ 25 °C). Om injektionsflaskorna och/eller de intravenösa påsarna är kylda måste de få uppnå rumstemperatur före användning.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2°C‑8°C).
Får ej frysas.
Förvaras i originalförpackningen för att skydda mot ljus.
För förvaringsförhållanden efter spädning av läkemedlet, se avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
HETRONIFLY infuusiokonsentraatti, liuosta varten
10 mg/ml (L:ei) 10 ml (1515,36 €)
PF-selosteen tieto
10 ml koncentrat i en 10 ml injektionsflaska av klart glas av typ I med klorbutylgummipropp och kombinationslock av aluminium‑plast som innehåller 100 mg serplulimab.
Förpackning med 1 injektionsflaska.
Läkemedlets utseende:
Färglös till svagt gul, klar till lätt opaliserande lösning, pH 5,2‑5,8, osmolalitet på cirka 280‑340 mOsm/kg.
Särskilda anvisningar för destruktion och övrig hantering
Förberedelse och administrering
-
Aseptisk hantering ska säkerställas under beredningen av infusionen.
-
Skaka inte injektionsflaskan.
-
Låt injektionsflaskan anta rumstemperatur (vid eller under 25°C).
-
Produkten ska inspekteras visuellt med avseende på partiklar och missfärgning före administrering. Koncentratet är en färglös till svagt gul, klar till lätt opaliserande lösning. Kassera injektionsflaskan om synliga partiklar observeras.
-
Bekräfta dosen av läkemedlet och beräkna den erforderliga volymen HETRONIFLY.
-
Dra upp en volym natriumklorid 9 mg/ml (0,9%) injektionslösning som motsvarar volymen av läkemedlet som ska infunderas från avsedd injektionspåse med hjälp av en steril spruta och kassera.
-
Använd en spruta för att dra upp den erforderliga volymen HETRONIFLY från injektionsflaskan och injicera den i natriumkloridlösningen 9 mg/ml (0,9 %) för injektion för att bereda en spädd lösning med en slutlig koncentration på 1,0 till 8,0 mg/ml. Blanda den spädda lösningen genom att försiktigt vända påsen.
-
Administrera infusionslösningen intravenöst med ett sterilt, icke‑pyrogent, låg‑proteinbindande 0,2 till 5,0 μm inbyggt eller monterat filter.
-
Ställ in den initiala infusionshastigheten till 100 ml per timme (25 droppar per minut rekommenderas). Infusionshastigheten kan justeras om infusion‑relaterade reaktioner inträffar (se avsnitt Dosering och administreringssätt). Om det inte uppkommer någon infusionsrelaterad biverkning under den första infusionen, kan varaktigheten av efterföljande administrering förkortas till 30 minuter (± 10 minuter).
-
Ur mikrobiologisk synvinkel bör läkemedlet, när det blivit spätt, användas omedelbart. Den spädda lösningen får inte frysas. Om den spädda lösningen inte används omedelbart kan den förvaras i 24 timmar vid 2°C till 8°C. Denna 24-timmarsperiod kan innefatta upp till 6 timmar i rumstemperatur (≤ 25°C). Om injektionsflaskorna och/eller intravenösa påsarna är kylda måste de få uppnå rumstemperatur före användning (se avsnitt Hållbarhet).
-
Vid slutet av infusionen ska infusionsröret spolas med natriumkloridlösning 9 mg/ml (0,9%) enligt sjukhusets rutinmässiga procedur.
-
Administrera inte andra läkemedel genom samma infusionsslang.
-
För att underlätta spårbarhet för biologiska läkemedel ska namn och tillverkningssatsnummer på det administrerade läkemedelt tydligt antecknas i patientjournalen.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
HETRONIFLY infuusiokonsentraatti, liuosta varten
10 mg/ml 10 ml
- Ei korvausta.
Atc-kod
L01FF12
Datum för översyn av produktresumén
19.06.2026
Yhteystiedot
 ACCORD HEALTHCARE OY
ACCORD HEALTHCARE OY Oksasenkatu 10 A 6
00100 Helsinki
Suomi
010 231 4180