
BEYONTTRA tabletti, kalvopäällysteinen 356 mg
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää akoramidiisihydrokloridia määrän, joka vastaa 356 mg akoramidiisia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
BEYONTTRA on tarkoitettu villityypin tai variantin transtyretiinivälitteisen amyloidoosin hoitoon aikuispotilaille, joilla on kardiomyopatia (ATTR-CM).
Ehto
Hoidon voi aloittaa lääkäri, joka on perehtynyt transtyretiinivälitteistä amyloidista kardiomyopatiaa (ATTR-CM) sairastavien potilaiden hoitoon.
Annostus ja antotapa
Hoidon voi aloittaa lääkäri, joka on perehtynyt transtyretiinivälitteistä amyloidista kardiomyopatiaa (ATTR-CM) sairastavien potilaiden hoitoon.
Annostus
Akoramidiisin suositeltu annos on 712 mg (kaksi 356 mg:n tablettia) suun kautta kahdesti vuorokaudessa, mikä vastaa 1 424 mg:n kokonaisvuorokausiannosta.
Tietoja tehosta New York Heart Association (NYHA) -luokan IV potilaiden hoidossa ei ole (ks. kohta Farmakodynamiikka).
Väliin jäänyt annos
Väliin jääneitä yksittäisiä annoksia ei pidä korvata kaksinkertaisella annoksella. Seuraava annos on otettava seuraavana tavanomaisena ajankohtana.
Erityisryhmät
Iäkkäät
Annostusta ei tarvitse muuttaa iäkkäitä potilaita hoidettaessa (≥ 65‑vuotiaat, ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Koska akoramidiisin munuaispuhdistuma on vähäistä, annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka). Tietoja vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastavista potilaista on saatavilla rajallisesti (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka), eikä tietoja dialyysipotilaista ole, minkä vuoksi akoramidiisia on käytettävä varoen tälle potilasryhmälle.
Maksan vajaatoiminta
Akoramidiisia ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla, joten sen käyttöä tälle potilasryhmälle ei suositella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää akoramidiisia pediatrisille potilaille käyttöaiheeseen ”villityypin tai variantin transtyretiinivälitteisen amyloidoosin aiheuttaman kardiomyopatian hoito”.
Antotapa
Suun kautta.
Kalvopäällysteiset tabletit niellään kokonaisina. BEYONTTRA voidaan ottaa veden kanssa ja joko ruoan kanssa tai ilman ruokaa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Maksan vajaatoiminta
Akoramidiisia ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla, joten sen käyttöä ei suositella tälle potilasryhmälle (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Tietoja vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastavista potilaista on saatavilla rajallisesti (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka), eikä tietoja dialyysipotilaista ole, minkä vuoksi akoramidiisia on käytettävä varoen tälle potilasryhmälle.
Hemodynaamiset munuaisparametrit
Akoramidiisihoitoa saaneilla potilailla esiintyi aluksi arvioidun glomerulusten suodatusnopeuden (eGFR) laskua hoidon ensimmäisen kuukauden aikana ja vastaavasti seerumin mitatun kreatiniiniarvon nousua (ks. kohta Farmakodynamiikka).
Tämä eGFR-arvon ja seerumin kreatiniiniarvon muutos ei ollut etenevä ja korjaantui potilailla, joiden hoito keskeytettiin. Siihen ei liittynyt munuaisvauriota, mikä on yhdenmukaista hemodynaamisen munuaisvaikutuksen kanssa.
Tietoa apuaineista
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan "natriumiton".
Yhteisvaikutukset
Akoramidiisin vaikutus muihin lääkevalmisteisiin
Transportterijärjestelmät
Terveillä vapaaehtoisilla aikuisilla tehdyn kliinisen tutkimuksen perusteella orgaanisten anionien kuljettajaproteiini (OAT) 1:n ja 3:n eston ei odoteta aiheuttavan kliinisesti merkittäviä lääkeyhteisvaikutuksia OAT1:n ja OAT3:n substraattien kanssa (esim. ei-steroidiset tulehduskipulääkkeet, bumetanidi, furosemidi, lamivudiini, metotreksaatti, oseltamiviiri, tenofoviiri, gansikloviiri, adefoviiri, sidofoviiri, tsidovudiini, tsalsitabiini).
In vitro -tutkimuksen perusteella ei ole odotettavissa lääkeyhteisvaikutuksia samanaikaisesti annettujen rintasyövän resistenssiproteiinin (BCRP) substraattien kanssa kliinisesti merkityksellisinä pitoisuuksina.
In vitro -tutkimusten perusteella akoramidiisi ei todennäköisesti aiheuta mitään uridiini-5’-difosfaatti (UDP) -glukuronyylitransferaasista tai sytokromi P450 ‑entsyymeistä riippuvia, kliinisesti merkityksellisiä yhteisvaikutuksia. Akoramidiisin todettiin kuitenkin olevan CYP2C8- ja CYP2C9-entsyymien estäjä in vitro. In vivo -tutkimuksia ei ole tehty, minkä vuoksi tulee noudattaa varovaisuutta käytettäessä kapean terapeuttisen indeksin omaavia CYP2C8:n ja CYP2C9:n substraatteja samanaikaisesti.
Muiden lääkevalmisteiden vaikutus akoramidiisiin
Diureetit
Populaatiofarmakokineettisen analyysin perusteella diureettien samanaikainen käyttö ei vaikuta akoramidiisin vakaan tilan plasmapitoisuuksiin.
Rintasyövän resistenssiproteiinin estäjät
Akoramidiisi on BCRP:n substraatti. In vitro ‑tutkimuksen perusteella ei ole odotettavissa kliinisesti merkityksellisiä lääkeyhteisvaikutuksia BCRP:n estäjien kanssa.
Mahahappoja vähentävät aineet
Erillistä in vivo -yhteisvaikutustutkimusta mahahappoa vähentävien aineiden kanssa ei tehty. Näin ollen mahahappoa vähentävien aineiden vaikutusta akoramidiisin farmakokinetiikkaan ei tunneta. Faasin III tutkimuksessa ei havaittu eroja systeemisessä altistumisessa akoramidiisille tai farmakodynaamisessa markkerissa (TTR:n stabilisaatio) mahahappoja vähentäviä aineita käyttävien ja niitä käyttämättömien potilaiden välillä siitä huolimatta, että akoramidiisin liukoisuus on vahvasti pH:sta riippuvainen fysiologisella pH-alueella.
Vaikutus laboratoriokokeisiin
Akoramidiisi voi pienentää vapaan tyroksiinin pitoisuuksia seerumissa ilman, että kilpirauhasta stimuloivan hormonin (TSH) pitoisuudet muuttuvat. Vastaavia kilpirauhasen toimintahäiriöön sopivia kliinisiä löydöksiä ei ole todettu.
Raskaus ja imetys
Raskaus
Tietoja akoramidiisin käytöstä raskaana oleville naisille ei ole olemassa.
Eläimillä tehdyissä tutkimuksissa on havaittu kehitystoksisuutta annoksella, joka oli toksinen myös emolle (ks. kohta Prekliiniset tiedot turvallisuudesta). Akoramidiisin käyttöä ei suositella raskauden aikana eikä naisille, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyvätkö akoramidiisi tai sen metaboliitit ihmisillä äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois (ks. kohta Prekliiniset tiedot turvallisuudesta). Akoramidiisia ei pidä käyttää imetyksen aikana.
Hedelmällisyys
Ihmisen hedelmällisyyttä koskevia tietoja ei ole saatavilla. Hedelmällisyyden heikkenemistä ei ole todettu ei-kliinisissä tutkimuksissa supraterapeuttisen altistuksen yhteydessä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
BEYONTTRA-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Kliinisen tutkimuksen perusteella yleisimmin raportoidut haittavaikutukset olivat ripuli (11,6 %) ja kihti (11,2 %).
Haittavaikutustaulukko
Turvallisuustiedot kattavat 421 tutkittavaa, joilla oli diagnosoitu ATTR‑CM ja jotka saivat 712 mg akoramidiisia (kaksi 356 mg:n tablettia) suun kautta kahdesti vuorokaudessa faasin III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa avaintutkimuksessa, jossa hoidon kestoksi oli määritetty 30 kuukautta.
Haittavaikutukset on lueteltu alla MedDRA-elinjärjestelmäluokan (SOC) ja yleisyysluokan mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10) ja melko harvinainen (≥ 1/1 000, < 1/100). Taulukossa luetellut haittavaikutukset ovat peräisin ATTR‑CM-potilailta kerätyistä kumulatiivisista kliinisistä tiedoista.
Taulukko 1: Luettelo haittavaikutuksista
| Elinjärjestelmäluokka | Hyvin yleinen |
| Ruoansulatuselimistö | Ripuli |
| Aineenvaihdunta ja ravitsemus | Kihti |
Valikoitujen haittavaikutusten kuvaus
Suurin osa ripuli- ja kihtitapahtumista eivät olleet vakavia ja olivat ohimeneviä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostuksesta ei ole kliinistä kokemusta.
Jos yliannostusta epäillään, hoidon pitää olla oireenmukaista ja elintoimintoja tukevaa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: sydänlääkkeet, muut sydänlääkkeet. ATC-koodi: C01EB25.
Vaikutusmekanismi
Transtyretiinivälitteinen amyloidinen kardiomyopatia kehittyy transtyretiini (TTR) -tetrameerin rakenteen hajotessa monomeereiksi. Ne laskostuvat virheellisesti ja kasautuvat oligomeerisiksi amyloidin esiasteiksi, jotka kertyvät sydämeen ja järjestäytyvät siellä amyloidifibrilleiksi.
Akoramidiisi on spesifinen TTR-stabilisaattori. Akoramidiisi suunniteltiin jäljittelemään sairaudelta suojaavaa geenivarianttia (T119M) siten, että se muodostaa vetysidoksia vierekkäisten seriinijäännösten kanssa tetrameerin molemmissa tyroksiinin sitoutumiskohdissa. Tämä vuorovaikutus vakauttaa tetrameerin estämällä sitä hajoamasta monomeereiksi, mikä hidastaa ATTR-CM:ään johtavaa amyloidogeenista prosessia.
Farmakodynaamiset vaikutukset
Akoramidiisi sai aikaan lähes täydellisen transtyretiinin stabilisaation villityypin ja kaikkien testattujen amyloidogeenisten varianttien genotyypeissä, mukaan lukien yleisimmät genotyypit V30M (p.V50M), T60A (p.T80A) ja V122I (p.V142I). ATTRibute‑CM-tutkimuksessa akoramidiisihoitoa (712 mg kahdesti vuorokaudessa) saaneilla potilailla (villityypin ATTR ja variantti ATTR) todettiin lähes täydellinen (≥ 90 %) TTR:n stabilisaatio ensimmäisessä hoidon aloittamisen jälkeisessä arvioinnissa (päivänä 28), ja vaikutus säilyi kuukauden 30 loppuun asti. Kaikissa lähtötilanteen jälkeisissä mittauksissa (päivästä 28 kuukauteen 30) TTR-arvo oli suurempi akoramidiisiryhmässä kuin lumeryhmässä (kuukauden 30 kohdalla keskimääräinen muutos lähtötilanteesta oli akoramidiisilla 9,1 mg/dl ja lumelääkkeellä 1,3 mg/dl).
ATTRibute-CM-tutkimuksessa B-tyypin natriureettisen peptidin N-terminaalisen prohormonin (NT-proBNP) pitoisuuden nousu kuukauden 30 kohdalla suosi akoramidiisia ja oli noin puolet lumeryhmässä todetusta noususta. Akoramidiisiryhmässä todettiin myös pienempi troponiini I ‑pitoisuuden nousu kuin lumeryhmässä.
ATTRibute-CM-tutkimuksessa seerumin kreatiniinipitoisuuden keskiarvo (ja arvioitu GFR-arvo [eGFR]) oli lähtötilanteessa 110,0 mikromol/l (eGFR: 60,9 ml/min/1,73 m2) akoramidiisiryhmässä ja 109,0 mikromol/l (eGFR: 61,0 ml/min/1,73 m2) lumeryhmässä. Päivänä 28 seerumin kreatiniinipitoisuuden (eGFR) keskiarvon muutos lähtötilanteesta oli suurempi akoramidiisiryhmässä (päivänä 28 todetut arvot, seerumin kreatiniini: 129,3 mikromol/l, eGFR: 52,4 ml/min/1,73 m2) kuin lumeryhmässä (päivänä 28 todetut arvot, seerumin kreatiniini: 110,6 mikromol/l, eGFR: 60,0 ml/min/1,73 m2). Päivän 28 jälkeen akoramidiisiryhmän seerumin kreatiniinipitoisuus (eGFR) pysyi vakaana tutkimuksen loppuajan. Lumeryhmässä seerumin kreatiniinipitoisuus suureni asteittain ja eGFR laski vastaavasti asteittain lähtötilanteesta kuukauden 30 loppuun. Kuukauden 30 kohdalla seerumin kreatiniinipitoisuus oli akoramidiisiryhmässä 123,4 mikromol/l (eGFR: 55,1 ml/min/1,73 m2) ja lumeryhmässä 117,2 mikromol/l (eGFR: 57,2 ml/min/1,73 m2). Akoramidiisihoitoa saaneilla potilailla todettu seerumin kreatiniinipitoisuuden suureneminen ja vastaava eGFR-arvon pieneneminen korjaantuivat, kun hoito keskeytettiin.
Sydämen elektrofysiologia
Akoramidiisin enimmäisannoksella 1 780 mg, jota tutkittiin terveille vapaaehtoisille aikuisille annettuna kerta-annoksena, ei ollut kliinisesti merkityksellistä vaikutusta sydämen johtumiseen tai repolarisaatioon (pitoisuus-QTc-vaikutusta ei todettu). Näiden havaintojen perusteella proarytmian riski on pieni.
Kliininen teho
ATTRibute‑CM oli kansainvälinen, satunnaistettu, kaksoissokkoutettu, lumekontrolloitu kliininen monikeskustutkimus, johon osallistuneilla 632 tutkittavalla oli villityypin ATTR‑CM tai variantti (perinnöllinen tai de novo) ATTR‑CM ja NYHA-luokan I–III sydämen vajaatoiminta ja senhetkisiä tai aiempia sydämen vajaatoiminnan oireita. Tutkittavat satunnaistettiin suhteessa 2:1 saamaan joko 712 mg akoramidiisia (n = 421) tai vastaavaa lumelääkettä (n = 211) kahdesti vuorokaudessa 30 kuukauden ajan. Hoitomääritys ositettiin sen mukaan, oliko tutkittavilla variantti ATTR‑CM (ATTRv‑CM) vai villityypin ATTR CM (ATTRwt-CM) sekä lähtötilanteen taudin vaikeusasteen, eli NT-proBNP-arvon ja eGFR-arvon perusteella määritellyn munuaisten toiminnan mukaan. Potilaat, joiden eGFR-arvo oli < 15 ml/min/1,73 m2, suljettiin pois tutkimuksesta.
Taulukko 2: Demografiset tiedot ja lähtötilanteen ominaisuudet (mITT-populaatio1)
| Ominaisuus | Akoramidiisi N = 409 | Lumelääke N = 202 |
| Ikä – vuotta | ||
| Keskiarvo (keskihajonta) | 77,3 (6,5) | 77,0 (6,7) |
| Sukupuoli – lukumäärä (%) | ||
| Mies | 374 (91,4) | 181 (89,6) |
| Nainen | 35 (8,6) | 21 (10,4) |
| TTR-genotyyppi2 – lukumäärä (%) | ||
| ATTRv | 39 (9,5) | 20 (9,9) |
| ATTRwt | 370 (90,5) | 182 (90,1) |
| NYHA-luokka – lukumäärä (%) | ||
| NYHA-luokka I | 51 (12,5) | 17 (8,4) |
| NYHA-luokka II | 288 (70,4) | 156 (77,2) |
| NYHA-luokka III | 70 (17,1) | 29 (14,4) |
| eGFR2 (ml/min/1,73 m2) – lukumäärä (%) | ||
| eGFR ≥ 45 | 344 (84,1) | 173 (85,6) |
| eGFR < 45 | 65 (15,9) | 29 (14,4) |
| NT-proBNP2 (pikog/ml) – lukumäärä (%) | ||
| ≤ 3 000 | 268 (65,5) | 133 (65,8) |
| > 3 000 | 141 (34,5) | 69 (34,2) |
| ATTR NAC -vaihe3 – lukumäärä (%) | ||
| I | 241 (58,9) | 120 (59,4) |
| II | 130 (31,8) | 66 (32,7) |
| III | 38 (9,3) | 16 (7,9) |
| Aiemmin asennettu pysyvä sydämentahdistin – lukumäärä (%) | 77 (18,8) | 38 (18,8) |
| Aiempi eteisvärinä – lukumäärä (%) | 236 (57,7) | 117 (57,9) |
Lyhenteet: ATTRv = variantti transtyretiiniamyloidi, ATTRwt = villityypin transtyretiiniamyloidi, NAC = National Amyloidosis Centre (Lontoo, Yhdistynyt kuningaskunta), NYHA = New York Heart Association, eGFR = arvioitu glomerulusten suodatusnopeus, NT‑proBNP = B‑tyypin natriureettisen peptidin N‑terminaalinen prohormoni, TTR = transtyretiini
1 mITT = modifioitu hoitoaikomus (lähtötilanteen eGFR ≥ 30 ml/min/1,73 m2).
2 Ositustekijät.
3 NAC-vaihe I (NT‑proBNP ≤ 3 000 pikog/ml ja eGFR ≥ 45 ml/min/1,73 m2), vaihe II (NT‑proBNP ≤ 3 000 pikog/ml ja eGFR < 45 ml/min/1,73 m2 tai NT‑proBNP > 3 000 pikog/ml ja eGFR ≥ 45 ml/min/1,73 m2), vaihe III (NT‑proBNP > 3 000 pikog/ml ja eGFR < 45 ml/min/1,73 m2).
Tutkittavat saivat aloittaa avoimen tafamidiisihoidon, jos sitä määrättiin samanaikaiseksi lääkehoidoksi tutkittavan osallistuttua tutkimukseen 12 kuukauden ajan. Yhteensä 107 tutkittavaa sai tafamidiisia, heistä 61 (14,9 %) akoramidiisiryhmässä ja 46 (22,8 %) lumeryhmässä.
Tutkimuksen ensisijaisena tavoitteena oli vahvistaa akoramidiisin paremmuus lumelääkkeeseen nähden hierarkkisessa päätetapahtumassa, joka kattoi mistä tahansa syystä johtuvan kuolleisuuden (ACM) ja sydän- ja verisuoniperäisten sairaalahoitojaksojen (CVH) kumulatiivisen esiintyvyyden. Toissijaisia tavoitteita olivat ACM:n arviointi, CVH:n arviointi, 6 minuutin kävelytesti, Kansas Cityn kardiomyopatiakyselyn (KCCQ) kokonaisyhteenvetopisteet (elämänlaadun mittari), seerumin TTR ja NT‑proBNP. Tärkeimmät tehon analyysit tehtiin modifioidun hoitoaikeen mukaisen (mITT) populaation 611 tutkittavalla, eikä avoimen tafamidiisihoidon aloituksen suhteen tehty mukautuksia.
Tehon analyysi
Tehon analyysissä ositettua Finkelstein-Schoenfeldin (F-S) testiä sovellettiin hierarkkisesti ACM:ään ja CVH:hon 30 kuukautta kestäneen tutkimuksen aikana. Menetelmässä jokaista tutkittavaa verrattiin pareittain kaikkiin muihin tutkittaviin kunkin komponentin osalta. Tässä hierarkkisessa menettelyssä kunkin parin tutkittavia verrattiin ensin ACM:n osalta ja sitten CVH:n osalta vain, jos ACM-vertailu päättyi tasapeliin. Tämän analyysin tulos oli tilastollisesti merkitsevä (taulukko 3).
Mistä tahansa syystä johtuvaa kuolleisuutta raportoitiin 19,3 %:lla akoramidiisiryhmän tutkittavista ja 25,7 %:lla lumeryhmän tutkittavista. Suurin osa (79 %) kuolemista liittyi sydän- ja verisuoniongelmiin, ja akoramidiisin osoitettiin pienentäneen sydän- ja verisuoniongelmiin liittyvän kuolleisuuden suhteellista riskiä 30 % lumelääkkeeseen verrattuna. Sydän- ja verisuoniongelmiin liittyviä kuolemia raportoitiin 14,9 %:lla akoramidiisiryhmän ja 21,3 %:lla lumeryhmän tutkittavista, riskisuhteen ollessa 0,709 (95 %:n luottamusväli: 0,476, 1,054, p = 0,0889, Coxin suhteellisen riskin malli).
Coxin regressioanalyysi osoitti mistä tahansa syystä johtuvan kuolleisuuden ja ensimmäisen sydän- ja verisuoniongelmista johtuvan sairaalahoidon yhdistetyn riskin pienentyneen 35,5 % (riskisuhde: 0,645 [95 %:n luottamusväli: 0,500, 0,832; p = 0,0008]).
Kaplan-Meier-käyrissä havaittiin eriytymistä kuukauden 3 kohdalla, ja eriytyminen jatkui tasaisesti kuukauteen 30 asti (kuva 1).
mITT-populaatiossa osoitetut ACM- ja CVH-tehotulokset todettiin myös ITT-populaatiossa (kaikki satunnaistetut tutkittavat lähtötilanteen eGFR-arvosta riippumatta).
Taulukko 3: Finkelstein-Schoenfeldin analyysin tehotulokset, mistä tahansa syystä johtuva kuolleisuus ja sydän- ja verisuoniongelmista johtuva sairaalahoito kuukauden 30 kohdalla ATTRibute-CM-tutkimuksessa (mITT-populaatio)
| Parametri | AkoramidiisiN = 409 | LumelääkeN = 202 |
ACM:n ja CVH:n kumulatiivisen esiintyvyyden yhdistelmä Voittosuhde (95 %:n luottamusväli) F‑S1 p‑arvo | 1,464 (1,067, 2,009) p = 0,0182 | |
| Elossa olevien tutkittavien lukumäärä (%) kuukauden 30 kohdalla2 | 330 (80,7 %) | 150 (74,3 %) |
CVH-tutkittavien lukumäärä (%) CVH-tapahtumien kokonaismäärä CVH:n esiintyvyys vuodessa tutkittavaa kohden (keskiarvo)3 Suhteellinen riskisuhde4 p-arvo | 109 (26,7 %) 182 0,29 | 86 (42,6 %) 170 0,55 |
0,496 p < 0,0001 | ||
Lyhenteet: F-S = Finkelstein-Schoenfeld; ACM = mistä tahansa syystä johtuva kuolleisuus; CVH = sydän- ja verisuoniongelmista johtuva sairaalahoito; mITT = modifioitu hoitoaikomus
1 F-S-menetelmässä jokaista tutkittavaa verrataan pareittain kussakin komponentissa hierarkkisesti, ACM:sta kuolleisuudesta alkaen. Jos ACM-parivertailun tulos on tasapeli, seuraavaksi arvioidaan CVH.
2 Sydämensiirto ja sydämen mekaanisen tukilaitteen implantaatio katsotaan lähestyvän loppuvaiheen indikaattoreiksi. Näin ollen näitä tapahtumia käsitellään analyysissä samoin kuin kuolleita. Tästä syystä nämä tutkittavat eivät sisälly "elossa olevien tutkittavien lukumäärä kuukauden 30 kohdalla" -kohtaan, vaikka he olisivatkin elossa kuukauden 30 elossaolon seuranta-arvioinnin perusteella. Kaikkien tutkittavien elossaolostatus kuukauden 30 kohdalla oli tiedossa.
3 CVH kunkin tutkittavan osalta lasketaan seuraavalla kaavalla: (tutkittavalla todettujen CVH-tapausten kokonaismäärä) / (seurannan kesto vuosina). Se sisältää kliinisen mielenkiinnon kohteena olevat tapahtumat (EOCI). Kliinisen mielenkiinnon kohteena olevat tapahtumat määritellään < 24 tuntia kestäviksi lääkärikäynneiksi (esim. päivystys- tai muu osasto, kiireellisen hoidon klinikka, päiväsairaala) sydämen dekompensaation takia annettavan laskimonsisäisen diureettihoidon vuoksi.
4 Peräisin negatiivisesta binomiregressiomallista.
Kuva 1: Aika mistä tahansa syystä johtuvaan kuolleisuuteen tai ensimmäiseen sydän- ja verisuoniongelmista johtuvaan sairaalahoitoon
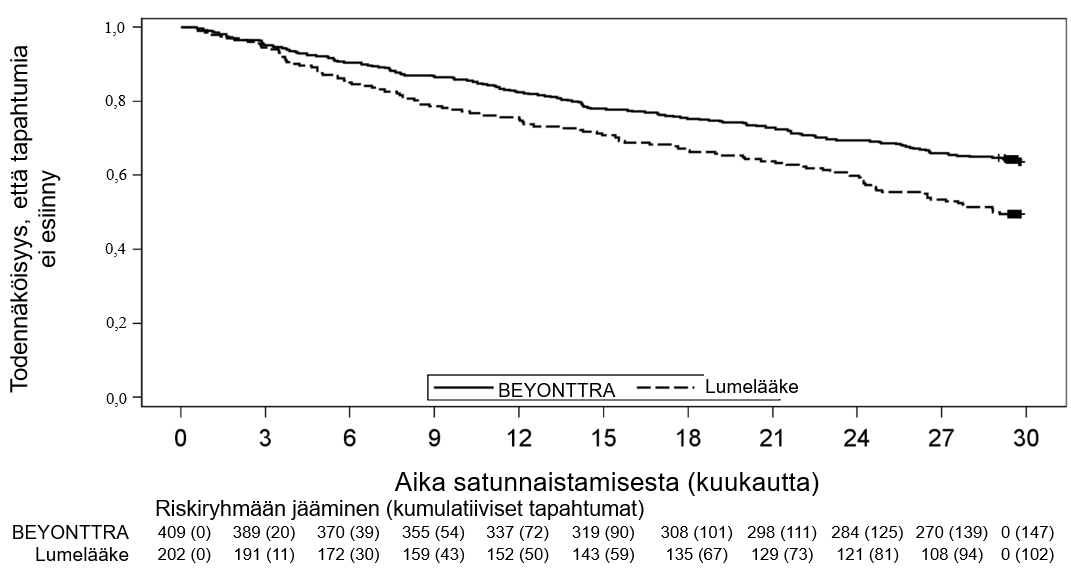
6 minuutin kävelytesti (6MWD) ja KCCQ
Akoramidiisin hoitovaikutusta toimintakykyyn ja terveydentilaan arvioitiin 6MWD-testillä ja KCCQ:n kokonaisyhteenvetopisteillä (KCCQ-OS); osa-alueina fyysiset rajoitukset, oireet, sosiaaliset rajoitukset ja elämänlaatu) (taulukko 4). Akoramidiisin hoitovaikutus lumelääkkeeseen verrattuna todettiin ensimmäisen kerran 6 minuutin kävelytestin tuloksissa kuukauden 18 kohdalla ja KCCQ-OS-kyselyn tuloksissa kuukauden 3 kohdalla, ja se säilyi kuukauden 30 loppuun asti.
Taulukko 4: 6MWD- ja KCCQ-OS-pisteet
| Päätetapahtumat* | Lähtötilanteen keskiarvo (SD) | Muutos lähtötilanteesta kuukauteen 30, LS-keskiarvo (SE) | Hoidon ero lumelääkkeen LS-keskiarvoon (96 %:n lv) | p-arvo | ||
Akoramidiisi N = 409 | Lumelääke N = 202 | Akoramidiisi N = 409 | Lumelääke N = 202 | |||
| 6 minuutin kävelytesti (metriä) | 362,78 (103,50) | 351,51 (93,83) | ‑64,65 (5,51) | ‑104,29 (7,77) | 39,64 (20,18, 59,10) | < 0,0001 |
| KCCQ-OS | 71,73 (19,37) | 70,48 (20,65) | ‑11,48 (1,18) | ‑21,42 (1,65) | 9,94 (5,79, 14,10) | < 0,0001 |
Lyhenteet: lv = luottamusväli; KCCQ-OS = Kansas Cityn kardiomyopatiakyselyn kokonaisyhteenvetopisteet, LS = pienin neliösumma, SD = keskihajonta, SE = keskivirhe
* Suuremmat arvot tarkoittavat parempaa terveydentilaa.
Alaryhmäanalyysi
ACM- ja CVH-komponentteihin sovelletun F‑S-testin tulokset (täydennettynä voittosuhteella) osoittivat johdonmukaisesti akoramidiisin olevan parempi lumelääkkeeseen verrattuna ositusparametrien (villityyppi tai variantti), NYHA-luokan ja National Amyloidosis Centren (NAC) ATTR -vaiheen alaryhmien osalta (kuva 2).
Kuva 2: Mistä tahansa syystä johtuvan kuolleisuuden ja sydän- ja verisuoniongelmista johtuvan sairaalahoidon hierarkkinen yhdistelmä, Finkelstein-Schoenfeld- ja voittosuhdetulokset kokonaisuutena ja alaryhmittäin (miTT-populaatio)1
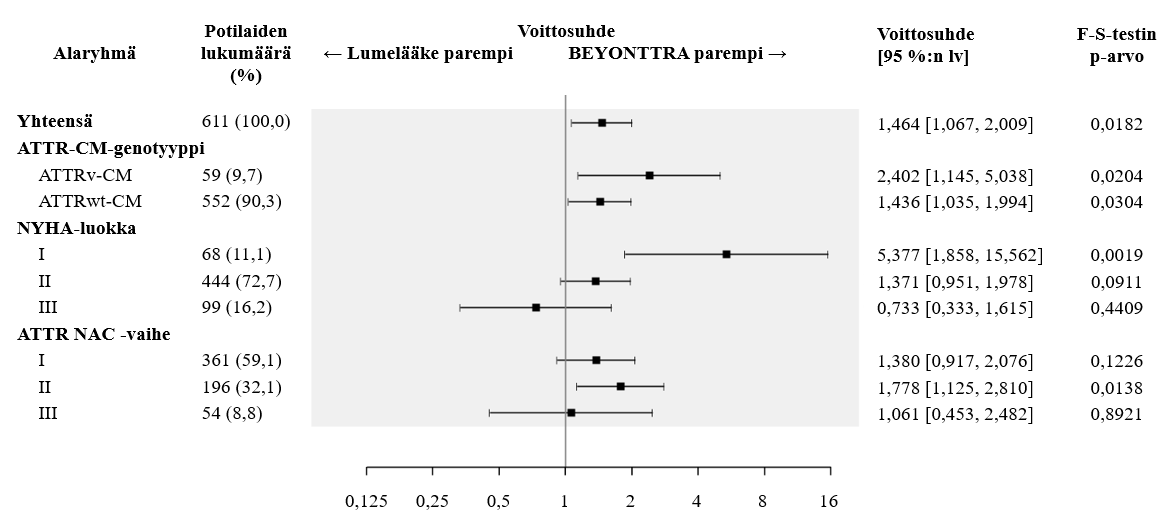
Lyhenteet: ACM = mistä tahansa syystä johtuva kuolleisuus; ATTRwt-CM = villityypin ATTR-CM; ATTRv‑CM = variantti ATTR-CM; CVH = sydän- ja verisuoniongelmista johtuva sairaalahoito; F‑S = Finkelstein-Schoenfeld; NAC = National Amyloidosis Centre (Lontoo, Yhdistynyt kuningaskunta); NYHA = New York Heart Association; NAC-vaihe I (NT-proBNP ≤ 3 000 pikog/ml ja eGFR ≥ 45 ml/min/1,73 m2), vaihe II (NT-proBNP ≤ 3 000 pikog/ml ja eGFR < 45 ml/min/1,73 m2 tai NT‑proBNP > 3 000 pikog/ml ja eGFR ≥ 45 ml/min/1,73 m2), vaihe III (NT-proBNP > 3 000 pikog/ml ja eGFR < 45 ml/min/1,73 m2)
1 Voittosuhde on niiden parien lukumäärä, joissa akoramidiisihoitoa saanut tutkittava "voitti", jaettuna niiden parien lukumäärällä, joissa lumehoitoa saanut tutkittava "voitti".
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset BEYONTTRA-valmisteen käytöstä ATTR‑CM:n hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Altistusparametrit (pitoisuus-aikakäyrän alle jäävä pinta-ala [AUC] ja enimmäispitoisuus [Cmax]) suurenivat vähemmän kuin suhteessa annokseen, kun lääkettä annettiin kerta-annoksia (enintään 1 780 mg) tai useita annoksia (enintään 712 mg) kahdesti vuorokaudessa.
Suun kautta annettu akoramidiisi imeytyy nopeasti, ja muuttumattomassa muodossa olevan akoramidiisin huippupitoisuus plasmassa saavutetaan yleensä 1 tunnin kuluessa. Plasman akoramidiisipitoisuuksien suurenemista todettiin annosvälillä 44,5 mg:sta kerran vuorokaudessa 712 mg:aan kerran vuorokaudessa. Plasman akoramidiisialtistukset näyttivät saturoituvan annosvälillä yli 712 mg:sta 1 068 mg:aan. Vakaa tila saavutetaan annostelun 10. päivään mennessä annoksella 712 mg kahdesti vuorokaudessa, ja toistuva annostelu aiheuttaa vähäistä (noin 1,3–1,6‑kertaista) akoramidiisin kertymistä.
Absoluuttista biologista hyötyosuutta ei tiedetä, mutta ihmisillä tehdyn ADME-tutkimuksen (imeytyminen, jakautuminen, metabolia, erittyminen) perusteella vähintään 75–80 % suun kautta annetusta 712 mg:n kerta-annoksesta imeytyy.
Ruoka ei vaikuta akoramidiisin kokonaisimeytymiseen.
Jakautuminen
Akoramidiisin näennäinen jakautumistilavuus vakaassa tilassa on 654 litraa annoksella 712 mg kahdesti vuorokaudessa. In vitro akoramidiisi sitoutuu ihmisen plasman proteiineihin 96,4‑prosenttisesti. Akoramidiisi sitoutuu ensisijaisesti TTR:ään.
Biotransformaatio
Akoramidiisin metaboliaa tutkittiin antamalla terveille vapaaehtoisille aikuisille suun kautta kerta-annos [14C]‑akoramidiisia. Akoramidiisi metaboloituu pääasiassa glukuronidaation kautta, ja pääasiallinen metaboliitti on akoramidiisi‑β‑D‑glukuronidi (akoramidiisi‑AG) (7,6 % verenkierrossa olevasta kokonaisradioaktiivisuudesta). Akoramidiisi‑AG on farmakologisesti noin 3 kertaa vähemmän aktiivinen kuin akoramidiisi, sen kovalenttinen sitoutuminen on vähäistä, eikä sillä ole farmakologisen aktiivisuuden kannalta merkityksellistä roolia.
Eliminaatio
Kerta-annoksen jälkeen akoramidiisin terminaalinen puoliintumisaika on noin 27 tuntia. Vakaassa tilassa akoramidiisin näennäinen oraalinen puhdistuma on 15,6 l/h.
Kun terveille vapaaehtoisille aikuisille annettiin suun kautta kerta-annos [14C]‑akoramidiisia, noin 34 % annoksen radioaktiivisuudesta erittyi ulosteeseen (pääasiassa akoramidiisina) ja noin 68 % virtsaan. Muuttumattomassa muodossa olevan akoramidiisin prosenttiosuus virtsassa oli < 10 %.
Erityisryhmät
Akoramidiisin farmakokinetiikassa ei todettu ikään (18,0–89,3 vuotta), rotuun / etniseen alkuperään (mukaan lukien japanilaiset ja ei-japanilaiset), sukupuoleen tai munuaisten vajaatoimintaan (eGFR 25,4–157 ml/min/1,73 m2) perustuvia kliinisesti merkittäviä eroja.
Populaatiofarmakokineettisen mallinnuksen perusteella akoramidiisin AUC-arvo oli vakaassa tilassa 37 % suurempi terveillä tutkittavilla kuin potilailla. Lisäksi valkoihoisiin tutkittaviin verrattuna vakaan tilan AUC-arvo oli 23 % suurempi tummaihoisilla tutkittavilla ja 38 % suurempi ei-valkoihoisilla, ei-tummaihoisilla tutkittavilla. Nämä vaikutukset ovat yksilöiden välisen vaihtelun rajoissa (CV = 38 %). Mallin ennusteen mukaan akoramidiisin farmakokinetiikassa ei myöskään ole painoon perustuvia kliinisesti merkittäviä eroja 50,9–133 kg:n painoalueella.
Erityisesti munuaisten vajaatoimintaa koskevaa tutkimusta ei ole tehty, koska akoramidiisi ei eliminoidu merkittävästi munuaisteitse. Vaikka päämetaboliitilla (akoramidiisi-AG) ei ole kliinisesti merkityksellistä vaikutusta farmakologiseen aktiivisuuteen tutkitussa populaatiossa, tiedot vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) ovat rajalliset, eikä tietoja ole lainkaan dialyysipotilaista. Vaikea munuaisten vajaatoiminta voi vaikuttaa akoramidiisimetaboliitin akoramidiisi-AG:n puhdistumaan, mikä voi johtaa mahdollisesti suurempaan akoramidiisi-AG:n systeemiseen altistumiseen. Vaikka tämän mahdollisesti suuremman akoramidiisi-AG:n altistuksen ei oleteta vaikuttavan kliinisesti merkityksellisesti farmakologiseen aktiivisuuteen, akoramidiisia tulee käyttää varoen vaikeaa munuaisten vajaatoimintaa sairastaville potilaille.
Akoramidiisia ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, karsinogeenisuutta sekä lisääntymis- ja kehitystoksisuutta (hedelmällisyyttä sekä alkion- ja sikiönkehitystä) koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Rotilla tehdyssä pre- ja postnataalista kehitystä koskevassa akoramidiisitutkimuksessa havaittiin poikasten eloonjäämisen heikkenemistä, poikasten painon laskua ja oppimishäiriöitä, kun emolle annettiin tiineyden ja imetyksen aikana akoramidiisia annoksena 1 000 mg/kg/vrk. Tällä annoksella havaittiin myös emoon kohdistuvaa vaikeaa toksisuutta, mukaan lukien kuolleisuutta ja painonlaskua organogeneesin aikana. Rotilla tehdyssä pre- ja postnataalista kehitystoksisuutta selvittäneessä tutkimuksessa tasoksi, jolla ei todettu haittavaikutuksia (NOAEL), määritettiin testattu akoramidiisiannos 350 mg/kg/vrk (AUC-arvot olivat noin 21‑kertaiset verrattuna ihmisen altistukseen kliinisellä akoramidiisiannoksella).
Eläimillä ei tehty istukan läpi kulkeutumista eikä maitoon erittymistä koskevia tutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa (E 460)
Kroskarmelloosinatrium (E 468)
Kolloidinen hydratoitu piidioksidi (E 551)
Magnesiumstearaatti (E 470b)
Kalvopäällyste
Makrogolipolyvinyylialkoholi-oksaskopolymeeri (E 1209)
Talkki (E 553b)
Titaanidioksidi (E 171)
Glyserolimonokaprylokapraatti, (tyyppi I) (E 471)
Polyvinyylialkoholi (E 1203)
Painomuste
Musta rautaoksidi (E 172)
Propyleeniglykoli (E 1520)
Hypromelloosi 2910 (E 464)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BEYONTTRA tabletti, kalvopäällysteinen
356 mg (L:ei) 120 fol (9828,10 €)
PF-selosteen tieto
Läpipainopakkaukset – lämpömuovatut kaksionteloiset PVC/PCTFE-läpipainoliuskat, joissa on alumiinifoliokansi.
Pakkauskoot: 120 tablettia 6 läpipainoliuskassa, joissa kussakin on 10 onteloa (2 tablettia onteloa kohden).
Valmisteen kuvaus:
Valkoisia, soikeita, kalvopäällysteisiä tabletteja, joiden koko on noin 15 mm × 7,5 mm ja joiden toiselle puolelle on painettu mustalla musteella ”BEYONTTRA”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BEYONTTRA tabletti, kalvopäällysteinen
356 mg 120 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Akoramidiisi ja tafamidiisi: Transtyretiinivälitteisen amyloidoosin hoito erityisin edellytyksin (3011).
ATC-koodi
C01EB25
Valmisteyhteenvedon muuttamispäivämäärä
03.11.2025
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi