
RUBRACA tabletti, kalvopäällysteinen 200 mg, 250 mg, 300 mg
Vaikuttavat aineet ja niiden määrät
Rubraca 200 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää rukaparibikamsylaattia määrän, joka vastaa 200 mg rukaparibia.
Rubraca 250 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää rukaparibikamsylaattia määrän, joka vastaa 250 mg rukaparibia.
Rubraca 300 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää rukaparibikamsylaattia määrän, joka vastaa 300 mg rukaparibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti.
Kliiniset tiedot
Käyttöaiheet
Rubraca on tarkoitettu pitkälle edenneen (FIGO-asteet III ja IV) korkea-asteisen epiteliaalisen munasarjasyövän, munanjohtimen syövän tai primaarisen vatsakalvosyövän ylläpitohoidoksi monoterapiana aikuispotilailla, jotka ovat saavuttaneet platinapohjaisen ensilinjan solunsalpaajahoidon päätyttyä (täydellisen tai osittaisen) hoitovasteen.
Rubraca on tarkoitettu platinaherkän uusiutuneen korkea-asteisen epiteliaalisen munasarjasyövän, munanjohtimen syövän tai primaarisen vatsakalvosyövän ylläpitohoidoksi monoterapiana aikuispotilailla, jotka ovat saavuttaneet platinapohjaisella solunsalpaajahoidolla (täydellisen tai osittaisen) hoitovasteen.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Rubraca-hoito on aloitettava ja sitä on valvottava lääkärin toimesta, joka on perehtynyt syöpälääkevalmisteiden käyttöön.
Annostus
Suositeltu annos Rubraca-valmistetta on 600 mg kahdesti vuorokaudessa, joka vastaa päivittäistä 1 200 mg:n annosta.
Potilaan tulisi aloittaa Rubraca-ylläpitohoito viimeistään 8 viikon kuluessa platinapohjaisen solunsalpaajahoidon viimeisen annoksen ottamisesta.
Hoidon kesto
Pitkälle edenneen munasarjasyövän ensilinjan ylläpitohoito:
Potilaan hoitoa voidaan jatkaa, kunnes tauti etenee tai ilmaantuu ei-hyväksyttävissä olevaa toksisuutta, tai kunnes hoitoa on annettu 2 vuoden ajan.
Platinaherkän uusiutuneen munasarjasyövän ylläpitohoito:
Potilaan hoitoa voidaan jatkaa, kunnes tauti etenee tai ilmaantuu ei-hyväksyttävissä olevaa toksisuutta.
Mikäli potilas oksentaa Rubraca-valmisteen ottamisen jälkeen, potilaan ei pidä ottaa annosta uudelleen, vaan hänen tulisi ottaa seuraava aikataulun mukainen annos.
Väliin jääneet annokset
Mikäli annos jää väliin, potilaan tulisi jatkaa Rubraca-valmisteen ottamista seuraavan aikataulun mukaisen annoksen kohdalla.
Annoksen muuttaminen haittavaikutusten myötä
Haittavaikutuksia voidaan hoitaa keskeyttämällä annostelu ja/tai vähentämällä annosta kohtalaisten tai vaikea-asteisten haittavaikutusten (CTCAE:n aste 3 tai 4), kuten neutropenian, anemian ja trombosytopenian, vuoksi.
Maksan transaminaasiarvojen kohoamista (aspartaattiaminotransferaasi (ASAT) ja/tai
alaniiniaminotransferaasi (ALAT)) esiintyy hoidon alkuvaiheessa ja se on yleensä ohimenevää. Asteen 1–3 ASAT/ALAT-arvojen kohoamista voidaan sietää ilman rukaparibiannoksen muuttamista, tai muuttamalla hoitoa (keskeyttämällä ja/tai annosta vähentämällä). Asteen 4 haittavaikutukset edellyttävät hoidon muuttamista (ks. taulukko 2).
Muita kohtalaisia tai vaikea-asteisia ei-hematologisia haittavaikutuksia, kuten pahoinvointia ja oksentelua, voidaan hallita keskeyttämällä annostelu ja/tai vähentämällä annosta, mikäli ne eivät ole riittävästi hallinnassa sopivalla oireenmukaisella hoidolla.
Taulukko 1. Suositellut annosmuutokset
Annoksen vähentäminen | Annos |
Aloitusannos | 600 mg kahdesti vuorokaudessa (kaksi 300 mg:n tablettia kahdesti vuorokaudessa) |
Ensimmäinen annoksen vähentäminen | 500 mg kahdesti vuorokaudessa (kaksi 250 mg:n tablettia kahdesti vuorokaudessa) |
Toinen annoksen vähentäminen | 400 mg kahdesti vuorokaudessa (kaksi 200 mg:n tablettia kahdesti vuorokaudessa) |
Kolmas annoksen vähentäminen | 300 mg kahdesti vuorokaudessa (yksi 300 mg:n tabletti kahdesti vuorokaudessa) |
Taulukko 2: Hoidosta aiheutuvien suurentuneiden ASAT-/ALAT-arvojen hoito
ASAT-/ALAT-arvojen kohoamisen aste | Hoito |
Aste 3, ilman muita merkkejä maksan vajaatoiminnasta | Maksan toimintaa seurataan viikoittain, kunnes aste on ≤ 2 Jatketaan rukaparibihoitoa edellyttäen, että bilirubiini on < ULN ja alkalinen fosfataasi on < 3 × ULN Hoito keskeytetään, jos ASAT-/ALAT-arvot eivät laske 2 viikon aikana asteeseen ≤ 2, minkä jälkeen rukaparibihoitoa jatketaan samalla tai pienemmällä annoksella |
Aste 4 | Rukaparibihoito keskeytetään, kunnes arvot palautuvat asteeseen ≤ 2, minkä jälkeen rukaparibihoitoa jatketaan pienemmällä annoksella ja maksan toimintaa seurataan viikoittain 3 viikon ajan |
Erityiset potilasryhmät
Iäkkäät
Aloitusannoksen muuttamista ei suositella iäkkäillä potilailla (≥ 65-vuotiaat, ks. kohdat Haittavaikutukset ja Farmakokinetiikka). Joidenkin iäkkäiden (≥ 65-vuotiaiden) potilaiden suurempaa herkkyyttä haittatapahtumille ei voida poissulkea. On olemassa vain vähän kliinisiä tietoja 75-vuotiaista ja sitä iäkkäämmistä potilaista.
Maksan vajaatoiminta
Aloitusannoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka). Potilaita, joilla on keskivaikea maksan vajaatoiminta, on seurattava tarkoin maksan toiminnan osalta ja haittavaikutusten varalta. Kliinisiä tietoja potilaista, joilla on vaikea-asteinen maksan vajaatoiminta (ts. kokonaisbilirubiini > 3 kertaa viitealueen yläraja [ULN]) ei ole olemassa. Sen vuoksi rukaparibin käyttöä ei suositella potilaille, joilla on vaikea-asteinen maksan vajaatoiminta.
Munuaisten vajaatoiminta
Aloitusannoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä tai kohtalainen munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka). Ei ole olemassa kliinisiä tietoja potilaista, joilla on vaikea-asteinen munuaisten vajaatoiminta (kreatiniinipuhdistuma alle 30 ml/min). Sen vuoksi rukaparibin käyttöä ei suositella potilaille, joilla on vaikea-asteinen munuaisten vajaatoiminta. Rukaparibia voidaan käyttää vaikea-asteista munuaisten vajaatoimintaa sairastaville potilaille vain siinä tapauksessa, että mahdollinen hyöty on haittaa suurempi. Potilaita, joilla on kohtalainen tai vaikea-asteinen munuaisten vajaatoiminta on seurattava tarkoin munuaisten toiminnan osalta ja haittavaikutusten varalta.
Pediatriset potilaat
Rubraca-valmisteen turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Rubraca on tarkoitettu käytettäväksi suun kautta ja se voidaan ottaa ruoan kanssa tai ilman ruokaa. Annokset tulisi ottaa noin 12 tunnin välein. Ks. kohta Farmakokinetiikka.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille. Imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Hematologinen toksisuus
Rukaparibihoidon aikana voidaan havaita myelosuppression löydöksiä (anemia, neutropenia, trombosytopenia) ja niitä havaitaan tyypillisesti ensimmäisen kerran 8–10 viikon rukaparibihoidon jälkeen. Nämä reaktiot ovat hallittavissa normaalilla lääkehoidolla ja/tai vaikea-asteisemmissa tapauksissa annosta muuttamalla. Täydellisen verenkuvan testausta suositellaan ennen Rubraca-hoidon aloittamista ja kuukausittain sen jälkeen. Potilaan ei pidä aloittaa Rubraca-hoitoa, ennen kuin hän on toipunut aiemman solunsalpaajahoidon aiheuttamasta hematologisesta toksisuudesta (≤ CTCAE:n aste
1).
Tukihoito on otettava käyttöön ja paikallisia ohjeita on noudatettava vähäisten verisolujen määrien, anemian ja neutropenian hoidossa. Rubraca-valmisteen käyttö on keskeytettävä tai annosta on vähennettävä taulukon 1 mukaisesti (ks. kohta Annostus ja antotapa) ja verisolujen määriä on seurattava viikoittain toipumiseen saakka. Mikäli pitoisuudet eivät ole palautuneet CTCAE:n asteelle 1 tai sitä parempaan tilanteeseen 4 viikon jälkeen, potilas on lähetettävä hematologin jatkotutkimuksiin.
Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia
Myelodysplastista oireyhtymää / akuuttia myelooista leukemiaa (MDS/AML), kuolemaan johtaneet tapaukset mukaan lukien, on ilmoitettu esiintyneen rukaparibia saaneilla potilailla. Rukaparibihoidon kesto potilailla, joille kehittyi MDS/AML, vaihteli < 2 kuukaudesta noin 6 vuoteen.
Mikäli MDS:ää/AML:aa epäillään, potilas on lähetettävä hematologin jatkotutkimuksiin, ml.
luuydintutkimukseen ja verinäytteen ottoon sytogeneettistä tutkimusta varten. Mikäli pitkittyneen hematologisen toksisuuden tutkimisen jälkeen todetaan MDS/AML, Rubraca-valmisteen käyttö on lopetettava.
Valoherkkyys
Valoherkkyyttä on havaittu rukaparibihoitoa saavilla potilailla. Potilaiden on vältettävä suorassa auringonvalossa oloa, koska he voivat palaa herkemmin rukaparibihoidon aikana. Ulkona ollessaan potilaiden on pidettävä päähinettä ja suojaavia vaatteita sekä käytettävä aurinkovoidetta ja huulivoidetta, joiden aurinkosuojakerron (SPF) on vähintään 50.
Ruoansulatuselimistön toksisuus
Ruoansulatuselimistön toksisuudesta (pahoinvointi ja oksentelu) on ilmoitettu usein rukaparibihoidon yhteydessä. Se on yleensä matala-asteista (CTCAE:n aste 1 tai 2), ja sitä voidaan hallita annosta vähentämällä (ks. taulukko 1) tai keskeyttämällä hoito; antiemeettejä, kuten 5-HT3:n antagonisteja, deksametasonia, aprepitanttia ja fosaprepitanttia, voidaan käyttää pahoinvoinnin/oksentelun hoitona ja niiden ennaltaehkäisevää käyttöä voidaan myös harkita ennen Rubraca-valmisteen käytön aloittamista. On tärkeää hoitaa nämä tapahtumat ennakoivasti, jotta vältytään pitkittyneeltä tai vaikeammalta pahoinvoinnilta/oksentelulta, jotka voivat mahdollisesti johtaa komplikaatioihin, kuten nestehukkaan tai sairaalahoitoon.
Suolitukos
Suolitukostapauksia on havaittu munasarjasyöpää sairastavilla potilailla rukaparibin kliinisissä tutkimuksissa; 3,5 %:lla rukaparibihoitoa saaneista potilaista ilmeni vakava suolitukostapahtuma.
Kuolemaan johtaneita tapauksia todettiin yhdellä rukaparibihoitoa saaneella potilaalla (alle 0,1 %).
Taustasairaus saattaa vaikuttaa suolitukoksen kehittymiseen munasarjasyöpää sairastavilla potilailla. Jos suolitukosta epäillään, diagnoosi on tehtävä viipymättä ja potilas hoidettava asianmukaisesti.
Alkio- ja sikiötoksisuus
Rubraca-valmiste voi aiheuttaa vaurioita sikiölle, kun sitä annetaan raskaana olevalle naiselle, sen vaikutusmekanismin ja eläinkokeiden löydösten perusteella. Eläimillä tehdyssä
lisääntymistutkimuksessa rukaparibin anto raskaana oleville rotille organogeneesin aikana aiheuttaa alkio- ja sikiötoksisuutta altistuksilla, jotka olivat pienempiä kuin suositeltua ihmisen annosta, 600 mg kahdesti vuorokaudessa, saavilla potilailla (ks. kohta Prekliiniset tiedot turvallisuudesta).
Raskaus/raskaudenehkäisy
Raskaana oleville naisille on kerrottava mahdollisesta sikiöön kohdistuvasta riskistä. Naisia, jotka voivat tulla raskaaksi, on kehotettava käyttämään tehokasta raskaudenehkäisyä hoidon aikana ja viimeisen Rubraca-annoksen jälkeisten 6 kuukauden aikana (ks. kohta Raskaus ja imetys). Lisääntymisikäisillä naisilla suositellaan raskaustestin tekemistä ennen hoidon aloittamista.
Apuaineet
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset rukaparibiin
Rukaparibin metaboliasta vastaavia entsyymejä ei ole yksilöity. In vitro-tietojen perusteella CYP2D6entsyymi, ja vähemmässä määrin CYP1A2- ja CYP3A4-entsyymit, pystyvät metaboloimaan rukaparibia. Vaikka CP3A4-välitteinen rukaparibin metabolia oli hidasta in vitro, merkittävää CYP3A4:n vaikutusta ei voida poissulkea in vivo. Varovaisuutta on noudatettava käytettäessä samanaikaisesti voimakkaita CYP3A4:n estäjiä tai induktoreita.
Rukaparibin osoitettiin olevan P-gp:n ja BCRP:n substraatti in vitro. P-gp:n ja BCRP:n estäjien vaikutusta rukaparibin farmakokinetiikkaan ei voida sulkea pois. Varovaisuutta suositellaan, kun rukaparibia annetaan samanaikaisesti sellaisten lääkevalmisteiden kanssa, jotka ovat voimakkaita Pgp:n estäjiä.
Rukaparibin vaikutukset muihin lääkevalmisteisiin
Syöpäpotilailla tehdyissä lääkevalmisteiden yhteisvaikutustutkimuksissa 600 mg:n kahdesti vuorokaudessa annetulla annoksella käytetyn vakaan tilan rukaparibin vaikutuksia CYP1A2-, CYP2C9-, CYP2C19- ja CYP3A-entsyymeihin, BCRP:hen sekä P-glykoproteiiniin arvioitiin suun kautta otettavilla herkillä koeaineilla (kofeiini, S-varfariini, omepratsoli, midatsolaami, rosuvastatiini ja digoksiini). Rukaparibin vaikutusta yhdistelmäehkäisytabletin (etinyyliestradiolin ja levonorgestreelin) farmakokinetiikkaan arvioitiin myös. Tiedot viittaavat siihen, että rukaparibi on kohtalainen CYP1A2-entsyymin estäjä ja vähäinen CYP2C9-, CYP2C19- ja CYP3A-entsyymien estäjä. Rukaparibi estää myös vähäisessä määrin P-glykoproteiinia ja on BCRP:n heikko estäjä suolessa.
CYP1A2:n substraatit
Rukaparibilla ei havaittu olevan vaikutusta kofeiinin Cmax-pitoisuuteen. Se nosti kohtalaisesti kofeiinin AUCinf-arvoa, 2,55-kertaiseksi (90 %:n luottamusväli: 2,12; 3,08). Kun CYP1A2:n metaboloimia lääkevalmisteita annetaan samanaikaisesti, varsinkin lääkkeitä, joilla on kapea terapeuttinen leveys (esim. titsanidiini, teofylliini), annoksen muuttamista voidaan harkita asianmukaisen kliinisen seurannan perusteella.
CYP2C9:n substraatit
Rukaparibi nosti S-varfariinin Cmax-pitoisuutta 1,05-kertaiseksi (90 %:n luottamusväli: 0,99–1,12) ja
AUC0-96 h-arvoa 1,49-kertaiseksi (90 %:n luottamusväli: 1,40–1,58). Annettaessa samanaikaisesti lääkevalmisteita, jotka ovat CYP2C9:n substraatteja ja joilla on kapea terapeuttinen leveys (esim. varfariini, fenytoiini), annoksen muuttamista voidaan harkita, jos se on kliinisesti aiheellista. Varovaisuutta on noudatettava ja ylimääräistä International Normalised Ratio (INR)-arvon seurantaa on harkittava annettaessa samanaikaisesti varfariinia. Fenytoiinin terapeuttisen lääkepitoisuuden seurantaa on harkittava, jos sitä käytetään samanaikaisesti rukaparibin kanssa.
CYP2C19:n substraatit
Rukaparibi nosti omepratsolin Cmax-pitoisuutta 1,09-kertaiseksi (90 %:n luottamusväli: 0,93–1,27) ja AUCinf-arvoa 1,55-kertaiseksi (90 %:n luottamusväli: 1,32–1,83). Yhtäaikaisesti annettujen protonipumppuestäjien kliinisesti merkittävän vaikutuksen riski on todennäköisesti pieni (ks. kohta Farmakokinetiikka). Annoksen muuttamisen ei katsota olevan tarpeen niiden samanaikaisesti annettavien lääkevalmisteiden osalta, jotka ovat CYP2C19:n substraatteja.
CYP3A:n substraatit
Rukaparibi nosti midatsolaamin Cmax-pitoisuutta 1,13-kertaiseksi (90 %:n luottamusväli: 0,95–1,36) ja AUCinf-arvoa 1,38-kertaiseksi (90 %:n luottamusväli: 1,13–1,69). Varovaisuutta suositellaan annettaessa samanaikaisesti lääkevalmisteita, jotka ovat CYP3A:n substraatteja ja joilla on kapea terapeuttinen leveys (esim. alfentaniili, astemitsoli, sisapridi, syklosporiini, dihydroergotamiini, ergotamiini, fentanyyli, pimotsidi, kinidiini, sirolimuusi, takrolimuusi, terfenadiini). Annoksen muuttamista voidaan harkita, jos se on kliinisesti aiheellista havaittujen haittavaikutusten perusteella.
Suun kautta otettavat ehkäisyvalmisteet
Rukaparibi nosti etinyyliestradiolin Cmax-pitoisuutta 1,09-kertaiseksi (90 %:n luottamusväli: 0,94– 1,27) ja AUClast-arvoa 1,43-kertaiseksi (90 %:n luottamusväli: 1,15–1,77). Rukaparibi nosti levonorgestreelin Cmax-pitoisuutta 1,19-kertaiseksi (90 %:n luottamusväli: 1,00–1,42) ja AUClast-arvoa 1,56-kertaiseksi (90 %:n luottamusväli: 1,33–1,83). Annoksen muuttamista ei suositella yhteiskäytössä suun kautta otettavien ehkäisyvalmisteiden kanssa.
BCRP:n substraatit
Rukaparibi nosti rosuvastatiinin Cmax-pitoisuutta 1,29-kertaiseksi (90 %:n luottamusväli: 1,07–1,55) ja AUCinf-arvoa 1,35-kertaiseksi (90 %:n luottamusväli: 1,17–1,57). Annoksen muuttamista ei suositella yhteiskäytössä lääkevalmisteiden kanssa, jotka ovat BCRP:n substraatteja.
P-gp:n substraatit
Rukaparibilla ei havaittu olevan vaikutusta digoksiinin Cmax-pitoisuuteen. Se nosti vähäisessä määrin AUC0-72 h-arvoa, 1,20-kertaiseksi (90 %:n luottamusväli: 1,12–1,29). Annoksen muuttamista ei suositella niiden samanaikaisesti annettavien lääkevalmisteiden osalta, jotka ovat P-gp:n substraatteja.
Rukaparibin yhteisvaikutusta muiden entsyymien ja kuljettajaproteiinin kanssa arvioitiin in vitro. Rukaparibi on heikko CYP2C8:n, CYP2D6:n ja UGT1A1:n estäjä. Rukaparibi vaimennussääteli CYP2B6-entsyymiä ihmisen hepatosyyteissä kliinisesti merkittävillä altistuksilla. Rukaparibi on voimakas MATE1:n ja MATE2-K:n estäjä, kohtalainen OCT1:n estäjä, ja heikko OCT2:n estäjä. Koska näiden kuljettajaproteiinien esto voi vähentää metformiinin eliminaatiota munuaisista ja vähentää metformiinin maksaan ottoa, varovaisuutta suositellaan annettaessa metformiinia samanaikaisesti rukaparibin kanssa. Rukaparibin aiheuttaman UGT1A1:n eston kliininen merkitys ei ole tiedossa. Varovaisuutta on noudatettava, kun rukaparibia annetaan samanaikaisesti UGT1A1:n substraattien (irinotekaani) kanssa potilailla, joilla on UGT1A1*28 (heikko metaboloija) mahdollisesta SN-38:lle (irinotekaanin aktiivinen metaboliitti) altistumisen lisääntymisestä ja siihen liittyvistä toksisuuksista johtuen.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/raskaudenehkäisy naisilla
Naisia, jotka voivat tulla raskaaksi, on neuvottava välttämään raskaaksi tulemista rukaparibia saadessaan. Potilaita on kehotettava käyttämään tehokasta raskaudenehkäisyä hoidon aikana ja 6 kuukauden ajan viimeisen rukaparibiannoksen jälkeen (ks. kohta Yhteisvaikutukset).
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja rukaparibin käytöstä raskaana olevilla naisilla. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Rukaparibin vaikutusmekanismin ja prekliinisten tietojen perusteella se voi vahingoittaa sikiötä annettaessa sitä raskaana olevalle naiselle. Rubraca-valmistetta ei pidä käyttää raskauden aikana, ellei naisen kliininen tila edellytä rukaparibihoitoa. Lisääntymisikäisillä naisilla suositellaan raskaustestin tekemistä ennen hoidon aloittamista.
Imetys
Rukaparibin erittymisestä rintamaitoon ei ole tehty eläinkokeita. Ei tiedetä, erittyykö/erittyvätkö rukaparibi tai sen metaboliitit ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Rubraca-valmistetta ei pidä käyttää imetyksen aikana.
Imetys on vasta-aiheista Rubraca-hoidon aikana ja kaksi viikkoa viimeisen annoksen jälkeen johtuen rukaparibin mahdollisista vakavista haittavaikutuksista imeväisikäisillä lapsilla (ks. kohta Vasta-aiheet).
Hedelmällisyys
Rukaparibin vaikutuksesta hedelmällisyyteen ei ole olemassa tietoja. Eläinkokeiden perusteella rukaparibin käyttöön liittyvää vaikutusta hedelmällisyyteen ei voida poissulkea (ks. kohta Prekliiniset tiedot turvallisuudesta). Tämän lisäksi rukaparibi voi vaikuttaa ihmisen hedelmällisyyteen vaikutusmekanisminsa perusteella.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Rubraca-valmisteella on vain vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Varovaisuutta suositellaan ajaessa tai käytettäessä koneita niiden potilaiden osalta, jotka ilmoittavat uupumuksesta, pahoinvoinnista tai heitehuimauksesta Rubraca-hoidon aikana (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Rukaparibin yleinen turvallisuusprofiili perustuu tietoihin kliinisistä tutkimuksista, joissa 1 594 munasarjasyöpää sairastavaa potilasta sai rukaparibia monoterapiana. Potilaiden rukaparibialtistuksen mediaanikesto oli 7,4 kuukautta.
Haittavaikutuksia, joita esiintyi ≥ 20 %:lla rukaparibia saaneista potilaista, olivat pahoinvointi, uupumus/astenia, oksentelu, anemia, vatsakipu, makuhäiriö, ALAT-arvon kohoaminen, ASAT-arvon kohoaminen, ruokahalun heikkeneminen, ripuli, neutropenia ja trombosytopenia. Suurin osa haittavaikutuksista oli lieviä tai kohtalaisia (aste 1 tai 2).
Vähintään asteen 3 haittavaikutuksia, joita esiintyi > 5 %:lla potilaista, olivat anemia (25 %), ALATarvon kohoaminen (10 %), neutropenia (10 %), uupumus/astenia (9 %) ja trombosytopenia (7 %). Ainoa vakava haittavaikutus, joka ilmeni > 2 %:lla potilaista, oli anemia (5 %).
Haittavaikutukset, jotka johtivat yleisimmin annoksen vähentämiseen tai hoidon keskeyttämiseen, olivat anemia (23 %), uupumus/astenia (15 %), pahoinvointi (14 %), trombosytopenia (14 %), neutropenia (10 %) ja ASAT-/ALAT-arvon kohoaminen (10 %). Haittavaikutuksia, jotka johtivat hoidon lopettamiseen, esiintyi 15 %:lla potilaista, ja niistä yleisimmin raportoituja olivat trombosytopenia, pahoinvointi, anemia ja uupumus/astenia.
Taulukoitu luettelo haittavaikutuksista
Haittavaikutusten esiintymistiheys on lueteltu MedDRA-järjestelmän elinjärjestelmäluokkien perusteella suositellun termin tasolla. Haittavaikutusten esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 3. Taulukoitu luettelo haittavaikutuksista MedDRA:n elinjärjestelmäluokan mukaan jaoteltuna
Haittavaikutukset | ||
MedDRA- elinjärjestelmäluokka | Kaikkien CTCAE-asteiden esiintymistiheys | CTCAE-asteen 3 ja sitä korkeampien asteiden esiintymistiheys |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Yleinen Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia a | Yleinen Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia a |
Veri ja imukudos | Hyvin yleinen Anemia b, trombosytopenia b, neutropenia b, leukopenia b Yleinen Lymfopenia b, kuumeinen neutropenia | Hyvin yleinen Anemia b, neutropenia b Yleinen Trombosytopenia b, kuumeinen neutropenia, leukopenia b, lymfopenia b |
Immuunijärjestelmä | Yleinen Yliherkkyys c | Melko harvinainen Yliherkkyys c |
Aineenvaihdunta ja ravitsemus | Hyvin yleinen Ruokahalun heikkeneminen, veren kreatiniiniarvon suureneminen b, hyperkolesterolemia b Yleinen Kuivuminen | Yleinen Ruokahalun heikkeneminen, kuivuminen, hyperkolesterolemia b Melko harvinainen Veren kreatiniiniarvon suureneminen b |
Hermosto | Hyvin yleinen Makuhäiriö, heitehuimaus | Melko harvinainen Makuhäiriö, heitehuimaus |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen Hengenahdistus | Melko harvinainen Hengenahdistus |
Ruoansulatuselimistö | Hyvin yleinen Pahoinvointi, oksentelu, ripuli, dyspepsia, vatsakipu Yleinen Suolitukos d, stomatiitti | Yleinen Pahoinvointi, oksentelu, ripuli, vatsakipu, suolitukos d Melko harvinainen Dyspepsia, stomatiitti |
Maksa ja sappi | Hyvin yleinen Alaniiniaminotransferaasiarvon suureneminen, aspartaattiaminotransferaasiarvon suureneminen Yleinen Transaminaasiarvojen suureneminen b | Hyvin yleinen Alaniiniaminotransferaasiarvon suureneminen, aspartaattiaminotransferaasiarvon suureneminen Melko harvinainen Transaminaasiarvojen suureneminen b |
Iho ja ihonalainen kudos | Hyvin yleinen Valoherkkyysreaktio, ihottuma Yleinen Makulopapulaarinen ihottuma, palmoplantaarisen erytrodysestesian oireyhtymä, eryteema | Melko harvinainen Valoherkkyysreaktio, ihottuma, makulopapulaarinen ihottuma, palmoplantaarisen erytrodysestesian oireyhtymä |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen Uupumus e, kuume | Yleinen Uupumus e Melko harvinainen Kuume |
- MDS:n/AML:n esiintyvyys perustuu niiden 3 025 potilaan kokonaispopulaatioon, jotka saivat vähintään yhden suun kautta otettavan rukaparibiannoksen.
- Sisältää laboratoriolöydökset c Yleisimmin havaittuja tapahtumia ovat yliherkkyys, lääkeyliherkkyys ja kasvojen ja silmien turvotus.
d Sisältää suolitukoksen, paksusuolen tukoksen ja ohutsuolen tukoksen e Sisältää uupumuksen, astenian ja letargian
Valikoitujen haittavaikutusten kuvaukset
Hematologinen toksisuus
Kaikilla CTCAE-asteilla olevia seuraavia hematologisia haittavaikutuksia ilmoitettiin: anemia (46 %), trombosytopenia (26 %) ja neutropenia (21 %). Anemia ja trombosytopenia johtivat hoidon lopettamiseen 2 %:lla ja 1 %:lla potilaista. Vähintään CTCAE:n asteella 3 olevia haittavaikutuksia esiintyi 25 %:lla (anemia), 10 %:lla (neutropenia) ja 7 %:lla (trombosytopenia) potilaista. Vähintään asteella 3 olevat myelosuppressiohaittavaikutukset alkoivat yleensä myöhemmässä hoidon vaiheessa (2 tai useamman kuukauden kuluttua). Katso riskin vähentämis- ja hoitotoimenpiteet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia
MDS/AML ovat vakavia haittavaikutuksia, joita esiintyi melko harvoin (0,5 %) hoitoa saavilla potilailla ja 28 vuorokauden turvallisuuden seurannassa ja yleisesti (1,1 %) kaikilla potilailla, mukaan lukien pitkän turvallisuuden seurannan aikana (esiintymistiheys on laskettu niiden 3 025 potilaan kokonaisturvallisuuspopulaation perusteella, jotka altistuivat vähintään yhdelle suun kautta otettavalle rukaparibiannokselle kaikissa kliinisissä tutkimuksissa). MDS:n/AML:n ilmaantuvuus hoidon aikana rukaparibia saaneilla potilailla oli 1,6 % lumekontrolloidussa faasin 3 tutkimuksessa ARIEL3 ja 0,5 % lumekontrolloidussa faasin 3 tutkimuksessa ATHENA-MONO. Vaikka hoidonaikaisista tapauksista ei ilmoitettu lumelääkettä saaneilla potilailla, kuudesta tapauksesta on ilmoitettu lumehoitoa saaneilla potilailla pitkän turvallisuuden seurannan aikana. Kaikilla potilailla oli mahdollisia MDS:n/AML:n kehittymisen myötävaikuttavia tekijöitä: kaikissa tapauksissa potilaat olivat saaneet aiemmin platinaa sisältävää solunsalpaajahoitoa ja/tai muita DNA:ta vaurioittavia aineita. Katso riskin vähentämis- ja hoitotoimenpiteet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Ruoansulatuselimistön toksisuus
Pahoinvointia on ilmoitettu 37 %:lla potilaista ja oksentelua 68 %:lla potilaista. Nämä ovat yleensä olleet matala-asteisia (CTCAE:n aste 1 tai 2). Vatsakipua (yhdistetty termi vatsakivulle, alavatsakivulle ja ylävatsakivulle) ilmoitettiin 39 %:lla rukaparibihoitoa saaneista potilaista, mutta se oli myös hyvin yleistä (34 %) lumelääkettä saaneilla potilailla, joten se todennäköisimmin liittyy perussairauteen. Katso riskin vähentämis- ja hoitotoimenpiteet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Valoherkkyys
Valoherkkyydestä on ilmoitettu 10 %:lla potilaista matala-asteisina ihoreaktioina (CTCAE:n aste 1 tai 2) ja 0,2 %:lla potilaista ≥ CTCAE:n aste 3 reaktioina. Katso riskin vähentämis- ja hoitotoimenpiteet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Seerumin aminotransferaasiarvojen suureneminen (ASAT/ALAT)
Alaniiniaminotransferaasi- (ALAT) tai aspartaattiaminotransferaasiarvojen (ASAT) suurenemiseen liittyviä tapahtumia on havaittu 39 %:lla (kaikki asteet) ja 10 %:lla (≥ CTCAE:n aste 3) potilaista.
Nämä tapahtumat ilmenivät muutaman ensimmäisen rukaparibihoitoviikon aikana, ne olivat kumoutuvia ja olivat harvoin yhteydessä bilirubiiniarvojen suurenemiseen. Suurentunut ALAT-arvo havaittiin 37 %:lla (kaikki asteet) ja 10 %:lla (≥ CTCAE:n aste 3) potilaista, suurentunut ASAT-arvo 33 %:lla (kaikki asteet) ja 3 %:lla (≥ CTCAE:n aste 3) potilaista ja sekä suurentuneet ALAT- että ASAT-arvot 31 %:lla (kaikki asteet) ja 3 %:lla (≥ CTCAE:n aste 3) potilaista. Mikään tapauksista ei täyttänyt Hy’s Law -kriteereitä lääkkeiden aiheuttamasta maksavauriosta. Kohonneet ASAT-/ALATarvot on ehkä hoidettava hoidon keskeytyksellä ja/tai annosta pienentämällä, kuten on kuvattu taulukossa 2 (ks. kohta Annostus ja antotapa). Useimmat potilaat pystyivät jatkamaan rukaparibihoitoa entisellään tai hoitoa muuttamalla ilman, että ≥ 3 asteen maksan toimintakokeeseen liittyviä poikkeamia esiintyi uudelleen.
Seerumin kreatiniiniarvon kohoaminen
Seerumin kreatiniiniarvon kohoamista, lähinnä lievää tai kohtalaista (CTCAE:n aste 1 tai 2), on havaittu 17 %:lla potilaista muutaman ensimmäisen rukaparibihoitoviikon aikana; 0,6 %:lla potilaista ilmoitettiin CTCAE:n aste 3 reaktioita. Rukaparibihoitoon liittyvät kreatiniiniarvon kohoamiset voivat johtua munuaisten kuljettajaproteiinien MATE1 ja MATE2-K estosta (ks. kohta Yhteisvaikutukset). Nämä seerumin kreatiniiniarvon suurenemiset olivat kliinisesti oireettomia.
Iäkkäät
≥ 75-vuotiailla potilailla joidenkin haittavaikutusten esiintymistiheydet suurenivat: veren kreatiniiniarvon suureneminen (33 %), heitehuimaus (19 %), kutina (16 %) ja muistin huononeminen (4 %) olivat korkeampia kuin alle 75-vuotiailla potilailla (veren kreatiniiniarvon suureneminen (16 %), heitehuimaus (14 %), kutina (11 %) ja muistin huononeminen (1 %)).
Munuaisten vajaatoiminta
Kohtalaista munuaisten vajaatoimintaa sairastavilla potilailla (kreatiniinipuhdistuma 30–59 ml/min) joidenkin vähintään asteen 3 haittavaikutusten esiintymistiheydet suurenivat: anemia (34 %), neutropenia (13 %), trombosytopenia (12 %), uupumus/astenia (12 %) ja sekä ALAT- että ASATarvon suureneminen (12 %) olivat korkeampia kuin potilailla, joiden munuaisten toiminta oli normaalia (kreatiniinipuhdistuma > 90 ml/min) (anemia (23 %), neutropenia (8 %), trombosytopenia (5 %), uupumus/astenia (7 %) ja sekä ALAT- että ASAT-arvon suureneminen (7 %)).
Pediatriset potilaat
Tutkimuksia ei ole tehty rukaparibin farmakokinetiikasta pediatrisilla potilailla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista liitteessä Vluetellun kansallisen ilmoitusjärjestelmän kautta.
Yliannostus
Rubraca-valmisteen yliannostukseen ei ole olemassa erityistä hoitoa. Yliannostuksen oireet eivät ole tiedossa. Epäillyn yliannostuksen sattuessa lääkärin on noudatettava yleisiä tukitoimenpiteitä ja hoidettava potilasta oireenmukaisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: muut antineoplastiset aineet, ATC-koodi: L01XK03
Vaikutusmekanismi ja farmakodynaamiset vaikutukset
Rukaparibi on DNA:n korjaamiseen osallistuvien poly(ADP-riboosi)polymeraasi (PARP)-entsyymien estäjä, mukaan lukien PARP-1, PARP-2, ja PARP-3. In vitro-tutkimukset ovat osoittaneet, että rukaparibin aikaansaamaan sytotoksisuuteen liittyy PARP:n entsymaattisen toiminnan estäminen ja PARP-DNA-kompleksien pidättäminen, joka saa aikaan lisääntynyttä DNA:n vaurioitumista, apoptoosia ja solukuolemaa.
Rukaparibilla on osoitettu in vitro ja in vivo olevan kasvaimia tuhoavaa vaikutusta BRCA:n mutanteissa solulinjoissa synteettisen letaalisuuden mekanismin kautta, jossa solukuolemaan tarvitaan kahden DNA:n korjausreitin katoaminen. Rukaparibin aiheuttaman sytotoksisuuden ja kasvaimia tuhoavan vaikutuksen lisääntymistä havaittiin kasvainsolulinjoissa, joissa oli BRCA1/2-geenien ja muiden DNA:ta korjaavien geenien virheitä. Rukaparibin on osoitettu vähentävän kasvaimen kasvua hiirillä tehdyissä ihmisen syövän ksenograftimalleissa, joissa BRCA-geenin virheitä joko on tai ei ole.
Kliininen teho
Pitkälle edenneen munasarjasyövän ensilinjan ylläpitohoito
Rukaparibin tehoa arvioitiin faasin 3 kaksoissokkoutetussa ATHENA-monikeskustutkimuksessa 538 potilaalla, joilla oli pitkälle edennyt munasarjasyöpä, munanjohtimen syöpä tai primaarinen vatsakalvosyöpä ja jotka olivat saavuttaneet vasteen ensilinjan platinapohjaiselle solunsalpaajahoidolle ja leikkaushoidolle. Vaste määriteltiin tilanteeksi, jossa potilaalla ei ollut näyttöä taudin etenemisestä radiologisesti tai CA-125-arvon nousun perusteella (Gynecological Cancer Intergroup
(GCIG) -suositusten mukaisesti) missään vaiheessa ensilinjan hoidon aikana; ja potilaalla ei joko ollut näyttöä mitattavissa olevasta taudista RECIST v.1.1 -kriteerien mukaisesti leikkauksen jälkeen, jos kasvain oli poistettu kokonaan, tai potilaalla oli (täydellinen tai osittainen) vaste, jos tauti oli mitattavissa leikkaushoidon jälkeen ja ennen solunsalpaajahoitoa, tai potilaalla oli GCIG CA-125 -vaste, jos tauti ei ollut mitattavissa samassa tilanteessa.
Kaikki potilaat olivat saaneet 4-8 sykliä platinapohjaista kaksoishoitoa (mukaan lukien ≥ 4 sykliä platinan ja taksaanin yhdistelmää). Bevasitsumabihoito sallittiin ensilinjan solunsalpaajahoidon aikana mutta ei rukaparibilla toteutetun ylläpitohoidon aikana. Kaikki potilaat satunnaistettiin 8 viikon sisällä viimeisen solunsalpaajasyklin ensimmäisestä päivästä.
Potilaat satunnaistettiin (4:1) saamaan rukaparibitabletteja 600 mg suun kautta kahdesti vuorokaudessa (n = 427) tai lumelääkettä (n = 111). Hoitoa jatkettiin taudin etenemiseen tai ei-hyväksyttävissä olevan toksisuuden ilmaantumiseen asti tai enintään 2 vuoden ajan. Satunnaistaminen ositettiin seuraavasti: taudin status solunsalpaajahoidon jälkeen (jäännöstauti vs. ei jäännöstautia), leikkaushoidon ajankohta (primaarileikkaus vs. kasvaimen pienennysleikkaus) ja kasvaimen biomarkkeristatus.
Biomarkkeristatuksen määrittämiseen käytettiin homologisen rekombinaation puutoksen (HRD) testiä:
kasvain määriteltiin biomarkkeripositiiviseksi, jos se oli HRD-positiivinen eli siinä oli haitallinen BRCA (tBRCA) -mutaatio tai villityypin tBRCA (tBRCAwt) / suuri genomien heterotsygotian menetys (LOHhigh), ja biomarkkerinegatiiviseksi, jos se oli HRD-negatiivinen tBRCAwt:n / pienen genomien heterotsygotian menetyksen (LOHlow) perusteella.
Ensisijainen tehoon liittyvä hoitotulos oli tutkijan arvioima etenemisvapaa elinaika (investigatorassessed progression-free survival, invPFS) Response Evaluation Criteria in Solid Tumours (RECIST) -kriteereiden version 1.1 mukaisesti. Keskeisiin toissijaisiin tehon päätetapahtumiin kuuluivat kokonaiselinaika (overall survival, OS) ja objektiivinen vasteprosentti (objective response rate, ORR) RECIST-kriteereiden version 1.1 mukaisesti. invPFS-, OS- ja ORR-testit tehtiin hierarkkisesti: ensin HRD-ryhmässä ja sen jälkeen ITT-potilasjoukossa. Hoitotulosmittarina oli lisäksi satunnaistamisesta taudin toiseen etenemiseen tai kuolemaan kulunut aika (PFS2).
Rukaparibihoitoa saaneiden potilaiden iän mediaani oli 61 vuotta (vaihteluväli: 30-83) ja lumelääkettä saaneiden potilaiden 62 vuotta (vaihteluväli: 31-80). Eastern Cooperative Oncology Group (ECOG) -toimintakykyluokka oli 0 kaikkiaan 69 %:lla rukaparibia saaneista potilaista ja 68 %:lla lumelääkettä saaneista potilaista. Rukaparibi- tai lumehoitoon satunnaistetuista 538 potilaasta 75 %:lla oli FIGO-asteen III tauti ja 25 %:lla oli FIGO-asteen IV tauti, ja 16 % oli saavuttanut täydellisen vasteen viimeisimmälle platinapohjaiselle hoidolleen. Rukaparibi- tai lumehoitoon satunnaistetuista 538 potilaasta 78 %:lla oli epiteliaalinen munasarjasyöpä, 13 %:lla oli munanjohtimen syöpä ja 9 %:lla oli primaarinen vatsakalvosyöpä, ja useimmilla potilailla (> 90 %) oli seroosi kasvaimen histologia. ITT-potilasjoukossa potilaat saivat 6 sykliä (mediaani) platinapohjaista kaksoissolunsalpaajahoitoa ja 17,8 % potilaista oli saanut bevasitsumabia ensilinjan solunsalpaajahoidon aikana. Primaari kasvaimen pienennysleikkaus oli tehty 48,1 %:lle potilaista, ja 51,9 % potilaista oli saanut neoadjuvanttia solunsalpaajahoitoa kasvaimen pienennysleikkauksen jälkeen.
Kaikkiaan 43 % potilaista oli HRD-positiivisia (21 %:lla oli haitallinen tBRCA-mutaatio ja 22 %:lla oli tBRCAwt / LOHhigh), 44 % oli HRD-negatiivisia (tBRCAwt / LOHlow) ja 12 %:n HRD-status oli tuntematon.
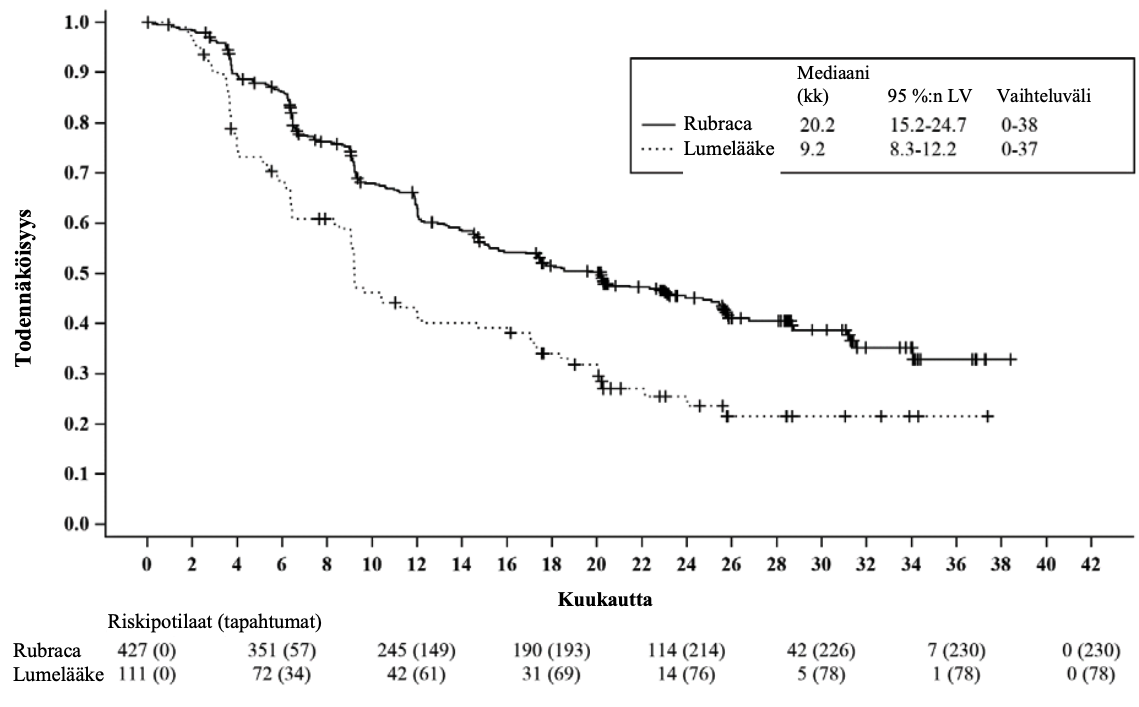
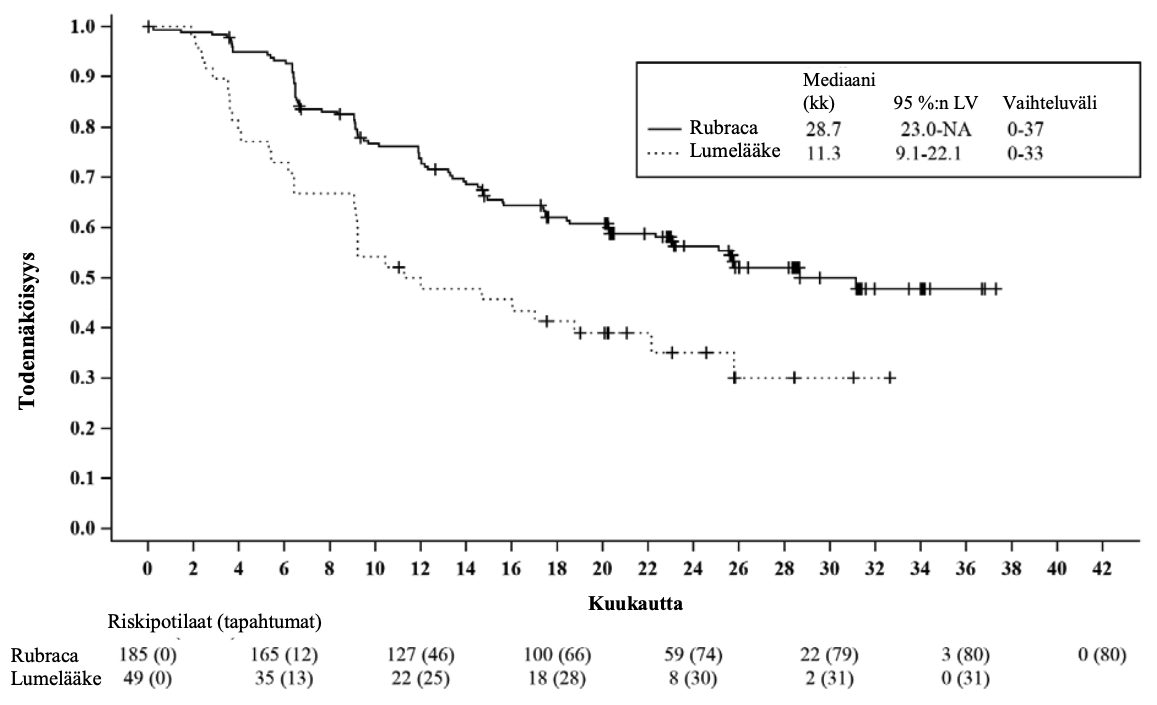
ATHENA-tutkimuksessa invPFS:n osoitettiin parantuneen tilastollisesti merkitsevästi rukaparibihoitoon satunnaistetuilla potilailla lumelääkkeeseen verrattuna HRD-ryhmässä ja ITTpotilasjoukossa. invPFS-tulokset olivat johdonmukaisia riippumatta siitä, sensuroitiinko potilaita uusien syöpähoitojen ja väliin jääneiden käyntien perusteella vai ei. Tehotulokset esitetään taulukossa 4, kuvassa 1 ja kuvassa 2.
Taulukko 4. Tehotulokset – ATHENA (tutkijan arviointi)
| HRD-ryhmäa | ITT-potilasjoukkob | |||||
Rubraca
| Lumelääke
| Rubraca
| Lumelääke
| |||
| PFSc-tapahtumat, n (%) | (43,2) | (63,3) | (53,9) | (70,3) | ||
| PFS, mediaani kuukausina (95 %:n luottamusväli) | 28,7 (23,0; NR) | 11,3 (9,1; 22,1) | 20,2 (15,2; 24,7) | 9,2 (8,3; 12,2) | ||
| Hasardisuhde (95 %:n luottamusväli) | 0,52 (0;40; 0,68) | |||||
| P-arvod | < 0,0001 | |||||
| OSe-tapahtumat, n (%) | 46 (24,9) | 12 (24,5) | 144 (33,7) | 42 (37,8) | ||
| OS, mediaani kuukausina | NR | NR | NR | 46,2 | ||
| Hasardisuhde (95 %:n luottamusväli) | ||||||
| P-arvod | ||||||
- Sisältää kaikki potilaat, joilla oli haitallinen tBRCA-mutaatio (N = 115) tai tBRCAwt / LOHhigh (N = 119).
- Kaikki satunnaistetut potilaat.
- Seuranta-ajan mediaani oli 26 kuukautta sekä rukaparibi- että lumeryhmissä.
- P-arvo perustuu ositettuun log rank -testiin.
- Toisen välianalyysin ajankohtana OS-tiedot eivät olleet valmiit (35 % potilaista oli kuollut); seuranta-ajan mediaani oli 37 kuukautta sekä rukaparibi- että lumeryhmissä. NR: Ei saavutettu.
Kuva 1. Kaplan-Meierin käyrä etenemisvapaasta elossaoloajasta ATHENA-tutkimuksessa tutkijan arvion perusteella: ITT-potilasjoukko
Kuva 2. Kaplan-Meierin käyrä etenemisvapaasta elossaoloajasta ATHENA-tutkimuksessa tutkijan arvion perusteella: HRD-potilasjoukko
Todennäköisyys
Alaryhmäanalyysi (PFS tutkijan arvioimana)
HRD-potilasjoukossa hasardisuhde 0,40 (95 %:n luottamusväli [0,21; 075]) todettiin alaryhmässä, jonka potilailla oli tBRCA-mutaatio (n = 115). Ei-tBRCA LOHhigh -alaryhmässä (n = 119) hasardisuhde oli 0,58 (95 %:n luottamusväli [0,33; 1,01]). HRD-negatiivisessa alaryhmässä (n = 238) todettu hasardisuhde oli 0,65 (95 %:n luottamusväli [0,45; 0,95]).
Uusiutuneen munasarjasyövän ylläpitohoito
Rukaparibin tehoa tutkittiin kaksoissokkoutetussa kliinisessä ARIEL3-monikeskustutkimuksessa 564 potilaalla, joilla oli uusiutunut epiteliaalinen munasarjasyöpä, munanjohtimen syöpä tai primaarinen vatsakalvosyöpä ja joilla oli vaste platinapohjaiselle solunsalpaajahoidolle. Potilaat satunnaistettiin (2:1) saamaan Rubraca-tabletteja 600 mg suun kautta kahdesti vuorokaudessa (n = 375) tai lumelääkettä (n = 189). Hoitoa jatkettiin taudin etenemiseen tai ei-hyväksyttävissä olevan toksisuuden ilmaantumiseen asti. Kaikki potilaat olivat saaneet vasteen (täydellisen tai osittaisen) heidän viimeisimmälle platinapohjaiselle solunsalpaajahoidolle ja heidän syöpäantigeeni 125 (CA-125) oli alle viitealueen ylärajan (ULN). Potilaat satunnaistettiin 8 viikon sisällä platinapohjaisen solunsalpaajahoidon päättymisestä eikä mitään väliin tulevaa ylläpitohoitoa sallittu. Potilaat eivät olleet voineet saada aiemmin rukaparibihoitoa tai muuta PARP-estäjähoitoa. Satunnaistaminen ositettiin seuraavasti: paras vaste viimeisimmälle platinahoidolle (täydellinen tai osittainen), aika etenemiseen toiseksi viimeisen platinahoidon jälkeen (6 – ≥ 12 kuukautta ja > 12 kuukautta) ja kasvaimen biomarkkeristatus (tBRCA, ei BRCA homologisen rekombinaation puutos [nbHRD] ja negatiivinen biomarkkeri).
Ensisijainen tehoon liittyvä hoitotulosmittari oli invPFS RECIST -kriteereiden version 1.1 mukaisesti. Sokkoutetun riippumattoman radiologin arvioima etenemisvapaa elinaika, PFS, oli keskeinen toissijainen tehon mittari. Toissijaisiin tehon päätetapahtumiin kuuluivat kokonaiselinaika (overall survival, OS).
Rukaparibia saaneiden potilaiden iän keskiarvo oli 61 vuotta (vaihteluväli 36–85 vuotta); suurin osa potilaista oli valkoihoisia (80 %), ja kaikki potilaat (100 %) kuuluivat ECOG-toimintakykyluokkaan 0 tai 1. Useimmilla potilailla primaarinen kasvain oli munasarjakasvain (84 %); useimmilla potilailla (95 %) oli seroosi histologia ja 4%:lla potilaista ilmoitettiin olevan endometrioidi histologia. Kaikki potilaat olivat saaneet vähintään kahta aiempaa platinapohjaista solunsalpaajahoitoa (vaihteluväli 2–6), ja 28 % potilaista oli saanut vähintään kolmea aiempaa platinapohjaista solunsalpaajahoitoa. Kaikkiaan 32 % potilaista oli saanut täydellisen vasteen (CR) heidän viimeisimmälle hoidolleen. Etenemisvapaa-aika viimeistä edelliseen platinahoitoon oli 6–12 kuukautta 39 %:lla potilaista ja > 12 kuukautta 61 %:lla potilaista. 22 % rukaparibia saaneista potilaista ja 23 % lumelääkettä saaneista potilaista oli saanut aiemmin bevasitsumabihoitoa. Demografiset tiedot, sairauden ominaisuudet lähtötilanteessa ja aiempi hoitohistoria olivat yleisesti ottaen hyvin tasapainossa rukaparibi- ja lumehoitoryhmien välillä.
Kukaan potilaista ei ollut saanut aiemmin PARP-estäjähoitoa. Rubracan tehoa potilailla, jotka ovat saaneet aiemmin PARP-estäjähoitoa, ei ole tutkittu ylläpitohoidon yhteydessä, eikä sitä voida ekstrapoloida saatavilla olevista tiedoista.
Kaikkien potilaiden (n = 564) kasvainkudosnäytteet testattiin keskitetysti positiivisen HRD:n (homologisen rekombinaation puutoksen) määrittämiseksi (positiivisen tuloksen määritelmänä on kasvainkudoksen haitallinen BRCA-mutaatio [tBRCA] tai suuri genomien heterotsygotian menetys). 94 % (186/196) tBRCA-potilaiden verinäytteistä tutkittiin ituradan BRCA (gBRCA) -testillä. Näiden tulosten perusteella 70 %:lla (130/186) tBRCA-potilaista oli gBRCA-mutaatio ja 30 %:lla (56/186) oli somaattinen BRCA-mutaatio.
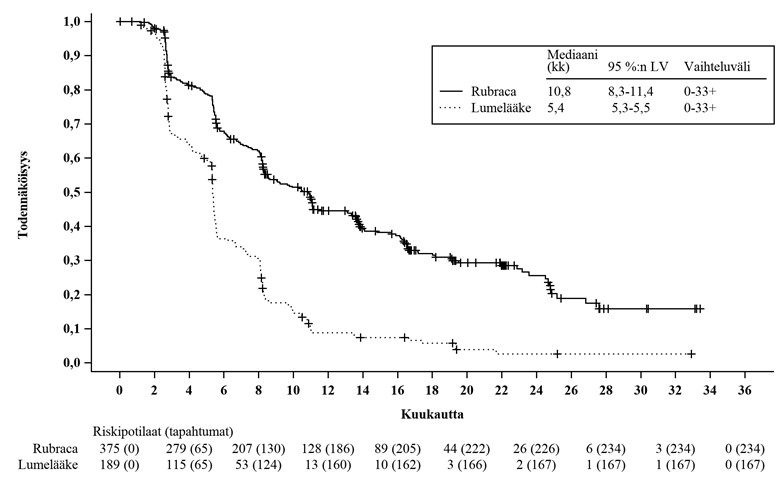
ARIEL3-tutkimus täytti ensisijaisen päätetapahtumansa ja osoitti invPFS:n tilastollisesti merkitsevän paranemisen niiden potilaiden osalta, jotka oli satunnaistettu saamaan rukaparibia verrattuna lumelääkettä saaneisiin potilaisiin ITT-potilasjoukossa ja HRD- ja tBRCA-ryhmissä. ITTpotilasjoukkoa koskeva riippumattoman radiologin arviointi (IRR) tuki ensisijaista päätetapahtumaa. PFS-tulosten yhteenveto on taulukossa 5 ja kuvassa 3.
Taulukko 5. ARIEL3-tutkimuksen tehoa koskevat tulokset (Yhteenveto ensisijaisen tavoitteen tuloksesta: PFS)
Muuttuja
| Tutkijan arvio | IRR | |||
| Rukaparibi | Lumelääke | Rukaparibi | Lumelääke | ||
| ITT-potilasjoukko a | |||||
| Potilaat, n | 375 | 189 | 375 | 189 | |
| PFS-tapahtumat, n (%) | 234 (62 %) | 167 (88 %) | 165 (44%) | 133 (70%) | |
| PFS, mediaani kuukausina (95 %:n luottamusväli) | 10,8 (8,3; 11,4) | 5,4 (5,3–5,5) | 13,7 (11,0, 19,1) | 5,4 (5,1, 5,5) | |
| HR (95 %:n luottamusväli) | 0,36 (0,30; 0,45) | 0,35 (0,28, 0,45) | |||
| p-arvo b | < 0,0001 | < 0,0001 | |||
| HRD-ryhmä c | |||||
| Potilaat, n | 236 | 118 | 236 | 118 | |
| PFS-tapahtumat, n (%) | 134 (57 %) | 101 (86 %) | 90 (38 %) | 74 (63 %) | |
| PFS, mediaani kuukausina (95 %:n luottamusväli) | 13,6 (10,9; 16,2) | 5,4 (5,1; 5,6) | 22,9 (16,2, NA) | 5,5 (5,1, 7,4) | |
| HR (95 %:n luottamusväli) | 0,32 (0,24; 0,42) | 0,34 (0,24, 0,47) | |||
| p-arvo b | < 0,0001 | < 0,0001 | |||
| tBRCA-ryhmä d | |||||
| Potilaat, n | 130 | 66 | 130 | 66 | |
| PFS-tapahtumat, n (%) | 67 (52 %) | 56 (85 %) | 42 (32 %) | 42 (64 %) | |
| PFS, mediaani kuukausina (95 %:n luottamusväli) | 16,6 (13,4; 22,9) | 5,4 (3,4; 6,7) | 26,8 (19,2, NA) | 5,4 (4,9, 8,1) | |
| HR (95 %:n luottamusväli) | 0,23 (0,16; 0,34) | 0,20 (0,13, 0,32) | |||
| p-arvo b | < 0,0001 | < 0,0001 | |||
| ei BRCA LOH+ -ryhmä | |||||
| Potilaat, n | 106 | 52 | 106 | 52 | |
| PFS-tapahtumat, n (%) | 67 (63 %) | 45 (87 %) | 48 (45 %) | 32 (62 %) | |
| PFS, mediaani kuukausina (95 %:n luottamusväli) | 9,7 (7,9, 13.1) | 5,4 (4,1, 5,7) | 11,1 (8,2, NA) | 5,6 (2,9, 8,2) | |
| HR kuukausina (95 %:n luottamusväli) | 0,44 (0,29, 0,66) | 0,554 (0,35, 0,89) | |||
| p-arvo b | < 0,0001 | 0,0135 | |||
| ei BRCA LOH- -ryhmä | |||||
| Potilaat, n | 107 | 54 | 107 | 54 | |
| PFS-tapahtumat, n (%) | 81 (73 %) | 50 (93 %) | 63 (59 %) | 46 (85 %) | |
| PFS, mediaani kuukausina (95 %:n luottamusväli) | 6,7 (5,4, 9,1) | 5,4 (5,3, 7,4) | 8,2 (5,6, 10,1) | 5,3 (2,8, 5,5) | |
| HR (95 %:n luottamusväli) | 0,58 (0,40, 0,85) | 0,47 (0,31, 0,71) | |||
| p-arvo b | 0,0049 | 0,0003 | |||
- Kaikki satunnaistetut potilaat.
- Kaksipuolinen p-arvo
- HRD käsittää kaikki potilaat, joilla oli haitallinen itulinjan tai somaattinen BRCA-mutaatio tai joilla ei ollut tBRCA:ta mutta joilla oli suuri genomien heterotsygotian menetys kliinisen tutkimuksen määrityksen (CTA) mukaan.
- tBRCA käsittää kaikki potilaat, joilla oli haitallinen itulinjan tai somaattinen BRCA-mutaatio kliinisen tutkimuksen määrityksen mukaan.
HR: hasardisuhde. Arvo < 1 puoltaa rukaparibia NA: Ei saavutettu
Kuva 3. Kaplan-Meierin käyrä etenemisvapaasta elossaoloajasta ARIEL 3 -tutkimuksessa tutkijan arvion perusteella: ITT-potilasjoukko
ITT-potilasjoukon OS-loppuanalyysissä (70 %:n maturiteetti) hasardisuhde (HR) oli 1,00 (95 %:n luottamusväli: 0,81; 1,22, mediaani 36 kuukautta rukaparibia saaneilla potilailla vs. 43,2 kuukautta lumelääkettä saaneilla potilailla). HRD-alaryhmässä raportoitu HR oli 1,01 (95 %:n luottamusväli: 0,77; 1,32, mediaani 40,5 kuukautta rukaparibia saaneilla potilailla vs. 47,8 kuukautta lumelääkettä saaneilla potilailla) ja tBRCA-alaryhmässä HR oli 0,83 (95 %:n luottamusväli: 0,58; 1,19, mediaani 45,9 kuukautta rukaparibia saaneilla potilailla vs. 47,8 kuukautta lumelääkettä saaneilla potilailla). Eksploratiivisissa alaryhmäanalyyseissa potilaille, joilla ei ollut tBRCA-mutaatiota (ei tutkimukseen upotetut potilasalajoukot, joissa ei ollut tBRCA:ta [LOH+, LOH–, LOH tuntematon]) OSpäätetapahtuman HR oli 1,084 (95 %:n luottamusväli: 0,841; 1,396, mediaani 32,2 kuukautta rukaparibia saaneilla potilailla vs. 38,3 kuukautta lumelääkettä saaneilla potilailla). Elinajan seurannan mediaani kaikille potilaille oli 77 kuukautta (6,4 vuotta) ja vaihteluväli oli 2 vuorokaudesta 93 kuukauteen (7,6 kuukautta).
Lopullisen analyysin ajankohtana 89 % lumelääkettä saaneista potilaista oli saanut vähintään yhtä myöhempää hoitoa, näistä 46 % sai PARP-estäjää. Rukaparibia saaneista potilaista 78 % oli saanut vähintään yhtä myöhempää hoitoa.
Sydämen elektrofysiologia
Pitoisuus-QTcF-välin pidentyminen -analyysi tehtiin 54 potilaan tiedoilla, joilla oli kiinteä kasvain ja joille annettu rukaparibi-annos vaihteli 40 mg:sta kerran vuorokaudessa 840 mg:aan kahdesti vuorokaudessa (1,4-kertaisesti myyntiluvan saanut suositeltu annos). Ennustetulla vakaan tilan Cmax – pitoisuuden mediaanilla 600 mg:n kahdesti vuorokaudessa annetun rukaparibiannoksen jälkeen QTcFvälin pidentyminen lähtötilanteesta oli 11,5 millisekuntia (90 %:n luottamusväli: 8,77–14,2 millisekuntia).Näin ollen kliinisesti merkittävän QTcF-välin pidentymisen riski lähtötilanteesta (> 20 millisekuntia) on vähäinen.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Rubracavalmisteen käytöstä munasarjasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Rukaparibin plasma-altistukset, kun mittarina käytetään Cmax-pitoisuutta ja AUC-arvoa, olivat suunnilleen suhteessa annokseen arvioiduilla annoksilla (40–500 mg kerran vuorokaudessa, 240–840 mg kahdesti vuorokaudessa). Vakaa tila saavutettiin 1 viikon annostelun jälkeen. Toistuvan, kahdesti vuorokaudessa tapahtuvan annostelun jälkeen kertyminen oli AUC-arvon perusteella 3,5–6,2-kertaista.
Imeytyminen
Syöpäpotilailla 600 mg:n kahdesti vuorokaudessa otetun rukaparibiannoksen jälkeen vakaan tilan
Cmax-pitoisuus oli 1940 ng/ml ja AUC0-12h-arvo 16900 h⋅ng/ml. t½max-aika oli 1,9 tuntia. Keskimääräinen absoluuttinen oraalinen biologinen hyötyosuus 12–120 mg:n rukaparibikertaannoksen jälkeen oli 36 %. Absoluuttista oraalista biologista hyötyosuutta ei ole määritetty 600 mg:n annoksella. Syöpäpotilailla hyvin rasvapitoisen aterian jälkeen Cmax-pitoisuus kohosi 20 %, AUC0-24harvo lisääntyi 38 %, ja t½max-aika piteni 2,5 tunnilla, verrattuna paastotilassa toteutettuun annosteluun. Ruoan vaikutuksen farmakokinetiikkaan ei katsottu olevan kliinisesti merkittävää. Rubraca voidaan antaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Proteiinin sitoutuminen rukaparibiin in vitro on 70,2-prosenttista ihmisen plasmassa terapeuttisilla pitoisuuksilla. Rukaparibi jakautui etusijaisesti punasoluihin. Veren ja plasman pitoisuussuhde oli 1,83. Syöpäpotilailla rukaparibin vakaan tilan jakautumistilavuus oli 113–262 litraa laskimonsisäisen 12–40 mg:n rukaparibikerta-annoksen jälkeen.
Biotransformaatio
Rukaparibia metaboloivat in vitro -ympäristössä pääasiassa CYP2D6-entsyymi, ja vähemmässä määrin CYP1A2- ja CYP3A4-entsyymit. Populaatiofarmakokineettisessä analyysissä ei havaittu kliinisesti merkittäviä eroja farmakokinetiikassa potilailla, joilla oli eri CYP2D6:n fenotyyppejä (mukaan lukien heikot metaboloijat, n=9; kohtalaiset metaboloijat, n=71; normaalit metaboloijat, n=76; ja erittäin nopeat metaboloijat, n=4) eikä potilailla, joilla oli eri CYP1A2:n fenotyyppejä (mukaan lukien normaalit metaboloijat, n=28; hyperindusoijat, n=136). Tuloksia on tulkittava varoen johtuen joidenkin alaryhmäfenotyyppien rajallisesta edustuksesta.
Muuttumattoman rukaparibin osuus oli 64,0 % plasmassa todetusta radioaktiivisuudesta sen jälkeen, kun potilaille, joilla oli kiinteitä kasvaimia, annettiin suun kautta [14C]-rukaparibin kerta-annos. Rukaparibin pääasialliset metaboliareitit olivat oksidaatio, N-demetylaatio, N-metylaatio, glukuronidaatio ja N-formylaatio. Runsaimmin esiintyvä metaboliitti oli M324, rukaparibin oksidatiivinen deaminaatiotuote, jonka osuus plasmassa todetusta radioaktiivisuudesta oli 18,6 %. M324 oli in vitro vähintään 30-kertaisesti heikompi PARP-1:tä, PARP-2:ta ja PARP-3:a vastaan kuin rukaparidi. Muiden vähäisten metaboliittien osuus plasman radioaktiivisuudesta oli 13,8 %.
Rukaparibin osuus virtsassa todetusta radioaktiivisuudesta oli 44,9 % ja ulosteessa 94,9 %, kun taas M324:n osuus virtsassa todetusta radioaktiivisuudesta oli 50,0 % ja ulosteessa 5,1 %.
Eliminaatio
Puhdistuman vaihteluväli oli 13,9–18,4 l/h, yhden laskimonsisäisen 12–40 mg:n rukaparibiannoksen jälkeen. Suun kautta potilaille annetun [14C]-rukaparibin 600 mg:n kerta-annoksen jälkeen radioaktiivisuuden keskimääräinen kokonaiskertymä oli 89,3 %, josta keskimääräinen kertymä ulosteessa oli 71,9 % ja virtsassa 17,4 % 288 tuntia annoksen jälkeen. Ulosteessa todetusta kertymästä 90 % saavutettiin 168 tuntia annoksen jälkeen. Rukaparibin keskimääräinen puoliintumisaika (t1/2) oli 25,9 tuntia.
Lääkevalmisteen yhteisvaikutukset
Rukaparibin osoitettiin olevan P-gp:n ja BCRP:n substraatti in vitro -ympäristössä, mutta ei munuaiseen oton kuljettajaproteiinien OAT1, OAT3 ja OCT2 eikä maksan kuljettajaproteiinien OAPT1B1 ja OATP1B3 substraatti. P-gp:n ja BCRP:n estäjien vaikutusta rukaparibin farmakokinetiikkaan ei voida poissulkea.
Rukaparibi esti kumoutuvasti CYP1A2-, CYP2C19-, CYP2C9- ja CYP3A-entsyymejä, sekä vähäisemmässä määrin CYP2C8-, CYP2D6- ja UGT1A1-entsyymejä in vitro- ympäristössä. Rukaparibi indusoi CYP1A2-entsyymiä ja vaimennussääteli CYP2B6- ja CYP3A4-entsyymejä ihmisen hepatosyyteissä kliinisesti merkittävillä altistuksilla.
Rukaparibi on voimakas MATE1:n ja MATE2-K:n estäjä, kohtalainen OCT1:n estäjä, ja heikko OCT2:n estäjä in vitro -ympäristössä. Kliinisillä altistuksilla rukaparibi ei estänyt sappihappojen suolojen poistopumppua (bile salt export pump BSEP), OATP1B1:tä, OATP1B3:a, OAT1:tä eikä OAT3:ta. Rukaparibin aiheuttamaa MRP4:n estoa ei voida täysin poissulkea kliinisillä altistuksilla. MRP2:n tai MRP3:n kanssa ei havaittu yhteisvaikutusta in vitro-ympäristössä rukaparibin kliinisellä altistuksella. Vähäistä kaksivaiheista MRP2:n aktivaatiota ja estoa sekä pitoisuusriippuvaista MRP3:n estoa havaittiin pitoisuuksilla, jotka olivat korkeampia kuin havaittu rukaparibin Cmax-pitoisuus plasmassa. Suolessa tapahtuvan MRP2- ja MRP3-yhteisvaikutuksen kliininen merkitys ei ole tiedossa.
Rukaparibi on BCRP:n ja P-gp:n ulosvirtauskuljettajaproteiinien estäjä in vitro -ympäristössä. Merkittävää P-gp:n estoa ei havaittu in vivo (ks. kohta Yhteisvaikutukset).
Populaatiofarmakokineettisen analyysin perusteella samanaikaisella protonipumpun estäjien (PPI:t) käytöllä ei todennäköisesti ole kliinisesti merkittävää vaikutusta rukaparibin farmakokinetiikkaan. Rukaparibin ja protonipumpun estäjien samanaikaisen annon vaikutuksesta ei voida tehdä vankkaa johtopäätöstä, koska protonipumpun estäjien annostasoa ja antoaikaa ei ole dokumentoitu yksityiskohtaisesti.
Farmakokinetiikka erityisillä potilasryhmillä
Ikä, rotu ja kehonpaino
Populaatiofarmakokineettisen analyysin perusteella ei havaittu kliinisesti merkittävää yhteyttä ennustetun vakaan tilan altistuksen ja potilaan iän, rodun ja kehonpainon välillä.
Populaatiofarmakokineettisessä tutkimuksessa mukana olevat potilaat olivat iältään 21–86-vuotiaita (58 % < 65-vuotiaita, 31 % 65–74-vuotiaita ja 11 % > 75-vuotiaita), 82 % oli kaukaasialaista rotua, ja kehonpaino oli välillä 41–171 kg (73 %:lla kehonpaino oli > 60 kg).
Maksan vajaatoiminta
Populaatiofarmakokineettinen analyysi toteutettiin maksan vajaatoiminnan rukaparibin puhdistumaan kohdistuvan vaikutuksen arvioimiseksi potilailla, jotka saivat rukaparibia 600 mg kahdesti vuorokaudessa. Kliinisesti merkittäviä eroja ei havaittu 34 lievää maksan vajaatoimintaa sairastavan potilaan (kokonaisbilirubiini ≤ ULN ja ASAT-arvo > viitealueen yläraja tai kokonaisbilirubiini > 1,0– 1,5 kertaa viitealueen yläraja ja mikä tahansa ASAT-arvo) ja 337 potilaan, joilla maksan toiminta oli normaalia, välillä. Rukaparibin farmakokinetiikkaa arvioivassa tutkimuksessa maksan vajaatoimintaa sairastavilla potilailla havaittiin, että keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden (N=8, National Cancer Institute - Organ Dysfunction Working Group -kriteerien mukaisesti; kokonaisbilirubiini > 1,5 - ≤ 3 kertaa viitealueen yläraja, ULN) rukaparibin AUC-arvo oli 600 mg:n kerta-annoksen jälkeen 45 % korkeampi verrattuna potilaisiin, joilla maksan toiminta oli normaali (N=8). Cmax ja Tmax olivat samankaltaisia ryhmien välillä. Tietoja ei ole olemassa potilaista, joilla on vaikea-asteinen maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Muodollisia tutkimuksia ei ole tehty rukaparibin käytöstä munuaisten vajaatoimintaa sairastaville potilaille. Populaatiofarmakokineettinen analyysi toteutettiin munuaisten vajaatoiminnan rukaparibin puhdistumaan kohdistuvan vaikutuksen arvioimiseksi potilailla, jotka saivat rukaparibia 600 mg kahdesti vuorokaudessa. Potilailla, joilla oli lievä munuaisten vajaatoiminta (N=149; kreatiniinipuhdistuma 60–89 ml/min, Cockcroft-Gaultin menetelmällä arvioituna) esiintyi noin 15 % korkeampi vakaan tilan AUC-arvo kuin potilailla, joilla munuaisten toiminta oli normaalia (N=147; kreatiniinipuhdistuma vähintään 90 ml/min). Kohtalaista munuaisten vajaatoimintaa sairastavilla potilailla (N=76; kreatiniinipuhdistuma 30–59 ml/min) vakaan tilan AUC-arvo oli 33 % korkeampi. Rukaparibin farmakokineettiset ominaisuudet potilailla, joilla kreatiniinipuhdistuma on alle 30 ml/min, tai dialyysissa olevilla potilailla, eivät ole tiedossa (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Yleinen toksisuus
Suun kautta otettavan rukaparibin ei-kliinisten toksisuustutkimusten tulokset olivat yleisesti yhdenmukaisia kliinisissä tutkimuksissa havaittujen haittatapahtumien kanssa. Enintään 3 kuukautta kestäneissä toistuvan annoksen toksisuustutkimuksissa, jotka toteutettiin rotilla ja koirilla, kohdeelimet olivat ruoansulatus-, hematopoieettinen ja lymfopoieettinen järjestelmä. Nämä tulokset ilmenivät altistuksilla, jotka olivat pienempiä kuin suositellulla annoksella hoidetuilla potilailla havaitut altistukset. Ne kumoutuivat suurelta osin 4 viikon sisällä annostelun lopettamisesta.
In vitro-ympäristössä rukaparibin IC50 –arvo ihmisen ether-à-go-go:hun liittyvään geeniin (hERG) nähden oli 22.6 µM. Tämä on suunnilleen 13-kertaa suurempi kuin Cmax –pitoisuus potilailla suositellulla annoksella.
Rukaparibin laskimoon anto rotilla ja koirilla aiheuttaa sydänvaikutuksia korkealla Cmax –pitoisuudella (5,4–7,3 kertaa korkeampi kuin potilailla), mutta ei matalammalla Cmax –pitoisuudella (1,3–3,8 kertaa suurempi kuin potilailla). Sydänvaikutuksia ei havaittu annettaessa rukaparibia suun kautta toistuvan annoksen toksisuustutkimuksissa, joissa rukaparibin Cmax –pitoisuus vastasi potilailla havaittua pitoisuutta. Vaikka sydänvaikutuksia ei havaittu suun kautta annon jälkeen, laskimoreittiä käyttävien tutkimusten tulosten ja turvallisuusmarginaalien perusteella sydänvaikutuksia potilailla ei voida poissulkea, kun rukaparibia annetaan suun kautta.
Karsinogeenisuus
Rukaparibin käyttöä koskien ei ole tehty karsinogeenisuustutkimuksia.
Genotoksisuus
Rukaparibi ei ollut mutageeninen bakteerien käänteismutaatiotestissä (Amesin testi). Rukaparibi indusoi rakenteellisia kromosomipoikkeamia ihmisen lymfosyyttien kromosomipoikkeavuuksien in vitro-määrityksessä.
Lisääntymistoksisuus
Rottien alkion ja sikiön kehityksen osalta rukaparibi oli yhteydessä implantaation jälkeiseen keskenmenoon altistuksilla, jotka olivat noin 0,04-kertaisia ihmisen AUC-arvoon nähden suositellulla annoksella.
Rukaparibin käyttöä koskien ei ole tehty hedelmällisyystutkimuksia. Koiraan ja naaraan hedelmällisyyteen kohdistuvia vaikutuksia ei havaittu 3 kuukauden pituisissa yleisissä toksisuustutkimuksissa rotilla ja koirilla altistuksilla, jotka olivat 0,09–0,3-kertaisia ihmisen AUCarvoon nähden suositellulla annoksella. Mahdollista riskiä ei voida poissulkea havaitun turvallisuusmarginaalin perusteella. Tämän lisäksi rukaparibin vaikutusmekanismin perusteella se voi kuitenkin mahdollisesti heikentää hedelmällisyyttä ihmisillä.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa
Natriumtärkkelysglykolaatti (tyyppi A)
Kolloidinen vedetön piidioksidi
Magnesiumstearaatti
Rubraca 200 mg kalvopäällysteiset tabletit
Tabletin päällyste
Polyvinyylialkoholi (E1203)
Titaanidioksidi (E171)
Makrogoli 4000 (E1521)
Talkki (E553b)
Briljanttisininen FCF-alumiinilakka (E133)
Indigokarmiinialumiinilakka (E132)
Rubraca 250 mg kalvopäällysteiset tabletit
Tabletin päällyste
Polyvinyylialkoholi (E1203)
Titaanidioksidi (E171)
Makrogoli 4000 (E1521)
Talkki (E553b)
Rubraca 300 mg kalvopäällysteiset tabletit
Tabletin päällyste
Polyvinyylialkoholi (E1203)
Titaanidioksidi (E171)
Makrogoli 4000 (E1521)
Talkki (E553b)
Keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
5 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RUBRACA tabletti, kalvopäällysteinen
200 mg (L:ei) 60 kpl (2223,60 €)
250 mg (L:ei) 60 kpl (2337,10 €)
300 mg (L:ei) 60 kpl (2433,58 €)
PF-selosteen tieto
HDPE-pullo, jossa on polypropyleeni (PP)-induktiotiivistesuljin, sisältää 60 tablettia. Yksi pahvipakkaus sisältää yhden pullon.
Valmisteen kuvaus:
Rubraca 200 mg kalvopäällysteinen tabletti
Sininen, 11 mm kokoinen, pyöreä kalvopäällysteinen tabletti, johon on kaiverrettu merkintä “C2”.
Rubraca 250 mg kalvopäällysteinen tabletti
Valkoinen, 11 × 15 mm kokoinen, vinoneliön muotoinen kalvopäällysteinen tabletti johon on kaiverrettu merkintä “C25”.
Rubraca 300 mg kalvopäällysteiset tabletit
Keltainen, 8 × 16 mm kokoinen, soikea kalvopäällysteinen tabletti, johon on kaiverrettu merkintä “C3”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
RUBRACA tabletti, kalvopäällysteinen
200 mg 60 kpl
250 mg 60 kpl
300 mg 60 kpl
- Ylempi erityiskorvaus (100 %). Niraparibi ja rukaparibi: Munasarjasyövän, munanjohtimen syövän tai primaarin vatsakalvon syövän hoito erityisin edellytyksin (1510).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Niraparibi ja rukaparibi: Monoterapiana uusiutunutta platinaherkkää korkean pahanlaatuisuusasteen seroosia epiteelistä munasarja-, munanjohdin- tai primaaria vatsakalvon syöpää sairastavien aikuispotilaiden hoito erityisin edellytyksin (3009).
ATC-koodi
L01XK03
Valmisteyhteenvedon muuttamispäivämäärä
Yhteystiedot
Flöjelbergsgatan 12
431 37 Mölndal
Sweden
https://www.ghnpharma.com