
WELIREG tabletti, kalvopäällysteinen 40 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 40 mg beltsutifaania (belzutifan).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
Munuaissolukarsinooma
WELIREG on tarkoitettu käytettäväksi monoterapiana aikuispotilailla, joilla on edennyt kirkassoluinen munuaissolukarsinooma ja joiden tauti on edennyt heidän saatuaan kaksi tai useamman hoitolinjan, joihin on sisältynyt PD-(L)1:n estäjä ja vähintään kaksi VEGF-kasvutekijään kohdennettua täsmähoitoa.
von Hippel-Lindaun (VHL) tautiin liittyvät kasvaimet
WELIREG on tarkoitettu käytettäväksi monoterapiana aikuispotilailla, joilla on von Hippel-Lindaun tauti ja jotka tarvitsevat hoitoa von Hippel-Lindaun tautiin liittyvän paikallisen munuaissolukarsinooman tai paikallisten von Hippel-Lindaun tautiin liittyvien keskushermoston hemangioblastoomien tai haiman neuroendokriinisten kasvainten (pNET) vuoksi ja joille paikalliset toimenpiteet eivät sovi.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Syövän hoitoon perehtyneen erikoislääkärin on aloitettava hoito ja valvottava sen toteuttamista.
Annostus
Suositeltu WELIREG-annos on 120 mg beltsutifaania (kolme 40 mg:n tablettia) kerran vuorokaudessa, samaan aikaan joka päivä.
Hoitoa on jatkettava, kunnes tauti etenee tai ilmenee toksisia vaikutuksia, joita ei voida hyväksyä.
Annoksen jääminen väliin
Jos WELIREG-annos jää väliin, se voidaan ottaa mahdollisimman pian samana päivänä. Seuraavana päivänä jatketaan tavanomaisen vuorokausiannoksen käyttöä. Väliin jääneen annoksen korvaamiseksi ei pidä ottaa ylimääräisiä tabletteja.
Jos potilas oksentaa milloin tahansa WELIREG-valmisteen ottamisen jälkeen, hänen ei pidä ottaa uutta annosta. Seuraava annos otetaan seuraavana päivänä.
Annosmuutokset
Taulukossa 1 on yhteenveto WELIREG-valmisteen suositelluista annosmuutoksista haittavaikutusten vuoksi.
Taulukko 1: Suositellut annosmuutokset
| Haittavaikutukset | Vaikeusaste* | Annoksen muuttaminen |
| Anemia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Aste 3 (hemoglobiini < 80 g/l; verensiirron tarve) |
|
| Aste 4 (hengenvaaralliset seuraukset tai kiireellisen toimenpiteen tarve) |
| |
| Hypoksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Aste 3 oireeton (pienentynyt happisaturaatio levossa [esim. pulssioksimetrilla mitattu arvo < 88 % tai PaO2 ≤ 55 mmHg]) |
|
| Aste 3 oireinen (pienentynyt happisaturaatio levossa [esim. pulssioksimetrilla mitattu arvo < 88 % tai PaO2 ≤ 55 mmHg]) |
| |
| Aste 4 (hengenvaarallinen hengitystien vaarantuminen; kiireellisen toimenpiteen [esim. trakeostomian tai intuboinnin] tarve) |
| |
| Muut haittavaikutukset (ks. kohta Haittavaikutukset) | Aste 3 |
|
| Aste 4 |
|
*Perustuu National Cancer Institute ‑organisaation (NCI) Common Terminology Criteria for Adverse Events ‑luokitukseen (CTCAE), versio 5.0
Erityisryhmät
Iäkkäät
Annoksen muuttamista ei suositella iäkkäiden potilaiden hoidossa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttamista ei suositella, jos potilaalla on munuaisten vajaatoiminta, mukaan lukien loppuvaiheen munuaissairaus (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttamista ei suositella, jos potilaalla on lievä (kokonaisbilirubiini ≤ viitealueen yläraja [ULN] ja aspartaattiaminotransferaasi [ASAT] > ULN tai kokonaisbilirubiini > 1 – 1,5 x ULN ja ASAT-arvo mikä tahansa) tai kohtalainen (kokonaisbilirubiini alueella > 1,5 x ULN ja ≤ 3 x ULN ja ASAT-arvo mikä tahansa tai Child–Pugh-luokka B) maksan vajaatoiminta. Beltsutifaania ei ole tutkittu potilailla, joilla on vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu (ks. kohta Farmakodynamiikka). Tietoja ei ole saatavilla.
Antotapa
WELIREG otetaan suun kautta.
Tabletit niellään kokonaisina, ja ne voidaan ottaa joko ruoan kanssa tai ilman ruokaa. Tabletteja ei saa jakaa, murskata eikä pureskella, sillä ei tiedetä, vaikuttaako tämä beltsutifaanin imeytymiseen.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Raskaus potilailla, joilla on VHL-tautiin liittyviä kasvaimia (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Anemia
Potilailla, jotka ovat saaneet beltsutifaania kliinisissä tutkimuksissa, on ilmoitettu anemiaa (ks. kohta Haittavaikutukset).
Potilaita on seurattava anemian varalta ennen beltsutifaanihoidon aloittamista ja säännöllisesti koko hoidon ajan. Jos potilaalle kehittyy asteen 3 anemia, beltsutifaanihoito on keskeytettävä ja potilasta on hoidettava tavanomaisen lääketieteellisen käytännön mukaisesti, muun muassa antamalla erytropoieesia stimuloivaa ainetta (ESA), kunnes anemia on korjautunut asteen ≤ 2 tasolle (lisätiedot, ks. erytropoieesia stimuloivien aineiden valmisteyhteenvedot). Jos asteen 3 anemia uusiutuu, beltsutifaanihoito on lopetettava. Jos potilaalle kehittyy asteen 4 anemia, beltsutifaanihoito on keskeytettävä; jos asteen 4 anemia uusiutuu, hoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Hypoksia
Potilailla, jotka ovat saaneet beltsutifaania kliinisissä tutkimuksissa, on ilmoitettu hypoksiaa (ks. kohta Haittavaikutukset). Potilaiden happisaturaatiota on seurattava pulssioksimetrialla ennen beltsutifaanihoidon aloittamista ja säännöllisesti koko hoidon ajan. Asteen 3 oireettomassa hypoksiassa on harkittava lisähapen antamista ja hoidon jatkamista tai keskeyttämistä. Jos hoito keskeytetään, beltsutifaanin antoa on jatkettava pienennetyllä annoksella. Jos potilaalla on asteen 3 oireinen hypoksia, beltsutifaanihoito on keskeytettävä, hypoksia on hoidettava ja beltsutifaanihoitoa on jatkettava pienennetyllä annoksella. Jos oireinen hypoksia uusiutuu edelleen, hoito on lopetettava. Asteen 4 hypoksian yhteydessä hoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Alkio- ja sikiötoksisuus: Naiset, jotka voivat tulla raskaaksi
Beltsutifaani saattaa aiheuttaa ihmisellä alkio- ja sikiövauriota, mukaan lukien sikiökuolemat (ks. kohdat Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta).
Jos potilas on nainen ja voi tulla raskaaksi, raskaustilanne on tarkastettava ennen beltsutifaanihoidon aloittamista.
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokkaita ehkäisymenetelmiä beltsutifaanihoidon aikana ja vähintään 1 viikon ajan viimeisen annoksen ottamisen jälkeen sikiöön kohdistuvan mahdollisen riskin vuoksi (ks. kohdat Yhteisvaikutukset ja Raskaus ja imetys).
Keskushermoston verenvuoto potilailla, joilla on VHL‑tautiin liittyviä keskushermoston hemangioblastoomia
Keskushermoston verenvuotoja, myös kuolemaan johtanut tapaus, on havaittu potilailla, joilla on VHL‑tautiin liittyviä keskushermoston hemangioblastoomia. Lääkärien on oltava varovaisia keskushermoston verenvuodon oireiden tai löydösten suhteen potilailla, joilla on VHL‑tautiin liittyviä keskushermoston hemangioblastoomia ja jotka saavat beltsutifaanihoitoa.
Tietoa apuaineista
Tämä lääkevalmiste sisältää natriumia alle 1 mmol (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
In vitro ‑tutkimukset ja farmakogenomiikan tutkimukset viittaavat siihen, että beltsutifaani metaboloituu UGT2B17:n ja CYP2C19:n välityksellä ja että beltsutifaani indusoi CYP3A4:n toimintaa pitoisuudesta riippuvaisella tavalla.
Beltsutifaanin vaikutus muihin lääkevalmisteisiin
Beltsutifaanin samanaikainen käyttö CYP3A4:n substraattien (kuten hormonaalisten ehkäisyvalmisteiden) kanssa pienentää CYP3A4:n substraattien pitoisuuksia, mikä saattaa huonontaa näiden substraattien tehoa. Tehon huononeminen saattaa olla voimakkaampaa potilailla, jotka ovat sekä UGT2B17:n että CYP2C19:n suhteen hitaita metaboloijia (ks. kohta Farmakokinetiikka). On vältettävä beltsutifaanin samanaikaista käyttöä sellaisten herkkien CYP3A4:n substraattien kanssa, joiden kohdalla hyvin vähäinen pitoisuuden pieneneminen saattaa johtaa substraatilla toteutetun hoidon epäonnistumiseen. Jos samanaikaista käyttöä ei voida välttää, herkän CYP3A4:n substraatin annosta suurennetaan kyseisen lääkkeen valmisteyhteenvedon mukaisesti.
Beltsutifaanin samanaikainen käyttö hormonaalisten ehkäisyvalmisteiden kanssa saattaa johtaa ehkäisyn pettämiseen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys) tai läpäisyvuotojen lisääntymiseen. Hormonaalisia ehkäisyvalmisteita käyttäviä potilaita on kehotettava käyttämään jotakin ei-hormonaalista ehkäisymenetelmää tai potilaiden miespuolisia kumppaneita on kehotettava käyttämään kondomia beltsutifaanihoidon aikana.
Eräässä kliinisessä tutkimuksessa 120 mg:n beltsutifaaniannosten toistuva anto kerran vuorokaudessa pienensi midatsolaamin AUC-arvoa (kuvaajan alle jäävä pinta‑ala) 40 %. Tämä viittaa siihen, että beltsutifaani on heikko CYP3A4:n indusori. Beltsutifaanin anto saattaa johtaa kohtalaiseen CYP3A4:n induktioon potilailla, joilla beltsutifaanialtistus plasmassa on suurempi (ks. kohta Farmakokinetiikka).
In vitro ‑tietojen perusteella beltsutifaanin odotetaan estävän MATE2‑K-toimintaa kliinisesti merkityksellisillä altistuksilla, eikä MATE1:n estoa voida sulkea pois.
Beltsutifaani on CYP2B6:n ja CYP2C8:n indusori in vitro. In vivo ‑tutkimuksia ei ole tehty. Samanaikainen käyttö beltsutifaanin kanssa saattaa pienentää herkkien CYP2B6:n ja/tai CYP2C8:n substraattien pitoisuuksia plasmassa kliinisesti merkittävästi.
Muiden lääkevalmisteiden vaikutukset beltsutifaaniin
Beltsutifaanin samanaikainen käyttö UGT2B17:n tai CYP2C19:n estäjien kanssa suurentaa beltsutifaanialtistusta plasmassa, mikä saattaa suurentaa beltsutifaaniin liittyvien haittavaikutusten ilmaantuvuutta ja vaikeusastetta. Potilaita on seurattava anemian ja hypoksian varalta, ja beltsutifaanin annosta on pienennettävä suositusten mukaisesti.
Voimakkaiden CYP2C19:n indusorien vaikutuksia beltsutifaanialtistukseen ei ole vielä tutkittu.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy miehillä ja naisilla
Jos potilas on nainen ja voi tulla raskaaksi, raskaustilanne on tarkistettava ennen beltsutifaanihoidon aloittamista.
Beltsutifaanin anto raskaana olevalle naiselle saattaa aiheuttaa alkio- ja sikiövauriota, mukaan lukien sikiökuolemat (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Prekliiniset tiedot turvallisuudesta). Naisille, jotka voivat tulla raskaaksi, on kerrottava sikiöön mahdollisesti kohdistuvasta riskistä.
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä beltsutifaanihoidon aikana ja vähintään 1 viikon ajan viimeisen annoksen ottamisen jälkeen. Beltsutifaanin käyttö saattaa huonontaa hormonaalisten ehkäisyvalmisteiden tehoa. Hormonaalisia ehkäisyvalmisteita käyttäviä potilaita on kehotettava käyttämään jotakin ei-hormonaalista ehkäisymenetelmää tai potilaiden miespuolisia kumppaneita on kehotettava käyttämään kondomia beltsutifaanihoidon aikana (ks. kohta Yhteisvaikutukset).
Raskaus
Beltsutifaanin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Munuaissolukarsinooma
Beltsutifaania ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tila edellytä hoitoa beltsutifaanilla.
von Hippel-Lindaun (VHL) tautiin liittyvät kasvaimet
Beltsutifaanin käyttö raskauden aikana on vasta-aiheista (ks. kohta Vasta-aiheet). Jos raskaus alkaa beltsutifaanihoidon aikana, hoito on lopetettava.
Imetys
Ei ole olemassa tietoja beltsutifaanin tai sen metaboliittien esiintymisestä ihmisen äidinmaidossa eikä niiden vaikutuksista imetettävään lapseen tai maidontuotantoon. On mahdollista, että imetettäville lapsille aiheutuu vakavia haittavaikutuksia, joten naisia on kehotettava olemaan imettämättä beltsutifaanihoidon aikana ja vähintään 1 viikon ajan viimeisen annoksen ottamisen jälkeen.
Hedelmällisyys
Eläimillä tehtyjen tutkimusten löydösten perusteella beltsutifaani saattaa heikentää lisääntymiskykyisten urosten ja naaraiden hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Potilaille on kerrottava tästä mahdollisesta riskistä. Ei tiedetä, onko hedelmällisyyteen kohdistuva vaikutus korjautuva.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Beltsutifaanilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Beltsutifaanin annon jälkeen saattaa esiintyä heitehuimausta ja väsymystä (ks. kohta Haittavaikutukset).
Potilaita on kehotettava olemaan ajamatta autoa ja käyttämättä koneita, kunnes he ovat kohtuullisen varmoja siitä, että beltsutifaanihoito ei vaikuta heihin haitallisesti.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Beltsutifaanin turvallisuutta on tutkittu 576 potilaalla, joilla oli edenneitä kiinteitä kasvaimia tai VHL-tautiin liittyviä paikallisia kasvaimia ja joille annettiin kliinisissä tutkimuksissa 120 mg beltsutifaania kerran vuorokaudessa. Beltsutifaanialtistuksen mediaanikesto oli 9,2 kuukautta (vaihteluväli 0,1–55,4 kuukautta).
Yleisimpiä beltsutifaanihoidon aikana esiintyneitä haittavaikutuksia olivat anemia (84,2 %), väsymys (42,7 %), pahoinvointi (24,1 %), hengenahdistus (21,4 %), heitehuimaus (17,9 %) ja hypoksia (16,3 %).
Yleisimpiä asteen 3 tai 4 haittavaikutuksia olivat anemia (28,8 %) ja hypoksia (12,2 %).
Yleisimpiä vakavia haittavaikutuksia olivat hypoksia (7,1 %), anemia (4,7 %) ja hengenahdistus (1,2 %).
Yleisimpiä beltsutifaanihoidon keskeyttämiseen johtaneita haittavaikutuksia olivat anemia (7,1 %), hypoksia (5,4 %), väsymys (2,6 %), pahoinvointi (2,4 %), hengenahdistus (1,7 %) ja heitehuimaus (1,6 %). Yleisimpiä haittavaikutuksia, jotka johtivat beltsutifaaniannoksen pienentämiseen, olivat hypoksia (6,3 %), anemia (3,8 %) ja väsymys (1,7 %). Yleisin beltsutifaanin käytön lopettamiseen johtanut haittavaikutus oli hypoksia (1,4 %).
Haittavaikutustaulukko
Taulukossa 2 luetellaan beltsutifaanihoitoa saaneita potilaita koskevassa yhdistetyssä aineistossa (n = 576) tai markkinoilletulon jälkeen ilmoitetut haittavaikutukset. Haittavaikutukset on esitetty elinjärjestelmän ja esiintymistiheyden mukaan. Esiintymistiheydet määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000) ja hyvin harvinainen (< 1/10 000).
Taulukko 2: Haittavaikutukset beltsutifaanihoitoa saaneilla potilailla*
| Elinjärjestelmä | Kaikki asteet | Aste 3–4 |
| Veri ja imukudos | ||
| Anemia† | Hyvin yleinen | Hyvin yleinen |
| Hermosto | ||
| Heitehuimaus | Hyvin yleinen | - |
| Verisuonisto | ||
| Verenvuoto‡# | Hyvin yleinen | Yleinen |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hengenahdistus | Hyvin yleinen | Yleinen |
| Hypoksia | Hyvin yleinen | Hyvin yleinen |
| Ruoansulatuselimistö | ||
| Pahoinvointi | Hyvin yleinen | Melko harvinainen |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Väsymys | Hyvin yleinen | Yleinen |
| Tutkimukset | ||
| Painon nousu | Yleinen | Yleinen |
*Taulukossa 2 esitetyt haittavaikutusten esiintymistiheydet saattavat johtua osittain perussairaudesta.
†Anemiaan sisältyy anemia ja hemoglobiiniarvon lasku.
‡Tähän sisältyy erilaisia verenvuototapahtumia eri alueilla. Tapahtumia ei ole lueteltu erikseen.
Seuraavia verenvuototermejä esiintyi vähintään viidellä beltsutifaanihoitoa saaneella potilaalla: verivirtsaisuus, veriyskä, kontuusio ja nenäverenvuoto (mikä tahansa aste) ja verivirtsaisuus (asteet 3–4).
#Tähän sisältyy keskushermoston verenvuoto (yksi tapaus johti kuolemaan) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Valikoitujen haittavaikutusten kuvaus
Anemia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Anemiaa esiintyi 83 %:lla beltsutifaania saaneista potilaista, joilla oli edennyt munuaissolukarsinooma. 32 %:lla oli asteen 3 anemia ja 0,5 %:lla oli asteen 4 anemia. Mediaaniaika anemian alkamiseen oli 29 päivää (vaihteluväli 1 päivä – 27 kuukautta). Anemiapotilaista 22 % sai vain verensiirtoja, 20 % sai vain erytropoieesia stimuloivia aineita ja 14 % sai sekä verensiirtoja että erytropoieesia stimuloivia aineita. Erytropoieesia stimuloivien aineiden annosten mediaanimäärä potilasta kohden oli 6,5 (vaihteluväli 1–87). Potilaat saivat erytropoieesia stimuloivaa ainetta hemoglobiinipitoisuuksien ja lääkärin harkinnan mukaisesti (ks. kohta Farmakodynamiikka).
Anemiaa esiintyi 90,2 %:lla potilaista, joilla oli VHL-tautiin liittyviä kasvaimia ja jotka saivat beltsutifaania. 11,5 %:lla potilaista oli asteen 3 anemia. Mediaaniaika anemian alkamiseen (kaikkien asteiden tapahtumat) oli 30 päivää (vaihteluväli 1 päivä – 8 kuukautta). Anemiapotilaista 1,8 % sai vain verensiirtoja, 16,4 % sai vain erytropoieesia stimuloivia aineita ja 9,1 % sai sekä verensiirtoja että erytropoieesia stimuloivia aineita. Erytropoieesia stimuloivien aineiden annosten mediaanimäärä potilasta kohden oli 5 (vaihteluväli 1–35). Potilaat saivat erytropoieesia stimuloivaa ainetta hemoglobiinipitoisuuksien ja lääkärin harkinnan mukaisesti (ks. kohta Farmakodynamiikka).
Asteen 3 anemian ilmaantuvuus suureni beltsutifaanialtistuksen suurentuessa potilailla, joilla hemoglobiinipitoisuus oli lähtötilanteessa < 120 g/l (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hypoksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Hypoksiaa esiintyi 15 %:lla beltsutifaania saaneista potilaista, joilla oli edennyt munuaissolukarsinooma. 10 %:lla potilaista oli asteen 3 hypoksia ja 0,3 %:lla asteen 4 hypoksia. Hypoksiapotilaista 70 % sai happihoitoa. Mediaaniaika hypoksian alkamiseen oli 31 päivää (vaihteluväli 1 päivä – 21 kuukautta).
Hypoksiaa (aste 3) ilmoitettiin 1,6 %:lla potilaista, joilla oli VHL-tautiin liittyviä kasvaimia ja jotka saivat beltsutifaania. Aika hypoksian alkamiseen oli 56 päivää.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Beltsutifaanin yliannostukseen ei ole spesifistä hoitoa. Epäillyn yliannostuksen yhteydessä on tarvittaessa keskeytettävä beltsutifaanihoito ja aloitettava tukihoito. Suurin kliinisesti tutkittu beltsutifaaniannos oli 240 mg:n kokonaisvuorokausiannos (120 mg kahdesti vuorokaudessa tai 240 mg kerran vuorokaudessa). Asteen 3 hypoksiaa esiintyi, kun annos oli 120 mg kahdesti vuorokaudessa. Asteen 4 trombosytopeniaa esiintyi, kun annos oli 240 mg kerran vuorokaudessa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet. ATC-koodi: L01XX74
Vaikutusmekanismi
Beltsutifaani on hypoksiassa indusoituva tekijä 2 alfa -transkriptiotekijän (HIF‑2α) estäjä. Kun happipitoisuus on normaali, VHL-proteiini hajottaa HIF‑2α-tekijää. VHL-proteiinin toiminnan heikentyminen johtaa HIF‑2α:n kumuloitumiseen. Tämän seurauksena HIF‑2α siirtyy tumaan ja säätelee sellaisten geenien ekspressiota, joilla on yhteys solujen proliferaatioon, angiogeneesiin ja kasvainten kasvuun. Beltsutifaani sitoutuu HIF‑2α-tekijään. Hypoksian tai VHL-proteiinin toiminnan heikentymisen yhteydessä beltsutifaani salpaa HIF‑2α:n ja HIF‑1β:n vuorovaikutuksen, mikä johtaa HIF‑2α:n kohdegeenien transkription ja ekspression vähenemiseen.
Farmakodynaamiset vaikutukset
Kiertävän erytropoietiinin (EPO) pitoisuuksia potilaiden plasmassa seurattiin HIF‑2α:n eston farmakodynaamisena markkerina. EPO-pitoisuuksien pienenemisen todettiin riippuvan annoksesta/altistuksesta, ja pitoisuuksien pienenemisen todettiin tasaantuvan, kun altistus oli sellainen kuin yli 120 mg:n annoksilla kerran vuorokaudessa saavutetaan. EPO:n suppressio oli voimakkaimmillaan, kun beltsutifaania oli annettu 2 perättäisen viikon ajan (pitoisuudet pienenivät lähtötasosta keskimäärin noin 60 %). Keskimääräiset EPO-pitoisuudet palautuivat vähitellen lähtötasolle 12 hoitoviikon jälkeen.
Beltsutifaanin suositellulla annoksella (120 mg kerran vuorokaudessa) ei havaittu kliinisesti merkityksellisiä vaikutuksia QTc-aikaan.
Kliininen teho
Kliininen tutkimus aikuispotilailla, joilla oli edennyt munuaissolukarsinooma
Beltsutifaanin tehoa arvioitiin LITESPARK‑005-tutkimuksessa, joka oli avoin, satunnaistettu, aktiivikontrolloitu vaiheen 3 kliininen tutkimus. Tutkimuksessa vertailtiin beltsutifaania ja everolimuusia 746 potilaalla, joilla oli leikkaukseen soveltumaton, paikallisesti edennyt tai etäpesäkkeinen kirkassoluinen munuaissolukarsinooma. Tauti oli edennyt sen jälkeen, kun potilaat olivat saaneet PD-1/L1-toimintaan kohdennettuja immuuniaktivaation vapauttajia ja VEGF-reseptoriin kohdennettuja täsmähoitoja joko peräkkäin tai yhdistelmähoitona. Potilaat olivat voineet saada aiemmin enintään kolme hoito-ohjelmaa, ja heidän tautinsa oli oltava mitattavissa RECIST v1.1 ‑kriteerien mukaisesti. Tutkimuksesta suljettiin pois potilaat, joilla oli hypoksiaa, aktiivisia keskushermostoetäpesäkkeitä tai kliinisesti merkittävä sydänsairaus. Potilaat satunnaistettiin suhteessa 1:1 saamaan 120 mg beltsutifaania tai 10 mg everolimuusia suun kautta kerran vuorokaudessa. Satunnaistamisessa käytettiin ositustekijöinä International Metastatic RCC Database Consortium (IMDC) riskiluokkia (suotuisa vs. kohtalainen vs. huono ennuste) ja aiempien VEGF-reseptoriin kohdennettujen täsmähoitojen määrää (1 vs. 2–3).
Potilaiden tilanne arvioitiin radiologisesti viikolla 9 satunnaistamisen jälkeen, sitten 8 viikon välein viikolle 49 asti ja tämän jälkeen 12 viikon välein.
LITESPARK‑005-tutkimuksen 746 potilaasta 369 potilasta oli saanut kahden tai useamman hoitolinjan hoitoa, ja hoitoihin oli sisältynyt PD‑(L)1:n estäjä ja vähintään kaksi VEGF-kasvutekijään kohdennettua täsmähoitoa. Kyseisten potilaiden tiedot lähtötilanteessa olivat seuraavat: iän mediaani 63 vuotta (vaihteluväli 33–82 vuotta), 40 % oli vähintään 65-vuotiaita; 11 % oli vähintään 75-vuotiaita; 79 % oli miehiä; 78 % oli valkoihoisia; 12 % oli aasialaisia; 1 % oli mustaihoisia tai afrikkalaisamerikkalaisia; 42 %:lla ECOG-toimintakykyluokka oli 0 ja 56 %:lla ECOG-toimintakykyluokka oli 1. Aiemmat hoitolinjat: 17 % potilaista oli saanut kahden, 81 % oli saanut kolmen ja 2 % oli saanut neljän aiemman hoitolinjan hoitoa. Potilaiden jakautuminen IMDC:n riskiluokkiin oli seuraava: 22 %:lla oli suotuisa ennuste, 66 %:lla kohtalainen ennuste ja 12 %:lla huono ennuste.
Ensisijaiset tehoa mittaavat tulosmuuttujat olivat elinaika ilman taudin etenemistä (PFS), jonka mittasi sokkoutettu, riippumaton keskitetty arviointitoimikunta (BICR) käyttäen RECIST v1.1 ‑kriteerejä, ja kokonaiselinaika (OS). Toissijaisiin tehoa mittaaviin lopputulosmuuttujiin kuuluivat objektiivisten vasteiden osuus (ORR) ja vasteen kesto (DOR), jotka arvioi BICR käyttäen RECIST v1.1 ‑kriteerejä.
Tutkimuksessa todettiin koko tutkimuspopulaatiossa tilastollisesti merkitsevästi parempi PFS (riskisuhde [HR]: 0,75 [95 % CI 0,63; 0,90], p‑arvo 0,00077) ja ORR (21,9 % vs. 3,5 %, p‑arvo < 0,00001) beltsutifaanihoitoon satunnaistetuilla potilailla kuin everolimuusihoitoon satunnaistetuilla ennalta määritellyn välianalyysin ajankohtana (seuranta-ajan mediaani 13,5 kuukautta [vaihteluväli 0,2–31,8 kuukautta]).
Taulukossa 3 on tiivistelmä LITESPARK‑005-tutkimuksen keskeisistä tehon mittareista niiden potilaiden alaryhmässä, jotka olivat saaneet kahden tai useamman hoitolinjan hoitoa ja joiden saamiin hoitoihin oli sisältynyt PD‑(L)1:n estäjä ja vähintään kaksi VEGF‑kasvutekijään kohdennettua täsmähoitoa. Kaplan–Meier-kuvaajat elinajalle ilman taudin etenemistä ja kokonaiselinajalle esitetään kuvissa 1 ja 2.
Taulukko 3: Tehoa koskevat tulokset LITESPARK‑005-tutkimuksessa potilailla, jotka olivat saaneet kahden tai useamman hoitolinjan hoitoa, joihin oli sisältynyt PD‑(L)1:n estäjä ja vähintään kaksi VEGF‑kasvutekijään kohdennettua täsmähoitoa
| Päätetapahtuma | Beltsutifaani n = 187 | Everolimuusi n = 182 | |
| PFS* | |||
| Tapahtumien määrä, n (%) | 127 (67,9 %) | 130 (71,4 %) | |
| PFS:n mediaani†, kk (95 % CI) | 4,6 (3,5; 7,3) | 5,4 (3,8; 6,5) | |
| Riskisuhde (HR)‡ (95 % CI) | 0,73 (0,57; 0,94) | ||
| OS¶ | |||
| Tapahtumien määrä, n (%) | 128 (68,1 %) | 125 (68,7 %) | |
| OS:n mediaani†, kk (95 % CI) | 21,8 (17,4; 25,8) | 18,1 (14,2; 23,9) | |
| Riskisuhde (HR)‡ (95 % CI) | 0,94 (0,74; 1,21) | ||
| ORR*, % (n) (95 % CI) | 24,1 % (18,1; 30,8) | 3,3 % (1,2; 7,0) | |
| Täydellinen vaste, n (%) | 5 (2,7 %) | 0 (0 %) | |
| Osittainen vaste, n (%) | 40 (21,4 %) | 6 (3,3 %) | |
| Vasteen kesto* | |||
| Mediaani, kk (vaihteluväli) | Ei saavutettu (1,9+; 23,1+) | 17,2 (3,8; 17,2) | |
| * Perustuu BICR:n arvioimaan ensimmäiseen ennalta määriteltyyn välianalyysiin (seuranta-ajan mediaani 13,2 kk). † Perustana rajatulomenetelmä (Kaplan–Meierin menetelmä) rajatuille tiedoille. ‡ Perustuu Coxin regressiomalliin. ¶ Perustuu lopulliseen analyysiin (seuranta-ajan mediaani 18,9 kk). + Tarkoittaa edelleen jatkuvaa vastetta CI = Luottamusväli | |||
LITESPARK‑005‑tutkimuksessa mediaaniaika vasteen saavuttamiseen (TTR) oli 3,7 kuukautta (vaihteluväli 1,7–16,6) beltsutifaanihaarassa ja 3,0 kuukautta (vaihteluväli 1,8–5,4) everolimuusihaarassa (seuranta‑ajan mediaani 13,5 kuukautta) alaryhmässä, jonka potilaat olivat saaneet kahden tai useamman hoitolinjan hoitoa ja joiden saamiin hoitoihin oli sisältynyt PD‑(L)1:n estäjä ja vähintään kaksi VEGF‑kasvutekijään kohdennettua täsmähoitoa.
Kuva 1: Kaplan–Meier-kuvaaja elinajalle ilman taudin etenemistä hoitohaaroittain LITESPARK‑005-tutkimuksessa potilailla, jotka olivat saaneet kahden tai useamman hoitolinjan hoitoa, joihin oli sisältynyt PD‑(L)1:n estäjä ja vähintään kaksi VEGF‑kasvutekijään kohdennettua täsmähoitoa*
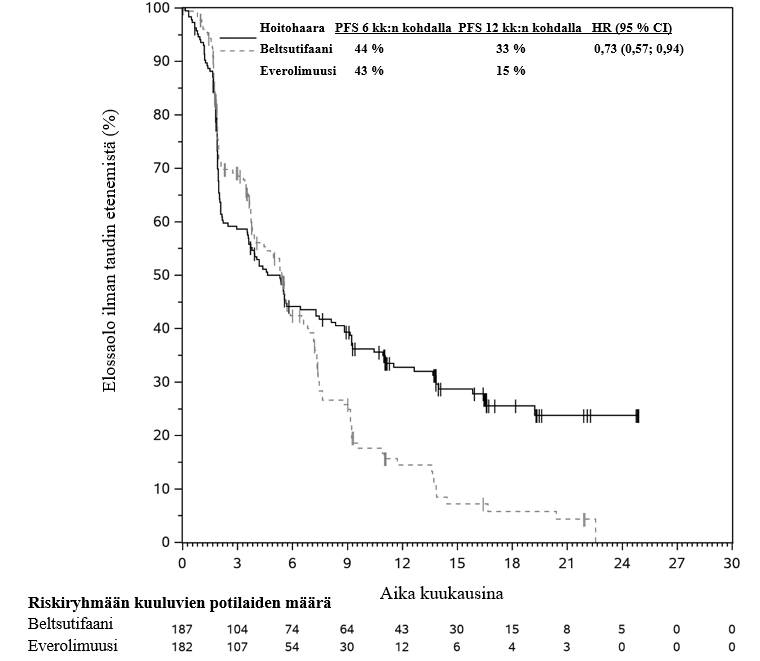
* Seuranta‑ajan mediaani 13,2 kk.
Kuva 2: Kaplan–Meier-kuvaaja kokonaiselinajalle hoitohaaroittain LITESPARK‑005-tutkimuksessa potilailla, jotka olivat saaneet kahden tai useamman hoitolinjan hoitoa joihin oli sisältynyt PD‑(L)1:n estäjä ja vähintään kaksi VEGF‑kasvutekijään kohdennettua täsmähoitoa*
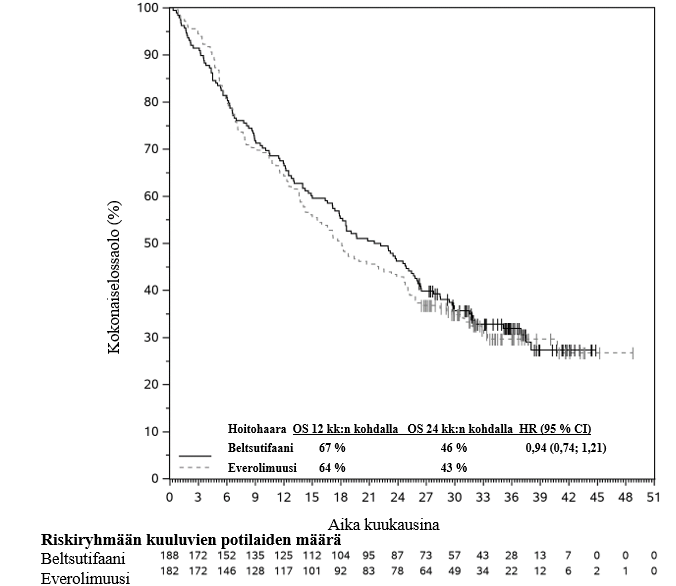
* Seuranta‑ajan mediaani 18,9 kk.
Kliininen tutkimus aikuispotilailla, joilla oli von Hippel-Lindaun (VHL) tautiin liittyviä kasvaimia
Beltsutifaanin tehoa tutkittiin LITESPARK‑004-tutkimuksessa, joka oli avoin vaiheen 2 kliininen tutkimus. Siihen osallistui 61 VHL-potilasta, joilla oli vähintään yksi mitattavissa oleva kiinteä kasvain (RECIST v1.1 ‑kriteerien mukaisesti määriteltynä) paikallisena munuaisissa ja jotka eivät tarvinneet välitöntä leikkaushoitoa. Potilailla saattoi olla myös muita VHL-tautiin liittyviä kasvaimia, kuten keskushermoston hemangioblastoomia tai pNET-kasvaimia. Potilaat saivat 120 mg beltsutifaania kerran vuorokaudessa. Potilaiden tilanne arvioitiin radiologisesti noin 12 viikon kuluttua hoidon aloittamisesta ja tämän jälkeen 12 viikon välein. Hoitoa jatkettiin, kunnes tauti eteni tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä. Potilaiden ECOG-toimintakykyluokan oli oltava 0 tai 1. Tutkimuksesta suljettiin pois potilaat, joilla oli mitä tahansa näyttöä etäpesäkkeisestä taudista (joko munuaissolukarsinoomasta tai muista VHL-tautiin liittyvistä kasvaimista), jos he tarvitsivat välittömästi kirurgisen toimenpiteen kasvaimensa hoitamiseksi, jos heille oli tehty mikä tahansa suuri kirurginen toimenpide tutkimukseenottoa edeltäneiden 4 viikon aikana, jos heillä oli ollut jokin merkittävä sydän- tai verisuonitapahtuma 6 kuukauden kuluessa ennen tutkimuslääkkeen antamista tai jos he olivat saaneet aiemmin systeemistä hoitoa VHL-tautiin liittyvän munuaissolukarsinooman vuoksi.
LITESPARK‑004-tutkimukseen osallistuneiden 61 potilaan populaation ominaisuudet olivat seuraavat: iän mediaani 41 vuotta, 3,3 % oli vähintään 65-vuotiaita; 52,5 % oli miehiä; 90,2 % oli valkoihoisia; 82,0 %:lla ECOG-toimintakykyluokka oli 0 ja 16,4 %:lla ECOG-toimintakykyluokka oli 1. 77 %:lle potilaista oli tehty aiemmin kirurginen toimenpide munuaissolukarsinooman vuoksi. Muita potilailla todettuja VHL-tautiin liittyviä kasvaimia olivat haimamuutokset (100,0 %), joista 36,1 % oli haiman neuroendokriinisia kasvaimia, sekä keskushermoston hemangioblastoomat (82,0 %) ja verkkokalvon angioomat (19,7 %).
Ensisijainen tehon päätetapahtuma VHL-tautiin liittyvän munuaissolukarsinooman hoidossa oli objektiivisten vasteiden osuus (ORR), joka mitattiin RECIST v1.1 ‑kriteerien mukaisesti radiologisen arvioinnin avulla ja jonka arvioi keskitetty, riippumaton arviointitoimikunta (IRC). Muihin tehon päätetapahtumiin kuuluivat vasteen kesto ja aika vasteen saavuttamiseen. Muihin VHL-tautiin liittyviin kasvaimiin liittyvä objektiivisten vasteiden osuus (ORR) ja näihin kasvaimiin liittyvä vasteen kesto arvioitiin toissijaisina tehon päätetapahtumina.
Taulukossa 4 on yhteenveto tehoa koskevista tuloksista VHL-tautiin liittyvien munuaissolukarsinoomakasvainten hoidossa LITESPARK‑004-tutkimuksessa. Tulokset perustuvat välianalyysiin, jonka toteutusajankohtana seuranta-ajan mediaani oli 49,7 kuukautta.
Taulukko 4: Tehoa koskevat tulokset VHL-tautiin liittyvien munuaissolukarsinoomakasvainten hoidossa LITESPARK‑004-tutkimuksessa
| Päätetapahtuma | Beltsutifaani n = 61 |
| ORR*, % (95 % CI) | 67,2 % (54,0; 78,7) |
| Täydellinen vaste | 11,5 % |
| Osittainen vaste | 55,7 % |
| Vasteen kesto† | |
| Mediaani, kk (vaihteluväli) | Ei saavutettu (8,6+; 44,4+) |
| Potilaat, joilla kesto ≥ 12 kk, % | 100,0 % |
| Aika vasteeseen | |
| Mediaani, kk (vaihteluväli) | 11,1 (2,7; 41,2) |
Tehoa koskevat tiedot, kun seuranta-ajan mediaani oli 49,7 kk (tiedonkeruun katkaisupäivä 3.4.2023)
* Vaste: paras objektiivinen vaste eli vahvistettu täydellinen tai osittainen vaste
† Perustuu Kaplan–Meier-estimaatteihin
+ Tarkoittaa edelleen jatkuvaa vastetta
Tehon päätetapahtumiin muiden VHL-tautiin liittyvien kasvainten hoidossa kuuluivat ORR ja vasteen kesto, jotka IRC arvioi RECIST v1.1 ‑kriteerien mukaisesti. Nämä tulokset esitetään taulukossa 5.
Taulukko 5: Beltsutifaanilla saavutetut tehoa koskevat tulokset muiden VHL-tautiin liittyvien kasvainten hoidossa
| Beltsutifaani n = 61 | ||
| Päätetapahtuma | Potilaat, joilla oli arviointikelpoinen keskushermoston hemangioblastooma n = 50 | Potilaat, joilla oli arviointikelpoinen haiman neuroendokriininen kasvain n = 22 |
| ORR*, % (95 % CI) | 48 % (33,7; 62,6) | 90,9 % (70,8; 98,9) |
| Täydellinen vaste | 8,0 % | 50,0 % |
| Osittainen vaste | 40,0 % | 40,9 % |
| Vasteen kesto† | ||
| Mediaani, kk (vaihteluväli) | Ei saavutettu (0,0+, 47,5+) | Ei saavutettu (11,0+, 48,3+) |
| Potilaat, joilla kesto ≥ 12 kk, % | 95,5 % | 100,0 % |
Tehoa koskevat tiedot, kun seuranta-ajan mediaani oli 49,7 kk (tiedonkeruun katkaisupäivä 3.4.2023)
* Vaste: paras objektiivinen vaste eli vahvistettu täydellinen tai osittainen vaste
† Perustuu Kaplan–Meier-estimaatteihin
+ Tarkoittaa edelleen jatkuvaa vastetta
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset WELIREG-valmisteen käytöstä munuaiskasvainten ja VHL-taudin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa tietoa käytöstä pediatristen potilaiden hoidossa).
Ehdollinen myyntilupa
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa. Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Beltsutifaanin farmakokinetiikka on samankaltainen terveillä tutkittavilla ja potilailla, joilla on kiinteitä kasvaimia, mukaan lukien edennyt munuaissolukarsinooma. Populaatiofarmakokineettisen analyysin perusteella simuloitu geometrinen keskiarvo vakaassa tilassa (CV %) on Cmax-arvon osalta 1,5 mikrog/ml (46 %) ja AUC0–24 h-arvon osalta 20,8 mikrog•h/ml (64 %) potilailla, joille annetaan 120 mg beltsutifaania. Vakaa tila saavutetaan noin 3 päivän kuluttua.
Imeytyminen
Kun beltsutifaania annettiin 120 mg:n kerta-annos suun kautta, beltsutifaanin huippupitoisuus plasmassa saavutettiin 1–2 tunnin kuluttua annoksen ottamisesta (mediaani Tmax).
Ruoan vaikutus
Runsasrasvainen, runsaskalorinen ateria viivästytti beltsutifaanin huippupitoisuuden saavuttamista noin 2 tuntia mutta ei vaikuttanut altistukseen (AUC). Cmax-arvo pieneni hieman (24 %) runsasrasvaisen, runsaskalorisen aterian nauttimisen jälkeen, mutta tämä ei ollut kliinisesti merkityksellistä. Beltsutifaani voidaan siis ottaa ruoasta riippumatta.
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella keskimääräinen jakautumistilavuus (CV%) on 120 l (28,2 %). 45 % beltsutifaanista sitoutuu plasman proteiineihin. Veren ja plasman beltsutifaanipitoisuuksien suhde on 0,88.
Biotransformaatio
Beltsutifaani metaboloituu pääasiassa UGT2B17:n ja CYP2C19:n välityksellä ja vähäisemmässä määrin CYP3A4:n välityksellä. Sekä UGT2B17:n että CYP2C19:n kohdalla esiintyy geneettistä polymorfismia (ks. Erityisryhmät – Sekä UGT2B17:n että CYP2C19:n suhteen hitaat metaboloijat).
Lääkkeiden yhteisvaikutusten arviointi in vitro
Beltsutifaani on UGT2B17:n, CYP2C19:n ja CYP3A4:n substraatti. Aktiivinen kuljetus ei vaikuta merkittävästi beltsutifaanin jakautumiseen. Beltsutifaani ei ole CYP-entsyymien, UGT-entsyymien eikä kuljettajaproteiinien estäjä lukuun ottamatta MATE2‑K:ta ja mahdollisesti MATE1:tä. Beltsutifaani ei indusoi CYP1A2-entsyymiä, mutta se indusoi CYP2B6‑, CYP2C8‑ ja CYP3A4-entsyymejä pitoisuudesta riippuvaisesti (ks. kohta Yhteisvaikutukset).
Eliminaatio
Populaatiofarmakokineettisen analyysin perusteella keskimääräinen puhdistuma (CV%) on 5,89 l/h (60,6 %) ja keskimääräinen eliminaation puoliintumisaika on noin 14 tuntia.
Kun radioaktiivisesti merkittyä beltsutifaania annettiin terveille tutkittaville suun kautta, noin 49,6 % annoksesta erittyi virtsaan ja 51,7 % ulosteeseen (lähinnä inaktiivisina metaboliitteina). Noin 6 % annoksesta erittyi kanta-aineena virtsaan.
Lineaarisuus
Plasmasta mitattu Cmax ja AUC suurenivat suhteessa annokseen annosvälillä 40 mg – 120 mg.
Erityisryhmät
Munuaisten vajaatoiminta
Terveillä tutkittavilla ja syöpäpotilailla tehdyn, beltsutifaania koskeneen populaatiofarmakokineettisen analyysin perusteella keskimääräisessä beltsutifaanialtistuksessa ei todettu kliinisesti merkittäviä eroja, kun tutkittavia, joiden munuaisten toiminta oli normaali, verrattiin tutkittaviin, joilla oli lievä tai keskivaikea munuaisten vajaatoiminta (glomerulusten laskennallisen suodatusnopeuden [eGFR:n] perusteella arvioituna). Asiaa koskeneessa farmakokineettisessä tutkimuksessa beltsutifaanialtistus (AUC0-INF) potilailla, joilla oli loppuvaiheen munuaissairaus, oli 6 % pienempi ennen hemodialyysia ja 14 % suurempi hemodialyysin jälkeen (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Terveillä tutkittavilla ja syöpäpotilailla tehdyn, beltsutifaania koskeneen populaatiofarmakokineettisen analyysin perusteella keskimääräisessä beltsutifaanialtistuksessa ei todettu kliinisesti merkittäviä eroja, kun tutkittavia, joiden maksan toiminta oli normaali (kokonaisbilirubiini ja ASAT ≤ ULN), verrattiin tutkittaviin, joilla oli lievä maksan vajaatoiminta (kokonaisbilirubiini ≤ ULN ja ASAT > ULN tai kokonaisbilirubiini > 1 – 1,5 x ULN ja ASAT-arvo mikä tahansa). Asiaa koskeneessa farmakokineettisessä tutkimuksessa beltsutifaanialtistus (AUC0-INF) oli 52 % suurempi potilailla, joilla oli kohtalainen maksan vajaatoiminta (Child–Pugh-luokka B). Potilaita, joilla on vaikea maksan vajaatoiminta, ei ole tutkittu (ks. kohta Annostus ja antotapa).
Sekä UGT2B17:n että CYP2C19:n suhteen hitaat metaboloijat
Potilailla, jotka ovat sekä UGT2B17:n että CYP2C19:n suhteen hitaita metaboloijia, on suurempi beltsutifaanialtistus. Tämä saattaa suurentaa beltsutifaanin haittavaikutusten ilmaantuvuutta ja vaikeusastetta, ja näitä potilaita on seurattava tarkoin (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Beltsutifaani metaboloituu pääasiassa UGT2B17:n ja CYP2C19:n välityksellä. Näiden entsyymien aktiivisuus vaihtelee henkilöillä, joilla on erilaisia geneettisiä variantteja. Tämä saattaa vaikuttaa beltsutifaanin pitoisuuksiin. Hitailla metaboloijilla tarkoitetaan henkilöitä, joilla katsotaan olevan vain vähän tai ei lainkaan entsyymiaktiivisuutta. Potilailla, jotka ovat sekä UGT2B17:n että CYP2C19:n suhteen hitaita metaboloijia, CYP3A4 voi olla merkittävä eliminaatioreitti.
Noin 15 % valkoihoisista, 11 % latinalaisamerikkalaisista, 6 % afrikkalaisamerikkalaisista, 38 % eteläaasialaisista ja 69 % itäaasialaisista on UGT2B17:n suhteen hitaita metaboloijia. Noin 2 % valkoihoisista, 1 % latinalaisamerikkalaisista, 5 % afrikkalaisamerikkalaisista, 8 % eteläaasialaisista ja 13 % itäaasialaisista on CYP2C19:n suhteen hitaita metaboloijia. Noin 0,4 % valkoihoisista, 0,1 % latinalaisamerikkalaisista, 0,3 % afrikkalaisamerikkalaisista, 3 % eteläaasialaisista ja 9 % itäaasialaisista on hitaita metaboloijia sekä UGT2B17:n että CYP2C19:n suhteen. Japanilaisessa väestössä oletetaan, että noin 77 % on UGT2B17:n suhteen hitaita metaboloijia, noin 19 % on CYP2C19:n suhteen hitaita metaboloijia ja noin 15 % on molempien suhteen hitaita metaboloijia. Yhdysvaltain väestössä oletetaan, että noin 16 % on UGT2B17:n suhteen hitaita metaboloijia, noin 3 % on CYP2C19:n suhteen hitaita metaboloijia ja noin 0,5 % on molempien suhteen hitaita metaboloijia, kun otetaan huomioon tärkeimpien rotuun ja etniseen taustaan perustuvien ryhmien ilmoitettu osuus Yhdysvaltain väestössä.
CYP2C19:n ja UGT2B17:n hitaan metabolian vaikutusta beltsutifaanialtistukseen arvioitiin populaatiofarmakokineettisessä analyysissä. Populaatiofarmakokineettisen mallin mukaan potilailla, jotka ovat CYP2C19:n ja/tai UGT2B17:n suhteen hitaita metaboloijia, on oletettavasti (vakaan tilan AUC0–24 h:n perusteella) 1,3-kertainen altistus (CYP2C19:n suhteen hitaat metaboloijat), 2,7-kertainen altistus (UGT2B17:n suhteen hitaat metaboloijat) tai 3,3-kertainen altistus (molempien suhteen hitaat metaboloijat) verrattuna tyypilliseen referenssipotilaaseen (joka on UGT2B17:n suhteen nopea metaboloija ja CYP2C19:n suhteen nopea tai keskitason metaboloija) suositusannosta käytettäessä. Tehoa, turvallisuutta ja riski-hyötyprofiilia koskevien altistusvasteanalyysien perusteella ei suositella annosmuutoksia.
Iän, sukupuolen, etnisen taustan, rodun ja painon vaikutukset
Populaatiofarmakokineettisen analyysin perusteella iällä (vaihteluväli 19–90 vuotta), sukupuolella, etnisellä taustalla, rodulla ja painolla (vaihteluväli 42,1–166 kg) ei ole kliinisesti merkityksellistä vaikutusta beltsutifaanin farmakokinetiikkaan. Altistuksessa saattaa olla eroja eri rotujen välillä, sillä metaboliaentsyymien esiintymistiheydet ovat erilaiset (ks. Erityisryhmät – Sekä UGT2B17:n että CYP2C19:n suhteen hitaat metaboloijat).
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttama toksisuus
Rotilla ja koirilla tehdyissä, enintään 3 kuukauden pituisissa tutkimuksissa, joissa arvioitiin toistuvan oraalisen altistuksen aiheuttamaa toksisuutta, havaittiin anemiaa kaikilla annostasoilla, myös ihmisen altistusta pienemmillä altistuksilla. Anemia oli korjautuvaa, mutta ilmiö on silti merkityksellinen ihmisten kohdalla.
Karsinogeneesi
Beltsutifaania arvioitiin 26 viikon pituisessa karsinogeenisuustutkimuksessa siirtogeenisillä rasH2‑hiirillä enintään annoksilla 600 mg/kg/vrk. Tällainen annos vastaa altistusta, joka on enintään 28‑kertainen verrattuna ihmisen altistukseen ohjeen mukaisella annoksella. Tutkimuksessa ei tehty beltsutifaaniin liittyviä neoplastisia löydöksiä millään annostasolla eikä todettu karsinogeenisuusriskiä.
Mutageneesi
Beltsutifaani ei ollut genotoksinen in vitro bakteereilla tehdyissä mutageneesi- ja mikrotumakokeissa eikä rotan mikrotumakokeessa in vivo, kun altistus oli 1,7-kertainen verrattuna ihmisen altistukseen.
Lisääntymistoksisuus
Beltsutifaanilla ei ole tehty hedelmällisyyttä koskevia tutkimuksia. Rotilla tehdyssä 3 kuukauden pituisessa tutkimuksessa, jossa arvioitiin toistuvan altistuksen aiheuttamaa toksisuutta, todettiin korjautumatonta kivesten atrofiaa/degeneraatiota ja oligotsoospermiaa altistuksilla, jotka olivat pienempiä kuin ihmisen altistus suositusannoksella 120 mg vuorokaudessa. Koirilla ei todettu kiveksiin kohdistuvaa toksisuutta, kun altistus oli samankaltainen kuin ihmisen altistus. Rotilla ja koirilla tehdyissä 3 kuukauden pituisissa toksisuustutkimuksissa ei todettu löydöksiä naaraiden lisääntymiselimissä, mutta HIF-2α:n toiminta hiiren kohdussa vaikuttaa alkion kiinnittymiseen ja tiineyden alkamiseen. Beltsutifaanille altistumisen aiheuttama HIF-2α:n esto voi häiritä alkion kiinnittymistä, jolloin naaraiden hedelmällisyys heikentyy.
Rotilla tehdyssä alkion- ja sikiönkehitystä koskeneessa tutkimuksessa beltsutifaanin anto organogeneesin aikana johti jopa 100-prosenttiseen alkio- ja sikiökuolleisuuteen, sikiöiden painon pienenemiseen ja sikiöiden luuston poikkeavuuksiin, kun altistus oli samankaltainen tai pienempi kuin ihmisen altistus suositellulla annoksella 120 mg vuorokaudessa.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Hypromelloosiasetaattisuksinaatti
Mikrokiteinen selluloosa (E460)
Mannitoli (E421)
Kroskarmelloosinatrium (E468)
Vedetön kolloidinen piidioksidi (E551)
Magnesiumstearaatti (E470b)
Kalvopäällyste
Polyvinyylialkoholi (E1203)
Titaanidioksidi (E171)
Makrogoli (E1521)
Talkki (E553b)
Indigokarmiinialumiinipigmentti (E132)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
WELIREG tabletti, kalvopäällysteinen
40 mg (L:ei) 90 fol (3x30 monipakkaus) (16638,10 €)
PF-selosteen tieto
Alumiini/alumiiniläpipainopakkaus.
Pakkaus, jossa 90 kalvopäällysteistä tablettia (kolme 30 tabletin koteloa).
Valmisteen kuvaus:
Sininen, soikea tabletti, jonka koko on noin 13 x 8 mm ja jonka toisella puolella on merkintä ”177”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
WELIREG tabletti, kalvopäällysteinen
40 mg 90 fol
- Ei korvausta.
ATC-koodi
L01XX74
Valmisteyhteenvedon muuttamispäivämäärä
08.01.2026
Yhteystiedot
 MSD FINLAND OY
MSD FINLAND OY Keilaniementie 1, PL 46
02151 Espoo
09 804 650
www.msd.fi
info@msd.fi