
ELAHERE infuusiokonsentraatti, liuosta varten 5 mg/ml
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
1 ml infuusiokonsentraattia sisältää 5 mg mirvetuksimabi-soravtansiinia.
Yksi injektiopullo sisältää 100 mg mirvetuksimabi-soravtansiinia 20 ml:ssa.
Mirvetuksimabi-soravtansiini on FRα-reseptoriin kohdennettu vasta-aine-lääkekonjugaatti (ADC). ADC koostuu FRα-reseptorin IgG1-alatyypin monoklonaalisesta vasta-aineesta, joka on valmistettu yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa ja liitetty pilkkoutuvalla linkkerillä (voihappo, 4-(2-pyridinyyliditio)-2-sulfo-1-(2,5-diokso-1-pyrrolidinyyli) -esteri) DM4-maytansinoidiin, joka on tubuliininestäjä. Mirvetuksimabi-soravtansiini sisältää keskimäärin 3,4 DM4-kantajamolekyyliä yhtä FRα-reseptorivasta-ainemolekyyliä kohden.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 2,11 mg polysorbaatti 20:tä per injektiopullo.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
ELAHERE on tarkoitettu monoterapiana folaattireseptori alfa (FRα) -positiivisen, platinaresistentin, korkean pahanlaatuisuusasteen seroosin epiteliaalisen munasarjasyövän, munanjohdinsyövän tai primaarin peritoneaalisen syövän hoitoon aikuispotilaille, jotka ovat saaneet 1–3:a aikaisempaa systeemistä hoitolinjaa (ks. kohta Annostus ja antotapa).
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Syöpälääkkeiden käyttöön perehtyneen lääkärin on aloitettava ELAHERE-hoito ja valvottava sitä.
Potilaiden valinta
Hoidettavilla potilailla on oltava positiivinen FRα-kasvainstatus, mikä määritellään solukalvon kohtalaiseksi (2+) ja/tai voimakkaaksi (3+) värjäytymiseksi ≥ 75 %:ssa elinkykyisistä kasvainsoluista immunohistokemiallisessa tutkimuksessa (IHC) tähän käyttöön tarkoitetulla CE-merkityllä in vitro -diagnostisella (IVD) laitteella arvioituna. Jos CE-merkittyä in vitro -diagnostista laitetta ei ole saatavilla, on käytettävä vaihtoehtoista validoitua testiä.
Annostus
Suositeltu ELAHERE-annos on 6 mg/kg potilaan korjatun ihannepainon (adjusted ideal body weight, AIBW) mukaan infuusiona laskimoon 3 viikon välein (21 päivän sykli), kunnes tauti etenee tai toksisuus ei ole hyväksyttävää. Korjattuun ihannepainoon perustuva annostus vähentää altistuksen vaihtelua ali- tai ylipainoisilla potilailla.
ELAHERE-kokonaisannos lasketaan kunkin potilaan korjatun ihannepainon (AIBW) perusteella seuraavan kaavan mukaan:
Naisten ihannepaino (kg) = 0,9 * pituus (cm) – 92
Korjattu ihannepaino (AIBW) = ihannepaino (kg) + 0,4 * (todellinen paino [kg] – ihannepaino)
Esimerkiksi naispotilaalle, jonka pituus on 165 cm ja paino 80 kg
| Laske ensin ihannepaino: | Ihannepaino = 0,9 * 165 – 92 = 56,5 kg |
| Laske sen jälkeen korjattu ihannepaino: | Korjattu ihannepaino = 56,5 + 0,4 * (80 – 56,5) = 65,9 kg |
Esilääkitys
Esilääkitys infuusioon liittyviin reaktioihin, pahoinvointiin ja oksenteluun
Anna taulukossa 1 luetellut esilääkkeet ennen kutakin ELAHERE-infuusiota infuusioon liittyvien reaktioiden, pahoinvoinnin ja oksentelun esiintymisen vähentämiseksi ja vaikeusasteen lievittämiseksi.
Taulukko 1: Esilääkitys ennen kutakin ELAHERE-infuusiota
| Esilääke | Antoreitti | Esimerkki (tai vastaava) | Annon ajoitus ennen ELAHERE-infuusiota |
| Kortikosteroidi | laskimoon | deksametasoni 10 mg | vähintään 30 minuuttia ennen |
| Antihistamiini | suun kautta tai laskimoon | difenhydramiini 25–50 mg | |
| Kuumelääke | suun kautta tai laskimoon | asetaminofeeni tai parasetamoli 325–650 mg | |
| Antiemeetti | suun kautta tai laskimoon | 5-HT3 -serotoniinireseptorin antagonisti tai muu asianmukainen vaihtoehto | ennen kutakin annosta muiden esilääkkeiden annon jälkeen |
Potilaille, joilla esiintyy pahoinvointia ja/tai oksentelua, voidaan tämän jälkeen harkita lisäantiemeettien antoa tarpeen mukaan.
Potilaille, joilla esiintyy asteen ≥ 2 infuusioon liittyvä reaktio, on harkittava lisäesilääkityksenä 8 mg:n deksametasoniannosta kaksi kertaa päivässä (tai vastaava) ELAHERE-valmisteen antoa edeltävänä päivänä.
Silmätutkimus ja esilääkitys
Silmätutkimus:Ennen ELAHERE-hoidon aloittamista on tehtävä silmätutkimus, joka sisältää näöntarkkuuden mittauksen ja rakolamppututkimuksen. Jos potilaalle ilmaantuu uusia silmäoireita tai ne pahenevat, tutkimus on toistettava ennen seuraavaa annosta. Potilaille, joilla esiintyy asteen ≥ 2 silmähaittavaikutuksia, silmien lisätutkimuksia on tehtävä vähintään joka toinen sykli sekä kliinisen tarpeen mukaan, kunnes oireet häviävät tai ne palautuvat lähtötasolle.
Silmiin paikallisesti käytettävät steroidit:Potilaille, joilla havaitaan rakolamppututkimuksessa merkkejä asteen ≥ 2 sarveiskalvon haittavaikutuksista (keratopatia), suositellaan sekundaarista profylaksiaa silmiin paikallisesti käytettävillä steroideilla seuraavissa ELAHERE-sykleissä, ellei potilaan silmälääkäri pidä tällaisen hoidon riskejä sen hyötyjä suurempina.
- Potilaita on neuvottava käyttämään steroidisilmätippoja infuusiopäivänä ja sitä seuraavina 7 päivänä jokaisessa seuraavassa ELAHERE-syklissä (ks. taulukko 3).
- Potilaita on ohjeistettava odottamaan vähintään 15 minuuttia silmiin paikallisesti käytettävän steroidin annostelun jälkeen ennen kosteuttavien silmätippojen tiputtamista.
Silmiin paikallisesti käytettävän steroidihoidon aikana silmänpaineen mittaus ja rakolamppututkimus on tehtävä säännöllisesti.
Kosteuttavat silmätipat:On suositeltavaa kehottaa potilaita käyttämään kosteuttavia silmätippoja koko ELAHERE-hoidon ajan.
Annosmuutokset
Ennen kunkin syklin alkamista potilasta on kehotettava ilmoittamaan kaikista uusista tai pahentuneista oireista hoitavalle lääkärille tai muulle terveydenhuollon ammattilaiselle.
Potilaille, joille ilmaantuu uusia tai pahentuneita silmäoireita, on tehtävä silmätutkimus ennen valmisteen antoa. Hoitavan lääkärin on arvioitava potilaan silmätutkimusraportti ennen antoa ja määritettävä ELAHERE-annos pahempioireisen silmän löydösten vaikeusasteen perusteella.
Taulukoissa 2 ja 3 on annettu ohjeet annoksen pienentämiseen ja annosmuutoksiin haittavaikutusten vuoksi. Antoaikataulu 3 viikon antoväleineen on pidettävä ennallaan.
Taulukko 2: Annoksen pienentäminen
| ELAHERE-hoidon annostaso | |
| Aloitusannos | 6 mg/kg korjatun ihannepainon mukaan |
| Ensimmäinen annoksen pienennys | 5 mg/kg korjatun ihannepainon mukaan |
| Toinen annoksen pienennys | 4 mg/kg korjatun ihannepainon mukaan* |
* Hoito on lopetettava pysyvästi potilailla, jotka eivät siedä annosta 4 mg/kg korjatun ihannepainon mukaan.
Taulukko 3: Annosmuutokset haittavaikutusten vuoksi
| Haittavaikutus | Haittavaikutuksen vaikeusaste* | Annosmuutos |
Sarveiskalvotulehdus/keratopatia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset) | Ei-konfluentti pinnallinen sarveiskalvotulehdus/keratopatia | Seuraa |
| Konfluentti pinnallinen sarveiskalvotulehdus/keratopatia, sarveiskalvon epiteelivaurio tai parhaan laseilla korjatun näöntarkkuuden heikentyminen vähintään 3 rivillä | Keskeytä hoito, kunnes pinnallinen sarveiskalvotulehdus/keratopatia on lievittynyt vähintään ei-konfluentiksi tai hävinnyt, ja jatka sen jälkeen samalla annostasolla. Harkitse annoksen pienentämistä potilaille, joilla konfluentti sarveiskalvotulehdus/keratopatia uusiutuu parhaasta tukihoidosta huolimatta, tai joiden silmätoksisuus jatkuu yli 14 päivän ajan. | |
| Sarveiskalvon haavauma tai stromaalinen sameus tai paras laseilla korjattu kaukonäöntarkkuus 6/60 tai huonompi | Keskeytä hoito, kunnes pinnallinen sarveiskalvotulehdus/keratopatia on lievittynyt vähintään ei-konfluentiksi tai hävinnyt, ja jatka sen jälkeen yhtä tasoa pienemmällä annoksella. | |
| Sarveiskalvon perforaatio | Lopeta hoito pysyvästi | |
Pneumoniitti (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset) | Aste 1 | Seuraa |
| Aste 2 | Keskeytä hoito, kunnes haittavaikutus on astetta 1 tai lievempi, ja jatka sen jälkeen samalla annostasolla tai harkitse annoksen pienentämistä, jos toistuu, kesto on yli 28 päivää, tai lääkärin harkinnan mukaan. | |
| Aste 3 tai 4 | Lopeta hoito pysyvästi | |
Perifeerinen neuropatia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset) | Aste 2 | Keskeytä hoito, kunnes aste 1 tai lievempi, ja jatka sen jälkeen yhtä tasoa pienemmällä annoksella. |
| Aste 3 tai 4 | Lopeta hoito pysyvästi | |
Infuusioon liittyvät reaktiot/ yliherkkyys (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset) | Aste 1 | Jatka samalla infuusionopeudella |
| Aste 2 |
| |
| Aste 3 tai 4 |
| |
Hematologiset haittavaikutukset (ks. kohta Haittavaikutukset.) | Aste 3 tai 4 | Keskeytä hoito, kunnes aste 1 tai lievempi, ja jatka sen jälkeen yhtä tasoa pienemmällä annoksella. |
Muut haittavaikutukset (ks. kohta Haittavaikutukset.) | Aste 3 | Keskeytä hoito, kunnes aste 1 tai lievempi, ja jatka sen jälkeen yhtä tasoa pienemmällä annoksella. |
| Aste 4 | Lopeta hoito pysyvästi |
*: NCI CTCAE -haittatapahtumatermistön (National Cancer Institute Common Terminology Criteria for Adverse Events) version 5.0 mukaan, ellei toisin ole mainittu.
Erityisryhmät
Pediatriset potilaat
Ei ole asianmukaista käyttää ELAHERE-valmistetta pediatrisille potilaille epiteliaalisen munasarjasyövän, munanjohdinsyövän tai primaarin peritoneaalisen syövän hoitoon (ks. kohta Farmakodynamiikka).
Iäkkäät
ELAHERE-annoksen muuttamista ≥ 65‑vuotiaille potilaille ei suositella (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
ELAHERE-annoksen muuttamista lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastaville potilaille (kreatiniinipuhdistuma [CLcr] 30 – < 90 ml/min) ei suositella. ELAHERE-hoitoa ei ole arvioitu potilailla, joilla on vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma [CLcr] 15 – < 30 ml/min) tai loppuvaiheen munuaissairaus, eikä mahdollista annoksen muuttamisen tarvetta näille potilaille voida määrittää (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
ELAHERE-annoksen muuttamista lievää maksan vajaatoimintaa sairastaville potilaille (kokonaisbilirubiini ≤ viitealueen yläraja [ULN] ja aspartaattiaminotransferaasi [ASAT] > ULN tai kokonaisbilirubiini > 1–1,5 × ULN ja mikä tahansa ASAT) ei suositella (ks. kohta Farmakokinetiikka).
ELAHERE-valmisteen käyttöä on vältettävä potilaille, joilla on keskivaikea tai vaikea maksan vajaatoiminta (kokonaisbilirubiini > 1,5 × ULN ja mikä tahansa ASAT).
Antotapa
ELAHERE annetaan infuusiona laskimoon nopeudella 1 mg/min. Jos infuusio on hyvin siedetty 30 minuutin jälkeen, infuusionopeutta voidaan lisätä 3 mg:aan/min. Jos infuusio on hyvin siedetty 30 minuutin jälkeen nopeudella 3 mg/min, infuusionopeutta voidaan lisätä 5 mg:aan/min.
Yhteensopimattomuudet, ks. kohta Yhteensopimattomuudet.
ELAHERE on laimennettava 5 %:n glukoosiliuoksella ennen antoa infuusiona laskimoon. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
ELAHERE-valmisteen saa antaa ainoastaan infuusiona laskimoon. Infuusioon on käytettävä 0,2 µm:n tai 0,22 µm:n polyeetterisulfonista (PES) valmistettua in-line-suodatinta (ks. erityiset käsittely- ja hävittämisohjeet kohdasta Käyttö- ja käsittelyohjeet).
Ennen lääkevalmisteen käsittelyä tai antamista tehtävät varotoimet
Tämä lääkevalmiste sisältää sytotoksisen komponentin, joka on kovalenttisesti sitoutunut monoklonaaliseen vasta-aineeseen (ks. erityiset käsittely- ja hävittämisohjeet kohdasta Käyttö- ja käsittelyohjeet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Silmähäiriöt
Mirvetuksimabi-soravtansiini voi aiheuttaa vaikeita silmähaittavaikutuksia, mukaan lukien näön heikentymistä (pääasiassa näön sumentumista), keratopatiaa (sarveiskalvon sairauksia), silmien kuivuutta, valonarkuutta ja silmäkipua (ks. kohdat Vaikutus ajokykyyn ja koneidenkäyttökykyyn ja Haittavaikutukset).
Potilaat on ohjattava silmälääkärille tai optometristille silmätutkimusta varten ennen mirvetuksimabi-soravtansiini-hoidon aloittamista.
Ennen kunkin syklin alkamista potilasta on kehotettava ilmoittamaan kaikista uusista tai pahentuneista silmäoireista hoitavalle lääkärille tai pätevälle terveydenhuollon ammattilaiselle.
Silmäoireiden ilmaantuessa potilaalle on tehtävä silmätutkimus, ja raportti silmätutkimuksesta on arvioitava, minkä jälkeen mirvetuksimabi-soravtansiinin annosta voidaan tarvittaessa muuttaa löydösten vaikeusasteen perusteella (ks. kohta Annostus ja antotapa).
Kosteuttavien silmätippojen käyttö mirvetuksimabi-soravtansiini-hoidon aikana on suositeltavaa. Potilaille, joille ilmaantuu asteen ≥ 2 sarveiskalvon haittavaikutuksia, suositellaan silmiin paikallisesti käytettäviä steroideja seuraavissa mirvetuksimabi-soravtansiini-sykleissä (ks. kohta Annostus ja antotapa).
Lääkärin on seurattava potilasta silmätoksisuuden varalta ja keskeytettävä mirvetuksimabi-soravtansiini-hoito, pienennettävä annosta tai lopetettava hoito pysyvästi silmähaittavaikutusten vaikeusasteen ja keston perusteella (ks. kohta Annostus ja antotapa).
Potilaita on neuvottava välttämään piilolinssien käyttöä mirvetuksimabi-soravtansiini-hoidon aikana, ellei terveydenhuollon ammattilainen kehota käyttämään niitä.
Pneumoniitti
Mirvetuksimabi-soravtansiinilla hoidettavilla potilailla voi esiintyä vaikeita, henkeä uhkaavia tai kuolemaan johtavia interstitiaalisia keuhkosairauksia (ILD), mukaan lukien pneumoniittia (ks. kohta Haittavaikutukset).
Potilaita on seurattava pneumoniitin keuhko-oireiden ja merkkien varalta. Niitä saattavat olla hypoksia, yskä, hengenahdistus tai radiologisessa tutkimuksessa havaittavat interstitiaaliset infiltraatit. Tällaisten oireiden johtuminen infektiosta, hyvän- tai pahanlaatuisesta kasvaimesta tai muista syistä on suljettava pois asianmukaisten tutkimusten avulla.
Jos potilaalle ilmaantuu pitkään jatkuva tai uusiutuva asteen 2 pneumoniitti, mirvetuksimabi-soravtansiini-hoito on keskeytettävä, kunnes oireet ovat lievittyneet asteeseen ≤ 1, ja annoksen pienentämistä on harkittava. Mirvetuksimabi-soravtansiini-hoito on lopetettava pysyvästi kaikilla potilailla, joilla on asteen 3 tai 4 pneumoniitti (ks. kohta Annostus ja antotapa). Oireettomien potilaiden mirvetuksimabi-soravtansiini-hoitoa voidaan jatkaa huolellisessa seurannassa.
Perifeerinen neuropatia
Mirvetuksimabi-soravtansiini-hoidon yhteydessä on esiintynyt perifeeristä neuropatiaa, mukaan lukien asteen ≥ 3 reaktioita (ks. kohta Haittavaikutukset).
Potilaita on seurattava neuropatian merkkien ja oireiden, kuten parestesian, kihelmöinnin tai polttavan tunteen, neuropaattisen kivun, lihasheikkouden tai tuntohäiriöiden, varalta. Jos potilaalle ilmaantuu uusi tai pahentunut perifeerinen neuropatia, mirvetuksimabi-soravtansiini-hoito on keskeytettävä, annosta on pienennettävä tai hoito on lopetettava pysyvästi perifeerisen neuropatian vaikeusasteen perusteella (ks. kohta Annostus ja antotapa).
Alkioon ja sikiöön kohdistuva toksisuus
Vaikutusmekanisminsa perusteella mirvetuksimabi-soravtansiini saattaa vahingoittaa alkiota ja sikiötä raskaana olevalle potilaalle annettuna, sillä se sisältää genotoksisen yhdisteen (DM4) ja vaikuttaa aktiivisesti jakautuviin soluihin.
Potilaiden, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä mirvetuksimabi-soravtansiini-hoidon aikana ja 7 kuukauden ajan viimeisen annoksen jälkeen (ks. kohta Raskaus ja imetys).
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää 2,11 mg polysorbaatti 20:tä per injektiopullo.
Yhteisvaikutukset
ELAHERE-valmisteella ei ole tehty kliinisiä lääkeyhteisvaikutustutkimuksia.
DM4 on CYP3A4:n substraatti. ELAHERE-valmisteen samanaikainen käyttö voimakkaiden CYP3A4:n estäjien kanssa saattaa lisätä altistusta konjugoimattomalle DM4:lle (ks. kohta Farmakokinetiikka), mikä saattaa lisätä ELAHERE-valmisteen haittavaikutusten riskiä (ks. kohta Haittavaikutukset). Jos samanaikaista käyttöä voimakkaiden CYP3A4:n estäjien (esim. seritinibi, klaritromysiini, kobisistaatti, idelalisibi, itrakonatsoli, ketokonatsoli, nefatsodoni, posakonatsoli, ritonaviiri, telitromysiini, vorikonatsoli) kanssa ei voida välttää, potilasta on seurattava huolellisesti haittavaikutusten varalta. Voimakkaat CYP3A4:n indusorit (esim. fenytoiini, rifampisiini, karbamatsepiini) saattavat vähentää altistusta konjugoimattomalle DM4:lle.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Raskausstatus on varmistettava potilailla, jotka voivat tulla raskaaksi, ennen mirvetuksimabi-soravtansiini-hoidon aloittamista.
Potilaiden, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä mirvetuksimabi-soravtansiini-hoidon aikana ja 7 kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Vaikutusmekanisminsa perusteella mirvetuksimabi-soravtansiini voi vahingoittaa alkiota ja sikiötä raskaana olevalle potilaalle annettuna, sillä se sisältää genotoksisen yhdisteen (DM4) ja vaikuttaa aktiivisesti jakautuviin soluihin (ks. kohdat Farmakodynamiikka ja Prekliiniset tiedot turvallisuudesta). Ihmisen immunoglobuliini G:n (IgG) tiedetään läpäisevän istukan, joten on mahdollista, että mirvetuksimabi-soravtansiini siirtyy raskaana olevasta potilaasta kehittyvään sikiöön. Mirvetuksimabi-soravtansiinin käytöstä raskaana oleville potilaille ei ole olemassa tietoja, eikä raskauden aikaiseen käyttöön liittyviä riskejä siten voida arvioida. Mirvetuksimabi-soravtansiinin lisääntymis- tai kehitystoksisuutta ei tutkittu eläimillä.
ELAHERE-valmisteen käyttöä raskaana oleville potilaille ei suositella, ja potilaille on kerrottava mahdollisista sikiöön kohdistuvista riskeistä siinä tapauksessa, että potilas tulee tai haluaa tulla raskaaksi. Potilaiden, jotka tulevat raskaaksi, on otettava välittömästi yhteyttä hoitavaan lääkäriinsä. Huolellinen seuranta on suositeltavaa, jos potilas tulee raskaaksi ELAHERE-hoidon aikana tai 7 kuukauden kuluessa viimeisen annoksen jälkeen.
Imetys
Ei tiedetä, erittyvätkö mirvetuksimabi-soravtansiini ja/tai sen metaboliitit ihmisillä rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida sulkea pois, sillä ihmisen immunoglobuliini G:n (IgG) tiedetään siirtyvän rintamaitoon. ELAHERE-valmisteen käytön aikana ei pidä imettää eikä 1 kuukauteen viimeisestä annoksesta.
Hedelmällisyys
Mirvetuksimabi-soravtansiinilla tai DM4:llä ei ole tehty hedelmällisyyttä koskevia tutkimuksia. ELAHERE-valmisteen vaikutuksesta ihmisen hedelmällisyyteen ei ole olemassa tietoja. Koska ELAHERE-valmisteen vaikutusmekanismi johtaa mikrotubulusten toiminnan häiriintymiseen ja nopeasti jakautuvien solujen kuolemaan, on kuitenkin mahdollista, että valmisteella on hedelmällisyyteen kohdistuvia vaikutuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
ELAHERE-valmisteella on kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Jos potilailla esiintyy näköhäiriöitä, perifeeristä neuropatiaa, väsymystä tai heitehuimausta mirvetuksimabi-soravtansiini-hoidon aikana, heitä on neuvottava olemaan ajamatta ja käyttämättä koneita, kunnes oireiden häviäminen kokonaan on vahvistettu.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimpiä mirvetuksimabi-soravtansiini-hoitoon liittyviä haittavaikutuksia olivat näön sumentuminen (43 %), pahoinvointi (41 %), ripuli (39 %), väsymys (35 %), vatsakipu (30 %), keratopatia (29 %), silmien kuivuus (27 %), ummetus (26 %), oksentelu (23 %), vähentynyt ruokahalu (22 %), perifeerinen neuropatia (20 %), päänsärky (19 %), voimattomuus (18 %), suurentunut ASAT-arvo (16 %) ja nivelkipu (16 %).
Yleisimmin raportoituja vakavia haittavaikutuksia olivat pneumoniitti (4 %), ohutsuolen tukos (3 %), suolitukos (3 %), pleuraeffuusio (2 %), vatsakipu (2 %), dehydraatio (1 %), ummetus (1 %), pahoinvointi (1 %), askites (1 %) ja trombosytopenia (< 1 %).
Yleisimpiä annoksen pienentämiseen tai viivästymiseen johtaneita haittavaikutuksia olivat näön sumentuminen (17 %), keratopatia (10 %), silmien kuivuus (5 %), neutropenia (5 %), sarveiskalvotulehdus (4 %), kaihi (3 %), heikentynyt näöntarkkuus (3 %), trombosytopenia (3 %), perifeerinen neuropatia (3 %) ja pneumoniitti (3 %).
Hoito lopetettiin pysyvästi haittavaikutuksen vuoksi 12 %:lla mirvetuksimabi-soravtansiini-hoitoa saaneista potilaista. Yleisimpiä hoidon pysyvään lopettamiseen johtaneita haittavaikutuksia olivat ruoansulatuselimistön häiriöt (4 %), hengityselimien, rintakehän ja välikarsinan häiriöt (3 %), veren ja imukudoksen häiriöt (1 %), hermoston häiriöt (1 %) ja silmähäiriöt (1 %).
Haittavaikutustaulukko
Haittavaikutusten esiintyvyydet perustuvat neljän kliinisen tutkimuksen yhdistettyihin tietoihin, jotka käsittivät 682 epiteliaalista munasarjasyöpää, munanjohdinsyöpää tai primaaria peritoneaalista syöpää (joihin viitataan jäljempänä yhdistetysti epiteliaalisen munasarjasyövän lyhenteellä EOC) sairastavaa potilasta, jotka saivat mirvetuksimabi-soravtansiinia annoksella 6 mg/kg korjatun ihannepainon mukaan 3 viikon välein. Mirvetuksimabi-soravtansiini-hoidon mediaanikesto oli 19,1 viikkoa (vaihteluväli: 3–132 viikkoa).
Haittavaikutusten esiintyvyydet kliinisissä tutkimuksissa perustuvat kaikista syistä johtuneiden haittatapahtumien esiintyvyyksiin. Syy-yhteys näiden haittatapahtumien ja lääkevalmisteen välillä on perusteellisen arvioinnin jälkeen vähintään kohtuullisen todennäköinen.
Esiintyvyysluokat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000). Haittavaikutukset on kussakin esiintyvyysluokassa esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 4: Luettelo kaikenasteisista haittavaikutuksista potilailla, jotka saivat mirvetuksimabi-soravtansiini-hoitoa kliinisissä tutkimuksissa
| Elinjärjestelmäluokka | Esiintyvyysluokka | Haittavaikutukset |
| Infektiot | Hyvin yleinen | Virtsatieinfektio |
| Veri ja imukudos | Hyvin yleinen | Anemia, trombosytopenia |
| Yleinen | Neutropenia | |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Vähentynyt ruokahalu, hypomagnesemia |
| Yleinen | Hypokalemia, dehydraatio | |
| Psyykkiset häiriöt | Yleinen | Unettomuus |
| Hermosto | Hyvin yleinen | Perifeerinen neuropatia1, päänsärky |
| Yleinen | Dysgeusia, heitehuimaus | |
| Silmät | Hyvin yleinen | Keratopatia2, kaihi3, näön sumentuminen4, valonarkuus, silmäkipu, silmien kuivuus5 |
| Yleinen | Epämukava tunne silmässä6 | |
| Verisuonisto | Yleinen | Hypertensio |
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Pneumoniitti7, hengenahdistus, yskä |
| Ruoansulatuselimistö | Hyvin yleinen | Ripuli, vatsakipu8, ummetus, vatsan turvotus, oksentelu, pahoinvointi |
| Yleinen | Askites, ruokatorven refluksitauti, suutulehdus, dyspepsia | |
| Maksa ja sappi | Yleinen | Hyperbilirubinemia |
| Iho ja ihonalainen kudos | Yleinen | Kutina |
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Nivelkipu |
| Yleinen | Lihaskipu, selkäkipu, raajakipu, lihasspasmit | |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Väsymys |
| Yleinen | Kuume | |
| Tutkimukset | Hyvin yleinen | Suurentunut aspartaattiaminotransferaasiarvo, suurentunut alaniiniaminotransferaasiarvo |
| Yleinen | Suurentunut veren alkalisen fosfataasin pitoisuus, suurentunut gammaglutamyylitransferaasiarvo, alentunut paino | |
| Vammat, myrkytykset ja hoitokomplikaatiot | Yleinen | Infuusioon liittyvä reaktio / yliherkkyys9 |
1 Perifeerinen neuropatia -ryhmätermi kattaa termit hypestesia, perifeerinen neuropatia, neurotoksisuus, parestesia, perifeerinen motorinen neuropatia, perifeerinen sensomotorinen neuropatia, perifeerinen sensorinen neuropatia ja polyneuropatia (ks. kohta Valikoitujen haittavaikutusten kuvaus).
2 Keratopatia-ryhmätermi kattaa termit sarveiskalvon kysta, sarveiskalvon kertymät, sarveiskalvon häiriö, sarveiskalvon epiteliaaliset mikrokystat, sarveiskalvon epiteelin vaurio, sarveiskalvon eroosio, sarveiskalvosamentuma, sarveiskalvon pigmentaatio, sarveiskalvotulehdus, interstitiaalinen sarveiskalvotulehdus, keratopatia, sarveiskalvon kantasolupuutos ja pisteinen sarveiskalvotulehdus (ks. kohta Valikoitujen haittavaikutusten kuvaus).
3 Kaihi-ryhmätermi kattaa termit kaihi, kuorikaihi ja tumakaihi (ks. kohta Valikoitujen haittavaikutusten kuvaus).
4 Näön sumentuminen -ryhmätermi kattaa termit silmän akkomodaatiohäiriö, kaksoiskuvat, kaukotaittoisuus, ikänäkö, taittovirhe, näön sumentuminen, näön heikentyminen, heikentynyt näöntarkkuus ja lasiaiskellujat (ks. kohta Valikoitujen haittavaikutusten kuvaus).
5 Silmien kuivuus -ryhmätermi kattaa termit silmien kuivuus ja vähentynyt kyynelnesteen eritys (ks. kohta Valikoitujen haittavaikutusten kuvaus).
6 Epämukava tunne silmässä -ryhmätermi kattaa termit silmän ärsytys, silmän kutina, vierasesineen tunne silmässä ja epämukava tunne silmässä (ks. kohta Valikoitujen haittavaikutusten kuvaus).
7 Pneumoniitti-ryhmätermi kattaa termit interstitiaalinen keuhkosairaus, organisoituva keuhkokuume, pneumoniitti, keuhkofibroosi ja hengitysvajaus (ks. kohta Valikoitujen haittavaikutusten kuvaus).
8 Vatsakipu-ryhmätermi kattaa termit epämukava tunne vatsassa, vatsakipu, alavatsakipu ja ylävatsakipu.
9 Infuusioon liittyvä reaktio / yliherkkyys -ryhmätermi kattaa SMQ:n ”Yliherkkyys” kapeat termit ja termit kasvojen punoitus, eryteema, silmäluomen eryteema.
Valikoitujen haittavaikutusten kuvaus
Silmähäiriöt
Silmähaittavaikutuksia (ryhmätermit) esiintyi 59 %:lla mirvetuksimabi-soravtansiini-hoitoa saaneista EOC-potilaista. Asteen 3 silmähaittavaikutuksia esiintyi 11 %:lla potilaista ja asteen 4 tapahtumia < 1 %:lla potilaista. Yleisimpiä asteen ≥ 3 silmähaittavaikutuksia olivat näön sumentuminen (5 %) ja keratopatia (5 %) (ryhmätermejä) sekä kaihi (4 %).
Mediaaniaika ensimmäisen silmähaittavaikutuksen ilmaantumiseen oli 5,1 viikkoa (vaihteluväli: 0,1–68,6 viikkoa). Silmähaittavaikutuksia saaneista potilaista 53 %:lla haittavaikutukset hävisivät kokonaan (aste 0) ja 38 %:lla ne paranivat osittain (määriteltiin vaikeusasteen lievittymisenä vähintään yhdellä asteella pahimmasta asteesta). Viimeisellä seurantakäynnillä 0,3 %:lla potilaista (2/682) oli ≥ asteen 3 silmähaittavaikutuksia (yhdellä potilaalla asteen 3 heikentynyt näöntarkkuus ja yhdellä potilaalla asteen 4 kaihi).
Silmähaittavaikutukset johtivat annoksen viivästymiseen 24%:lla potilaista ja annoksen pienenemiseen 15%:lla potilaista. Mirvetuksimabi-soravtansiini-hoito lopetettiin pysyvästi silmähaittavaikutusten vuoksi 1 %:lla potilaista.
Pneumoniitti
Pneumoniittia (ryhmätermit) esiintyi 10 %:lla mirvetuksimabi-soravtansiini-hoitoa saaneista EOC-potilaista. Asteen 3 tapahtumia esiintyi 0,9 %:lla potilaista (6/682) ja asteen 4 tapahtumia 0,2 %:lla potilaista (1/682). Kaksi potilasta (0,3 %) kuoli hengitysvajauksen takia. Toisella näistä potilaista (0,2 %) oli samanaikainen asteen 1 pneumoniitti sekä ruumiinavauksessa vahvistettuja keuhkometastaaseja. Toisella näistä potilaista (0,2 %) ei ollut samanaikaista pneumoniittia ja hengitysvajauksen etiologia oli epäselvä.
Mediaaniaika pneumoniitin ilmaantumiseen oli 18,1 viikkoa (vaihteluväli: 1,6–97,0 viikkoa). Pneumoniitti johti mirvetuksimabi-soravtansiinin annosten siirtämiseen 3 %:lla potilaista, annosten pienentämiseen 1 %:lla potilaista ja hoidon pysyvään lopettamiseen 3 %:lla potilaista.
Perifeerinen neuropatia
Perifeeristä neuropatiaa (ryhmätermit) esiintyi 36 %:lla mirvetuksimabi-soravtansiini-hoitoa kliinisissä tutkimuksissa saaneista EOC-potilaista. Asteen 3 perifeeristä neuropatiaa esiintyi 3 %:lla potilaista.
Mediaaniaika perifeerisen neuropatian ilmaantumiseen oli 5,9 viikkoa (vaihteluväli: 0,1–126,7 viikkoa). Perifeerinen neuropatia johti mirvetuksimabi-soravtansiinin annosten siirtämiseen 2 %:lla potilaista, annosten pienentämiseen 4 %:lla potilaista ja hoidon pysyvään lopettamiseen 0,7 %:lla potilaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Mirvetuksimabi-soravtansiinin yliannostukseen ei ole tunnettua hoitoa/vastalääkettä. Yliannostustapauksessa potilasta on tarkkailtava huolellisesti haittavaikutusten merkkien ja oireiden varalta, ja oireenmukainen hoito on aloitettava tarvittaessa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Syöpälääkkeet ja immuunivasteen muuntajat, monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, muut monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet. ATC-koodi: L01FX26
Vaikutusmekanismi
Mirvetuksimabi-soravtansiini on vasta-aine-lääkekonjugaatti. Vasta-aine on muunneltu immunoglobuliini G1, joka on kohdennettu folaattireseptori alfaan (FRα). Vasta-ainekomponentin tarkoitus on sitoutua munasarjasyöpäsolujen pinnalla ilmentyvään FRα-reseptoriin. DM4 on mikrotubulusten estäjä, joka on liitetty vasta-aineeseen pilkkoutuvalla linkkerillä. FRα-reseptoriin sitouduttuaan mirvetuksimabi-soravtansiini internalisoituu, mikä johtaa DM4:n solunsisäiseen vapautumiseen proteolyyttisen pilkkoutumisen seurauksena. DM4 häiritsee solunsisäisen mikrotubulusverkoston toimintaa, mikä pysäyttää solusyklin ja aiheuttaa apoptoottisen solukuoleman.
Farmakodynaamiset vaikutukset
Sydämen sähköfysiologia
Hyväksytyllä suositellulla annoksella mirvetuksimabi-soravtansiini ei aiheuttanut QTc-ajan keskimääräistä pidentymistä > 10 millisekunnilla pitoisuus-QTc-analyysin perusteella.
Kliininen teho ja turvallisuus
Tutkimus IMGN853‑0416 (MIRASOL)
Mirvetuksimabi-soravtansiinin tehoa ja turvallisuutta arvioitiin tutkimuksessa IMGN853‑0416, joka oli avoin, aktiivikontrolloitu, satunnaistettu, kaksi hoitoryhmää käsittävä vaiheen 3 monikeskustutkimus platinaresistenttiä, pitkälle edennyttä, korkean pahanlaatuisuusasteen seroosia epiteliaalista munasarjasyöpää, primaaria peritoneaalista syöpää tai munanjohdinsyöpää sairastavilla potilailla, joiden kasvaimet (mukaan lukien arkistoitu kudosnäyte) olivat FRα-positiivisia FOLR1 (FOLR1-2.1) RxDx -testillä määritettynä (solukalvon kohtalainen [2] ja/tai voimakas [3] värjäytyminen ≥ 75 %:ssa elinkykyisistä kasvainsoluista immunohistokemiallisessa tutkimuksessa [IHC]).
Platinaresistentti tauti määriteltiin EOC:ksi, joka uusiutui 6 kuukauden kuluessa viimeisestä platina-annoksesta.
Tutkimukseen ei otettu potilaita, joilla oli primaari platinarefraktaarinen tauti, potilaita, joiden ECOG-toimintakykyluokka oli ≥ 2, tai potilaita, joilla oli aktiivisia tai kroonisia sarveiskalvon häiriöitä, jatkuvaa hoitoa vaativia silmäsairauksia, asteen ≥ 2 perifeerinen neuropatia tai ei‑infektioosi interstitiaalinen keuhkosairaus (ILD)/pneumoniitti.
Potilaat satunnaistettiin suhteessa 1:1 saamaan joko ELAHERE-valmistetta annoksella 6 mg/kg korjatun ihannepainon mukaan laskimoon (N = 227) kunkin 3 viikon syklin päivänä 1 tai jotakin seuraavista, tutkijan ennen satunnaistamista valitsemista solunsalpaajahoidoista (N = 226):
- paklitakseli (Pac) annoksella 80 mg/m2 kerran viikossa 4 viikon syklinä
- pegyloitu liposomaalinen doksorubisiini (PLD) annoksella 40 mg/m2 neljän viikon välein
- topotekaani (Topo) annoksella 4 mg/m2 neljän viikon syklien päivinä 1, 8 ja 15 tai annoksella 1,25 mg/m2 kunkin 21 vuorokauden syklin päivinä 1–5.
Satunnaistaminen ositettiin aikaisempien hoitolinjojen lukumäärän (1, 2 tai 3) ja tutkijan valitseman solunsalpaajahoidon (Pac, PLD tai Topo) mukaan. Hoitoa annettiin taudin etenemiseen, kuolemaan, suostumuksen perumiseen tai ei-hyväksyttävän toksisuuden ilmaantumiseen asti.
Ensisijainen tehon tulosmittari oli elossaoloaika ilman taudin etenemistä (PFS), joka perustui tutkijan RECIST 1.1 -kriteerien mukaiseen arvioon. Objektiivinen vasteosuus (ORR) ja kokonaiselossaoloaika (OS) olivat keskeisiä toissijaisia tehon tulosmittareita.
Yhteensä 453 potilasta satunnaistettiin. Potilaiden mediaani-ikä oli 63 vuotta (vaihteluväli: 29–88 vuotta), ja suurin osa potilaista (66 %) oli valkoihoisia (12 % oli aasialaisia). Useimmilla potilailla (80 %) oli epiteliaalinen munasarjasyöpä. Munanjohdinsyöpää sairasti 11 % potilaista ja primaaria peritoneaalista syöpää 8 %. Kaikki syövät (100 %) olivat histologisesti korkean pahanlaatuisuusasteen serooseja. Noin puolet potilaista (47 %) oli saanut kolmea, 39 % potilaista kahta ja 14 % potilaista yhtä aikaisempaa systeemistä hoitoa. Suurin osa potilaista oli aikaisemmin saanut poly(ADP-riboosi)polymeraasin (PARP) estäjää (55 %) ja bevasitsumabia (62 %). Viimeisimmän hoitolinjan jälkeisen platinattoman jakson pituus oli 41 %:lla potilaista ≤ 3 kuukautta ja 58 %:lla potilaista 3–6 kuukautta. ECOG-toimintakykyluokka oli 55 %:lla potilaista 0 ja 44 %:lla potilaista 1.
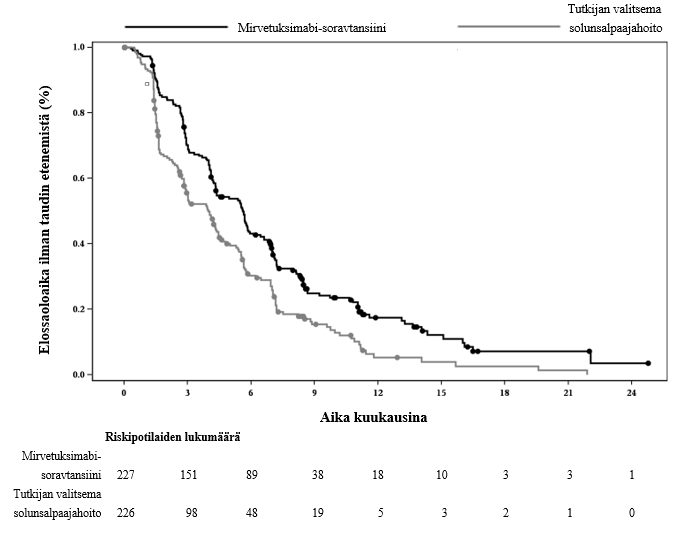
Ensisijainen analyysi osoitti tilastollisesti merkitsevän paraneman elossaoloajassa ilman taudin etenemistä ja kokonaiselossaoloajassa ELAHERE-ryhmään satunnaistetuilla potilailla verrattuna tutkijan valitsemaan solunsalpaajahoitoon.
Taulukko 5 sisältää yhteenvedon tehoa koskevista tuloksista tutkimuksessa IMGN853‑0416 (MIRASOL).
Taulukko 5: Tehoa koskevat tulokset tutkimuksessa IMGN853‑0416
| Tehomuuttuja | ELAHERE N = 227 | Tutkijan valitsemat solunsalpaajahoidot N = 226 |
| Tutkijan arvioima elossaoloaika ilman taudin etenemistä (PFS) | ||
| Tapahtumien lukumäärä (%) | 176 (77,5) | 166 (73,5) |
| Mediaani, kuukautta (95 %:n lv) | 5,62 (4,34; 5,95) | 3,98 (2,86; 4,47) |
| Riskisuhde (95 %:n lv) | 0,65 (0,521; 0,808) | |
| p‑arvo | < 0,0001 | |
| Kokonaiselossaoloaika (OS) | ||
| Tapahtumien lukumäärä (%) | 90 (39,6) | 114 (50,4) |
| Mediaani, kuukautta (95 %:n lv) | 16,46 (14,46; 24,57) | 12,75 (10,91; 14,36) |
| Riskisuhde (95 %:n lv) | 0,67 (0,504; 0,885) | |
| p‑arvo | 0,0046* | |
Tiedonkeruun katkaisuajankohta 6. maaliskuuta 2023
*: ennalta määritetty tehoraja = 0,01313, kaksisuuntainen (korjattu havaittujen kuolemien määrän 204 mukaan).
Kaplan-Meierin käyrät tutkijan arvioimalle elossaoloajalle ilman taudin etenemistä (seuranta-ajan mediaani 11,2 kuukautta) ja kokonaiselossaoloajalle (seuranta-ajan mediaani 13,1 kuukautta) on esitetty kuvissa 1 ja 2.
Kuva 1: Kaplan-Meierin käyrä hoitoryhmän mukaiselle elossaoloajalle ilman taudin etenemistä MIRASOL-tutkimuksessa (hoitoaikeen mukainen populaatio)
Kuva 2: Kaplan-Meierin käyrä hoitoryhmän mukaiselle kokonaiselossaoloajalle MIRASOL-tutkimuksessa (hoitoaikeen mukainen populaatio)
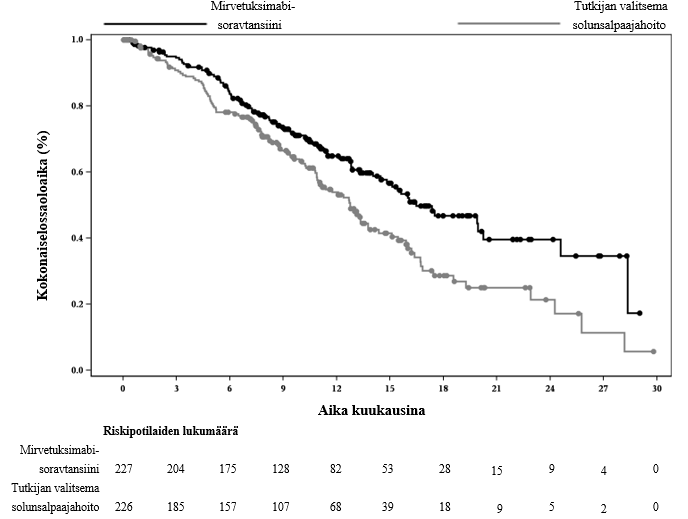
Kuvailevassa lisäanalyysissa, jossa seuranta-ajan mediaani oli 20,3 kuukautta, kokonaiselossaoloaikaa koskevat tulokset olivat yhdenmukaisia ensisijaisen analyysin tulosten kanssa.
Immunogeenisuus
Lääkevasta-aineita (anti-drug antibodies, ADA) havaittiin yleisesti. Lääkevasta-aineiden ei havaittu vaikuttavan farmakokinetiikkaan, tehoon tai turvallisuuteen, mutta tietoja on toistaiseksi vain vähän.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset ELAHERE-valmisteen käytöstä munasarjasyövän, munanjohdinsyövän ja peritoneaalisen syövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Farmakokinetiikkaa arvioitiin potilailla, joilla annettiin mirvetuksimabi-soravtansiinia annoksilla 0,161–8,71 mg/kg korjatun ihannepainon mukaan (0,0268–1,45 kertaa hyväksytty suositeltu annos 6 mg/kg korjatun ihannepainon mukaan), jollei toisin mainita.
Taulukossa 6 on yhteenveto potilaiden mirvetuksimabi-soravtansiinille sekä konjugoimattomalle DM4:lle ja sen S‑metyyli‑DM4-metaboliitille altistuksen parametreista ensimmäisen mirvetuksimabi-soravtansiini-syklin (3 viikkoa) jälkeen annoksella 6 mg/kg. Mirvetuksimabi-soravtansiinin huippupitoisuudet havaittiin lähellä laskimoon annetun infuusion päättymistä, konjugoimattoman DM4:n huippupitoisuudet toisena mirvetuksimabi-soravtansiini-infuusion jälkeisenä päivänä ja S‑metyyli‑DM4:n huippupitoisuudet noin 3 päivää mirvetuksimabi-soravtansiini-infuusion jälkeen. Mirvetuksimabi-soravtansiinin, DM4:n ja S‑metyyli‑DM4:n vakaan tilan pitoisuudet saavutettiin yhden hoitosyklin jälkeen. Mirvetuksimabi-soravtansiinin, DM4:n ja S‑metyyli‑DM4:n kumuloituminen oli minimaalista mirvetuksimabi-soravtansiinin toistuvassa annossa.
Taulukko 6: Mirvetuksimabi-soravtansiinille, konjugoimattomalle DM4:lle ja S‑metyyli‑DM4:lle altistuksen parametrit ensimmäisen mirvetuksimabi-soravtansiini-hoitosyklin jälkeen annoksella 6 mg/kg
| Mirvetuksimabi-soravtansiini Keskiarvo (±keskihajonta) | Konjugoimaton DM4 Keskiarvo (±keskihajonta) | S‑metyyli‑DM4 Keskiarvo (±keskihajonta) | |
| Cmax | 137,3 (±62,3) µg/ml | 4,11 (±2,29) ng/ml | 6,98 (±6,79) ng/ml |
| AUCtau | 20,65 (±6,84) h*mg/ml | 530 (±245) h*ng/ml | 1 848 (±1 585) h*ng/ml |
Cmax = huippupitoisuus, AUCtau = pitoisuus-aikakäyrän alle jäävä pinta-ala antovälin (21 päivää) aikana.
Imeytyminen
Mirvetuksimabi-soravtansiini annetaan infuusiona laskimoon. Muita antoreittejä koskevia tutkimuksia ei ole tehty.
Jakautuminen
Mirvetuksimabi-soravtansiinin vakaan tilan jakautumistilavuuden keskiarvo (±keskihajonta) oli 2,63 (±2,98) litraa. DM4 ja S‑metyyli‑DM4 sitoutuivat > 99‑prosenttisesti ihmisen plasman proteiineihin in vitro.
Biotransformaatio
Mirvetuksimabi-soravtansiinin monoklonaalisen vasta-ainekomponentin odotetaan metaboloituvan pieniksi peptideiksi katabolisten reittien kautta. Konjugoimaton DM4 ja S‑metyyli‑DM4 metaboloituvat CYP3A4:n välityksellä. DM4 ja S‑metyyli‑DM4 olivat pääasialliset kiertävät metaboliitit ihmisen plasmassa, ja niiden osuudet mirvetuksimabi-soravtansiinin AUC-arvosta olivat noin 0,4 % ja 1,4 %.
Eliminaatio
Mirvetuksimabi-soravtansiinin kokonaispuhdistuman keskiarvo (± keskihajonta) plasmasta oli 18,9 (± 9,8) ml/h. Mirvetuksimabi-soravtansiinin terminaalisen puoliintumisajan keskiarvo ensimmäisen annoksen jälkeen oli 4,9 päivää. Konjugoimattoman DM4:n kokonaispuhdistuman keskiarvo (± keskihajonta) plasmasta oli 14,5 (± 4,5) l/h ja terminaalisen puoliintumisajan keskiarvo 2,8 päivää. S‑metyyli‑DM4:n kokonaispuhdistuman keskiarvo (± keskihajonta) plasmasta oli 5,3 (± 3,4) l/h ja terminaalisen puoliintumisajan keskiarvo 5,1 päivää. In vitro -tutkimusten ja ei-kliinisten in vivo -tutkimusten mukaan DM4 ja S‑metyyli‑DM4 metaboloituvat pääasiassa CYP3A4:n vaikutuksesta ja eliminoituvat sapen mukana ulosteeseen.
Erityisryhmät
Mirvetuksimabi-soravtansiinin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja seuraavien perusteella: ikä (32–89 vuotta), rotu (valkoihoinen, tummaihoinen tai aasialainen), paino (36–136 kg), lievä maksan vajaatoiminta (kokonaisbilirubiini ≤ viitealueen yläraja ULN ja ASAT > ULN tai kokonaisbilirubiini > 1–1,5 × ULN ja mikä tahansa ASAT) tai lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma [CLcr] ≥ 30 – < 90 ml/min).
Mirvetuksimabi-soravtansiinin farmakokinetiikkaa ei ole tutkittu potilailla, joilla on keskivaikea tai vaikea maksan vajaatoiminta (kokonaisbilirubiini > 1,5 × ULN ja mikä tahansa ASAT) tai vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma [CLcr] 15–30 ml/min).
Yhteisvaikutustutkimukset
In vitro -tutkimukset
Sytokromi P450 (CYP) -entsyymit: Konjugoimaton DM4 on aikariippuvainen CYP3A4:n estäjä. Konjugoimaton DM4 ja S‑metyyli‑DM4 eivät ole seuraavien suoria estäjiä: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ja CYP3A. DM4 ja S‑metyyli‑DM4 eivät ole seuraavien indusoijia: CYP1A2, CYP2B6 ja CYP3A4.
Kuljettajaproteiinit:Konjugoimaton DM4 ja S‑metyyli‑DM4 ovat P‑gp:n substraatteja, mutta ne eivät ole P‑gp:n estäjiä.
Prekliiniset tiedot turvallisuudesta
Jaavanmakakeille annetun mirvetuksimabi-soravtansiinin kerta-annoksen jälkeiset löydökset rajoittuivat ihoon sekä luuytimen ja imukudoksen solukatoon. Toistuvassa annossa jaavanmakakeille ja hollantilaisille kaniineille havaittiin lisäksi silmälöydöksiä, mukaan lukien sarveiskalvon mikrokystoja, sarveiskalvon epiteelin pigmentaatiota, heikentymistä ja rappeutumista / nekroosia. Nämä löydökset olivat riippuvaisia annosintensiteetistä (annos ja aikataulu), ja 3 viikon välein annossa (kliininen annosaikataulu) löydöksiä oli vähemmän ja niiden havaittiin korjaantuvan.
Mirvetuksimabi-soravtansiinilla tai DM4:llä ei ole tehty karsinogeenisuustutkimuksia.
DM4 ja S‑metyyli‑DM4 eivät olleet mutageenisia bakteereilla tehdyssä takaisinmutaatiotestissä (Amesin testissä). DM4 ja S‑metyyli‑DM4 aiheuttivat mikrotumaisuutta polykromaattisissa punasoluissa.
Mirvetuksimabi-soravtansiinin lisääntymis- tai kehitystoksisuutta ei tutkittu eläimillä.
Mirvetuksimabi-soravtansiinilla tai DM4:llä ei ole tehty hedelmällisyyttä koskevia tutkimuksia. ELAHERE-valmisteen vaikutuksesta ihmisen hedelmällisyyteen ei ole olemassa tietoja. Koska ELAHERE-valmisteen vaikutusmekanismi johtaa mikrotubulusten toiminnan häiriintymiseen ja nopeasti jakautuvien solujen kuolemaan, on kuitenkin mahdollista, että valmisteella on hedelmällisyyteen kohdistuvia vaikutuksia.
Farmaseuttiset tiedot
Apuaineet
Etikkahappo, väkevä (E260)
Natriumasetaatti (E262)
Sakkaroosi
Polysorbaatti 20 (E432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
ELAHERE ei ole yhteensopiva natriumkloridi 9 mg/ml (0,9 %) infuusionesteen kanssa. Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
5 vuotta
Laimennettu liuos
Laimentamisen jälkeen pitoisuudeltaan 1–2 mg/ml olevan liuoksen kemiallisen ja fysikaalisen säilyvyyden on osoitettu olevan 8 tuntia 15 °C – 25 °C:n lämpötilassa tai 24 tuntia 2 °C – 8 °C:n lämpötilassa ja sen jälkeen 8 tuntia 15 °C – 25 °C:n lämpötilassa.
Mikrobiologiselta kannalta valmiste on käytettävä välittömästi, ellei laimentamiseen käytettävä menetelmä poissulje mikrobiologisen kontaminaation riskiä. Jos liuosta ei käytetä heti, säilytysaika ja säilytysolosuhteet ennen käyttöä ovat käyttäjän vastuulla.
Säilytys
Säilytä pystyasennossa jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ELAHERE infuusiokonsentraatti, liuosta varten
5 mg/ml (L:ei) 20 ml (3629,33 €)
PF-selosteen tieto
Tyypin I lasia oleva injektiopullo, jossa on butyylikumitulppa, alumiinisinetti ja syvänsininen polypropyleenia oleva repäisykorkki ja joka sisältää 20 ml infuusiokonsentraattia.
Pakkauskoko: 1 injektiopullo.
Valmisteen kuvaus:
Kirkas tai hieman opalisoiva, väritön liuos.
Käyttö- ja käsittelyohjeet
ELAHERE on sytotoksinen lääkevalmiste. Noudata asiaankuuluvia käsittely- ja hävittämisohjeita.
Valmistelu
- Laske annos (mg) (potilaan korjatun ihannepainon mukaan), tarvittava liuoksen kokonaismäärä (ml) ja tarvittava ELAHERE-injektiopullojen lukumäärä (ks. kohta Annostus ja antotapa). Täyteen annokseen tarvitaan useampi kuin yksi injektiopullo.
- Ota ELAHERE-injektiopullot jääkaapista ja anna niiden lämmetä huoneenlämpöisiksi.
- Parenteraaliset lääkevalmisteet on ennen antamista tarkastettava silmämääräisesti hiukkasten ja poikkeavan värin varalta aina silloin, kun liuos ja säilytysastia mahdollistavat sen. ELAHERE on kirkas tai hieman opalisoiva, väritön liuos.
- Lääkevalmistetta ei saa käyttää, jos se on poikkeavan väristä tai sameaa tai jos siinä näkyy hiukkasia.
- Pyöritä kutakin injektiopulloa varovasti ja tarkasta pullo, ennen kuin vedät pullosta laskettuun annokseen tarvittavan määrän ELAHERE-valmistetta jatkolaimentamista varten. Älä ravista injektiopulloa.
- Vedä pullosta laskettuun annokseen tarvittava määrä ELAHERE-valmistetta jatkolaimentamista varten aseptista tekniikkaa käyttäen. Jokaisessa pullossa on ylitäyttö, joka mahdollistaa merkityn määrän vetämisen.
- ELAHERE ei sisällä säilytysaineita, ja se on tarkoitettu vain kerta-annokseen. Hävitä injektiopullossa oleva käyttämättä jäänyt liuos.
Laimentaminen
- ELAHERE on ennen antamista laimennettava 5 %:n glukoosiliuoksella 1–2 mg/ml:n lopulliseen pitoisuuteen.
- ELAHERE ei ole yhteensopiva natriumkloridi 9 mg/ml (0,9 %) infuusionesteen kanssa. ELAHERE-valmistetta ei saa sekoittaa muiden lääkevalmisteiden tai laskimoon annettavien nesteiden kanssa.
- Laske tarvittava 5 %:n glukoosiliuoksen määrä, jolla saavutetaan lopullinen, laimennettu vaikuttavan aineen pitoisuus. Poista ylimääräinen 5 %:n glukoosiliuos esitäytetystä infuusiopussista tai lisää laskettu määrä 5 %:n glukoosiliuosta steriiliin tyhjään infuusiopussiin. Lisää sen jälkeen laskettuun annokseen tarvittava määrä ELAHERE-valmistetta infuusiopussiin.
- Varmista, että laimennettu liuos sekoittuu tasaisesti kääntelemämällä pussia hitaasti ylösalaisin useita kertoja. Älä ravista tai liikuttele voimakkaasti.
- Jos laimennettua infuusioliuosta ei käytetä heti, säilytä liuosta kohdan Kestoaika ohjeiden mukaisesti. Jos liuosta säilytetään jääkaapissa, anna infuusiopussin lämmetä huoneenlämpöiseksi ennen antamista. Jääkaapista ottamisen jälkeen laimennettu infuusioliuos on annettava 8 tunnin kuluessa (infuusioon kuluva aika mukaan lukien).
- Älä pakasta valmisteltua infuusioliuosta.
Antaminen
- Tarkasta ELAHERE-infuusiopussi ennen antamista silmämääräisesti hiukkasten ja poikkeavan värin varalta.
- Anna esilääkkeet ennen ELAHERE-valmisteen antamista (ks. kohta Annostus ja antotapa).
- ELAHERE-valmisteen saa antaa ainoastaan infuusiona laskimoon. Infuusioon on käytettävä 0,2 µm:n tai 0,22 µm:n polyeetterisulfonista (PES) valmistettua in-line-suodatinta. Älä käytä muista materiaaleista valmistettuja suodatinkalvoja.
- Di-2-etyyliheksyyliftalaattia (DEHP) sisältävien antolaitteiden käyttöä on vältettävä.
- Anna ensimmäinen annos infuusiona laskimoon nopeudella 1 mg/min. Jos infuusio on hyvin siedetty 30 minuutin jälkeen nopeudella 1 mg/min, infuusionopeutta voidaan lisätä 3 mg:aan/min. Jos infuusio on hyvin siedetty 30 minuutin jälkeen nopeudella 3 mg/min, infuusionopeutta voidaan lisätä 5 mg:aan/min.
- Jos edellisen annoksen yhteydessä ei ole esiintynyt infuusioon liittyviä reaktioita, seuraavien annosten infuusiot on aloitettava suurimmalla siedetyllä nopeudella, minkä jälkeen nopeutta voidaan lisätä enintään 5 mg:aan/min (suurin infuusionopeus) potilaan sietokyvyn mukaan.
- Huuhtele infuusion jälkeen infuusioletku 5 %:n glukoosiliuoksella, jotta potilas saa varmasti koko annoksen. Älä käytä huuhtelemiseen muita laskimoon annettavia nesteitä.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ELAHERE infuusiokonsentraatti, liuosta varten
5 mg/ml 20 ml
- Ei korvausta.
ATC-koodi
L01FX26
Valmisteyhteenvedon muuttamispäivämäärä
09.01.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi