
RYBREVANT injektionsvätska, lösning 1600 mg, 2240 mg, 2400 mg, 3520 mg
Observera
▼ Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Rybrevant 1 600 mg injektionsvätska, lösning
En ml med injektionsvätska, lösning innehåller 160 mg amivantamab.
En 10 ml injektionsflaska med injektionsvätska, lösning innehåller 1 600 mg amivantamab.
Rybrevant 2 240 mg injektionsvätska, lösning
En ml med injektionsvätska, lösning innehåller 160 mg amivantamab.
En 14 ml injektionsflaska med injektionsvätska, lösning innehåller 2 240 mg amivantamab.
Rybrevant 2 400 mg injektionsvätska, lösning
En ml med injektionsvätska, lösning innehåller 160 mg amivantamab.
En 15 ml injektionsflaska med injektionsvätska, lösning innehåller 2 400 mg amivantamab.
Rybrevant 3 520 mg injektionsvätska, lösning
En ml med injektionsvätska, lösning innehåller 160 mg amivantamab.
En 22 ml injektionsflaska med injektionsvätska, lösning innehåller 3 520 mg amivantamab.
Amivantamab är en helt human immunglobulin-G1 (IgG1)-baserad bispecifik antikropp riktad mot receptorerna för epidermal tillväxtfaktor (EGF) och mesenkymal epitelövergång (MET), tillverkad i en däggdjurscellinje (kinesisk hamsterovarie [CHO]) med rekombinant DNA-teknik.
Hjälpämne med känd effekt:
En ml lösning innehåller 0,6 mg polysorbat 80.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Injektionsvätska, lösning.
Kliniska uppgifter
Terapeutiska indikationer
Rybrevant subkutan formulering är avsedd:
- i kombination med lazertinib för första linjens behandling av vuxna patienter med avancerad icke-småcellig lungcancer (NSCLC) med EGFR exon 19-deletion eller substitutionsmutation L858R i exon 21.
-
i kombination med karboplatin och pemetrexed för behandling av vuxna patienter med avancerad NSCLC med EGFR exon 19-deletion eller substitutionsmutation L858R i exon 21 efter svikt på tidigare behandling, inkluderande en EGFR-tyrosinkinashämmare (TKI).
-
i kombination med karboplatin och pemetrexed för första linjens behandling av vuxna patienter med avancerad NSCLC med aktiverande insertionsmutationer i EGFR exon 20.
- som monoterapi för behandling av vuxna patienter med avancerad NSCLC med aktiverande insertionsmutationer i EGFR exon 20, efter svikt på platinumbaserad kemoterapi.
Villkor
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Dosering och administreringssätt
Behandling med Rybrevant subkutan formulering ska sättas in och övervakas av läkare med erfarenhet av användning av cancerläkemedel.
Innan behandling med Rybrevant subkutan formulering inleds, måste EGFR-mutationsstatus i tumörvävnad eller plasmaprover fastställas med hjälp av en validerad testmetod. Om ingen mutation detekteras i ett plasmaprov ska tumörvävnad testas, om sådan finns tillgänglig i tillräcklig mängd och kvalitet, på grund av risken för falskt negativa resultat med ett plasmatest. Test behöver inte upprepas när EFGR-mutationsstatus har fastställts (se avsnitt Farmakodynamiska egenskaper).
Rybrevant subkutan formulering ska administreras av hälso- och sjukvårdspersonal med tillgång till lämpligt medicinskt stöd för hantering av administreringsrelaterade reaktioner, om sådana skulle uppträda.
För patienter som för närvarande får amivantamab intravenös formulering kan Rybrevant subkutan formulering användas som ett alternativ till den intravenösa amivantamab-formuleringen med början vid nästa schemalagda dos.
För patienter som för närvarande får Rybrevant subkutan formulering med en doseringsregim varannan vecka kan en doseringsregim var fjärde vecka användas som ett alternativ med början vid nästa schemalagda dos.
Dosering
Premedicinering ska administreras för att minska risken för administreringsrelaterade reaktioner vid användning av Rybrevant subkutan formulering (se nedan ”Dosändringar” och ”Rekommenderade samtidiga läkemedel”).
Rybrevant subkutan formulering i kombination med lazertinib eller som monoterapi
Rekommenderade doser av Rybrevant subkutan formulering i kombination med lazertinib eller som monoterapi, baserat på kroppsvikt vid behandlingsstart anges i tabell 1 (dosering var fjärde vecka) och tabell 2 (dosering varannan vecka).
| Tabell 1: Rekommenderad dos av Rybrevant subkutan formulering i kombination med lazertinib eller som monoterapi (dosering var fjärde vecka) | ||
| Kroppsvikt vid behandlingsstart* | Rekommenderad dos | Doseringsschema |
| Mindre än 80 kg | 1 600 mg |
|
| 3 520 mg |
| |
| 80 kg eller mer | 2 240 mg |
|
| 4 640 mg |
| |
| * Dosjustering är ej nödvändig vid efterföljande förändringar i kroppsvikt. | ||
| Tabell 2: Rekommenderad dos av Rybrevant subkutan formulering i kombination med lazertinib eller som monoterapi (dosering varannan vecka) | ||
| Kroppsvikt vid behandlingsstart* | Rekommenderad dos | Doseringsschema |
| Mindre än 80 kg | 1 600 mg |
|
| 80 kg eller mer | 2 240 mg |
|
| * Dosjustering är ej nödvändig vid efterföljande förändringar i kroppsvikt. | ||
Vid användning i kombination med lazertinib rekommenderas att Rybrevant subkutan formulering administreras när som helst efter lazertinib när det ges samma dag. Se avsnitt Dosering och administreringssätt i produktresumén för lazertinib för rekommenderade doseringsanvisningar för lazertinib.
Rybrevant subkutan formulering i kombination med karboplatin och pemetrexed
Rekommenderade doser av Rybrevant subkutan formulering när det används i kombination med karboplatin och pemetrexed, baserat på kroppsvikt vid behandlingsstart, anges i tabell 3.
| Tabell 3: Rekommenderad dos av Rybrevant subkutan formulering i kombination med karboplatin och pemetrexed (dosering var tredje vecka) | ||
| Kroppsvikt vid behandlingsstart* | Rekommenderad dos | Doseringsschema |
| Mindre än 80 kg | 1 600 mg | Första dosen vecka 1 dag 1 |
| 2 400 mg |
| |
| 80 kg eller mer | 2 240 mg | Första dosen vecka 1 dag 1 |
| 3 360 mg |
| |
| * Dosjustering är ej nödvändig vid efterföljande förändringar i kroppsvikt. | ||
Vid användning i kombination med karboplatin och pemetrexed ska Rybrevant subkutan formulering administreras efter karboplatin och pemetrexed i följande ordning: pemetrexed, karboplatin och därefter Rybrevant. Se avsnitt Farmakodynamiska egenskaper och tillverkarens förskrivningsinformation för doseringsanvisningar för karboplatin och pemetrexed.
Behandlingstid
Det rekommenderas att patienter behandlas med Rybrevant subkutan formulering till sjukdomsprogression eller tills oacceptabel toxicitet uppträder.
Missad dos
Vid dosering var fjärde eller varannan vecka: Om en dos av Rybrevant subkutan formulering missas mellan vecka 1 och 4 ska den administreras inom 24 timmar. Om en dos av Rybrevant subkutan formulering missas från vecka 5 och framåt ska den administreras inom 7 dagar.
Vid dosering var tredje vecka: Om en dos av Rybrevant subkutan formulering missas mellan vecka 1 och 3 ska den administreras inom 24 timmar. Om en dos av Rybrevant subkutan formulering missas från vecka 4 och framåt ska den administreras inom 7 dagar.
Om den missade dosen inte administreras enligt dessa riktlinjer ska den missade dosen inte administreras och nästa dos ska administreras enligt det vanliga doseringsschemat.
Dosändringar
Vid förekomst av biverkningar av grad 3 eller 4 ska behandlingen avbrytas till dess att biverkningen gått tillbaka till ≤ grad 1 eller till utgångsläget. Återuppta behandlingen med aktuell dos om avbrottet varar i 7 dagar eller mindre. Om avbrottet varar i mer än 7 dagar, bör behandlingen återupptas med en reducerad dos enligt tabell 4. För specifika dosjusteringar i samband med specifika biverkningar, se information efter tabell 4.
Om det används i kombination med lazertinib, se avsnitt Dosering och administreringssätt i lazertinibs produktresumé för information om dosändringar.
| Tabell 4: Rekommenderade dosändringar vid biverkningar | |||
| Dos* | Dos efter 1:a avbrottet vid biverkning | Dos efter 2:a avbrottet vid biverkning | Dos efter 3:e avbrottet vid biverkning |
| 1 600 mg | 1 050 mg | 700 mg | Sätt ut Rybrevant subkutan formulering |
| 2 240 mg | 1 600 mg | 1 050 mg | |
| 2 400 mg | 1 600 mg | 1 050 mg | |
| 3 360 mg | 2 240 mg | 1 600 mg | |
| 3 520 mg | 2 400 mg | 1 600 mg | |
| 4 640 mg | 3 360 mg | 2 240 mg | |
| * Dos vid vilken biverkningen uppstod. | |||
Administreringsrelaterade reaktioner
Premedicinering ska administreras för att minska risken för administreringsrelaterade reaktioner med Rybrevant subkutan formulering (se ”Rekommenderade samtidiga läkemedel”). Injektionerna ska avbrytas vid första tecknet på administreringsrelaterade reaktioner. Om det är kliniskt indicerat ska understödjande läkemedel (t.ex. ytterligare glukokortikoider, antihistamin, febernedsättande medel och antiemetika) administreras (se avsnitt Varningar och försiktighet).
- Grad 1–3 (lätt–allvarlig): När symtomen har gått tillbaka ska injektionerna med Rybrevant subkutan formulering återupptas. Samtidig medicinering ska administreras vid nästa dostillfälle, inklusive dexametason (20 mg) eller motsvarande (se tabell 5).
- Återkommande grad 3 eller grad 4 (livshotande): Sätt ut Rybrevant permanent.
Venösa tromboemboliska (VTE) händelser vid samtidig användning med lazertinib
När behandlingen inleds ska profylaktiska antikoagulantia administreras för att förebygga VTE-händelser hos patienter som får Rybrevant subkutan formulering i kombination med lazertinib. I enlighet med kliniska riktlinjer ska patienterna få profylaktisk dosering av antingen en direktverkande oral antikoagulant (DOAC) eller ett lågmolekylärt heparin (LMWH). Användning av vitamin K-antagonister rekommenderas inte.
Vid VTE-händelser i samband med klinisk instabilitet (t.ex. andningssvikt eller hjärtdysfunktion) ska båda läkemedlen sättas ut tills patienten är kliniskt stabil. Därefter kan båda läkemedlen återinsättas med samma dos. Vid återfall trots lämplig antikoagulation ska Rybrevant sättas ut. Behandlingen kan fortsätta med lazertinib i samma dos (se avsnitt Varningar och försiktighet).
Hud- och nagelreaktioner
Profylaktisk behandling med orala och topikala antibiotika rekommenderas för att minska risken för och allvarlighetsgraden av hud- och nagelreaktioner hos patienter som får Rybrevant. Icke-komedogen fuktkräm för huden (ceramidbaserad eller andra formuleringar som ger långvarig återfuktning av huden och utan torkmedel är att föredra) i ansiktet och på hela kroppen (utom hårbotten) och klorhexidinlösning för att tvätta händer och fötter rekommenderas också. Patienter ska instrueras att begränsa solexponering under och i 2 månader efter Rybrevant-behandlingen. För ytterligare information om profylax för hud- och nagelreaktioner, se avsnitt Varningar och försiktighet.
Om patienten utvecklar en hud- eller nagelreaktion av grad 1–2 ska stödjande behandling inledas om det är kliniskt indicerat. Om det inte sker någon förbättring inom 2 veckor ska dosreduktion övervägas för svåra utslag av grad 2 (se tabell 4). Om patienten utvecklar en hud- eller nagelreaktion av grad 3 ska stödjande behandling inledas om det är kliniskt indicerat, och behandlingsavbrott bör övervägas tills biverkningen förbättrats. Efter återhämtning från hud- eller nagelreaktionen till ≤ grad 2 ska behandling med Rybrevant subkutan formulering återupptas med reducerad dos. Om patienten utvecklar hudreaktioner av grad 4, sätt ut Rybrevant permanent (se avsnitt Varningar och försiktighet).
Interstitiell lungsjukdom
Vid misstanke om interstitiell lungsjukdom (ILD) eller ILD-liknande biverkningar (t.ex. pneumonit) ska behandling med Rybrevant subkutan formulering sättas ut tillfälligt. Om patienten bekräftas ha ILD eller ILD-liknande biverkningar (t.ex. pneumonit), sätt ut Rybrevant permanent (se avsnitt Varningar och försiktighet).
Rekommenderade samtidiga läkemedel
Före den initiala dosen (vecka 1, dag 1) ska antihistaminer, febernedsättande medel och glukokortikoider administreras för att minska risken för administreringsrelaterade reaktioner (se tabell 5). Inför efterföljande doser krävs administrering av antihistaminer och febernedsättande medel. Glukokortikoider ska också återinsättas efter långvariga dosavbrott. Antiemetika ska administreras efter behov.
| Tabell 5: Doseringsschema för premedicinering | |||
| Premedicinering | Dos | Administreringsväg | Rekommenderad doseringstid före administrering av Rybrevant subkutan formulering |
| Antihistamin* | Difenhydramin (25 till 50 mg) eller motsvarande | Intravenös | 15 till 30 minuter |
| Oral | 30 till 60 minuter | ||
| Febernedsättande* | Paracetamol/acetaminofen (650 till 1 000 mg) eller motsvarande | Intravenös | 15 till 30 minuter |
| Oral | 30 till 60 minuter | ||
| Glukokortikoid† | Dexametason (20 mg) eller motsvarande | Intravenös | 45 till 60 minuter |
| Oral | Minst 60 minuter | ||
| Glukokortikoid‡ | Dexametason (10 mg) eller motsvarande | Intravenös | 45 till 60 minuter |
| Oral | 60 till 90 minuter | ||
| * Krävs vid alla doser. † Krävs vid startdos (vecka 1, dag 1), eller vid nästa efterföljande dos i händelse av en administreringsrelaterad reaktion. ‡ Valfritt vid efterföljande doser. | |||
Särskilda populationer
Pediatrisk population
Det finns ingen relevant användning av amivantamab för en pediatrisk population vid behandling av icke-småcellig lungcancer.
Äldre
Inga dosjusteringar är nödvändiga (se avsnitt Biverkningar, avsnitt Farmakodynamiska egenskaper och avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Inga formella studier av amivantamab har utförts på patienter med nedsatt njurfunktion. Baserat på farmakokinetiska populationsanalyser behövs ingen dosjustering hos patienter med lätt eller måttligt nedsatt njurfunktion. Försiktighet ska iakttas hos patienter med kraftigt nedsatt njurfunktion, eftersom amivantamab inte har studerats i denna patientpopulation (se avsnitt Farmakokinetiska egenskaper) Om behandling inleds ska patienterna övervakas avseende biverkningar och dosjusteringar göras enligt rekommendationerna ovan.
Nedsatt leverfunktion
Inga formella studier av amivantamab har utförts på patienter med nedsatt leverfunktion. Baserat på farmakokinetiska populationsanalyser behövs ingen dosjustering hos patienter med lätt nedsatt leverfunktion. Försiktighet ska iakttas hos patienter med måttligt eller kraftigt nedsatt leverfunktion, eftersom amivantamab inte har studerats i dessa patientpopulationer (se avsnitt Farmakokinetiska egenskaper) Om behandling inleds ska patienterna övervakas avseende biverkningar och dosjusteringar göras enligt rekommendationerna ovan.
Administreringssätt
Rybrevant injektionsvätska, lösning är endast avsedd för subkutan användning.
Rybrevant subkutan formulering är inte avsedd för intravenös administrering och ska endast ges genom subkutan injektion med de doser som anges. Anvisningar om hantering av läkemedlet före administrering finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Injicera den volym som krävs av Rybrevant subkutan formulering i den subkutana vävnaden i buken under cirka 5 minuter. Administrera inte på andra ställen i kroppen eftersom inga data finns tillgängliga.
Pausa eller sänk administreringshastigheten om patienten upplever smärta. Om smärtan inte lindras genom att pausa eller sänka leveranshastigheten kan ett andra injektionsställe väljas på motsatt sida av buken för att leverera resterande del av dosen.
Vid administrering med ett subkutant infusionsset, säkerställ att hela dosen tillförs genom infusionssetet. Natriumkloridlösning 9 mg/ml (0,9 %) kan användas för att spola ut resterande läkemedel genom slangen.
Injicera inte i tatueringar eller ärr eller områden där huden är röd, har blåmärken, är öm, hård, inte intakt eller inom 5 cm runt det periumbilikala området.
Injektionsställena ska växlas för injektioner som följer på varandra.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Administreringsrelaterade reaktioner
Administreringsrelaterade reaktioner inträffade hos patienter som behandlades med Rybrevant subkutan formulering (se avsnitt Biverkningar).
Före den första injektionen (vecka 1, dag 1) ska antihistaminer, febernedsättande medel och glukokortikoider administreras för att minska risken för administreringsrelaterade reaktioner. För efterföljande doser ska antihistaminer och febernedsättande medel administreras.
Behandlingen ska ges på en klinik med tillgång till lämpligt medicinskt stöd för behandling av administreringsrelaterade reaktioner. Pågående injektionen ska avbrytas vid första tecken på administreringsrelaterade reaktioner oavsett och efterföljande läkemedel ska administreras så som kliniskt indicerat. När symtomen har försvunnit ska injektionen återupptas. För administreringsrelaterade reaktioner av grad 4 eller återkommande av grad 3 ska Rybrevant sättas ut permanent (se avsnitt Dosering och administreringssätt).
Interstitiell lungsjukdom
Interstitiell lungsjukdom (ILD) eller ILD-liknande biverkningar (t.ex. pneumonit) har rapporterats hos patienter behandlade med amivantamab, inklusive händelser med dödlig utgång (se avsnitt Biverkningar). Patienter ska övervakas avseende symtom som tyder på ILD/pneumonit (t.ex. andnöd, hosta, feber). Om symtom uppträder ska behandlingen med Rybrevant avbrytas i väntan på utredning av dessa symtom. Misstänkt ILD eller ILD-liknande biverkningar ska utvärderas och lämplig behandling ska påbörjas efter behov. Rybrevant ska sättas ut permanent hos patienter med bekräftad ILD eller ILD-liknande biverkningar (se avsnitt Dosering och administreringssätt).
Venösa tromboemboliska (VTE) händelser vid samtidig användning med lazertinib
Hos patienter som fick amivantamab i kombination med lazertinib rapporterades VTE-händelser, inklusive djup ventrombos (DVT) och lungemboli (PE) (se avsnitt Biverkningar). Dödsfall observerades för amivantamab intravenös formulering.
I enlighet med kliniska riktlinjer ska patienterna få profylaktisk dosering av antingen ett direktverkande oralt antikoagulantium (DOAC) eller ett lågmolekylärt heparin (LMWH). Användning av vitamin K-antagonister rekommenderas inte.
Tecken och symtom på VTE-händelser ska övervakas. Patienter med VTE-händelser ska behandlas med antikoagulation enligt klinisk indikation. Vid VTE-händelser i samband med klinisk instabilitet ska behandlingen sättas ut tillfälligt tills patienten är kliniskt stabil. Därefter kan behandlingen med båda läkemedlen återinsättas med samma dos.
I händelse av återfall trots lämplig antikoagulation ska Rybrevant sättas ut. Behandlingen kan fortsätta med lazertinib i samma dos (se avsnitt Dosering och administreringssätt).
Hud- och nagelreaktioner
Utslag (inklusive akneliknande dermatit), klåda, torr hud och hudsår har förekommit hos patienter behandlade med amivantamab (se avsnitt Biverkningar). Patienter ska instrueras att begränsa sin exponering för sol under behandling och i 2 månader efter behandling med Rybrevant. Skyddande kläder och användning av solskyddsmedel som skyddar mot både UVA och UVB rekommenderas. Ett profylaktiskt tillvägagångssätt för att förhindra utslag rekommenderas. Detta inkluderar profylaktisk behandling vid behandlingsstart med ett oralt antibiotikum (t.ex. doxycyklin eller minocyklin, 100 mg två gånger dagligen) med början dag 1 under de första 12 behandlingsveckorna och efter avslutad oral antibiotikabehandling, en lokalverkande antibiotika i form av lotion i hårbotten (t.ex. klindamycin 1 %) under de följande 9 behandlingsmånaderna. En icke-komedogen fuktkräm som inte täpper porerna (ceramidbaserad eller andra formuleringar som ger långvarig återfuktning av huden och utan torkmedel är att föredra) för ansiktet och hela kroppen (utom hårbotten) och klorhexidinlösning till att tvätta händer och fötter med rekommenderas, med början dag 1 och fortsatt under hela behandlingstiden.
Det rekommenderas att recept på ytterligare topikala och/eller orala antibiotika och topikala kortikosteroider finns tillhands vid tidpunkten för den första dosen. Detta för att minimera eventuella fördröjningar i den profylaktiska behandlingen om utslag förekommer trots förebyggande åtgärder. Om hudreaktioner uppträder ska stödjande behandling, topikala kortikosteroider och topikala och/eller orala antibiotika administreras. Vid förekomst av hudreaktioner av grad 3 eller dåligt tolererade reaktioner av grad 2 ska även systemiska antibiotika och orala steroider administreras. Patienter med svåra utslag som har ett atypiskt utseende eller atypisk utbredning eller som inte förbättras inom 2 veckor ska omedelbart remitteras till en hudläkare. Behandling med Rybrevant ska dosreduceras, avbrytas eller sättas ut permanent baserat på allvarlighetsgrad (se avsnitt Dosering och administreringssätt).
Toxisk epidermal nekrolys (TEN) har rapporterats. Behandling med detta läkemedel ska sättas ut om TEN bekräftas.
Ögonsjukdomar
Ögonsjukdomar, inklusive keratit, förekom hos patienter som behandlades med amivantamab (se avsnitt Biverkningar). Patienter som uppvisar förvärrade ögonsymtom ska omedelbart remitteras till ögonläkare och sluta använda kontaktlinser tills symtomen har utvärderats. Se avsnitt Dosering och administreringssätt för dosändringar vid ögonsjukdomar av grad 3 eller 4.
Natriuminnehåll
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per dos, d.v.s. är näst intill ”natriumfritt” (se avsnitt Särskilda anvisningar för destruktion och övrig hantering).
Polysorbatinnehåll
Detta läkemedel innehåller 0,6 mg polysorbat 80 per ml motsvarande 6 mg per 10 ml‑injektionsflaska, 8,4 mg per 14 ml‑injektionsflaska, 9 mg per 15 ml‑injektionsflaska eller 13,2 mg per 22 ml‑injektionsflaska. Polysorbater kan orsaka allergiska reaktioner.
Interaktioner
Inga studier avseende läkemedelsinteraktioner har utförts. Som monoklonal IgG1-antikropp är det osannolikt att renal utsöndring och hepatisk enzymmedierad metabolism av intakt amivantamab är stora elimineringsvägar. Därför förväntas inte variationer i läkemedelsmetaboliserande enzymer påverka elimineringen av amivantamab. På grund av den höga affiniteten till en unik epitop på EGFR respektive MET, förutses inte amivantamab förändra läkemedelsmetaboliserande enzymer.
Vacciner
Det finns inga tillgängliga data avseende effekt och säkerhet av vaccinationer hos patienter som tar amivantamab. Undvik att använda levande eller levande försvagade vacciner, när patienter tar amivantamab.
Fertilitet, graviditet och amning
Fertila kvinnor/Preventivmetod
Fertila kvinnor ska använda en effektiv preventivmetod under behandlingen och i 3 månader efter avslutad behandling med amivantamab.
Graviditet
Eventuella risker med amivantamab under graviditet kan inte bedömas, eftersom data från människa saknas och inga reproduktionsstudier på djur har utförts för att undersöka en läkemedelsrelaterad risk. Administrering av EGFR- och MET-hämmande molekyler till dräktiga djur resulterade i en ökad incidens av försämrad embryofetal utveckling, embryodödlighet och spontanabort. Baserat på verkningsmekanismen och fynd i djurmodeller, kan således amivantamab orsaka fosterskada vid administrering till gravida kvinnor. Amivantamab ska inte ges under graviditet, om inte nyttan med behandlingen av kvinnan anses uppväga potentiella risker för fostret. Om patienten blir gravid under behandlingen, ska hon informeras om den eventuella risken för fostret (se avsnitt Prekliniska säkerhetsuppgifter).
Amning
Det är okänt om amivantamab utsöndras i bröstmjölk. Det är känt att humana IgG-antikroppar utsöndras i bröstmjölk under de första dagarna efter förlossningen, för att sedan sjunka till en låg koncentration strax efteråt. En risk för det ammade barnet kan inte uteslutas under denna korta period strax efter födseln, även om IgG-antikropparna sannolikt bryts ner i magtarmkanalen hos det ammade barnet och inte absorberas. Ett beslut måste fattas om amning ska avbrytas eller om behandling med amivantamab ska avbrytas/avstås, efter att hänsyn tagits till fördelen med amning för barnet och fördelen med behandling för kvinnan.
Fertilitet
Det finns inga data om effekten av amivantamab på fertiliteten hos människa. Effekterna på manlig och kvinnlig fertilitet har inte utvärderats i djurstudier.
Effekter på förmågan att framföra fordon och använda maskiner
Rybrevant kan ha måttlig effekt på förmågan att framföra fordon och använda maskiner. Se avsnitt Biverkningar (t.ex. yrsel, trötthet, synrubbningar). Om patienter upplever behandlingsrelaterade symtom som påverkar koncentrations- och reaktionsförmågan, inklusive synrelaterade biverkningar, bör de inte framföra fordon eller använda maskiner förrän symtomen försvunnit.
Biverkningar
Sammanfattning av säkerhetsprofilen
Rybrevant som monoterapi
I datasetet för Rybrevant intravenös formulering som monoterapi (N = 380) var de mest förekommande biverkningarna i alla grader utslag (76 %), infusionsrelaterade reaktioner (67 %), nageltoxicitet (47 %), hypoalbuminemi (31 %), ödem (26 %), trötthet (26 %), stomatit (24 %), illamående (23 %) och förstoppning (23 %). Allvarliga biverkningar innefattade ILD (1,3 %), infusionsrelaterade reaktioner (1,1 %) och utslag (1,1 %). Tre procent av patienterna avslutade behandlingen med Rybrevant på grund av biverkningar. De mest förekommande biverkningarna som ledde till avslutande av behandling var infusionsrelaterade reaktioner (1,1 %), ILD (0,5 %) samt nageltoxicitet (0,5 %).
Biverkningar i tabellform
Tabell 6 sammanfattar de biverkningar som uppträdde hos patienter som fick Rybrevant som monoterapi.
Dessa data återspeglar exponering för Rybrevant intravenös formulering hos 380 patienter med lokalt avancerad eller metastatisk icke-småcellig lungcancer efter behandlingssvikt med platinumbaserad kemoterapi. Patienter fick amivantamab 1 050 mg (till patienter < 80 kg) eller 1 400 mg (till patienter ≥ 80 kg). Medianexponeringen för amivantamab var 4,1 månader (intervall: 0,0 till 39,7 månader).
Biverkningar observerade under kliniska studier listas nedan efter frekvenskategori. Frekvenskategorier definieras enligt följande: mycket vanliga (≥ 1/10); vanliga (≥ 1/100, < 1/10); mindre vanliga (≥ 1/1 000, < 1/100); sällsynta (≥ 1/10 000, < 1/1 000); mycket sällsynta (< 1/10 000); samt ingen känd frekvens (kan inte beräknas från tillgängliga data).
Inom varje frekvenskategori presenteras biverkningarna i fallande allvarlighetsgrad.
Tabell 6: Biverkningar hos patienter som fått Rybrevant som monoterapi (N = 380) | |||
Organsystemklass | Frekvenskategori | Alla grader (%) | Grad 3‑4 (%) |
Metabolism och nutrition | |||
Hypoalbuminemi* (se avsnitt Farmakodynamiska egenskaper) | Mycket vanliga | 31 | 2† |
Minskad aptit | 16 | 0,5† | |
Hypokalcemi | 10 | 0,3† | |
Hypokalemi | Vanliga | 9 | 2 |
Hypomagnesemi | 8 | 0 | |
Centrala och perifera nervsystemet | |||
Yrsel* | Mycket vanliga | 13 | 0,3† |
Ögon | |||
Synrubbningar* | Vanliga | 3 | 0 |
Tillväxt av ögonfransar* | 1 | 0 | |
Andra ögonsjukdomar* | 6 | 0 | |
Keratit | Mindre vanliga | 0,5 | 0 |
Uveit | 0,3 | 0 | |
Andningsvägar, bröstkorg och mediastinum | |||
Interstitiell lungsjukdom* | Vanliga | 3 | 0,5† |
Magtarmkanalen | |||
Diarré | Mycket vanliga | 11 | 2† |
Stomatit* | 24 | 0,5† | |
Illamående | 23 | 0,5† | |
Förstoppning | 23 | 0 | |
Kräkningar | 12 | 0,5† | |
Buksmärta* | Vanliga | 9 | 0,8† |
Hemorrojder | 3,7 | 0 | |
Lever och gallvägar | |||
Ökat alaninaminotransferas | Mycket vanliga | 15 | 2 |
Ökat aspartataminotransferas | 13 | 1 | |
Ökat alkaliskt fosfatas i blodet | 12 | 0,5† | |
Hud och subkutan vävnad | |||
Utslag* | Mycket vanliga | 76 | 3† |
Nageltoxicitet* | 47 | 2† | |
Torr hud* | 19 | 0 | |
Klåda | 18 | 0 | |
Hudsår | Mindre vanliga | 0,8 | 0 |
Toxisk epidermal nekrolys | 0,3 | 0,3† | |
Muskuloskeletala systemet och bindväv | |||
Myalgi | Mycket vanliga | 11 | 0,3† |
Allmänna symtom och/eller symtom vid administreringsstället | |||
Ödem* | Mycket vanliga | 26 | 0,8† |
Trötthet* | 26 | 0,8† | |
Pyrexi | 11 | 0 | |
Skador, förgiftningar och behandlingskomplikationer | |||
Infusionsrelaterad reaktion | Mycket vanliga | 67 | 2 |
* Grupperade termer | |||
Rybrevant i kombination med lazertinib
Sammantaget överensstämde säkerhetsprofilen för Rybrevant subkutan formulering med den etablerade säkerhetsprofilen för Rybrevant intravenös formulering, med en lägre incidens av administreringsrelaterade reaktioner och VTE observerade med den subkutana formuleringen jämfört med den intravenösa formuleringen.
I datasetet för Rybrevant (antingen intravenösa eller subkutana formuleringar) i kombination med lazertinib (N = 829) var de vanligaste biverkningarna av någon grad (≥ 20 % av patienterna) utslag (87 %), nageltoxicitet (68 %) hypoalbuminemi (49 %), hepatotoxicitet (43 %), stomatit (43 %), ödem (42 %), trötthet (34 %), parestesi (29 %), diarré (26 %), förstoppning (25 %), torr hud (25 %), illamående (24 %), klåda (24 %) och minskad aptit (23 %).
Kliniskt relevanta skillnader mellan de intravenösa och subkutana formuleringarna, när de gavs i kombination med lazertinib, observerades för administreringsrelaterade reaktioner (63 % för intravenösa jämfört med 14 % för subkutana) och VTE (37 % för intravenösa jämfört med 11 % för subkutana).
Allvarliga biverkningar rapporterades hos 14 % av de patienter som fick Rybrevant subkutan formulering i kombination med lazertinib, inklusive ILD (4,2 %), VTE (2,2 %), hepatotoxicitet (2,2 %) och trötthet (1,5 %). Sju procent av patienterna avbröt behandlingen med Rybrevant subkutan formulering på grund av biverkningar. Hos patienter som behandlades med Rybrevant subkutan formulering i kombination med lazertinib var de vanligaste biverkningarna av någon grad (≥ 1 % av patienterna) som ledde till utsättning av Rybrevant subkutan formulering ILD (3,7 %), utslag (1,2 %) och nageltoxicitet (1,0 %).
Biverkningar i tabellform
Biverkningarna för Rybrevant (antingen intravenös eller subkutan formulering) när det ges i kombination med lazertinib sammanfattas i tabell 7.
Säkerhetsdatan nedan återspeglar exponering för Rybrevant (antingen intravenös eller subkutan formulering) i kombination med lazertinib hos 829 patienter med lokalt avancerad eller metastaserad NSCLC, inklusive 421 patienter i MARIPOSA, 202 patienter i PALOMA-2 kohort 1, 5 och 6 och 206 patienter i PALOMA-3 subkutan arm. Patienterna fick Rybrevant (antingen intravenös eller subkutan formulering) tills sjukdomsprogression eller oacceptabel toxicitet. Mediantiden för behandling med amivantamab totalt för både intravenösa och subkutana formuleringar var 9,1 månader (intervall: 0,1 till 31,4 månader). Mediantiden för behandling med den subkutana formuleringen var 6,1 månader (intervall: 0,1 till 13,2 månader) medan mediantiden för behandling med den intravenösa formuleringen var 18,5 månader (intervall: 0,2 till 31,4 månader).
Biverkningar observerade under kliniska studier listas nedan efter frekvenskategori. Frekvenskategorier definieras enligt följande: mycket vanliga (≥ 1/10); vanliga (≥ 1/100, < 1/10); mindre vanliga (≥ 1/1 000, < 1/100); sällsynta (≥ 1/10 000, < 1/1 000); mycket sällsynta (< 1/10 000); samt ingen känd frekvens (kan inte beräknas från tillgängliga data).
Tabell 7: Biverkningar hos patienter som fått Rybrevant (antingen intravenös eller subkutan formulering) i kombination med lazertinib (N = 829) | |||
Organsystemklass | Frekvenskategori | Alla grader | Grad 3‑4 |
Metabolism och nutrition | |||
Hypoalbuminemi* | Mycket vanliga | 49 | 4,6 |
Minskad aptit | 23 | 0,7 | |
Hypokalcemi | 18 | 1,1 | |
Hypokalemi | 12 | 2,7 | |
Hypomagnesemi | Vanliga | 6 | 0 |
Centrala och perifera nervsystemet | |||
Parestesi*, a | Mycket vanliga | 29 | 1,2 |
Yrsel* | 12 | 0 | |
Ögon | |||
Andra ögonsjukdomar* | Mycket vanliga | 19 | 0,5 |
Synrubbningar* | Vanliga | 3,6 | 0 |
Keratit | 1,9 | 0,2 | |
Tillväxt av ögonfransar* | 1,8 | 0 | |
Blodkärl | |||
Venös tromboembolism | |||
Amivantamab intravenös*, b | Mycket vanliga | 37 | 11 |
Amivantamab subkutan*, c | Mycket vanliga | 11 | 0,7 |
Andningsvägar, bröstkorg och mediastinum | |||
Interstitiell lungsjukdom* | Vanliga | 3,6 | 1,7 |
Magtarmkanalen | |||
Stomatit* | Mycket vanliga | 43 | 2,2 |
Diarré | 26 | 1,8 | |
Förstoppning | 25 | 0 | |
Illamående | 24 | 0,7 | |
Kräkningar | 15 | 0,5 | |
Buksmärta* | Vanliga | 9 | 0,1 |
Hemorrojder | 8 | 0,1 | |
Lever och gallvägar | |||
Hepatotoxicitet* | Mycket vanliga | 43 | 7 |
Hud och subkutan vävnad | |||
Utslag* | Mycket vanliga | 87 | 22 |
Nageltoxicitet* | 68 | 8 | |
Torr hud* | 25 | 0,7 | |
Klåda | 24 | 0,4 | |
Hudsår | Vanliga | 3,7 | 0,5 |
Palmar-plantar erytrodysestesi syndrom | 3,5 | 0,1 | |
Urtikaria | 1,6 | 0 | |
Muskuloskeletala systemet och bindväv | |||
Myalgi | Mycket vanliga | 15 | 0,5 |
Muskelspasmer | 13 | 0,4 | |
Allmänna symtom och/eller symtom vid administreringsstället | |||
Ödem* | Mycket vanliga | 42 | 2,4 |
Trötthet* | 34 | 3,4 | |
Pyrexi | 11 | 0 | |
Reaktioner vid injektionsstället*, c, d | Vanliga | 6 | 0 |
Skador, förgiftningar och behandlingskomplikationer | |||
Infusions‑/Administreringsrelaterade reaktioner | |||
Amivantamab intravenöstb, e | Mycket vanliga | 63 | 6 |
Amivantamab subkutantc, f | Mycket vanliga | 14 | 0,5 |
* Grupperade termer. | |||
Rybrevant i kombination med karboplatin och pemetrexed
I datasetet för Rybrevant (antingen intravenösa eller subkutana formuleringar) i kombination med karboplatin och pemetrexed (N = 444) var de vanligaste biverkningarna av någon grad (≥ 20 % av patienterna) utslag (83 %), nageltoxicitet (57 %), neutropeni (56 %), trötthet (46 %), illamående (44 %), stomatit (42 %), ödem (41 %), trombocytopeni (40 %), förstoppning (39 %), hypoalbuminemi (36 %), minskad aptit (32 %), ökat alaninaminotransferas (28 %), ökat aspartataminotransferas (24 %) och kräkningar (23 %).
Allvarliga biverkningar rapporterades hos 19 % av de patienter som fick Rybrevant subkutan formulering i kombination med karboplatin och pemetrexed, inklusive kräkningar (2,8 %), ILD (2,8 %), illamående (2,1 %), trötthet (2,1 %), VTE (2,1 %), neutropeni (2,1 %), diarré (1,4 %) och hypokalcemi (1,4 %). 6 % av patienterna avbröt behandlingen med Rybrevant subkutan formulering på grund av biverkningar. Hos patienter som behandlades med Rybrevant subkutan formulering i kombination med karboplatin och pemetrexed var de vanligaste biverkningarna av någon grad (≥ 1 % av patienterna) som ledde till utsättning av Rybrevant subkutan formulering ILD (2,8 %) och utslag (1,4 %).
Biverkningar i tabellform
Biverkningarna för Rybrevant (antingen intravenös eller subkutan formulering) när det ges i kombination med karboplatin och pemetrexed sammanfattas i tabell 8.
Säkerhetsdatan nedan återspeglar exponering för Rybrevant (antingen intravenös eller subkutan formulering) i kombination med karboplatin och pemetrexed hos 444 patienter med lokalt avancerad eller metastaserad NSCLC, inklusive 151 patienter i PAPILLON, 130 patienter i MARIPOSA-2, 20 patienter i CHRYSALIS och 143 patienter i PALOMA‑2 kohort 2 och 3b. Patienterna fick Rybrevant (antingen intravenös eller subkutan formulering) tills sjukdomsprogression eller oacceptabel toxicitet. Mediantiden för behandling med amivantamab totalt för både intravenösa och subkutana formuleringar var 7,4 månader (intervall: 0,0 till 28,1 månader). Mediantiden för behandling med den subkutana formuleringen var 6,9 månader (intervall: 0,0 till 15,4 månader) medan mediantiden för behandling med den intravenösa formuleringen var 7,7 månader (intervall: 0,0 till 28,1 månader).
Biverkningar observerade under kliniska studier listas nedan efter frekvenskategori. Frekvenskategorier definieras enligt följande: mycket vanliga (≥ 1/10); vanliga (≥ 1/100, < 1/10); mindre vanliga (≥ 1/1 000, < 1/100); sällsynta (≥ 1/10 000, < 1/1 000); mycket sällsynta (< 1/10 000); samt ingen känd frekvens (kan inte beräknas från tillgängliga data).
Tabell 8: Biverkningar hos patienter som fått Rybrevant (antingen intravenös eller subkutan formulering) i kombination med karboplatin och pemetrexed (N = 444) | |||
Organsystemklass | Frekvenskategori | Alla grader | Grad 3‑4 |
Blodet och lymfsystemet | |||
Neutropeni | Mycket vanliga | 56 | 36 |
Trombocytopeni | 40 | 13 | |
Metabolism och nutrition | |||
Hypoalbuminemi* | Mycket vanliga | 36 | 5 |
Minskad aptit | 32 | 1,1 | |
Hypokalemi | 19 | 7 | |
Hypokalcemi | 14 | 2,0 | |
Hypomagnesemi | 11 | 1,8 | |
Centrala och perifera nervsystemet | |||
Yrsel* | Mycket vanliga | 10 | 0,5 |
Ögon | |||
Andra ögonsjukdomar* | Mycket vanliga | 15 | 0 |
Synrubbningar* | Vanliga | 2,9 | 0 |
Tillväxt av ögonfransar | Mindre vanliga | 0,7 | 0 |
Keratit | 0,2 | 0 | |
Uveit | 0,2 | 0 | |
Blodkärl | |||
Venös tromboembolism | |||
Amivantamab intravenös*, a | Mycket vanliga | 14 | 3,0 |
Amivantamab subkutan*, b | Mycket vanliga | 18 | 2,8 |
Andningsvägar, bröstkorg och mediastinum | |||
Interstitiell lungsjukdom* | Vanliga | 2,5 | 1,4 |
Magtarmkanalen | |||
Illamående | Mycket vanliga | 44 | 1,4 |
Stomatit* | 42 | 4,3 | |
Förstoppning | 39 | 0,2 | |
Kräkningar | 23 | 2,9 | |
Diarré | 19 | 2,5 | |
Buksmärta* | 12 | 0,5 | |
Hemorrojder | Vanliga | 8 | 0,5 |
Hud och subkutan vävnad | |||
Utslag* | Mycket vanliga | 83 | 14 |
Nageltoxicitet* | 57 | 4,3 | |
Torr hud* | 14 | 0,2 | |
Klåda | 12 | 0 | |
Hudsår | Vanliga | 3,2 | 0,7 |
Muskuloskeletala systemet och bindväv | |||
Myalgi | Vanliga | 6 | 0,5 |
Allmänna symtom och/eller symtom vid administreringsstället | |||
Trötthet* | Mycket vanliga | 46 | 6 |
Ödem* | 41 | 1,4 | |
Pyrexi | 13 | 0,2 | |
Reaktioner vid injektionsstället*, b, c | Vanliga | 3,5 | 0 |
Undersökningar och provtagningar | |||
Ökat alaninaminotransferas | Mycket vanliga | 28 | 4,1 |
Ökat aspartataminotransferas | 24 | 1,4 | |
Ökat alkaliskt fosfatas i blodet | Vanliga | 8 | 0,2 |
Skador, förgiftningar och behandlingskomplikationer | |||
Infusions‑/Administreringsrelaterade reaktioner | |||
Amivantamab intravenösta, d | Mycket vanliga | 51 | 3,0 |
Amivantamab subkutantb, e | Vanliga | 7 | 0 |
* Grupperade termer. | |||
Beskrivning av utvalda biverkningar
Administreringsrelaterade reaktioner
Administreringsrelaterade reaktioner rapporterades hos 14 % av patienterna som behandlades med Rybrevant subkutan formulering i kombination med lazertinib och 7 % av patienterna som behandlades med Rybrevant subkutan formulering i kombination med karboplatin och pemetrexed. De vanligaste tecknen och symtomen på administrationsrelaterade reaktioner inkluderar dyspné, rodnad, feber, frossa, illamående och obehag i bröstet. Hos patienter som behandlades med Rybrevant subkutan formulering i kombination med lazertinib var mediantiden till debut av de första administrationsrelaterade reaktionerna 1,9 timmar (intervall: 0,0 till 176,5 timmar) och de flesta administreringsrelaterade reaktionerna (96 %) var av allvarlighetsgrad 1 eller 2. Hos patienter som behandlades med Rybrevant subkutan formulering i kombination med karboplatin och pemetrexed var mediantiden till första administreringsrelaterade reaktion 2,1 timmar (intervall: 0,7 till 3,1 timmar), och alla administreringsrelaterade reaktioner var av allvarlighetsgrad 1 eller 2. I PALOMA‑3 rapporterades administreringsrelaterade reaktioner hos 13 % av patienterna som behandlades med Rybrevant subkutan formulering i kombination med lazertinib jämfört med 66 % när de behandlades med Rybrevant intravenös formulering i kombination med lazertinib.
Reaktioner på injektionsstället
Reaktioner på injektionsstället förekom hos 6 % av patienterna som behandlades med Rybrevant subkutan formulering i kombination med lazertinib och 3,5 % av patienterna som behandlades med Rybrevant subkutan formulering i kombination med karboplatin och pemetrexed. Alla reaktioner på injektionsstället var av allvarlighetsgrad 1 eller 2. Det vanligaste symtomet på reaktioner på injektionsstället var erytem.
Interstitiell lungsjukdom
Interstitiell lungsjukdom (ILD) eller ILD-liknande biverkningar har rapporterats vid användning av amivantamab liksom med andra EGFR-hämmare. ILD rapporterades hos 3,6 % av patienterna som behandlades med Rybrevant (antingen intravenös eller subkutan formulering) i kombination med lazertinib, inklusive 2 (0,2 %) patienter med en dödlig reaktion och 2,5 % av patienterna som behandlades med Rybrevant (antingen intravenös eller subkutan formulering) i kombination med karboplatin och pemetrexed. Patienter med ILD i sjukdomshistorien, inklusive läkemedelsinducerad ILD eller strålningspneumonit, uteslöts från PALOMA-2 och PALOMA-3.
Venösa tromboemboliska (VTE) händelser vid samtidig användning med lazertinib
VTE-händelser, inklusive djup ventrombos (DVT) och lungemboli (PE), rapporterades hos 11 % av patienterna som fick Rybrevant subkutan formulering i kombination med lazertinib i PALOMA-2 och PALOMA-3. De flesta fallen var av grad 1 eller 2, medan händelser av grad 3 inträffade hos 3 (0,7 %) patienter. Dessutom tog 336 (82 %) av dessa 408 patienter som fick Rybrevant subkutan formulering profylaktiska antikoagulantia med en direkt oral antikoagulant eller lågmolekylärt heparin under de första fyra månaderna av studiebehandlingen.
I PALOMA-3, för direkt jämförelse mellan armarna, var förekomsten av VTE-händelser 9 % för patienter som behandlades med Rybrevant subkutan formulering i kombination med lazertinib, jämfört med 14 % för patienter som behandlades med Rybrevant intravenös formulering i kombination med lazertinib, med liknande frekvenser av profylaktisk antikoagulantiabehandling i båda behandlingsarmarna (80 % i den subkutana armen jämfört med 81 % i den intravenösa armen). För patienter som inte fick profylaktiska antikoagulantia var den totala incidensen av VTE-händelser 17 % för patienter som behandlades med Rybrevant subkutan formulering i kombination med lazertinib, med alla VTE-händelser rapporterade som grad 1–2 och allvarliga VTE-händelser rapporterade hos 4,8 % av dessa patienter, jämfört med en total incidens på 26 % för patienter som behandlades med Rybrevant intravenös formulering i kombination med lazertinib, med VTE-händelser av grad 3 och grad 4 rapporterade hos 10 % respektive 2,6 % av patienterna och allvarliga VTE-händelser rapporterade hos 10 % av dessa patienter.
Hud- och nagelreaktioner
Utslag (inklusive akneiform dermatit), klåda och torr hud har förekommit hos patienter som behandlats med Rybrevant (antingen intravenös eller subkutan formulering). Utslag förekom hos 87 % av patienterna som behandlades med Rybrevant i kombination med lazertinib, vilket ledde till utsättning av Rybrevant hos 0,6 % av patienterna, och hos 83 % av patienterna som behandlades med Rybrevant i kombination med karboplatin och pemetrexed, vilket ledde till utsättning av Rybrevant hos 0,5 % av patienterna. Bland patienterna som behandlades med Rybrevant i kombination med lazertinib var de flesta fallen av grad 1 eller 2, medan reaktioner av grad 3 och 4 förekom hos 22 % respektive 0,1 % av patienterna. Bland patienterna som behandlades med Rybrevant i kombination med karboplatin och pemetrexed var de flesta fallen av grad 1 eller 2, medan reaktioner av grad 3 förekom hos 14 % av patienterna.
En fas 2-studie på patienter som behandlades med Rybrevant i kombination med lazertinib genomfördes för att utvärdera användningen av profylaktisk behandling med ett oralt antibiotikum, ett topikalt antibiotikum i hårbotten, en fuktighetskräm i ansiktet och på hela kroppen (utom hårbotten) samt ett antiseptiskt medel på händer och fötter (se avsnitt Dosering och administreringssätt och Varningar och försiktighet). En minskning av förekomsten av dermatologiska biverkningar av ≥ grad 2 under de första 12 veckorna av behandlingen påvisades, jämfört med den dermatologiska standardbehandling som används enligt klinisk praxis (38,6 % jämfört med 76,5 %, p < 0,0001). Dessutom sågs en minskning av biverkningar av ≥ grad 2 i hårbotten under de första 12 veckorna av behandlingen (8,6 % jämfört med 29,4 %) samt en lägre förekomst av dosreduktioner (7,1 % jämfört med 19,1 %), dosavbrott (15,7 % jämfört med 33,8 %) och behandlingsavbrott (1,4 % jämfört med 4,4 %) på grund av dermatologiska biverkningar.
Ögonsjukdomar
Ögonsjukdomar förekom hos patienter som behandlades med Rybrevant (antingen intravenös eller subkutan formulering), inklusive keratit hos 1,9 % av patienterna som behandlades med Rybrevant i kombination med lazertinib och 0,2 % av patienterna som behandlades med Rybrevant i kombination med karboplatin och pemetrexed. Andra rapporterade biverkningar inkluderade tillväxt av ögonfransar, synrubbningar och andra ögonsjukdomar.
Särskilda populationer
Äldre
Det finns begränsade kliniska data för amivantamab hos patienter som är 75 år och äldre (se avsnitt Farmakodynamiska egenskaper). Det har inte observerats några övergripande skillnader i säkerhet mellan patienter som är ≥ 65 år och patienter som är < 65 år.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning til
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Det finns ingen information om överdosering med Rybrevant subkutan formulering och ingen känd specifik antidot mot överdosering. Vid överdosering ska behandlingen med Rybrevant stoppas och patienten ska övervakas med avseende på tecken eller symtom på biverkningar. Lämpliga allmänna stödåtgärder ska omedelbart vidtas tills den kliniska toxiciteten har minskat eller försvunnit.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Monoklonala antikroppar och antikroppskonjugat, ATC-kod: L01FX18.
Rybrevant subkutan formulering innehåller rekombinant humant hyaluronidas (rHuPH20). rHuPH20 verkar lokalt och övergående för att bryta ned hyaluronan ((HA), en naturligt förekommande glykoaminoglykan som finns i hela kroppen) i det extracellulära matrix i det subkutana utrymmet, genom att klyva kopplingen mellan de två sockerarterna (N-acetylglukosamin och glukuronsyra) som utgör HA.
Verkningsmekanism
Amivantamab är en helt human IgG1-baserad bispecifik antikropp mot EGFR-MET med lågt fukosinnehåll och immuncellstyrande aktivitet, som riktar sig mot tumörer med aktiverande mutationer i EGFR-genen såsom exon 19-deletion, substitutionsmutation L858R i exon 21 och insertionsmutationer i exon 20. Amivantamab binds till de extracellulära domänerna på EGFR och MET.
Amivantamab stör signalfunktioner hos EGFR och MET genom att blockera ligandbindning och öka nedbrytningen av EGFR och MET, och förhindrar på så sätt tumörtillväxt och progression. Närvaron av EGFR och MET på tumörcellernas yta möjliggör även inriktning (”targeting”) mot dessa celler för destruktion genom immuneffektorceller, såsom NK-celler (natural killer cells) och makrofager, med antikroppsberoende cellmedierad cytotoxicitet (ADCC) respektive trogocytosmekanismer.
Farmakodynamisk effekt
Efter den första fulla dosen av Rybrevant subkutan formulering minskade de genomsnittliga EGFR- och MET-koncentrationerna i serum avsevärt och förblev dämpade under hela behandlingstiden för alla studerade doser.
Albumin
Rybrevant subkutan formulering minskade koncentrationen av serumalbumin, en farmakodynamisk effekt av MET-hämning, vanligtvis under de första 8 veckorna (se avsnitt Biverkningar). Därefter stabiliserades albuminkoncentrationen under återstående delen av behandlingen med amivantamab.
Klinisk erfarenhet av Rybrevant subkutan formulering
Effekten av Rybrevant subkutan formulering hos patienter med EGFR-muterad lokalt avancerad eller metastaserad NSCLC baseras på att en PK-exponering uppnåddes som inte var sämre än intravenös amivantamab i non-inferiority-studien PALOMA-3 (se avsnitt Farmakokinetiska egenskaper). Studien visade att subkutan amivantamab inte var sämre än intravenös amivantamab i kombination med lazertinib hos patienter med EGFR-muterad lokalt avancerad eller metastaserad NSCLC vars sjukdom hade progredierat under eller efter behandling med osimertinib och platinabaserad kemoterapi.
Klinisk erfarenhet av Rybrevant intravenös formulering
Tidigare obehandlad NSCLC med EGFR exon 19-deletion eller substitutionsmutation L858R i exon 21 (MARIPOSA)
NSC3003 (MARIPOSA) är en randomiserad, öppen, multicenter-, fas 3-studie med aktiv kontroll som utvärderar effekt och säkerhet för Rybrevant intravenös formulering i kombination med lazertinib jämfört med osimertinib som monoterapi vid första linjens behandling av patienter med EGFR-muterad lokalt avancerad eller metastaserad icke-småcellig lungcancer (NSCLC) som inte är mottaglig för kurativ behandling. Patientproverna måste ha en av de två vanliga EGFR-mutationerna (exon 19-deletion eller substitutionsmutation L858R i exon 21) som identifierats med lokala tester. Prover på tumörvävnad (94 %) och/eller plasma (6 %) från alla patienter testades lokalt för att fastställa EGFR exon 19-deletion och/eller substitutionsmutation L858R i exon 21 med hjälp av Polymerase Chain Reaction (PCR) hos 65 % av patienterna och Next generation Sequencing (NGS) hos 35 % av patienterna.
Totalt 1 074 patienter randomiserades (2:2:1) till att få Rybrevant intravenös formulering i kombination med lazertinib, osimertinib monoterapi eller lazertinib monoterapi fram till sjukdomsprogression eller oacceptabel toxicitet. Rybrevant intravenös formulering administrerades intravenöst med 1 050 mg (för patienter < 80 kg) eller 1 400 mg (för patienter ≥ 80 kg) en gång i veckan i 4 veckor, därefter varannan vecka med början vecka 5. Lazertinib administrerades med 240 mg oralt en gång dagligen. Osimertinib administrerades oralt i en dos på 80 mg en gång dagligen. Randomiseringen stratifierades efter EGFR-mutationstyp (exon 19-deletion eller L858R i exon 21), etnicitet (asiatisk eller icke-asiatisk) och tidigare hjärnmetastaser (ja eller nej).
Demografiska data och sjukdomskarakteristika vid baseline var balanserade mellan behandlingsarmarna. Medianåldern var 63 år (intervall: 25 –88) med 45 % av patienterna ≥ 65 år; 62 % var kvinnor; 59 % var asiater och 38 % var vita. Vid baseline var funktionsstatus 0 (34 %) eller 1 (66 %) enligt Eastern Cooperative Oncology Group (ECOG); 69 % hade aldrig rökt; 41 % hade tidigare haft hjärnmetastaser; och 90 % hade cancer i stadium IV vid första diagnos. När det gäller EGFR-mutationsstatus var 60 % exon 19-deletioner och 40 % substitutionsmutation L858R i exon 21.
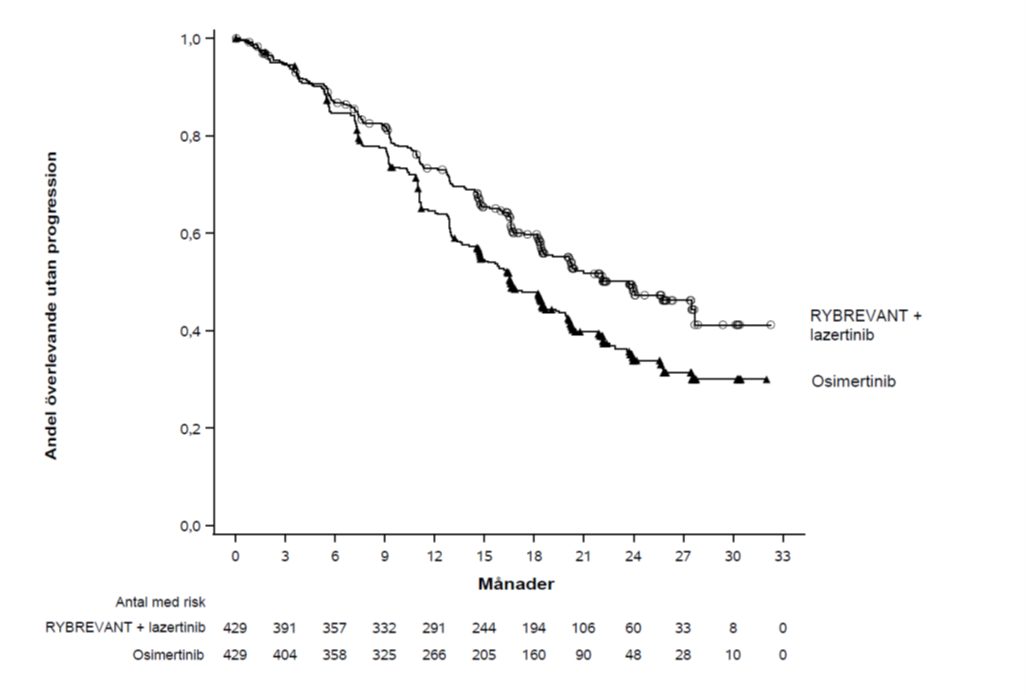
Rybrevant intravenös formulering i kombination med lazertinib uppvisade en statistiskt signifikant förbättring av progressionsfri överlevnad (PFS) enligt BICR-bedömning.
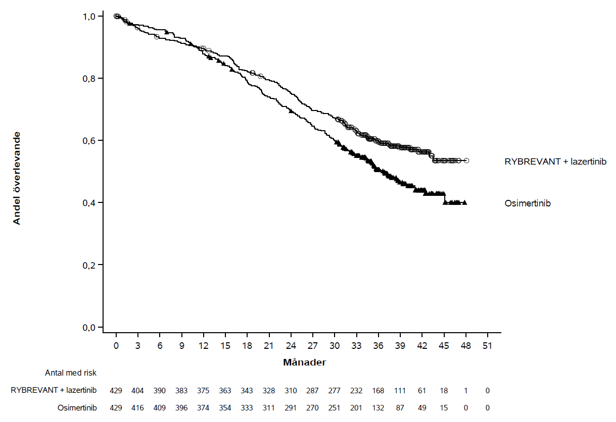
Den slutliga analysen av OS visade en statistiskt signifikant förbättring i OS för Rybrevant intravenös sammansättning i kombination med lazertinib jämfört med osimertinib (se tabell 9 och figur 2).
| Tabell 9: Effektresultat i MARIPOSA | ||
| Rybrevant intravenös formulering + lazertinib (N = 429) | Osimertinib (N = 429) | |
| Progressionfri överlevnad (PFS)a | ||
| Antal händelser | 192 (45 %) | 252 (59 %) |
| Median, månader (95 % KI) | 23,7 (19,1, 27,7) | 16,6 (14,8, 18,5) |
| HR (95 % KI); p‑värde | 0,70 (0,58, 0,85); p = 0,0002 | |
| Total överlevnad (OS) | ||
| Antal händelser | 173 (40 %) | 217 (51 %) |
| Median, månader (95 % KI) | NE (42,9, NE) | 36,7 (33,4, 41,0) |
| HR (95 % KI); p‑värde | 0,75 (0,61, 0,92); p = 0,0048 | |
| Objektiv svarsfrekvens (ORR)a,b | ||
| ORR % (95 % KI) | 80 % (76 %, 84 %) | 77 % (72 %, 81 %) |
| Svarets varaktighet (DOR)a,b | ||
| Median (95 % KI), månader | 25,8 (20,3, 33,9) | 18,1 (14,8, 20,1) |
| BICR = blindad oberoende granskning; KI = konfidensintervall; NE = kan inte uppskattas. PFS-resultat är från datagränsvärde 11 augusti 2023 med en medianuppföljning på 22,0 månader. DOR- och ORR-resultat är från datagränsvärde 13 maj 2024 med mediantid för uppföljning på 31,3 månader. OS-resultat är från datagränsvärde 4 december 2024 med en medianuppföljning på 37,8 månader. a BICR enligt RECIST v1.1. b Baserat på bekräftade respondenter. | ||
Figur 1: Kaplan-Meier-kurva av PFS hos tidigare obehandlade patienter med NSCLC via BICR-bedömning
Figur 2: Kaplan-Meier-kurva av OS hos tidigare obehandlade patienter med NSCLC
Intrakraniell ORR och DOR enligt BICR var förutbestämda effektmått i MARIPOSA. I undergruppen av patienter med intrakraniella lesioner vid baseline visade kombinationen av Rybrevant intravenös formulering och lazertinib liknande intrakraniell ORR som kontrollgruppen. Enligt protokoll genomgick alla patienter i MARIPOSA på varandra följande magnetkameraundersökningar (MR) av hjärnan för att bedöma intrakraniellt svar och varaktighet. Resultaten sammanfattas i tabell 10.
| Tabell 10: Intrakraniell ORR och DOR enligt BICR-bedömning hos försökspersoner med intrakraneilla lesioner vid baseline – MARIPOSA | ||
Rybrevant intravenös formulering + lazertinib (N = 180) | Osimertinib (N = 186) | |
| Intrakraniell tumörresponsbedömning | ||
| Intrakraniell ORR (CR + PR), % (95 % KI) | 78 % (71 %, 84 %) | 77 % (71 %, 83 %) |
| Komplett svar | 64 % | 59 % |
| Intrakraniell DOR | ||
| Antal respondenter | 140 | 144 |
| Median, månader (95 % KI) | 35,0 (20,4, NE) | 25,1 (22,1, 31,2) |
| KI = konfidensintervall NE = kan inte uppskattas Intrakraniella ORR- och DOR-resultat är från datagränsvärde 4 december 2024 med en medianuppföljning på 37,8 månader. | ||
Tidigare behandlad NSCLC med EGFR exon 19-deletion eller substitutionsmutation L858R i exon 21 (MARIPOSA‑2)
MARIPOSA‑2 är en randomiserad (2:2:1), öppen, multicenter-, fas 3-studie som genomförs på patienter med lokalt avancerad eller metastaserande NSCLC med EGFR exon 19-deletion eller substitionsmutation L858R i exon 21 (mutationstestning kan ha utförts vid eller efter tidpunkten för diagnos av lokalt avancerad eller metastaserad sjukdom. Testningen behövde inte upprepas vid tidpunkten för studiestart när EGFR-mutationsstatus tidigare hade fastställts) efter svikt på tidigare behandling inklusive tredje generationens EGFR-tyrosinkinashämmare (TKI). Totalt 657 patienter randomiserades i studien, där 263 fick karboplatin och pemetrexed (CP); och 131 fick Rybrevant intravenös formulering i kombination med karboplatin och pemetrexed (Rybrevant intravenös formulering-CP). Dessutom randomiserades 263 patienter till att få Rybrevant intravenös formulering i kombination med lazertinib, karboplatin och pemetrexed i en separat arm av studien. Rybrevant intravenös formulering administrerades intravenöst med 1 400 mg (hos patienter < 80 kg) eller 1 750 mg (hos patienter ≥ 80 kg) en gång i veckan under 4 veckor, därefter var tredje vecka med en dos på 1 750 mg (hos patienter < 80 kg) eller 2 100 mg (hos patienter ≥ 80 kg), med start i vecka 7 och fram till sjukdomsprogression eller oacceptabel toxicitet. Karboplatin administrerades intravenöst med en area under koncentrationskurvan på 5 mg/ml per minut (AUC 5) en gång var tredje vecka i upp till 12 veckor. Pemetrexed adminstrerades intravenöst med 500 mg/m2 en gång var tredje vecka tills sjudomsprogression eller oacceptabel toxicitet uppstod.
Patienter stratifierades efter behandlingslinje med osimertinib (första linjen eller andra linjen), förekomst av hjärnmetastaser (ja eller nej) och asiatiskt ursprung (ja eller nej).
Av de 394 patienterna som randomiserades till Rybrevant intravenös formulering-CP-armen eller CP‑armen var medianåldern 62 (intervall 31–85) år, där 38 % av patienterna var ≥ 65 år; 60 % var kvinnor, 48 % var asiater och 46 % var vita. Vid behandlingsstart var Eastern Cooperative Oncology Groups (ECOG)-funktionsstatus 0 (40 %) eller 1 (60 %); 66 % hade aldrig rökt; 45 % hade tidigare haft hjärnmetastaser, och 92 % hade stadie IV‑cancer vid första diagnosen.
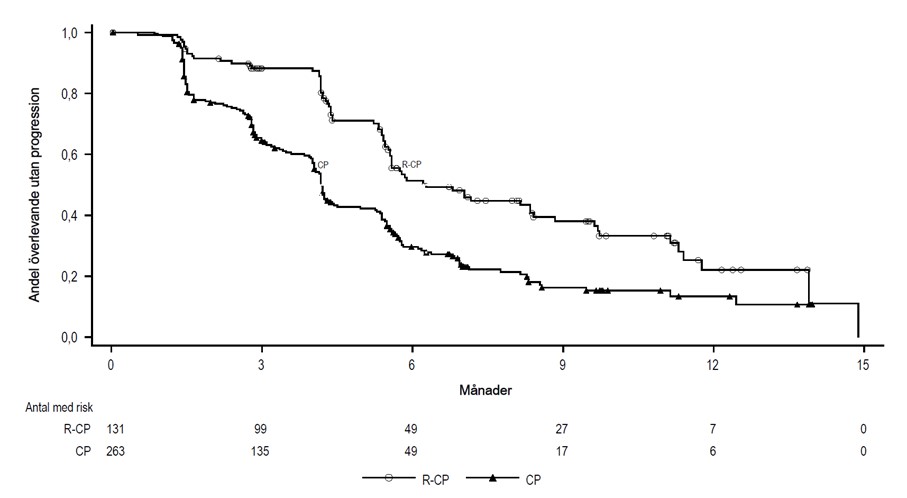
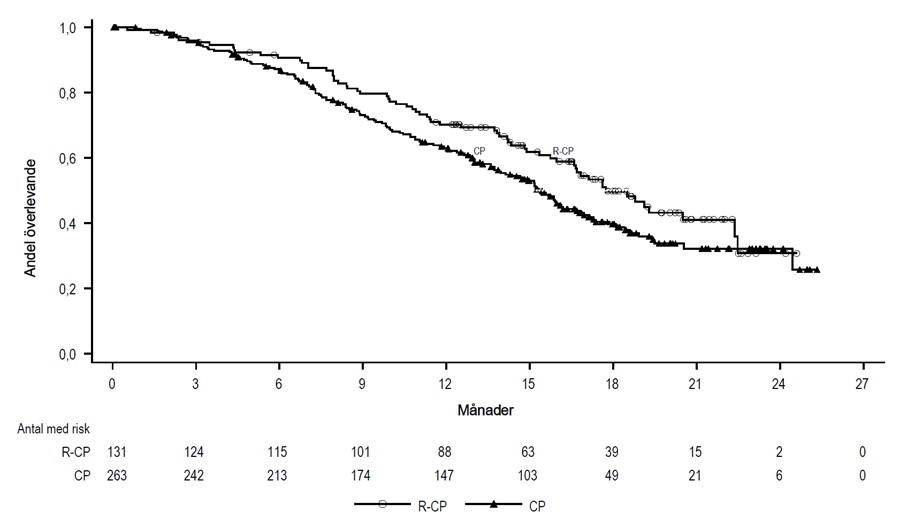
Rybrevant intravenös formulering i kombination med karboplatin och pemetrexed visade en statistiskt signifikant förbättring i progressionsfri överlevnad (PFS) jämfört med karboplatin och pemetrexed, med en HR på 0,48 (95 % KI: 0,36; 0,64; p < 0,0001). Vid tiden för den andra interimanalysen för OS, med en medianuppföljning på cirka 18,6 månader för Rybrevant intravenös formulering-CP och cirka 17,8 månader för CP, var OS HR 0,73 (95 % KI: 0,54; 0,99; p = 0,0386). Detta var inte statistiskt signifikant (testat med en förspecificerad signifikansnivå på 0,0142).
Effektresultat sammanfattas i tabell 11.
| Tabell 11: Effektresultat i MARIPOSA-2 | ||
| Rybrevant intravenös formulering + karboplatin + pemetrexed (N = 131) | karboplatin + pemetrexed (N = 263) | |
| Progressionfri överlevnad (PFS)a | ||
| Antal händelser (%) | 74 (57) | 171 (65) |
| Median, månader (95 % KI) | 6,3 (5,6; 8,4) | 4,2 (4,0; 4,4) |
| HR (95 % KI); p-värde | 0,48 (0,36; 0,64); p < 0,0001 | |
| Total överlevnad (OS) | ||
| Antal händelser (%) | 65 (50) | 143 (54) |
| Median, månader (95 % KI) | 17,7 (16,0; 22,4) | 15,3 (13,7; 16,8) |
| HR (95 % KI); p-värdeb | 0,73 (0,54; 0,99); p = 0,0386 | |
| Objektiv svarsfrekvensa | ||
| ORR, % (95 % KI) | 64 % (55 %; 72 %) | 36 % (30 %; 42 %) |
| Odds-kvot (95 % KI); p-värde | 3,10 (2,00; 4,80); p < 0,0001 | |
| Svarets varaktighet (DOR)a | ||
| Median (95 % KI), månader | 6,90 (5,52; NE) | 5,55 (4,17; 9,56) |
| Patienter med DOR ≥ 6 månader | 31,9 % | 20,0 % |
| KI = konfidensintervall NE = kan inte uppskattas PFS- DOR- och ORR-resultat är från datagränsvärde10 juli 2023, när hypotestestning och slutlig analys av dessa effektmått utfördes. OS-resultat är från datagränsvärde 26 april 2024 från den andra interim-OS-analysen. a Hämtad från BICR b P-värdet jämförs med en 2-sidig signifikansnivå på 0,0142. OS-resultaten är således inte signifikanta från och med den andra interimsanalysen. | ||
Figur 3: Kaplan-Meier-kurva av PFS hos tidigare behandlade patienter med NSCLC via BICR-bedömning
PFS-fördelen med Rybrevant intravenös formulering-CP jämfört med CP var konsekvent i alla fördefinierade subgrupper som analyserades, inklusive etnicitet, ålder, kön, historik av rökning och CNS-metastaser vid studiens början.
Figur 4: Kaplan-Meier-kurva av OS hos tidigare behandlade patienter med NSCLC
Effektdata intrakraniella metastaser
Patienter med asymtomatiska eller tidigare behandlade och stabila intrakraniella metastaser var kvalificerade att bli randomiserade i MARIPOSA‑2. Behandling med Rybrevant intravenös formulering-CP förknippades med en numerisk ökning i intrakraniell ORR (23,3 % för Rybrevant intravenös formulering-CP jämfört med 16,7 % för CP, oddskvot på 1,52; 95 % KI (0,51; 4,50)), och intrakraniell DOR (13,3 månader; 95 % KI (1,4; NE) i armen med Rybrevant intravenös formulering-CP jämfört med 2,2 månader; 95 % KI (1,4; NE) i CP-armen). Medianuppföljningstiden för Rybrevant intravenös formulering-CP var cirka 18,6 månader.
Tidigare obehandlad icke-småcellig lungcancer (NSCLC) med insertionsmutationer i exon 20 (PAPILLON)
PAPILLON är en randomiserad, öppen, multicenter-, fas 3-studie som jämför behandling med Rybrevant intravenös formulering i kombination med karboplatin och pemetrexed med enbart kemoterapi (karboplatin och pemetrexed) hos patienter med behandlingsnaiv, lokalt avancerad eller metastaserad NSCLC med aktiverande insertionsmutationer i EGFR-genens exon 20. Tumörvävnadsprover (92,2 %) och/eller plasmaprover (7,8 %) från samtliga 308 patienter testades lokalt för att fastställa mutationsstatus för insertion i EGFR-genens exon 20 med hjälp av Next generation Sequencing (NGS) hos 55,5 % av patienterna och/eller Polymerase Chain Reaction (PCR) hos 44,5 % av patienterna. Central testning utfördes också med hjälp av AmoyDx® LC10-vävnadstest, Thermo Fisher Oncomine Dx Target Test och Guardant 360® CDx-plasmatest.
Patienter med hjärnmetastaser vid screening kunde rekryteras till studien när de var definitivt behandlade, kliniskt stabila, symtomfria och utan kortikosteroidbehandling i minst 2 veckor före randomiseringen.
Rybrevant intravenös formulering administrerades intravenöst med 1 400 mg (hos patienter < 80 kg) eller 1 750 mg (hos patienter ≥ 80 kg) en gång i veckan under 4 veckor, därefter var tredje vecka med en dos på 1 750 mg (hos patienter < 80 kg) eller 2 100 mg (hos patienter ≥ 80 kg) med start vecka 7 tills sjukdomsprogression eller oacceptabel toxicitet. Karboplatin administrerades intravenöst med en area under koncentrationskurvan på 5 mg/ml per minut (AUC 5) en gång var tredje vecka i upp till 12 veckor. Pemetrexed administrerades intravenöst med 500 mg/m2 en gång var tredje vecka tills sjukdomsprogression eller oacceptabel toxicitet uppstod. Randomiseringen stratifierades efter ECOG‑funktionsstatus (0 eller 1), och tidigare hjärnmetastaser (ja eller nej). Patienter som randomiserats till karboplatin och pemetrexed-armen och som hade bekräftad sjukdomsprogression fick gå över till att få Rybrevant intravenös formulering som monoterapi.
Totalt 308 försökspersoner randomiserades (1:1) till Rybrevant intravenös formulering i kombination med karboplatin och pemetrexed (N = 153) eller karboplatin och pemetrexed (N = 155). Medianåldern var 62 år (intervall: 27 till 92), med 39 % av deltagarna ≥ 65 år; 58 % var kvinnor; 61 % var asiater och 36 % var vita. Vid behandlingsstart var Eastern Cooperative Oncology Groups (ECOG)‑funktionsstatus 0 (35 %) eller 1 (64 %), 58 % hade aldrig rökt, 23 % hade tidigare haft hjärnmetastaser och 84 % hade stadie IV‑cancer vid första diagnosen.
Det primära effektresultatet för PAPILLON var progressionsfri överlevnad (PFS), utvärderat enligt BICR. Medianuppföljningen var 14,9 månader (intervall: 0,3 till 27,0).
Effektresultaten är sammanfattade i tabell 12.
| Tabell 12: Effektresultat i PAPILLON | |||
| Rybrevant intravenös formulering + karboplatin + pemetrexed (N = 153) | Karboplatin + pemetrexed (N = 155) | ||
| Progressionsfri överlevnad (PFS)a | |||
| Antal händelser | 84 (55 %) | 132 (85 %) | |
| Median, månader (95 % KI) | 11,4 (9,8; 13,7) | 6,7 (5,6; 7,3) | |
| HR (95 % KI); p-värde | 0,395 (0,29; 0,52); p < 0,0001 | ||
| Objektiv svarsfrekvensa, b | |||
| ORR, % (95 % KI) | 73 % (65 %; 80 %) | 47 % (39 %; 56 %) | |
| Oddskvot (95 % KI); p-värde | 3,0 (1,8; 4,8); p < 0,0001 | ||
| Komplett svar | 3,9 % | 0,7 % | |
| Partiellt svar | 69 % | 47 % | |
| Total överlevnad (OS)c | |||
| Antal händelser | 40 | 52 | |
| Median OS, månader (95 % KI) | NE (28,3; NE) | 28,6 (24,4; NE) | |
| HR (95 % KI); p-värde | 0,756 (0,50; 1,14); p = 0,1825 | ||
| KI = konfidensintervall NE = kan inte uppskattas a Blindad oberoende central granskning enligt RECIST v1.1 b Baserat på Kaplan-Meier-skattning. c Baserat på resultaten av ett uppdaterad OS med en medianuppföljning på 20,9 månader. OS-analysen justerades inte för de potentiellt störande effekterna av behandlingsbyte (78 [50,3 %] patienter i karboplatin + pemetrexed-armen som fick efterföljande monoterapibehandling med Rybrevant intravenös formulering). | |||
Figur 5: Kaplan-Meier-kurva för PFS hos tidigare obehandlade patienter med NSCLC enligt BICR-bedömning
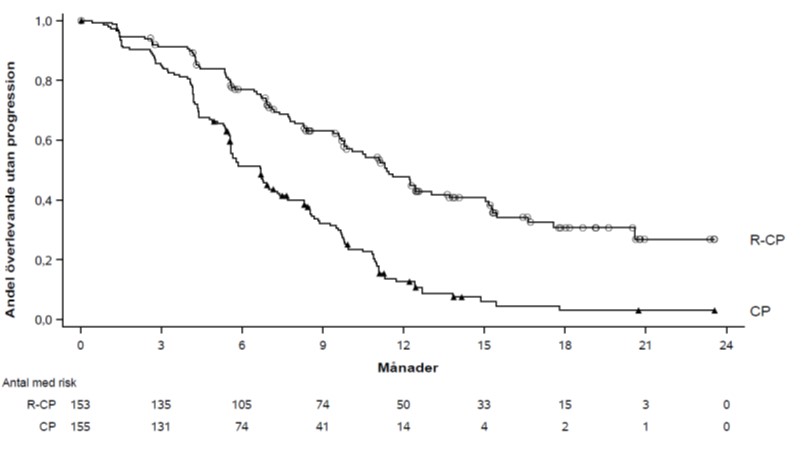
PFS-fördelen med Rybrevant intravenös formulering i kombination med karboplatin och pemetrexed jämfört med karboplatin och pemetrexed var enhetlig för alla fördefinierade undergrupper med hjärnmetastaser vid studiestart (ja eller nej), ålder (< 65 eller ≥ 65), kön (man eller kvinna), ras (asiatisk eller icke-asiatisk), vikt (< 80 kg eller ≥ 80 kg), ECOG-funktionsstatus (0 eller 1) och rökhistorik (ja eller nej).
Figur 6: Kaplan-Meier-kurva för OS hos tidigare obehandlade NSCLC-patienter
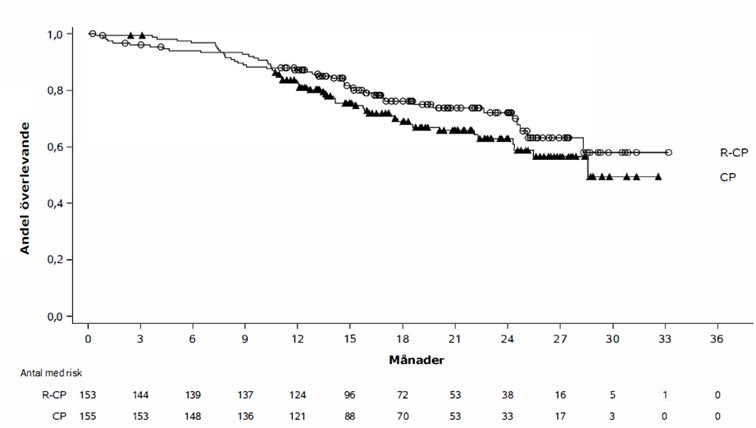
Tidigare behandlad icke-småcellig lungcancer (NSCLC) med insertionsmutationer i exon 20 (CHRYSALIS)
CHRYSALIS är en öppen multicenterstudie med flera kohorter utförd för att utvärdera säkerheten och effekten av Rybrevant intravenös formulering hos patienter med lokalt avancerad eller metastatisk icke-småcellig lungcancer (NSCLC). Effekten utvärderades hos 114 patienter med lokalt avancerad eller metastatisk NSCLC med insertionsmutationer i EGFR-genens exon 20, som progredierat under eller efter platinumbaserad kemoterapi. Mediantiden för uppföljning var 12,5 månader. Prover på tumörvävnad (93 %) och/eller plasma (10 %) för alla patienter testades lokalt för att fastställa status för insertionsmutation i EGFRgenens exon 20 med hjälp av Next Generation Sequencing (NGS) hos 46 % av patienterna och/eller Polymerase Chain Reaction (PCR) hos 41 % av patienterna; för 4 % av patienterna var testmetoderna inte specificerade. Patienter med obehandlade hjärnmetastaser eller en anamnes på ILD som krävt behandling med långverkande steroider eller andra immunhämmande medel under de senaste 2 åren rekryterades inte till studien. Rybrevant intravenös formulering administrerades intravenöst med 1 050 mg till patienter som vägde < 80 kg eller 1 400 mg till patienter som vägde ≥ 80 kg, en gång i veckan i 4 veckor och därefter varannan vecka från vecka 5 till förlust av klinisk nytta eller uppkomst av oacceptabel toxicitet. Det primära effektmåttet var baserat på prövarens bedömning av total svarsfrekvens (ORR), definierad som bekräftat komplett svar (CR) eller partiellt svar (PR) baserat på RECIST v1.1. Dessutom bedömdes det primära effektmåttet genom en blindad oberoende central granskning (BICR). Sekundära effektmått inkluderade svarets varaktighet (DOR).
Medianåldern var 62 år (intervall: 36–84 år); 41 % av patienterna var ≥ 65 år; 61 % var kvinnor; 52 % var asiater och 37 % var vita. Medianantalet av tidigare behandlingar var 2 (intervall: 1 till 7 behandlingar). Vid baseline hade 29 % funktionsstatus 0 enligt Eastern Cooperative Oncology Group (ECOG) och 70 % hade ECOG-funktionsstatus 1; 57 % hade aldrig rökt; 100 % hade cancer i stadium IV; och 25 % hade tidigare behandlats för hjärnmetastaser. Insertioner i exon 20 observerades vid 8 olika platser; de vanligaste var A767 (22 %), S768 (16 %), D770 (12 %) och N771 (11 %).
Effektresultaten är sammanfattade i tabell 13.
| Tabell 13: Effektresultat i CHRYSALIS | |
| Prövarens bedömning (N = 114) | |
| Total svarsfrekvensa, b(95 % KI) | 37 % (28 %, 46 %) |
| Komplett svar | 0 % |
| Partiellt svar | 37 % |
| Svarets varaktighet | |
| Medianc (95 % KI), månader | 12,5 (6,5,; 16,1) |
| Patienter med DOR ≥ 6 månader | 64 % |
| KI = Konfidensintervall a Bekräftat svar b ORR- och DOR-resultat enligt prövarens bedömning var enhetliga med de som rapporterades efter den blindade oberoende granskningen (BICR). ORR var enligt BICR-granskningen 43 % (34 %, 53 %) med en komplett svarsfrekvens på 3 % och en partiell svarsfrekvens på 40 %, medianvärdet på DOR var enligt BICR-granskningen 10,8 månader (95 % KI: 6,9; 15,0), och patienter med DOR ≥ 6 månader var enligt BICR-granskningen 55 %. c Baserat på uppskattning enligt Kaplan-Meier. | |
Antitumöraktivitet observerades i samtliga studerade subtyper av mutationer.
Immunogenicitet
Antikroppar mot läkemedel upptäcktes i sällsynta fall efter behandling Rybrevant subkutan formulering. Ingen påverkan av antikroppar mot läkemedel observerades på farmakokinetik, effekt eller säkerhet. Bland de 741 deltagare som fick Rybrevant subkutan formulering som monoterapi eller som en del av kombinationsbehandling, var 66 deltagare (9 %) positiva för behandlingsframkallande antikroppar mot rHuPH20. Den immunogenicitet mot rHuPH20 som observerades hos dessa deltagare påverkade inte farmakokinetiken för amivantamab.
Äldre
Inga övergripande skillnader i effekt observerades mellan patienter ≥ 65 år och patienter < 65 år.
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för Rybrevant för alla grupper av den pediatriska populationen för icke-småcellig lungcancer (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption
Efter subkutan administrering är det geometriska medelvärdet (%CV) för biotillgängligheten av amivantamab 66,6 % (14,9 %) med en mediantid för att nå maximal koncentration på 3 dagar, baserat på de individuella PK-parameteruppskattningarna för amivantamab för deltagare som fick subkutan administrering i PK-analysen av populationen.
För den subkutana doseringsregimen varannan vecka var det geometriska medelvärdet (%CV) för maximal dalvärdekoncentration av amivantamab efter den 4:e veckodosen 335 µg/ml (32,7 %). Medelvärdet för AUC1 vecka ökade 3,5-faldigt från den första dosen till cykel 2 dag 1. Maximal dalkoncentration av amivantamab efter subkutan administrering som monoterapi och i kombination med lazertinib observeras vanligen i slutet av veckodoseringen (cykel 2 dag 1). Amivantamabs steady state-koncentration uppnås ungefär vecka 13. Det geometriska medelvärdet (%CV) för steady state-dalkoncentrationen av amivantamab vid cykel 4 dag 1 var 206 µg/ml (39,1 %).
Tabell 14 visar observerade geometriska medelvärden (%CV) för maximal dalkoncentration (cykel 2 dag 1 Cdal) och cykel 2 area under koncentrationstidskurvan (AUCdag 1–15) efter de rekommenderade doserna av amivantamab administrerat subkutant och intravenöst hos patienter med NSCLC. Dessa PK-effektmått låg till grund för påvisandet av non-inferiority som stöder övergången från intravenös till subkutan behandling.
| Tabell 14: Sammanfattning av farmakokinetiska parametrar i serum för amivantamab hos patienter med NSCLC (PALOMA-3-studien) | ||
| Parameter | Rybrevant subkutan formulering 1 600 mg (2 240 mg för kroppsvikt ≥ 80 kg) | Rybrevant intravenös formulering 1 050 mg (1 400 mg för kroppsvikt ≥ 80 kg) |
| Geometriskt medelvärde (%CV) | ||
| Cykel 2 dag 1 Cdal (µg/ml) | 335 (32,7 %) | 293 (31,7 %) |
| Cykel 2 AUC(dag1-15) (µg/ml) | 135 861 (30,7 %) | 131 704 (24,0 %) |
För den subkutana doseringsregimen var 3:e vecka var det geometriska medelvärdet (%CV) för maximal dalvärdekoncentration av amivantamab efter den 3:e veckodosen 438 µg/ml (26,6 %). Det geometriska medelvärdet (%CV) för steady state-dalkoncentrationen av amivantamab var 208 µg/ml (35,6 %).
För den subkutana doseringsregimen var 4:e vecka var det geometriska medelvärdet (%CV) för maximal dalvärdekoncentration av amivantamab efter den 4:e veckodosen 350 µg/ml (30,5 %). Det geometriska medelvärdet (%CV) för steady state-dalkoncentrationen av amivantamab var 131 µg/ml (55,9 %).
Distribution
Baserat på individuella uppskattningar av PK-parametrar för amivantamab för deltagare som fick subkutan administrering i PK-populationsanalysen, är det geometriska medelvärdet (%CV) för den totala distributionsvolymen för amivantamab som administreras subkutant 5,69 l (23,8 %).
Eliminering
Baserat på individuella uppskattningar av PK-parametrar för amivantamab för deltagare som fick subkutan administrering i PK-populationsanalysen, är det uppskattade geometriska medelvärdet (%CV) för linjär CL och tillhörande terminal halveringstid 0,224 l/dag (26,0 %) respektive 18,8 dagar (34,3 %).
Särskilda populationer
Äldre
Inga kliniskt signifikanta skillnader i farmakokinetiken för amivantamab observerades baserat på ålder (21–88 år).
Nedsatt njurfunktion
Ingen kliniskt signifikant effekt på amivantamabs farmakokinetik observerades hos patienter med lätt (60 ≤ kreatininclearance [CrCl] < 90 ml/min), måttligt (29 ≤ CrCl < 60 ml/min) eller kraftigt (15 ≤ CrCl < 29 ml/min) nedsatt njurfunktion. Data för patienter med kraftigt nedsatt njurfunktion är begränsade (n = 1), men det finns inga belägg för att dosjustering skulle krävas för dessa patienter. Effekten av terminal njursjukdom (CrCl < 15 ml/min) på amivantamabs farmakokinetik är okänd.
Nedsatt leverfunktion
Det är osannolikt att förändringar i leverfunktionen har någon effekt på elimineringen av amivantamab eftersom IgG1-baserade molekyler som amivantamab inte metaboliseras genom levern.
Ingen kliniskt signifikant effekt på amivantamabs farmakokinetik observerades baserat på lätt [(totalt bilirubin ≤ ULN och AST > ULN) eller (ULN < totalt bilirubin ≤ 1,5 x ULN)] eller måttligt (1,5 x ULN < totalt bilirubin ≤ 3 x ULN och något AST) nedsatt leverfunktion. Data för patienter med måttligt nedsatt leverfunktion är begränsade (n = 1), men det finns inget som tyder på att dosjustering krävs för dessa patienter. Effekten av kraftigt (totalt bilirubin > 3 gånger ULN) nedsatt leverfunktion på amivantamabs farmakokinetik är okänd.
Pediatrisk population
Farmakokinetiken för Rybrevant hos barn har inte undersökts.
Prekliniska säkerhetsuppgifter
Gängse studier avseende allmäntoxicitet visade inte några särskilda risker för människa.
Karcinogenicitet och mutagenicitet
Inga djurstudier har utförts för att fastställa den karcinogenicitet hos amivantamab. Rutinmässiga genotoxicitets- och karcinogenicitetsstudier är i allmänhet inte tillämpliga på biologiska läkemedel, eftersom stora proteiner inte kan diffundera in i celler och inte kan interagera med DNA eller kromosomalt material.
Reproduktionstoxikologi
Inga djurstudier har utförts för att utvärdera effekterna på reproduktion och fosterutveckling, men baserat på dess verkningsmekanism kan amivantamab orsaka fosterskador eller utvecklingsanomalier. Som rapporteras i litteraturen kan reduktion, eliminering eller störning av embryofetal eller maternell EGFR-signalering förhindra implantation, orsaka embryofetal förlust under olika stadier av graviditeten (genom effekter på placentans utveckling), orsaka utvecklingsanomalier i flera organ eller tidig död hos överlevande foster. På liknande sätt var utslagning av MET, eller dess ligand – hepatocyttillväxtfaktor (HGF), embryonalt dödlig på grund av allvarliga defekter i placentans utveckling, och foster uppvisade defekter i muskelutvecklingen i flera organ. Humant IgG1 är känt för att passera placentan, och därför har amivantamab potential att överföras från modern till fostret under dess utveckling.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Rekombinant humant hyaluronidas (rHuPH20)
Dinatriumedetat (dihydrat)
Ättiksyra, koncentrerad
L-Metionin
Polysorbat 80 (E433)
Natriumacetattrihydrat
Sackaros
Vatten för injektionsvätskor
Inkompatibiliteter
Detta läkemedel får inte blandas med andra läkemedel förutom de som nämns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Hållbarhet
Oöppnad injektionsflaska
2år
Efter spädning
Kemisk och fysisk stabilitet vid användning har visats upp till 24 timmar vid förvaring i 2 °C till 8 °C följt av upp till 24 timmar i 15 °C till 30 °C. Ur ett mikrobiologiskt perspektiv ska produkten användas omedelbart, såvida inte spädningsmetoden utesluter risk för mikrobiell kontaminering. Om produkten inte används omedelbart, är förvaringstider och förvaringsbetingelser användarens ansvar.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C - 8 °C).
Får ej frysas.
Förvaras i originalförpackningen. Ljuskänsligt.
Förvaringsanvisningar för läkemedlet efter beredning finns i avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
RYBREVANT injektioneste, liuos
1600 mg (L:ei) 1 kpl (10 ml (160 mg/ml)) (4532,57 €)
2240 mg (L:ei) 1 kpl (14 ml (160 mg/ml)) (6273,35 €)
2400 mg (L:ei) 1 kpl (15 ml (160 mg/ml)) (6708,55 €)
3520 mg (L:ei) 1 kpl (22 ml (160 mg/ml)) (9754,93 €)
PF-selosteen tieto
10 ml lösning i en injektionsflaska av typ 1-glas med elastomerisk förslutning och en aluminiumförsegling med snäpplock innehållande 1 600 mg amivantamab. Förpackningsstorlek med 1 injektionsflaska.
14 ml lösning i en injektionsflaska av typ 1-glas med elastomerisk förslutning och en aluminiumförsegling med snäpplock innehållande 2 240 mg amivantamab. Förpackningsstorlek med 1 injektionsflaska.
15 ml lösning i en injektionsflaska av typ 1-glas med elastomerisk förslutning och en aluminiumförsegling med snäpplock innehållande 2 400 mg amivantamab. Förpackningsstorlek med 1 injektionsflaska.
22 ml lösning i en injektionsflaska av typ 1-glas med elastomerisk förslutning och en aluminiumförsegling med snäpplock innehållande 3 520 mg amivantamab. Förpackningsstorlek med 1 injektionsflaska.
Läkemedlets utseende:
Lösningen är färglös till svagt gul.
Särskilda anvisningar för destruktion och övrig hantering
Rybrevant subkutan formulering är endast avsedd för engångsbruk och är färdig att användas.
Förbered injektionslösningen med aseptisk teknik enligt följande:
Beredning
- Bestäm dosen som krävs och lämplig(a) injektionsflaska(or) av Rybrevant subkutan formulering som behövs baserat på patientens vikt vid behandlingsstart (se avsnitt Dosering och administreringssätt).
- För dosering var fjärde vecka får patienter < 80 kg 1 600 mg varje vecka från vecka 1 till 4 och därefter 3 520 mg var fjärde vecka med start vecka 5 och framåt. Patienter ≥ 80 kg får 2 240 mg varje vecka från vecka 1 till 4 och därefter 4 640 mg var fjärde vecka med start vecka 5 och framåt.
- För dosering varannan vecka får patienter < 80 kg 1 600 mg och patienter ≥ 80 kg får 2 240 mg varje vecka från vecka 1 till 4 och därefter varannan vecka med start vecka 5 och framåt.
- För dosering var tredje vecka får patienter < 80 kg 1 600 mg vecka 1 dag 1, sedan 2 400 mg varje vecka från vecka 2 till 4 och därefter var tredje vecka med start vecka 7 och framåt. Patienter ≥ 80 kg får 2 240 mg vecka 1 dag 1, sedan 3 360 mg varje vecka från vecka 2 till 4 och därefter var tredje vecka med start vecka 7 och framåt.
-
Ta ut lämplig(a) injektionsflaska(or) med Rybrevant subkutan formulering från kylförvaring (2 °C - 8 °C).
- Kontrollera att Rybrevant-lösningen är färglös till svagt gul. Använd inte om ogenomskinliga partiklar, missfärgningar eller andra främmande partiklar förekommer.
- Se till att Rybrevant subkutan formulering uppnår rumstemperatur (15 °C - 30 °C) i minst 15 minuter. Rybrevant subkutan formulering får inte värmas på något annat sätt. Skaka inte.
- Dra upp den injektionsvolym som krävs av Rybrevant subkutan formulering från injektionsflaskan till en spruta av lämplig storlek med hjälp av en överföringsnål. Mindre sprutor kräver mindre kraft under beredning och administrering.
-
Varje injektionsvolym får inte överstiga 15 ml. Dela upp doser som kräver mer än 15 ml i ungefär lika stora volymer i flera sprutor.
- Rybrevant subkutan formulering är kompatibel med injektionsnålar av rostfritt stål och sprutor av polypropen och polykarbonat samt subkutana infusionsset av polyeten, polyuretan och polyvinylklorid. En natriumkloridlösning 9 mg/ml (0,9 %) kan också användas för att spola ett infusionsset vid behov.
- Byt ut överföringsnålen mot lämpliga tillbehör för transport eller administrering. Användning av en 21G till 23G nål eller infusionsset rekommenderas för att säkerställa enkel administrering.
Förvaring av förberedd spruta
Den förberedda sprutan ska administreras omedelbart. Om omedelbar administrering inte är möjlig, förvara den förberedda sprutan i kylskåp vid 2 °C - 8 °C i upp till 24 timmar följt av i rumstemperatur vid 15 °C - 30 °C i upp till 24 timmar. Den förberedda sprutan ska kasseras om den förvaras i mer än 24 timmar i kylskåp eller mer än 24 timmar i rumstemperatur. Om lösningen förvaras i kylskåp ska den uppnå rumstemperatur före administrering.
Destruktion
Detta läkemedel är endast för engångsbruk. Ej använt läkemedel ska kasseras enligt gällande anvisningar.
Ersättning
RYBREVANT injektioneste, liuos
1600 mg 1 kpl
2240 mg 1 kpl
2400 mg 1 kpl
3520 mg 1 kpl
- Ei korvausta.
Atc-kod
L01FX18
Datum för översyn av produktresumén
20.02.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com