
IQIRVO tabletti, kalvopäällysteinen 80 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 80 mg elafibranoria.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
Iqirvo on tarkoitettu primaarisen biliaarisen kolangiitin hoitoon yhdessä ursodeoksikoolihapon kanssa aikuisille, jotka eivät ole saaneet riittävää vastetta ursodeoksikoolihapolle, tai monoterapiana potilaille, joille ursodeoksikoolihappo ei sovi.
Annostus ja antotapa
Annostus
Suositeltu annos on 80 mg kerran päivässä.
Väliin jääneet annokset
Jos elafibranoriannos jää väliin, potilas ei saa ottaa väliin jäänyttä annosta, vaan hänen pitää ottaa seuraava annos seuraavana aikataulun mukaisena ottoajankohtana. Potilas ei saa ottaa kaksinkertaista annosta korvatakseen unohtuneen annoksen.
Iäkkäät potilaat
Annostusta ei tarvitse muuttaa yli 65-vuotiaille potilaille (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää elafibranoria pediatrisille potilaille (alle 18 vuoden ikäisille) primaarisen biliaarisen kolangiitin hoitoon.
Munuaisten vajaatoiminta
Annostusta ei tarvitse muuttaa potilaille, joilla on munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annostusta ei tarvitse muuttaa potilaille, joilla on lievä (Child–Pugh A) tai keskivaikea (Child–Pugh B) maksan vajaatoiminta.
Elafibranorin tehoa ja turvallisuutta sellaisten primaarista biliaarista kolangiittia sairastavien potilaiden hoidossa, joilla on vaikea maksan vajaatoiminta, ei ole varmistettu. Käyttöä potilaille, joilla on vaikea maksan vajaatoiminta (Child–Pugh C), ei suositella (ks. kohta Farmakokinetiikka).
Antotapa
Suun kautta.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Tiedossa oleva tai epäilty raskaus sekä käyttö naisille, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Maksaan liittyvät tapahtumat
Elafibranoria saaneilla potilailla on ilmoitettu suurentuneita arvoja maksan biokemiallisissa kokeissa, transaminaasi- ja bilirubiinipitoisuudet mukaan lukien.
Maksan toiminta on arvioitava kliinisesti ja laboratoriotutkimuksin ennen elafibranorihoidon aloittamista sekä sen jälkeen tavanomaisen hoitokäytännön mukaisesti.
Jos maksan biokemiallisissa tutkimuksissa todetaan kohonneita arvoja ja/tai todetaan maksan toimintahäiriöitä, syy on suositeltavaa selvittää viipymättä ja elafibranorihoidon keskeyttämistä on harkittava.
Kohonnut veren kreatiinikinaasi ja lihasvauriot
Elafibranoria saaneilla tutkittavilla on ilmoitettu veren kreatiinikinaasipitoisuuden kohoamista (ks. kohta Haittavaikutukset). Kreatiinikinaasipitoisuus on tarkistettava ennen elafibranorihoidon aloittamista sekä sen jälkeen tavanomaisen hoitokäytännön mukaisesti. Elafibranorihoitoa aloitettaessa voidaan harkita säännöllisiä kreatiinikinaasimittauksia, etenkin HMG-CoA-reduktaasin estäjiä käyttäville potilaille. Jos todetaan kohonneita kreatiinikinaasiarvoja tai selittämättömiä lihasvaurion oireita ja löydöksiä, syy on suositeltavaa selvittää viipymättä ja elafibranorihoidon keskeyttämistä on harkittava (ks. kohta Haittavaikutukset).
Alkio- ja sikiötoksisuus
Eläimillä tehdyistä tutkimuksista saatujen tietojen perusteella elafibranorin epäillään aiheuttavan synnynnäisiä epämuodostumia ja vähentävän sikiöiden eloonjääntiä, kun sitä annetaan raskaana oleville naisille (ks. kohta Raskaus ja imetys). Tästä syystä elafibranori on vasta-aiheista naisille, joiden tiedetään tai epäillään olevan raskaana, sekä naisille, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä (ks. kohta Vasta-aiheet). Tästä on kerrottava naisille, jotka voivat tulla raskaaksi.
Apuaineet
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
In vitro- ja in vivo ‑tutkimusten perusteella elafibranorilla ei oletettavasti ole kliinisesti oleellisia yhteisvaikutuksia muiden samanaikaisesti annettujen lääkevalmisteiden kanssa (ks. kohta Farmakokinetiikka).
Raskaus ja imetys
Hedelmällisyys
Elafibranorin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoja. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria eikä epäsuoria vaikutuksia hedelmällisyyteen tai lisääntymiskykyyn (ks. kohta Prekliiniset tiedot turvallisuudesta).
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 3 viikkoa viimeisen elafibranoriannoksen jälkeen. Potilaiden, jotka voivat tulla raskaaksi, täytyy tehdä raskaustesti mahdollisen raskauden selvittämiseksi ennen elafibranorihoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Elafibranorin käytöstä raskaana oleville naisille on vain vähän tietoja.
Eläimillä tehdyissä tutkimuksissa, joissa elafibranoria annettiin tiineille eläimille, on havaittu lisääntymistoksisuutta (sikiöiden menetyksiä, epämuodostumia, poikasten syntymistä kuolleena ja/tai perinataalikuolemia) kliinisesti oleellisella altistuksella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Prekliiniset tiedot turvallisuudesta).
Elafibranorin käyttö raskauden aikana on vasta-aiheista (ks. kohta Vasta-aiheet). Jos potilas tulee raskaaksi, elafibranorihoito on lopetettava.
Imetys
Ei tiedetä, erittyvätkö elafibranori tai sen metaboliitit ihmisillä äidinmaitoon. Ei ole tietoa elafibranorin tai sen metaboliittien erittymisestä maitoon koe-eläimillä. Jälkeläisiin kohdistuvia haittavaikutuksia on kuitenkin todettu, kun elafibranoria on annettu naarasrotille tiineyden aikana (ks. kohta Prekliiniset tiedot turvallisuudesta) ja imetyksen aikana kliinisesti oleellisella altistuksella.
Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois.
Elafibranoria ei pidä käyttää imetyksen aikana, eikä lasta saa imettää vähintään 3 viikkoon viimeisen elafibranoriannoksen jälkeen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Elafibranorilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät elafibranorihoitoon (n = 108) liittyvät haittavaikutukset, joita ilmeni yli 10 %:lla tutkittavista ja joiden ilmaantuvuus oli suurempi kuin lumeryhmässä (n = 53; ero > 1 %), olivat vatsakipu (11,1 % vs. 5,7 %), ripuli (11,1 % vs. 9,4 %), pahoinvointi (11,1 % vs. 5,7 %) ja oksentelu (11,1 % vs. 1,9 %). Nämä haittavaikutukset eivät olleet vakavia, ja ne olivat lieviä tai keskivaikeita, ilmenivät hoidon varhaisvaiheessa ja yleensä hävisivät muutamassa päivässä tai muutamassa viikossa ilman annoksen muuttamista tai tukitoimia.
Yleisin hoidon keskeyttämiseen johtanut haittavaikutus oli suurentunut veren kreatiinikinaasipitoisuus (3,7 %).
Haittavaikutustaulukko
Haittavaikutukset on lueteltu elinjärjestelmäluokittain ja esiintyvyyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen |
| Hermosto | Päänsärky | ||
| Ruoansulatuselimistö | Vatsakipua Ripuli Pahoinvointi Oksentelu | Ummetus | |
| Maksa ja sappi | Sappikivitauti | ||
| Iho ja ihonalainen kudos | Kutiseva ihottuma | ||
| Luusto, lihakset ja sidekudos | Lihaskipu | ||
| Tutkimukset | Suurentunut veren kreatiinikinaasipitoisuus | Suurentunut veren kreatiniinipitoisuus |
a Sisältää ylävatsakivun ja alavatsakivun
Valikoitujen haittavaikutusten kuvaus
Päänsärky
Vaiheen 3 avaintutkimuksessa (ELATIVE) päänsärkyä ilmeni elafibranoriryhmässä 9 tutkittavalla (8,3 %) ja lumeryhmässä 6 tutkittavalla (11,3 %). Kuitenkin tutkimushoidon 10 ensimmäisen päivän aikana päänsärkyä ilmeni elafibranoriryhmässä useammalla tutkittavalla kuin lumeryhmässä (3,7 %:lla vs. 0 %:lla).
Suurentunut veren kreatiinikinaasipitoisuus
Vaiheen 3 avaintutkimuksessa (ELATIVE) veren kreatiinikinaasipitoisuuden kliinisesti merkittävää suurenemista, mikä johti hoidon lopettamiseen, todettiin elafibranoriryhmässä 4 tutkittavalla (3,7 %), mutta lumeryhmässä ei yhdelläkään tutkittavalla. Kahdella näistä 4 tutkittavasta veren kreatiinikinaasipitoisuus oli > 5 kertaa viitealueen ylärajaa (ULN) suurempi. Yksikään tapahtumista ei ollut vakava, ja ne kaikki olivat vaikeusasteeltaan lieviä tai keskivaikeita. Kahdella tutkittavista tapahtumaan liittyi oireena lihaskipua. Lähtötilanteessa keskimääräiset kreatiinikinaasipitoisuudet olivat molemmissa hoitoryhmissä samankaltaiset ja normaalialueella, ja viikolla 52 arvot olivat edelleen normaalialueella molemmissa ryhmissä. Keskimääräinen muutos lähtötilanteesta viikolle 52 oli elafibranoriryhmässä 6,2 (keskihajonta 38,1) U/l ja lumeryhmässä 12,3 (keskihajonta 67,0) U/l.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostuksen tapahtuessa potilasta on seurattava tarkkaan ja aloitettava asianmukainen oireenmukainen hoito sekä tukitoimet.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Sapeneritystä lisäävät valmisteet ja maksan suoja-aineet, muut sappilääkkeet
ATC-koodi: A05AX06
Vaikutusmekanismi
Elafibranori ja sen pääasiallinen aktiivinen metaboliitti GFT1007 ovat peroksisomiproliferaattorin aktivoimien alfa- ja deltareseptorien (PPAR-α/δ) kaksoisagonisteja.
PPAR-α/δ-reseptorien arvellaan olevan keskeisiä sappihappojen homeostaasin, tulehduksen ja fibroosin säätelijöitä. PPAR-α- ja PPAR-δ-reseptorien aktivoituminen moduloi sappihappojen synteesiä, detoksikaatiota ja kuljettajaproteiineja ja vähentää siten sapen toksisuutta ja kolestaasia.
PPAR-α- ja PPAR-δ-reseptorien aktivoitumisella on myös anti-inflammatorinen vaikutus eri reitteihin kohdistuvan vaikutuksen kautta.
Farmakodynaamiset vaikutukset
Vaiheen 3 avaintutkimuksessa (ELATIVE) elafibranorihoito vähensi huomattavasti alkalisen fosfataasin pitoisuutta lähtötilanteesta jo 4 viikossa, ja tulos säilyi viikolle 52 saakka. Sappihappojen synteesin biomarkkereiden, mukaan lukien sappihappojen esiaste 7-alfa-hydroksi-4-kolesteeni-3-oni (C4) ja sappihappojen synteesiä säätelevä fibroblastikasvutekijä 19 (FGF-19), vähenemisen todettiin olevan suurempaa elafibranoriryhmässä, mikä oli yhdenmukaista todetun biokemiallisen vasteen kanssa.
Sydämen elektrofysiologia
Perusteellinen QT-analyysi osoitti, että elafibranorilla ei ollut QT-aikaa / korjattua QT-aikaa (QTc) pidentävää vaikutusta, kun sitä annettiin toistuvasti enintään 300 mg:n annoksina 14 päivän ajan.
Kliinisissä tutkimuksissa elafibranorihoitoa saaneilla ei todettu kliinisesti merkitseviä muutoksia vitaalimerkeissä tai sydänsähkökäyrässä (EKG) (QTc-väli mukaan lukien).
Kliininen teho
Elafibranorin tehoa ja turvallisuutta arvioitiin vaiheen 3 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa GFT505B-319-1 (ELATIVE), johon osallistui 161 primaarista biliaarista kolangiittia sairastavaa aikuista, jotka eivät olleet saaneet riittävää vastetta ursodeoksikoolihapolle tai joille ursodeoksikoolihappo ei sopinut. Tutkittavat stratifioitiin kahden tekijän suhteen (AFOS > 3 x ULN tai kokonaisbilirubiinipitoisuus > ULN ja PBC Worst Itch Numeric Rating Scale (WI‑NRS) ‑kutinamittarin pistemäärä ≥ 4) ja satunnaistettiin suhteessa 2:1 saamaan joko elafibranoria 80 mg tai lumevalmistetta kerran päivässä vähintään 52 viikon ajan. Ursodeoksikoolihappoa ennen tutkimusta käyttäneet tutkittavat jatkoivat käyttöä samalla annostuksella. Tutkittavat otettiin mukaan tutkimukseen, jos AFOS oli ≥ 1,67 x ULN ja kokonaisbilirubiinipitoisuus oli ≤ 2 x ULN. Tutkittavia, joilla oli kompensoitumaton kirroosi tai joiden maksasairaus johtui muusta syystä, ei otettu mukaan tutkimukseen.
Keski-ikä oli 57,1 vuotta ja keskimääräinen paino 70,8 kg. Valtaosa tutkimuspopulaatiosta oli naisia (96 %) ja valkoihoisia (91 %). Keskimääräinen AFOS-arvo lähtötilanteessa oli 321,9 U/l, 39 %:lla tutkittavista AFOS-arvo oli lähtötilanteessa > 3 x ULN ja 35 %:lla tutkittavista oli lähtötilanteessa pitkälle edennyt tauti, jonka määritelmänä oli maksan jäykkyys > 10 kPa ja/tai histologisessa tutkimuksessa bridging-tyyppinen fibroosi tai kirroosi.
Altistuksen keston mediaani oli elafibranoriryhmässä 63,07 viikkoa ja lumeryhmässä 61,00 viikkoa.
Kokonaisbilirubiinipitoisuuksien keskiarvo lähtötilanteessa oli 9,6 µmol/l, ja 96 %:lla tutkittavista kokonaisbilirubiinipitoisuus oli lähtötilanteessa viitealueen ylärajan alapuolella tai ylärajalla. Keskimääräinen maksan jäykkyys lähtötilanteessa transientilla elastografialla mitattuna oli 10,1 kPa. Lähtötilanteessa keskimääräinen PBC WI‑NRS ‑pistemäärä oli 3,3, ja 41 %:lla oli lähtötilanteessa keskivaikeaa tai vaikeaa kutinaa (PBC WI‑NRS ‑pistemäärä ≥ 4); tutkittavilla, joilla kutina oli keskivaikeaa tai vaikeaa, lähtötilanteen keskimääräinen PBC Worst Itch NRS ‑pistemäärä oli elafibranoria 80 mg saaneessa ryhmässä 6,2 ja lumeryhmässä 6,3. Valtaosa (95 %) tutkittavista sai hoitoa yhdistelmähoitona ursodeoksikoolihapon kanssa; 5 % tutkittavista, joille ursodeoksikoolihappo ei sopinut, sai monoterapiaa.
Ensisijainen päätetapahtuma oli kolestaasivaste viikolla 52; kolestaasivasteen määritelmänä oli yhdistetty päätetapahtuma: AFOS < 1,67 x ULN ja kokonaisbilirubiinipitoisuus ≤ ULN ja AFOS-arvon pienenemä ≥ 15 %. Keskeiset toissijaiset päätetapahtumat olivat AFOS-arvon normalisoituminen viikolla 52 ja kutinan muutos lähtötilanteesta viikolle 52 sekä viikolle 24 PBC WI-NRS ‑pistemäärällä mitattuna potilailla, joilla oli lähtötilanteessa keskivaikeaa tai vaikeaa kutinaa.
Ensisijainen yhdistetty päätetapahtuma (kolestaasivaste) sekä keskeinen toissijainen päätetapahtuma (AFOS-arvon normalisoituminen) esitetään taulukossa 1.
Taulukko 1.Ensisijaisen tehoa koskevan yhdistetyn päätetapahtuman (kolestaasivaste) ja keskeisen toissijaisen tehoa koskevan päätetapahtuman (AFOS-arvon normalisoituminen) viikolla 52 saavuttaneiden primaarista biliaarista kolangiittia sairastavien aikuisten tutkittavien prosentuaalinen osuus
| Analyysin populaatio | Elafibranori 80 mg (N = 108) | Lumehoito (N = 53) | Hoitojen ero (95 %:n luottamusväli)[3] | Kerroinsuhde (95 %:n luottamusväli)[4] | P-arvo[4] |
| Ensisijainen yhdistetty päätetapahtuma: kolestaasivaste[1] | |||||
| ITT | 51 % | 4 % | 47 % (32, 57) | 37,6 (7,6, 302,2) | < 0,0001 |
Ensimmäinen keskeinen toissijainen päätetapahtuma: AFOS-arvon normalisoituminen[2] | |||||
| ITT | 15 % | 0 | 15 % (6, 23) | Ääretön (2,8, ääretön) | 0,0019 |
ITT: Intention-to-treat, hoitoaikeen mukainen
[1] Kolestaasivasteen määritelmänä on AFOS < 1,67 x ULN ja kokonaisbilirubiinipitoisuus ≤ ULN ja AFOS-arvon pienenemä lähtötilanteesta ≥ 15 % viikolla 52. Tutkittavat, jotka keskeyttivät tutkimushoidon ennenaikaisesti (keskeyttävä tapahtuma 1) tai käyttivät primaariseen biliaariseen kolangiittiin salvage-hoitoa (keskeyttävä tapahtuma 2) ennen viikon 52 arviointia, luokiteltiin hoitoon reagoimattomiksi. Jos tutkittavasta ei ollut tietoja viikolta 52 eikä tutkittavalla ollut keskeyttävää tapahtumaa, huomioitiin kaksoissokkoutetulta jaksolta ajallisesti lähin arviointi, josta tutkittavasta oli tietoja.
[2] AFOS-arvon normalisoituminen viikolla 52 määriteltiin niiden potilaiden suhteellisena osuutena, joilla AFOS oli ≤ 1,0 x ULN. Keskeyttäviä tapahtumia tai puuttuvia tietoja käsiteltiin samoin kuin ensisijaisen päätetapahtuman osalta.
[3] Hoitoryhmien väliset erot vasteosuuksissa ja 95 %:n luottamusvälit on laskettu Newcomben menetelmällä stratifioituna satunnaistamisen stratifiointiryhmien mukaan kolestaasivasteen suhteen ja stratifioimatta AFOS-arvon normalisoitumisen suhteen.
[4] Hoitojen vertailussa käytetyt vasteen kerroinsuhteet ja p-arvot ovat peräisin Cochran-Mantel-Haenszelin (CMH) eksaktista testistä, joka oli ositettu satunnaistamisen stratifiointiryhmien mukaan.
AFOS-arvon merkitsevää pienenemistä lähtötilanteesta todettiin elafibranoriryhmässä lumeeseen verrattuna jo viikolla 4, ja se pysyi 52 viikkoa kestäneen hoidon ajan (kuva 1).
Kuva 1. AFOS-arvon keskimääräinen (pienimpien neliösummien keskiarvo, 95 %:n luottamusväli) muutos lähtötilanteesta ajan mittaan – ITT-analyysijoukko
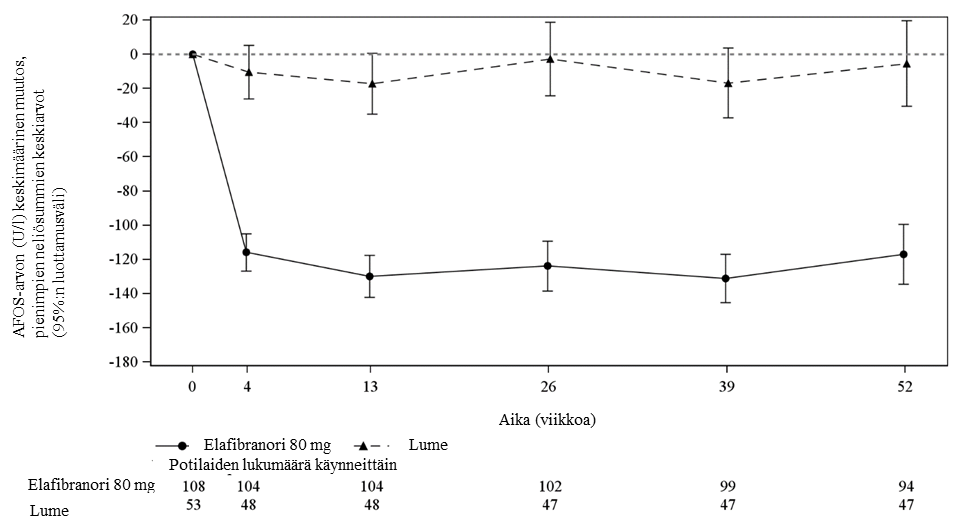
Tutkittavista, joilla lähtötilanteen AFOS-arvo oli ≤ 3 x ULN tai kokonaisbilirubiinipitoisuus oli < ULN, 71 % elafibranoriryhmässä ja 6 % lumeryhmässä saavutti ensisijaisen päätetapahtuman (kolestaasivaste). Tutkittavista, jolla AFOS-arvo oli > 3 x ULN tai kokonaisbilirubiinipitoisuus > ULN, kolestaasivasteen saaneiden osuus oli 21 % elafibranoriryhmässä ja 0 % lumeryhmässä.
Pitkälle edennyttä tautia sairastaneista 54 tutkittavasta 16/35 (46 %) tutkittavaa elafibranoriryhmässä ja 0/19 (0 %) tutkittavaa lumeryhmässä saavutti ensisijaisen päätetapahtuman eli kolestaasivasteen. Pitkälle edennyttä tautia sairastavien tutkittavien lukumäärä on pieni, joten tuloksia on tulkittava varoen.
Potilaan raportoimat tulokset
Tutkittavilla, joilla on lähtötilanteessa keskivaikeaa tai vaikeaa kutinaa, PBC WI‑NRS ‑pistemäärä pieneni lähtötilanteeseen verrattuna viikolla 52 ja viikolla 24 enemmän elafibranoriryhmään satunnaistetuilla tutkittavilla kuin lumeryhmään satunnaistetuilla tutkittavilla, mutta ero ei ollut tilastollisesti merkitsevä (taulukko 2).
Taulukko 2. Muutos kutinassa lähtötilanteesta viikolla 52 ja viikolla 24 PBC WI-NRS ‑mittarilla mitattuna tutkittavilla, joilla oli lähtötilanteessa keskivaikeaa tai vaikeaa kutinaa
Elafibranori 80 mg (N = 44) | Lumehoito (N = 22) | Hoitojen ero | P-arvo | |
| Toinen keskeinen toissijainen päätetapahtuma: muutos viikolle 52[1] | ||||
| Pienimpien neliösummien keskiarvo (95 %:n luottamusväli) | -1,9 (-2,6, -1,3) | -1,1 (-2,1, -0,2) | -0,8 (-2,0, 0,4) | 0,1970 |
| Kolmas keskeinen toissijainen päätetapahtuma: muutos viikolle 24[1] | ||||
| Pienimpien neliösummien keskiarvo (95 %:n luottamusväli) | -1,6 (-2,2, -1,0) | -1,3 (-2,2, -0,3) | -0,3 (-1,5, 0,8) | - |
[1] Analyysissa käytettiin toistettujen mittausten sekamallia, jossa kiinteitä tekijöitä olivat hoito, 4 viikon hoitojakso sekä hoidon ja 4 viikon hoitojakson yhdysvaikutus, korjattuna lähtötilanteen PBC WI‑NRS ‑pistemäärän ja stratifiointitekijän AFOS > 3 x ULN tai kokonaisbilirubiinipitoisuus > ULN suhteen. Käytettyä korrelaatiorakennetta ei ollut strukturoitu. Hoitovaikutus viikolle 52 saakka on kolmentoista 4 viikon hoitojakson NRS-pistemäärän muutoksen keskiarvo lähtötilanteesta. Hoitovaikutus viikolle 52 on kolmentoista ensimmäisen 4 viikon hoitojakson NRS-pistemäärän muutoksen keskiarvo lähtötilanteesta ja hoitovaikutus viikolle 24 on kuuden ensimmäisen 4 viikon hoitojakson NRS-pistemäärän muutoksen keskiarvo. PBC WI‑NRS ‑pistemäärien arvioinnissa tiedot katsotaan puuttuviksi sen jälkeen, kun tutkittavat keskeyttivät tutkimuksen ennenaikaisesti tai käyttivät kutinaan salvage-hoitoa.
Elafibranorihoitoon liittyi kutinan paraneminen, mitä osoitti PBC-40 Itch- ja 5‑D Itch ‑kokonaispistemäärien pieneneminen lumevalmisteeseen verrattuna viikolla 52 (taulukko 3).
Taulukko 3. Muutos kutinassa viikolla 52 verrattuna lähtötilanteeseen PBC-40 Itch- ja 5‑D Itch ‑kokonaispistemäärinä mitattuna tutkittavilla, joilla oli lähtötilanteessa keskivaikeaa tai vaikeaa kutinaa
Elafibranori 80 mg (N = 44) | Lumehoito (N = 22) | Hoitojen ero | ||
| PBC-40 Itch ‑kokonaispistemäärä: muutos viikolla 52[1] | ||||
| Pienimpien neliösummien keskiarvo (95 %:n luottamusväli) | -2,5 (-3,4, -1,6) | -0,1 (-1,6, 1,3) | -2,3 (-4,0, -0,7) | |
| 5-D Itch ‑kokonaispistemäärä: muutos viikolla 52[1] | ||||
| Pienimpien neliösummien keskiarvo (95 %:n luottamusväli) | -4,2 (-5,6, -2,9) | -1,2 (-3,3, 0,9) | -3,0 (-5,5, -0,5) | |
[1] Analyysissa käytettiin toistettujen mittausten sekamallia, jossa kiinteitä tekijöitä olivat hoito, hoitokäynnit (viikolle 52 saakka) ja hoito x hoitokäynti ‑yhdysvaikutus, korjattuna lähtötilanteen pistemäärän ja stratifiointitekijän AFOS > 3 x ULN tai kokonaisbilirubiinipitoisuus > ULN suhteen.
Lipidiparametrit
Elafibranorilla on osoitettu olevan suotuisa vaikutus lipidiparametreihin. VLDL-kolesterolin ja triglyseridien pitoisuudet pienenivät elafibranoriryhmässä keskimäärin enemmän kuin lumeryhmässä viikolla 52. Pienimpien neliösummien keskiarvon ero lumeryhmään verrattuna oli VLDL-kolesterolin osalta -0,1 mmol/l [(95 % luottamusväli: -0,2, -0,1); p < 0,001] ja triglyseridien osalta -0,3 mmol/l [(95 %:n luottamusväli: -0,4, -0,1)]; p < 0,001]. HDL-kolesterolipitoisuus pysyi elafibranorihoitoa saavilla vakaana.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Iqirvo-valmisteen käytöstä primaarisen biliaarisen kolangiitin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa.
Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Elafibranorin pitoisuus plasmassa (AUC) suurenee suhteellisesti 50 mg:sta 360 mg:aan (0,6–4,5 kertaa suositeltu annos). Vakaa tila saavutetaan 14. päivään mennessä, kun elafibranoria otetaan kerran päivässä. Kun elafibranoria annettiin toistuvasti 16 päivän ajan, elafibranorin ja sen pääasiallisen aktiivisen metaboliitin GFT1007:n farmakokinetiikan todettiin olevan ajasta riippumatonta. Elafibranorin ja sen aktiivisen metaboliitin pitoisuudet primaarista biliaarista kolangiittia sairastavilla tutkittavilla esitetään taulukossa 4.
Taulukko 4. Elafibranori- ja GFT1007-pitoisuudet vakaassa tilassa primaarista biliaarista kolangiittia sairastavilla tutkittavilla, jotka saivat elafibranoria 80 mg kerran päivässä
| Cmax,ss (ng/ml) | AUC0-24 (ng • h/ml) | Kumulaatiosuhde | |
| Elafibranori | 802 | 3 758 | 2,9 |
| GFT1007 | 2 058 | 11 985 | 1,3 |
Imeytyminen
Annettaessa elafibranoria 80 mg toistuvasti suun kautta primaarista biliaarista kolangiittia sairastaville tutkittaville elafibranorin ja GFT1007:n huippupitoisuudet (mediaani) plasmassa saavutettiin 1,25 tunnissa.
Elafibranorin anto runsasrasvaisen ja runsasenergisen aterian yhteydessä viivästytti elafibranorin Tmax‑aikaa 30 minuuttia ja GFT1007:n Tmax‑aikaa 1 tunnin verrattuna antoon paasto-olosuhteissa. Elafibranorin pitoisuus plasmassa (AUC) pieneni 15 %. GFT1007:n pitoisuuteen plasmassa perustuva AUC pysyi samana. Kun otetaan huomioon, että farmakologisesti aktiivisen GFT1007-metaboliitin pitoisuus plasmassa on suurempi kuin elafibranorin pitoisuus, ruokailulla katsottiin olevan vain vähäinen kliininen merkitys kanta-aineen ja aktiivisen metaboliitin yhteenlaskettujen pitoisuuksien perusteella.
Jakautuminen
Noin 99,7 % sekä elafibranorista että GFT1007:stä sitoutuu plasmaan (pääasiassa seerumin albumiiniin). Elafibranorin näennäinen jakautumistilavuus (Vd/F) ihmiselle paasto-olosuhteissa annetun 80 mg:n kerta-annoksen jälkeen on keskimäärin 4 731 litraa.
Biotransformaatio
Elafibranori metaboloituu 15-ketoprostaglandini 13-Δ-reduktaasin (PTGR1) välityksellä in vitro. Elafibranori ja GFT1007 eivät kumpikaan metaboloidu merkittävästi pääasiallisten sytokromi-P450:n (CYP) ja uridiini-difosfaatti-glukurosyylitransferaasin (UGT) isoformien välityksellä.
Suun kautta annettu 14C-merkitty elafibranori hydrolysoituu nopeasti aktiiviseksi metaboliitiksi GFT1007:ksi. Plasmasta on löydetty kaksi pääasiallista metaboliittia, GFT1007 (aktiivinen metaboliitti) ja glukuronidikonjugaatteja (inaktiivisia metaboliitteja).
Eliminaatio
Paasto-olosuhteissa annetun 80 mg:n kerta-annoksen annon jälkeen elafibranorin keskimääräinen eliminaation puoliintumisaika on 68,2 tuntia ja GFT1007-metaboliitin keskimääräinen eliminaation puoliintumisaika on 15,4 tuntia. Paasto-olosuhteissa annetun 80 mg:n kerta-annoksen annon jälkeen elafibranorin keskimääräinen näennäinen kokonaispuhdistuma (CL/F) on 50,0 l/h.
Erittyminen
Kun terveille vapaaehtoisille annettiin suun kautta 120 mg:n kerta-annos 14C-merkittyä elafibranoria, noin 77,1 % annoksesta erittyi ulosteisiin, pääasiassa elafibranorina (56,7 % annetusta annoksesta) ja sen aktiivisena metaboliittina GFT1007:nä (6,08 % annetusta annoksesta). Noin 19,3 % erittyi virtsaan, pääasiassa glukuronidikonjugaatteina.
Erityiset potilasryhmät
Ei ole näyttöä siitä, että ikä (18–80 vuotta), sukupuoli, etnisyys, painoindeksi (BMI) ja munuaisstatus vaikuttaisivat kliinisesti merkitsevästi elafibranorin ja GFT1007:n farmakokinetiikkaan.
Maksan vajaatoiminta
Kokonaisaltistumisessa kanta-aineelle ja aktiiviselle metaboliitille ei ollut merkittäviä eroja niiden tutkittavien, joiden maksan toiminta oli normaali, ja maksan vajaatoimintaa (Child–Pugh A, B ja C) sairastavien tutkittavien välillä. Annostusta ei tarvitse muuttaa potilaille, joilla on lievä (Child–Pugh A) tai keskivaikea (Child–Pugh B) maksan vajaatoiminta. Elafibranorin ja GFT1007:n sitoutumattomat osuudet suurenevat noin kolminkertaisiksi tutkittavilla, joilla on vaikea maksan vajaatoiminta (Child–Pugh C). Elafibranoria ei suositella potilaille, joilla on vaikea maksan vajaatoiminta (Child–Pugh C).
Lääkeyhteisvaikutukset
In vitro ‑tutkimusten perusteella CYP- ja UGT-entsyymit eivät vaikuta merkittävästi elafibranorin metaboliaan. Lääkeyhteisvaikutukset CYP- tai UGT-entsyymien aktiivisuutta merkittävästi muuttavien lääkeaineiden kanssa ovat oletettavasti vähäisiä.
Kliiniset tutkimukset
Varfariini (CYP2C9:n substraatti):
Elafibranorin samanaikainen anto varfariinin kanssa ei suurentanut varfariinin pitoisuutta (AUC, Cmax) eikä INR (international normalized ratio) ‑arvossa ollut eroja verrattuna varfariiniin yksinään.
Simvastatiini (CYP3A:n, BRCP:n [Breast Cancer Resistance Protein], orgaanisia anioneja kuljettavien polypeptidien OATP1B1:n ja OATP1B3:n substraatti) ja atorvastatiini (CYP3A:n, orgaanisia anioneja kuljettavien polypeptidien OATP1B1:n ja OATP1B3:n substraatit):
Elafibranorin toistuva anto samanaikaisesti simvastatiinin tai atorvastatiinin kanssa ei suurentanut simvastatiinin tai sen beetahydroksihappometaboliitin eikä atorvastatiinin pitoisuuksia (AUC, Cmax).
Sitagliptiini (dipeptidyylipeptidaasi IV:n (DPP-IV:n) estäjä):
Annettaessa samanaikaisesti 100 mg elafibranoria lääkeyhteisvaikutuksia aiheuttavana aineena kerran päivässä 15 päivän ajan ja 100 mg:n kerta-annos sitagliptiinia ateriakokeen aikana ei havaittu kliinisesti merkitsevää vaikutusta GLP-1:n pitoisuuksiin.
In vitro ‑tutkimukset
Sytokromi P450:n (CYP) esto ja induktio:
Elafibranoria ja GFT1007:ää ei pidetty pääasiallisten CYP-entsyymien estäjinä. Ajasta riippuvaa CYP:n estoa ei todettu.
Elafibranori ja GFT1007 eivät aiheuttaneet CYP1A2:n, CYP2B6:n tai CYP3A4:n induktiota.
UGT:n esto:
In vitro ‑tulosten perusteella elafibranori ja GFT1007 eivät oletettavasti estä pääasiallisia UGT-entsyymejä kliinisesti merkitsevinä pitoisuuksina.
Kuljetusmekanismit:
Elafibranori on OATP1B3:n ja BCRP:n estäjä. Simvastatiinilla ja atorvastatiinilla tehtyjen in vivo ‑tutkimusten perusteella OATP1B3:n ja BCRP:n estosta ei oletettavasti aiheudu kliinisiä seurauksia.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Lisääntymis- ja kehitystoksisuus
Elafibranorin kehitystoksisuudesta on saatu näyttöä sekä rotalla että kaniineilla. Rotalla tehdyssä pre- ja postnataalitutkimuksessa emon altistuminen elafibranorille (altistuksen ollessa vähintään 2‑kertainen suurimmasta ihmiselle suositellusta annoksesta aiheutuvaan altistukseen (AUC) verrattuna) johti poikasten eloonjäännin vähenemiseen, kehityksen viivästymiseen tai tromboosiin.
Tiineillä kaniineilla emon altistus (3-kertainen suurimmasta ihmiselle suositellusta annoksesta aiheutuvaan altistukseen (AUC) verrattuna) elafibranori aiheutti huomattavaa emoon kohdistuvaa toksisuutta, suurentunutta alkiokuolleisuutta, pienentynyttä sikiöpainoa sekä vähäistä sikiöiden epämuodostumien ilmaantuvuutta.
Farmaseuttiset tiedot
Apuaineet
Tabletin sisällys
Mikrokiteinen selluloosa
Povidoni
Kroskarmelloosinatrium
Vedetön kolloidinen piidioksidi
Magnesiumstearaatti
Kalvopäällyste
Polyvinyylialkoholi, osittain hydrolysoitu
Titaanidioksidi (E171)
Makrogoli
Talkki
Keltainen rautaoksidi (E172)
Punainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
IQIRVO tabletti, kalvopäällysteinen
80 mg (L:ei) 30 kpl (5591,28 €)
PF-selosteen tieto
40 ml:n suurtiheyspolyeteenistä (HDPE) valmistettu purkki, jossa on polypropeeninen kiertämällä avattava turvakorkki.
Yksi purkki sisältää 30 kalvopäällysteistä tablettia.
Saatavana on seuraavia pakkauskokoja: 30 kalvopäällysteistä tablettia ja kerrannaispakkaus, jossa on 90 (3 x 30) kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Oransseja, pyöreitä, halkaisijaltaan noin 8 mm:n kokoisia tabletteja, joiden toisella puolella on tunniste ’ELA 80’.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
IQIRVO tabletti, kalvopäällysteinen
80 mg 30 kpl
- Ei korvausta.
ATC-koodi
A05AX06
Valmisteyhteenvedon muuttamispäivämäärä
01.07.2025
Yhteystiedot
Kista Science Tower, Färögatan 33
SE-164 51 Kista
Sweden
+46 8 451 60 00
www.ipsen.com
info.se@ipsen.com