
FABHALTA kapsel, hård 200 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Potilaan turvakortti - Tärkeää turvallisuustietoa FABHALTA-valmistetta käyttäville potilaille
Terveydenhuollon ammattilainen
Terveydenhuollon ammattilaisen opas FABHALTA iptakopaani
Yleinen
Opas potilaalle/potilasta hoitavalle, Fabhalta iptakopaani
Observera
▼ Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
En kapsel innehåller iptakopanhydrokloridmonohydrat motsvarande 200 mg iptakopan (iptacopan).
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Hård kapsel (kapsel)
Kliniska uppgifter
Terapeutiska indikationer
Paroxysmal nokturn hemoglobinuri
FABHALTA är indicerat som monoterapi vid behandling av paroxysmal nokturn hemoglobinuri (PNH) hos vuxna patienter med hemolytisk anemi.
Komplement faktor 3-glomerulopati
FABHALTA är avsett för behandling av vuxna patienter med komplementfaktor 3-glomerulopati (C3G), i kombination med en hämmare av renin-angiotensinsystemet (RAS-hämmare), eller till patienter som är intoleranta mot RAS-hämmare, eller för vilka RAS-hämmare är kontraindicerat (se avsnitt Farmakodynamiska egenskaper).
Dosering och administreringssätt
Dosering
Rekommenderad dos är 200 mg oralt två gånger om dagen.
Hälso- och sjukvårdspersonal ska informera patienterna om vikten av att följa doseringsschemat. Det är viktigt att patienter med PNH följer schemat för att minimera risken för hemolys (se avsnitt Varningar och försiktighet).
Patienter som har missat en eller flera doser ska uppmanas att ta en dos så fort som möjligt (även om det inom kort är dags för nästa planerade dos) och sedan fortsätta följa det vanliga doseringsschemat. Patienter med PNH som har missat flera på varandra följande doser ska monitoreras avseende tecken och symtom på hemolys.
PNH är en sjukdom som kräver långtidsbehandling. Utsättning av behandlingen rekommenderas inte, såvida det inte är kliniskt motiverat (se avsnitt Varningar och försiktighet).
Patienter med PNH som övergår till iptakopan från behandling med en C5 ‑ hämmare (ekulizumab, ravulizumab) eller andra PNH ‑behandlingar
För att minska den potentiella risken för hemolys i samband med abrupt utsättning av behandlingen:
- För patienter som övergår från behandling med ekulizumab ska iptakopan sättas in senast 1 vecka efter den sista dosen ekulizumab.
- För patienter som övergår från behandling med ravulizumab ska iptakopan sättas in senast 6 veckor efter den sista dosen ravulizumab.
Övergång från andra komplementhämmare än ekulizumab och ravulizumab har inte studerats.
Patienter med C3G efter njurtransplantation (recidiverande C3G)
Diagnos av recidiverande C3G ställs genom påvisande av C3-deposition i glomeruli vid histologisk undersökning av den transplanterade njuren. C3-deposition kan påvisas i en rutinbiopsi efter transplantation; annars ska en biopsi utföras vid kliniska tecken till recidiverande C3G. Behandling med iptakopan kan påbörjas innan kliniska symtom som minskning av estimerad glomerulär filtrationshastighet (eGFR) eller ökning av urinprotein/kreatinin-kvoten (UPCR) uppstår, som det gjordes i studie X2202 (se avsnitt Farmakodynamiska egenskaper). I kliniska studier är erfarenheten begränsad vad gäller användningen av iptakopan hos patienter med recidiverande C3G efter transplantation (se avsnitt Farmakodynamiska egenskaper).
Särskilda patientgrupper
Äldre
Ingen dosjustering krävs för patienter i åldern 65 år och äldre (se avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Ingen dosjustering krävs för patienter med lätt (eGFR 60–< 90 ml/min) eller måttligt (eGFR 30–< 60 ml/min) njurfunktionsnedsättning. Inga data finns för närvarande tillgängliga om patienter som har svår njurfunktionsnedsättning eller genomgår dialys, och inga doseringsrekommendationer kan ges (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Användning av iptakopan rekommenderas inte till patienter med svår leverfunktionsnedsättning (Child‑Pugh‑klass C). Ingen dosjustering krävs för patienter med lindrig (Child‑Pugh‑klass A) eller medelsvår (Child‑Pugh‑klass B) leverfunktionsnedsättning (se avsnitt Farmakokinetiska egenskaper).
Pediatrisk population
Säkerhet och effekt för iptakopan för barn under 18 år har inte fastställts. Inga data finns tillgängliga.
Administreringssätt
Oral användning.
Detta läkemedel kan tas med eller utan mat (se avsnitt Farmakokinetiska egenskaper).
Kontraindikationer
- Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
- Patienter som för närvarande inte är vaccinerade mot Neisseria meningitidis och Streptococcuspneumoniae, såvida inte risken med en senarelagd behandlingsstart överstiger risken för att utveckla en infektion orsakad av dessa inkapslade bakterier (se avsnitt Varningar och försiktighet).
- Patienter med en pågående infektion orsakad av inkapslade bakterier, såsom Neisseria meningitidis, Streptococcus pneumoniae eller Haemophilus influenzae typ B, vid behandlingsstarten.
Varningar och försiktighet
Allvarliga infektioner orsakade av inkapslade bakterier
Användning av komplementhämmare som iptakopan kan predisponera personen för allvarliga, livshotande eller dödliga infektioner orsakade av inkapslade bakterier. För att minska risken för infektion måste alla patienter vaccineras mot inkapslade bakterier, såsom Neisseria meningitidis och Streptococcus pneumoniae. Också vaccination mot Haemophilus influenzae typ B rekommenderas, om vaccinet är tillgängligt. Hälso- och sjukvårdspersonal ska kontrollera lokala vaccinationsrekommendationer.
Vaccinerna ska ges minst två veckor före den första dosen iptakopan. Om behandlingen måste sättas in före vaccinationen ska patienten vaccineras så snart som möjligt och få profylaktisk antibiotikabehandling fram till två veckor efter vaccinationen.
Vid behov kan patienterna revaccineras i enlighet med lokala vaccinationsrekommendationer.
Vaccination minskar risken för allvarlig infektion men eliminerar den inte helt. En allvarlig infektion kan snabbt framskrida och bli livshotande eller dödlig om den inte identifieras och behandlas i ett tidigt skede. Patienterna ska informeras om tidiga tecken och symtom på allvarlig infektion och monitoreras avseende sådana. Vid misstanke om infektion ska patientens tillstånd bedömas och behandling sättas in omedelbart. Användning av iptakopan under behandling av en allvarlig infektion kan övervägas efter en nytta‑riskbedömning (se avsnitt Biverkningar).
PNH laboratorieövervakning
Patienter med PNH som får iptakopan bör övervakas regelbundet för tecken och symtom på hemolys, inklusive mätning av laktatdehydrogenas (LDH)-nivåer.
Monitorering av manifestationer av PNH efter utsättning av behandlingen
Om behandlingen måste sättas ut ska patienter med PNH monitoreras noga avseende tecken och symtom på hemolys i minst två veckor efter den sista dosen. Exempel på sådana tecken och symtom är förhöjda nivåer av LDH förenat med plötslig minskning av hemoglobin eller PNH‑klonstorlek, trötthet, hemoglobinuri, buksmärta, dyspné, dysfagi, erektil dysfunktion eller större vaskulära händelser, inklusive venös eller arteriell trombos. Om utsättning av behandlingen är nödvändigt ska en alternativ behandling övervägas.
Om hemolys utvecklas efter utsättning av iptakopan ska återinsättning av behandlingen övervägas.
Samtidig användning med andra läkemedel
Samtidig användning av iptakopan och starka inducerare av CYP2C8, UGT1A1, PgP, BCRP och OATP1B1/3 har inte studerats kliniskt; och rekommenderas därför inte att användas samtidigt på grund av risken för minskad effekt av iptakopan (se avsnitt Interaktioner). Om samtidig användning inte kan undvikas, ska patienter med PNH övervakas med avseende på tecken och symtom på hemolys.
Behandling av patienter med C3G
Patienter med C3G som behandlas med immunsuppressiva läkemedel kan visa en måttlig minskning av proteinuri med iptakopan, vilket sannolikt är kopplat till en mer behandlingsresistent karaktär av C3G hos dessa patienter.
Det finns ingen erfarenhet av användning av iptakopan hos patienter med C3G i nativ njure som har proteinuri under 1 g/g vid behandlingsstart.
Utbildningsmaterial
Alla läkare som har för avsikt att förskriva FABHALTA måste säkerställa att de har fått utbildningsmaterialet som är avsett för läkare och satt sig in i det. Läkarna måste förklara nyttan och riskerna med behandling med FABHALTA och diskutera dessa med patienten samt ge dem informationspaketet för patienter. Patienten ska instrueras att söka vård omedelbart om de upplever tecken eller symtom på allvarlig infektion eller allvarlig hemolys (patienter med PNH) efter utsättning av behandling.
Interaktioner
Effekter av andra läkemedel på iptakopan
Starka inducerare av CYP2C8, UGT1A1, PgP, BCRP och OATP1B1/3
Även om samtidig administrering av iptakopan med starka inducerare av CYP2C8, UGT1A1, PgP, BCRP och OATP1B1/3, som exempelvis rifampicin, inte har studerats kliniskt så rekommenderas inte samtidig användning på grund av potentiellt minskad effekt av iptakopan (se avsnitt Varningar och försiktighet).
Effekter av iptakopan på andra läkemedel
CYP3A4 -substrat
In vitro-data visade att iptakopan har potential för induktion av CYP3A4 och kan minska exponeringen av känsliga CYP3A4-substrat. Samtidig användning av iptakopan och känsliga CYP3A4-substrat har inte studerats kliniskt. Försiktighet bör iakttas om samtidig användning av iptakopan med känsliga CYP3A4-substrat krävs, särskilt för dem med ett smalt terapeutiskt index (t.ex. karbamazepin, ciklosporin, ergotamin, fentanyl, pimozid, kinidin, sirolimus, takrolimus).
CYP2C8-substrat
In vitro-data visade att iptakopan har en potential för tidsberoende hämning av CYP2C8 och kan öka exponeringen av känsliga CYP2C8-substrat, såsom repaglinid, dasabuvir eller paklitaxel. Samtidig användning av iptakopan och känsliga CYP2C8-substrat har inte studerats kliniskt. Försiktighet bör iakttas om samtidig administrering av iptakopan med känsliga CYP2C8-substrat krävs.
Fertilitet, graviditet och amning
Graviditet
Det finns inga eller begränsad mängd data från användningen av iptakopan hos gravida kvinnor. Data från djurstudier visar inga direkta eller indirekta reproduktionstoxikologiska effekter vid exponeringar mellan 2- och 8-gånger den mänskliga exponeringen vid den maximala rekommenderade humandosen (MRHD) (se avsnitt Prekliniska säkerhetsuppgifter).
PNH under graviditet är förknippad med skadliga effekter som drabbar modern, inklusive förvärrade cytopenier, trombotiska händelser, infektioner, blödningar, missfall och ökad mödradödlighet, samt skadliga effekter som drabbar fostret, såsom fosterdöd och prematura födslar.
C3G under graviditet kan vara förknippat med skadliga effekter som drabbar modern, i synnerhet preeklampsi och missfall, samt skadliga effekter som drabbar fostret, såsom förtidig födsel och låg födelsevikt.
Användning av iptakopan hos kvinnor som är gravida eller planerar att bli gravida kan övervägas, endast vid behov, efter en noggrann nytta‑riskbedömning.
Amning
Det är okänt om iptakopan utsöndras i bröstmjölk. Det finns inga data om effekterna av iptakopan på ammade nyfödda/spädbarn eller på mjölkproduktionen.
En risk för det nyfödda barnet/spädbarnet kan inte uteslutas. Ett beslut måste fattas om man ska avbryta amningen eller avbryta/avstå från behandling med FABHALTA efter att man tagit hänsyn till fördelen med amning för barnet och nyttan med behandling för kvinnan.
Fertilitet
Det finns inga data om effekten av iptakopan på fertilitet hos människa. Tillgängliga icke‑kliniska data tyder inte på att behandling med iptakopan har någon effekt på fertilitet (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
FABHALTA har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligaste rapporterade biverkningarna hos vuxna patienter med PNH var övre luftvägsinfektion (18,9 %), huvudvärk (18,3 %) och diarré (11,0 %). Den vanligaste rapporterade allvarliga biverkningen var urinvägsinfektion (1,2 %).
Den vanligaste rapporterade biverkningen hos vuxna patienter med C3G var övre luftvägsinfektion (12,9 %). Den vanligaste rapporterade allvarliga biverkningen var pneumokockinfektion (1 %).
Tabell över biverkningar
I tabell 1 presenteras biverkningarna som observerats i kliniska studier med iptakopan hos patienter med PNH och C3G. Biverkningarna listas enligt MedDRA:s klassificering av organsystem och frekvens i enlighet med följande konvention: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), sällsynta (≥ 1/10 000, < 1/1 000) eller mycket sällsynta (< 1/10 000).
Inom varje frekvensgrupp presenteras biverkningarna efter fallande allvarlighetsgrad.
Tabell 1 Biverkningar
Organsystem Biverkning | Frekvenskategori | |
PNH | C3G | |
Infektioner och infestationer | ||
Övre luftvägsinfektion1 | Mycket vanliga | Mycket vanliga |
Urinvägsinfektion2 | Vanliga | |
Bronkit3 | Vanliga | |
Pneumkockinfektion4 | Vanliga | |
Lunginflammation, bakteriell | Mindre vanliga | |
Blodet och lymfsystemet | ||
Minskat antal trombocyter | Vanliga | |
Centrala och perifera nervsystemet | ||
Huvudvärk5 | Mycket vanliga | |
Yrsel | Vanliga | |
Magtarmkanalen | ||
Diarré | Mycket vanliga | |
Buksmärta6 | Vanliga | |
Illamående | Vanliga | |
Hud och subkutan vävnad | ||
Urtikaria | Mindre vanliga | |
Muskuloskeletala systemet och bindväv | ||
Artralgi | Vanliga | |
1 Övre luftvägsinfektion innefattar de rekommenderade termerna influensa, nasofaryngit, faryngit, rinit, sinuit, övre luftvägsinfektion och virusinfektion i övre luftvägarna. 2 Urinvägsinfektion innefattar de rekommenderade termerna urinvägsinfektion och cystit orsakad av Escherichia coli. 3 Bronkit innefattar de rekommenderade termerna bronkit, Haemophilus-bronkit, och bakteriell bronkit. 4 Pneumokockinfektion innefattar de rekommenderade termerna pneumokockpneumoni och pneumokocksepsis. 5 Huvudvärk innefattar de rekommenderade termerna huvudvärk och obehag i huvudet. 6 Buksmärta innefattar de rekommenderade termerna buksmärta, högt sittande buksmärta, bukömhet och obehag i buken. | ||
Beskrivning av utvalda biverkningar
Infektioner
I PNH kliniska studier rapporterades allvarlig bakteriell pneumoni hos 1 av 164 patienter (0,6 %) med PNH under behandling med iptakopan. Patienten hade vaccinerats mot Neisseria meningitidis, Streptococcus pneumoniae och Haemophilus influenzae typ B och tillfrisknade efter behandling med antibiotika, samtidigt som behandlingen med iptakopan fortsatte.
I slutförda C3G kliniska studier rapporterades allvarlig pneumokockinfektion med pneumoni och sepsis hos 1 patient med C3G under behandling med iptakopan. Patienten hade vaccinerats mot Neisseria meningitidis, Streptococcus pneumoniae och Haemophilus influenzae typ B och tillfrisknade efter behandling med antibiotika. Iptakopanbehandlingen avbröts tillfälligt och återupptogs efter tillfrisknandet.
Minskat antal trombocyter hos patienter med PNH
En minskning i antalet trombocyter rapporterades hos 12 av 164 patienter (7 %) med PNH. Bland dessa var biverkningen lindrig hos 5 patienter, medelsvår hos 5 patienter och svår hos 2 patienter. Patienter med en svår biverkning hade samtidiga antikroppar mot trombocyter eller idiopatisk benmärgsaplasi med befintlig trombocytopeni. Biverkningarna uppkom inom de 2 första månaderna av iptakopanbehandling hos 7 av 12 patienter och efter längre exponering (111–951 dagar) hos 5 av 12 patienter. Vid tidpunkten för cut‑off hade 7 patienter (58 %) återhämtat sig, alternativt höll biverkningarna på att gå tillbaka, och iptakopanbehandlingen fortsatte hela tiden för alla patienter.
Förhöjt kolesterolvärde och blodtryc k hos patienter med PNH
Hos patienter som behandlades med iptakopan 200 mg två gånger per dag i kliniska PNH‑studier sågs genomsnittliga ökningar om ca 0,7 mmol/l i totalkolesterol och LDL‑kolesterol vid månad 6 jämfört med utgångsläget (baseline). De genomsnittliga värdena kvarstod inom de normala intervallen. Förhöjningar av blodtrycket observerades, särskilt i det diastoliska blodtrycket (genomsnittlig förhöjning 4,7 mmHg vid månad 6). Det genomsnittliga diastoliska blodtrycket översteg inte 80 mmHg. Förhöjningar av totalkolesterol, LDL‑kolesterol och diastoliskt blodtryck korrelerade med en ökning av hemoglobinvärdet (lindring av anemi) hos patienter med PNH (se avsnitt Farmakodynamiska egenskaper).
Hos patienter som behandlades med iptakopan 200 mg två gånger per dag i den kliniska C3G-studien sågs inga kliniskt relevanta skillnader i totalkolesterol, LDL-kolesterol eller blodtryck i jämförelse med placebo.
Pulsminskning hos patienter med PNH
Hos patienter som behandlats med iptakopan 200 mg två gånger per dag i kliniska studier med PNH, sågs en genomsnittlig minskning av hjärtfrekvensen på cirka 5 slag per minut vid månad 6 (medelvärde på 68 slag per minut).
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta‑riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via det nationella rapporteringssystemet listat i bilaga V.
Överdosering
Under kliniska studier tog några patienter iptakopan i en dos om högst 800 mg per dag, och denna dos var väl tolererad. Hos friska frivilliga var den högsta dosen 1 200 mg givet som en enkeldos, och denna dos var väl tolererad.
Allmänna understödjande åtgärder och symtomatisk behandling ska sättas in vid misstanke om överdosering.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Immunsuppressiva medel, komplementhämmare, ATC‑kod: L04AJ08
Verkningsmekanism
Iptakopan är en proximal komplementhämmare som riktar sig mot faktor B (FB) och selektivt hämmar den alternativa signalvägen. Vid PNH förhindrar hämning av faktor B i komplementkaskadens alternativa signalväg aktiveringen av C3‑konvertas och det efterföljande bildandet av C5‑konvertas och kontrollerar därmed både C3‑medierad extravaskulär hemolys (EVH) och terminal komplementmedierad intravaskulär hemolys (IVH).
Vid C3G leder överaktivering av komplementsystemet via den alternativa vägen till deponering av C3 i glomeruli, vilket utlöser inflammation, glomerulusskador samt njurfibros. Iptakopan blockerar selektivt överaktiveringen av den alternativa vägen genom att hämma den alternativa vägen som är kopplad till aktivitet hos C3-konvertas. Detta leder till minskad klyvning av C3 och minskad C3-deponering i njurarna.
Farmakodynamisk effekt
Utifrån en ex vivo alternativ signalvägsanalys samt en analys av Bb‑nivåerna (b‑fragmentet av faktor B) och plasmakoncentrationerna av C5b‑9 började hämningen av den alternativa signalvägen ≤ 2 timmar efter att en enkeldos iptakopan getts till friska frivilliga.
En jämförbar effekt av iptakopan sågs både hos PNH‑patienter som tidigare exponerats för C5‑hämmare och hos tidigare obehandlade PNH‑patienter.
Hos tidigare obehandlade PNH‑patienter minskade iptakopan i en dos om 200 mg två gånger per dag LDH‑koncentrationen med > 60% jämfört med utgångsläget efter 12 veckor, och effekten kvarstod fram till slutet av studien.
Hos patienter med C3G hade genomsnittlig C3-nivå i serum på dag 14 med iptakopanbehandling ökat med 249 % jämfört med utgångsläget, vilket visar på hämning av patologisk C3-klyvning. Lösligt C5b-9 i plasma respektive i urin hade vid den första observationen på dag 30 med iptakopan 200 mg två gånger per dag minskat med respektive 71,8 % och 92,1 % jämfört med utgångsläget. Effekten kvarstod under den 12 månader långa observationsperioden. Efter 6 månader observerades även en minskad deponering av C3 i glomeruli, baserat på förändringen av deposit score för C3.
Hjärtats elektrofysiologi
I en klinisk QTc‑studie observerades ingen effekt på hjärtats repolarisation eller QT‑tid när friska frivilliga fick supraterapeutiska enkeldoser iptakopan på högst 1 200 mg (vilket resulterade i en mer än fyrfaldig exponering jämfört med dosen 200 mg två gånger per dag).
Klinisk effekt och säkerhet
Paroxysmal nokturn hemoglobinuri
Effekten och säkerheten av iptakopan hos vuxna patienter med PNH utvärderades i två öppna, 24 veckor långa multicenterstudier i fas III: en kontrollerad studie med aktivt jämförelseläkemedel (APPLY‑PNH) och en enarmad studie (APPOINT‑PNH).
APPLY‑PNH: patienter med PNH som tidigare fått behandling med C5‑hämmare
I APPLY‑PNH deltog vuxna PNH‑patienter (RBC‑klonstorlek ≥ 10 %) med kvarstående anemi (hemoglobin < 10 g/dl) trots tidigare behandling med en stabil dos av C5‑hämmare (antingen ekulizumab eller ravulizumab) i minst 6 månader före randomiseringen.
Patienterna (N = 97) randomiserades i förhållandet 8:5 till att antingen få iptakopan 200 mg oralt två gånger per dag (N = 62) eller fortsätta behandlingen med C5‑hämmare (ekulizumab N = 23; eller ravulizumab N = 12) under hela den 24 veckor långa randomiserade, kontrollerade perioden. Randomiseringen stratifierades utifrån tidigare behandling med C5‑hämmare och transfusioner under de senaste 6 månaderna.
Behandlingsgrupperna var generellt väl balanserade gällande demografiska egenskaper och sjukdomskarakteristika vid utgångsläget. Vid baslinjen hade patienterna en medelålder (standardavvikelse [SD]) på 51,7 (16,9) år (intervall 22–84) och 49,8 (16,7) år (intervall 20–82) i iptakopan respektive anti-C5-grupperna och 69 % av patienterna var kvinnor i båda grupperna. Medelvärdet (SD) hemoglobin var 8,9 (0,7) g/dl och 8,9 (0,9) g/dl, i iptakopan respektive anti-C5-gruppen. 57 % (iptakopan-gruppen) och 60 % (anti-C5-gruppen) av patienterna fick minst en transfusion under 6 månaderna före randomiseringen. Bland dessa var det genomsnittliga (SD) antalet transfusioner 3,1 (2,6) och 4,0 (4,3) i iptakopan respektive anti-C5-gruppen. Den genomsnittliga (SD) LDH-nivån var 269,1 (70,1) U/l i iptakopan-gruppen och 272,7 (84,8) U/l i anti-C5-gruppen. Det genomsnittliga (SD) absoluta retikulocytantalet var 193,2 (83,6) 109/l i iptakopan-gruppen och 190,6 (80,9) 109/l i anti-C5-gruppen. Den genomsnittliga (SD) totala PNH RBC-klonstorleken (typ II+III) var 64,6 % (27,5 %) i iptakopan-gruppen och 57,4 % (29,7 %) i anti-C5-gruppen.
Under den randomiserade, kontrollerade perioden avbröt 1 patient i iptakopangruppen sin behandling till följd av graviditet; i anti‑C5‑gruppen avbröt ingen behandlingen.
Överlägsenhet för iptakopan gentemot C5‑hämmare i fråga om att uppnå hematologisk respons efter 24 veckors behandling utan behov av transfusioner påvisades på basis av två primära effektmått genom att bedöma andelen patienter som uppvisade: 1) en varaktig förhöjning av hemoglobinvärdet på ≥ 2 g/dl jämfört med utgångsläget (förbättrat hemoglobinvärde) och/eller 2) ett varaktigt hemoglobinvärde på ≥ 12 g/dl.
Iptakopan visade överlägsenhet gentemot C5‑hämmare beträffande de två primära resultatmåtten, liksom också beträffande flera sekundära resultatmått, inklusive undvikande av transfusion, förändringar i hemoglobinvärden jämfört med utgångsläget, FACIT‑Fatigue‑poäng (funktionell utvärdering av trötthet vid behandling av kronisk sjukdom), absolut retikulocytantal (ARC) och det årliga antalet fall av kliniskt fastställd genombrottshemolys (se tabell 2).
Iptakopanbehandlingens effekt på hemoglobinvärdena sågs så tidigt som på dag 7 och kvarstod under studien (se figur 1).
Tabell 2 Effektresultat under den 24 veckor långa randomiserade behandlingsperioden i APPLY-PNH
| Resultatmått | Iptakopan (N = 62) | Anti‑C5 (N = 35) | Skillnad (95 % KI) p‑värde |
| Primära resultatmått | |||
| Antal patienter vars hemoglobinvärde förbättrades (varaktig förhöjning av hemoglobinvärdet på ≥ 2 g/dl jämfört med vid baslinjena utan transfusioner) | 51/60b | 0/35b | |
| Responsandelc (%) | 82,3 | 2,0 | 80,2 (71,2; 87,6) < 0,0001 |
| Antal patienter som uppnådde ett varaktigt hemoglobinvärde på ≥ 12 g/dla utan transfusioner | 42/60b | 0/35b | |
| Responsandelc (%) | 68,8 | 1,8 | 67,0 (56,4; 76,9) < 0,0001 |
| Sekundära resultatmått | |||
| Antal patienter som inte behövde transfusionerd,e | 59/62b | 14/35b | |
| Andel undvikande av transfusionc (%) | 94,8 | 25,9 | 68,9 (51,4; 83,9) < 0,0001 |
| Förändring i hemoglobinvärdet jämfört med utgångsläget (g/dl) (justerat medelvärdef) | 3,60 | -0,06 | 3,66 (3,20; 4,12) < 0,0001 |
| Förändring i FACIT‑Fatigue-poäng jämfört med utgångsläget (justerat medelvärdeg) | 8,59 | 0,31 | 8,29 (5,28; 11,29) < 0,0001 |
| Kliniskt fastställd genombrottshemolysh,i, % (n/N) | 3,2 (2/62) | 17,1 (6/35) | |
| Årligt antal fall av kliniskt fastställd genombrottshemolys | 0,07 | 0,67 | RR = 0,10 (0,02; 0,61) 0,01 |
| Förändring i absolut antal retikulocyter jämfört med utgångsläget (109/l) (justerat medelvärdeg) | -115,8 | 0,3 | -116,2 (-132,0; -100,3) < 0,0001 |
| LDH‑förhållande till utgångsläget (justerat geometriskt medelvärdeg) | 0,96 | 0,98 | Kvot = 0,99 (0,89; 1,10) 0,84 |
Större vaskulära händelserh % (n/N) | 1,6 (1/62) | 0 | |
| Årligt antal större vaskulära händelserh | 0,03 | 0 | 0,03 (-0,03; 0,10) 0,32 |
RR: frekvenskvot (rate ratio); LDH: laktatdehydrogenas a,d,h Bedömt mellan: dagarna 126 och 168(a), dagarna 14 och 168(d), dagarna 1 och 168(h). b Baserat på observationsdata om evaluerbara patienter. (Hos 2 patienter med delvis saknade centrala hemoglobindata mellan dag 126 och 168 kunde det hematologiska svaret inte fastställas entydigt. Det hematologiska svaret härleddes med hjälp av multipel imputation. Dessa patienter avbröt inte behandlingen.) c Responsandelen återspeglar den uppskattade modellandelen. e Definition av undvikande av transfusion: patienten har inte fått erytrocyttransfusioner eller uppfyllt transfusionskriterierna mellan dagarna 14 och 168. f,g Justerat medelvärde fastställt mellan dagarna 126 och 168, värdena som erhållits inom 30 dagar efter en transfusion exkluderades i(f)/inkluderades ur(g) analysen. i Definition av kliniskt fastställd genombrottshemolys: uppfyllande av de kliniska kriterierna (minskning av hemoglobinvärdet på ≥ 2 g/dl jämfört med den senaste analysen eller inom 15 dagar, eller tecken eller symtom på betydande hemoglobinuri, smärtkris, dysfagi eller andra betydande PNH‑relaterade kliniska tecken eller symtom) och uppfyllande av laboratoriekriterierna (LDH > 1,5 x ULN och högre än de två senaste uppmätta värdena). | |||
Figur 1 Genomsnittligt hemoglobinvärde* (g/dl) under den 24 veckor långa randomiserade behandlingsperioden i APPLY-PNH
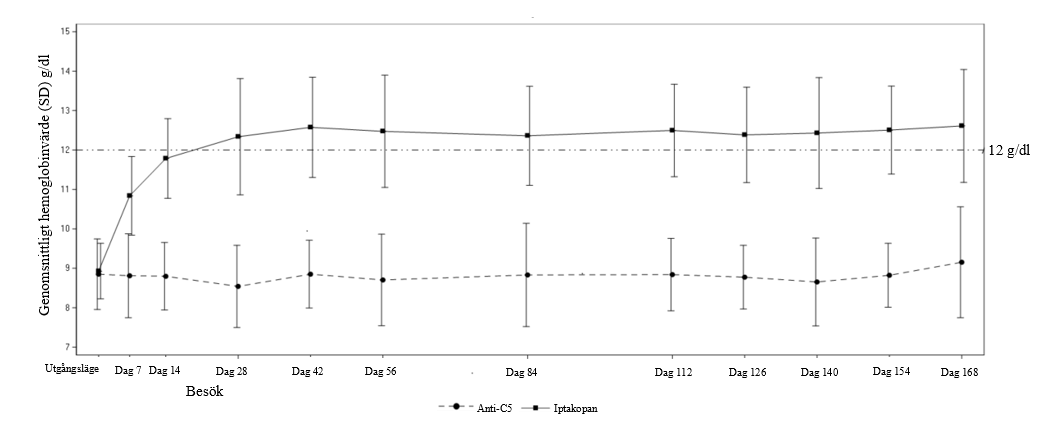
*Observera: Figuren innefattar alla data om hemoglobinvärden som samlats in under studien, inklusive de värden som uppmättes inom 30 dagar efter en erytrocyttransfusion.
Behandlingsförlängning
Totalt 95 APPLY-PNH-patienter påbörjade den 24 veckor långa förlängningsperioden av behandlingen, där samtliga patienter fick iptakopan, vilket resulterade i en total exponering på upp till 48 veckor. Effektresultaten vid vecka 48 överensstämde med resultaten vid vecka 24 och visade på bibehållen effekt av iptakopan behandling.
APPOINT-PNH: patienter som inte tidigare fått behandling med komplementhämmare
APPOINT‑PNH var en enarmad studie med 40 vuxna PNH‑patienter (RBC‑klonstorlek ≥ 10 %), som hade ett hemoglobinvärde på < 10 g/dl och LDH > 1,5 x ULN och som inte tidigare fått behandling med komplementhämmare. Alla 40 patienter fick iptakopan oralt i en dos om 200 mg två gånger per dag under den 24 veckor långa öppna, behandlingsperioden.
Vid baslinjen hade patienterna en medelålder (SD) på 42,1 (15,9) år (intervall 18–81) och 43 % var kvinnor. Medelvärdet (SD) hemoglobin var 8,2 (1,1) g/dl. 70 % av patienterna fick minst en transfusion under 6 månaderna före behandlingen. Bland dessa var medelantalet (SD) transfusioner 3,1 (2,1). Den genomsnittliga (SD) LDH-nivån var 1 698,8 (683,3) U/l, och det genomsnittliga (SD) absoluta retikulocytantalet var 154,3 (63,7) 109/l. Den genomsnittliga (SD) totala PNH RBC klonstorleken (typ II + III) var 42,7 % (21,2 %). Ingen av patienterna avbröt behandlingen i studiens behandlingsfas.
Bedömningen av iptakopanbehandlingens effekt baserades på det primära effektmåttet mätt som andelen patienter vars hemoglobinvärde förbättrades (varaktig förhöjning av hemoglobinvärdet på ≥ 2 g/dl jämfört med utgångsläget utan behov av erytrocyttransfusion vid vecka 24).
I tabell 3 presenteras detaljerade uppgifter om effekt och i figur 2 förändringen i det genomsnittliga LDH‑värdet under den 24 veckor långa behandlingsperioden.
Tabell 3 Effektresultat under den 24 veckor långa egentliga behandlingsperioden i APPOINT-PNH
| Resultatmått | Iptakopan (N = 40) 95 % KI |
| Primärt resultatmått | |
| Antal patienter vars hemoglobinvärde förbättrades (varaktig förhöjning av hemoglobinvärdet på ≥ 2 g/dl jämfört med utgångslägeta utan transfusioner) | 31/33b |
| Responsandelc (%) | 92,2 (82,5; 100,0)d |
| Sekundära resultatmått | |
| Antal patienter som uppnådde ett varaktigt hemoglobinvärde på ≥ 12 g/dla utan transfusioner | 19/33b |
| Responsandelc (%) | 62,8 (47,5; 77,5) |
| Antal patienter som inte behövde transfusionere,f | 40/40b |
| Andel undvikande av transfusionc (%) | 97,6 (92,5; 100,0) |
Förändring i hemoglobinvärdet jämfört med utgångsläget (g/dl) (justerat medelvärdeg) | +4,3 (3,9; 4,7) |
| Kliniskt fastställd genombrottshemolysi,j, % (n/N) | 0/40 |
| Årligt antal fall av kliniskt fastställd genombrottshemolys | 0,0 (0,0; 0,2) |
| Förändring i absolut antal retikulocyter jämfört med utgångsläget (109/l) (justerat medelvärdeh) | -82,5 (-89,3; -75,6) |
Procentuell förändring i LDH‑värdet jämfört med utgångsläget (justerat medelvärdeh) | -83,6 (-84,9; -82,1) |
| Procentandel patienter med större vaskulära händelserh | 0,0 |
a,e,j Bedömt mellan: dagarna 126 och 168(a), dagarna 14 och 168(e), dagarna 1 och 168(h). b Baserat på observationsdata om evaluerbara patienter. (Hos 7 patienter med delvis saknade centrala hemoglobindata mellan dag 126 och 168 kunde det hematologiska svaret inte fastställas entydigt. Det hematologiska svaret härleddes med hjälp av multipel imputation. Dessa patienter avbröt inte behandlingen.) c Responsandelen återspeglar den uppskattade modell andelen. d Tröskelvärdet för påvisande av nytta var 15 %, som motsvarar nivån som skulle ha förväntats vid behandling med C5‑hämmare. f Definition av undvikande av transfusion: patienten har inte fått erytrocyttransfusioner eller uppfyllt transfusionskriterierna mellan dagarna 14 och 168. g,h Justerat medelvärde bedömt mellan dag 126 och 168, värden inom 30 dagar efter transfusion exkluderades(g)/inkluderades(h) i analysen. i Definition av kliniskt fastställd genombrottshemolys: uppfyllande av de kliniska kriterierna (minskning av hemoglobinvärdet på ≥ 2 g/dl jämfört med den senaste analysen eller inom 15 dagar, eller tecken eller symtom på betydande hemoglobinuri, smärtkris, dysfagi eller andra betydande PNH‑relaterade kliniska tecken eller symtom) och uppfyllande av laboratoriekriterierna (LDH > 1,5 x ULN och högre än de två senaste uppmätta värdena). | |
Figur 2 Genomsnittligt LDH‑värde (U/l) under den 24 veckor långa behandlingsperioden i APPOINT‑PNH
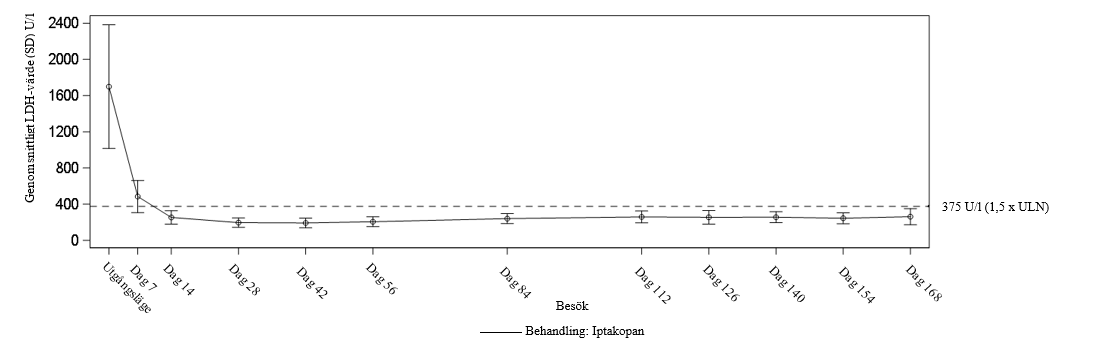
Behandlingsförlängning
Alla 40 APPOINT-PNH-patienter påbörjade den 24 veckor långa förlängningsperioden för behandling, där samtliga patienter fortsatte med iptakopan behandling, vilket resulterade i en total exponering på upp till 48 veckor. Effektresultaten vid vecka 48 överensstämde med resultaten vid vecka 24, vilket visade på bibehållen effekt av iptakopan behandling.
Komplementfaktor 3-glomerulopati
Effekten och säkerheten för iptakopan behandling av C3G utvärderades på totalt 101 patienter med C3G i en pivotal fas III-studie (APPEAR-C3G, hos patienter med nativ njure, N=74) och två stödjande öppna studier (studie X2202 hos patienter med nativ njure (N=16) och patienter med recidiverande C3G (N=11) och en roll-over-förlängningsstudie).
APPEAR-C3G
APPEAR-C3G, en multicenter, randomiserad, dubbelblind, placebokontrollerad studie, inkluderade 74 vuxna patienter med biopsibekräftad C3G, (UPCR) på ≥1 g/g och eGFR på ≥30 ml/min/1,73 m2.
Patienterna randomiserades (1:1) till att få antingen iptakopan 200 mg peroralt två gånger per dag (N=38) eller placebo (N=36) i 6 månader, följt av en 6 månaders öppen behandlingsperiod med iptakopan 200 mg peroralt två gånger per dag. Samtliga 74 patienter fullföljde hela den dubbelblinda perioden och 73 patienter fullföljde den öppna perioden med iptakopan.
Patienterna stod på en stabil maximal tolererad dos RAS-hämmare (renin-angiotensinsystem-hämmare). Randomiseringen stratifierades efter samtidig behandling med immunsuppressiva medel (dvs. kortikosteroid och/eller mykofenolatmofetil/-natrium [MMF/MPS] respektive ingen sådan behandling). Alla dessa behandlingar (RAS-hämmare, kortikosteroider och MMF/MPS) krävde stabila doser i 90 dagar före randomiseringen och under hela studien.
I utgångsläget var patienternas ålder i genomsnitt (standardavvikelse [SD]) 26,1 (10,4) år (intervall 18‑52) i iptakopangruppen och 29,8 (10,8) år (intervall 18‑60) i placebogruppen. 40 % (iptakopan) och 17 % (placebo) av patienterna var < 18 år när de fick diagnosen C3G. 29 % (iptakopan) och 44 % (placebo) av patienterna var kvinnor. Geometriskt medelvärde för UPCR var 3,33 g/g i iptakopangruppen och 2,58 g/g i placebogruppen. Den genomsnittliga modellerade historiska eGFR-lutningen före randomisering var -10,75 jämfört med -7,64 ml/min/1,73 m2 per år i respektive iptakopan- och placebo-armarna. Genomsnittligt (SD) eGFR var 89,3 (35,2) ml/min/1,73 m2 i iptakopangruppen och 99,2 (26,9) ml/min/1,73 m2 i placebogruppen. Undertyper var C3-glomerulonefrit (C3GN) hos 68 % (iptakopan) och 89 % (placebo) av patienterna och dense deposit disease (DDD) var 23,7 % (iptakopan) och 2,8 % (placebo). En stabil dos av immunsuppresiv behandling med kortikosteroider och/eller MMF/MPS gavs till 42 % (iptakopan) och 47 % (placebo) av patienterna.
Primärt effektmått var procentuell minskning av 24-timmars UPCR efter 6 månaders behandling, jämfört med utgångsläget.
Iptakopan var överlägset placebo med en statistiskt signifikant minskning av 24-timmars UPCR på 35,1 % (95 % KI, 13,8 %; 51,1 %, 1-sidigt p=0,0014) från utgångsläget jämfört med placebo efter 6 månaders behandling (-30,2 % och +7,6 % för iptakopan respektive placebo). Effekten av iptakopanbehandlingen på 24-timmars UPCR kvarstod i upp till 12 månader (-40,0 % från utgångsläget). Hos patienterna som bytte från placebo till iptakopan under den 6 månader långa öppna behandlingsperioden sjönk 24‑timmars UPCR med 31,0 % från månad 6 till månad 12. Första morgonurinen (FMV)-UPCR är beskrivet i figur 3.
I en post hoc-analys ledde behandling med iptakopan till en minskning av procentandelen patienter med nefrotisk proteinuri (definierat som UPCR ≥3 g/g) från 55,3 % i utgångsläget till 31,6 % månad 6 och 36,8 % månad 12. Andelen patienter som randomiserats till placebo med nefrotisk proteinuri ökade från 30,6 % i utgångsläget till 41,7 % månad 6. Efter att ha bytt till iptakopan minskade andelen till 27,8 % månad 12.
Figur 3Geometriskt medelvärde för procentuell förändring från utgångsläget av FMV UPCR i upp till 12 månader (APPEAR-C3G)
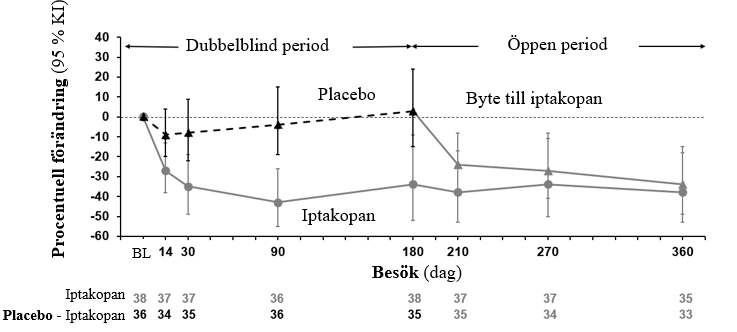
Iptakopanbehandling i 6 månader resulterade i en numerisk förbättring på 2,2 ml/min/1,73 m2 (95 % KI: -2,7; 7,1, 1-sidigt p=0,3241) av eGFR från utgångsläget jämfört med placebo (1,3 respektive ‑0,9 ml/min/1,73 m2 för iptakopan och placebo). eGFR var stabilt under de 12 studiemånaderna i gruppen som fick iptakopan (+0,4 ml/min/1,73 m2 från utgångsläget).
Iptakopanbehandling i 6 månader resulterade i en genomsnittlig förändring avdeponering av C3 i glomeruli på 1,9 (95 % KI: -3,3; 0,5, nominellt 1-sidigt p=0,0053) från utgångslägen jämfört med placebo. Med iptakopan sågs en förändring på -0,78 (95 % KI: -1,81; 0,25) från utgångsläget jämfört med en ökning på 1,09 (95 % KI: 0,11; 2,08) med placebo.
X2202 och r oll-over-förlängningsstudie
Effekten av iptakopan hos vuxna med C3G understöds av en öppen fas 2-studie X2202 på patienter med C3G i nativa njurar (N=16) och patienter med recidiverande C3G efter njurtransplantation (N=11) i 3 månader.
Diagnos av recidiverande C3G krävde histologisk bedömning av glomerulär C3-färgningsintensitet på en nyligen genomförd biopsi av den transplanterade njuren. Medelåldern vid baslinjen var 35 år (intervall 18–70), det geometriska medelvärdet för UPCR var 0,32 g/g, medelvärdet för (SD) eGFR var 52,2 (17,29) ml/min/1,73 m2 och medianvärdet för C3-avsättning var 3 på en skala från 0–12 vid utgångsläget. Alla patienter fick MMF/MPS och/eller kortikosteroider i tillägg till kalcineurinhämmare.
Hos patienter med nativ njure resulterade iptakopan i en statistiskt signifikant minskning av 24-timmars UPCR på 45 % (-162,6 g/mol p=0,0003) vid 3 månader. Hos patienter med recidiverande C3G minskade iptakopan signifikant poängen för histologisk C3-deponering med 2,50 (p=0,0313) vid 3 månader.
De flesta patienter (n=26) i studien gick över till en pågående roll-over-förlängningsstudie där de fick iptakopan 200 mg två gånger per dag i upp till 39 månader. Genomsnittlig UPCR och eGFR kvarstod stabila under studien hos de 16 patienterna med C3G i nativa njurar. Bland de 10 patienter med recidiverande C3G efter transplantation hoppade 2 patienter av på grund av försämrad njurfunktion. Hos de andra 8 deltagarna förblev eGFR och UPCR i huvudsak konstanta till slutet av observationsperioden (upp till 48 månader).
Pediatrisk population
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för FABHALTA för en eller flera grupper av den pediatriska populationen för PNH och C3G (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption
Vid oral administrering av iptakopan uppnåddes maximala plasmakoncentrationer ca 2 timmar efter intaget. Med den rekommenderade dosen 200 mg två gånger per dag uppnås steady state inom ca 5 dagar med liten ackumulering (1,4‑faldig). Hos friska frivilliga var steady-state Cmax,ss (geo-medelvärde (%CV)) var 4 020 ng/ml (23,8 %) och AUCtau,ss var 25 400 ng*hr/ml (15,2 %). Den inter- och intraindividuella variabiliteten i iptakopans farmakokinetik är ringa eller måttlig.
På basis av resultat från en studie med friska frivilliga, där man undersökte effekten av födointag med en fettrik och kaloririk måltid, verkade inte mat påverka Cmax och arean under kurvan (AUC) för iptakopan. Därmed kan iptakopan tas med eller utan mat.
Distribution
Iptakopan uppvisade koncentrationsberoende plasmaproteinbindning till följd av bindning till målet, faktor B, i den systemiska cirkulationen. Iptakopan var proteinbundet till 75–93 % in vitro vid relevanta kliniska plasmakoncentrationer. Efter administrering av iptakopan i en dos om 200 mg två gånger per dag, var geo-medelvärde, den skenbara distributionsvolymen vid steady state ca 265 liter.
Metabolism
Den huvudsakliga elimineringsvägen för iptakopan är metabolism, och ca 50 % av dosen metaboliseras via oxidativa vägar. Iptakopans metabolism innefattar N‑dealkylering, O‑deetylering, oxidation och dehydrogenering, främst via CYP2C8 och i liten utsträckning också via CYP2D6. En mindre metabolismväg är direkt glukuronidering (via enzymerna UGT1A1, UGT1A3 och UGT1A8). I plasma var iptakopan den huvudsakliga komponenten, som stod för 83 % av AUC0‑48 h. De enda metaboliterna som detekterades i plasma var två acylglukuronider som förekom i liten utsträckning, motsvarande 8 % respektive 5 % av AUC0‑48 h. Iptakopans metaboliter anses inte vara farmakologiskt aktiva.
Eliminering
I en studie på friska frivilliga skedde i genomsnitt 71,5 % av den totala utsöndringen av radioaktivitet (iptakopan och metaboliter) via feces och 24,8 % via urinen efter en oral enkeldos om 100 mg [14C]‑märkt iptakopan. 17,9 % av dosen utsöndrades som oförändrat iptakopan i urinen och 16,8 % i feces. Skenbart clearance (CL/F) vid steady state efter administrering av iptakopan i en dos om 200 mg två gånger per dag är 7 960 ml/timme. Halveringstiden (t½) för iptakopan vid steady state är ca 25 timmar efter administrering av 200 mg två gånger per dag.
Linjäritet/icke‑linjäritet
Vid doser om 25–100 mg två gånger per dag var farmakokinetiken för iptakopan totalt sett mindre än dosproportionell. Men orala doser om 100 mg–200 mg var ungefär dosproportionella. Icke‑linjäritet ansågs främst bero på den mättnadsbara bindningen av iptakopan till faktor B i plasma.
Läkemedelsinteraktioner
I en interaktionsstudie där iptakopan administrerades tillsammans med andra läkemedel till friska frivilliga sågs inga kliniskt relevanta interaktioner.
Iptakopan som substrat
CYP2C8-inducerare
Vid samtidig administrering av klopidogrel (en måttlig CYP2C8‑hämmare) ökade Cmax och arean under kurvan (AUC) för iptakopan med 5 % respektive 36 %.
OATP 1B1/1B3‑hämmare
Vid samtidig administrering av ciklosporin (en stark OATP 1B1/1B3 hämmare och en PgP- och BCRP hämmare) ökade Cmax och AUC för iptakopan med 41% respektive 50%.
Iptakopan som hämmare
PgP-substrat
I närvaro av iptakopan ökade Cmax för digoxin (ett PgP substrat) med 8 % medan AUC förblev oförändrat.
OATP‑substrat
I närvaro av iptakopan förblev Cmax och AUC för rosuvastatin (ett OATP substrat) oförändrade.
Särskilda patientgrupper
En populationsfarmakokinetisk (PK) analys gjordes utifrån data från 234 patienter. Ålder (18–84 år), kroppsvikt, eGFR, etnisk bakgrund och kön hade ingen signifikant inverkan på iptakopans farmakokinetik. Studier som innefattade asiatiska patienter visade att farmakokinetiken för iptakopan hos dem motsvarade den hos kaukasiska (vita) patienter.
Nedsatt njurfunktion
Effekten av nedsatt njurfunktion på clearance av iptakopan utvärderades genom en populationsfarmakokinetisk analys. Inga kliniskt betydelsefulla skillnader i clearance av iptakopan förekom mellan patienter med normal njurfunktion och patienter med lindrig (eGFR 60–<90 ml/min) eller medelsvår (eGFR 30–<60 ml/min) njurfunktionsnedsättning, och ingen dosjustering krävs (se avsnitt Dosering och administreringssätt). Patienter som har svår njurfunktionsnedsättning eller som genomgår dialys har inte studerats.
Nedsatt leverfunktion
I en studie hade lindrig (Child‑Pugh A, n = 8), medelsvår (Child‑Pugh B, n = 8) eller svår (Child‑Pugh C, n = 6) leverfunktionsnedsättning en försumbar effekt på den totala systemiska exponeringen av iptakopan jämfört med normal leverfunktion. Cmax för obundet iptakopan ökade 1,4-, 1,7- och 2,1‑faldigt, och AUCinf för obundet iptakopan ökade 1,5-, 1,6- och 3,7‑faldigt hos patienter med lindrig, medelsvår respektive svår leverfunktionsnedsättning (se avsnitt Dosering och administreringssätt).
Prekliniska säkerhetsuppgifter
Gängse studier avseende säkerhetsfarmakologi, allmäntoxicitet, gentoxicitet, karcinogenicitet, reproduktionseffekter och effekter på utveckling visade inte några särskilda risker för människa.
Reproduktionstoxicitet
I fertilitetsstudier på djur med orala doser påverkade iptakopan inte fertiliteten hos hanråttor ens vid den högsta testade dosen (750 mg/kg/dag), vilket motsvarar 6 gånger den maximala rekommenderade dosen för människa på basis av AUC. I studier avseende toxicitet vid upprepad dosering observerades reversibla effekter på reproduktionssystemet hos hanar (tubulär degeneration i testiklarna och hypospermatogenes) när råttor och hundar gavs orala doser som motsvarade > 3 gånger den maximala rekommenderade dosen för människa på basis av AUC. Inga märkbara effekter på spermiernas antal, morfologi eller rörlighet eller på djurens fertilitet observerades.
I en studie på råtta avseende fertilitet hos honor och tidig embryonal utveckling var iptakopanrelaterade fynd begränsade till ökade förluster före och efter implantation och därmed minskat antal levande embryon. Detta förekom endast vid den högsta orala dosen som var 1 000 mg/kg/dag, vilket motsvarar ca 5 gånger den maximala rekommenderade dosen för människa på basis av totalt AUC. Nivån utan observerade biverkningar (NOAEL) är 300 mg/kg/dag, vilket motsvarar ca 2 gånger den maximala rekommenderade dosen för människa på basis av AUC.
I reproduktionsstudier på råtta och kanin konstaterades att oral administrering av iptakopan under organogenesen inte gav upphov till skadlig embryo- eller fetotoxicitet ens vid de högsta doserna, som på basis av AUC motsvarade 5 gånger (för råtta) respektive 8 gånger (för kanin) den maximala rekommenderade dosen för människa på 200 mg två gånger per dag.
I en pre- och postnatal utvecklingsstudie på råtta, där iptakopan gavs oralt till moderdjur under dräktighet, nedkomst och digivning (från dräktighetsdag 6 till digivningsdag 21), observerades inga biverkningar hos dräktiga moderdjur eller avkomma ens vid den högsta testade dosen 1 000 mg/kg/dag (uppskattad till 5 gånger den maximala rekommenderade dosen för människa på basis av AUC).
Toxicitet vid upprepad dosering
I den kroniska toxicitetsstudien avlivades en hanhund vid den högsta testade dosen (marginal till klinisk exponering nära 20-faldig) 103 dagar efter avslutad administrering av iptakopan på grund av irreversibel icke-regenerativ svår anemi associerad med benmärgsfibros. Under behandlingsfasen observerades hematologiska fynd som tyder på inflammation och dyserytropoes. Ingen mekanism för de observerade fynden har identifierats och ett samband med behandling kan inte uteslutas.
Mutagenicitet och karcinogenicitet
Iptakopan var inte genotoxiskt eller mutagent i en serie in vitro- och in vivo‑analyser.
I karcinogenicitetsstudier med mus och råtta där iptakopan administrerades oralt observerades ingen karcinogen potential. De högsta doserna iptakopan som studerades hos mus (1 000 mg/kg/dag) och råtta (750 mg/kg/dag) motsvarade ca 4 gånger respektive 12 gånger den maximala rekommenderade dosen för människa på basis av AUC.
Fototoxicitet
In vitro och in vivo fototoxicitetstester var tvetydiga. I fototoxicitetsstudien in vivo, med iptakopan i doser mellan 100 och 1 000 mg/kg (motsvarande 38 gånger det totala humana Cmax vid MRHD), visade några möss ett icke-dos-responsmönster av övergående minimalt erytem, sårskorpor och torrhet och en lätt ökning av den genomsnittliga öronvikten efter bestrålning.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Kapselhölje
Gelatin
Röd järnoxid (E172)
Titandioxid (E171)
Gul järnoxid (E172)
Trycksvärta
Svart järnoxid (E172)
Ammoniaklösning, koncentrerad (E527)
Kaliumhydroxid (E525)
Propylenglykol (E1520)
Shellack (E904)
Inkompatibiliteter
Ej relevant.
Hållbarhet
3 år.
Särskilda förvaringsanvisningar
Inga särskilda förvaringsanvisningar.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
FABHALTA kapseli, kova
200 mg (L:ei) 56 fol (28066,51 €)
PF-selosteen tieto
FABHALTA tillhandahålls i PVC/PE/PVDC‑blister med baksida av aluminiumfolie.
Förpackningar med 28 eller 56 hårda kapslar.
Flerpack med 168 (3 förpackningar om 56) hårda kapslar.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
Ljusgul, ogenomskinlig hård kapsel av storlek 0 (21,2–22,2 mm) märkt med ”LNP200” på kapselkroppen och ”NVR” på kapsellocket, innehållande vitt eller nästan vitt till ljust rosaviolett pulver.
Särskilda anvisningar för destruktion och övrig hantering
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
FABHALTA kapseli, kova
200 mg 56 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Iptakopaani ja pegsetakoplaani: Aikuisten kohtauksittaisen yöllisen hemoglobiinivirtsaisuuden (PNH) hoito erityisin edellytyksin (3078).
Atc-kod
L04AJ08
Datum för översyn av produktresumén
19.12.2025
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com