
UPLIZNA infuusiokonsentraatti, liuosta varten 100 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 100 mg inebilitsumabia 10 ml:ssa (10 mg/ml). Lopullinen pitoisuus laimentamisen jälkeen on 1,0 mg/ml.
Inebilitsumabi on humanisoitu monoklonaalinen vasta-aine, joka tuotetaan kiinanhamsterin munasarjasolulinjassa yhdistelmä-DNA-tekniikalla.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 16,1 mg natriumia yhdessä injektiopullossa.
Tämä lääkevalmiste sisältää 1 mg polysorbaatti 80:tä per injektiopullo, mikä vastaa 0,1 mg:aa/ml.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Neuromyelitis optica ‑kirjon häiriö (NMOSD)
Uplizna on tarkoitettu käytettäväksi monoterapiana sellaisten aikuisten potilaiden hoitoon, joilla on NMOSD ja jotka ovat akvaporiini-4:n immunoglobuliini G (AQP4-IgG) ‑vasta-aineseropositiivisia (ks. kohta Farmakodynamiikka).
Immunoglobuliini G4:ään liittyvä sairaus (IgG4‑tauti)
Uplizna on tarkoitettu käytettäväksi sellaisten aikuisten potilaiden hoitoon, joilla on aktiivinen IgG4‑tauti (ks. kohta Farmakodynamiikka).
Yleistynyt myasthenia gravis (MG)
Uplizna on tarkoitettu käytettäväksi tavanomaisen hoidon lisänä sellaisten aikuisten potilaiden hoitoon, joilla on yleistynyt myasthenia gravis ja jotka ovat AChR (asetyylikoliinireseptori) - tai MuSK (lihasperäinen tyrosiinikinaasi) -vasta-ainepositiivisia (ks. kohta Farmakodynamiikka).
Ehto
Hoito on aloitettava sellaisen lääkärin valvonnassa, jolla on kokemusta NMOSD:n tai IgG4-taudin tai yleistyneen myasthenia graviksen hoidosta ja jonka saatavilla on asianmukainen lääketieteellinen tuki mahdollisten vaikeiden haittavaikutusten, kuten vakavien infuusioon liittyvien reaktioiden, hoitamiseen.
Annostus ja antotapa
Hoito on aloitettava sellaisen lääkärin valvonnassa, jolla on kokemusta NMOSD:n tai IgG4‑taudin tai yleistyneen myasthenia graviksen hoidosta ja jonka saatavilla on asianmukainen lääketieteellinen tuki mahdollisten vaikeiden haittavaikutusten, kuten vakavien infuusioon liittyvien reaktioiden, hoitamiseen.
Potilasta on tarkkailtava mahdollisten infuusioon liittyvien reaktioiden varalta infuusion aikana ja vähintään tunnin ajan infuusion lopettamisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Arvioinnit ennen ensimmäistä inebilitsumabiannosta
Ennen hoidon aloittamista on suoritettava seuraavat testit:
-
Kvantitatiiviset seerumin immunoglobuliinit, B‑solumäärä ja täydellinen verenkuva (TVK) mukaan lukien erittelylaskennat (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet)
-
Hepatiitti B ‑virustesti (HBV) (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet)
-
Hepatiitti C ‑virustesti (HCV) ja -hoito, joka aloitetaan ennen inebilitsumabihoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
-
Aktiivisen tuberkuloosin arviointi ja latentin infektion testaus (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet)
Kaikki elävät rokotteet tai elävät heikennetyt rokotteet on annettava immunisaatio-ohjeiden mukaisesti vähintään neljä viikkoa ennen inebilitsumabihoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos tehon menetyksen ajatellaan johtuvan immunogeenisuudesta, lääkärin tulee seurata B-solumääriä kliinisen vaikutuksen suorana mittana (ks. kohta Farmakodynamiikka).
Annostus
Aloitusannokset
Suositeltu aloitusannos on 300 mg (kolme 100 mg:n injektiopulloa) infuusiona laskimoon, jonka jälkeen annetaan kahden viikon kuluttua toinen 300 mg:n infuusio laskimoon.
Ylläpitoannokset
Suositeltu ylläpitoannos on 300 mg:n infuusio laskimoon kuuden kuukauden välein. Inebilitsumabi on tarkoitettu pitkäaikaishoitoon.
IgG4‑tauti on luonteeltaan krooninen sairaus, ja hoidon jatkamisen 52 viikkoa pidempään on perustuttava sairauden aktiivisuuteen, lääkärin harkintaan ja potilaan valintaan.
Annoksen viivästyminen tai antamatta jättäminen
Jos jokin inebilitsumabi-infuusio jää antamatta, se tulee antaa mahdollisimman pian odottamatta seuraavaan suunniteltuun antoajankohtaan.
Infuusioon liittyvien reaktioiden esilääkitys
Infektion arviointi
Ennen jokaista inebilitsumabi-infuusiota on määritettävä, onko potilaalla kliinisesti merkittävä infektio. Jos potilaalla on infektio, inebilitsumabi-infuusiota on viivytettävä, kunnes infektio paranee.
Tarvittava esilääkitys
Esilääkitys kortikosteroidilla (esim. 80–125 mg metyyliprednisolonia laskimoon, tai vastaavaa) pitää antaa noin 30 minuuttia ennen jokaista inebilitsumabi-infuusiota, sekä antihistamiinilla (esim. 25–50 mg difenhydramiinia suun kautta, tai vastaavaa) ja kuumetta alentavalla lääkkeellä (esim. 500–650 mg parasetamolia suun kautta, tai vastaavaa) noin 30–60 minuuttia ennen jokaista inebilitsumabi-infuusiota (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erityiset potilasryhmät
Iäkkäät
Inebilitsumabia on annettu 72 iäkkäälle potilaalle (≥ 65-vuotiaille) kliinisissä tutkimuksissa. Saatavilla olevien tietojen perusteella annoksen muuttamista ei pidetä tarpeellisena yli 65-vuotiaille potilaille (ks. kohta Farmakokinetiikka).
Munuaisten ja maksan vajaatoiminta
Inebilitsumabia ei ole tutkittu potilailla, joilla on vaikea munuaisten tai maksan vajaatoiminta. Annoksen muuttaminen munuaisten tai maksan toiminnan perusteella ei kuitenkaan ole aiheellista, koska immunoglobuliini (Ig) G-luokan monoklonaaliset vasta-aineet eivät poistu ensisijaisesti munuaisten tai maksan kautta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Inebilitsumabin turvallisuutta ja tehoa 0–18-vuotiailla lapsilla ja nuorilla ei ole vielä määritetty. Tietoja ei ole saatavilla.
Antotapa
Laskimoon.
Injektiopulloja ei saa ravistaa.
Injektiopulloja on säilytettävä pystyasennossa.
Valmisteltu liuos on annettava laskimoon infuusiopumpulla vähitellen lisääntyvällä nopeudella loppuun asti (noin 90 minuuttia) infuusioletkun kautta, jossa on steriili, niukasti proteiineja sitova 0,2 tai 0,22 mikronin suodatin, taulukossa 1 esitetyn aikataulun mukaisesti.
Taulukko 1. Suositeltu infuusionopeus laimennettuna 250 ml:n infuusiopussissa
Kulunut aika (minuutteina) | Infuusionopeus (ml/tunti) |
0–30 | 42 |
31–60 | 125 |
61–lopetus | 333 |
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle (vaikuttaville aineille) tai kohdassa Apuaineet mainituille apuaineille
- vaikea aktiivinen infektio, mukaan lukien aktiivinen krooninen infektio, kuten hepatiitti B
- aktiivinen tai hoitamaton latentti tuberkuloosi
- aiempi progressiivinen multifokaalinen leukoenkefalopatia (PML)
- vaikea immuunivajavuustila
- aktiiviset maligniteetit.
Varoitukset ja käyttöön liittyvät varotoimet
Potilaille annettavat ohjeet lääkkeen määräämisen yhteydessä
Uplizna-valmisteella hoidettaville potilaille annetaan potilaskortti, jossa on tietoa siitä, että inebilitsumabihoito saattaa lisätä infektioiden, myös vakavien infektioiden, virusten reaktivaation, opportunististen infektioiden ja progressiivisen multifokaalisen leukoenkefalopatian (PML) riskiä, sekä siitä, miten hakeutua varhain lääkärin hoitoon infektion tai PML:n oireiden ja löydösten ilmetessä.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infuusioon liittyvät reaktiot ja yliherkkyys
Inebilitsumabi voi aiheuttaa infuusioon liittyviä reaktioita ja yliherkkyysreaktioita, joita voivat olla päänsärky, pahoinvointi, uneliaisuus, hengenahdistus, kuume, lihaskipu, ihottuma, sydämentykytykset tai muut oireet. Infuusioon liittyvät reaktiot olivat yleisimpiä ensimmäisen infuusion yhteydessä, mutta niitä havaittiin myös myöhempien infuusioiden aikana. Inebilitsumabin kliinisissä tutkimuksissa ilmeni vakavia infuusioon liittyviä reaktioita, vaikka ne olivatkin harvinaisia (ks. kohta Haittavaikutukset).
Ennen infuusiota
Esilääkitys pitää antaa kortikosteroidilla (esim. 80–125 mg metyyliprednisolonia laskimoon, tai vastaavaa), antihistamiinilla (esim. 25–50 mg difenhydramiinia suun kautta, tai vastaavaa) ja kuumetta alentavalla lääkkeellä (esim. 500–650 mg parasetamolia suun kautta, tai vastaavaa) (ks. kohta Annostus ja antotapa).
Infuusion aikana
Potilasta on tarkkailtava infuusioon liittyvien reaktioiden varalta. Infuusioon liittyvien reaktioiden hoitosuositukset riippuvat reaktion tyypistä ja vaikeusasteesta. Jos ilmenee hengenvaarallisia infuusioreaktioita, hoito on lopetettava välittömästi ja pysyvästi ja potilaalle on annettava asianmukaista elintoimintoja tukevaa hoitoa. Jos ilmenee vähemmän vaikeita infuusioreaktioita, hoitoon saattaa kuulua infuusion tilapäinen keskeyttäminen, infuusionopeuden vähentäminen ja/tai oireenmukaisen hoidon antaminen.
Infuusion jälkeen
Potilasta on tarkkailtava mahdollisten infuusioon liittyvien reaktioiden varalta vähintään tunnin ajan infuusion lopettamisen jälkeen.
Infektiot
Inebilitsumabi vähentää perifeerisen veren lymfosyyttimääriä ja Ig-pitoisuuksia B-soluja vähentävän vaikutusmekanismin mukaisesti. Myös neutrofiilimäärien vähenemistä ilmoitettiin. Siksi inebilitsumabi saattaa lisätä infektioherkkyyttä (ks. kohta Haittavaikutukset).
Enintään kuusi kuukautta ennen inebilitsumabihoidon aloittamista on otettava täydellinen verenkuva (TVK) mukaan lukien erittelylaskennat ja immunoglobuliinit. Täydellisen verenkuvan, mukaan lukien erittelylaskennat ja immunoglobuliinit, arviointien tekemistä suositellaan myös säännöllisesti hoidon aikana ja sen päättymisen jälkeen B-solujen palautumiseen asti. Ennen jokaista inebilitsumabi-infuusiota on määritettävä, onko potilaalla kliinisesti merkittävä infektio. Jos potilaalla on infektio, inebilitsumabi-infuusiota on viivytettävä, kunnes infektio paranee. Potilaita on neuvottava ilmoittamaan lääkärille välittömästi infektion oireista. Hoidon lopettamista pitää harkita, jos potilaalle tulee vakava opportunistinen infektio tai toistuvia infektioita, mikäli Ig-tasot osoittavat immuunijärjestelmän heikentymistä.
Yleisimpiä infektioita, joita inebilitsumabilla hoidetut NMOSD-potilaat ilmoittivat koko satunnaistetun, kontrolloidun jakson (RCP) ja avoimen tutkimusjakson (OLP) aikana, olivat virtsatieinfektiot (26,2 %), nenänielutulehdus (20,9 %), ylähengitystieinfektiot (15,6 %), influenssa (8,9 %) ja keuhkoputkentulehdus (6,7 %). IgG4‑taudin yhteydessä satunnaistetun, kontrolloidun jakson (RCP) ja avoimen tutkimusjakson (OLP) aikana yleisimpiä infektioita, joita inebilitsumabilla hoidetut potilaat ilmoittivat, olivat ylähengitystieinfektio (10,7 %) nenänielutulehdus (9,8 %), virtsatieinfektio (8,9 %) ja influenssa (6,3 %). Yleistyneen myasthenia graviksen yhteydessä satunnaistetun, kontrolloidun jakson (RCP) ja avoimen tutkimusjakson (OLP) aikana yleisimpiä infektioita, joita inebilitsumabilla hoidetut potilaat ilmoittivat, olivat nenänielutulehdus (6,9 %) ja ylähengitystieinfektio (6,9 %).
Hepatiitti B -viruksen uudelleenaktivoituminen
HBV:n uudelleenaktivoitumisen riskiä on havaittu muilla B-soluja vähentävillä vasta-aineilla. Yleistynyttä myastenia gravista koskeneen kliinisen tutkimuksen yhteydessä havaittiin HBV:n uudelleenaktivoituminen yhdellä inebilitsumabilla hoidetulla potilaalla. Potilaat, joilla oli krooninen HBV, suljettiin pois inebilitsumabin kliinisistä tutkimuksista. HBV-testit pitää tehdä kaikille potilaille ennen inebilitsumabihoidon aloittamista. Inebilitsumabia ei saa antaa potilaille, joilla on HBV:n aiheuttama aktiivinen hepatiitti ja jotka ovat hepatiitti B:n pinta-antigeeni (HBsAg)- tai hepatiitti B:n ydinvasta-aine (HBcAb) -positiivisia. Potilaiden, jotka ovat HBV:n [HBsAg+] kroonisia kantajia, pitää keskustella maksatautien asiantuntijan kanssa ennen hoidon aloittamista ja sen aikana (ks. kohta Vasta-aiheet).
Hepatiitti C -virus
Potilaat, jotka olivat HCV-positiivisia, suljettiin pois inebilitsumabin kliinisistä tutkimuksista. Lähtötilanteen HCV-testaus on tarpeen viruksen havaitsemiseksi ja hoidon aloittamiseksi ennen inebilitsumabihoidon aloittamista.
Tuberkuloosi
Ennen inebilitsumabihoidon aloittamista potilaille on tehtävä aktiivisen tuberkuloosin arviointi ja latentin infektion testaus. Ennen inebilitsumabihoidon aloittamista on otettava yhteyttä infektiosairauksien asiantuntijaan, jos potilaalla on aktiivinen tuberkuloosi tai positiivinen tuberkuloositesti eikä hän ole saanut asianmukaista hoitoa.
Progressiivinen multifokaalinen leukoenkefalopatia (PML)
PML on John Cunninghamin viruksen (JCV) aiheuttama aivojen opportunistivirusinfektio, joka ilmenee tavallisesti immuunivajepotilailla. Se saattaa johtaa kuolemaan tai vakavaan vammaan. Progressiiviseen multifokaaliseen leukoenkefalopatiaan johtavaa JC-virustartuntaa on havaittu potilailla, joita hoidetaan muilla B-solujen määrää vähentävillä vasta-aineilla.
Inebilitsumabin kliinisissä tutkimuksissa ei todettu yhtään vahvistettua PML-tapausta. Inebilitsumabin kliinisissä tutkimuksissa yksi tutkittava (NMOSD-tutkimus) kuoli sen jälkeen, kun hänelle oli kehittynyt uusia aivoleesioita, joille ei voitu määrittää lopullista diagnoosia. Erotusdiagnoosi käsitti kuitenkin epätyypillisen NMOSD-pahenemisvaiheen, progressiivisen multifokaalisen leukoenkefalopatian tai akuutin disseminoituneen enkefalomyeliitin.
Lääkärien on tarkkailtava huolellisesti kliinisiä oireita tai magneettikuvauksen tuloksia, jotka saattavat viitata progressiiviseen multifokaaliseen leukoenkefalopatiaan. Magneettikuvaustulokset saattavat antaa viitteitä ennen kliinisten merkkien ja oireiden ilmaantumista. Progressiiviseen multifokaaliseen leukoenkefalopatiaan tavallisesti liittyvät oireet ovat moninaisia, niiden eteneminen voi kestää päivistä viikkoihin, ja niihin kuuluu progressiivinen heikkous kehon toisella puolella tai raajojen kömpelyys, näköhäiriö sekä muutokset ajattelussa, muistissa ja orientoitumisessa, mitkä johtavat sekavuuteen ja persoonallisuuden muutoksiin.
Ensimmäisten progressiiviseen multifokaaliseen leukoenkefalopatiaan viittaavien merkkien tai oireiden ilmetessä inebilitsumabihoito on keskeytettävä, kunnes progressiivinen multifokaalinen leukoenkefalopatia on suljettu pois. Lisäarviointeja, mukaan lukien neurologin konsultointi, magneettikuvaus mieluiten varjoaineen kanssa, JC-viruksen DNA:n tutkiminen aivo-selkäydinnesteestä sekä toistuvat neurologiset arvioinnit, tulisi harkita. Jos sairaus vahvistetaan, inebilitsumabihoito pitää lopettaa.
Myöhäinen neutropenia
Myöhäisessä vaiheessa alkavaa neutropeniaa on raportoitu (ks. kohta Haittavaikutukset). Vaikka osa niistä oli 3. asteen tapauksia, suurin osa oli 1. tai 2. asteen tapauksia. Myöhäisessä vaiheessa alkavaa neutropeniaa on raportoitu vähintään neljä viikkoa viimeisimmän inebilitsumabi-infuusion jälkeen. Potilaille, joilla on infektion oireita ja löydöksiä, suositellaan veren neutrofiilimäärien mittaamista.
Vaikeaa immuunivajavuutta sairastavien potilaiden hoito
Potilaita, joilla on vaikea immuunivajavuustila, ei saa hoitaa valmisteella, ennen kuin tila paranee (ks. kohta Vasta-aiheet).
Jos inebilitsumabi yhdistetään toiseen immuunisalpaushoitoon, lisääntyneiden immuunisalpausvaikutusten mahdollisuus on otettava huomioon.
Potilaita, joilla on tunnettu synnynnäinen tai hankittu immuunivajavuustila, mukaan lukien HIV-infektio tai splenektomia, ei ole tutkittu.
Rokotukset
Kaikki rokotteet on annettava immunisaatio-ohjeiden mukaisesti vähintään neljä viikkoa ennen inebilitsumabihoidon aloittamista. Inebilitsumabihoitoa seuraavan immunisaation tehoa ja turvallisuutta elävillä rokotteilla tai elävillä heikennetyillä rokotteilla ei ole tutkittu, eikä rokottamista elävillä heikennetyillä tai elävillä rokotteilla suositella hoidon aikana eikä ennen B-solujen palautumista.
Inebilitsumabille raskauden aikana altistuneiden äitien lapsille ei saa antaa eläviä tai eläviä heikennettyjä rokotteita, ennen kuin on varmistettu lapsen B-solumäärän palautuminen. B-solujen väheneminen näillä altistuneilla lapsilla saattaa lisätä elävien tai elävien heikennettyjen rokotteiden riskejä. Ei-eläviä rokotteita voidaan antaa mainitulla tavalla ennen B-solumäärän ja Ig-tason palautumista niiden vähenemisen jälkeen, mutta pätevän asiantuntijan konsultoimista pitää harkita sen arvioimiseksi, saatiinko suojaava immuunivaste.
B-solujen palautumisaika
B‑solujen palautumiseen kuluvaa aikaa inebilitsumabin antamisen jälkeen ei tunneta (ks. kohta Farmakodynamiikka).
Raskaus
Varmuuden vuoksi inebilitsumabin käyttöä on suositeltavaa välttää raskauden aikana sekä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä (ks. kohta Raskaus ja imetys). Potilaita on neuvottavailmoittamaan terveydenhuollon ammattilaiselle, jos he ovat raskaana tai suunnittelevat raskautta inebilitsumabihoitoa saadessaan. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä (menetelmiä, joiden käyttäjistä alle 1 % tulee raskaaksi) Uplizna-valmistetta saadessaan ja kuuden kuukauden ajan viimeisimmän Uplizna-annoksen jälkeen.
Maligniteetti
Immuunivastetta säätelevät lääkevalmisteet saattavat lisätä maligniteetin riskiä. Nykyiset tiedot eivät viittaa maligniteetin lisääntyneeseen riskiin; tässä vaiheessa ei kuitenkaan voida sulkea pois mahdollista kiinteiden kasvaimien kehittymisen riskiä (ks. kohta Haittavaikutukset).
Yleistynyttä myasthenia gravista sairastavat potilaat, joiden Myasthenia Gravis Foundation of America (MGFA) -luokka on V
Inebilitsumabia ei ole tutkittu yleistynyttä myasthenia gravista sairastavilla potilailla, joiden MGFA-luokka on V.
Apuaineet, joiden vaikutus tunnetaan
Natriumpitoisuus
Tämä lääkevalmiste sisältää 48,3 mg natriumia per annos, mikä vastaa 2 %:a WHO:n aikuisille suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisilla.
Polysorbaatti
Tämä lääkevalmiste sisältää 1 mg polysorbaatti 80:tä per injektiopullo, mikä vastaa 0,1 mg:aa/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Ensisijainen terapeuttisten vasta-aineiden poistumisreitti on puhdistuma retikuloendoteliaalijärjestelmän kautta. Sytokromi P450 -entsyymit, effluksipumput ja proteiineja sitovat mekanismit eivät ole mukana terapeuttisten vasta-aineiden puhdistumassa. Siksi mahdollinen inebilitsumabin ja muiden lääkevalmisteiden välisten farmakokineettisten interaktioiden riski on pieni.
Rokotukset
Inebilitsumabihoitoa seuraavan immunisaation tehoa ja turvallisuutta ei ole tutkittu elävillä rokotteilla tai elävillä heikennetyillä rokotteilla. Rokotusvaste saattaa heikentyä, jos B-solujen määrä vähenee. On suositeltavaa, että potilaat ottavat kaikki tarvittavat rokotteet ennen inebilitsumabihoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immuunisalpaajat
NMOSD-potilailla tehdyn keskeisen tutkimuksen satunnaistetun, kontrolloidun jakson aikana kaikille tutkittaville annettiin kaksi viikkoa kestävä, suun kautta otettava kortikosteroidilääkitys (ja yhden viikon asteittain vähenevä lääkitys) inebilitsumabin ensimmäisen antokerran jälkeen. IgG4‑tautia sairastavilla tehdyn keskeisen tutkimuksen satunnaistetun, kontrolloidun jakson aikana tutkittavat saivat yhtenäistä glukokortikoidiannosta inebilitsumabihoidon aloitushetkellä. Tämän jälkeen glukokortikoidiannosta vähennettiin ennalta määritellysti asteittain, kunnes lääkitys lopetettiin 8 viikon kuluttua. Yleistynyttä myasthenia gravista sairastavilla tehdyssä keskeisessä tutkimuksessa tutkittavat saivat joko suun kautta otettavaa kortikosteroidilääkitystä tai jotakin muuta immuunisalpaajaa vakaana annoksena inebilitsumabihoidon aloitushetkellä. Tutkittavat aloittivat ennalta määritellysti kortikosteroidin vähentämisen asteittain annokseen 5 mg/vrk viikosta 4 viikkoon 24, mutta saattoivat edelleen saada muita immuunisalpaajia inebilitsumabihoidon aikana (ks. kohta Farmakodynamiikka).
Inebilitsumabin samanaikainen käyttö immuunisalpaajien kanssa, mukaan lukien systeemiset kortikosteroidit, saattaa lisätä infektion riskiä. Inebilitsumabin vaikutus B-soluihin ja immunoglobuliineihin saattaa jatkua kuuden kuukauden tai pitemmän ajan annostelun jälkeen.
Kun inebilitsumabi aloitetaan muiden immuunisalpaushoitojen jälkeen, joilla on pitkäaikainen vaikutus immuunijärjestelmään, tai kun inebilitsumabin jälkeen aloitetaan muita immuunisalpaushoitoja, joilla on pitkäaikainen vaikutus immuunijärjestelmään, näiden lääkevalmisteiden vaikutuksen kesto ja vaikutustapa pitää ottaa huomioon mahdollisten additiivisten immuunisalpausvaikutusten tähden (ks. kohta Farmakodynamiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä (menetelmiä, joiden käyttäjistä alle 1 % tulee raskaaksi) Upliznaa saadessaan ja kuuden kuukauden ajan viimeisimmän Uplizna-annoksen jälkeen.
Raskaus
Inebilitsumabin käytöstä raskaana oleville naisille on saatavilla rajallisesti tietoja. Inebilitsumabi on humanisoitu monoklonaalinen IgG1-vasta-aine, ja immunoglobuliinien tiedetään läpäisevän istukan. Ohimenevää perifeeristä B-solujen vähenemistä ja lymfosytopeniaa on ilmoitettu lapsilla, jotka ovat syntyneet äideille, jotka altistuivat muille B-soluja vähentäville vasta-aineille raskauden aikana.
Eläinkokeet eivät osoita suoria tai epäsuoria haitallisia lisääntymistoksisia vaikutuksia; ne ovat kuitenkin osoittaneet B-solujen vähenemistä jälkeläisten maksassa sikiöasteella (ks. kohta Prekliiniset tiedot turvallisuudesta).
Inebilitsumabihoitoa on vältettävä raskauden aikana, ellei äidille mahdollisesti koituva hyöty ole suurempi kuin sikiölle mahdollisesti koituva haitta.
Jos äiti altistuu raskauden aikana, vastasyntyneillä on odotettavissa B-solujen vähenemistä valmisteen farmakologisten ominaisuuksien ja eläinkokeiden tulosten perusteella (ks. kohta Prekliiniset tiedot turvallisuudesta). Kliinisissä tutkimuksissa ei ole tutkittu B‑solujen määriä imeväisikäisillä, kun äiti on altistunut inebilitsumabille. B-solujen vähenemisen kestoa lapsilla, jotka altistuvat inebilitsumabille in utero, ja B-solujen vähenemisen vaikutusta rokotteiden turvallisuuteen ja tehoon ei tunneta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka). Tämän vuoksi vastasyntyneiden B-solujen vähenemistä pitää tarkkailla ja rokotuksia eläviä viruksia sisältävillä rokotteilla, kuten Bacillus Calmette-Guérin (BCG) -rokote, pitää siirtää, kunnes lapsen B-solumäärä on palautunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Inebilitsumabin käyttöä naisille imetyksen aikana ei ole tutkittu. Ei tiedetä, erittyykö inebilitsumabi ihmisen rintamaitoon. Ihmisillä IgG-vasta-aineiden erittymistä maitoon ilmenee muutaman ensimmäisen päivän aikana syntymän jälkeen, ja se vähenee alhaisiin pitoisuuksiin pian tämän jälkeen.
Sen vuoksi riskiä imetettävälle lapselle ei voida sulkea pois tämän lyhyen jakson aikana. Tämän jälkeen Upliznaa voidaan käyttää imetyksen aikana, jos se on kliinisesti tarpeen. Jos potilasta kuitenkin hoidettiin Uplizna-valmisteella raskauden viimeisiin kuukausiin asti, imetys voidaan aloittaa heti syntymän jälkeen.
Hedelmällisyys
Inebilitsumabin vaikutuksesta ihmisen hedelmällisyyteen on saatavilla rajallisesti tietoja; eläintutkimukset ovat kuitenkin osoittaneet hedelmällisyyden vähentymistä. Näiden ei-kliinisten tulosten kliinistä merkitystä ei tunneta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tähän mennessä ilmoitetut farmakologiset vaikutukset ja haittavaikutukset viittaavat siihen, että inebilitsumabilla ei ole haitallista vaikutusta tai on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kolmessa keskeisessä kliinisessä tutkimuksessa NMOSD-tautia, IgG4‑tautia ja yleistynyttä myasthenia gravista sairastavilla potiailla yleisimpiä inebilitsumabilla hoidetuilla potilailla ilmoitettuja haittavaikutuksia koko satunnaistetun, kontrolloidun jakson (RCP) ja avoimen jakson (OLP) aikana olivat virtsatieinfektiot (26,2 %), nenänielutulehdus (20,9 %), ylähengitystieinfektio (15,6 %), nivelkipu (17,3 %), selkäkipu (13,8 %), lymfopenia (10,7 %) ja päänsärky (10,3 %).
Yleisimpiä inebilitsumabilla hoidetuilla potilailla ilmoitettuja vakavia haittavaikutuksia koko satunnaistetun, kontrolloidun jakson ja avoimen jakson aikana olivat infektiot (11,1 %) (mukaan lukien virtsatieinfektiot (4,0 %), keuhkokuume (1,8 %)) ja NMOSD (1,8 %).
Haittavaikutustaulukko
Inebilitsumabihoidon yhteydessä kliinisissä tutkimuksissa ja markkinoille saattamisen jälkeen raportoidut haittavaikutukset on esitetty taulukossa 2 seuraavan esiintyvyysluokituksen mukaisesti: hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1 000, <1/100), harvinainen (≥1/10 000, < 1/1 000), hyvin harvinainen (<1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 2. Haittavaikutukset, joita raportoitiin inebilitsumabin kliinisissä tutkimuksissa NMOSD-tautia, yleistynyttä myasthenia gravista ja IgG4-tautia sairastavilla potilailla sekä markkinoille saattamisen jälkeen
MedDRA-elinjärjestelmä | Hyvin yleinen (≥1/10) | Yleinen (≥1/100, <1/10) | Melko harvinainen (≥1/1 000, <1/100) |
Infektiot | Virtsatieinfektio, hengitystieinfektio, nenänielutulehdus, influenssa | Keuhkokuume, selluliitti, herpes zoster, sinuiitti | Sepsis, ihonalainen märkäpesäke, bronkioliitti |
Veri ja imukudos | Lymfopenia* | Neutropenia, myöhään ilmenevä neutropenia |
|
Luusto, lihakset ja sidekudos | Nivelkipu, selkäkipu | Lihaskipu |
|
Yleisoireet ja antopaikassa todettavat haitat |
| Kuume |
|
Hermosto | Päänsärky |
|
|
Tutkimukset | Vähentyneet immunoglobuliinit |
|
|
Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioon liittyvä reaktio |
|
|
Hengityselimet, rintakehä ja välikarsina |
| Yskä |
|
* Lymfopenia sisältää pienentyneen lymfosyyttimäärän | |||
Valikoitujen haittavaikutusten kuvaus
Infuusioon liittyvät reaktiot
Inebilitsumabi voi aiheuttaa infuusioon liittyviä reaktioita, joita voivat olla päänsärky, pahoinvointi, uneliaisuus, hengenahdistus, kuume, lihaskipu, ihottuma, sydämentykytykset tai muut oireet. Kaikille potilaille annettiin esilääkitystä. Infuusioon liittyviä reaktioita havaittiin 9,2 %:lla NMOSD-potilaista inebilitsumabin aloitusjakson aikana verrattuna 10,7 %:iin lumelääkettä saaneista potilaista. Infuusioon liittyviä reaktioita havaittiin inebilitsumabin yhteydessä 7,4 %:lla IgG4‑tautia sairastavista potilaista verrattuna 14,9 %:iin lumelääkettä saaneista potilaista satunnaistetun, kontrolloidun jakson aikana. Infuusioon liittyviä reaktioita havaittiin inebilitsumabin yhteydessä 10,1 %:lla yleistynyttä myasthenia gravista sairastavista potilaista verrattuna 5,9 %:iin lumelääkettä saaneista potilaista satunnaistetun, kontrolloidun jakson aikana. Infuusioon liittyvät reaktiot olivat yleisimpiä ensimmäisessä infuusiossa, mutta niitä havaittiin myös myöhempien infuusioiden aikana. Suurin osa inebilitsumabilla hoidetuilla potilailla ilmoitetuista infuusioon liittyvistä reaktioista oli joko lieviä tai keskivaikeita.
Infektiot
Kliinisissä tutkimuksissa 74,7 %:lla inebilitsumabilla hoidetuista NMOSD-potilaista,70,5 %:lla IgG4‑tautia sairastavista potilaista ja 42,9 %:lla yleistynyttä myasthenia gravista sairastavista potilaista ilmoitettiin infektio koko satunnaistetun, kontrolloidun jakson ja avoimen jakson aikana. Yleisimpiä infektioita NMOSD-potilailla olivat virtsatieinfektio (26,2 %), nenänielutulehdus (20,9 %), ylähengitystieinfektio (15,6 %), influenssa (8,9 %) ja keuhkoputkentulehdus (6,7 %). Vakavia infektioita, joista ilmoitti useampi kuin yksi inebilitsumabilla hoidettu NMOSD-potilas, olivat virtsatieinfektio (4,0 %) ja keuhkokuume (1,8 %). Yleisimpiä infektioita IgG4‑tautia sairastavilla potilailla olivat ylähengitystieinfektio (10,7 %), nenänielutulehdus (9,8 %), virtsatieinfektio (8,9 %) ja influenssa (6,3 %). Vakava infektio, josta ilmoitti useampi kuin yksi inebilitsumabilla hoidettu IgG4‑tautia sairastava potilas, oli keuhkokuume (1,8 %). Yleisimpiä infektioita yleistynyttä myasthenia gravista sairastavilla potilailla olivat nenänielutulehdus (6,9 %) ja ylähengitystieinfektio (6,9 %). Vakava infektio, josta ilmoitti useampi kuin yksi inebilitsumabilla hoidettu yleistynyttä myasthenia gravista sairastava potilas, oli keuhkokuume (1,5 %). Katso kohdasta Varoitukset ja käyttöön liittyvät varotoimet, miten infektion tapauksessa pitää toimia.
Opportunistiset ja vakavat infektiot
NMOSD-potilailla tehdyssä tutkimuksessa ei satunnaistetun, kontrolloidun jakson aikana ilmennyt yhtään opportunistista infektiota kummassakaan hoitoryhmässä, ja vain yksi 4. asteen infektiivinen haittavaikutus (epätyypillinen keuhkokuume) ilmeni inebilitsumabilla hoidetulla potilaalla. Avoimen tutkimusjakson aikana kahdella inebilitsumabilla hoidetulla potilaalla (0,9 %) esiintyi opportunistinen infektio (näistä toista ei vahvistettu) ja kolmella inebilitsumabilla hoidetulla potilaalla (1,4 %) esiintyi 4. asteen infektiivinen haittavaikutus. Katso kohdasta Varoitukset ja käyttöön liittyvät varotoimet, miten infektion tapauksessa pitää toimia. IgG4‑tautia sairastavilla potilailla tehdyssä tutkimuksessa kolmella inebilitsumabilla hoidetulla potilaalla (2,7 %) esiintyi opportunistinen infektio (kaikilla herpes zoster, joka ei ollut kenelläkään vakava) koko satunnaistetun, kontrolloidun jakson ja avoimen tutkimusjakson aikana. Yleistynyttä myasthenia gravista sairastavilla potilailla tehdyssä tutkimuksessa yhdellä inebilitsumabilla hoidetulla potilaalla (0,8 %) esiintyi vakava 3. asteen tapahtuma (disseminoitunut herpes zoster) satunnaistetun, kontrolloidun tutkimusjakson aikana.
Laboratorioarvojen poikkeavuudet
Vähentyneet immunoglobuliinit
Inebilitsumabin vaikutusmekanismin mukaisesti keskimääräiset immunoglobuliinipitoisuudet pienenivät lääkkeen käytön myötä. NMOSD-potilailla tehdyn tutkimuksen kuuden ja puolen kuukauden satunnaistetun, kontrolloidun jakson lopussa niiden potilaiden osuudet, joiden pitoisuudet olivat pienemmät kuin viitealueen alaraja, olivat seuraavat: IgA 9,8 % inebilitsumabiryhmässä ja 3,1 % lumelääkeryhmässä, IgE 10,6 % inebilitsumabiryhmässä ja 12,5 % lumelääkeryhmässä, IgG 3,8 % inebilitsumabiryhmässä ja 9,4 % lumelääkeryhmässä ja IgM 29,3 % inebilitsumabiryhmässä ja 15,6 % lumelääkeryhmässä. Yksi haittavaikutus, IgG-tason väheneminen, ilmoitettiin (2. aste, avoimen tutkimusjakson aikana). Niiden inebilitsumabilla hoidettujen potilaiden osuus, joiden IgG-pitoisuudet alittivat viitealueen alarajan, oli vuoden 1 kohdalla 7,4 % ja vuoden 2 kohdalla 9,9 %. Kun mediaanialtistus oli 3,2 vuotta, kohtalaisen IgG-pitoisuuksien vähenemisen (300 – < 500 mg/dl) esiintymistiheys oli 14,2 % ja vaikean IgG-tasojen vähenemisen (< 300 mg/dl) esiintymistiheys oli 3,6 %. IgG4‑tautia sairastavilla potilailla tehdyn tutkimuksen 12 kuukauden satunnaistetun, kontrolloidun jakson lopussa immunoglobuliinien kokonaispitoisuus oli pienentynyt lähtötasosta noin 12 % inebilitsumabilla hoidetuilla potilailla verrattuna pitoisuuden kasvuun 21 %:lla lumelääkettä saaneilla potilailla. Immunoglobuliini G (IgG) väheni lähtötasosta keskimäärin noin 9 % ja immunoglobuliini M (IgM) noin 32 % inebilitsumabilla hoidetuilla potilailla, kun lumelääkettä saaneilla potilailla IgG lisääntyi 26 %:lla ja IgM noin 3 %:lla. Yleistynyttä myasthenia gravista sairastavilla potilailla tehdyn tutkimuksen 6 kuukauden satunnaistetun, kontrolloidun jakson lopussa immunoglobuliinien kokonaispitoisuus oli pienentynyt lähtötasosta 13,3 % inebilitsumabilla hoidetuilla potilailla verrattuna pitoisuuden kasvuun 14,5 %:lla lumelääkettä saaneilla potilailla. Immunoglobuliini G (IgG) väheni lähtötasosta keskimäärin 8,4 % ja immunoglobuliini M (IgM) keskimäärin 30 % inebilitsumabilla hoidetuilla potilailla, kun lumelääkettä saaneilla potilailla IgG lisääntyi 17,8 %:lla ja IgM noin 4,9 %:lla.
Vähentyneet neutrofiilimäärät
NMOSD-potilailla tehdyn tutkimuksen kuuden ja puolen kuukauden hoidon jälkeen neutrofiilimäärät 1,0–1,5 × 109/l (2. aste) havaittiin 7,5 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 1,8 %:iin lumelääkettä saaneista potilaista. Neutrofiilimäärät 0,5–1,0 × 109/l (3. aste) havaittiin 1,7 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 0 %:iin lumelääkettä saaneista potilaista. IgG4‑tautia sairastavilla potilailla tehdyn tutkimuksen 12 kuukauden satunnaistetun, kontrolloidun jakson aikana neutrofiilimäärät 1,0–1,5 × 109/l havaittiin 7,5 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 3 %:iin lumelääkettä saaneista potilaista. Neutrofiilimäärät 0,5–1,0 × 109/l havaittiin 0 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 1,5 %:iin lumelääkettä saaneista potilaista. Neutropenia oli yleensä ohimenevä eikä se ollut yhteydessä vakaviin infektioihin.
Vähentyneet lymfosyyttimäärät
NMOSD-potilailla tehdyn tutkimuksen kuuden ja puolen kuukauden hoidon aikana inebilitsumabilla hoidetuilla potilailla havaittiin yleisemmin lymfosyyttimäärien vähenemistä kuin lumelääkettä saaneilla potilailla: lymfosyyttimäärät 500 ‑ < 800/mm3 (2. aste) havaittiin 21,4 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 12,5 %:iin lumelääkettä saaneista potilaista. Lymfosyyttimäärät 200 ‑ < 500/mm3 (3. aste) havaittiin 2,9 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 1,8 %:iin lumelääkettä saaneista potilaista. IgG4‑tautia sairastavilla potilailla tehdyn tutkimuksen 12 kuukauden satunnaistetun, kontrolloidun jakson aikana inebilitsumabilla hoidetuilla potilailla havaittiin yleisemmin lymfosyyttimäärien vähenemistä kuin lumelääkettä saaneilla potilailla: Lymfosyyttimäärät 500 ‑ < 800/mm3 (2. aste) havaittiin 26,9 %:lla sekä inebilitsumabilla hoidetuista potilaista että lumelääkettä saaneista potilaista. Lymfosyyttimäärät 200 ‑ < 500/mm3 (3. aste) havaittiin 10,4 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 3,0 %:iin lumelääkettä saaneista potilaista. Yleistynyttä myasthenia gravista sairastavilla potilailla tehdyn tutkimuksen 6 kuukauden satunnaistetun, kontrolloidun jakson lopussa havaittiin lymfosyyttimäärien vähenemistä 35 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 30,8 %:iin lumelääkettä saaneista potilaista. Lymfosyyttimäärät 500 ‑ < 800/mm3 (2. aste) havaittiin 21,4 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 12,8 %:iin lumelääkettä saaneista potilaista. Lymfosyyttimäärät 200 ‑ < 500/mm3 (3. aste) havaittiin 10,3 %:lla inebilitsumabilla hoidetuista potilaista verrattuna 7,7 %:iin lumelääkettä saaneista potilaista. Tämä tulos on B-soluja vähentävän vaikutusmekanismin mukainen, koska B-solut ovat lymfosyyttipopulaation alaryhmä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta: www.fimea.fi.
Yliannostus
Suurin autoimmuunipotilailla testattu inebilitsumabiannos oli 1 200 mg, joka annettiin kahtena 600 mg:n infuusiona laskimoon kahden viikon välein. Haittavaikutukset olivat samanlaisia kuin inebilitsumabin pivotaalitutkimuksessa havaitut vaikutukset.
Yliannostustapauksessa ei ole käytettävissä erityistä vasta-ainetta; infuusio pitää keskeyttää välittömästi ja potilasta tarkkailla infuusioon liittyvien reaktioiden varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Potilaita on tarkkailtava huolellisesti haittavaikutusten merkkien tai oireiden varalta ja tarpeen mukaan potilaalle on annettava asianmukaista elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunosuppressantit, monoklonaaliset vasta-aineet, ATC-koodi: L04AG10
Vaikutusmekanismi
Inebilitsumabi on monoklonaalinen vasta-aine, joka sitoutuu CD19‑molekyyliin, solukalvon antigeeniin, jota esiintyy pre‑B- ja kypsissä B‑solulymfosyyteissä, mukaan lukien plasmablastit ja jotkin plasmasolut. Sitouduttuaan B-lymfosyyttien solukalvoon inebilitsumabi tukee vasta-ainevälitteistä solutuhoa (ADCC) ja vasta-ainevälitteistä fagosytoosia (ADCP). B-solujen uskotaan olevan keskeisessä asemassa NMOSD:n, IgG4‑taudin ja yleistyneen myasthenia graviksen patogeneesissä. Inebilitsumabin vaikutusmekanismin näiden tautien hoidossa oletetaan liittyvän B‑solujen vähentämiseen ja se saattaa käsittää vasta-aine-erityksen, antigeenien esittelyn, B‑ ja T‑solujen välisen interaktion sekä tulehduksellisten välittäjien tuottamisen heikentämisen.
Farmakodynaamiset vaikutukset
Inebilitsumabin farmakodynamiikkaa arvioitiin CD20+ B-solukokeella, koska inebilitsumabi voi häiritä CD19+ B‑solukoetta. Inebilitsumabihoito vähentää CD20+ B‑solujen määrää veressä kahdeksassa päivässä infuusion jälkeen. Kliinisessä tutkimuksessa, johon osallistui 174 NMOSD-potilasta, CD20+ B‑solujen määrä väheni viitealueen alarajan alapuolelle neljässä viikossa 100 %:lla inebilitsumabilla hoidetuista potilaista ja pysyi viitealueen alarajan alapuolella 94 %:lla potilaista 28 viikkoa hoidon aloittamisen jälkeen. Kliinisessä tutkimuksessa, johon osallistui 68 IgG4‑tautia sairastavaa potilasta, CD20+ B‑solujen määrä väheni viitealueen alarajan alapuolelle viikkoon 2 mennessä 100 %:lla inebilitsumabilla hoidetuista potilaista ja pysyi viitealueen alarajan alapuolella 82 %:lla potilaista viikon 26 kohdalla ja 79 %:lla viikon 52 kohdalla, kun hoitoväli oli 6 kuukautta. Yleistynyttä myasthenia gravista sairastavilla potilailla tehdyssä tutkimuksessa CD20+ B‑solujen määrä väheni viitealueen alarajan alapuolelle viikkoon 4 mennessä koko tutkimusryhmässä ≥ 99 %:lla inebilitsumabilla hoidetuista potilaista (AChR‑ ja MuSK‑vasta-ainepositiiviset). B‑solujen määrät pysyivät viitealueen alarajan alapuolella ≥ 96 %:lla koko tutkimusryhmän potilaista 6 kuukauden satunnaistetun, kontrolloidun tutkimusjakson (RCP) lopussa. Yhden vuoden inebilitsumabihoidon jälkeen B‑solujen määrät olivat viitealueen alarajan alapuolella 92 %:lla AChR‑vasta-ainepositiivisista yleistynyttä myasthenia gravista sairastavista potilaista. B-solujen palautumiseen kuluvaa aikaa inebilitsumabin antamisen jälkeen ei tunneta.
ADA (lääkevasta-aineet) -positiivisella tilalla ei näyttänyt olevan kliinisesti merkittävää vaikutusta PK- ja PD (B-solu) -parametreihin, eikä se vaikuttanut pitkäaikaiseen turvallisuusprofiiliin. ADA-tilalla ei ollut ilmeistä vaikutusta tehoon. Tätä vaikutusta ei voida kuitenkaan arvioida täysin inebilitsumabihoitoon liittyvien lääkevasta-aineiden vähäisen ilmaantumisen tähden.
Kliininen teho ja turvallisuus
Neuromyelitis optica -kirjon häiriö (NMOSD)
Inebilitsumabin tehokkuutta NMOSD-taudin hoidossa tutkittiin satunnaistetussa (3:1), kaksoissokkoutetussa, lumelääkekontrolloidussa kliinisessä tutkimuksessa aikuisilla, joilla oli AQP4-IgG-seropositiivinen tai AQP4-IgG-seronegatiivinen NMOSD. Tutkimukseen osallistui potilaita, joilla oli ollut edeltävänä vuonna vähintään yksi akuutti NMOSD-pahenemisvaihe tai kahtena edeltävänä vuonna vähintään kaksi pahenemisvaihetta, joihin tarvittiin varahoitoa (esim. steroideja, plasmanvaihto, immunoglobuliini laskimoon), ja joilla EDSS-asteikon (toimintakyvyn arviointiasteikko, Expanded Disability Severity Scale) pistemäärä oli ≤ 7,5 (potilaat, joiden pistemäärä oli 8,0, saivat osallistua tutkimukseen, jos he pystyivät kohtuudella osallistumaan siihen). Potilaat suljettiin pois, jos heitä oli aiemmin hoidettu immuunisalpaajahoidoilla kullekin hoidolle määritetyn aikavälin sisällä. Peruslääkitystä immunosuppressanttihoidoilla NMOSD-pahenemisvaiheiden estämiseksi ei sallittu. Potilaalle annettiin kaksi viikkoa kestävä, suun kautta otettava kortikosteroidilääkitys (ja yhden viikon asteittain vähenevä lääkitys) inebilitsumabihoidon alussa pivotaalitutkimuksessa.
Potilaita hoidettiin 300 mg:n inebilitsumabi-infuusioilla laskimoon päivänä 1 ja päivänä 15 tai vastaavalla lumelääkkeellä, minkä jälkeen heitä seurattiin 197 vuorokauden ajan tai määritettyyn pahenemisvaiheeseen asti, mistä käytettiin nimitystä satunnaistettu, kontrolloitu jakso (RCP). Sokkoutettu ja riippumaton asiantuntijakomitea arvioi kaikki mahdolliset pahenemisvaiheet ja määritti, täyttikö pahenemisvaihe tutkimussuunnitelmassa määritetyt kriteerit. Pahenemisvaiheen kriteereissä otettiin huomioon kaikki alueet, joihin NMOSD vaikuttaa (näköhermotulehdus, myeliitti, aivot ja aivorunko), ja ne sisälsivät kriteereitä, jotka perustuivat vain merkittäviin kliinisiin merkkeihin, sekä kriteereitä, jotka täydensivät vähäisempiä kliinisiä merkkejä magneettikuvauksella (ks. taulukko 3).
Taulukko 3. Yleiskuva tutkimussuunnitelmassa määritetyistä NMOSD-pahenemisvaiheen kriteereistä
Alue | Tyypilliset oireet | Vain kliiniset tulokset | Kliiniset JA radiologiset tulokset |
Näköhermo | Näön sumeneminen Näön heikkeneminen Silmäkipu | Kahdeksan kriteeriä, jotka perustuvat näöntarkkuuden muutoksiin tai relatiiviseen afferenttiin mustuaisdefektiin (RAPD) | Kolme kriteeriä, jotka perustuvat näöntarkkuuden muutoksiin tai relatiiviseen afferenttiin mustuaisdefektiin (RAPD) ja vastaavan näköhermon magneettikuvaustuloksiin |
Selkäydin | Syvä tai hermojuurikipu Raajan parestesia Heikkous Sulkijalihaksen toimintahäiriö Lhermitten oire (ei yksinään) | Kaksi kriteeriä, jotka perustuvat pyramidiradan, rakon/suoliston tai aistien toiminnan pistemääriin | Kaksi kriteeriä, jotka perustuvat pyramidiradan, rakon/suoliston tai aistien toiminnan pistemääriin JA vastaaviin selkäytimen magneettikuvaustuloksiin |
Aivorunko | Pahoinvointi Vaikea oksentelu Vaikea hikka Muut neurologiset merkit (esim. kahtena näkeminen, dysartria, dysfagia, huimaus, okulomotorinen halvaus, heikkous, silmävärve, muu kraniaalihermon poikkeavuus) | Ei mitään | Kaksi kriteeriä, jotka perustuvat aivorungon/pikkuaivojen oireiden tai toiminnan muutosten pistemääriin JA vastaaviin aivorungon magneettikuvaustuloksiin |
Aivot | Enkefalopatia Hypotalamuksen toimintahäiriö | Ei mitään | Yksi kriteeri, joka perustuu aivojen/aistien/pyramidiradan toiminnan pistemääriin JA vastaaviin aivojen magneettikuvaustuloksiin |
Potilaat, joilla oli asiantuntijakomitean määrittämä pahenemisvaihe satunnaistetulla, kontrolloidulla jaksolla (RCP) tai joilla ei ollut pahenemisvaihetta päivän 197 käyntiin mennessä, poistuivat RCP-jaksosta ja heille annettiin mahdollisuus rekisteröityä OLP-jatkotutkimukseen ja aloittaa inebilitsumabihoito tai jatkaa sitä.
Tutkimukseen rekisteröitiin kaikkiaan 230 potilasta: 213 potilasta oli AQP4‑IgG-seropositiivisia potilaita ja 17 oli seronegatiivisia potilaita; 174 potilasta hoidettiin inebilitsumabilla ja 56 potilasta sai lumelääkettä tutkimuksen RCP-jaksolla. Näistä 213:sta AQP4-IgG-seropositiivisesta potilaasta 161:tä hoidettiin inebilitsumabilla ja 52 potilasta sai lumelääkettä tutkimuksen RCP-jaksolla. Lähtötilanne- ja tehotulokset esitetään AQP4‑IgG-seropositiivisille potilaille.
Lähtötilanteen demografiset ja tautiin liittyvät ominaisuudet tasapainotettiin kahden hoitoryhmän välillä (ks. taulukko 4).
Taulukko 4. AQP4-IgG-seropositiivisten NMOSD-potilaiden demografiset ja lähtötilanneominaisuudet
Ominaisuus | Lumelääke N = 52 | Inebilitsumabi N = 161 | Yhteensä N = 213 |
Ikä (vuotta): keskiarvo (keskihajonta [SD]) | 42,4 (14,3) | 43,2 (11,6) | 43,0 (12,3) |
Ikä ≥ 65 vuotta, n (%) | 4 (7,7) | 6 (3,7) | 10 (4,7) |
Sukupuoli: Mies, n (%) | 3 (5,8) | 10 (6,2) | 13 (6,1) |
Sukupuoli: Nainen, n (%) | 49 (94,2) | 151 (93,8) | 200 (93,9) |
EDSS-asteikko (toimintakyvyn arviointiasteikko): keskiarvo (SD) | 4,35 (1,63) | 3,81 (1,77) | 3,94 (1,75) |
Taudin kesto (vuotta): keskiarvo (SD) | 2,92 (3,54) | 2,49 (3,39) | 2,59 (3,42) |
Aiempien relapsien lukumäärä: ≥ 2, n (%) | 39 (75,0) | 137 (85,1) | 176 (82,6) |
Vuotuinen relapsimäärä: keskiarvo (SD) | 1,456 (1,360) | 1,682 (1,490) | 1,627 (1,459) |
Varahoito aloitettiin tarpeen mukaan NMOSD-pahenemisvaiheissa. Kaikki potilaat saivat esilääkitystä ennen tutkimuslääkkeen antamista infuusioon liittyvien reaktioiden riskin vähentämiseksi.
Ensisijainen tehon päätetapahtuma oli aika (päivinä) päivästä 1 asiantuntijakomitean määrittämän NMOSD-pahenemisvaiheen alkuun päivänä 197 tai sitä ennen. Tärkeitä toissijaisia lisäpäätetapahtumia olivat EDSS-pistemäärän huononeminen lähtötilanteesta RCP-jakson viimeisellä käynnillä, muutos lähtötilanteesta alhaisen kontrastin silmien yhteisnäöntarkkuuden pistemäärässä, joka mitattiin alhaisen kontrastin Landolt C Broken Ring -menetelmällä RCP-jakson viimeisellä käynnillä, sekä NMOSD-tautiin liittyvien sairaalajaksojen lukumäärä. Potilaan EDSS-pistemäärän katsottiin huonontuneen, jos jokin seuraavista kriteereistä täyttyi: (1) Kahden tai useamman pisteen huononeminen EDSS-pistemäärässä potilailla, joiden lähtötilanteen pistemäärä oli 0; (2) yhden tai useamman pisteen huononeminen EDSS-pistemäärässä potilailla, joiden lähtötilanteen pistemäärä oli 1–5; (3) 0,5:n tai useamman pisteen huononeminen EDSS-pistemäärässä potilailla, joiden lähtötilanteen pistemäärä oli vähintään 5,5. Vaikka vertailuryhmää ei ollut käytettävissä avoimen tutkimusjakson aikana, vuotuinen pahenemisvaiheiden määrä koko satunnaistetun, kontrolloidun jakson ja avoimen jakson aikana määritettiin.
AQP4‑IgG-seropositiivisten potilaiden tulokset esitetään taulukossa 5 ja kuvassa 1. Tässä tutkimuksessa inebilitsumabihoito vähensi tilastollisesti merkitsevästi asiantuntijakomitean määrittämän NMOSD-pahenemisvaiheen riskiä verrattuna lumelääkehoitoon (riskisuhde: 0,227, p < 0,0001; 77,3 %:n väheneminen asiantuntijakomitean määrittämän NMOSD-pahenemisvaiheen riskissä) AQP4‑IgG-seropositiivisilla potilailla. AQP4-IgG-seronegatiivisilla potilailla ei havaittu hoidosta saatua hyötyä.
Inebilitsumabiryhmässä EDSS-pistemäärä huononi merkittävästi harvemmin kuin lumelääkeryhmässä (14,9 % vs. 34,6 % tutkittavista). Tutkimusryhmien välillä ei havaittu eroja alhaisen kontrastin silmien yhteisnäöntarkkuuden pistemäärässä). Magneettikuvauksessa havaittujen aktiivisten leesioiden keskimääräinen yhteislukumäärä (1,7 vs. 2,3) ja keskimääräinen NMOSD-tautiin liittyvien sairaalassaolojen yhteislukumäärä (1,0 vs. 1,4) olivat pienemmät tutkimuksen inebilitsumabiryhmässä.
Taulukko 5. Keskeisen tutkimuksen tehotulokset AQP4-IgG-seropositiivisessa NMOSD-taudissa
| Hoitoryhmä | |
Lumelääke N = 52 | Inebilitsumabi N = 161 | |
Aika asiantuntijakomitean määrittämään pahenemisvaiheeseen (ensisijainen tehon päätetapahtuma) | ||
Pahenemisvaiheen saaneiden potilaiden määrä (%) | 22 (42,3 %) | 18 (11,2 %) |
Riskisuhde (95 %:n luottamusväli)a | 0,227 (0,1214, 0,4232) | |
p-arvoa | < 0,0001 | |
a Coxin regressiomenetelmä, kun lumelääke on viiteryhmänä. | ||
Kuva 1. Kaplan-Meier-kuvaaja ajasta ensimmäiseen asiantuntijakomitean määrittämään NMOSD-pahenemisvaiheeseen RCP-jakson aikana AQP4-IgG-seropositiivisilla potilailla
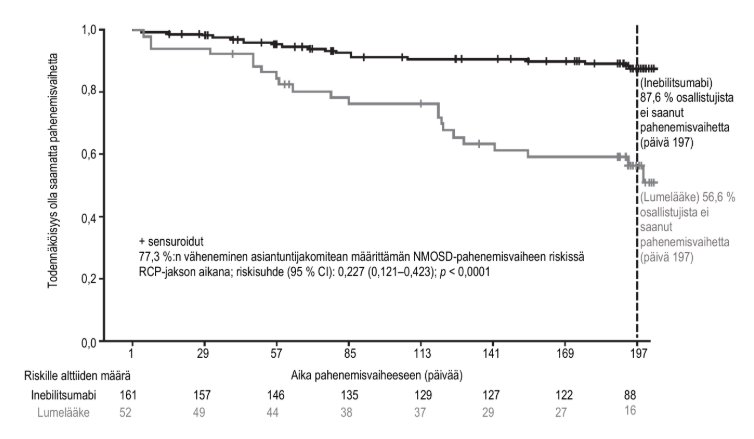
AQP4-IgG = akvaporiini‑4 immunoglobuliini G -vasta-aine; CI = luottamusväli; NMOSD = neuromyelitis optica -kirjon häiriöt; RCP = satunnaistettu, kontrolloitu jakso.
Koko RCP- ja OLP-jakson aikana vuotuinen asiantuntijakomitean määrittämien NMOSD-pahenemisvaiheiden määrä analysoitiin toissijaisena päätetapahtumana, ja inebilitsumabilla hoidettujen AQP4‑IgG-seropositiivisten potilaiden tulos oli 0,09.
Immunoglobuliini G4:ään liittyvä sairaus (IgG4‑tauti)
Inebilitsumabin tehoa IgG4‑taudin hoidossa tutkittiin satunnaistetussa (1:1), kaksoissokkoutetussa, 52 viikon lumelääkekontrolloidussa kliinisessä monikeskustutkimuksessa, johon otettiin mukaan 135 aikuispotilasta, joilla oli aktiivinen IgG4‑tauti. Potilailla oli aktiivinen tauti, joka oli määritetty kliinisten tai kuvantamis-, laboratorio- tai biopsialöydösten perusteella ja joka lääkärin arvion mukaan tarvitsi hoitoa. Tutkimukseen soveltuvilla potilailla oli äskettäin diagnosoitu tai uusiutunut IgG4-tauti, jota hoidettiin seulontavaiheessa glukokortikoideilla. Tauti oli jossain vaiheessa esiintynyt jossain ennalta määritellyssä elimessä ja täytti 2019 ACR/EULAR ‑luokituskriteerit.
Tutkija arvioi kaikki tutkimuksen aikana tapahtuneet mahdolliset taudin pahenemiset (flare) ja tämän jälkeen ne arvioi sokkoutettu ja riippumaton asiantuntijakomitea, joka määritti, täyttikö taudin paheneminen yhden tai useamman tutkimussuunnitelmassa määritetyistä, elinkohtaisista taudin pahenemisen diagnostisista kriteereistä. Taudin pahenemiseksi katsottiin uudet/pahenevat oireet tai löydökset, jotka asiantuntijakomitea vahvisti ja jotka tutkijan arvion mukaan vaativat hoitoa. Edellytyksenä oli vaihtoehtoisten diagnoosien puuttuminen.
Potilaat saivat 300 mg inebilitsumabia tai lumelääkettä laskimoon satunnaistetun, kontrolloidun jakson päivänä 1, päivänä 15 ja päivänä 183. Potilaat saivat yhtenäistä glukokortikoidiannosta (vastasi 20 mg prednisonia vuorokaudessa) satunnaistamishetkellä. Tämän jälkeen vuorokausiannosta vähennettiin ennalta määritellysti 5 mg 2 viikon välein, kunnes lääkitys lopetettiin 8 viikon kuluttua. Tutkimuksenaikainen glukokortikoidien käyttö sallittiin IgG4‑taudin pahenemisen hoitamiseen ja muihin tarkoituksiin, kuten tutkimushoidon esilääkityksenä, suun kautta otettavana glukokortikoidihoitona enintään 2 viikon ajan tai lisämunuaisen vajaatoiminnan hoitoon enintään 2,5 mg prednisonia tai vastaavaa vuorokaudessa. Biologisten ja muiden kuin biologisten immuunisalpaajien samanaikainen käyttö tutkimuksen aikana oli kielletty. Potilaille, jotka olivat osallistuneet tutkimukseen satunnaistetun, kontrolloidun jakson loppuun saakka, tarjottiin mahdollisuutta osallistua OLP-jatkotutkimukseen ja aloittaa inebilitsumabihoito tai jatkaa sitä.
Tutkimusta varten seulottiin 227 potilasta. Tutkimukseen otettiin mukaan 135 IgG4‑tautia sairastavaa potilasta, joista 68 satunnaistettiin saamaan inebilitsumabia ja 67 lumelääkettä. IgG4‑tautia sairastavien potilaiden lähtötilanteen demografiset ja tautiin liittyvät ominaisuudet satunnaistetun, kontrolloidun jakson aikana olivat tasapainossa hoitoryhmien välillä (ks. taulukko 6). Vaikka vertailuryhmää ei ollut käytettävissä avoimen tutkimusjakson aikana, avoimen tutkimusjakson aikana hoidetut ja asiantuntijakomitean määrittämät taudin pahenemiset määritettiin.
Taulukko 6. IgG4‑tautia sairastavien potilaiden demografiset ja muut ominaisuudet lähtötilanteessa
Ominaisuus | Lumelääke N = 67 | Inebilitsumabi N = 68 | Yhteensä N = 135 |
Ikä (vuotta): keskiarvo (keskihajonta [SD]) | 58,2 (12,2) | 58,2 (11,5) | 58,2 (11,8) |
Ikä ≥ 65 vuotta, n (%) | 21 (31,3 %) | 21 (30,9 %) | 42 (31,1 %) |
Sukupuoli: Mies, n (%) | 49 (73,1 %) | 39 (57,4 %) | 88 (65,2 %) |
Taudin kesto (vuotta): keskiarvo (SD) | 2,54 (3,06) | 2,64 (3,73) | 2,59 (3,40) |
IgG4‑taudin manifestaatio Diagnosoitu äskettäin | 31 (46,3 %) | 31 (45,6 %) | 62 (45,9 %) |
ACR/EULAR-luokituspisteet Keskiarvo (SD) | 38,3 (11,7) | 40,1 (12,1) | 39,2 (11,9) |
Aiempi IgG4‑taudin muu kuin glukokortikoidihoito Kyllä | 20 (29,9 %) | 17 (25,0 %) | 37 (27,4 %) |
IgG4‑taudin RI (Responder Index) -pisteet lähtötilanteessa Keskiarvo (SD) | 6,0 (4,0) | 5,4 (4,0) | 5,7 (4,0) |
IgG4‑tautia sairastavien potilaiden tulokset esitetään kuvassa 2 ja taulukossa 7.
Tutkimuksessa saavutettiin ensisijainen tehon päätetapahtuma eli aika ensimmäiseen hoidettuun ja asiantuntijakomitean määrittämään IgG4‑taudin pahenemiseen (flare), joka oli inebilitsumabiryhmässä pidempi kuin lumelääkeryhmässä (riskisuhde (HR): 0,13; p < 0,0001; ks. kuva 2). Myös tärkeät toissijaiset päätetapahtumat saavutettiin tilastollisesti merkitsevästi (ks. taulukko 7).
Kuva 2. Ensisijainen päätetapahtuma – Kaplan-Meier-kuvaaja ajasta ensimmäiseen hoidettuun ja asiantuntijakomitean määrittämään IgG4‑taudin pahenemiseen (flare) satunnaistetun, kontrolloidun jakson aikana
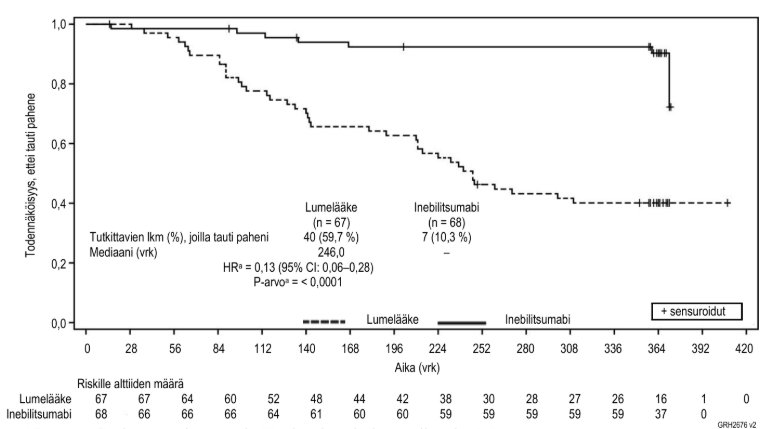
a Perustuu Coxin regressiomenetelmään, kun lumelääke on viiteryhmänä.
Potilaat, jotka eivät osallistuneet tutkimukseen satunnaistetun, kontrolloidun jakson loppuun saakka ja joilla ei esiintynyt hoidettua ja asiantuntijakomitean määrittämää taudin pahenemista (flare) satunnaistetun, kontrolloidun jakson aikana, sensuroitiin, kun hoito lopetettiin.
Taulukko 7. IgG4‑tautia sairastavien potilaiden tärkeät toissijaiset tehotulokset
| Hoitoryhmä | |
Inebilitsumabi N = 68 | Lumelääke N = 67 | |
Hoidettujen ja asiantuntijakomitean määrittämien IgG4‑taudin pahenemisten vuotuinen määrä | 0,10 | 0,71 |
Määrien suhde (95 %:n luottamusväli)a | 0,14 (0,06–0,31) | |
p‑arvoa | < 0,0001 | |
Täydellisen remission, jossa tauti ei vaadi hoitoa eikä ole pahentunut, viikolla 52 saavuttaneiden tutkittavien osuusb | 39 (57,4 %) | 15 (22,4 %) |
Ristitulosuhde (95 %:n luottamusväli)c | 4,68 (2,21–9,91) | |
p‑arvoc | < 0,0001 | |
Täydellisen remission, jossa tauti ei vaadi kortikosteroidihoitoa eikä ole pahentunut, viikolla 52 saavuttaneiden tutkittavien osuusd | 40 (58,8 %) | 15 (22,4 %) |
Ristitulosuhde (95 %:n luottamusväli)c | 4,96 (2,34–10,52) | |
p‑arvoc | < 0,0001 | |
a Arvioitu negatiivisen binomiaalisen regression perusteella, kun lumelääke on viiteryhmänä. b Määritelty ilmeisen taudin aktiivisuuden puuttumisena (IgG4‑taudin RI = 0 tai tutkijan päätös) viikolla 52, ei asiantuntijakomitean määrittämää taudin pahenemista (flare) RCP-jakson aikana eikä muuta taudin pahenemisen tai itse taudin hoitoa kuin vaadittu 8 viikon asteittain vähenevä glukokortikoidilääkitys. c Perustuu logistiseen regressiomalliin, kun lumelääke on viiteryhmänä. d Määritelty ilmeisen taudin aktiivisuuden puuttumisena (IgG4‑taudin RI = 0 tai tutkijan päätös) viikolla 52, ei asiantuntijakomitean määrittämää taudin pahenemista (flare) RCP-jakson aikana eikä muuta taudin pahenemisen tai itse taudin kortikosteroidihoitoa kuin vaadittu 8 viikon asteittain vähenevä glukokortikoidilääkitys. | ||
IgG4‑taudin hoitoon käytetty glukokortikoidien keskimääräinen (SD) potilaskohtainen kokonaismäärä oli inebilitsumabiryhmässä pienempi kuin lumelääkeryhmässä: inebilitsumabiryhmässä käyttö prednisoniekvivalenttina oli keskimäärin (SD) 118,25 (438,97) mg ja lumelääkeryhmässä 1 384,53 (1 723,26) mg satunnaistetun, kontrolloidun jakson aikana. Glukokortikoidien keskimääräinen (SD) päivittäin käytetty määrä satunnaistetun, kontrolloidun jakson aikana oli glukokortikoideja käyttävää potilasta kohden inebilitsumabiryhmässä 3,34 (2,09) mg prednisoniekvivalenttina ja lumelääkeryhmässä 5,97 (4,20) mg prednisoniekvivalenttina. Glukokortikoidien keskimääräinen (SD) käytetty kokonaismäärä satunnaistetun, kontrolloidun jakson aikana oli glukokortikoideja käyttävää potilasta kohden inebilitsumabiryhmässä 1 148,71 (877,92) mg prednisoniekvivalenttina ja lumelääkeryhmässä 2 208,65 (1 707,56) mg prednisoniekvivalenttina.
Avoimesta jatkotutkimusjaksosta, jossa potilaat jatkoivat inebilitsumabihoitoa, saadut tiedot tukevat inebilitsumabin pitkäaikaista hoitovaikutusta.
Yleistynyt myasthenia gravis (MG)
Inebilitsumabin tehoa yleistyneen myasthenia graviksen hoidossa tutkittiin satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa kliinisessä monikeskustutkimuksessa. Satunnaistettu, kontrolloitu jakso oli 52 viikkoa tutkimusryhmälle, jotka olivat AChR‑vasta-ainepositiivisia ja jotka saivat 300 mg inebilitsumabia tai lumelääkettä laskimoon päivänä 1, päivänä 15 ja päivänä 183. Satunnaistettu, kontrolloitu jakso oli 26 viikkoa tutkimusryhmälle, jotka olivat MuSK‑vasta-ainepositiivisia ja jotka saivat 300 mg inebilitsumabia tai lumelääkettä laskimoon päivänä 1 ja päivänä 15. Ensisijainen analyysi tehtiin viikon 26 jälkeen molemmille tutkimusryhmille.
Potilaat täyttivät seuraavat kelpoisuuskriteerit:
-
AChR‑ tai MuSK‑autovasta-aineita todettu
-
MGFA‑luokka II–IV (kliininen Myasthenia Gravis Foundation of America -luokitus)
-
MG‑ADL (Myasthenia Gravis-Activities of Daily Living) ‑asteikon pistemäärä 6–10, josta > 50 % tuli asteikon muista kuin silmiin liittyvistä kohdista, tai MG‑ADL-pistemäärä ≥ 11
-
Kvantitatiivinen myasthenia gravis (QMG) -pistemäärä ≥ 11
-
Satunnaistamista edeltänyt hoito vakaalla kortikosteroidiannoksella tai jollakin määritellyllä ei-steroidaalisella immuunisalpaajalla (NSIST) tai näiden yhdistelmällä.
Kortikosteroidin vakaa annos (> 5 mg/vrk prednisonia tai vastaavaa) vähennettiin asteittain 5 mg:aan/vrk (prednisonia tai vastaavaa) viikosta 4 viikkoon 24. Varahoitoon sisältyi IVIg ja plasmanvaihto.
Inebilitsumabia annettiin suositusannostuksen mukaisesti (ks. kohta Annostus ja antotapa).
Tutkimukseen otetut 238 yleistynyttä myasthenia gravista sairastavaa potilasta satunnaistettiin suhteessa 1:1 seuraavasti: inebilitsumabihoito 95 potilaalle, jotka olivat AChR‑vasta-ainepositiivisia ja 24 potilaalle, jotka olivat MuSK‑vasta-ainepositiivisia ja lumelääke 95 potilaalle, jotka olivat AChR‑vasta-ainepositiivisia ja 24 potilaalle, jotka olivat MuSK‑vasta-ainepositiivisia.
Yleistynyttä myasthenia gravista sairastavien potilaiden lähtötilanteen demografiset ja tautiin liittyvät ominaisuudet satunnaistetun, kontrolloidun jakson aikana olivat tasapainossa hoitoryhmien välillä (ks. taulukko 8).
Taulukko 8. Yleistynyttä myasthenia gravista sairastavien potilaiden demografiset ja muut ominaisuudet lähtötilanteessa koko tutkimusryhmässä
Ominaisuus | Lumelääke N = 117 | Inebilitsumabi N = 119 | Yhteensä N = 236 |
Ikä (vuotta): keskiarvo (keskihajonta [SD]) | 47,9 (15,0) | 47,1 (15,7) | 47,5 (15,3) |
Ikä ≥ 65 vuotta, n (%) | 16 (13,7) | 22 (18,5) | 38 (16,1) |
Sukupuoli: Mies, n (%) | 52 (44,4) | 40 (33,6) | 92 (39,0) |
Sukupuoli: Nainen, n (%) | 65 (55,6) | 79 (66,4) | 144 (61,0) |
Rotu, (%) |
|
|
|
Aasialainen | 46,2 | 38,7 | 42,4 |
Mustaihoinen tai afroamerikkalainen | 2,6 | 1,7 | 2,1 |
Valkoihoinen | 47,9 | 58 | 53 |
Taudin kesto (vuotta): keskiarvo (SD) | 6,73 (7,28) | 5,94 (6,96) | 6,34 (7,12) |
Lähtötilanteen MG‑ADL-pistemäärä: keskiarvo (SD) |
|
|
|
Koko tutkimusryhmä | 9,1 (2,8) | 9,0 (2,8) | 9,1 (2,8) |
AChR+‑ryhmä | 9,3 (2,8) | 9,1 (2,7) | 9,2 (2,7) |
MuSK+‑ryhmä | 8,3 (2,5) | 8,8 (3,1) | 8,5 (2,8) |
Lähtötilanteen QMG‑pistemäärä: keskiarvo (SD) |
|
|
|
Koko tutkimusryhmä | 17,3 (4,2) | 16,7 (4,2) | 17,0 (4,2) |
AChR+‑ryhmä | 17,4 (4,3) | 16,9 (4,1) | 17,1 (4,2) |
MuSK+‑ryhmä | 17,2 (3,9) | 16,1 (4,8) | 16,7 (4,3) |
Lähtötilanteessa vakaata immuunisalpaajahoitoa saaneet potilaat: n (%) |
|
|
|
Vain kortikosteroidi | 68 (58,1) | 82 (68,9) | 150 (63,6) |
Vain NSIST | 9 (7,7) | 8 (6,7) | 17 (7,2) |
Kortikosteroidi ja 1 NSIST | 39 (33,3) | 29 (24,4) | 68 (28,8) |
Asetyylikoliiniesteraasin estäjä: n (%) | 93 (79,5) | 94 (79,0) | 187 (79,2) |
Ensisijainen päätetapahtuma ja tärkeät toissijaiset päätetapahtumat on esitetty taulukossa 9 ja kuvissa 3–5.
Inebilitsumabin tehoa mitattiin MG‑ADL-asteikolla. Siinä yleistyneen myasthenia graviksen vaikutusta arvioidaan 8‑kohtaisella kyselylomakkeella MG‑potilaalle merkityksellisten oireiden ja päivittäisiin toimintoihin liittyvän toimintakyvyn suhteen. Kutakin kohtaa arvioidaan 4‑portaisella asteikolla, jossa pistemäärä 0 tarkoittaa normaalia toimintaa ja pistemäärä 3 taudin vaikutusta vaikeimmillaan. MG‑ADL-kokonaispistemäärän vaihteluväli on 0–24; suurempi pistemäärä viittaa suurempaan toiminnanvajaukseen.
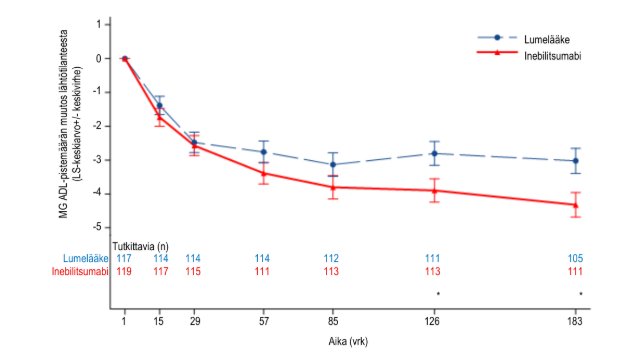
Ensisijainen tehon päätetapahtuma oli MG‑ADL-pistemäärän muutos lähtötilanteesta viikolla 26 koko tutkimusryhmässä. MG‑ADL-kokonaispistemäärän keskimääräisessä muutoksessa lähtötilanteesta havaittiin inebilitsumabia suosiva tilastollisesti merkitsevä ero (−4,3 inebilitsumabilla vs. −3,0 lumelääkkeellä, ero −1,3, 95 %:n luottamusväli: −2,2 – −0,4; p‑arvo: 0,0067). Niiden potilaiden osuus, jotka saivat varahoitoa viikkoon 26 mennessä, oli pienempi inebilitsumabiryhmässä lumelääkeryhmään verrattuna (8,4 % inebilitsumabilla vs. 23,9 % lumelääkkeellä).
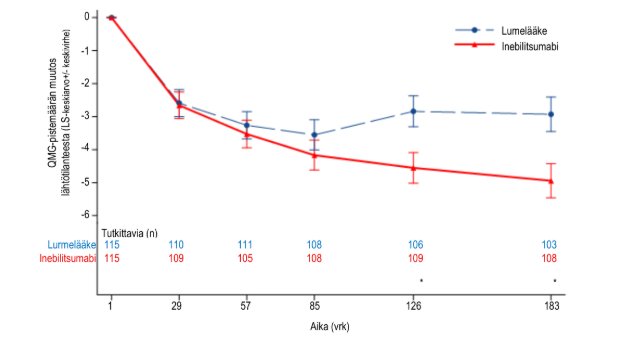
Tärkeä toissijainen päätetapahtuma oli QMG‑pistemäärän muutos lähtötilanteesta viikolla 26 koko tutkimusryhmässä. QMG‑pistemäärä saadaan 13 kohdan luokitusjärjestelmästä, joka arvioi taudin aiheuttamaa haittaa kvantitatiivisesti pääasiassa lihasheikkouden suhteen. Kutakin kohtaa arvioidaan 4‑portaisella asteikolla, jossa pistemäärä 0 tarkoittaa, ettei tauti aiheuta haittaa ja pistemäärä 3 tarkoittaa vaikea-asteista haittaa. Mahdollisen kokonaispistemäärän vaihteluväli on 0–39; suurempi pistemäärä viittaa taudin vaikea-asteisempaan pahenemiseen. QMG‑kokonaispistemäärän keskimääräisessä muutoksessa lähtötilanteesta havaittiin inebilitsumabia suosiva tilastollisesti merkitsevä ero (−4,9 inebilitsumabilla vs. −2,9 lumelääkkeellä, ero −2,0, 95 %:n luottamusväli: −3,3 – −0,7).
Taulukko 9. MG‑ADL‑ ja QMG‑pistemäärien muutos lähtötilanteesta viikolla 26 aikuisilla yleistynyttä myasthenia gravista sairastavilla AChR‑ tai MuSK‑vasta-ainepositiivisilla potilailla
| Koko tutkimusryhmä | |
| Inebilitsumabi N = 119 | Lumelääke N = 117 |
MG‑ADL-pistemäärä | ||
LS‑keskiarvo | −4,3 | −3,0 |
Ero | −1,3 | |
95 %:n luottamusväli | (−2,2 – −0,4) | |
p‑arvo | 0,0067 | |
QMG‑pistemäärä | ||
LS‑keskiarvo | −4,9 | −2,9 |
Ero | −2,0 | |
95 %:n luottamusväli | (−3,3 – −0,7) | |
p‑arvo | 0,0028 | |
LS = pienin neliösumma | ||
Inebilitsumabiryhmässä 68,7 %:lla ja lumelääkeryhmässä 48,2 %:lla potilaista MG‑ADL-pistemäärä parani ≥ 3‑pistettä viikolla 26 ilman varahoidon käyttöä päivän 28 ja viikon 26 välillä.
Kuva 3. MG‑ADL-pistemäärän keskimääräinen muutos lähtötilanteesta viikkoon 26 mennessä koko tutkimusryhmässä
Kuva 4. QMG‑pistemäärän keskimääräinen muutos lähtötilanteesta viikkoon 26 mennessä koko tutkimusryhmässä
Yleistynyttä myasthenia gravista sairastavat potilaat, jotka olivat AChR‑vasta-ainepositiivisia, jatkoivat satunnaistetussa, kontrolloidussa tutkimusjaksossa viikkoon 52 asti. Näiden potilaiden tulokset osoittavat, että inebilitsumabia suosiva ero suureni ajan myötä lumelääkkeeseen verrattuna. Viikolla 52 MG‑ADL-pistemäärän keskimääräinen muutos lähtötilanteesta potilailla, jotka olivat AChR‑vasta-ainepositiivisia, oli −5,1 inebilitsumabiryhmässä ja −3,0 lumelääkeryhmässä.
Kuva 5. MG‑ADL-pistemäärän keskimääräinen muutos lähtötilanteesta viikkoon 52 mennessä potilailla, jotka olivat AChR‑vasta-ainepositiivisia
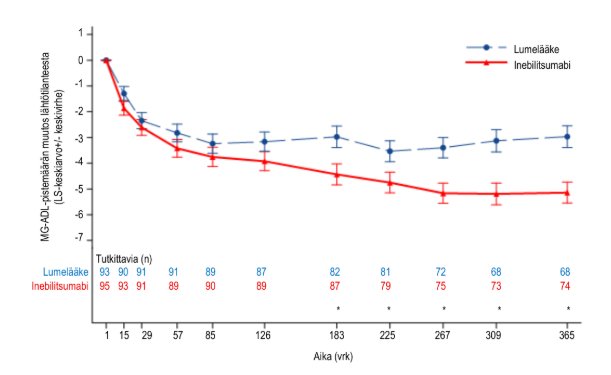
Ensisijaisen analyysin tekohetkellä yhteensä 94 potilasta (79,0 %) inebilitsumabiryhmässä ja 84 potilasta (70,6 %) lumelääkeryhmässä olivat saaneet vähintään yhden inebilitsumabiannoksen avoimen tutkimusjakson aikana. Niillä potilailla, jotka saivat aluksi inebilitsumabia satunnaistetun, kontrolloidun tutkimusjakson aikana, MG‑ADL‑ ja QMG‑pistemäärien havaittiin paranevan edelleen avoimen tutkimusjakson viikon 78 loppuun asti AChR‑vasta-ainepositiivisten alaryhmässä ja MuSK‑vasta-ainepositiivisten alaryhmässä. Niillä potilailla, jotka saivat aluksi lumelääkettä ja aloittivat inebilitsumabihoidon avoimen tutkimusjakson aikana, MG‑ADL‑ ja QMG‑pistemäärät paranivat edelleen avoimen tutkimusjakson viikon 78 loppuun asti AChR‑vasta-ainepositiivisten alaryhmässä ja MuSK‑vasta-ainepositiivisten alaryhmässä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset inebilitsumabin käytöstä NMOSD-taudin, IgG4‑taudin ja yleistyneen myasthenia graviksen hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Inebilitsumabi annetaan infuusiona laskimoon. NMOSD‑potilailla tehdyssä tutkimuksessa keskimääräinen maksimikonsentraatio oli 108 μg/ml (300 mg, toinen annos päivänä 15) ja kumulatiivinen käyrän alapuolelle jäävä alue (AUC) oli 2 980 µg×vrk/ml 26 viikkoa kestäneellä hoitojaksolla, jonka aikana NMOSD‑potilaat saivat kaksi annosta laskimonsisäisesti 2 viikon välein. IgG4‑tautia sairastavilla potilailla tehdyssä tutkimuksessa keskimääräinen maksimikonsentraatio oli 127 μg/ml (300 mg, toinen annos päivänä 15) ja kumulatiivinen käyrän alapuolelle jäävä alue (AUC) oli 4 290 µg×vrk/ml 52 viikkoa kestäneellä hoitojaksolla, jonka aikana IgG4‑tautia sairastavat potilaat saivat kaksi annosta laskimonsisäisesti 2 viikon välein ja kolmannen annoksen viikolla 26.
Yleistynyttä myasthenia gravista sairastavilla potilailla tehdyssä tutkimuksessa keskimääräinen maksimikonsentraatio oli 126 μg/ml (300 mg, toinen annos päivänä 15) AChR‑vasta-ainepositiivisten alaryhmässä (n = 90) ja 159 μg/ml MuSK‑vasta-ainepositiivisten alaryhmässä (n = 20). Kumulatiivinen AUC oli 3 120 µg×vrk/ml 26 viikkoa kestäneellä hoitojaksolla, jonka aikana AChR‑vasta-ainepositiiviset potilaat (n = 82) saivat kaksi annosta laskimonsisäisesti 2 viikon välein. Kumulatiivinen AUC oli 3 740 µg×vrk/ml 26 viikkoa kestäneellä hoitojaksolla, jonka aikana MuSK‑vasta-ainepositiiviset potilaat (n = 17) saivat kaksi annosta laskimonsisäisesti 2 viikon välein. Lisäksi kumulatiivinen AUC oli 4 240 µg×vrk/ml 52 viikkoa kestäneellä hoitojaksolla, jonka aikana AChR‑vasta-ainepositiiviset potilaat (n = 84) saivat kaksi annosta laskimonsisäisesti 2 viikon välein ja kolmannen annoksen viikolla 26.
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella inebilitsumabin arvioitu tyypillinen sentraalinen jakautumistilavuus oli 2,95 l ja perifeerinen 2,57 l.
Biotransformaatio
Inebilitsumabi on humanisoitu monoklonaalinen IgG1-luokan vasta-aine, joka hajoaa elimistöön laajalti jakautuneiden proteolyyttisten entsyymien vaikutuksesta.
Eliminaatio
Aikuisilla NMOSD-tautia, IgG4‑tautia ja yleistynyttä myasthenia gravista sairastavilla potilailla eliminaation terminaalinen puoliintumisaika oli noin 18 vuorokautta. Populaatiofarmakokineettisen analyysin perusteella arvioitu inebilitsumabin systeeminen puhdistuma oli 0,19 l/vrk (ensimmäisen asteen puhdistuma). Pienillä pitoisuuksilla inebilitsumabiin kohdistui todennäköisesti reseptori (CD19) ‑välitteinen puhdistuma, joka väheni aikaa myöten oletettavasti inebilitsumabihoidon aiheuttaman B-soluvajeen tähden.
Erityisryhmät
Pediatriset potilaat
Inebilitsumabia ei ole tutkittu nuorilla eikä lapsilla.
Iäkkäät
Populaatiofarmakokineettisen analyysin perusteella ikä ei vaikuttanut inebilitsumabin puhdistumaan.
Sukupuoli, rotu
Populaatiofarmakokineettinen analyysi osoitti, ettei sukupuolella tai rodulla ollut merkittävää vaikutusta inebilitsumabin puhdistumaan.
Munuaisten vajaatoiminta
Varsinaisia kliinisiä tutkimuksia ei ole suoritettu sen tutkimiseksi, kuinka munuaisten vajaatoiminta vaikuttaa inebilitsumabiin. Monoklonaalisen IgG-luokan vasta-aineen suuren molekyylipainon ja hydrodynaamisen koon tähden inebilitsumabin ei odoteta suodattuvan glomeruluksen läpi. Populaatiofarmakokineettisen analyysin perusteella inebilitsumabin puhdistuma potilailla, joilla oli eriasteista munuaisten vajaatoimintaa, oli verrattavissa potilaisiin, joilla oli normaali arvioitu glomerulusten suodatusnopeus.
Maksan vajaatoiminta
Varsinaisia kliinisiä tutkimuksia ei ole suoritettu sen tutkimiseksi, kuinka maksan vajaatoiminta vaikuttaa inebilitsumabiin. Kliinisissä tutkimuksissa inebilitsumabille ei ole altistettu vaikeaa maksan vajaatoimintaa sairastavia tutkittavia. Monoklonaaliset IgG-luokan vasta-aineet eivät ensisijaisesti poistu maksan kautta; siksi maksan toiminnan muutosten ei odoteta vaikuttavan inebilitsumabin puhdistumaan. Populaatiofarmakokineettisen analyysin perusteella lähtötilanteen maksantoiminnan biomarkkereilla (ASAT, alkalinen fosfataasi ja bilirubiini) ei ollut kliinisesti merkittävää vaikutusta inebilitsumabin puhdistumaan.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Inebilitsumabi arvioitiin yhdistetyssä hedelmällisyys- ja alkion ja sikiön kehitystutkimuksessa naaras- ja urospuolisilla huCD19 Tg -hiirillä 3 ja 30 mg/kg laskimonsisäisillä annoksilla. Valmisteella ei ollut vaikutusta alkion tai sikiön kehitykseen, mutta molemmilla testatuilla annoksilla ilmeni hoitoon liittyvä hedelmällisyysindeksin pieneneminen. Tämän tuloksen merkitystä ihmisille ei tunneta. Lisäksi ilmeni B-solupopulaatioiden vähenemistä B-solukehityskohdassa hiiren sikiöissä, jotka syntyivät inebilitsumabilla hoidetuille eläimille, verrattuna kontrollieläinten jälkeläisiin, mikä viittaa siihen, että inebilitsumabi läpäisee istukan ja vähentää B-solumäärää.
Yhdistetyssä hedelmällisyys- ja alkion ja sikiön kehitystutkimuksessa kerättiin vain niukasti toksikokineettisiä näytteitä; ensimmäisen annoksen maksimipitoisuuden (Cmax) perusteella altistus 300 mg:n kliiniseen hoitoannokseen verrattuna oli naaraspuolisilla huCD19 Tg -hiirillä 0,4‑kertainen annoksella 3 mg/kg ja 4-kertainen annoksella 30 mg/kg.
Synnytystä edeltävässä ja sen jälkeisessä kehitystutkimuksessa siirtogeenisillä hiirillä inebilitsumabin antaminen emoille tiineyspäivästä 6 imetyspäivään 20 vähensi jälkeläisten B-solupopulaatioita synnytyksen jälkeisen päivän 50 kohdalla. Jälkeläisten B-solupopulaatiot palautuivat synnytyksen jälkeiseen päivään 357 mennessä. Inebilitsumabia saaneiden eläinten jälkeläisten immuunivaste neoantigeenille oli pienempi kuin kontrollieläinten jälkeläisillä, mikä viittaa normaalin B-solutoiminnan häiriintymiseen.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Histidiinihydrokloridimonohydraatti
Natriumkloridi
Trehaloosidihydraatti
Polysorbaatti 80 [E433]
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
5 vuotta
Kestoaika laimentamisen jälkeen
Valmisteltu infuusioliuos pitää antaa välittömästi. Jos liuosta ei anneta välittömästi, säilytä sitä enintään 24 tuntia jääkaapissa 2–8 °C:ssa tai 4 tuntia huoneenlämmössä ennen infuusion aloittamista.
Säilytys
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
UPLIZNA infuusiokonsentraatti, liuosta varten
100 mg (L:ei) 3 x 10 ml (10 mg/ml) (59077,99 €)
PF-selosteen tieto
10 ml konsentraattia injektiopullossa, joka on tyypin 1 lasia ja jossa on elastomeerinen korkki ja sumunharmaa alumiininen repäisysuojus.
Pakkauskoko 3 injektiopulloa.
Valmisteen kuvaus:
Kirkas tai hieman opalisoiva, väritön tai hieman kellertävä liuos. Liuoksen pH on noin 6,0 ja osmolaliteetti noin 280 mOsm/kg.
Käyttö- ja käsittelyohjeet
Infuusioliuoksen valmistelu
Ennen laskimoinfuusion aloittamista valmistellun infuusioliuoksen pitää olla huoneenlämmössä 20–25 °C:ssa.
Konsentraatti on tutkittava silmämääräisesti hiukkasten ja värinmuutosten varalta. Injektiopullo on hävitettävä, jos liuos on samea, siinä on värinmuutoksia tai se sisältää selviä vierashiukkasia.
-
Injektiopulloa ei saa ravistaa.
-
Injektiopulloa on säilytettävä pystyasennossa.
-
Hanki infuusiopussi, jossa on 250 ml natriumkloridi-injektioliuosta 9 mg/ml (0,9 %). Älä käytä muita laimentimia inebilitsumabin laimentamiseen, koska niiden käyttöä ei ole testattu.
-
Käytä injektiopullon korkin läpäisemiseen vain 21 G:n tai halkaisijaltaan pienempää neulaa (esim. 22 tai 23 G). Älä käytä injektiopullon piikkiä tai suljettuja siirtolaitteita, sillä ne voivat vaurioittaa korkkia ja vaarantaa injektiopullon eheyden.
-
Vedä 10 ml Uplizna-valmistetta kustakin pakkauksen kolmesta injektiopullosta ja siirrä yhteensä 30 ml valmistetta 250 ml:n infuusiopussiin. Sekoita laimennettu liuos varovasti ylösalaisin kääntämällä. Älä ravista liuosta.
Hävitys
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
UPLIZNA infuusiokonsentraatti, liuosta varten
100 mg 3 x 10 ml
- Ei korvausta.
ATC-koodi
L04AG10
Valmisteyhteenvedon muuttamispäivämäärä
22.05.2026
Yhteystiedot
Keilaranta 10, PL 86
02101 Espoo
09 5490 0500