
IQIRVO filmdragerad tablett 80 mg
Observera
▼Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Varje filmdragerad tablett innehåller 80 mg elafibranor.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Filmdragerad tablett (tablett)
Kliniska uppgifter
Terapeutiska indikationer
Iqirvo är indicerat för behandling av primär biliär kolangit (PBC) i kombination med ursodeoxicholsyra (UDCA) hos vuxna med otillräckligt svar på UDCA, eller som monoterapi hos patienter som inte kan tolerera UDCA.
Dosering och administreringssätt
Dosering
Rekommenderad dos är 80 mg en gång dagligen.
Missad dos
Om en dos av elafibranor missas ska patienten inte ta den missade dosen utan i stället ta sin efterföljande dos vid nästa schemalagda tidpunkt. Patienten ska inte ta en dubbel dos för att kompensera för den missade dosen.
Äldre
Ingen dosjustering krävs för patienter som är 65 år eller äldre (se avsnitt Farmakokinetiska egenskaper).
Pediatrisk population
Det finns ingen relevant användning av elafibranor för en pediatrisk population (under 18 år) för indikationen PBC.
Nedsatt njurfunktion
Ingen dosjustering krävs för patienter med nedsatt njurfunktion (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Ingen dosjustering krävs för patienter med lindrigt (Child-Pugh A) eller måttligt (Child-Pugh B) nedsatt leverfunktion.
Säkerheten och effekten av elafibranor har inte fastställts hos patienter med PBC med gravt nedsatt leverfunktion. Användning hos patienter med gravt nedsatt leverfunktion (Child-Pugh C) rekommenderas inte (se avsnitt Farmakokinetiska egenskaper).
Administreringssätt
För oral användning.
Kontraindikationer
- Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
- Känd eller misstänkt graviditet och hos fertila kvinnor som inte använder preventivmedel (se avsnitt Fertilitet, graviditet och amning).
Varningar och försiktighet
Leverrelaterade händelser
Förhöjda levervärden i biokemiska tester inklusive transaminaser och bilirubinnivåer har rapporterats hos patienter som fått elafibranor.
Leverfunktionen bör bedömas kliniskt och laboratorievärden utvärderas innan behandling med elafibranor påbörjas och därefter enligt rutinmässig patientbehandling.
Om förhöjda biokemiska leverprover och/eller leverdysfunktion observeras, rekommenderas omedelbar utredning av orsaken och avbrytande av behandlingen med elafibranor bör övervägas.
Förhöjt blodkreatinfosfokinas och muskelskada
Ökning av blodkreatinfosfokinas (CPK) har rapporterats hos patienter som fått elafibranor (se avsnitt Biverkningar). CPK bör utvärderas innan behandling med elafibranor påbörjas och därefter enligt rutinmässig patientbehandling. Periodiska CPK-mätningar kan övervägas hos patienter som påbörjar behandling med elafibranor, särskilt hos de som samtidigt behandlas med HMG-CoA-reduktashämmare. Om ökning av CPK eller oförklarliga tecken och symtom på muskelskada observeras, rekommenderas omedelbar utredning av orsaken och avbrytande av behandlingen med elafibranor bör övervägas (se avsnitt Biverkningar).
Embryo- och fostertoxicitet
Baserat på data från djurstudier, misstänks elafibranor orsaka medfödda missbildningar och lägre överlevnad hos foster när det administeras till gravida kvinnor (se avsnitt Fertilitet, graviditet och amning). Elafibranor är därför kontraindicerat hos kvinnor med känd eller misstänkt graviditet och hos fertila kvinnor som inte använder preventivmedel (se avsnitt Kontraindikationer). Fertila kvinnor ska informeras om detta.
Hjälpämnen
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per tablett, d.v.s. är näst intill "natriumfritt".
Interaktioner
Baserat på in vitro- och in vivo-studier förväntas ingen kliniskt relevant läkemedelsinteraktion vid samtidig administrering av elafibranor med andra läkemedel (se avsnitt Farmakokinetiska egenskaper).
Fertilitet, graviditet och amning
Fertilitet
Inga humandata om effekten av elafibranor på fertiliteten finns tillgängliga. Djurstudier tyder inte på några direkta eller indirekta effekter på fertiliteten eller förmågan att fortplanta sig (se avsnitt Prekliniska säkerhetsuppgifter).
Kvinnor i fertil ålder/preventivmedel
Fertila kvinnor ska använda effektiv preventivmetod under behandlingen och upp till minst tre veckor efter den sista dosen av elafibranor. Graviditetsstatusen för fertila patienter bör kontrolleras innan behandling med elafibranor påbörjas (se avsnitt Varningar och försiktighet).
Graviditet
Det finns begränsad mängd data från användning av elafibranor hos gravida kvinnor.
Studier på dräktiga djur med elafibranor har visat reproduktionstoxikologiska effekter (förlust av foster, missbildningar, dödfödslar och/eller perinatala dödsfall) vid kliniskt relevant exponering (se avsnitt Varningar och försiktighet och Prekliniska säkerhetsuppgifter).
Elafibranor är kontraindicerat under graviditet (se avsnitt Kontraindikationer). Om en patient blir gravid ska behandlingen med elafibranor avbrytas.
Amning
Det är okänt om elafibranor eller dess metaboliter utsöndras i bröstmjölk. Det finns ingen information om elafibranor eller dess metaboliter utsöndras i mjölk från djur, men biverkningar sågs hos avkomma när elafibranor administrerades till honråttor under dräktighet (se avsnitt Prekliniska säkerhetsuppgifter) och amning vid kliniskt relevant exponering.
En risk för det ammade barnet kan inte uteslutas.
Elafibranor ska inte användas under amning och barnet får inte ammas fram till minst tre veckor efter den sista dosen av elafibranor.
Effekter på förmågan att framföra fordon och använda maskiner
Elafibranor har ingen effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligaste rapporterade biverkningarna i samband med behandling med elafibranor (n = 108) som inträffade hos mer än 10 % av patienterna och med en högre incidens än i placebogruppen (n = 53; skillnad > 1 %) var buksmärta (11,1 % mot 5,7 %), diarré (11,1 % mot 9,4 %), illamående (11,1 % mot 5,7 %) och kräkningar (11,1 % mot 1,9 %). Dessa var inte allvarliga, milda till måttliga, inträffade tidigt i behandlingen och tenderade att försvinna inom några dagar till några veckor utan någon dosändring eller stödjande åtgärder.
Den vanligaste biverkningen som ledde till att behandlingen avbröts var en ökning av CPK i blodet (3,7 %).
Tabell över biverkningar
Inom varje organsystem klassificeras biverkningarna efter frekvens, i följande kategorier: mycket vanliga (≥ 1/10), vanliga (≥ 1/100 till < 1/10), mindre vanliga (≥ 1/1 000 till < 1/100), sällsynta (≥ 1/10 000 till < 1/1 000), mycket sällsynta (< 1/10 000) och ingen känd frekvens (kan inte beräknas från tillgängliga data).
| Organsystemklass | Mycket vanliga | Vanliga | Mindre vanliga |
| Centrala och perifera nervsystemet | Huvudvärk | ||
| Magtarmkanalen | Buksmärtaa Diarré Illamående Kräkningar | Förstoppning | |
| Lever och gallvägar | Kolelitiasis | ||
| Hud och subkutan vävnad | Hudutslag, kliande | ||
| Muskuloskeletala systemet och bindväv | Myalgi | ||
| Undersökningar och provtagningar | Förhöjt kreatinfosfokinas (CPK) i blod | Förhöjt blodkreatinin |
a inkluderar högt sittande buksmärta och lågt sittande buksmärta
Beskrivning av utvalda biverkningar
Huvudvärk
I den pivotala fas 3-studien, ELATIVE, upplevde 9 (8,3 %) deltagare i elafibranorgruppen och 6 (11,3 %) deltagare i placebogruppen huvudvärk. Inom de första 10 dagarna av behandlingen i den kliniska studien upplevde dock fler deltagare i elafibranorgruppen huvudvärk jämfört med placebogruppen (3,7 % respektive 0 %).
Förhöjt CPK i blod
I den pivotala fas 3-studien, ELATIVE, hade 4 (3,7 %) deltagare i elafibranorgruppen och inga deltagare i placebogruppen en kliniskt signifikant ökning av CPK i blodet, vilket ledde till att behandlingen med läkemedlet avbröts. Hos 2 av de 4 deltagarna var CPK > 5 x övre normalgränsen (ULN). Alla händelser var icke allvarliga och milda till måttliga i intensitet. Två av deltagarna upplevde också associerade symtom på myalgi. Vid studiestart var medelvärdena av CPK likartade mellan behandlingsgrupperna och inom normalområdet; värdena vid vecka 52 förblev inom normalområdet i båda grupperna. Den genomsnittliga förändringen (standardavvikelsen) från baslinjen vid vecka 52 var 6,2 (38,1) E/l i elafibranorgruppen och 12,3 (67,0) E/l i placebogruppen.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till:
Finland:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Sverige:
Läkemedelsverket
Box 26
751 03 Uppsala
Överdosering
I händelse av misstänkt överdosering ska patienterna observeras noggrant och lämplig symtomatisk och understödjande behandling bör inledas.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Gall- och leverterapi, övriga medel vid gallterapi, ATC-kod: A05AX06
Verkningsmekanism
Elafibranor och dess huvudsakliga aktiva metabolit GFT1007 är dubbla peroxisomproliferatoraktiverade receptor-(PPAR)α/δ-agonister.
PPARα/δ tros vara den viktigaste regulatorn för gallsyrahomeostas, inflammation och fibros. Aktivering av PPARα och PPARδ minskar galltoxicitet och kolestas genom att modulera gallsyrasyntes, avgiftning och transportörer.
Aktivering av PPARα och PPARδ har också antiinflammatoriska effekter genom olika signalvägar.
Farmakodynamisk effekt
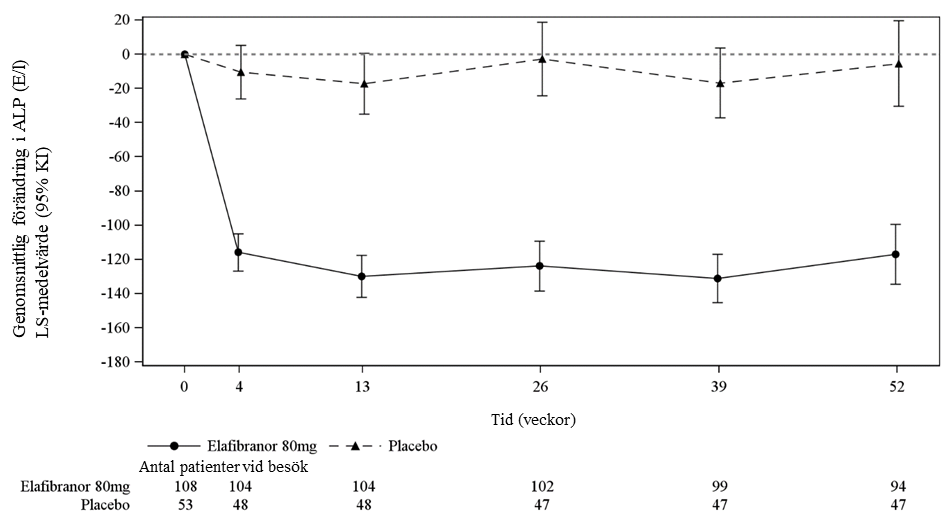
I den pivotala fas 3-studien, ELATIVE, resulterade behandling med elafibranor i en markant minskning av alkaliskt fosfatas (ALP) från utgångsvärdet så tidigt som efter 4 veckor, vilket kvarstod till och med vecka 52. I linje med det observerade biokemiska svaret, sågs vid behandlingen med elafibranor en större minskning av biomarkörer för gallsyrasyntes inklusive gallsyraprekursorn 7 alfa-hydroxi-4-cholesten-3-on (C4) och fibroblasttillväxtfaktor-19 (FGF-19), en gallsyrasyntesregulator.
Hjärt ats elektrofysiologi
Grundlig QT (TQT)-analys uteslöt all effekt av elafibranor på förlängning av QT/korrigerat QT (QTc)-intervall vid upprepade doser på upp till 300 mg under 14 dagar.
I kliniska studier har inga kliniskt betydelsefulla förändringar i vitala tecken eller i elektrokardiogram (EKG) (inklusive QTc-intervall) observerats hos deltagare som behandlats med elafibranor.
Klinisk effekt
Effekten av elafibranor utvärderades i studie GFT505B-319-1 (ELATIVE), en randomiserad, dubbelblind (DB), placebokontrollerad fas 3-studie med 161 vuxna med PBC som hade ett otillräckligt svar eller intolerans mot UDCA. Deltagarna randomiserades i ett 2:1-förhållande stratifierat efter två faktorer (ALP > 3 x ULN eller totalt bilirubin (TB) > ULN och PBC Worst Itch Numeric Rating Scale (WI-NRS)-poäng ≥ 4) till att få elafibranor 80 mg eller placebo en gång dagligen i minst 52 veckor. I förekommande fall fortsatte deltagarna med dosen av UDCA från förstudien genom hela studien. Deltagarna inkluderades i studien om deras ALP var ≥ 1,67 x ULN och TB var ≤ 2 x ULN. Deltagare med dekompenserad cirros eller andra orsaker till leversjukdom uteslöts.
Totalt sett var medelåldern 57,1 år och medelvikten var 70,8 kg. Studiepopulationen bestod övervägande av kvinnor (96 %) och vita (91 %). Den genomsnittliga ALP-koncentrationen vid baslinjen var 321,9 E/l, 39 % av deltagarna hade en ALP-koncentration > 3 x ULN vid baslinjen och 35 % av deltagarna hade avancerad sjukdom vid studiestart, definierad som leverstelhet > 10 kPa och/eller överbryggande fibros eller cirros vid histologi.
Mediantiden för exponeringen var 63,07 och 61,00 veckor i elafibranor- respektive i placebogruppen.
Den genomsnittliga TB-koncentrationen vid baslinjen var 9,6 µmol/l och 96 % av deltagarna hade en baslinjekoncentration av TB som var mindre än eller lika med ULN. Det genomsnittliga värdet vid baslinjen för leverstelhet, mätt genom transient elastografi, var 10,1 kPa. Genomsnittligt PBC WI-NRS-poäng vid baslinjen var 3,3 och 41 % hade måttlig till svår klåda vid studiestart (PBC WI-NRS-poäng ≥ 4). För dem med måttlig till svår klåda var den genomsnittliga PBC WI-NRS-poängen 6,2 vid studiestart för deltagare i elafibranor 80 mg-gruppen och 6,3 för deltagare i placebogruppen. Majoriteten (95 %) av deltagarna fick behandling i kombination med UDCA, eller som monoterapi till 5 % av deltagarna som inte tolererade UDCA.
Det primära effektmåttet var kolestasrespons vid vecka 52, definierat som det sammansatta effektmåttet: ALP < 1,67 x ULN och TB ≤ ULN och ALP-minskning ≥ 15 %. De viktigaste sekundära effektmåtten var ALP-normalisering vid vecka 52 och förändringen i klåda från baslinjen till och med vecka 52 och till och med vecka 24 baserat på PBC WI-NRS-poäng hos deltagare med måttlig till svår klåda vid baslinjen.
Tabell 1 visar det primära sammansatta effektmåttet för kolestasrespons och det viktigaste sekundära effektmåttet för ALP-normalisering.
Tabell 1. Andelen vuxna deltagare med PBC som uppnådde det primära sammansatta effektmåttet för kolestasrespons och det viktigaste sekundära effektmåttet för ALP-normalisering vid vecka 52
| Populationsanalys | Elafibranor 80 mg (N = 108) | Placebo (N = 53) | Behandlingsskillnad (95 % KI)[3] | Oddskvot (95 % KI)[4] | P-värde[4] |
| Primära sammansatta effektmåttet: kolestasrespons[1] | |||||
| ITT | 51 % | 4 % | 47 % (32, 57) | 37,6 (7,6, 302,2) | < 0,0001 |
| Första viktigaste sekundära effektmåttet: ALP-normalisering[2] | |||||
| ITT | 15 % | 0 | 15 % (6, 23) | Oändlighet (2,8, oändlig) | 0,0019 |
ITT: Intention-to-treat
[1] Kolestasrespons definieras som ALP < 1,67 x ULN och TB ≤ ULN och ALP-minskning från baslinjen ≥ 15 % vid vecka 52. Deltagare som avbröt studiebehandlingen i förtid (interkurrent händelse 1) eller använde räddningsterapi för PBC (interkurrent händelse 2) före vecka 52 bedömdes som icke-svarare. Vid uteblivna data vid vecka 52 för deltagare utan interkurrent händelse, togs den närmaste icke-saknade bedömningen från DB-behandlingsperioden i beaktande.
[2] Normalisering av ALP vid vecka 52 definierat som andelen deltagare med ALP ≤ 1,0 × ULN. Tillvägagångssättet för att hantera interkurrenta händelser eller saknad data är detsamma som för det primära effektmåttet.
[3] Skillnaden i responsfrekvens mellan behandlingsgrupperna och 95 % KI beräknades med hjälp av Newcombe-metoden stratifierad efter randomiseringsstrata för kolestasrespons och ostratifierad för ALP-normalisering.
[4] Oddskvoter för respons och p-värden för att jämföra behandlingar var från det exakta Cochran-Mantel-Haenszel (CMH)-testet stratifierat efter randomiseringsstrata.
En signifikant minskning av ALP från utgångsvärdet sågs så tidigt som i vecka 4 och var ihållande under 52 veckors behandling i elafibranorgruppen jämfört med placebo (Figur 1).
Figur 1. Genomsnittlig (minsta kvadrat (LS)-medelvärde med 95 % KI) förändring från utgångsvärdet i ALP över tid – ITT analysset
Det primära effektmåttet för kolestasrespons hos deltagare med utgångsvärdet ALP ≤ 3 x ULN eller TB < ULN uppnåddes hos 71 % av deltagarna som fick elafibranor mot 6 % av deltagarna som fick placebo, jämfört med dem med ALP > 3 x ULN eller TB > ULN där kolestasrespons uppnåddes hos 21 % av deltagarna som fick elafibranor mot 0 % som fick placebo.
Bland de 54 deltagarna med avancerad sjukdom uppnådde 16/35 (46 %) deltagare som fick elafibranor det primära effektmåttet för kolestasrespons, mot 0/19 (0 %) deltagare som fick placebo. På grund av det begränsade antalet deltagare med avancerad sjukdom bör dessa resultat tolkas med försiktighet.
Patientrapporterat utfall
Hos deltagare med måttlig till svår klåda vid studiestart observerades en större minskning från utgångsvärdet i PBC WI-NRS-poäng till och med vecka 52 och vecka 24 hos deltagare som randomiserats till elafibranor jämfört med placebo, men detta nådde inte statistisk signifikans (tabell 2).
Tabell 2. Förändring i klåda från studiestart till och med vecka 52 och vecka 24 mätt med PBC WI-NRS hos de med måttlig till svår klåda vid studiestart
Elafibranor 80 mg (N = 44) | Placebo (N = 22) | Behandlingsskillnad | P-värde | |
| Andra viktigaste sekundära effektmåttet: förändring till och med vecka 52[1] | ||||
Minsta kvadratmedelvärde (95 % KI) | -1,9 (-2,6, -1,3) | -1,1 (-2,1, -0,2) | -0,8 (-2,0, 0,4) | 0,1970 |
| Tredje viktigaste sekundära effektmåttet: förändring till och med vecka 24[1] | ||||
Minsta kvadratmedelvärde (95 % KI) | -1,6 (-2,2, -1,0) | -1,3 (-2,2, -0,3) | -0,3 (-1,5, 0,8) | - |
[1] Analysen använde den blandade modellen för upprepade mätningar (Mixed Model for Repeated Measures, MMRM) med behandling, 4-veckorsperiod och behandling med 4-veckorsperiodinteraktion som fasta faktorer och justering för baslinje-PBC WI-NRS och stratifieringsfaktorn ALP > 3 x ULN eller TB > ULN. En ostrukturerad korrelationsstruktur användes. Behandlingseffekten till och med vecka 52 är genomsnittet av förändring i NRS-poäng från baslinjen för de tretton 4-veckorsperioderna. Behandlingseffekten till och med vecka 52 och vecka 24 är den genomsnittliga behandlingseffekten av förändring i NRS-poäng från baslinjen under de första tretton 4-veckorsperioderna respektive de första sex 4-veckorsperioderna. Bedömning av PBC WI-NRS-poäng efter att deltagarna avbröt studiebehandlingen i förtid eller fick en räddningsterapi för klåda, ansågs som saknad.
Behandling med elafibranor var förknippad med en förbättring av klåda, vilket framgår av en minskning av totalpoängen för PBC-40-klåda och 5-D-klåda jämfört med placebo vid vecka 52 (tabell 3).
Tabell 3. Förändring av klåda från studiestart till vecka 52 i totalpoäng för PBC-40-klåda och 5-D-klåda hos de med måttlig till svår klåda vid studiestart
Elafibranor 80 mg (N = 44) | Placebo (N = 22) | Behandlingsskillnad | |
| PBC-40-klåda, total poäng: förändring vid vecka 52[1] | |||
| Minsta kvadratmedelvärde (95 % KI) | -2,5 (-3,4, -1,6) | -0,1 (-1,6, 1,3) | -2,3 (-4,0, -0,7) |
| 5-D-klåda, total poäng: förändring vid vecka 52[1] | |||
| Minsta kvadratmedelvärde (95 % KI) | -4,2 (-5,6, -2,9) | -1,2 (-3,3, 0,9) | -3,0 (-5,5, -0,5) |
[1] Analysen använde den blandade modellen för upprepade mätningar (Mixed Model for Repeated Measures, MMRM) med behandling, återbesök (till vecka 52) och behandling vid besöksinteraktion som fasta faktorer och justering för poäng vid studiestart och stratifieringsfaktorn ALP > 3 x ULN eller TB > ULN.
Lipidparametrar
Elafibranor visade en gynnsam effekt på lipidparametrar. Den genomsnittliga minskningen av VLDL-kolesterol (Very low-density lipoprotein-cholesterol, VLDL-C) och triglycerider var större hos deltagare som behandlades med elafibranor jämfört med placebo vid vecka 52. Genomsnittlig skillnad i minsta kvadratmedelvärdet från placebo i VLDL-C var -0,1 mmol/l [(95 % KI: -0,2, -0,1); p < 0,001] och för triglycerider var -0,3 mmol/l [(95 % KI: -0,4, -0,1)]; p < 0,001]. HDL-kolesterol (High-density lipoprotein-cholesterol, HDL-C) förblev stabilt vid behandling med elafibranor.
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för Iqirvo för alla grupper av den pediatriska populationen för primär biliär kolangit (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Detta läkemedel har godkänts enligt reglerna om ”villkorat godkännande för försäljning”. Detta innebär att det ska inkomma ytterligare evidens för detta läkemedel.
Europeiska läkemedelsmyndigheten går igenom ny information om detta läkemedel minst varje år och uppdaterar denna produktresumé när så behövs.
Farmakokinetiska egenskaper
Elafibranors plasmaexponering (AUC) ökar proportionellt från 50 till 360 mg (0,6 till 4,5 gånger den rekommenderade dosen). Steady state uppnås dag 14 vid dosering en gång dagligen. Farmakokinetiken (PK) för elafibranor och dess huvudsakliga aktiva metabolit GFT1007 visade sig vara tidsoberoende efter 16 dagars upprepad administrering. Exponering av elafibranor och dess aktiva metabolit hos deltagare med PBC listas i tabell 4.
Tabell 4. Exponering av elafibranor och GFT1007 hos deltagare med PBC vid steady state efter 80 mg QD (en gång dagligen)
| Cmax,ss (ng/ml) | AUC0-24 (ng • tim/ml) | Ackumuleringsförhållande | |
| Elafibranor | 802 | 3758 | 2,9 |
| GFT1007 | 2058 | 11985 | 1,3 |
Absorption
Efter upprepad oral administrering till deltagare med PBC uppnås median maximal plasmanivå av elafibranor och GFT1007 vid doser på 80 mg inom 1,25 timmar.
Vid administrering tillsammans med en fettrik måltid med högt kaloriinnehåll, var det en 30-minuters fördröjning i Tmax för elafibranor och en fördröjning på 1 timme för GFT1007 jämfört med fastande tillstånd. Plasmaexponeringen (AUC) av elafibranor minskade med 15 % och AUC i plasma för GFT1007 påverkades inte. Med tanke på de högre cirkulerande plasmanivåerna av den farmakologiskt aktiva metaboliten GFT1007 jämfört med elafibranor, ansågs födointag ha begränsad klinisk effekt baserat på den totala exponeringen av modersubstansen och dess aktiva metabolit.
Distribution
Graden av plasmaproteinbindning för både elafibranor och GFT1007 är cirka 99,7 % (främst till serumalbumin). Den genomsnittliga skenbara distributionsvolymen (Vd/F) av elafibranor hos människor är 4731 liter efter en engångsdos av elafibranor på 80 mg under fasta.
Metabolism
In vitro metaboliseras elafibranor av 15-ketoprostaglandin 13-Δ-reduktas (PTGR1). In vitro visar varken elafibranor eller GFT1007 någon större metabolism av de primära cytokrom P450 (CYP)- och uridindifosfat (UDP)-glukuronosyltransferas (UGT)-isoformerna.
Efter oral administrering av 14C-radiomärkt elafibranor hydrolyserades det snabbt till den aktiva metaboliten GFT1007. Två huvudmetaboliter identifierades i plasma, GFT1007 (aktiv metabolit) och glukuronidkonjugat (inaktiv metabolit).
Eliminering
Efter en engångsdos på 80 mg under fasta, är den genomsnittliga elimineringshalveringstiden 68,2 timmar för elafibranor och 15,4 timmar för metaboliten GFT1007. Elafibranors genomsnittliga skenbara totala clearance (CL/F) var 50,0 l/timme efter en engångsdos på 80 mg under fasta.
Utsöndring
Efter en oral engångsdos på 120 mg av 14C-radiomärkt elafibranor hos friska frivilliga återfanns cirka 77,1 % av dosen i feces, främst som elafibranor (56,7 % av den administrerade dosen) och dess aktiva metabolit GFT1007 (6,08 % av den administrerade dosen). Cirka 19,3 % återfanns i urin, främst som glukuronidkonjugat.
Särskilda patientgrupper
Det fanns inga bevis för att ålder (från 18 till 80 år), kön, etnicitet, Body Mass Index (BMI) eller njurstatus, hade någon kliniskt betydelsefull inverkan på elafibranor och GFT1007 PK.
Nedsatt leverfunktion
Den totala läkemedelsexponeringen av modersubstansen och den aktiva metaboliten skilde sig inte signifikant mellan deltagare med normal leverfunktion och deltagare med nedsatt leverfunktion (Child-Pugh A, B och C). Dosjustering är inte nödvändig för patienter med lindrigt (Child-Pugh A) eller måttligt (Child-Pugh B) nedsatt leverfunktion. Den obundna fraktionen av elafibranor och GFT1007 ökade dock med cirka 3 gånger hos deltagare med gravt (Child-Pugh C) nedsatt leverfunktion. Elafibranor rekommenderas inte till patienter med gravt nedsatt leverfunktion (Child-Pugh C).
Läkemedelsinteraktioner
Baserat på in vitro-studier visade sig CYP- och UGT-enzymer inte spela någon större roll i metabolismen av elafibranor. Läkemedelsinteraktioner förväntas vara minimala med läkemedel som signifikant förändrar CYP- eller UGT-aktiviteten.
Kliniska studier
Warfarin (CYP2C9-substrat):
Samtidig administrering av elafibranor och warfarin resulterade inte i någon ökning av exponeringen (AUC, Cmax) av warfarin och ingen skillnad i internationellt normaliserat ratio (INR) jämfört med enbart warfarin.
Simvastatin (CYP3A, bröstcancerresistensprotein (BCRP), organiska anjontransporterande polypeptider 1B1 (OATP1B1) och OATP1B3-substrat) och atorvastatin (CYP3A, organiska anjontransporterande polypeptider 1B1 (OATP1B1) och OATP1B3-substrat):
Samtidig administrering av upprepade doser av elafibranor med simvastatin eller atorvastatin resulterade inte i någon ökning av exponeringen (AUC, Cmax) av simvastatin eller dess metabolit β-hydroxisyra eller atorvastatin.
Sitagliptin (dipeptidylpeptidas-IV (DPP-IV)-hämmare):
Inga kliniskt signifikanta effekter på blodnivåerna av GLP-1 observerades vid samtidig administrering av 100 mg elafibranor, som utövare av läkemedelsinteraktion, en gång dagligen i 15 dagar med en oral engångsdos på 100 mg sitagliptin under ett måltidstest.
In vitro-studier
Cytokrom P450 (CYP) hämning och induktion:
Elafibranor och GFT1007 ansågs inte vara hämmare av de viktigaste CYP-enzymerna. Ingen tidsberoende CYP-hämning observerades.
Elafibranor och GFT1007 orsakade inte induktion av CYP1A2, CYP2B6 och CYP3A4.
UGT-hämning:
Baserat på in vitro-data förväntades inte elafibranor och GFT1007 hämma de viktigaste UGT-enzymerna vid kliniskt signifikanta koncentrationer.
Transportörsystem:
Elafibranor var en hämmare av OATP1B3 och BCRP. Baserat på in vivo-studierna med simvastatin och atorvastatin, förväntas inga kliniska konsekvenser av hämningen av OATP1B3 och BCRP.
Prekliniska säkerhetsuppgifter
Gängse studier avseende säkerhetsfarmakologi, allmäntoxicitet, gentoxicitet och karcinogenicitet visade inte några särskilda risker för människa.
Reproduktions- och utvecklingstoxicitet
Elafibranor har visat tecken på utvecklingstoxicitet hos både råttor och kaniner. I pre- och postnatal studie på råtta, ledde maternell exponering (på eller över 2 gånger AUC-exponeringen vid maximal rekommenderad human dos) för elafibranor till minskad överlevnad hos ungar, försenad utveckling eller trombos. Hos dräktiga kaniner orsakade maternell exponering (3 gånger AUC-exponeringen vid maximal rekommenderad human dos) för elafibranor markant maternell toxicitet, förhöjd embryodödlighet, minskad fostervikt och en låg förekomst av missbildningar hos foster.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Tablett innehåll
Mikrokristallin cellulosa
Povidon
Kroskarmellosnatrium
Kolloidal vattenfri kiseldioxid
Magnesiumstearat
Film dragering
Polyvinylalkohol, partiellt hydrolyserad
Titandioxid (E171)
Makrogol
Talk
Järnoxid gul (E172)
Järnoxid röd (E172)
Inkompatibiliteter
Ej relevant.
Hållbarhet
3 år
Särskilda förvaringsanvisningar
Inga särskilda förvaringsanvisningar.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
IQIRVO tabletti, kalvopäällysteinen
80 mg (L:ei) 30 kpl (5591,28 €)
PF-selosteen tieto
40 ml burk av högdensitetspolyeten (HDPE) med barnskyddande skruvlock av polypropen.
Varje burk innehåller 30 filmdragerade tabletter.
Följande förpackningsstorlekar finns tillgängliga: 30 filmdragerade tabletter och flerpack med 90 (3 förpackningar om 30) filmdragerade tabletter.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
Tabletterna är runda, orangea, cirka 8 mm i diameter, präglade med ”ELA 80” på ena sidan.
Särskilda anvisningar för destruktion och övrig hantering
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
IQIRVO tabletti, kalvopäällysteinen
80 mg 30 kpl
- Ei korvausta.
Atc-kod
A05AX06
Datum för översyn av produktresumén
01.07.2025
Yhteystiedot
Kista Science Tower, Färögatan 33
SE-164 51 Kista
Sweden
+46 8 451 60 00
www.ipsen.com
info.se@ipsen.com