
VYVGART injektionsvätska, lösning 1000 mg
Observera

Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Varje injektionsflaska innehåller 1 000 mg efgartigimod alfa i 5,6 ml (180 mg/ml).
Efgartigimod alfa är ett humant, rekombinant Fc-fragment som härrör från immunglobulin G1 (IgG1), vilket framställts i äggstocksceller från kinesisk hamster (CHO-celler) med hybrid-DNA-teknik.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Injektionsvätska, lösning
Kliniska uppgifter
Terapeutiska indikationer
Vyvgart är avsett som
- ett tillägg till standardterapi för behandling av vuxna patienter med generaliserad myastenia gravis (gMG) som har antikroppar mot acetylkolinreceptorer (AChR).
- monoterapi för behandling av vuxna patienter med progressiv eller recidiverande aktiv kronisk inflammatorisk demyeliniserande polyneuropati (CIDP) efter tidigare behandling med kortikosteroider eller immunoglobuliner.
Villkor
Valmistetta saa antaa vain terveydenhoitoalan ammattilainen hyväksytyn käyttöaiheen hoitoon perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Behandling måste initieras och övervakas av en läkare med erfarenhet av behandling av patienter med neuromuskulära sjukdomar.
Dosering
Generaliserad myastenia gravis
Den första behandlingscykeln och den första administreringen i den andra behandlingscykeln måste administreras antingen av eller under överinseende av sjukvårdspersonal. Efterföljande behandling ska administreras av sjukvårdspersonal eller kan administreras hemma av en patient eller vårdare som fått adekvat utbildning i subkutan injektionsteknik.
Rekommenderad dos är 1 000 mg vilken ska administreras subkutant i cykler med injektion en gång i veckan under 4 veckor. Efterföljande behandlingscykler ska administreras efter klinisk bedömning. Frekvensen för behandlingscykler kan variera för olika patienter (se avsnitt Farmakodynamiska egenskaper).
I det kliniska utvecklingsprogrammet kunde påföljande behandlingscykel tidigast påbörjas 7 veckor efter första infusionen i föregående cykel.
För patienter som för närvarande får efgartigimod alfa intravenöst kan subkutan injektion, lösning, användas som ett alternativ. Det rekommenderas att byta mellan formuleringar i början av en ny behandlingscykel. Det finns inga data om säkerhet och effekt tillgängliga för patienter som byter formulering under samma cykel.
Kronisk inflammatorisk demyeliniserande polyneuropati
De första 4 injektionerna måste administreras antingen av eller under överinseende av sjukvårdspersonal. Efterföljande injektioner ska administreras av sjukvårdspersonal eller kan administreras hemma av en patient eller vårdare som fått adekvat utbildning i subkutan injektionsteknik.
Rekommenderad dos är 1 000 mg som administreras som en subkutan injektion en gång i veckan.
Behandlingen inleds med en veckovis dosregim och kan justeras till varannan vecka baserat på klinisk utvärdering. Vid försämring av symtomen ska administreringen av injektioner en gång i veckan återupptas.
För patienter som övergår från sina nuvarande CIDP-behandlingar ska behandlingen med Vyvgart helst inledas innan den kliniska effekten av dessa tidigare behandlingar börjar avta.
Kliniskt svar uppnås vanligen inom 3 månader efter påbörjad behandling med efgartigimod alfa subkutant. Klinisk utvärdering ska övervägas 3 till 6 månader efter behandlingsstart för att bedöma behandlingseffekten och därefter med regelbundna intervall.
Missad dos
Ett intervall på minst 3 dagar ska tillämpas mellan två på varandra följande administreringar. Om administreringarna inte kan göras vid den inplanerade tidpunkten ska de göras så snart som möjligt och minst 3 dagar före nästa administrering. Om det är mindre än 3 dagar till nästa administrering ska den missade dosen hoppas över och nästa dos ska administreras vid den inplanerade tidpunkten.
Särskilda populationer
Äldre
Det krävs ingen dosjustering för patienter i åldern 65 år och uppåt (se avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Det finns begränsat med data om säkerhet och effekt för patienter med lätt nedsatt njurfunktion – ingen dosjustering krävs för patienter med lätt nedsatt njurfunktion. Det finns mycket begränsat med data om säkerhet och effekt för patienter med måttligt eller svårt nedsatt njurfunktion (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Det finns inga tillgängliga data för patienter med nedsatt leverfunktion. Ingen dosjustering krävs för patienter med nedsatt leverfunktion (se avsnitt Farmakokinetiska egenskaper).
Pediatrisk population
Säkerhet och effekt för efgartigimod alfa för den pediatriska populationen har ännu inte fastställts. Inga data finns tillgängliga.
Administreringssätt
Detta läkemedel får endast administreras genom subkutan injektion. Får ej administreras intravenöst.
Efter att ha tagit ut injektionsflaskan ur kylskåpet, vänta i minst 15 minuter innan du injicerar så att lösningen når rumstemperatur. Använd aseptisk teknik vid beredning och administrering av läkemedelslösningen. Skaka inte injektionsflaskan.
Injektionsvätska, lösning kan administreras med en polypropenspruta, överföringsnålar av rostfritt stål och infusionsset med fjärilsnål av polyvinylklorid med en maximal fyllningsvolym på 0,4 ml.
- Dra upp hela innehållet i injektionsflaskan med efgartigimod alfa-lösningen med hjälp av en överföringsnål.
- Byt ut nålen på sprutan till fjärilsnålen.
- Före administrering ska volymen i sprutan justeras till 5,6 ml.
Under de initiala administreringarna av efgartigimod alfa (se avsnitt Dosering och administreringssätt) bör lämplig behandling finnas tillgänglig för användning i händelse av en injektions- eller överkänslighetsreaktion (se avsnitt Varningar och försiktighet). De rekommenderade injektionsställena (buken) ska växlas och injektioner ska aldrig ges i födelsemärken, ärr eller områden där huden är öm, har blåmärken, är röd eller hård. Volymen på 5,6 ml ska injiceras under 30 till 90 sekunder. Injektionen kan administreras långsammare om patienten upplever obehag.
Den första självadministreringen ska alltid ske under överinseende av sjukvårdspersonal. Efter adekvat utbildning i subkutan injektionsteknik kan patienter eller vårdare injicera läkemedlet hemma om sjukvårdspersonal bedömer att det är lämpligt. Patienter eller vårdare ska instrueras att injicera Vyvgart enligt anvisningarna i bipacksedeln.
Utförliga instruktioner för administrering av läkemedlet finns i bruksanvisningen i bipacksedeln.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Patienter i MGFA-klass V (Myasthenia Gravis Foundation of America)
Behandlingen med efgartigimod alfa har inte studerats hos patienter i MGFA-klass V (d.v.s. myasten kris), definierat som intubering med eller utan mekanisk ventilering förutom i samband med rutinmässig postoperativ vård. Sekvensen för inledning av behandling mellan etablerade behandlingar mot myasten kris och efgartigimod alfa samt potentiella interaktioner bör övervägas (se avsnitt Interaktioner).
Infektioner
Eftersom efgartigimod alfa orsakar en övergående minskning av IgG-halten kan risken för infektion öka (se avsnitt Biverkningar och Farmakodynamiska egenskaper). De vanligaste infektionerna som observerats i kliniska studier är övre luftvägsinfektion och urinvägsinfektion (se avsnitt Biverkningar). Under behandlingen med Vyvgart ska patienterna övervakas med avseende på kliniska tecken och symtom på infektion. Hos patienter med en aktiv infektion bör nyttan/risken med att bibehålla eller avbryta behandlingen med efgartigimod alfa övervägas tills infektionen har gått tillbaka. Om allvarliga infektioner uppstår bör man överväga att skjuta upp behandlingen med efgartigimod alfa tills infektionen har försvunnit.
Injektionsreaktioneroch överkänslighetsreaktioner
Injektionsreaktioner såsom utslag och klåda rapporterades i de kliniska studierna (se avsnitt Biverkningar). Dessa var lindriga till måttliga. Fall av anafylaktiska reaktioner har rapporterats med efgartigimod alfa intravenöst efter marknadsintroduktion. De första administreringarna av Vyvgart måste utföras under överinseende av sjukvårdspersonal (se avsnitt Dosering och administreringssätt). Patienterna bör övervakas med avseende på kliniska tecken och symtom på injektionsreaktioner under 30 minuter efter administrering. Om en reaktion uppstår och beroende på reaktionens svårighetsgrad ska lämpliga stödåtgärder initieras. Efterföljande injektioner kan administreras försiktigt baserat på en klinisk utvärdering.
Om en anafylaktisk reaktion inträffar ska administreringen av Vyvgart omedelbart avbrytas och lämplig medicinsk behandling påbörjas. Patienter bör informeras om tecken och symtom på överkänslighet och anafylaktiska reaktioner och rådas att omedelbart kontakta sjukvårdspersonal om dessa skulle uppstå.
Immunisering
Alla vacciner ska administreras i enlighet med riktlinjerna för immunisering.
Säkerheten vid immunisering med levande eller levande försvagade vacciner och reaktionen på immunisering med dessa vacciner under behandling med efgartigimod alfa är okänd. För patienter som behandlas med efgartigimod alfa rekommenderas i allmänhet inte vaccination med levande eller levande försvagade vacciner. Om vaccination med levande eller levande försvagade vacciner krävs, ska dessa vacciner administreras minst 4 veckor innan behandling och minst 2 veckor efter sista dosen efgartigimod alfa.
Andra vacciner kan administreras vid behov när som helst under behandling med efgartigimod alfa.
Immunogenicitet
I den aktivt kontrollerade studien ARGX‑113‑2001 påvisades redan befintliga antikroppar som binds till efgartigimod alfa hos 12 av 110 (11 %) patienter med gMG. Antikroppar mot anti-efgartigimod alfa påvisades hos 19 av 55 (35 %) patienter som behandlades med efgartigimod alfa subkutant jämfört med 11 av 55 (20 %) patienter som behandlades med den intravenösa formuleringen. Neutraliserande antikroppar påvisades hos 2 (4 %) patienter som behandlades med efgartigimod alfa intravenöst.
I studien ARGX‑113‑1802 påvisades redan befintliga antikroppar som binds till efgartigimod alfa hos 13 av 317 (4,1 %) patienter med CIDP. Antikroppar mot anti-efgartigimod alfa påvisades hos 20 av 317 (6,3 %) patienter som behandlades i den öppna delen av studien (steg A) och hos 2 av 111 (1,8 %) patienter som behandlades i den placebokontrollerade delen (steg B). Neutraliserande antikroppar påvisades hos 1 (0,3 %) patient i den öppna delen av studien (se avsnitt Farmakodynamiska egenskaper).
Inverkan av antikropparna mot efgartigimod alfa på den kliniska effekten och säkerheten, på farmakokinetik och farmakodynamik kan inte bedömas med tanke på den låga förekomsten av neutraliserande antikroppar.
Behandling med immunsuppressiva läkemedel och antikolinesteraser
När en behandling med steroidfria immunsuppressiva läkemedel, kortikosteroider eller antikolinesteraser sänks eller sätts ut ska patienterna övervakas noggrant med avseende på tecken på förvärrad sjukdom.
Hjälpämnen med känd effekt
Natrium
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per injektionsflaska, d.v.s. är näst intill ”natriumfritt”.
Polysorbater
Detta läkemedel innehåller 2,7 mg polysorbat 20 per injektionsflaska motsvarande 0,4 mg/ml. Polysorbater kan orsaka allergiska reaktioner.
Interaktioner
Inga interaktionsstudier har utförts.
Efgartigimod alfa kan minska koncentrationen av kemiska föreningar som binds till den humana neonatala Fc-receptorn (FcRn), d.v.s. immunglobulinläkemedel, monoklonala antikroppar och antikroppsderivat som innehåller den humana Fc-domänen i IgG-underklassen. Det rekommenderas att inledningen av behandlingen med dessa läkemedel om möjligt skjuts upp till 2 veckor efter sista dosen av Vyvgart. De patienter som får Vyvgart under pågående behandling med dessa läkemedel ska som en försiktighetsåtgärd övervakas noggrant med avseende på avsedd reaktion på de läkemedlen.
Vid plasmabyte, immunadsorption och plasmaferes kan halten av efgartigimod alfa i blodcirkulationen minska.
Alla vacciner ska administreras i enlighet med riktlinjerna för immunisering.
Den potentiella interaktionen med vacciner har studerats i en icke-klinisk modell med användning av KLH (keyhole limpet hemocyanin) som antigen. Den veckovisa administreringen om 100 mg/kg till apor påverkade inte den immunologiska reaktionen på KLH-immuniseringen.
För patienter som behandlas med efgartigimod alfa rekommenderas i allmänhet inte vaccination med levande eller levande försvagade vacciner. Om vaccination med levande eller levande försvagade vacciner krävs, ska dessa vacciner administreras minst 4 veckor innan behandling och minst 2 veckor efter sista dosen av efgartigimod alfa (se avsnitt Varningar och försiktighet).
Fertilitet, graviditet och amning
Graviditet
Det finns inga tillgängliga data om användning av efgartigimod alfa under graviditet. Det är känt att antikroppar, däribland terapeutiska monoklonala antikroppar, aktivt transporteras över placentan efter 30 veckors graviditet genom att de binds till FcRn.
Efgartigimod alfa kan överföras från modern till fostret som är under utveckling. Eftersom efgartigimod alfa förväntas sänka halten av antikroppar från modern och även förväntas hämma överföringen av antikroppar från modern till fostret, förväntas det nyfödda barnet ha ett minskat passivt skydd. Därför måste nyttan och risken med att administrera levande eller levande, försvagade vacciner till spädbarn som exponerats för efgartigimod alfa in utero beaktas (se avsnitt Varningar och försiktighet).
Behandling med Vyvgart bör endast övervägas för gravida kvinnor om den kliniska nyttan överväger riskerna.
Amning
Det finns ingen information gällande förekomsten av efgartigimod alfa i bröstmjölk, effekterna på barn som ammas eller effekterna på mjölkbildningen. Det har inte utförts några djurstudier av övergången av efgartigimod alfa till bröstmjölk och därför kan inte utsöndring i modersmjölken uteslutas. Det är känt att maternellt IgG finns i bröstmjölk. Behandling med efgartigimod alfa bör endast övervägas för ammande kvinnor om den kliniska nyttan överväger riskerna.
Fertilitet
Det finns inga tillgängliga data om effekten av efgartigimod alfa på fertiliteten hos människor. Djurstudier har inte visat någon påverkan av efgartigimod alfa på manliga och kvinnliga fertilitetsparametrar (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
Vyvgart har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligaste biverkningarna som observerades var reaktioner på injektionsstället (33 %), övre luftvägsinfektion (10,7 %) och urinvägsinfektion (9,5 %).
Den övergripande säkerhetsprofilen för Vyvgart subkutant för både cykliska och kontinuerliga dosregimer överensstämde med den kända säkerhetsprofilen för den intravenösa formuleringen.
Lista över biverkningar i tabellform
Biverkningarna som beskrivs i detta avsnitt har identifierats i kliniska studier och från rapporter efter marknadsföring. Dessa reaktioner presenteras efter organsystem och föredragen term. Frekvenskategorierna definieras som: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), sällsynta (≥ 1/10 000, < 1/1 000) eller ingen känd frekvens (kan inte beräknas från tillgängliga data). I varje frekvensgrupp presenteras biverkningarna i fallande allvarlighetsgrad.
Tabell 1. Biverkningar
| Organsystem | Biverkning | Frekvenskategori |
| Infektioner och infestationer* | Övre luftvägsinfektion | Mycket vanliga |
| Urinvägsinfektion | Vanliga | |
| Bronkit | Vanliga | |
| Immunsystemet | Anafylaktisk reaktion a | Ingen känd frekvens |
| Magtarmkanalen | Illamående b | Vanliga |
| Muskuloskeletala systemet och bindväv | Myalgi | Vanliga |
| Allmänna symtom och/eller symtom vid administreringsstället* | Reaktioner på injektionsstället c, d | Mycket vanliga |
| Skador, förgiftningar och behandlingskomplikationer* | Procedurrelaterad huvudvärk e | Vanliga |
* Se avsnittet ”Beskrivning av utvalda biverkningar”
a Från spontan rapportering efter godkännande för försäljning för produkten med intravenöst administreringssätt
b Från spontan rapportering efter godkännande för försäljning
c Endast subkutan administrering
d (t.ex. utslag på injektionsstället, erytem på injektionsstället, klåda på injektionsstället, smärta på injektionsstället)
e Endast intravenös administrering.
Beskrivning av utvalda biverkningar
Reaktioner på injektionsstället
I den sammanslagna datamängden från två kliniska studier på gMG med efgartigimod alfa subkutant (n = 168) var alla reaktioner på injektionsstället lindriga till måttliga i svårighetsgrad och ledde inte till att behandlingen avbröts. 44,0 % (n = 74) av patienterna upplevde en reaktion på injektionsstället. Reaktioner på injektionsstället inträffade inom 24 timmar efter administrering hos 78,4 % (58 av 74) av patienterna och försvann utan behandling hos 85,1 % (63 av 74) av patienterna. Incidensen av reaktioner på injektionsstället var högst under den första behandlingscykeln, rapporterad hos 36,3 % (61 av 168) av patienterna under den första behandlingscykeln och minskade till 20,1 % (30 av 149), 15,4 % (18 av 117) och 12,5 % (10 av 80) av patienterna med den andra, tredje respektive fjärde behandlingscykeln. I den sammanslagna datamängden från två kliniska studier på patienter med CIDP som fick kontinuerlig administrering av efgartigimod alfa subkutant var incidensen av reaktioner på injektionsstället 26 % (61 av 235). Analys av 3-månadersintervall visade att andelen deltagare med reaktioner på injektionsstället var högst under de första 3 månaderna av behandlingen (73 [22,2 %] deltagare) och minskade under efterföljande 3-månadersintervall (intervall: 0 till 17 [6,8 %] deltagare).
Infektioner
I den placebokontrollerade studien ARGX‑113‑1704 på gMG med efgartigimod alfa intravenöst var de vanligaste rapporterade biverkningarna var infektioner och de mest rapporterade infektionerna var övre luftvägsinfektion (10,7 % [n = 9] av patienterna som behandlades med efgartigimod alfa intravenöst och 4,8 % [n = 4] av patienterna som behandlades med placebo) och urinvägsinfektion (9,5 % [n = 8] av patienterna som behandlades med efgartigimod alfa intravenöst och 4,8 % [n = 4] av patienterna som behandlades med placebo). Dessa infektioner var lindriga till måttliga i svårighetsgrad hos de patienter som fick efgartigimod alfa intravenöst (≤ grad 2 enligt Common Terminology Criteria for Adverse Events). Totalt sett rapporterades infektioner som uppkommit i samband med behandlingen hos 46,4 % (n = 39) av patienterna som behandlades med efgartigimod alfa intravenöst och 37,3 % (n = 31) av patienterna som behandlades med placebo. Mediantiden från behandlingsstart till uppkomsten av infektion var 6 veckor. Förekomsten av infektioner ökade inte med påföljande behandlingscykler. Behandlingen sattes ut eller avbröts tillfälligt på grund av infektion hos mindre än 2 % av patienterna. I den placebokontrollerade delen av studien ARGX-113-1802 på patienter med CIDP var kontinuerlig administrering av efgartigimod alfa subkutant inte förknippad med någon ökning av förekomsten av infektioner (31,5 % [35 av 111] i gruppen med efgartigimod alfa subkutant och 33,6 % [37 av 110] i placebogruppen) (se avsnitt Farmakodynamiska egenskaper).
Procedurrelaterad huvudvärk
Procedurrelaterad huvudvärk rapporterades hos 4,8 % av patienterna som behandlades med efgartigimod alfa intravenöst och 1,2 % av patienterna som behandlades med placebo. Procedurrelaterad huvudvärk rapporterades när huvudvärken bedömdes vara tidsmässigt relaterad tillden intravenösa infusionen av efgartigimod alfa. I samtliga fall var huvudvärken lindrig eller måttlig förutom i ett fall då den rapporterades som allvarlig (grad 3).
Övriga biverkningar var lindriga eller måttliga, med undantag av ett fall av myalgi (grad 3).
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för
läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Det finns inga kända specifika tecken och symtom på överdosering med efgartigimod alfa. I händelse av överdosering förväntas biverkningarna som kan uppträda inte vara annorlunda än de som kan observeras med den rekommenderade dosen. Patienterna bör övervakas med avseende på biverkningar och lämplig symtomatisk och understödjande behandling påbörjas. Det finns inget specifikt motgift vid överdosering med efgartigimod alfa.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Immunsuppressiva medel, selektiva immunsuppressiva medel, ATC‑kod: L04AA58
Verkningsmekanism
Efgartigimod alfa är ett humant IgG1-antikroppsfragment som är framtaget för ökad affinitet med den neonatala Fc-receptorn (FcRn) Efgartigimod alfa binds till FcRn, vilket resulterar i en minskad halt IgG, inklusive patogena IgG-autoantikroppar, i blodcirkulationen. Efgartigimod alfa påverkar inte halten av övriga immunglobuliner (IgA, IgD, IgE eller IgM) och sänker inte halten av albumin.
IgG-autoantikropparna är den bakomliggande orsaken till patogenesen för IgG-medierade autoimmuna sjukdomar.
Vid MG försämrar dessa den neuromuskulära signalöverföringen genom att de binds till acetylkolinreceptorer (AChR), muskelspecifikt tyrosinkinas (MuSK) eller LRP4 (lipoprotein receptor-related protein 4 – lipoproteinreceptorrelaterat protein 4).
Vid CIDP finns det flera bevis som pekar på IgG-autoantikropparnas nyckelroll i patogenesen för sjukdomen. Detta inkluderar påvisandet av autoreaktiva IgG-antikroppar mot komponenter i myeliniserade nerver, passiv överföring av CIDP-symtom till djurmodeller med hjälp av sera eller IgG från patienter med CIDP, samt den terapeutiska effekten av plasmabyte och immunadsorption för behandling av patienter med CIDP.
Farmakodynamisk effekt
Intravenös formulering
I den dubbelblinda, placebokontrollerade studien ARGX‑113‑1704 av patienter med gMG minskade efgartigimod alfa 10 mg/kg administrerat en gång i veckan under 4 veckor IgG‑halten och halten av AChR‑autoantikroppar (AChR‑ak) i serum. Den maximala, genomsnittliga procentuella minskningen av den totala IgG‑halten jämfört med baslinjen, uppgick till 61 % en vecka efter sista infusionen i den inledande behandlingscykeln och återgick till baslinjehalten 9 veckor efter sista infusionen. Liknande effekter observerades också för alla undertyper av IgG. Minskningen av halten av AChR‑ak följde ett liknande tidsförlopp, med en maximal genomsnittlig, procentuell minskning på 58 % en vecka efter sista infusionen och återgång till baslinjehalten 7 veckor efter sista infusionen. Liknande förändringar observerades under den andra cykeln i studien.
Subkutan formulering
I studien ARGX‑113‑2001 följde minskningar av AChR‑ak ett jämförbart tidsförlopp som totala IgG‑nivåer och var liknande mellan grupperna efgartigimod alfa subkutant och intravenöst. Maximala genomsnittliga procentuella minskningar av AChR‑ak på 62,2 % och 59,6 % observerades en vecka efter den senaste administreringen i grupperna efgartigimod alfa subkutant respektive intravenöst. För både grupperna efgartigimod alfa subkutant respektive intravenöst var minskningen av totala halter av IgG och AChR‑ak förknippade med ett kliniskt svar, mätt genom förändringen från baslinjen i MG‑ADL totalpoäng (se bild 1).
Bild 1. Samband mellan totalt IgG och AChR-ak och MG-ADL totalpoäng i AChR‑ak‑seropositiv population behandlad med efgartigimod alfa subkutant (1A) och efgartigimod alfa intravenöst (1B) (studie ARGX‑113‑2001)
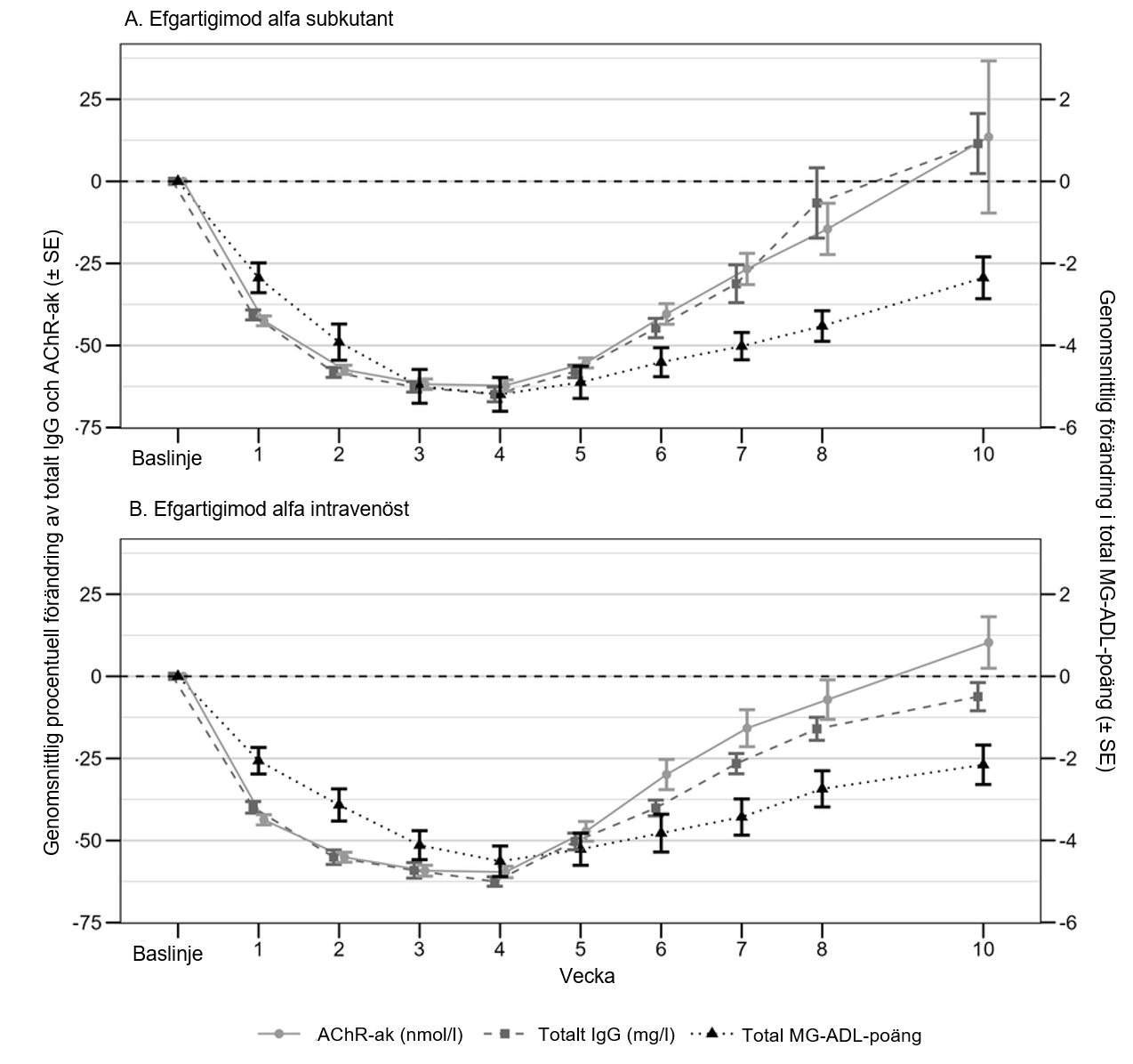
I studien ARGX‑113‑1802 på patienter med CIDP som fick kontinuerlig administrering en gång i veckan av efgartigimod alfa subkutant med dosen 1 000 mg, bibehölls den genomsnittliga procentuella förändringen från baslinjen av totala IgG-nivåer från vecka 4 under hela behandlingsperioden (genomsnittlig procentuell minskning från baslinjen varierade mellan 66,8 och 71,6 %).
Klinisk effekt och säkerhet
Generaliserad myastenia gravis
Intravenös formulering
Effekten av efgartigimod alfa för behandling av vuxna med generaliserad myastenia gravis (gMG) har studerats i en 26 veckor lång randomiserad, dubbelblind, placebokontrollerad multicenterstudie (ARGX‑113‑1704).
I denna studie var det nödvändigt att patienterna uppfyllde följande huvudkriterier vid screeningen:
- Klinisk klass II, III eller IV enligt MGFA (Myasthenia Gravis Foundation of America)
- Patient med antingen positivt eller negativt serologiskt test för antikroppar mot AChR
- Totalpoäng ≥ 5 på MG‑ADL-skalan (MG-Activities of Daily Living – ADL-aktiviteter vid myastenia gravis)
- På stabil dos av läkemedelsbehandling mot MG före screeningen, vilket omfattade acetylkolinesterashämmare (AChE-hämmare), steroider eller steroidfria immunsuppressiva läkemedel (NSIST), antingen i kombination eller för sig [till NSIST-preparaten hörde bland annat azatioprin, metotrexat, cyklosporin, takrolimus, mykofenolatmofetil och cyklofosfamid]
- IgG‑halt på minst 6 g/l
Patienter med gMG i MGFA-klass V, patienter med dokumenterad brist på kliniskt svar på PLEX, patienter som behandlats med PLEX, intravenös immunglobulinbehandling en månad innan behandlingsstart och monoklonala antikroppar sex månader innan behandlingsstart samt patienter med en aktiv (akut eller kronisk) hepatit B-infektion, seropositivitet för hepatit C och diagnosen AIDS exkluderades från studierna.
Totalt 167 patienter inkluderades i studien och randomiserades till antingen efgartigimod alfa intravenöst (n = 84) eller placebo (n = 83). Baslinjeegenskaperna var likartade mellan behandlingsgrupperna, däribland medianåldern när diagnosen ställdes [45 (19‑81) år], könstillhörighet [de flesta var kvinnor, 75 % (efgartigimod alfa) jämfört med 66 % (placebo)], rastillhörighet [de flesta patienter var vita, 84,4 %] och mediantiden sedan diagnosen hade ställts [8,2 år (efgartigimod alfa) och 6,9 år (placebo)].
Majoriteten av patienterna (77 % i varje grupp) hade ett positivt provsvar för antikroppar mot AChR (AChR-ak) och 23 % av patienterna hade ett negativt provsvar för AChR-ak.
Under studien fick över 80 % av patienterna i varje grupp AChE-hämmare, över 70 % i varje behandlingsgrupp fick steroider och ungefär 60 % i varje behandlingsgrupp fick NSIST-preparat i stabil dos. Cirka 30 % av patienterna i varje behandlingsgrupp hade inte tidigare exponerats för NSIST-preparat när de gick med i studien.
Medianvärdet för totalpoängen på MG-ADL-skalan var 9,0 i båda behandlingsgrupperna och medianvärdet för totalpoängen på QMG-skalan (Quantitative Myasthenia Gravis) var 17 respektive 16 i gruppen med efgartigimod alfa respektive med placebo.
Patienterna behandlades med efgartigimod alfa intravenöst 10 mg/kg administrerat en gång i veckan under 4 veckor och fick maximalt 3 behandlingscykler.
Effekten av efgartigimod alfa mättes med hjälp av MG‑ADL-skalan (Myasthenia Gravis-Specific Activities of Daily Living – ADL-aktiviteter vid myastenia gravis) med vilken effekten av gMG på dagliga funktioner bedöms. Totalpoängintervallet sträcker sig från 0 till 24, där de högre poängen tyder på en mer omfattande funktionsnedsättning. I denna studie ansågs en patient svara på behandlingen enligt MG‑ADL-skalan om hans/hennes totala MG‑ADL-poäng minskade med ≥ 2 poäng jämfört med baslinjen i behandlingscykeln under minst 4 veckor i rad, där den första minskningen skedde senast 1 vecka efter sista infusionen i cykeln.
Effekten av efgartigimod alfa mättes också med totalpoängen på QMG-skalan, ett graderingssystem för bedömning av muskelsvaghet med en möjlig totalpoäng på 0 till 39 där de högre poängen tyder på svårare funktionsnedsättning. I denna studie ansågs en patient svara på behandlingen enligt QMG‑skalan om hans/hennes totala OMG-poäng minskade med ≥ 3 poäng jämfört med baslinjen i behandlingscykeln under minst 4 veckor i rad, där den första minskningen skedde senast 1 vecka efter sista infusionen i cykeln.
Det primära effektmåttet på effekten var jämförelsen av andelen patienter som svarade på behandlingen enligt MG‑ADL-skalan under den första behandlingscykeln (C1) mellan behandlingsgrupperna i populationen med seropositivitet för AChR‑ak.
Ett viktigt sekundärt effektmått var jämförelsen av andelen patienter som svarade på behandlingen enligt QMG-skalan under C1 mellan båda behandlingsgrupperna med patienter med seropositivitet för AChR‑ak.
Tabell 2. Patienter i populationen med seropositivitet för AChR-ak (analysuppsättning för mITT) som svarade på behandlingen enligt MG-ADL- och QMG-skalan under cykel 1
| Population | Efgartigimod alfa n/N (%) | Placebo n/N (%) | P-värde | Skillnad efgartigimod alfa-placebo (95 % KI) | |
| MG-ADL | Seropositivitet för AChR-ak | 44/65 (67,7) | 19/64 (29,7) | < 0,0001 | 38,0 (22,1‑54,0) |
| QMG | Seropositivitet för AChR-ak | 41/65 (63,1) | 9/64 (14,1) | < 0,0001 | 49,0 (34,5‑63,5) |
AChR-ak = antikropp mot acetylkolinreceptorer, MG-ADL = Myasthenia Gravis Activities of Daily Living – ADL-aktivitet vid myastenia gravis, QMG = QMG-skalan (Quantitative Myasthenia Gravis), mITT = modifierad intention-to-treat (avsikt att behandla), n = antal patienter för vilka observationen rapporterades, N = antal patienter i analysuppsättningen, KI = konfidensintervall
Logistisk regression, stratifierad för AChR-ak-status (om tillämpligt), japaner/icke-japaner och standardvård, med MG-ADL-poängen vid baslinjen som kovariat/QMG som kovariat
Dubbelsidigt, exakt p-värde
Analyserna visar att andelarna patienter som svarade på behandlingen enligt MG-ADL-skalan under den andra behandlingscykeln liknande dem under den första behandlingscykeln (se tabell 3).
Tabell 3. Patienter i populationen med seropositivitet för AChR-ak (analysuppsättning för mITT) som svarade på behandlingen enligt MG-ADL-skalan och QMG-skalan under cykel 2
| Population | Efgartigimod alfa n/N (%) | Placebo n/N (%) | |
| MG-ADL | Seropositivitet för AChR-ak | 36/51 (70,6) | 11/43 (25,6) |
| QMG | Seropositivitet för AChR-ak | 24/51 (47,1) | 5/43 (11,6) |
AChR-ak = antikroppar mot acetylkolinreceptorer, MG-ADL = Myasthenia Gravis Activities of Daily Living – ADL-aktiviteter vid myastenia gravis, QMG = QMG-skalan (Quantitative Myasthenia Gravis), mITT = modifierad intention-to-treat (avsikt att behandla), n = antal patienter för vilka observationen rapporterades, N = antal patienter i analysuppsättningen
Explorativa data visar att begynnande behandlingssvar observerades inom 2 veckor efter den inledande infusionen hos 37 av 44 (84 %) patienter som behandlats med efgartigimod alfa intravenöst bland de patienter med seropositivitet för AChR-ak som svarade på behandlingen enligt MG‑ADL‑skalan.
I den dubbelblinda, placebokontrollerade studien (ARGX‑113‑1704) var den tidigaste möjliga tidpunkten för att inleda påföljande behandlingscykel 8 veckor efter den inledande infusionen i första behandlingscykeln. I den totala populationen var den genomsnittliga tiden till andra behandlingscykeln i gruppen med efgartigimod alfa intravenöst 13 veckor (SD 5,5 veckor) och mediantiden var 10 veckor (8‑26 veckor) från den inledande infusionen i första behandlingscykeln. I den öppna förlängningsstudien (ARGX‑113‑1705) var den tidigaste möjliga tidpunkten för att inleda påföljande behandlingscykler 7 veckor.
Bland patienterna som svarade på behandlingen varade den kliniska förbättringen i 5 veckor hos 5 av 44 (11 %) av patienterna, 6‑7 veckor hos 14 av 44 (32 %) av patienterna, 8‑11 veckor hos 10 av 44 (23 %) av patienterna och 12 veckor eller mer hos 15 av 44 (34 %) av patienterna.
Subkutan formulering
En 10 veckor lång, randomiserad, öppen, parallellgrupperad multicenterstudie (ARGX‑113‑2001) genomfördes på vuxna patienter med gMG för att utvärdera icke-underlägsenhet av den farmakodynamiska effekten av efgartigimod alfa subkutant jämfört med efgartigimod alfa intravenöst. De huvudsakliga inklusions- och exklusionskriterierna var desamma som i studie ARGX‑113‑1704.
Totalt 110 patienter randomiserades och fick en behandlingscykel med administrering en gång i veckan under 4 veckor av antingen efgartigimod alfa subkutant 1 000 mg (n = 55) eller efgartigimod alfa intravenöst 10 mg/kg (n = 55). Majoriteten av patienterna var positiva för antikroppar mot AChR (AChR‑ak): 45 patienter (82 %) i gruppen med efgartigimod alfa subkutant och 46 patienter (84 %) i gruppen med efgartigimod alfa intravenöst. Alla patienter fick stabila doser av MG-behandling före screening som inkluderade AChE-hämmare, steroider eller NSIST, antingen i kombination eller enskilt.
Baslinjeegenskaperna var likartade mellan behandlingsgrupperna.
Under studien fick över 80 % av patienterna i varje grupp AChE-hämmare, över 60 % av patienterna i varje grupp fick steroider och cirka 40 % i varje behandlingsgrupp fick NSIST i stabila doser. När studien påbörjades hade cirka 56 % av patienterna i varje behandlingsgrupp ingen tidigare exponering för NSIST.
Det primära effektmåttet var jämförelsen av den procentuella minskningen av totala halter av IgG från baslinjen vid dag 29 mellan behandlingsgrupper i den totala populationen. Resultaten i den AChR‑ak‑seropositiva populationen visar icke-underlägsenhet för efgartigimod alfa subkutant jämfört med efgartigimod alfa intravenöst (se tabell 4).
Tabell 4. ANCOVA-analys av procentuell förändring från baslinjen i total IgG vid dag 29 i AChR-ak-seropositiv population (mITT-analysuppsättning)
| Efgartigimod alfa SK | Efgartigimod alfa IV | Skillnad efgartigimod alfa SK- efgartigimod alfa IV | ||||||
| N | LS medelvärde | 95 % KI | N | LS medelvärde | 95 % KI | LS av medel-skillnad | 95 % KI | p-värde |
| 41 | -66,9 | -69,78, -64,02 | 43 | -62,4 | -65,22, -59,59 | -4,5 | -8,53, -0,46 | < 0,0001 |
AChR-ak = antikroppar mot acetylkolinreceptorer, ANCOVA = analys av kovarians, KI = konfidensintervall, SK = subkutant, IV = intravenöst, LS = minsta kvadrater, mITT = modifierad intention-to-treat (avsikt att behandla), N = antal patienter per grupp som ingick i ANCOVA-analysen
Sekundära effektmått var jämförelser av andelen MG‑ADL- och QMG-svarare, enligt definition i studie ARGX‑113‑1704, mellan båda behandlingsgrupperna. Resultaten i AChR-ak-seropositiv population presenteras i tabell 5.
Tabell 5. MG-ADL- och QMG-svarare dag 29 i AChR-ak-seropositiv population (mITT‑analysuppsättning)
| Efgartigimod alfa SK n/N (%) | Efgartigimod alfa IV n/N (%) | Skillnad efgartigimod alfa SK-efgartigimod alfa IV (95 % KI) | |
| MG-ADL | 32/45 (71,1) | 33/46 (71,7) | -0,6 (-19,2 till 17,9) |
| QMG | 31/45 (68,9) | 24/45 (53,3) | 15,6 (-4,3 till 35,4) |
AChR-ak = antikroppar mot acetylkolinreceptorer, MG-ADL = Myasthenia Gravis Activities of Daily Living, QMG = Quantitative Myasthenia Gravis, SK = subkutant, IV = intravenöst, mITT = modifierad intention-to-treat (avsikt att behandla), n = antal patienter för vilka observationen rapporterades, N = antal patienter i analysuppsättningen, KI = konfidensintervall
Undersökande data visar att svarsstart observerades inom 2 veckor efter initial administrering hos 28 av 32 (88 %) patienter behandlade med efgartigimod alfa subkutant och 27 av 33 (82 %) patienter behandlade med efgartigimod alfa intravenöst i AChR‑ak-seropositiva MG‑ADL-svarare.
Kronisk inflammatorisk demyeliniserande polyneuropati
Effekten av efgartigimod alfa subkutant för behandling av vuxna med CIDP studerades i en prospektiv multicenterstudie, ARGX-113-1802, som genomfördes i två behandlingssteg: ett öppet steg A och ett dubbelblindat, placebokontrollerat steg B med randomiserad utsättning.
Patienterna hade antingen fått eller inte fått CIDP-behandling under de 6 månader som föregick studiestarten. De som tidigare behandlats för CIDP såväl som de som inte behandlats för CIDP utan några dokumenterade tecken på försämring av CIDP under den senaste tiden, gick in i en behandlingsfri inkörningsperiod, och patienter som uppvisade tecken på kliniskt betydelsefull försämring gick sedan in i steg A av studien. De som inte behandlades för CIDP och som nyligen hade dokumenterade tecken på försämring av CIDP hoppade över inkörningsperioden och gick direkt in i steg A.
Totalt 322 patienter inkluderades i steg A. Patienterna fick upp till 12 injektioner en gång i veckan av efgartigimod alfa subkutant 1 000 mg tills bevis på klinisk förbättring (ECI) uppträdde vid två på varandra följande studiebesök. Därefter gick patienterna med bekräftad ECI in i steg B av studien och randomiserades till att få veckovisa administreringar av antingen efgartigimod alfa subkutant (111 patienter) eller placebo (110 patienter). ECI definierades som klinisk förbättring på den justerade aINCAT-skalan (Inflammatory Neuropathy Cause and Treatment) eller förbättring på I-RODS-skalan (Inflammatory Rasch-built Overall Disability Scale)/greppstyrka hos patienter som försämrats på dessa skalor endast före steg A.
I steg A hade patienterna en medianålder på 54 år (intervall: 20 till 82 år), en mediantid sedan CIDP-diagnos på 2,8 år och median-INCAT-poäng på 4,0. Sextiofem procent var män och 66 % var vita. I steg B hade patienterna en medianålder på 55 år (intervall: 20 till 82 år), en mediantid sedan CIDP-diagnos på 2,2 år och median-INCAT-poäng på 3,0. Sextiofyra procent var män och 65 % var vita. Baslinjeegenskaperna för steg B var likartade mellan behandlingsgrupperna.
I steg A var det primära effektmåttet andelen patienter som svarade på behandlingen, definierat som patienter som uppnådde bekräftad ECI. Det primära effektmåttet uppnåddes hos 66,5 % av patienterna; ytterligare detaljer presenteras i tabell 6.
Ett sekundärt effektmått i steg A var tiden till den första bekräftade ECI. Vecka 4 var den tidigaste tidpunkten då kriterierna för ECI kunde uppfyllas. Vid denna tidpunkt nådde upp till 40 % av patienterna ECI. Baserat på en ytterligare förspecificerad analys visade 25 % av patienterna kliniskt relevant förbättring efter 9 dagar i minst en av 3 parametrar (aINCAT, I-RODS eller greppstyrka).
Majoriteten av patienterna uppnådde bekräftad ECI i samtliga tidigare CIDP-läkemedelsgrupper.
Tabell 6. Bevis på klinisk förbättring hos patienter med CIDP i ARGX-113-1802 steg A
| ECI-svarare och tid till första bekräftade ECI | Steg A |
| Efgartigimod alfa SK (N = 322) | |
ECI-svarare (patienter med bekräftad klinisk förbättring) n/N (%) (95 % KI) | 214/322 (66,5 %) (61,0; 71,6) |
Tid till första bekräftade ECI i dagar median (95 % KI) | 43,0 (31,0; 51,0) |
n = antal patienter för vilka observationen rapporterades; N = antal patienter i analysuppsättningen
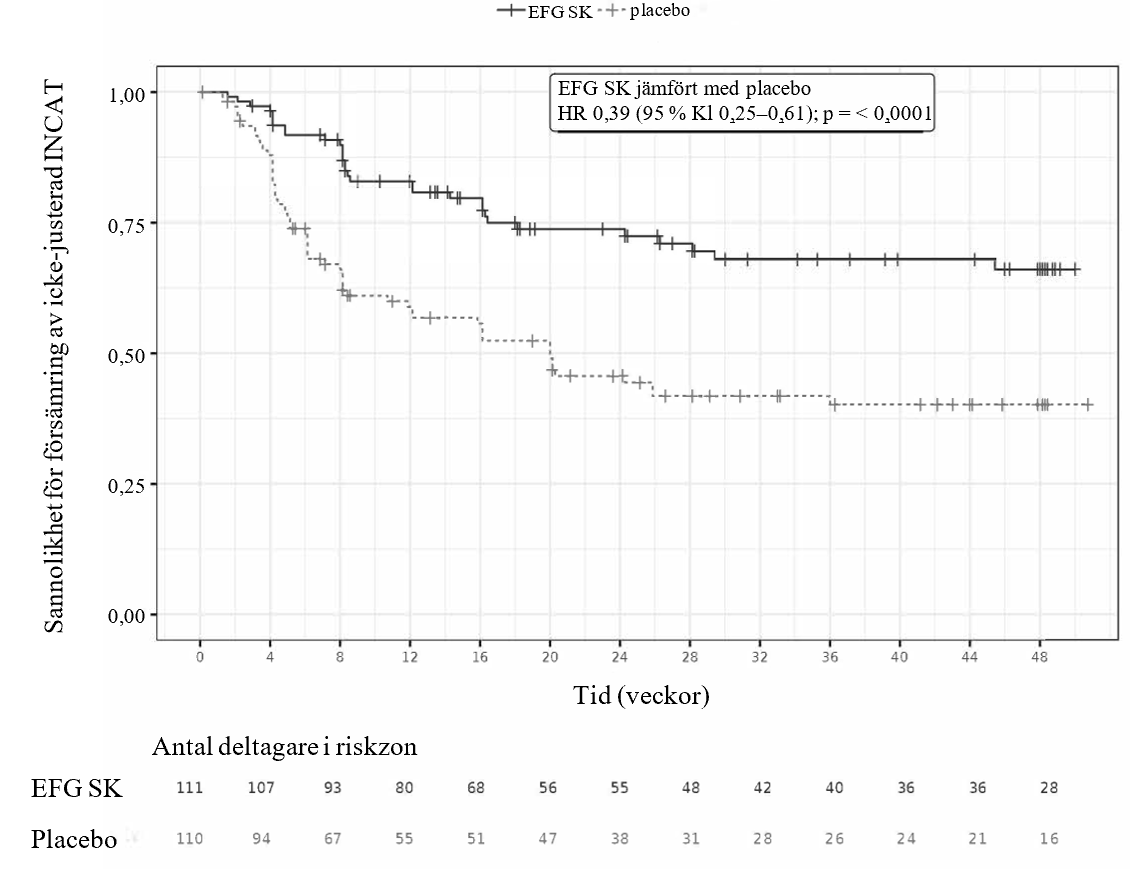
I steg B definierades det primära effektmåttet som tiden till förekomsten av det första beviset på klinisk försämring (en 1-poängs ökning av aINCAT jämfört med baslinjen i steg B, vilket bekräftades vid ett påföljande besök efter den första 1-poängsökningen av aINCAT eller en ≥ 2-poängs ökning av aINCAT jämfört med baslinjen i steg B). Patienter som fick efgartigimod alfa subkutant förblev återfallsfria (dvs. ingen klinisk försämring) signifikant längre än patienter som fick placebo, vilket visades av riskkvoten på 0,394 [95 % KI (0,253; 0,614)]. 31 av 111 (27,9 %) av patienterna som fick efgartigimod alfa subkutant under steg B i studien fick återfall jämfört med 59 av 110 (53,6 %) av patienterna som fick placebo. Resultaten presenteras i tabell 7 och bild 2.
Tabell 7. Första beviset på klinisk försämring hos patienter med CIDP i studie ARGX‑113‑1802 steg B
| Tid till första aINCAT-ökning (klinisk försämring) | Steg B | |
Efgartigimod alfa SK (N = 111) | Placebo (N = 110) | |
| Riskkvot (95 % KI) | 0,394 (0,253; 0,614) p-värde < 0,0001 | |
| Mediantid i dagar (95 % KI) | NC (NC; NC) | 140,0 (75,0; NC) |
NC = ej beräknat; N = antal patienter i analysuppsättningen; aINCAT = justerad Inflammatory Neuropathy Cause and Treatment
Bild 2. Tid till den första försämringen av aINCAT (Kaplan-Meier-kurva) hos patienter med CIDP i studie ARGX-113-1802 steg B
Pediatrisk population
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för Vyvgart för en eller flera grupper av den pediatriska populationen för myasthenia gravis (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för Vyvgart för alla grupper av den pediatriska populationen för CIDP (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption
Baserat på en analys av farmakokinetiska data från en population är den uppskattade biotillgängligheten med efgartigimod alfa 1 000 mg subkutant 77 %.
Medelvärdet för Ctrough efter 4 administreringar en gång i veckan med efgartigimod alfa 1 000 mg subkutant och efgartigimod alfa 10 mg/kg intravenöst var 22,0 µg/ml (37 % CV) respektive 14,9 µg/ml (43 % CV). AUC0-168h för efgartigimod alfa efter administrering av en behandlingscykel med 1 000 mg subkutant och 10 mg/kg intravenöst var jämförbara.
Hos patienter som fick kontinuerlig subkutan administrering av efgartigimod alfa 1 000 mg en gång i veckan varierade medelvärdet för Ctrough mellan 14,9 och 20,1 µg/ml.
Distribution
Baserat på en analys av farmakokinetiska data från en population med friska deltagare och patienter är distributionsvolymen 18 liter.
Metabolism
Efgartigimod alfa förväntas brytas ned av proteolytiska enzymer till små peptider och aminosyror.
Eliminering
Den slutliga halveringstiden är 80 till 120 timmar (3 till 5 dagar). Baserat på en analys av farmakokinetiska data från en population är clearance 0,128 l/h. Molekylvikten på efgartigimod alfa är cirka 54 kDa, vilket är gränsen för molekyler som filtreras genom njurarna.
Linjäritet/icke-linjäritet
Den farmakokinetiska profilen för efgartigimod alfa är linjär, oberoende av dos och tid med minimal ackumulering.
Särskilda populationer
Ålder, könstillhörighet, rastillhörighet och kroppsvikt
Farmakokinetiken för efgartigimod alfa påverkades varken av ålder (19‑84 år), könstillhörighet, rastillhörighet eller kroppsvikt.
Nedsatt njurfunktion
Inga specifika farmakokinetiska studier på patienter med nedsatt njurfunktion har utförts.
Effekten av njurfunktionsmarkören estimerad glomerulär filtrationshastighet (eGFR) som kovariat i en farmakokinetisk populationsanalys visade på en exponeringsökning (11 % till 21 %) hos patienterna med lätt nedsatt njurfunktion (eGFR 60‑89 ml/min/1,73 m2). Ingen specifik dosjustering rekommenderas för patienter med lätt nedsatt njurfunktion.
Det finns otillräckligt med data om effekten av måttligt nedsatt njurfunktion (eGFR 30‑59 ml/min/1,73 m2) och svårt nedsatt njurfunktion (eGFR < 30 ml/min/1,73 m2) på de farmakokinetiska parametrarna för efgartigimod alfa.
Nedsatt leverfunktion
Inga specifika farmakokinetiska studier har utförts på patienter med nedsatt leverfunktion.
Effekten av leverfunktionsmarkörer som kovariater i en farmakokinetisk populationsanalys visade ingen påverkan på farmakokinetiken hos efgartigimod alfa.
Prekliniska säkerhetsuppgifter
Gängse studier avseende säkerhetsfarmakologi och allmäntoxicitet visade inte några särskilda risker för människa.
I reproduktionsstudier på råttor och kaniner resulterade intravenös administrering av efgartigimod alfa inte i några biverkningar på fertilitet och graviditet och inte heller observerades några teratogena effekter vid dosnivåer på upp till motsvarade 11 gånger (råttor) och 56 gånger (kaniner) exponeringen på 10 mg/kg hos människa baserat på AUC.
Karcinogenicitet och gentoxicitet
Inga studier har utförts för att bedöma den karcinogena och gentoxiska potentialen hos efgartigimod alfa.
Hyaluronidas finns i de flesta vävnader i människokroppen. Gängse studier för rekombinant humant hyaluronidas avseende allmäntoxicitet, inklusive säkerhetsfarmakologiska effektmått, visade inte några särskilda risker för människa. Reproduktionstoxikologiska studier med rHuPH20 visade embryofetal toxicitet hos möss vid hög systemisk exponering men visade ingen teratogen potential.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Rekombinant humant hyaluronidas (rHuPH20)
L-histidin
L-histidinhydrokloridmonohydrat
L-metionin
Polysorbat 20 (E432)
Natriumklorid
Sackaros
Vatten för injektion
Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
18 månader
Vid behov kan oöppnade injektionsflaskor förvaras i rumstemperatur (högst 30 °C) i upp till 3 dagar. Efter förvaring i rumstemperatur kan oöppnade injektionsflaskor återföras till kylskåpet. Om de förvaras utanför och sedan återförs till kylning bör den totala sammanlagda tiden utanför kylskåpet inte överstiga 3 dagar.
Ur mikrobiell synvinkel ska läkemedlet användas omedelbart, såvida inte risken för mikrobiell kontaminering med metoden för beredning av sprutan är utesluten. Om den inte används omedelbart är förvaringstiden och förvaringsförhållandena vid öppnad förpackning användarens ansvar.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C‑8 °C).
Får ej frysas.
Förvaras i originalförpackningen. Ljuskänsligt.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Ei markkinoilla olevia pakkauksia.
PF-selosteen tieto
5,6 ml lösning i en 6 ml injektionsflaska av glas typ I med gummipropp, aluminiumförsegling och snäpplock av polypropen.
Förpackningar med 1 injektionsflaska.
Läkemedlets utseende:
Gulaktigt, klart till opaliserande, pH 6,0.
Särskilda anvisningar för destruktion och övrig hantering
Vyvgart levereras som en färdig lösning i engångsinjektionsflaska. Läkemedlet behöver inte spädas.
Kontrollera visuellt att innehållet i injektionsflaskan är en gulaktig, klar till opaliserande lösning och utan partiklar. Injektionsflaskan får inte användas om synliga partiklar observeras.
Efter att du tagit ut injektionsflaskan ur kylen, vänta minst 15 minuter innan du injicerar för att låta lösningen uppnå rumstemperatur (se avsnitt Hållbarhet).
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
VYVGART injektioneste, liuos
1000 mg 1 kpl
- Ei korvausta.
Atc-kod
L04AA58
Datum för översyn av produktresumén
05.01.2026
Yhteystiedot
Industriepark-Zwijnaarde 7
9052 Gent
Belgium
0800 412838
medinfofi@argenx.com