
TREMFYA ONEPRESS injektioneste, liuos, esitäytetty kynä 100 mg, TREMFYA PUSHPEN injektioneste, liuos, esitäytetty kynä 100 mg
Vaikuttavat aineet ja niiden määrät
Tremfya 100 mg OnePress injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 100 mg guselkumabia 1 ml:ssa liuosta.
Tremfya 100 mg PushPen injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 100 mg guselkumabia 1 ml:ssa liuosta.
Guselkumabi on ihmisen immunoglobuliini-G1-lambdan (IgG1λ) monoklonaalinen vasta-aine, joka tuotetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa.
Apuaine(et), joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 0,5 mg polysorbaatti 80:tä (E433) per esitäytetty kynä, mikä vastaa 0,5 mg:aa/ml.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Aikuisiän läiskäpsoriaasi
Tremfya on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon aikuisille, joille harkitaan systeemistä hoitoa.
Nivelpsoriaasi
Tremfya on yksinään tai yhdistelmänä metotreksaatin kanssa tarkoitettu aktiivisen nivelpsoriaasin hoitoon aikuisille potilaille, joiden vaste aiempaan taudin kulkuun vaikuttavaan reumalääkkeeseen (DMARD-lääkkeeseen) on ollut riittämätön tai jotka eivät ole sietäneet tällaista hoitoa (ks. kohta Farmakodynamiikka).
Haavainen paksusuolitulehdus
Tremfya on tarkoitettu keskivaikean tai vaikean aktiivisen haavaisen paksusuolitulehduksen hoitoon aikuisille potilaille, jotka eivät ole saaneet riittävää vastetta tavanomaiseen tai biologiseen hoitoon, joilla vaste on hävinnyt tai jotka eivät ole sietäneet tällaista hoitoa.
Crohnin tauti
Tremfya on tarkoitettu keskivaikean tai vaikean aktiivisen Crohnin taudin hoitoon aikuisille potilaille, jotka eivät ole saaneet riittävää vastetta tavanomaiseen tai biologiseen hoitoon, joilla vaste on hävinnyt tai jotka eivät ole sietäneet tällaista hoitoa.
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Tämä lääkevalmiste on tarkoitettu käytettäväksi sen käyttöaiheisiin kuuluvien sairauksien diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus
Läiskäpsoriaasi
Suositeltu annos on 100 mg injektiona ihon alle viikoilla 0 ja 4, minkä jälkeen hoitoa jatketaan kahdeksan viikon välein annettavilla ylläpitoannoksilla.
Jos potilaalla ei todeta vastetta 16 viikon hoidon jälkeen, hoidon lopettamista on harkittava.
Nivelpsoriaasi
Suositeltu annos on 100 mg injektiona ihon alle viikoilla 0 ja 4, minkä jälkeen hoitoa jatketaan kahdeksan viikon välein annettavilla ylläpitoannoksilla. Jos potilaalla on kliinisen arvion perusteella suuri nivelvaurioriski, voidaan harkita annoksen 100 mg antamista neljän viikon välein (ks. kohta Farmakodynamiikka).
Jos potilaalla ei todeta vastetta 24 viikon hoidon jälkeen, hoidon lopettamista on harkittava.
Haavainen paksusuolitulehdus
Kumpaa tahansa seuraavista kahdesta induktiohoito-ohjelmasta suositellaan:
- 200 mg infuusiona laskimoon viikolla 0, viikolla 4 ja viikolla 8. Ks. Tremfya 200 mg infuusiokonsentraatin, liuosta varten, valmisteyhteenveto.
tai
- 400 mg injektiona ihon alle (kahtena peräkkäisenä injektiona, joista kumpikin on 200 mg) viikolla 0, viikolla 4 ja viikolla 8. Ks. Tremfya 200 mg injektionesteen, liuoksen, valmisteyhteenveto.
Induktiohoito-ohjelman päättymisen jälkeen suositeltu ylläpitoannos viikosta 16 alkaen on 100 mg injektiona ihon alle kahdeksan viikon välein. Jos potilaalla ei todeta kliinisen arvion perusteella riittävää hoidollista hyötyä induktiohoidosta, voidaan vaihtoehtoisesti harkita ylläpitoannosta 200 mg injektiona ihon alle viikosta 12 alkaen ja sen jälkeen neljän viikon välein (ks. kohta Farmakodynamiikka). Ks. 200 mg:n annosta koskevat tiedot Tremfya 200 mg injektionesteen, liuoksen, valmisteyhteenvedosta.
Immunomodulaattoreiden ja/tai kortikosteroidien käyttöä voidaan jatkaa guselkumabihoidon aikana. Jos potilas on saanut vasteen guselkumabihoitoon, kortikosteroidien käyttöä voidaan vähentää tai kortikosteroidihoito voidaan lopettaa tavanomaisen hoitokäytännön mukaisesti.
Jos potilaalla ei ole havaittu hoidollista hyötyä 24 viikon jälkeen, hoidon lopettamista pitää harkita.
Crohnin tauti
Kumpaa tahansa seuraavista kahdesta induktiohoito-ohjelmasta suositellaan:
- 200 mg infuusiona laskimoon viikolla 0, viikolla 4 ja viikolla 8. Ks. Tremfya 200 mg infuusiokonsentraatin, liuosta varten, valmisteyhteenveto.
tai
- 400 mg injektiona ihon alle (kahtena peräkkäisenä injektiona, joista kumpikin on 200 mg) viikolla 0, viikolla 4 ja viikolla 8.Ks. Tremfya 200 mg injektionesteen, liuoksen, valmisteyhteenveto.
Induktiohoito-ohjelman päättymisen jälkeen suositeltu ylläpitoannos viikosta 16 alkaen on 100 mg injektiona ihon alle kahdeksan viikon välein. Jos potilaalla ei todeta kliinisen arvion perusteella riittävää hoidollista hyötyä induktiohoidosta, voidaan vaihtoehtoisesti harkita ylläpitoannosta 200 mg injektiona ihon alle viikosta 12 alkaen ja sen jälkeen neljän viikon välein (ks. kohta Farmakodynamiikka). Ks. 200 mg:n annosta koskevat tiedot Tremfya 200 mg injektionesteen, liuoksen, valmisteyhteenvedosta.
Immunomodulaattoreiden ja/tai kortikosteroidien käyttöä voidaan jatkaa guselkumabihoidon aikana. Jos potilas on saanut vasteen guselkumabihoitoon, kortikosteroidien käyttöä voidaan vähentää tai kortikosteroidihoito voidaan lopettaa tavanomaisen hoitokäytännön mukaisesti.
Jos potilaalla ei ole havaittu hoidollista hyötyä 24 viikon jälkeen, hoidon lopettamista pitää harkita.
Väliin jäänyt annos
Jos annos on jäänyt väliin, se on annettava mahdollisimman pian. Sen jälkeen hoitoa jatketaan tavanomaisen hoitoaikataulun mukaisesti.
Erityiset potilasryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka).
Iältään ≥ 65-vuotiaista potilaista on vähän tietoja, ja iältään ≥ 75-vuotiaista potilaista on hyvin vähän tietoja (ks. kohta Farmakokinetiikka).
Munuaisten tai maksan vajaatoiminta
Tremfya‑valmistetta ei ole tutkittu näillä potilasryhmillä. Näiden sairauksien ei yleisesti oleteta vaikuttavan merkittävästi monoklonaalisten vasta-aineiden farmakokinetiikkaan eikä annoksen muuttamista katsota tarpeelliseksi. Lisätietoja guselkumabin eliminaatiosta, ks. kohta Farmakokinetiikka.
Pediatriset potilaat
Tremfya‑valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten potilaiden haavaisen paksusuolitulehduksen, Crohnin taudin ja nivelpsoriaasin hoidossa ja alle 6 vuoden ikäisten potilaiden psoriaasin hoidossa ei ole varmistettu. Tietoja ei ole saatavilla. Tremfya 100 mg injektionestettä, liuosta, esitäytettyä kynää ei suositella alle 18 vuoden ikäisten lasten hoitoon, koska tiedot tehosta ja turvallisuudesta ovat riittämättömät. Saatavissa olevat tiedot muista valmistemuodoista on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka.
Antotapa
Vain ihon alle. Injektiokohtia ovat vatsa, reisi ja olkavarren takaosa. Tremfya-valmistetta ei pidä injisoida ihoalueille, missä on aristusta, mustelma, punoitusta, kovettuma, paksuuntumista tai hilseilyä. Jos mahdollista, injektiokohtina ei pidä käyttää ihoalueita, joilla on psoriaasia.
Kun potilas on saanut asianmukaisen opastuksen ihon alle annettavien injektioiden injektiotekniikkaan, hän voi injisoida Tremfya‑injektiot itse, jos lääkäri katsoo sen tarkoituksenmukaiseksi. Lääkärin on kuitenkin varmistettava potilaan asianmukainen seuranta. Potilasta on neuvottava injisoimaan koko liuosmäärä kotelon sisältämien Käyttöohjeiden mukaisesti.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen valmistelusta ennen lääkkeen antoa.
Vasta-aiheet
Vakava yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Kliinisesti tärkeät aktiiviset infektiot (esim. aktiivinen tuberkuloosi, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Guselkumabi saattaa lisätä infektioiden riskiä. Jos potilaalla on kliinisesti tärkeä aktiivinen infektio, hoitoa ei saa aloittaa ennen kuin infektio on parantunut tai sitä on hoidettu riittävästi.
Guselkumabihoitoa saavia potilaita pitää kehottaa hakeutumaan lääkäriin, jos heille ilmaantuu kliinisesti tärkeän kroonisen tai akuutin infektion oireita tai löydöksiä. Jos potilaalle kehittyy kliinisesti tärkeä tai vakava infektio tai potilas ei saa hoitovastetta tavanomaiseen hoitoon, potilaan tilaa on seurattava tarkoin ja hoito on keskeytettävä, kunnes infektio on parantunut.
Tuberkuloosin tutkiminen ennen hoitoa
Potilailta on tutkittava tuberkuloosi-infektio ennen hoidon aloittamista. Guselkumabihoitoa saavia potilaita pitää seurata hoidon aikana ja sen jälkeen aktiivisen tuberkuloosin oireiden ja löydösten havaitsemiseksi. Jos potilaalla on aiemmin ollut piilevä tai aktiivinen tuberkuloosi, jonka riittävästä hoidosta ei voida varmistua, tuberkuloosihoidon antamista on harkittava ennen hoidon aloittamista.
Yliherkkyys
Valmisteen markkinoille tulon jälkeen on raportoitu vakavia yliherkkyysreaktioita, mukaan lukien anafylaksiaa (ks. kohta Haittavaikutukset). Osa vakavista yliherkkyysreaktioista ilmeni useita päiviä guselkumabihoidon jälkeen, mukaan lukien tapaukset, joihin liittyi urtikariaa ja hengenahdistusta. Jos ilmaantuu vakava yliherkkyysreaktio, guselkumabin antaminen on lopetettava heti, ja asianmukainen hoito on aloitettava.
Kohonnut maksan transaminaasipitoisuus
Nivelpsoriaasia koskeneissa kliinisissä tutkimuksissa suurentuneiden maksaentsyymipitoisuuksien ilmaantuvuus oli guselkumabihoitoa neljän viikon välein saaneilla potilailla suurempi kuin guselkumabihoitoa kahdeksan viikon välein tai lumehoitoa saaneilla potilailla (ks. kohta Haittavaikutukset).
Määrättäessä guselkumabia neljän viikon välein nivelpsoriaasin hoitoon maksaentsyymipitoisuus suositellaan arvioimaan hoitoa aloitettaessa ja sen jälkeen potilaan tavanomaisen hoidon mukaisesti. Jos alaniiniaminotransferaasiarvon (ALAT) tai aspartaattiaminotransferaasiarvon (ASAT) havaitaan suurentuneen ja epäillään lääkeaineen aiheuttamaa maksavauriota, hoito pitää keskeyttää tilapäisesti, kunnes diagnoosi on suljettu pois.
Rokotukset
Kaikkien asianmukaisten rokotusten antamista voimassa olevien rokotusohjeiden mukaisesti on harkittava ennen hoidon aloittamista. Guselkumabihoitoa saaville potilaille ei saa antaa samanaikaisesti eläviä taudinaiheuttajia sisältäviä rokotteita. Vasteesta eläville tai inaktivoiduille rokotteille ei ole tietoja.
Ennen eläviä viruksia tai eläviä bakteereja sisältävien rokotusten antamista hoito pitää keskeyttää vähintään 12 viikoksi viimeisen annoksen antamisen jälkeen, ja hoitoa voidaan jatkaa aikaisintaan 2 viikon kuluttua rokotuksesta. Lääkkeen määräävän lääkärin pitää tarkistaa kyseisen rokotteen valmisteyhteenvedosta lisätiedot ja ohjeet immunosuppressiivisten aineiden samanaikaisesta käytöstä rokotuksen jälkeen.
Apuaineet, joiden vaikutus tunnetaan
Polysorbaatti 80 -pitoisuus
Tämä lääkevalmiste sisältää 0,5 mg polysorbaatti 80:tä (E433) per esitäytetty kynä, mikä vastaa 0,5 mg:aa/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutukset CYP450:n substraattien kanssa
Keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavilla potilailla tehdyssä vaiheen I tutkimuksessa muutokset systeemisessä altistuksessa (Cmax ja AUCinf) midatsolaamille, S‑varfariinille, omepratsolille, dekstrometorfaanille ja kofeiinille eivät olleet guselkumabikerta-annoksen jälkeen kliinisesti oleellisia, mikä osoittaa, että yhteisvaikutukset guselkumabin ja eri CYP-entsyymien substraattien (CYP3A4, CYP2C9, CYP2C19, CYP2D6 ja CYP1A2) välillä eivät ole todennäköisiä. Annosmuutokset eivät ole tarpeen, jos guselkumabia ja CYP450:n substraatteja annetaan samanaikaisesti.
Samanaikainen immunosuppressiivinen hoito tai valohoito
Guselkumabin tehoa ja turvallisuutta yhdistelmänä immunosuppressiivisten lääkeaineiden, mukaan lukien biologisten lääkkeiden, tai valohoidon kanssa ei ole tutkittu psoriaasia koskevissa tutkimuksissa. Nivelpsoriaasitutkimuksissa metotreksaatin samanaikainen käyttö ei näyttänyt vaikuttavan guselkumabin turvallisuuteen tai tehoon.
Haavaista paksusuolitulehdusta ja Crohnin tautia koskeneissa tutkimuksissa immunomodulaattoreiden (esim. atsatiopriini, 6-merkaptopuriini) tai kortikosteroidien samanaikainen käyttö ei näyttänyt vaikuttavan guselkumabin turvallisuuteen tai tehoon.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisymenetelmää hoidon aikana ja vähintään 12 viikon ajan hoidon päättymisen jälkeen.
Raskaus
Guselkumabin käytöstä raskaana oleville naisille on vain vähän tietoja. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria eikä epäsuoria haitallisia vaikutuksia raskauteen, alkion tai sikiön kehitykseen, synnytykseen tai syntymänjälkeiseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Tremfya‑valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö guselkumabi ihmisillä äidinmaitoon. Ihmisen IgG:t erittyvät tunnetusti äidinmaitoon muutaman päivän ajan synnytyksen jälkeen. Pian sen jälkeen pitoisuudet pienenevät vähäisiksi. Näin ollen imetettävälle lapselle tänä ajanjaksona aiheutuvaa riskiä ei voida sulkea pois. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö Tremfya-hoidosta ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille. Ks. kohdasta Prekliiniset tiedot turvallisuudesta tietoja guselkumabin erittymisestä eläinten (cynomolgus-apinoiden) maitoon.
Hedelmällisyys
Guselkumabin vaikutusta ihmisen hedelmällisyyteen ei ole tutkittu. Eläinkokeissa ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tremfya‑valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisin haittavaikutus oli hengitystieinfektiot (kliinisissä tutkimuksissa esiintyvyys noin 8 % haavaista paksusuolitulehdusta sairastavilla potilailla, 11 % Crohnin tautia sairastavilla potilailla ja 15 % psoriaasia ja nivelpsoriaasia sairastavilla potilailla).
Tremfya-hoitoa saaneiden psoriaasia, nivelpsoriaasia, haavaista paksusuolitulehdusta tai Crohnin tautia sairastavien potilaiden kokonaisturvallisuusprofiili on samankaltainen.
Haittavaikutustaulukko
Taulukossa 1 luetellaan haittavaikutukset, joita esiintyi psoriaasia, nivelpsoriaasia, haavaista paksusuolitulehdusta ja Crohnin tautia koskeneissa kliinisissä tutkimuksissa sekä valmisteen markkinoille tulon jälkeen raportoidut haittavaikutukset. Haittavaikutukset on luokiteltu MedDRA-elinjärjestelmän ja esiintymistiheyden mukaan seuraavaa käytäntöä noudattaen: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 1. Haittavaikutusluettelo | ||
| Elinjärjestelmä | Esiintymistiheys | Haittavaikutukset |
| Infektiot | Hyvin yleinen | Hengitystieinfektiot |
| Melko harvinainen | Herpes simplex ‑infektiot | |
| Melko harvinainen | Silsainfektiot | |
| Melko harvinainen | Gastroenteriitti | |
| Immuunijärjestelmä | Harvinainen | Yliherkkyys |
| Harvinainen | Anafylaksia | |
| Hermosto | Yleinen | Päänsärky |
| Ruoansulatuselimistö | Yleinen | Ripuli |
| Iho ja ihonalainen kudos | Yleinen | Ihottuma |
| Melko harvinainen | Urtikaria | |
| Luusto, lihakset ja sidekudos | Yleinen | Nivelkipu |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Injektiokohdan reaktiot |
| Tutkimukset | Yleinen | Suurentunut transaminaasipitoisuus |
| Melko harvinainen | Pienentynyt neutrofiilien määrä | |
Valikoitujen haittavaikutusten kuvaus
Suurentunut transaminaasipitoisuus
Kahdessa vaiheen III kliinisessä nivelpsoriaasitutkimuksessa lumekontrolloitujen jaksojen aikana raportoitiin haittavaikutuksina suurentuneita transaminaasipitoisuuksia (joita olivat suurentunut ALAT-arvo, suurentunut ASAT-arvo, suurentunut maksaentsyymipitoisuus, suurentunut transaminaasipitoisuus, poikkeavuudet maksan toimintakokeissa, hypertransaminasemia) guselkumabihoitoa saaneissa ryhmissä (100 mg ihon alle neljän viikon välein saaneessa ryhmässä 8,6 % ja 100 mg ihon alle kahdeksan viikon välein saaneessa ryhmässä 8,3 %) yleisemmin kuin lumeryhmässä (4,6 %). Suurentuneita transaminaasiarvoja (kuten edellä) raportoitiin 1. vuonna haittavaikutuksina 12,9 %:lla potilaista hoitoa neljän viikon välein saaneessa ryhmässä ja 11,7 %:lla potilaista hoitoa kahdeksan viikon välein saaneessa ryhmässä.
Transaminaasipitoisuuden (ASAT ja ALAT) suureneminen oli laboratoriotutkimusten perusteella useimmiten ≤ 3 x ULN (ULN = upper limit of normal, viitearvojen yläraja). Transaminaasipitoisuus suureni harvoin tasoille > 3 – ≤ 5 x ULN ja > 5 x ULN, ja tällaista havaittiin guselkumabihoitoa neljän viikon välein saaneessa ryhmässä useammin kuin guselkumabihoitoa kahdeksan viikon välein saaneessa ryhmässä (taulukko 2). Kaksi vuotta kestäneen vaiheen III kliinisen nivelpsoriaasitutkimuksen loppuun mennessä havaittiin, että esiintyvyys vaikeusasteittain ja hoitoryhmittäin oli samankaltainen.
| Taulukko 2. Suurentuneen transaminaasipitoisuuden esiintyvyys lähtötilanteen jälkeen kahdessa vaiheen III kliinisessä nivelpsoriaasitutkimuksessa | |||||
| 24 viikon aikanaa | 1 vuoden aikanab | ||||
Lumelääke N = 370c | guselkumabi 100 mg 8 viikon välein N = 373c | guselkumabi 100 mg 4 viikon välein N = 371c | guselkumabi 100 mg 8 viikon välein N = 373c | guselkumabi 100 mg 4 viikon välein N = 371c | |
| ALAT | |||||
| > 1 – ≤ 3 x ULN | 30,0 % | 28,2 % | 35,0 % | 33,5 % | 41,2 % |
| > 3 – ≤ 5 x ULN | 1,4 % | 1,1 % | 2,7 % | 1,6 % | 4,6 % |
| > 5 x ULN | 0,8 % | 0,8 % | 1,1 % | 1,1 % | 1,1 % |
| ASAT | |||||
| > 1 – ≤ 3 x ULN | 20,0 % | 18,8 % | 21,6 % | 22,8 % | 27,8 % |
| > 3 – ≤ 5 x ULN | 0,5 % | 1,6 % | 1,6 % | 2,9 % | 3,8 % |
| > 5 x ULN | 1,1 % | 0,5 % | 1,6 % | 0,5 % | 1,6 % |
a lumekontrolloitu jakso b mukana ei ole lumehoitoon lähtötilanteessa satunnaistettuja ja myöhemmin guselkumabihoitoon siirtyneitä potilaita c niiden potilaiden lukumäärä, joista ajanjakson aikana vähintään yksi lähtötilanteen jälkeen tehty tietyn laboratoriokokeen tulos | |||||
Kliinisissä psoriaasitutkimuksissa ensimmäisen vuoden aikana suurentuneen transaminaasipitoisuuden (ALAT ja ASAT) esiintyvyys oli guselkumabihoitoa kahdeksan viikon välein saaneessa ryhmässä samankaltainen kuin kliinisissä nivelpsoriaasitutkimuksissa guselkumabihoitoa kahdeksan viikon välein saaneessa ryhmässä. Suurentuneiden transaminaasipitoisuuksien ilmaantuvuus ei lisääntynyt viiden vuoden aikana kutakin guselkumabihoitovuotta kohden arvioituna. Transaminaasipitoisuuksien suureneminen oli useimmiten ≤ 3 x ULN.
Transaminaasipitoisuuden suureneminen oli useimmiten ohimenevää eikä johtanut hoidon lopettamiseen.
Yhdistetyissä vaiheen II ja vaiheen III kliinisissä Crohnin tautia koskeneissa tutkimuksissa suurentuneita transaminaasipitoisuuksia (käsittää suurentuneen ALAT-arvon, suurentuneen ASAT-arvon, suurentuneen maksaentsyymipitoisuuden, suurentuneen transaminaasipitoisuuden ja suurentuneet maksan toimintakokeiden tulokset) koskevia haittavaikutuksia raportoitiin koko lumekontrolloidun induktiojakson (viikot 0–12) aikana yleisemmin guselkumabihoitoa saaneissa ryhmissä (1,7 %:lla potilaista) kuin lumeryhmässä (0,6 %:lla potilaista). Yhdistetyissä vaiheen II ja vaiheen III kliinisissä Crohnin tautia koskeneissa tutkimuksissa suurentuneita transaminaasipitoisuuksia (käsittää suurentuneen ALAT-arvon, suurentuneen ASAT-arvon, suurentuneen maksaentsyymipitoisuuden, suurentuneen transaminaasipitoisuuden, poikkeavan maksan toiminnan ja suurentuneet maksan toimintakokeiden tulokset) koskevia haittavaikutuksia raportoitiin noin yhden vuoden raportointijakson aikana 3,4 %:lla potilaista 200 mg guselkumabia ihon alle neljän viikon välein saaneessa hoitoryhmässä ja 4,1 %:lla potilaista 100 mg guselkumabia ihon alle kahdeksan viikon välein saaneessa hoitoryhmässä verrattuna 2,4 %:iin lumeryhmässä.
Yhdistetyissä vaiheen II ja vaiheen III kliinisissä Crohnin tautia koskeneissa tutkimuksissa tehtyjen laboratoriotutkimusten perusteella kohonneiden ALAT- tai ASAT-arvojen yleisyys oli pienempi kuin nivelpsoriaasia koskeneissa vaiheen III kliinisissä tutkimuksissa. Yhdistettyjen vaiheen II ja vaiheen III kliinisten Crohnin tautia koskeneiden tutkimusten lumekontrolloidun jakson (viikko 12) aikana guselkumabihoitoa saaneilla potilailla raportoitiin tasolle ≥ 3 x ULN kohonneita ALAT-arvoja (< 1 %:lla potilaista) ja ASAT-arvoja (< 1 %:lla potilaista). Yhdistetyissä vaiheen II ja vaiheen III kliinisissä Crohnin tautia koskeneissa tutkimuksissa tasolle ≥ 3 x ULN kohonneita ALAT-arvoja ja/tai ASAT-arvoja raportoitiin noin yhden vuoden raportointijakson aikana 2,7 %:lla potilaista 200 mg guselkumabia ihon alle neljän viikon välein saaneessa ryhmässä ja 2,6 %:lla potilaista 100 mg guselkumabia ihon alle kahdeksan viikon välein saaneessa ryhmässä verrattuna 1,9 %:iin lumeryhmässä. Transaminaasipitoisuuden suureneminen oli useimmiten ohimenevää eikä johtanut hoidon keskeyttämiseen.
Pienentynyt neutrofiilimäärä
Pienentyneitä neutrofiilimääriä raportoitiin kahdessa vaiheen III kliinisessä nivelpsoriaasitutkimuksessa lumekontrolloidun jakson aikana haittavaikutuksena guselkumabihoitoa saaneessa ryhmässä (0,9 %) yleisemmin kuin lumeryhmässä (0 %). Pienentyneitä neutrofiilimääriä raportoitiin 1. vuonna haittavaikutuksena 0,9 %:lla guselkumabihoitoa saaneista potilaista. Veren neutrofiilimäärän väheneminen oli useimmiten lievää, ohimenevää, infektioon liittymätöntä, eikä se johtanut hoidon keskeyttämiseen.
Gastroenteriitti
Gastroenteriittiä esiintyi kahden vaiheen III kliinisen psoriaasitutkimuksen lumekontrolloidun jakson aikana yleisemmin guselkumabihoitoa saaneessa ryhmässä (1,1 %) kuin lumeryhmässä (0,7 %). Viikkoon 264 mennessä 5,8 % kaikista guselkumabihoitoa saaneista potilaista raportoi gastroenteriittiä. Haittavaikutuksena esiintyneet gastroenteriitit eivät olleet vakavia eivätkä johtaneet viikkoon 264 mennessä guselkumabihoidon keskeyttämiseen. Gastroenteriitin esiintyvyyden havaittiin olleen kliinisten nivelpsoriaasitutkimusten lumekontrolloidun jakson aikana samankaltainen kuin kliinisissä psoriaasitutkimuksissa.
Injektiokohdan reaktiot
Injektiokohdan reaktioita liittyi kahden vaiheen III kliinisen psoriaasitutkimuksen viikkoon 48 mennessä 0,7 %:iin guselkumabi‑injektioista ja 0,3 %:iin lumeinjektioista. Viikkoon 264 mennessä 0,4 %:iin guselkumabi-injektioista liittyi injektiokohdan reaktioita. Injektiokohdan reaktiot olivat yleensä vaikeusasteeltaan lieviä tai keskivaikeita, yksikään tapauksista ei ollut vakava; yksi tapaus johti guselkumabihoidon keskeyttämiseen.
Kahdessa vaiheen III kliinisessä nivelpsoriaasitutkimuksessa viikkoon 24 mennessä yhden tai useampia injektiokohdan reaktioita raportoineiden potilaiden lukumäärä oli pieni ja guselkumabiryhmissä hieman suurempi kuin lumeryhmässä: guselkumabihoitoa kahdeksan viikon välein saaneessa ryhmässä 5 (1,3 %) potilasta, guselkumabihoitoa neljän viikon välein saaneessa ryhmässä 4 (1,1 %) potilasta ja lumeryhmässä 1 (0,3 %) potilas. Yksi potilas lopetti guselkumabihoidon kliinisen nivelpsoriaasitutkimuksen lumekontrolloidun jakson aikana injektiokohdan reaktion vuoksi. Yhden tai useamman injektiokohdan reaktion raportoineiden potilaiden osuus 1. vuonna oli 1,6 % guselkumabihoitoa kahdeksan viikon välein saaneessa ryhmässä ja 2,4 % guselkumabihoitoa neljän viikon välein saaneessa ryhmässä. Sellaisten injektioiden lukumäärän, joihin liittyi injektiokohdan reaktioita, havaittiin olleen kliinisten nivelpsoriaasitutkimusten lumekontrolloidun jakson aikana yleisesti samankaltainen kuin kliinisissä psoriaasitutkimuksissa.
Vaiheen III kliinisessä haavaisen paksusuolitulehduksen ylläpitohoitoa koskeneessa tutkimuksessa viikkoon 44 mennessä yhden tai useampia guselkumabista aiheutuneita injektiokohdan reaktioita raportoineiden osuus oli 200 mg guselkumabia ihon alle neljän viikon välein (haavaisen paksusuolitulehduksen ylläpitohoitoa koskeneessa vaiheen III kliinisessä tutkimuksessa 200 mg guselkumabia annettiin kahtena 100 mg:n injektiona) saaneiden potilaiden ryhmässä 7,9 % (2,5 %:ssa injektioista). 100 mg guselkumabia ihon alle kahdeksan viikon välein saaneessa ryhmässä ei raportoitu injektiokohdan reaktioita. Valtaosa injektiokohdan reaktioista oli lieviä eikä yksikään niistä ollut vakava.
Vaiheen II ja vaiheen III kliinisissä Crohnin tautia koskeneissa tutkimuksissa viikkoon 48 mennessä yhden tai useampia guselkumabista aiheutuneita injektiokohdan reaktioita raportoineiden osuus oli 4,1 % (0,8 %:ssa injektioista) hoitoryhmässä, joka sai 200 mg guselkumabia laskimoon induktiohoitona ja sen jälkeen 200 mg guselkumabia ihon alle neljän viikon välein, ja 1,4 % potilaista (0,6 %:ssa injektioista) ryhmässä, joka sai 200 mg guselkumabia laskimoon induktiohoitona ja sen jälkeen 100 mg ihon alle kahdeksan viikon välein. Injektiokohdan reaktiot olivat kaiken kaikkiaan lieviä eikä yksikään niistä ollut vakava.
Vaiheen III kliinisessä Crohnin tautia koskeneessa tutkimuksessa viikkoon 48 mennessä yhden tai useampia guselkumabista aiheutuneita injektiokohdan reaktioita raportoineiden osuus oli 7 % (1,3 %:ssa injektioista) hoitoryhmässä, joka sai 400 mg ihon alle induktiohoitona ja sen jälkeen 200 mg ihon alle neljän viikon välein, ja 4,3 % potilaista (0,7 %:ssa injektioista) ryhmässä, joka sai 400 mg guselkumabia ihon alle induktiohoitona ja sen jälkeen 100 mg ihon alle kahdeksan viikon välein. Valtaosa injektiokohdan reaktioista oli lieviä eikä yksikään niistä ollut vakava.
Immunogeenisuus
Guselkumabin immunogeenisuutta tutkittiin herkän ja lääkettä sietävän immunomäärityksen avulla.
Vaiheen II ja vaiheen III yhdistetyissä psoriaasi- ja nivelpsoriaasipotilaiden analyyseissä 5 %:lle (n = 145) guselkumabihoitoa saaneista potilaista kehittyi lääkevasta-aineita enintään 52 viikon hoidon aikana. Niistä potilaista, joille lääkevasta-aineita kehittyi, noin 8 %:lla (n = 12) vasta-aineet luokiteltiin neutraloiviksi, mikä vastaa 0,4 %:a kaikista guselkumabihoitoa saaneista potilaista. Vaiheen III yhdistetyissä psoriaasipotilaiden analyyseissa todettiin, että hoitoviikkoon 264 mennessä lääkevasta-aineita oli kehittynyt noin 15 %:lle guselkumabihoitoa saaneista potilaista. Niistä potilaista, joille lääkevasta-aineita kehittyi, noin 5 %:lla vasta-aineet luokiteltiin neutraloiviksi, mikä vastaa 0,76 %:a kaikista guselkumabihoitoa saaneista potilaista. Lääkevasta-aineisiin ei liittynyt hoidon tehon heikkenemistä eikä injektiokohdan reaktioiden kehittymistä.
Vaiheen II ja vaiheen III yhdistetyissä analyyseissä niistä haavaista paksusuolitulehdusta sairastavista potilaista, jotka saivat induktiohoidon laskimoon ja sen jälkeen ylläpitohoitoa ihon alle, noin 12 %:lle (n = 58) guselkumabihoitoa enintään 56 viikon ajan saaneista potilaista kehittyi lääkevasta-aineita. Niistä potilaista, joille lääkevasta-aineita kehittyi, noin 16 %:lla (n = 9) oli neutraloiviksi luokiteltavia vasta-aineita, mikä vastaa 2 %:a kaikista guselkumabihoitoa saaneista potilaista. Vaiheen III analyysissa viikkoon 24 saakka, induktiohoidon ihon alle ja sen jälkeen ylläpitohoitoa ihon alle saaneista haavaista paksusuolitulehdusta sairastavista potilaista noin 9 %:lle (n = 24) guselkumabihoitoa saaneista potilaista kehittyi lääkevasta-aineita. Niistä potilaista, joille lääkevasta-aineita kehittyi, 13 %:lla (n = 3) oli neutraloiviksi luokiteltavia vasta-aineita, mikä vastaa 1 %:a guselkumabihoitoa saaneista potilaista. Lääkevasta-aineisiin ei liittynyt heikompaa tehoa eikä injektiokohdan reaktioiden kehittymistä.
Vaiheen II ja vaiheen III yhdistetyissä Crohnin tautia sairastavien potilaiden analyyseissä viikkoon 48 saakka guselkumabia laskimoon annettavana induktiohoitona ja sen jälkeen ihon alle annettavana ylläpitohoitona saaneista potilaista noin 5 %:lle (n = 30) kehittyi lääkevasta-aineita. Niistä potilaista, joille lääkevasta-aineita kehittyi, noin 7 %:lla (n = 2) oli neutraloiviksi luokiteltavia vasta-aineita, mikä vastaa 0,3 %:a guselkumabihoitoa saaneista potilaista. Vaiheen III analyysissä viikkoon 48 saakka Crohnin tautia sairastavista potilaista, jotka saivat guselkumabihoitoa ihon alle annettavana induktiohoitona ja sen jälkeen ihon alle annettavana ylläpitohoitona, noin 9 %:lle (n = 24) kehittyi lääkevasta-aineita. Näistä potilaista 13 %:lla (n = 3) oli vasta-aineita, jotka luokiteltiin neutraloiviksi vasta-aineiksi, mikä vastaa 1 %:a guselkumabihoitoa saaneista potilaista. Lääkevasta-aineisiin ei liittynyt hoidon tehon heikkenemistä eikä injektiokohdan reaktioiden kehittymistä.
Pediatriset potilaat
Läiskäpsoriaasi
Guselkumabin turvallisuutta arvioitiin vaiheen III lumevalmisteella ja vaikuttavalla aineella kontrolloidussa tutkimuksessa pediatrisilla potilailla, joilla oli keskivaikea tai vaikea läiskäpsoriaasi. Tässä kliinisessä tutkimuksessa turvallisuutta arvioitiin viikkoon 52 saakka 120 potilaalla, jotka olivat iältään 6–17-vuotiaita. Ihon alle annettujen guselkumabi-injektioiden turvallisuusprofiili käytettäessä 6–17-vuotiaille pediatrisille potilaille 45 mg/0,45 ml esitäytettyä kynää tai 100 mg:n esitäytettyä ruiskua oli yhdenmukainen aikuisiän läiskäpsoriaasia koskeneissa tutkimuksissa raportoidun turvallisuusprofiilin kanssa (ks. kohta Annostus ja antotapa).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa guselkumabia on annettu laskimoon enintään annoksina 1 200 mg sekä ihon alle enintään annoksina 400 mg yhdellä lääkkeen antoon liittyneellä käynnillä, eikä annosta rajoittavaa toksisuutta esiintynyt. Yliannostapauksessa potilasta on seurattava haittavaikutusten oireiden ja löydösten havaitsemiseksi, ja tarkoituksenmukaista oireenmukaista hoitoa on annettava heti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC16.
Vaikutusmekanismi
Guselkumabi on ihmisen IgG1λ monoklonaalinen vasta-aine, joka sitoutuu erittäin spesifisesti ja suurella affiniteetilla antigeenin sitoutumiskohdan kautta selektiivisesti interleukiini-23-proteiiniin (IL‑23). IL-23 on sytokiini, joka osallistuu inflammatorisiin ja immuunivasteisiin. Guselkumabi estää IL-23:n sitoutumisen reseptoriinsa ja estää siten IL-23-riippuvaista solusignalointia ja tulehdusta edistävien sytokiinien vapautumista.
Läiskäpsoriaasia sairastavan potilaan ihossa IL‑23:n pitoisuus on koholla. Haavaista paksusuolitulehdusta tai Crohnin tautia sairastavilla potilailla koolonkudoksen IL-23-pitoisuus on koholla. Guselkumabin osoitettiin in vitro ‑malleissa estävän IL‑23:n biologista aktiivisuutta salpaamalla sen vuorovaikutuksen solun pinnalla olevan IL‑23-reseptorin kanssa ja keskeyttämällä siten IL‑23-välitteisen signaalinvälityksen, aktivaation ja sytokiinikaskadit. Guselkumabin kliiniset hoitovaikutukset läiskäpsoriaasissa, nivelpsoriaasissa, haavaisessa paksusuolitulehduksessa ja Crohnin taudissa perustuvat IL‑23:n sytokiinireitin salpaukseen.
Psoriaasissa, haavaisessa paksusuolitulehduksessa ja Crohnin taudissa Fc-gamma-reseptoria 1 (CD64) ilmentävien myeloidisolujen on osoitettu olevan tulehtuneessa kudoksessa IL-23:n pääasiallinen lähde. Guselkumabin on osoitettu in vitro salpaavan IL-23:a ja sitoutuvan CD64:ään. Nämä tulokset viittaavat siihen, että guselkumabi kykenee neutraloimaan IL-23:n tulehduksen lähdesoluissa.
Farmakodynaamiset vaikutukset
Guselkumabihoito johti vaiheen I tutkimuksessa IL-23/Th17-reitin geenien ilmentymisen ja psoriaasiin liittyvien geenien ilmentymisprofiilien vähenemiseen. Tämä osoitettiin läiskäpsoriaasia sairastavien potilaiden leesioista otettujen ihon biopsianäytteiden mRNA-analyyseillä viikolla 12 lähtötilanteeseen verrattuna. Guselkumabihoito johti samassa vaiheen I tutkimuksessa psoriaasin histologisten mittareiden paranemiseen viikolla 12, mukaan lukien epidermiksen ohenemiseen ja T‑solutiheyden vähenemiseen. Guselkumabihoitoa saaneilla potilailla havaittiin lisäksi vaiheen II ja vaiheen III läiskäpsoriaasitutkimuksissa seerumin IL-17A-, IL-17F- ja IL-22-pitoisuuksien pienenemistä verrattuna lumelääkkeeseen. Nämä tulokset ovat yhdenmukaisia läiskäpsoriaasia sairastavilla potilailla guselkumabihoidosta havaitun kliinisen hyödyn kanssa.
Vaiheen III nivelpsoriaasitutkimuksissa akuutin vaiheen proteiinien C-reaktiivisen proteiinin, seerumin amyloidi A:n ja IL-6:n sekä Th17-efektorisytokiinien IL-17A, IL-17F ja IL-22 pitoisuus seerumissa oli lähtötilanteessa koholla. Guselkumabi pienensi näiden proteiinien pitoisuutta 4 viikon kuluessa hoidon aloittamisesta. Guselkumabi lisäksi pienensi näiden proteiinien pitoisuutta viikkoon 24 mennessä lähtötilanteeseen ja myös lumelääkkeeseen verrattuna.
Haavaista paksusuolitulehdusta tai Crohnin tautia sairastavilla potilailla guselkumabihoito pienensi tulehdusmerkkiaineiden, mukaan lukien C-reaktiivisen proteiinin (CRP) ja ulosteen kalprotektiinin, pitoisuutta induktiohoitoviikkoon 12 saakka, ja pitoisuudet pysyivät pienentyneinä yhden ylläpitohoitovuoden ajan. IL-17A-, IL-22- ja IFNγ ‑proteiinien pitoisuus seerumissa oli pienentynyt jo viikolla 4, ja pitoisuudet pienenivät edelleen induktiohoitoviikkoon 12 saakka. Guselkumabi pienensi myös koolonin limakalvobiopsiasta mitattujen IL-17A:n, IL-22:n ja IFNγ:n RNA-pitoisuuksia viikon 12 aikapisteessä.
Kliininen teho ja turvallisuus
Läiskäpsoriaasi
Guselkumabin tehoa ja turvallisuutta arvioitiin kolmessa satunnaistetussa, kaksoissokkoutetussa, aktiivisella aineella kontrolloidussa vaiheen III tutkimuksessa aikuispotilailla, joilla oli keskivaikea tai vaikea läiskäpsoriaasi ja joille harkittiin valohoitoa tai systeemistä hoitoa.
VOYAGE 1 ja VOYAGE 2
Kahdessa tutkimuksessa (VOYAGE 1 ja VOYAGE 2) tutkittiin guselkumabin tehoa ja turvallisuutta 1829 aikuispotilaalla lumehoitoon ja adalimumabiin verrattuna. Guselkumabihoitoon satunnaistetut potilaat (N = 825) saivat 100 mg:n annoksia viikoilla 0 ja 4, ja sen jälkeen 8 viikon välein viikkoon 48 saakka (VOYAGE 1) ja viikkoon20 saakka (VOYAGE 2). Adalimumabihoitoon satunnaistetut potilaat (N = 582) saivat 80 mg:n annoksen viikolla 0 ja 40 mg:n annoksen viikolla 1, ja sen jälkeen 40 mg joka toinen viikko viikkoon 48 saakka (VOYAGE 1) ja viikkoon23 saakka (VOYAGE 2). Kummassakin tutkimuksessa lumehoitoon satunnaistetut potilaat (N = 422) saivat guselkumabia 100 mg:n annoksina viikoilla 16 ja 20 sekä sen jälkeen 8 viikon välein. VOYAGE 1 -tutkimuksessa kaikki potilaat, myös adalimumabihoitoon viikolla 0 satunnaistetut potilaat, aloittivat avoimen vaiheen guselkumabihoidon (8 viikon välein) viikolla 52. Tutkimuksessa VOYAGE 2 guselkumabihoitoon viikolla 0 satunnaistetut potilaat, jotka olivat saaneet PASI 90 (Psoriasis Area and Severity Index) ‑vasteen viikolla 28, satunnaistettiin uudelleen joko jatkamaan guselkumabihoitoa kahdeksan viikon välein (ylläpitohoito) tai saamaan lumehoitoa (hoidon lopettaminen). Hoidon lopettamiseen satunnaistetut potilaat aloittivat guselkumabihoidon uudestaan (hoitoa annettiin hoidon uudestaan aloittamisajankohtana, 4 viikon kuluttua ja sen jälkeen 8 viikon välein), kun vähintään 50 % viikolla 28 todetusta PASI-vasteen paranemisesta oli hävinnyt. Adalimumabihoitoon viikolla 0 satunnaistetut potilaat, jotka eivät olleet saaneet PASI 90 ‑vastetta, saivat guselkumabia viikoilla 28 ja 32 ja sen jälkeen 8 viikon välein. VOYAGE 2 -tutkimuksessa kaikki potilaat aloittivat avoimen vaiheen guselkumabihoidon (8 viikon välein) viikolla 76.
Sairauden ominaisuudet lähtötilanteessa olivat tutkimusten VOYAGE 1 ja 2 potilasjoukoissa yhdenmukaiset: kehon pinta-alan (body surface area, BSA) mediaani oli 22 % (VOYAGE 1) ja 24 % (VOYAGE 2), PASI-pisteiden mediaani lähtötilanteessa oli 19 kummassakin tutkimuksessa, DLQI (dermatology quality of life index) -pisteiden mediaani lähtötilanteessa oli 14 (VOYAGE 1) ja 14,5 (VOYAGE 2), lähtötilanteen IGA (investigator global assessment) -pisteet osoittivat vaikea-asteista sairautta 25 %:lla (VOYAGE 1) ja 23 %:lla (VOYAGE 2) potilaista ja nivelpsoriaasia oli aiemmin sairastanut 19 % (VOYAGE 1) ja 18 % (VOYAGE 2) potilaista.
Kaikista VOYAGE 1- ja VOYAGE 2 -tutkimuksissa mukana olleista potilaista 32 % (VOYAGE 1) ja 29 % (VOYAGE 2) ei ollut aiemmin saanut tavanomaista systeemistä tai biologista hoitoa, 54 % (VOYAGE 1) ja 57 % (VOYAGE 2) oli aiemmin saanut valohoitoa ja 62 % (VOYAGE 1) ja 64 % (VOYAGE 2) oli aiemmin saanut tavanomaista systeemistä hoitoa. Kummassakin tutkimuksessa 21 % oli saanut aiemmin biologista hoitoa: 11 % oli saanut vähintään yhtä tuumorinekroositekijäalfasalpaajaa (TNFα-salpaaja) ja noin 10 % oli saanut IL-12/IL-23-salpaajaa.
Guselkumabin tehoa arvioitiin ihosairauden kokonaistilanteen, sairauden esiintymisalueen (päänahka, kädet ja jalkaterät sekä kynnet) ja elämänlaadun sekä potilaan raportoiman hoitotuloksen suhteen. VOYAGE 1 ja 2 –tutkimuksissa yhdistetty ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat parantumista tai minimaalista sairautta osoittavat IGA-pisteet (IGA0/1) ja PASI 90 ‑vasteen viikolla 16 lumehoitoon verrattuna (ks. taulukko 3).
Ihosairauden kokonaistilanne
Guselkumabihoito paransi taudin aktiivisuutta osoittavia mittareita merkittävästi lume- ja adalimumabihoitoon verrattuna viikolla 16 sekä adalimumabihoitoon verrattuna viikoilla 24 ja 48. Ensisijaisen ja tärkeimmän toissijaisen tutkimuksen päätetapahtumien keskeiset tehoa koskevat tulokset esitetään jäljempänä taulukossa 3.
| Taulukko 3. Yhteenveto kliinisistä vasteista tutkimuksissa VOYAGE 1 ja VOYAGE 2 | ||||||
| Potilaiden lukumäärä (%) | ||||||
| VOYAGE 1 | VOYAGE 2 | |||||
Lumelääke (N = 174) | guselkumabi (N = 329) | adalimumabi (N = 334) | Lumelääke (N = 248) | guselkumabi (N = 496) | adalimumabi (N = 248) | |
| Viikko16 | ||||||
| PASI 75 | 10 (5,7) | 300 (91,2)a | 244 (73,1)b | 20 (8,1) | 428 (86,3)a | 170 (68,5)b |
| PASI 90 | 5 (2,9) | 241 (73,3)c | 166 (49,7)b | 6 (2,4) | 347 (70,0)c | 116 (46,8)b |
| PASI 100 | 1 (0,6) | 123 (37,4)a | 57 (17,1)d | 2 (0,8) | 169 (34,1)a | 51 (20,6)d |
| IGA 0/1 | 12 (6,9) | 280 (85,1)c | 220 (65,9)b | 21 (8,5) | 417 (84,1)c | 168 (67,7)b |
| IGA 0 | 2 (1,1) | 157 (47,7)a | 88 (26,3)d | 2 (0,8) | 215 (43,3)a | 71 (28,6)d |
| Viikko24 | ||||||
| PASI 75 | - | 300 (91,2) | 241 (72,2)e | - | 442 (89,1) | 176 (71,0)e |
| PASI 90 | - | 264 (80,2) | 177 (53,0)b | - | 373 (75,2) | 136 (54,8)b |
| PASI 100 | - | 146 (44,4) | 83 (24,9)e | - | 219 (44,2) | 66 (26,6)e |
| IGA 0/1 | - | 277 (84,2) | 206 (61,7)b | - | 414 (83,5) | 161 (64,9)b |
| IGA 0 | - | 173 (52,6) | 98 (29,3)b | - | 257 (51,8) | 78 (31,5)b |
| Viikko48 | ||||||
| PASI 75 | - | 289 (87,8) | 209 (62,6)e | - | - | - |
| PASI 90 | - | 251 (76,3) | 160 (47,9)b | - | - | - |
| PASI 100 | - | 156 (47,4) | 78 (23,4)e | - | - | - |
| IGA 0/1 | - | 265 (80,5) | 185 (55,4)b | - | - | - |
| IGA 0 | - | 166 (50,5) | 86 (25,7)b | - | - | - |
a p < 0,001 guselkumabin ja lumehoidon vertailulle. b p < 0,001 guselkumabin ja adalimumabin tärkeimpien toissijaisten päätetapahtumien vertailulle. c p < 0,001 guselkumabin ja lumehoidon yhdistetyn ensisijaisen päätetapahtuman vertailulle. d guselkumabin ja adalimumabin välillä ei tehty vertailuja. e p < 0,001 guselkumabin ja adalimumabin väliselle vertailulle. | ||||||
Ajan mittaan todettu vaste
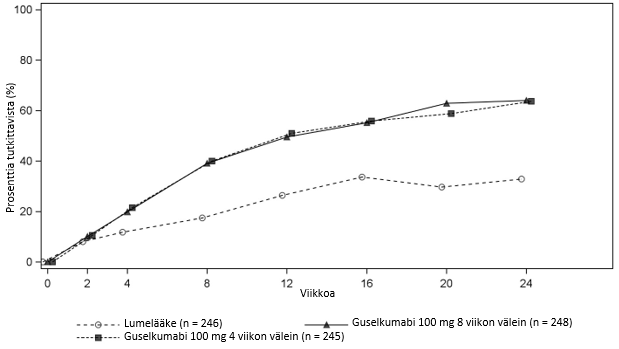
Guselkumabin vaikutuksen todettiin alkavan nopeasti, ja PASI-indeksin paranemisprosentti oli merkittävästi suurempi lumehoitoon verrattuna jo viikolla 2 (p < 0,001). PASI 90 -vasteen saavuttaneiden potilaiden prosenttiosuus oli guselkumabihoidossa numeerisesti suurempi kuin adalimumabihoidossa, mikä oli todettavissa alkaen viikosta 8, ja ero oli suurimmillaan noin viikolla 20 (VOYAGE 1 ja 2) ja säilyi viikkoon 48 saakka (VOYAGE 1) (ks. kuva 1).
Kuva 1. PASI 90 ‑vasteen viikkoon 48 saakka saavuttaneiden potilaiden prosenttiosuus tutkimuksen VOYAGE 1 tutkimuskäynneittäin (potilaat satunnaistettu viikolla 0)
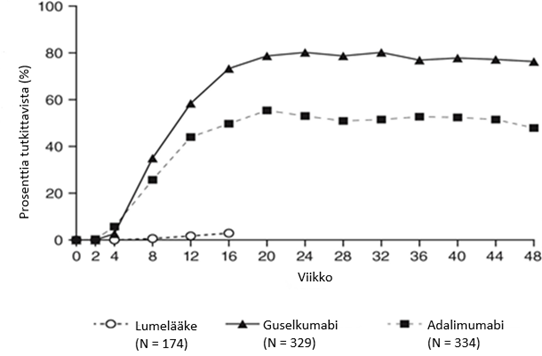
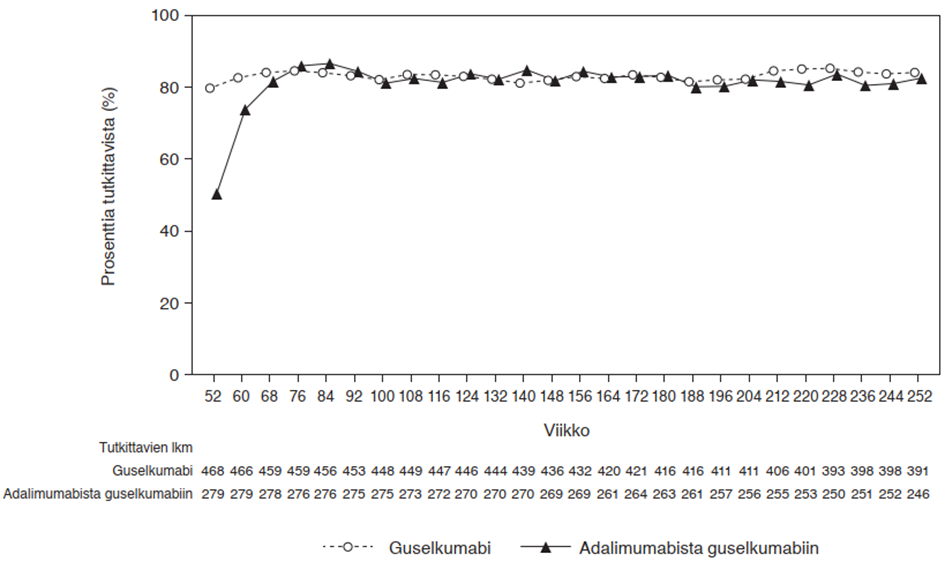
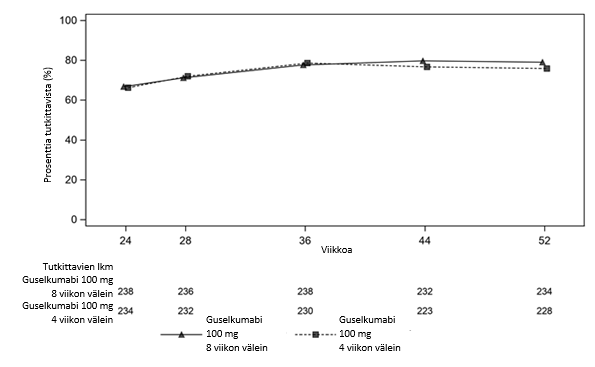
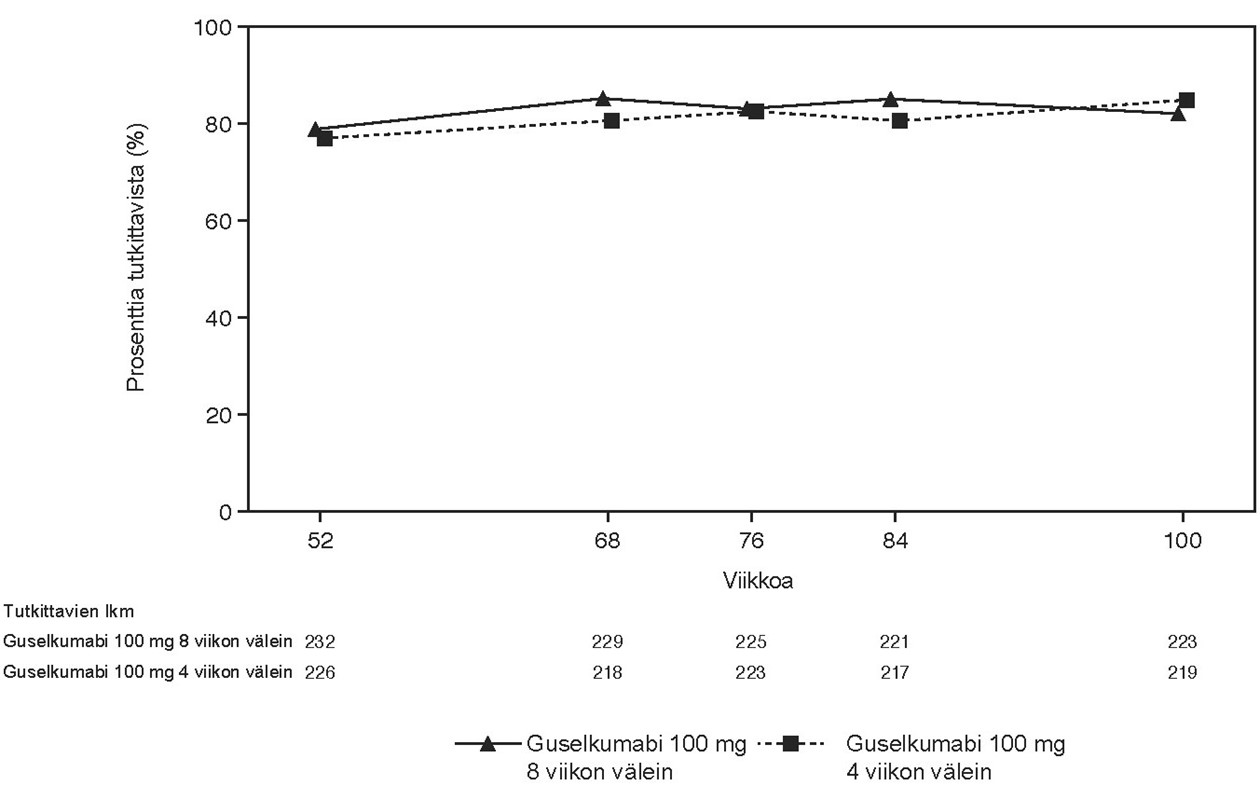
VOYAGE 1 -tutkimuksessa jatkuvaa guselkumabihoitoa saavien tutkittavien PASI 90 -vaste säilyi viikosta 52 viikkoon 252. Potilailla, jotka satunnaistettiin viikolla 0 adalimumabihoitoon ja siirtyivät viikolla 52 guselkumabihoitoon, PASI 90 -vasteluku suureni viikosta 52 viikkoon 76 ja säilyi sen jälkeen viikkoon 252 saakka (ks. kuva 2).
Kuva 2. PASI 90 -vasteen saavuttaneiden
potilaiden
prosenttiosuus tutkimuksen VOYAGE 1 avoimessa vaiheessa tutkimuskäynneittäin
Guselkumabin teho ja turvallisuus osoitettiin riippumatta iästä, sukupuolesta, rodusta, painosta, läiskien sijainnista, sairauden vaikeusasteesta lähtötilanteessa PASI-pisteiden perusteella, samanaikaisesta nivelpsoriaasista ja aiemmasta biologisesta hoidosta. Guselkumabin teho todettiin potilailla, jotka eivät olleet aiemmin saaneet tavanomaista systeemistä tai biologista hoitoa, sekä potilailla, jotka olivat aiemmin saaneet biologista hoitoa.
Tutkimuksessa VOYAGE 2 guselkumabia ylläpitohoitoon viikolla 48 saaneista potilaista 88,6 % oli saanut PASI 90 ‑vasteen verrattuna 36,8 %:iin potilaista, joiden hoito lopetettiin viikolla 28 (p < 0,001). PASI 90 ‑vasteen häviäminen todettiin jo 4 viikkoa guselkumabihoidon lopettamisen jälkeen, ja ajan mediaani PASI 90 ‑vasteen häviämiseen oli noin 15 viikkoa. Niistä potilaista, joiden hoito lopetettiin ja joille sen jälkeen aloitettiin guselkumabihoito uudestaan, 80 % oli saanut PASI 90 -vasteen uudelleen, kun vastetta arvioitiin 20 viikkoa hoidon uudestaan aloittamisen jälkeen.
VOYAGE 2 -tutkimuksessa adalimumabihoitoon satunnaistetuista 112 potilaasta, jotka eivät olleet saavuttaneet PASI 90 ‑vastetta viikolla 28, 66 % saavutti PASI 90 ‑vasteen 20 guselkumabihoitoviikon jälkeen, ja 76 % saavutti PASI 90 -vasteen 44 guselkumabihoitoviikon jälkeen. Lisäksi guselkumabihoitoon satunnaistetuista 95 potilaasta, jotka eivät olleet saavuttaneet PASI 90 -vastetta viikolla 28, 36 % saavutti PASI 90 -vasteen vielä 20 viikon ajan jatketun guselkumabihoidon jälkeen, ja 41 % saavutti PASI 90 -vasteen vielä 44 viikon ajan jatketun guselkumabihoidon jälkeen. Adalimumabihoidosta guselkumabihoitoon siirtyneillä potilailla ei havaittu uusia turvallisuutta koskevia löydöksiä.
Erityisalueiden sairaus
Tutkimuksissa VOYAGE 1 ja 2 päänahassa, käsissä ja jalkaterissä sekä kynsipsoriaasissa (mitattiin päänahkaspesifisellä tutkijan kokonaisarviolla [Scalp-specific Investigator Global Assessment, ss-IGA], lääkärin kokonaisarviolla käsistä ja/tai jalkateristä [Physician’s Global Assessment of Hands and/or Feet, hf-PGA], lääkärin kokonaisarviolla kynsistä [Fingernail Physician’s Global Assessment, f-PGA] ja kynsipsoriaasin vaikeusasteindeksillä [Nail Psoriasis Severity Index, NAPSI]) todettiin viikolla 16 merkittävää paranemista guselkumabihoitoa saaneilla potilailla verrattuna lumehoitoa saaneisiin potilaisiin (p < 0,001, taulukko 4). Guselkumabi osoitettiin adalimumabia paremmaksi päänahan sekä käsien ja jalkaterien psoriaasin osalta viikolla 24 (VOYAGE 1 ja 2) sekä viikolla 48 (VOYAGE 1) (p ≤ 0,001, lukuun ottamatta käsien ja jalkaterien psoriaasia viikolla 24 [VOYAGE 2] ja viikolla 48 [VOYAGE 1], p < 0,05).
| Taulukko 4. Yhteenveto erityisalueiden sairauden vasteista tutkimuksissa VOYAGE 1 ja VOYAGE 2 | ||||||
| VOYAGE 1 | VOYAGE 2 | |||||
| Lumelääke | guselkumabi | adalimumabi | Lumelääke | guselkumabi | adalimumabi | |
| ss-IGA (N)a | 145 | 277 | 286 | 202 | 408 | 194 |
| ss-IGA0/1b, n (%) | ||||||
| Viikko 16 | 21 (14,5) | 231 (83,4)c | 201 (70,3)d | 22 (10,9) | 329 (80,6)c | 130 (67,0)d |
| hf-PGA (N)a | 43 | 90 | 95 | 63 | 114 | 56 |
| hf-PGA0/1b, n (%) | ||||||
| Viikko 16 | 6 (14,0) | 66 (73,3)e | 53 (55,8)d | 9 (14,3) | 88 (77,2)e | 40 (71,4)d |
| f-PGA (N)a | 88 | 174 | 173 | 123 | 246 | 124 |
| f-PGA0/1, n (%) | ||||||
| Viikko 16 | 14 (15,9) | 68 (39,1)e | 88 (50,9)d | 18 (14,6) | 128 (52,0)e | 74 (59,7)d |
| NAPSI (N)a | 99 | 194 | 191 | 140 | 280 | 140 |
| Paraneminen prosenttia, keskiarvo (keskihajonta) | ||||||
| Viikko 16 | -0,9 (57,9) | 34,4 (42,4)e | 38,0 (53,9)d | 1,8 (53,8) | 39,6 (45,6)e | 46,9 (48,1)d |
a käsittää vain potilaat, joiden lähtötilanteen ss-IGA-, f-PGA-, hf-PGA-pisteet ≥ 2 tai lähtötilanteen NAPSI-pisteet > 0. b käsittää vain potilaat, jotka saavuttivat ss-IGA- ja/tai hf-PGA-arvioinneissa ≥ 2 yksikön paranemisen lähtötilanteesta. c p < 0,001 guselkumabin ja lumehoidon vertailulle tärkeimmän toissijaisen päätetapahtuman osalta. d guselkumabin ja adalimumabin välillä ei tehty vertailuja. e p < 0,001 guselkumabin ja lumehoidon vertailulle. | ||||||
Terveyteen liittyvä elämänlaatu / potilaiden raportoimat hoitotulokset
Tutkimuksissa VOYAGE 1 ja 2 todettiin guselkumabihoitoa saaneilla potilailla lumehoitoa saaneisiin potilaisiin verrattuna viikolla 16 merkittävästi suurempaa terveyteen liittyvän elämänlaadun paranemista. Tämä mitattiin ihoon liittyvällä elämänlaatuindeksillä (Dermatology Life Quality Index, DLQI) sekä potilaan raportoimilla psoriaasin oireilla (kutina, kipu, kirvely, pistely ja ihon kiristäminen) ja löydöksillä (ihon kuivuus, halkeilu, suomuisuus, irtoaminen tai hilseily, punoitus ja verenvuoto), joita mitattiin psoriaasin oireita ja löydöksiä koskevalla päiväkirjalla (Psoriasis Symptoms and Signs Diary, PSSD) (taulukko 5). Potilaiden raportoimien hoitotulosten perusteella oireiden paraneminen säilyi viikkoon 24 (VOYAGE 1 ja 2) ja viikkoon 48 (VOYAGE 1) saakka. VOYAGE 1 -tutkimuksessa oireiden paraneminen säilyi jatkuvaa guselkumabihoitoa saaneilla potilailla avoimessa vaiheessa viikkoon 252 saakka (taulukko 6).
| Taulukko 5. Yhteenveto potilaiden viikolla 16 raportoimista hoitotuloksista tutkimuksissa VOYAGE 1 ja VOYAGE 2 | ||||||
| VOYAGE 1 | VOYAGE 2 | |||||
| Lumelääke | guselkumabi | adalimumabi | Lumelääke | guselkumabi | adalimumabi | |
| DLQI, potilaiden lähtötilanteen pisteet | 170 | 322 | 328 | 248 | 495 | 247 |
| Muutos lähtötilanteesta, keskiarvo (keskihajonta) | ||||||
| Viikko16 | -0,6 (6,4) | -11,2 (7,2)c | -9,3 (7,8)b | -2,6 (6,9) | -11,3 (6,8)c | -9,7 (6,8)b |
| PSSD-oireita koskevat pisteet, potilaiden lähtötilanteen pisteet > 0 | 129 | 248 | 273 | 198 | 410 | 200 |
| Oireita koskevat pisteet = 0, n (%) | ||||||
| Viikko16 | 1 (0,8) | 67 (27,0)a | 45 (16,5)b | 0 | 112 (27,3)a | 30 (15,0)b |
| PSSD-löydöksiä koskevat pisteet, potilaiden lähtötilanteen pisteet > 0 | 129 | 248 | 274 | 198 | 411 | 201 |
| Löydöksiä koskevat pisteet = 0, n (%) | ||||||
| Viikko16 | 0 | 50 (20,2)a | 32 (11,7)b | 0 | 86 (20,9)a | 21 (10,4)b |
a p < 0,001 guselkumabin ja lumehoidon vertailulle. b guselkumabin ja adalimumabin välillä ei tehty vertailuja. c p < 0,001 guselkumabin ja lumehoidon vertailulle tärkeimmän toissijaisen päätetapahtuman osalta. | ||||||
| Taulukko 6. Yhteenveto potilaiden raportoimista hoitotuloksista tutkimuksen VOYAGE 1 avoimessa vaiheessa | ||||||
| guselkumabi | adalimumabi-guselkumabi | |||||
| Viikko 76 | Viikko 156 | Viikko 252 | Viikko 76 | Viikko 156 | Viikko 252 | |
| DLQI-pisteet > 1 lähtötilanteessa, n | 445 | 420 | 374 | 264 | 255 | 235 |
| Potilaita, joiden DLQI-pisteet0/1 | 337 (75,7 %) | 308 (73,3 %) | 272 (72,7 %) | 198 (75,0 %) | 190 (74,5 %) | 174 (74,0 %) |
| PSSD-oireita koskevat pisteet, potilaiden lähtötilanteen pisteet > 0 | 347 | 327 | 297 | 227 | 218 | 200 |
| Oireita koskevat pisteet = 0, n (%) | 136 (39,2 %) | 130 (39,8 %) | 126 (42,4 %) | 99 (43,6 %) | 96 (44,0 %) | 96 (48,0 %) |
| PSSD-löydöksiä koskevat pisteet, potilaiden lähtötilanteen pisteet > 0 | 347 | 327 | 297 | 228 | 219 | 201 |
| Löydöksiä koskevat pisteet = 0, n (%) | 102 (29,4 %) | 94 (28,7 %) | 98 (33,0 %) | 71 (31,1 %) | 69 (31,5 %) | 76 (37,8 %) |
Tutkimuksessa VOYAGE 2 guselkumabihoitoa saaneilla potilailla todettiin viikolla 16 terveyteen liittyvässä elämänlaadussa, ahdistuneisuudessa ja masennuksessa sekä työkyvyn rajoittumisessa merkittävästi suurempaa paranemista lähtötilanteesta lumehoitoon verrattuna. Näitä mitattiin 36‑kohtaisella terveyskyselyllä (Short Form health survey questionnaire, SF-36), sairaalassa tehtävällä ahdistuneisuuden ja masennuksen pisteytyksellä (Hospital Anxiety and Depression Scale, HADS) sekä työkyvyn rajoittumista koskevalla kyselyllä (Work Limitations Questionnaire, WLQ). SF-36-, HADS- ja WLQ-indekseissä todettu paraneminen säilyi viikolla 28 ylläpitohoitoon satunnaistetuilla potilailla viikkoon 48 saakka ja avoimessa vaiheessa viikkoon 252 saakka.
NAVIGATE
NAVIGATE-tutkimuksessa selvitettiin guselkumabin tehoa potilailla, joiden vaste ustekinumabiin oli viikolla 16 riittämätön (eli jotka eivät olleet saavuttaneet vastetta ”parantunut” tai ”minimaalinen” sairaus, joksi oli määritelty IGA≥ 2). Kaikki potilaat (N = 871) saivat avoimessa tutkimuksessa ustekinumabia (45 mg ≤ 100 kg ja 90 mg > 100 kg) viikoilla 0 ja 4. 268 potilasta, joiden IGA-pisteet olivat ≥ 2, satunnaistettiin viikolla 16 joko jatkamaan ustekinumabihoitoa (N = 133) 12 viikon välein tai aloittamaan guselkumabihoito (N = 135) viikoilla16 ja 20 ja sen jälkeen 8 viikon välein. Satunnaistettujen potilaiden ominaisuudet olivat lähtötilanteessa samankaltaiset kuin tutkimuksissa VOYAGE 1 ja 2.
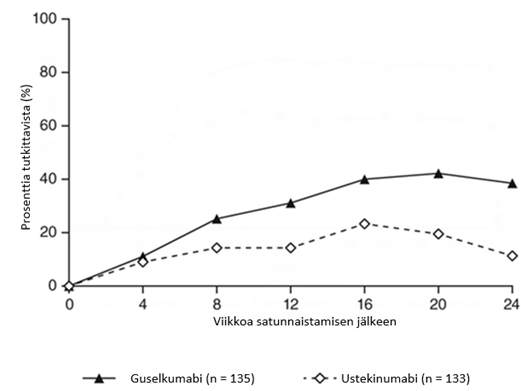
Satunnaistamisen jälkeen ensisijainen päätetapahtuma oli satunnaistamisen jälkeisten tutkimuskäyntien lukumäärä viikkojen 12 ja 24 välillä, jolloin potilaat saavuttivat IGA-pisteet0/1 ja ≥ 2yksikön paranemisen. Potilaat tutkittiin neljän viikon välein yhteensä neljällä tutkimuskäynnillä. Niistä potilaista, joilla vaste ustekinumabiin oli satunnaistamisajankohtana riittämätön, havaittiin merkittävästi suurempaa tehon paranemista siinä potilasjoukossa, joka siirtyi guselkumabihoitoon, verrattuna ustekinumabihoitoa jatkaneisiin potilaisiin. Satunnaistamisen jälkeisten viikkojen 12 ja 24 välillä guselkumabipotilaat saavuttivat IGA-pisteet0/1 ja ≥ 2yksikön paranemisen kaksi kertaa niin usein kuin ustekinumabia saaneet potilaat (keskiarvo1,5 [guselkumabi] vs 0,7 [ustekinumabi] tutkimuskäyntiä, p < 0,001). Lisäksi 12 viikon kuluttua satunnaistamisesta suurempi osuus guselkumabia saaneista potilaista verrattuna ustekinumabia saaneisiin potilaisiin saavutti IGA-pisteet0/1 ja ≥ 2yksikön paranemisen (31,1 % [guselkumabi] vs. 14,3 % [ustekinumabi], p = 0,001) ja PASI 90 ‑vasteen (48 % [guselkumabi] vs 23 % [ustekinumabi], p < 0,001). Erot guselkumabi- ja ustekinumabihoitoa saaneiden potilaiden vasteluvuissa havaittiin jo 4 viikkoa satunnaistamisen jälkeen (11,1 % [guselkumabi] ja 9,0 % [ustekinumabi]), ja ne olivat suurimmillaan 24 viikkoa satunnaistamisen jälkeen (ks. kuva 3). Ustekinumabihoidosta guselkumabihoitoon siirtyneillä potilailla ei havaittu uusia turvallisuutta koskevia löydöksiä.
Kuva 3. IGA-pisteet parantunut (0) tai minimaalinen sairaus (1) ja IGA-pisteiden vähintään 2yksikön paranemisen viikosta 0 viikkoon 24 saakka saavuttaneiden potilaiden prosenttiosuus tutkimuskäynneittäin tutkimuksen NAVIGATE satunnaistamisen jälkeen
ECLIPSE
Guselkumabin tehoa ja turvallisuutta tutkittiin myös kaksoissokkotutkimuksessa, jossa sitä verrattiin sekukinumabiin. Potilaat satunnaistettiin saamaan guselkumabia (N = 534; 100 mg viikoilla 0 ja 4 ja sen jälkeen kerran 8 viikossa) tai sekukinumabia (N = 514; 300 mg viikoilla 0, 1, 2, 3 ja 4 ja sen jälkeen kerran 4 viikossa). Kummassakin hoitoryhmässä viimeinen annos annettiin viikolla 44.
Sairauden ominaisuudet olivat keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavilla potilailla lähtötilanteessa yhdenmukaiset: ihottumaa oli 20 %:ssa (mediaani) kehon pinta-alasta, PASI-pisteiden mediaani oli 18, ja 24 %:lla potilaista oli vaikea-asteisen sairauden osoittavat IGA-pisteet.
Ensisijaisella päätetapahtumalla (PASI 90 -vaste viikolla 48) mitattuna guselkumabi oli sekukinumabia parempi (84,5 % versus 70,0 %, p < 0,001). PASI-vastelukujen vertailu esitetään taulukossa 7.
| Taulukko 7. PASI-vasteluvut ECLIPSE-tutkimuksessa | ||
| Potilaiden lukumäärä (%) | ||
| guselkumabi (N = 534) | sekukinumabi (N = 514) | |
| Ensisijainen päätetapahtuma | ||
| PASI 90 ‑vaste viikolla 48 | 451 (84,5 %)a | 360 (70,0 %) |
| Tärkeimmät toissijaiset päätetapahtumat | ||
| PASI 75 ‑vaste sekä viikolla 12 että viikolla 48 | 452 (84,6 %)b | 412 (80,2 %) |
| PASI 75 ‑vaste viikolla 12 | 477 (89,3 %)c | 471 (91,6 %) |
| PASI 90 ‑vaste viikolla 12 | 369 (69,1 %)c | 391 (76,1 %) |
| PASI 100 ‑vaste viikolla 48 | 311 (58,2 %)c | 249 (48,4 %) |
a p< 0,001 paremmuuden osalta b p< 0,001 vertailukelpoisuuden osalta, p= 0,062 paremmuuden osalta c varsinaista tilastollista testausta ei tehty | ||
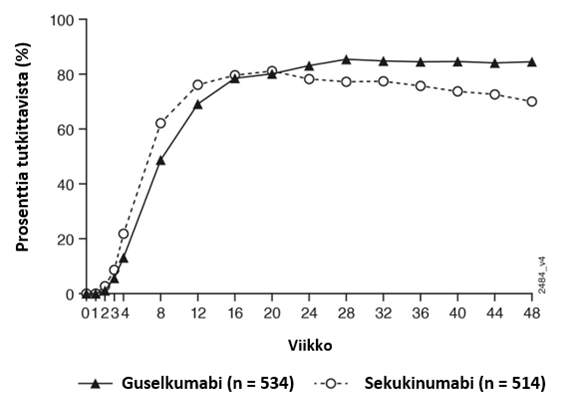
PASI 90 -vasteluvut guselkumabi- ja sekukinumabihoidossa viikkoon 48 saakka esitetään kuvassa 4.
Kuva 4.
PASI 90 ‑vasteen viikkoon 48 saakka saavuttaneiden potilaiden prosenttiosuus tutkimuksen ECLIPSE tutkimuskäynneittäin (potilaat satunnaistettu viikolla 0)
Nivelpsoriaasi
Guselkumabin on osoitettu vähentävän aktiivista nivelpsoriaasia sairastavien aikuisten potilaiden oireita ja löydöksiä, parantavan fyysistä toimintakykyä ja terveyteen liittyvää elämänlaatua sekä hidastavan raajojen nivelvaurioiden etenemistä.
DISCOVER 1 ja DISCOVER 2
Kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen III tutkimuksessa (DISCOVER 1 ja DISCOVER 2) verrattiin guselkumabin ja lumehoidon tehoa ja turvallisuutta aikuisilla potilailla, joilla oli aktiivinen nivelpsoriaasi (DISCOVER 1 ‑tutkimuksessa ≥ 3 turvonnutta ja ≥ 3 aristavaa niveltä ja C‑reaktiivisen proteiinin [CRP] pitoisuus ≥ 0,3 mg/dl, ja DISCOVER 2 ‑tutkimuksessa ≥ 5 turvonnutta ja ≥ 5 aristavaa niveltä ja CRP-pitoisuus ≥ 0,6 mg/dl) huolimatta hoidosta tavanomaisella synteettisellä (cs)DMARD-lääkkeellä, apremilastilla tai tulehduskipulääkkeillä. Näissä tutkimuksissa potilailla oli ollut CASPAR (Classification criteria for Psoriatic Arthritis) ‑luokittelukriteeristöön perustuva nivelpsoriaasidiagnoosi 4 vuoden (mediaani) ajan. Kumpaankin tutkimukseen otettiin mukaan nivelpsoriaasin eri alatyyppejä sairastavia potilaita; alatyyppejä olivat monen nivelen niveltulehdus ilman reumakyhmyjä (40 %), spondyliitti, johon liittyy raajojen niveltulehduksia (30 %), epäsymmetriset raajojen niveltulehdukset (23 %), kärkinivelten oireet (7 %) ja mutiloiva niveltulehdus (1 %). Yli 65 %:lla potilaista oli lähtötilanteessa entesiitti, ja yli 42 %:lla potilaista oli lähtötilanteessa daktyliitti, ja yli 75 %:lla potilaista oli psoriaasin oireita iholla ≥ 3 %:n alueella kehon pinta-alasta. DISCOVER 1 ‑tutkimuksessa arvioitiin 381 potilasta ja DISCOVER 2 ‑tutkimuksessa arvioitiin 739 potilasta, jotka saivat 100 mg guselkumabia viikoilla 0 ja 4 ja sen jälkeen 8 viikon välein tai 100 mg guselkumabia 4 viikon välein tai lumehoitoa. Kummassakin tutkimuksessa lumehoitoa saaneet potilaat siirtyivät viikolla 24 guselkumabihoitoon 100 mg:n annoksina neljän viikon välein. Kummassakin tutkimuksessa noin 58 % potilaista jatkoi metotreksaattihoitoa vakaina annoksina (≤ 25 mg/viikko).
Kummassakin tutkimuksessa yli 90 % potilaista oli aiemmin käyttänyt (cs)DMARD-lääkettä. DISCOVER 1 ‑tutkimuksessa 31 % potilaista oli saanut aiemmin TNFα:n estäjähoitoa. DISCOVER 2 ‑tutkimuksessa potilaat eivät olleet saaneet aiemmin biologista lääkehoitoa.
Oireet ja löydökset
Guselkumabihoito paransi viikolla 24 merkittävästi taudin aktiivisuutta osoittavia mittareita lumehoitoon verrattuna. Kummankin tutkimuksen ensisijainen päätetapahtuma oli niiden potilaiden prosenttiosuus, jotka saavuttivat ACR 20 (American College of Rheumatology) ‑vasteen viikolla 24. Keskeiset tehon tulokset esitetään taulukossa 8.
| Taulukko 8. Kliiniset vasteet DISCOVER 1- ja DISCOVER 2 ‑tutkimuksissa | ||||||
| DISCOVER 1 | DISCOVER 2 | |||||
Lumelääke (N = 126) | guselkumabi 100 mg 8 viikon välein (N = 127) | guselkumabi 100 mg 4 viikon välein (N = 128) | Lumelääke (N = 246) | guselkumabi 100 mg 8 viikon välein (N = 248) | guselkumabi 100 mg 4 viikon välein (N = 245) | |
| ACR 20 ‑vaste | ||||||
| Viikko 16 | 25,4 % | 52,0 %b | 60,2 %b | 33,7 % | 55,2 %g | 55,9 %c |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | 26,7 (15,3; 38,1) | 34,8 (23,5; 46,0) | - | 21,5 (13,1; 30,0) | 22,2 (13,7; 30,7) |
| Viikko 24 | 22,2 % | 52,0 %a | 59,4 %a | 32,9 % | 64,1 %a | 63,7 %a |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | 29,8 (18,6; 41,1) | 37,1 (26,1; 48,2) | - | 31,2 (22,9; 39,5) | 30,8 (22,4; 39,1) |
| ACR 50 ‑vaste | ||||||
| Viikko 16 | 12,7 % | 22,8 %d | 26,6 %c | 9,3 % | 28,6 %g | 20,8 %c |
| Ero lumelääkkeeseen (95 %:n luottamusväli)) | - | 10,2 (1,0; 19,3) | 13,9 (4,4; 23,4) | - | 19,3 (12,6; 25,9) | 11,5 (5,2; 17,7) |
| Viikko 24 | 8,7 % | 29,9 %b | 35,9 %b | 14,2 % | 31,5 %g | 33,1 %c |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | 21,4 (12,1; 30,7) | 27,2 (17,6; 36,8) | - | 17,2 (10,0; 24,4) | 18,8 (11,5; 26,1) |
| ACR 70 ‑vaste | ||||||
| Viikko 24 | 5,6 % | 11,8 %d | 20,3 %b | 4,1 % | 18,5 %g | 13,1 %c |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | 6,4 (-0,3; 13,1) | 14,8 (6,9; 22,7) | - | 14,5 (9,1; 19,9) | 9,0 (4,1; 13,8) |
| DAS 28 (CRP) -muutos lähtötilanteesta, pienimmän neliösumman keskiarvo | ||||||
| Viikko 24 c | -0,70 | -1,43b | -1,61b | -0,97 | -1,59b | -1,62b |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | -0,73 (-0,98; -0,48) | -0,91 (-1,16; -0,66) | - | -0,61 (-0,80; -0,43) | -0,65 (-0,83; -0,47) |
| Minimaalinen taudin aktiivisuus (minimal disease activity, MDA) | ||||||
| Viikko 24 | 11,1 % | 22,8 %f | 30,5 %e | 6,1 % | 25,0 %e | 18,8 %e |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | 11,9 (2,9; 20,9) | 19,3 (9,7; 28,9) | - | 18,9 (12,8; 25,0) | 12,7 (7,0; 18,4) |
| Potilaita, joilla ≥ 3 % kehon pinta-alasta ja IGA-pisteet ≥ 2 | ||||||
| n = 78 | n = 82 | n = 89 | n = 183 | n = 176 | n = 184 | |
| IGA-vaste h | ||||||
| Viikko 24 | 15,4 % | 57,3 %b | 75,3 %b | 19,1 % | 70,5 %b | 68,5 %b |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | 42,0 (28,9; 55,1) | 60,0 (48,3; 71,8) | - | 50,9 (42,2; 59,7) | 49,8 (41,2; 58,4) |
| PASI 90 ‑vaste | ||||||
| Viikko 16 | 10,3 % | 45,1 %e | 52,8 %e | 8,2 % | 55,1 %e | 53,8 %e |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | 34,9 (22,2; 47,6) | 42,6 (30,5; 54,8) | - | 46,6 (38,4; 54,8) | 45,6 (37,6; 53,6) |
| Viikko 24 | 11,5 % | 50,0 %e | 62,9 %e | 9,8 % | 68,8 %e | 60,9 %e |
| Ero lumelääkkeeseen (95 %:n luottamusväli) | - | 38,6 (25,8; 51,4) | 51,7 (39,7; 63,7) | - | 58,6 (50,6; 66,6) | 51,3 (43,2; 59,3) |
a p < 0,001 (ensisijainen päätetapahtuma) b p < 0,001 (pääasiallinen toissijainen päätetapahtuma) c p = 0,006 (pääasiallinen toissijainen päätetapahtuma) d ei tilastollisesti merkitsevä p = 0,086 (pääasiallinen toissijainen päätetapahtuma) e nimellinen p < 0,001 f nimellinen p = 0,012 g ei varsinaisesti testattu hierarkkisella testausmenetelmällä, nimellinen p < 0,001 (pääasiallinen toissijainen päätetapahtuma) h määritelty IGA-vasteeksi 0 (parantunut) tai 1 (minimaalinen) ja psoriaasia koskevassa IGA-arvioinnissa ≥ 2 yksikön paraneminen lähtötilanteesta | ||||||
ACR 20/50/70-, DAS 28 (CRP)-, MDA-, IGA- ja PASI 90 ‑vastelukujen perusteella arvioitu kliininen vaste säilyi DISCOVER 1- ja
DISCOVER 2 ‑tutkimuksissa viikkoon 52 saakka (ks. taulukko 9).
| Taulukko 9. Kliiniset vasteet DISCOVER 1- ja DISCOVER 2 ‑tutkimuksissa viikolla 52a | ||||
| DISCOVER 1 | DISCOVER 2 | |||
guselkumabi 100 mg 8 viikon välein | guselkumabi 100 mg 4 viikon välein | guselkumabi 100 mg 8 viikon välein | guselkumabi 100 mg 4 viikon välein | |
| ACR 20 | ||||
| Nb | 112 | 124 | 234 | 228 |
| Vaste (%) | 67.9 % | 75,8 % | 79,1 % | 75,9 % |
| ACR 50 | ||||
| Nb | 113 | 124 | 234 | 228 |
| Vaste (%) | 43,4 % | 55,6 % | 51,3 % | 49,1 % |
| ACR 70 | ||||
| Nb | 114 | 124 | 234 | 228 |
| Vaste (%) | 28,9 % | 29,8 % | 29,5 % | 28,1 % |
| DAS 28 (CRP) ‑muutos lähtötilanteesta | ||||
| Nc | 112 | 123 | 234 | 227 |
| Keskiarvo (keskihajonta) | -2,03 (1,250) | -1,99 (1,062) | -2,08 (1,121) | -2,11 (1,128) |
| Minimaalinen taudin aktiivisuus (minimal disease activity, MDA) | ||||
| Nb | 112 | 124 | 234 | 228 |
| Vaste (%) | 33,9 % | 40,3 % | 32,9 % | 36,8 % |
| Potilaita, joilla ≥ 3 % kehon pinta-alasta ja IGA lähtötilanteessa ≥ 2 | ||||
| IGA-vaste | ||||
| Nb | 75 | 88 | 170 | 173 |
| Vaste (%) | 69,3 % | 83,0 % | 77,1 % | 84,4 % |
| PASI 90 | ||||
| Nb | 75 | 88 | 170 | 173 |
| Vaste (%) | 66,7 % | 76,1 % | 77,1 % | 81,5 % |
a Viikon 24 jälkeen ei ollut lumeryhmää. b Arvioitavissa olevat potilaat, joilla havaittiin vaste. cPotilailla havaittava muutos lähtötilanteesta. | ||||
ACR 20/50/70-, DAS 28 (CRP)-, MDA-, IGA- ja PASI 90 ‑vastelukujen perusteella arvioitu kliininen vaste säilyi DISCOVER 2 ‑tutkimuksessa viikkoon 100 saakka (ks. taulukko 10).
| Taulukko 10. Kliiniset vasteet DISCOVER 2 ‑tutkimuksessa viikolla 100a | ||
guselkumabi 100 mg 8 viikon välein | guselkumabi 100 mg 4 viikon välein | |
| ACR 20 | ||
| Nb | 223 | 219 |
| Vaste (%) | 82,1 % | 84,9 % |
| ACR 50 | ||
| Nb | 224 | 220 |
| Vaste (%) | 60,7 % | 62,3 % |
| ACR 70 | ||
| Nb | 224 | 220 |
| Vaste (%) | 39,3 % | 38,6 % |
| DAS 28 (CRP) -muutos lähtötilanteesta | ||
| Nc | 223 | 219 |
| Keskiarvo (keskihajonta) | -2,37 (1,215) | -2,36 (1,120) |
| Vähäinen taudin aktiivisuus (minimal disease activity, MDA) | ||
| Nb | 224 | 220 |
| Vaste (%) | 44,6 % | 42,7 % |
| Potilaita, joilla ≥ 3 % kehon pinta-alasta ja IGA lähtötilanteessa ≥ 2 | ||
| IGA-vaste | ||
| Nb | 165 | 170 |
| Vaste (%) | 76,4 % | 82,4 % |
| PASI 90 | ||
| Nb | 164 | 170 |
| Vaste (%) | 75,0 % | 80,0 % |
a Viikon 24 jälkeen ei ollut lumeryhmää. b Arvioitavissa olevat potilaat, joilla havaittiin vaste. c Potilailla havaittava muutos lähtötilanteesta. | ||
Vaste ajan kuluessa
DISCOVER 2 ‑tutkimuksessa havaittiin jo viikolla 4 suurempi ACR 20 ‑vaste kummassakin guselkumabiryhmässä verrattuna lumehoitoon, ja hoidon jatkuessa ero suureni edelleen viikkoon 24 saakka (kuva 5).
Kuva 5. ACR 20 ‑vaste DISCOVER 2 ‑tutkimuksessa käynneittäin viikkoon 24 saakka
DISCOVER 2 ‑tutkimuksessa guselkumabihoitoa jatkuvasti viikolla 24 saaneiden potilaiden ACR 20 ‑vaste säilyi viikosta 24 viikkoon 52 (ks. kuva 6). Guselkumabihoitoa jatkuvasti viikolla 52 saaneiden potilaiden ACR 20 ‑vaste säilyi viikosta 52 viikkoon 100 (ks. kuva 7).
Kuva 6. ACR 20 ‑vaste DISCOVER 2 ‑tutkimuksessa käynneittäin viikosta 24 viikkoon 52
Kuva 7. ACR 20 ‑vaste DISCOVER 2 ‑tutkimuksessa käynneittäin viikosta 52 viikkoon 100
Guselkumabiryhmissä havaitut vasteet olivat samankaltaisia riippumatta csDMARD-lääkkeiden, mukaan lukien metotreksaatin, käytöstä (DISCOVER 1 ja 2). Lisäksi tarkasteltaessa ikää, sukupuolta, etnistä taustaa, painoa ja aiempaa csDMARD-lääkkeiden käyttöä (DISCOVER 1 ja 2) sekä aiempaa TNFα:n estäjien käyttöä (DISCOVER 1) näiden alaryhmien guselkumabivasteessa ei tunnistettu eroja.
DISCOVER 1- ja 2 ‑tutkimuksissa osoitettiin kaikkien ACR-pisteiden osa-alueiden, myös potilaan kipuarvion parantuneen. Kummankin tutkimuksen viikolla 24 niiden potilaiden osuus, joilla todettiin nivelpsoriaasin aktiivisuuden muokatun mittarin (PsARC) perusteella vaste, oli guselkumabiryhmissä suurempi kuin lumeryhmässä. PsARC-vasteet säilyivät DISCOVER 1 -tutkimuksessa viikosta 24 viikkoon 52 ja DISCOVER 2 -tutkimuksessa viikkoon 100.
Daktyliittiä ja entesiittiä arvioitiin DISCOVER 1- ja 2 -tutkimusten yhdistettyjen tietojen perusteella. Niistä potilaista, joilla oli lähtötilanteessa daktyliitti, viikolla 24 daktyliitti oli hävinnyt suuremmalla osalla potilaista guselkumabia 8 viikon välein saaneessa ryhmässä (59,4 %, nimellinen p < 0,001) ja 4 viikon välein saaneessa ryhmässä (63,5 %, p = 0,006) kuin lumehoidossa (42,2 %). Niistä potilaista, joilla oli lähtötilanteessa entesiitti, viikolla 24 entesiitti oli hävinnyt suuremmalla osalla potilaista guselkumabia 8 viikon välein saaneessa ryhmässä (49,6 %, nimellinen p < 0,001) ja 4 viikon välein saaneessa ryhmässä (44,9 %, p = 0,006) kuin lumehoidossa (29,4 %). Viikolla 52 niiden potilaiden osuus, joilla daktyliitti oli hävinnyt (81,2 % hoitoa 8 viikon välein saaneessa ryhmässä ja 80,4 % hoitoa 4 viikon välein saaneessa ryhmässä) ja entesiitti oli hävinnyt (62,7 % hoitoa 8 viikon välein saaneessa ryhmässä ja 60,9 % hoitoa 4 viikon välein saaneessa ryhmässä), oli säilynyt ennallaan. DISCOVER 2 ‑tutkimuksessa potilaista, joilla oli lähtötilanteessa daktyliitti ja entesiitti, niiden potilaiden osuus, joilla daktyliitti oli hävinnyt (91,1 % hoitoa 8 viikon välein saaneessa ryhmässä ja 82,9 % hoitoa 4 viikon välein saaneessa ryhmässä) ja entesiitti oli hävinnyt (77,5 % hoitoa 8 viikon välein saaneessa ryhmässä ja 67,7 % hoitoa 4 viikon välein saaneessa ryhmässä), oli säilynyt viikolla 100.
DISCOVER 1- ja 2 ‑tutkimuksissa niistä guselkumabihoitoa saaneista potilaista, joilla sairauden ensisijainen ilmenemismuoto oli spondyliitti ja raajojen niveltulehduksia, taudin aktiivisuutta spondyloartropatioissa mittaava BASDAI-indeksi (Bath Ankylosing Spondylitis Disease Activity Index) parani viikolla 24 merkittävämmin kuin lumehoidossa. BASDAI-indeksin paraneminen säilyi DISCOVER 1 -tutkimuksessa viikosta 24 viikkoon 52 ja DISCOVER 2 -tutkimuksessa viikkoon 100.
Radiologinen vaste
DISCOVER 2 -tutkimuksessa mitattiin radiologisesti rakennevaurion etenemisen estymistä, mikä ilmaistiin muokattujen van der Heijde-Sharp (vdH-S) -kokonaispisteiden keskimuutoksena lähtötilanteesta. Guselkumabia 4 viikon välein saaneessa ryhmässä osoitettiin viikolla 24 tilastollisesti merkitsevästi vähemmän radiologista etenemistä, ja guselkumabia 8 viikon välein saaneessa ryhmässä osoitettiin numeerisesti vähemmän etenemistä kuin lumehoidossa (taulukko 11). Neljän viikon välein annettavasta guselkumabihoito-ohjelmasta havaittu hyöty radiologisen etenemisen estämisessä (eli hoitoa 4 viikon välein saaneessa ryhmässä muokattujen van der Heijde-Sharp -kokonaispisteiden pienempi keskimuutos lähtötilanteesta lumehoitoon verrattuna) oli merkittävämpää potilailla, joilla oli lähtötilanteessa sekä suuri C-reaktiivisen proteiinin pitoisuus että eroosiota useammissa nivelissä.
| Taulukko 11. Muokattujen van der Heijde-Sharp ‑kokonaispisteiden muutos lähtötilanteesta DISCOVER 2 ‑tutkimuksen viikolla 24 | ||
| N | Muokattujen van der Heijde-Sharp ‑kokonaispisteiden pienimmän neliösumman keskiarvon muutos (95 %:n luottamusväli) lähtötilanteesta viikkoon 24 | |
| Lumelääke | 246 | 0,95 (0,61; 1,29) |
| guselkumabi 100 mg 8 viikon välein | 248 | 0,52a (0,18; 0,86) |
| guselkumabi 100 mg 4 viikon välein | 245 | 0,29b (-0,05; 0,63) |
a ei tilastollisesti merkitsevä p = 0,068 (pääasiallinen toissijainen päätetapahtuma) b p = 0,006 (pääasiallinen toissijainen päätetapahtuma) | ||
Muokattujen van der Heijde-Sharp ‑kokonaispisteiden keskimuutos lähtötilanteesta viikolla 52 ja viikolla 100 oli guselkumabia 8 viikon välein ja 4 viikon välein saaneissa ryhmissä samankaltainen (taulukko 12).
| Taulukko 12. Muokattujen van der Heijde-Sharp ‑kokonaispisteiden muutos lähtötilanteesta DISCOVER 2 ‑tutkimuksen viikolla 52 ja viikolla 100 | ||
| Na | Muokattujen van der Heijde-Sharp ‑kokonaispisteiden keskimuutos (keskihajonta) lähtötilanteesta | |
| Viikko 52 | ||
| guselkumabi 100 mg 8 viikon välein | 235 | 0,97 (3,623) |
| guselkumabi 100 mg 4 viikon välein | 229 | 1,07 (3,843) |
| Viikko 100 | ||
| guselkumabi 100 mg 8 viikon välein | 216 | 1,50 (4,393) |
| guselkumabi 100 mg 4 viikon välein | 211 | 1,68 (7,018) |
a arvioitavissa olevia potilaita, joilla havaittiin muutos kyseisenä ajanjaksona Huom.: ei lumeryhmää viikon 24 jälkeen | ||
Fyysinen toimintakyky ja terveyteen liittyvä elämänlaatu
DISCOVER 1- ja 2 ‑tutkimuksissa guselkumabihoitoa saaneiden potilaiden fyysisen toimintakyvyn osoitettiin parantuneen merkittävästi (p < 0,001) lumehoitoon verrattuna, kun sitä arvioitiin viikolla 24 HAQ-DI (Health Assessment Questionnaire-Disability Index) ‑toimintakykymittarilla. HAQ-DI-toimintakykymittarilla todettu paraneminen säilyi DISCOVER 1 -tutkimuksessa viikosta 24 viikkoon 52 ja DISCOVER 2 -tutkimuksessa viikkoon 100.
SF-36-kyselyn fyysistä toimintakykyä selvittävän osion (Physical Component Summary, PCS) pisteissä havaittiin guselkumabihoitoa saaneilla potilailla merkittävästi suurempi paraneminen lähtötilanteesta lumehoitoa saaneisiin verrattuna sekä DISCOVER 1 ‑tutkimuksen (kummankin annosryhmän p < 0,001) että DISCOVER 2 ‑tutkimuksen (4 viikon välein hoitoa saaneen ryhmän p = 0,006) viikolla 24. FACIT-F (Functional Assessment of Chronic Illness Therapy-Fatigue) ‑mittarilla havaittiin kummassakin tutkimuksessa viikolla 24 suurempi pisteiden lisäys guselkumabihoitoa saaneilla potilailla lumehoitoa saaneisiin potilaisiin verrattuna. DISCOVER 2 ‑tutkimuksessa havaittiin viikolla 24 ihotaudeissa elämänlaadun mittarina käytetyllä DLQI (Dermatology Life Quality Index) ‑kysymyssarjalla mitattuna terveyteen liittyvän elämänlaadun parantuneen enemmän kuin lumehoidossa. SF-36 PCS ‑pisteissä, FACIT-F-mittarilla ja DLQI-kysymyssarjalla havaittu paraneminen säilyi DISCOVER 1 -tutkimuksessa viikosta 24 viikkoon 52 ja DISCOVER 2 -tutkimuksessa viikkoon 100.
Haavainen paksusuolitulehdus
Guselkumabin tehoa ja turvallisuutta arvioitiin kolmessa vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (QUASAR-tutkimus laskimoon annettavasta induktiohoidosta, QUASAR-tutkimus ylläpitohoidosta ja ASTRO-tutkimus ihon alle annettavasta induktiohoidosta) aikuisilla potilailla, joilla oli keskivaikea tai vaikea aktiivinen haavainen paksusuolitulehdus ja joiden vaste kortikosteroideihin, tavanomaisiin immunomodulaattoreihin (atsatiopriini, merkaptopuriini), biologiseen hoitoon (TNF-estäjät, vedolitsumabi), januskinaasin (JAK) estäjään ja/tai sfingosiini-1-fosfaattireseptorin muuntajiin (koskee vain ASTRO-tutkimusta), oli ollut riittämätön, joiden vaste näihin oli hävinnyt tai jotka eivät olleet sietäneet tällaisia hoitoja. Guselkumabin tehoa ja turvallisuutta arvioitiin lisäksi satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen IIb induktiohoidon annoshakututkimuksessa (QUASAR- induktiohoidon annoshakututkimus), johon otettiin mukaan samankaltainen haavaista paksusuolitulehdusta sairastava potilasjoukko kuin vaiheen III induktiohoitotutkimukseen.
Sairauden aktiivisuutta arvioitiin modifioidulla Mayo-pisteytyksellä (mMS). Se on kolmen osa-alueen Mayo-pisteytys (0–9), joka koostuu seuraavien osa-alueiden pisteiden summasta (jokaisen osa-alueen pisteet 0–3): ulostustiheys, peräsuoliverenvuoto ja keskitetysti arvioidut endoskopialöydökset. Keskivaikean tai vaikean aktiivisen haavaisen paksusuolitulehduksen määritelmä täyttyi jos: modifioidut Mayo-pisteet 5–9, peräsuoliverenvuodon osa-alueen pisteet ≥ 1 ja endoskopialöydösten osa-alueen pisteet 2 (määritelty merkittäväksi punoitukseksi, verisuonikuvioituksen puuttumiseksi, kosketusverenvuodoksi ja/tai eroosioiksi) tai endoskopialöydösten osa-alueen pisteet 3 (määritelty spontaaniksi verenvuodoksi ja haavautumiseksi).
Induktiohoitotutkimus: QUASAR IS
QUASAR IS ‑induktiohoitotutkimuksessa potilaat satunnaistettiin suhteessa 3:2 saamaan joko 200 mg guselkumabia tai lumehoitoa infuusiona laskimoon viikolla 0, viikolla 4 ja viikolla 8. Yhteensä 701 potilasta arvioitiin. Modifioitujen Mayo-pisteiden mediaani oli lähtötilanteessa 7, ja 35,5 %:lla potilaista lähtötilanteen modifioidut Mayo-pisteet olivat 5–6, 64,5 %:lla modifioidut Mayo-pisteet olivat 7–9 ja 67,9 %:lla potilaista lähtötilanteen endoskopialöydösten osa-alueen pisteet olivat 3. Iän mediaani oli 39 vuotta (vaihteluväli 18–79 vuotta); 43,1 % oli naisia, ja 72,5 % identifioitui valkoihoisiksi, 21,4 % aasialaisiksi ja 1 % mustaihoisiksi.
Tutkimukseen mukaan otettujen potilaiden oli sallittua käyttää vakaina annoksina suun kautta otettavia aminosalisylaatteja, metotreksaattia, merkaptopuriinia, atsatiopriinia ja/tai suun kautta otettavia kortikosteroideja. Lähtötilanteessa 72,5 % potilaista sai aminosalisylaatteja, 20,8 % potilaista sai immunomodulaattoreita (metotreksaatti, merkaptopuriini tai atsatiopriini) ja 43,1 % potilaista sai kortikosteroideja. Samanaikaiset biologiset hoidot tai JAK-estäjät eivät olleet sallittuja.
Yhteensä 49,1 %:lla potilaista vähintään yksi aiempi biologinen hoito ja/tai hoito JAK-estäjällä oli epäonnistunut. Näistä potilaista 87,5 %:lla oli aiemmin epäonnistunut TNF-estäjähoito, 54,1 %:lla vedolitsumabihoito ja 18 %:lla JAK-estäjähoito, ja 47,4 %:lla potilaista näistä hoidoista oli epäonnistunut vähintään kaksi. Yhteensä 48,4 % potilaista ei ollut aiemmin saanut biologista hoitoa tai JAK-estäjähoitoa, ja 2,6 % oli aiemmin saanut biologista hoitoa tai JAK-estäjähoitoa eikä hoito ollut epäonnistunut.
Ensisijainen päätetapahtuma oli kliininen remissio, joka määriteltiin modifioidulla Mayo-pisteytyksellä viikon 12 aikapisteessä. Toissijaisia päätetapahtumia viikon 12 aikapisteessä olivat oireenmukainen remissio, endoskooppinen paraneminen, kliininen vaste, histologis-endoskooppinen limakalvon paraneminen, uupumuksen suhteen todettu vaste ja remissio tulehduksellista suolistosairautta koskevan IBDQ (Inflammatory Bowel Disease Questionnaire) ‑kyselyn perusteella (taulukko 13).
Viikon 12 aikapisteessä merkittävästi suurempi osuus guselkumabihoitoa saaneen ryhmän kuin lumeryhmän potilaista oli kliinisessä remissiossa.
| Taulukko 13. Tehon päätetapahtumat QUASAR IS ‑induktiohoitotutkimuksen viikon 12 aikapisteessä täyttäneiden potilaiden osuus | |||
| Päätetapahtuma | Lumelääke % | 200 mg guselkumabia laskimoon induktiohoitonaa % | Hoitojen ero (95 %:n luottamusväli) |
| Kliininen remissiob | |||
| Kokonaispotilasjoukko | 8 % (N = 280) | 23 % (N = 421) | 15 % (10 %; 20 %)c |
| Ei aiempaa biologista tai JAK-estäjähoitoad | 12 % (N = 137) | 32 % (N = 202) | 20 % (12 %; 28 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunute | 4 % (N = 136) | 13 % (N = 208) | 9 % (3 %; 14 %) |
| Oireenmukainen remissiof | |||
| Kokonaispotilasjoukko | 21 % (N = 280) | 50 % (N = 421) | 29 % (23 %; 36 %)c |
| Ei aiempaa biologista tai JAK-estäjähoitoad | 26 % (N = 137) | 60 % (N = 202) | 34 % (24 %; 44 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunute | 14 % (N = 136) | 38 % (N = 208) | 24 % (16 %; 33 %) |
| Endoskooppinen paranemineng | |||
| Kokonaispotilasjoukko | 11 % (N = 280) | 27 % (N = 421) | 16 % (10 %; 21 %)c |
| Ei aiempaa biologista tai JAK-estäjähoitoad | 17 % (N = 137) | 38 % (N = 202) | 21 % (12 %; 30 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunute | 5 % (N = 136) | 15 % (N = 208) | 10 % (4 %; 16 %) |
| Kliininen vasteh | |||
| Kokonaispotilasjoukko | 28 % (N = 280) | 62 % (N = 421) | 34 % (27 %; 41 %)c |
| Ei aiempaa biologista tai JAK-estäjähoitoad | 35 % (N = 137) | 71 % (N = 202) | 36 % (26 %; 46 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunute | 20 % (N = 136) | 51 % (N = 208) | 32 % (22 %; 41 %) |
| Histologis-endoskooppinen limakalvon paranemineni | |||
| Kokonaispotilasjoukko | 8 % (N = 280) | 24 % (N = 421) | 16 % (11 %; 21 %)c |
| Ei aiempaa biologista tai JAK-estäjähoitoad | 11 % (N = 137) | 33 % (N = 202) | 22 % (13 %; 30 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunute | 4 % (N = 136) | 13 % (N = 208) | 9 % (3 %; 15 %) |
| Uupumuksen suhteen todettu vastej | |||
| Kokonaispotilasjoukko | 21 % (N = 280) | 41 % (N = 421) | 20 % (13 %; 26 %)c |
| Ei aiempaa biologista tai JAK-estäjähoitoad | 29 % (N = 137) | 42 % (N = 202) | 12 % (2 %; 23 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunute | 13 % (N = 136) | 38 % (N = 208) | 25 % (17 %; 34 %) |
| IBDQ-kyselyyn perustuva remissiok | |||
| Kokonaispotilasjoukko | 30 % (N = 280) | 51 % (N = 421) | 22 % (15 %; 29 %)c |
| Ei aiempaa biologista tai JAK-estäjähoitoad | 34 % (N = 137) | 62 % (N = 202) | 28 % (18 %; 38 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunute | 24 % (N = 136) | 39 % (N = 208) | 15 % (5 %; 25 %) |
a 200 mg guselkumabia laskimoon induktiohoitona viikolla 0, viikolla 4 ja viikolla 8. bUlostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eivätkä suurentuneet lähtötilanteesta, peräsuoliverenvuotoa koskevan osa-alueen pisteet 0 ja endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa. c p < 0,001, Cochran–Mantel–Haenszelin menetelmään perustuva (korjattu ositustekijöiden suhteen: biologisen ja/tai JAK-estäjähoidon epäonnistumisstatus ja kortikosteroidien samanaikainen käyttö lähtötilanteessa) korjattu hoitojen ero (95 %:n luottamusväli). d Lisäksi 7 potilasta lumeryhmässä ja 11 potilasta guselkumabiryhmässä oli aiemmin altistunut biologiselle hoidolle tai JAK-estäjälle eikä hoito ollut epäonnistunut. e Sisältää riittämättömän vasteen haavaiseen paksusuolitulehdukseen annettuun biologiseen hoitoon (TNF-estäjät, vedolitsumabi) ja/tai JAK-estäjään sekä vasteen häviämisen näille hoidoille tai kyvyttömyyden sietää näitä hoitoja. f Ulostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eivätkä suurentuneet induktiohoidon lähtötilanteesta sekä peräsuoliverenvuotoa koskevan osa-alueen pisteet 0. g Endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa. h Modifioitujen Mayo-pisteiden pieneneminen induktiohoidon lähtötilanteesta ≥ 30 % ja ≥ 2 pistettä ja joko peräsuoliverenvuotoa koskevassa osa-alueessa ≥ 1 pisteen pieneneminen lähtötilanteesta tai peräsuoliverenvuotoa koskevan osa-alueen pisteet 0 tai 1. i Seuraavien yhdistelmä: histologinen paraneminen (Geboes-pisteytyksen mukaan neutrofiilien infiltraatiota < 5 %:ssa kryptoista, ei kryptojen tuhoutumista eikä eroosioita, haavaumia eikä granulaatiokudosta) ja endoskooppinen paraneminen, kuten edellä määritelty. j Uupumusta arvioitiin PROMIS-Fatigue Short form 7a ‑kyselyllä. Uupumusta koskevaksi vasteeksi määriteltiin ≥ 7 pisteen paraneminen lähtötilanteesta, mikä katsotaan kliinisesti merkittäväksi. k IBDQ‑kyselyn kokonaispisteet ≥ 170. | |||
QUASAR IS ‑induktiohoitotutkimukseen ja QUASAR- induktiohoidon annoshakututkimukseen otettiin mukaan myös 48 potilasta, joiden lähtötilanteen modifioidut Mayo-pisteet olivat 4, mukaan lukien endoskopialöydösten osa-alueen pisteet 2 tai 3 ja peräsuoliverenvuoto ≥ 1. Potilailla, joiden lähtötilanteen modifioidut Mayo-pisteet olivat 4, guselkumabin teho lumehoitoon verrattuna oli kliinisellä remissiolla, kliinisellä vasteella ja endoskooppisella paranemisella viikon 12 aikapisteessä mitattuna yhdenmukainen koko keskivaikeaa tai vaikeaa aktiivista haavaista paksusuolitulehdusta sairastavan potilasjoukon kanssa.
Peräsuoliverenvuotoa ja ulostamistiheyttä koskevien osa-alueiden pisteet
Peräsuoliverenvuotoa ja ulostamistiheyttä koskevien osa-alueiden pisteiden havaittiin pienentyneen guselkumabihoitoa saaneilla potilailla jo viikolla 2 ja väheneminen jatkui viikkoon 12 saakka.
Ylläpitohoitotutkimus: QUASAR MS
QUASAR MS ‑ylläpitohoitotutkimuksessa arvioitiin 568 potilasta, joilla todettiin kliininen vaste 12 viikon aikapisteessä sen jälkeen, kun guselkumabia oli annettu laskimoon joko QUASAR IS ‑induktiohoitotutkimuksessa tai QUASAR- induktiohoidon annoshakututkimuksessa. QUASAR MS ‑ylläpitohoitotutkimuksessa nämä potilaat satunnaistettiin saamaan ihon alle annettavana ylläpitohoitona joko 100 mg guselkumabia 8 viikon välein, 200 mg guselkumabia 4 viikon välein tai lumehoitoa 44 viikon ajan.
Ensisijainen päätetapahtuma oli kliininen remissio, joka määriteltiin viikon 44 aikapisteen modifioiduilla Mayo-pisteillä. Toissijaisia päätetapahtumia viikon 44 aikapisteessä olivat mm. oireenmukainen remissio, endoskooppinen paraneminen, kliininen remissio ilman kortikosteroideja, histologis-endoskooppinen limakalvon paraneminen, uupumuksen suhteen todettu vaste ja IBDQ-kyselyyn perustuva remissio (taulukko 14).
Viikon 44 aikapisteessä merkittävästi suurempi osuus kummankin guselkumabihoitoa saaneen ryhmän potilaista kuin lumehoitoa saaneen ryhmän potilaista oli kliinisessä remissiossa.
| Taulukko 14. Tehon päätetapahtumat QUASAR MS ‑ylläpitotutkimuksen viikon 44 aikapisteessä täyttäneiden potilaiden osuus | |||||
| Päätetapahtuma | Lumelääke % | 100 mg guselkumabia injektiona ihon alle 8 viikon väleina % | 200 mg guselkumabia injektiona ihon alle 4 viikon väleinb % | Hoitojen ero (95 %:n luottamusväli) | |
| 100 mg guselkumabia | 200 mg guselkumabia | ||||
| Kliininen remissioc | |||||
| Kokonaispotilasjoukkod | 19 % (N = 190) | 45 % (N = 188) | 50 % (N = 190) | 25 % (16 %; 34 %)e | 30 % (21 %; 38 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 26 % (N = 108) | 50 % (N = 105) | 58 % (N = 96) | 24 % (12 %; 36 %) | 29 % (17 %; 41 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 8 % (N = 75) | 40 % (N = 77) | 40 % (N = 88) | 30 % (19 %; 42 %) | 32 % (21 %; 44 %) |
| Oireenmukainen remissioh | |||||
| Kokonaispotilasjoukkod | 37 % (N = 190) | 70 % (N = 188) | 69 % (N = 190) | 32 % (23 %; 41 %)e | 31 % (21 %; 40 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 46 % (N = 108) | 74 % (N = 105) | 76 % (N = 96) | 28 % (15 %; 40 %) | 28 % (15 %; 41 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 24 % (N = 75) | 65 % (N = 77) | 60 % (N = 88) | 39 % (26 %; 52 %) | 37 % (23 %; 50 %) |
| Kliininen remissio ilman kortikosteroidejai | |||||
| Kokonaispotilasjoukkod | 18 % (N = 190) | 45 % (N = 188) | 49 % (N = 190) | 26 % (17 %; 34 %)e | 29 % (20 %; 38 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 26 % (N = 108) | 50 % (N = 105) | 56 % (N = 96) | 24 % (12 %; 36 %) | 27 % (14 %; 39 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 7 % (N = 75) | 40 % (N = 77) | 40 % (N = 88) | 32 % (21 %; 43 %) | 34 % (23 %; 45 %) |
| Endoskooppinen paraneminenj | |||||
| Kokonaispotilasjoukkod | 19 % (N = 190) | 49 % (N = 188) | 52 % (N = 190) | 30 % (21 %; 38 %)e | 31 % (22 %; 40 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 26 % (N = 108) | 53 % (N = 105) | 59 % (N = 96) | 27 % (15 %; 40 %) | 30 % (18 %; 42 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 8 % (N = 75) | 45 % (N = 77) | 42 % (N = 88) | 36 % (24 %; 48 %) | 35 % (23 %; 46 %) |
| Histologis-endoskooppinen limakalvon paraneminenk | |||||
| Kokonaispotilasjoukkod | 17 % (N = 190) | 44 % (N = 188) | 48 % (N = 190) | 26 % (17 %; 34 %)e | 30 % (21 %; 38 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 23 % (N = 108) | 50 % (N = 105) | 56 % (N = 96) | 26 % (14 %; 38 %) | 30 % (17 %; 42 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 8 % (N = 75) | 38 % (N = 77) | 39 % (N = 88) | 28 % (16 %; 39 %) | 31 % (20 %; 43 %) |
| Kliininen vastel | |||||
| Kokonaispotilasjoukkod | 43 % (N = 190) | 78 % (N = 188) | 75 % (N = 190) | 34 % (25 %; 43 %)e | 31 % (21 %; 40 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 54 % (N = 108) | 83 % (N = 105) | 81 % (N = 96) | 29 % (17 %; 41 %) | 26 % (14 %; 39 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 28 % (N = 75) | 70 % (N = 77) | 67 % (N = 88) | 41 % (27 %; 54 %) | 39 % (26 %; 53 %) |
| Kliinisen remission säilyminen viikon 44 aikapisteessä potilailla, joilla todettiin kliininen remissio 12 viikkoa induktiohoidon jälkeen | |||||
| Kokonaispotilasjoukkoq | 34 % (N = 59) | 61 % (N = 66) | 72 % (N = 69) | 26 % (9 %; 43 %)m | 38 % (23 %; 54 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoar | 34 % (N = 41) | 65 % (N = 43) | 79 % (N = 48) | 31 % (9 %; 51 %) | 45 % (25 %; 62 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 27 % (N = 15) | 60 % (N = 20) | 56 % (N = 18) | 33 % (-1 %; 62 %) | 29 % (-6 %; 59 %) |
| Endoskooppinen normalisoituminenn | |||||
| Kokonaispotilasjoukkod | 15 % (N = 190) | 35 % (N = 188) | 34 % (N = 190) | 18 % (10 %; 27 %)e | 17 % (9 %; 25 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 20 % (N = 108) | 38 % (N = 105) | 42 % (N = 96) | 17 % (6 %; 29 %) | 17 % (6 %; 29 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 8 % (N = 75) | 31 % (N = 77) | 24 % (N = 88) | 21 % (10 %; 33 %) | 16 % (6 %; 26 %) |
| Uupumuksessa todettu vasteo | |||||
| Kokonaispotilasjoukkod | 29 % (N = 190) | 51 % (N = 188) | 43 % (N = 190) | 20 % (11 %; 29 %)e | 13 % (3 %; 22 %)m |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 36 % (N = 108) | 51 % (N = 105) | 53 % (N = 96) | 15 % (2 %; 28 %) | 16 % (3 %; 29 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 19 % (N = 75) | 47 % (N = 77) | 32 % (N = 88) | 27 % (13 %; 40 %) | 13 % (1 %; 26 %) |
| IBDQ-kyselyyn perustuva remissiop | |||||
| Kokonaispotilasjoukkod | 37 % (N = 190) | 64 % (N = 188) | 64 % (N = 190) | 26 % (17 %; 36 %)e | 26 % (16 %; 35 %)e |
| Ei aiempaa biologista tai JAK-estäjähoitoaf | 49 % (N = 108) | 68 % (N = 105) | 74 % (N = 96) | 19 % (6 %; 32 %) | 24 % (11 %; 37 %) |
| Aiempi biologinen ja/tai JAK-estäjähoito epäonnistunutg | 19 % (N = 75) | 58 % (N = 77) | 53 % (N = 88) | 38 % (26 %; 50 %) | 35 % (23 %; 48 %) |
a 100 mg guselkumabia injektiona ihon alle 8 viikon välein induktiohoito-ohjelman jälkeen. b 200 mg guselkumabia injektiona ihon alle 4 viikon välein induktiohoito-ohjelman jälkeen. c Ulostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eivätkä suurentuneet lähtötilanteesta, peräsuoliverenvuotoa koskevan osa-alueen pisteet 0 ja endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa. d Potilaat, joilla todettiin kliininen vaste 12 viikon kuluttua guselkumabin antamisesta laskimoon joko QUASAR-induktiotutkimuksessa tai QUASAR- induktiohoidon annoshakututkimuksessa. e p < 0,001, satunnaistamisen ositustekijöillä korjattuun Cochran–Mantel–Haenszelin menetelmään perustuva korjattu hoitojen ero (95 %:n luottamusväli). f Lisäksi 7 potilasta lumeryhmässä, 6 potilasta 100 mg guselkumabia saaneessa ryhmässä ja 6 potilasta 200 mg guselkumabia saaneessa ryhmässä oli aiemmin altistunut jollekin biologiselle hoidolle tai JAK-estäjälle eivätkä nämä hoidot olleet epäonnistuneet. g Sisältää riittämättömän vasteen haavaiseen paksusuolitulehdukseen annettuun biologiseen hoitoon (TNF-estäjät, vedolitsumabi) ja/tai JAK-estäjään sekä vasteen häviämiseen näille hoidoille tai kyvyttömyyden sietää näitä hoitoja. h Ulostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eivätkä suurentuneet induktiohoidon lähtötilanteesta sekä peräsuoliverenvuotoa koskevan osa-alueen pisteet 0. i Ei vaatinut kortikosteroidihoitoa vähintään 8 viikkoon ennen viikkoa 44 ja täytti myös kliinisen remission kriteerit viikon 44 aikapisteessä. j Endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa. k Seuraavien yhdistelmä: histologinen paraneminen (Geboes-pisteytyksen mukaan neutrofiilien infiltraatiota < 5 %:ssa kryptoista, ei kryptojen tuhoutumista eikä eroosioita, haavaumia eikä granulaatiokudosta) ja endoskooppinen paraneminen, kuten edellä määritelty. l Modifioitujen Mayo-pisteiden pieneneminen induktiohoidon lähtötilanteesta ≥ 30 % ja ≥ 2 pistettä ja joko peräsuoliverenvuotoa koskevassa osa-alueessa ≥ 1 pisteen pieneneminen lähtötilanteesta tai peräsuoliverenvuotoa koskevan osa-alueen pisteet 0 tai 1. m p < 0,01, satunnaistamisen ositustekijöillä korjattuun Cochran–Mantel–Haenszelin menetelmään perustuva korjattu hoitojen ero (95 %:n luottamusväli) n Endoskopialöydöksiä koskevan osa-alueen pisteet 0. o Uupumusta arvioitiin PROMIS-Fatigue Short form 7a ‑kyselyllä. Uupumusta koskevaksi vasteeksi määriteltiin ≥ 7 pisteen paraneminen induktiohoidon lähtötilanteesta, mikä katsotaan kliinisesti merkittäväksi. p IBDQ‑kyselyn kokonaispisteet ≥ 170. q Tutkittavat, joilla todettiin kliininen remissio 12 viikon kuluttua guselkumabin antamisesta laskimoon joko QUASAR-induktiotutkimuksessa tai QUASAR- induktiohoidon annoshakututkimuksessa. r Lisäksi 3 potilasta lumeryhmässä, 3 potilasta 100 mg guselkumabia saaneessa ryhmässä ja 3 potilasta 200 mg guselkumabia saaneessa ryhmässä oli aiemmin altistunut jollekin biologiselle hoidolle tai JAK-estäjälle eivätkä nämä hoidot olleet epäonnistuneet. | |||||
QUASAR IS ‑induktiohoitotutkimuksessa ja QUASAR MS ‑ylläpitohoitotutkimuksessa guselkumabin teho ja turvallisuus osoitettiin yhdenmukaisesti riippumatta iästä, sukupuolesta, etnisestä taustasta, painosta ja aiemmasta hoidosta biologisella valmisteella tai JAK-estäjällä.
QUASAR MS ‑ylläpitohoitotutkimuksessa potilaat, joilla oli suuri tulehdustaakka induktiohoidon päättymisen jälkeen, saivat lisähyötyä 200 mg:n guselkumabiannoksista ihon alle 4 viikon välein verrattuna 100 mg ihon alle 8 viikon välein annostukseen. Näiden kahden guselkumabiannosryhmän välillä havaittiin viikon 44 aikapisteessä seuraavien päätetapahtumien osalta kliinisesti merkittäviä > 15 %:n numeerisia eroja potilailla, joiden CRP-pitoisuus oli > 3 mg/l induktiohoidon päättymisen jälkeen: kliininen remissio (48 % annoksen 200 mg 4 viikon välein yhteydessä vs. 30 % annoksen 100 mg 8 viikon välein yhteydessä), kliinisen remission säilyminen (88 % annoksen 200 mg 4 viikon välein yhteydessä vs. 50 % annoksen 100 mg 8 viikon välein yhteydessä), kliininen remissio ilman kortikosteroideja (46 % annoksen 200 mg 4 viikon välein yhteydessä vs. 30 % annoksen 100 mg 8 viikon välein yhteydessä), endoskooppinen paraneminen (52 % annoksen 200 mg 4 viikon välein yhteydessä vs. 35 % annoksen 100 mg 8 viikon välein yhteydessä) ja histologis-endoskooppinen limakalvon paraneminen (46 % annoksen 200 mg 4 viikon välein yhteydessä vs. 29 % annoksen 100 mg 8 viikon välein yhteydessä).
QUASAR MS ‑ylläpitohoitotutkimukseen otettiin mukaan 31 potilasta, joilla induktiohoidon lähtötilanteen modifioidut Mayo-pisteet olivat 4, mukaan lukien endoskopialöydösten osa-alueen pisteet 2 tai 3 ja peräsuoliverenvuotoa koskevat pisteet ≥ 1 ja joilla todettiin kliininen vaste 12 viikon kuluttua guselkumabin antamisesta laskimoon QUASAR IS ‑induktiohoitotutkimuksessa tai QUASAR- induktiohoidon annoshakututkimuksessa. Näillä potilailla guselkumabin teho lumelääkkeeseen verrattuna viikon 44 aikapisteessä oli kliinisellä remissiolla, kliinisellä vasteella ja endoskooppisella paranemisella mitattuna yhdenmukainen kokonaispotilasjoukon kanssa.
Oireenmukainen remissio ajan kuluessa
QUASAR MS ‑ylläpitohoitotutkimuksessa oireenmukainen remissio, joksi määriteltiin ulostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eikä pisteiden suurenemista induktiohoidon lähtötilanteesta, ja peräsuoliverenvuotoa koskevan osa-alueen pisteet 0, säilyi kummassakin guselkumabihoitoryhmässä viikkoon 44 saakka, kun taas lumelääkeryhmässä näissä havaittiin huononemista (kuva 8):
Kuva 8. Oireenmukaisessa remissiossa olevien potilaiden osuus QUASAR MS ‑ylläpitohoitotutkimuksen viikkoon 44 mennessä
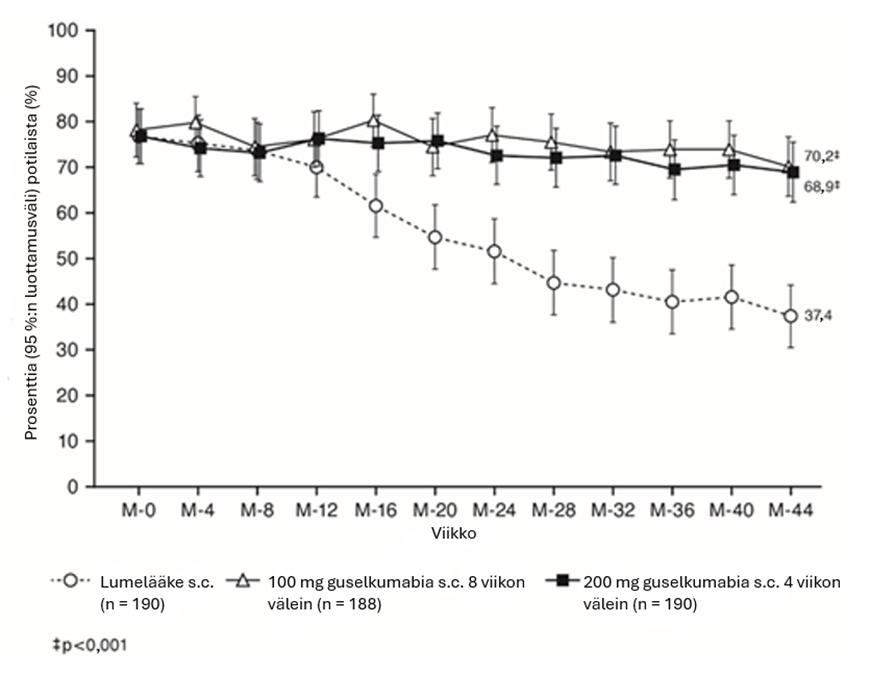
Pidennettyyn guselkumabihoitoon viikolla 24 vasteen saaneet
Guselkumabihoitoa saaneet potilaat, jotka eivät olleet saavuttaneet kliinistä vastetta induktiohoidon viikolla 12, saivat 200 mg guselkumabia ihon alle viikoilla 12, 16 ja 20. QUASAR IS ‑induktiohoitotutkimuksessa 120 guselkumabihoitoa saaneesta potilaasta, joilla ei ollut kliinistä vastetta induktiohoidon viikolla 12, 66:lla (55 %) todettiin kliininen vaste viikolla 24. Potilaat, joilla oli vaste guselkumabihoitoon viikolla 24, otettiin mukaan QUASAR MS ‑ylläpitohoitotutkimukseen, jossa he saivat 200 mg guselkumabia ihon alle 4 viikon välein. Näistä QUASAR MS ‑ylläpitohoitotutkimuksen 123 potilaasta 83:lla (67 %) kliininen vaste oli säilynyt ja 37:llä (30 %) todettiin kliininen remissio viikon 44 aikapisteessä.
Tehon saavuttaminen uudelleen guselkumabihoitovasteen hävittyä
Yhdeksäntoista potilasta, jotka saivat 100 mg guselkumabia ihon alle 8 viikon välein ja joilla vaste hävisi ensimmäisen kerran (10 %) QUASAR MS ‑ylläpitohoitotutkimuksen viikkojen 8 ja 32 välillä, sai sokkoutetun guselkumabiannoksen 200 mg guselkumabia ihon alle 4 viikon välein. Yhdellätoista näistä potilaista (58 %) todettiin oireenmukainen vaste, ja 5 potilaalla (26 %) todettiin oireenmukainen remissio 12 viikon jälkeen.
Histologinen ja endoskooppinen arviointi
Histologiseksi remissioksi määriteltiin Geboesin histologiset pisteet ≤ 2 B.0 (Geboes-pisteytyksen mukaan limakalvoilla [sekä limakalvon tukikerroksessa että epiteelissä] ei neutrofiilejä, ei kryptojen tuhoutumista eikä eroosioita, haavaumia eikä granulaatiokudosta). QUASAR IS ‑induktiohoitotutkimuksessa histologinen remissio todettiin viikon 12 aikapisteessä 40 %:lla guselkumabihoitoa saaneista potilaista ja 19 %:lla lumeryhmän potilaista. QUASAR MS ‑ylläpitohoitotutkimuksessa histologinen remissio todettiin viikon 44 aikapisteessä 59 %:lla 100 mg guselkumabia ihon alle 8 viikon välein saaneista potilaista ja 61 %:lla 200 mg guselkumabia ihon alle 4 viikon välein saaneista potilaista sekä 27 %:lla lumeryhmän potilaista.
Limakalvon endoskooppiseksi normalisoitumiseksi määriteltiin endoskopialöydöksiä koskevan osa-alueen pisteet 0. QUASAR IS ‑induktiohoitotutkimuksessa endoskooppinen normalisoituminen todettiin viikon 12 aikapisteessä 15 %:lla guselkumabihoitoa saaneista potilaista ja 5 %:lla lumeryhmän potilaista.
Limakalvon yhdistetyt histologis-endoskooppiset päätetapahtumat
Oireenmukaisen remission, endoskooppisen normalisoitumisen, histologisen remission ja ulosteen kalprotektiinipitoisuuden ≤ 250 mg/kg yhdistelmä todettiin viikon 44 aikapisteessä suuremmalla osalla 100 mg guselkumabia ihon alle 8 viikon välein tai 200 mg guselkumabia ihon alle 4 viikon välein saaneista kuin lumelääkettä saaneista potilaista (22 %:lla 100 mg guselkumabia ihon alle 8 viikon välein saaneista, 28 %:lla 200 mg guselkumabia ihon alle 4 viikon välein saaneista vs. 9 %:lla lumelääkettä saaneista potilaista).
Terveyteen liittyvä elämänlaatu
QUASAR IS ‑induktiohoitotutkimuksessa guselkumabia saaneilla potilailla todettiin viikon 12 aikapisteessä tulehduksellisen suolistosairauden (IBD) suhteen spesifisessä elämänlaadussa suurempaa ja kliinisesti merkittävää paranemista lähtötilanteesta kuin lumelääkettä saaneilla. Tulehduksellisen suolistosairauden suhteen spesifistä elämänlaatua arvioitiin IBDQ-kokonaispisteiden ja kaikkien IBDQ-osa-alueiden (suolisto-oireet, mukaan lukien vatsakipu ja ulostamispakko, systeemiset toiminnot, emotionaaliset toiminnot ja sosiaaliset toiminnot) perusteella. Tämä paraneminen säilyi guselkumabihoitoa QUASAR MS ‑ylläpitohoitotutkimuksessa saaneilla potilailla viikkoon 44 saakka.
Haavaiseen paksusuolitulehdukseen liittyvä sairaalahoito
QUASAR IS ‑induktiohoitotutkimuksen viikkoon 12 mennessä pienempi osuus guselkumabiryhmän kuin lumeryhmän potilaista oli joutunut sairaalahoitoon haavaisen paksusuolitulehduksen vuoksi (guselkumabiryhmässä 1,9 %, 8/421 vs. lumeryhmässä 5,4 %, 15/280).
ASTRO
ASTRO-tutkimuksessa potilaat satunnaistettiin suhteessa 1:1:1 saamaan induktiohoitona 400 mg guselkumabia ihon alle viikoilla 0, 4 ja 8 ja sen jälkeen ylläpitohoitona 100 mg guselkumabia ihon alle 8 viikon välein tai induktiohoitona 400 mg guselkumabia ihon alle viikoilla 0, 4 ja 8 ja sen jälkeen ylläpitohoitona 200 mg guselkumabia ihon alle 4 viikon välein tai lumehoitoa.
Yhteensä 418 potilasta arvioitiin. Potilaiden iän mediaani oli 40 vuotta (vaihteluväli 18–80 vuotta); 38,8 % oli naisia ja 64,6 % identifioitui valkoihoisiksi, 28,9 % aasialaisiksi ja 3,1 % mustaihoisiksi.
Tutkimukseen mukaan otettujen potilaiden oli sallittua käyttää vakaina annoksina suun kautta otettavia aminosalisylaatteja, immunomodulaattoreita (atsatiopriini, 6‑merkaptopuriini, metotreksaatti) ja/tai suun kautta otettavia kortikosteroideja (enintään 20 mg/vrk prednisonia tai vastaavaa). Lähtötilanteessa 77,3 % potilaista sai aminosalisylaatteja, 20,1 % potilaista sai immunomodulaattoreita ja 32,8 % potilaista sai kortikosteroideja. Samanaikaiset biologiset hoidot, JAK-estäjät tai sfingosiini-1-fosfaatti (S1P) ‑reseptorin muuntajat eivät olleet sallittuja. Yhteensä 40,2 %:lla potilaista vähintään yksi aiempi biologinen hoito, hoito JAK-estäjällä ja/tai S1P-reseptorin muuntajalla oli epäonnistunut. 58,1 % ei ollut aiemmin saanut biologista hoitoa, JAK-estäjää tai S1P-reseptorin muuntajaa, ja 1,7 % oli aiemmin saanut jotakin biologista hoitoa, JAK-estäjää tai S1P-reseptorin muuntajaa eikä hoito ollut epäonnistunut.
ASTRO-tutkimuksen ensisijainen päätetapahtuma oli kliininen remissio viikon 12 aikapisteessä modifioidulla Mayo-pisteytyksellä määriteltynä. Toissijaisia päätetapahtumia viikon 12 aikapisteessä olivat oireenmukainen remissio, endoskooppinen paraneminen, kliininen vaste ja histologis-endoskooppinen limakalvon paraneminen (ks. taulukko 15). Toissijaisia päätetapahtumia viikon 24 aikapisteessä olivat kliininen remissio ja endoskooppinen paraneminen (ks. taulukko 16).
| Taulukko 15. Tehon päätetapahtumat ASTRO-tutkimuksen viikon 12 aikapisteessä täyttäneiden potilaiden osuus | |||
| Päätetapahtuma | Lumelääke % | 400 mg guselkumabia ihon alle induktiohoitonaa % | Hoidon ero lumehoitoon verrattuna (95 %:n luottamusväli)b |
| Kliininen remissioc | |||
| Kokonaispotilasjoukko | 6 % (N = 139) | 28 % (N = 279) | 21 % (15 %; 28 %)e |
| Ei aiempaa biologista hoitoa, JAK-estäjähoitoa tai hoitoa S1P-reseptorin muuntajallaf | 9 % (N = 79) | 36 % (N = 164) | 27 % (18 %; 37 %) |
| Aiempi biologinen hoito, JAK-estäjähoito ja/tai hoito S1P-reseptorin muuntajalla epäonnistunutg | 4 % (N = 56) | 16 % (N = 112) | 12 % (3 %; 20 %) |
| Oireenmukainen remissiod | |||
| Kokonaispotilasjoukko | 21 % (N = 139) | 51 % (N = 279) | 30 % (22 %; 39 %)e |
| Ei aiempaa biologista hoitoa, JAK-estäjähoitoa tai hoitoa S1P-reseptorin muuntajallaf | 25 % (N = 79) | 59 % (N = 164) | 34 % (22 %; 46 %) |
| Aiempi biologinen hoito, JAK-estäjähoito ja/tai hoito S1P-reseptorin muuntajalla epäonnistunutg | 14 % (N = 56) | 41 % (N = 112) | 26 % (13 %; 39 %) |
| Endoskooppinen paraneminenh | |||
| Kokonaispotilasjoukko | 13 % (N = 139) | 37 % (N = 279) | 24 % (17 %; 32 %)e |
| Ei aiempaa biologista hoitoa, JAK-estäjähoitoa tai hoitoa S1P-reseptorin muuntajallaf | 18 % (N = 79) | 46 % (N = 164) | 28 % (17 %; 40 %) |
| Aiempi biologinen hoito, JAK-estäjähoito ja/tai hoito S1P-reseptorin muuntajalla epäonnistunutg | 7 % (N = 56) | 24 % (N = 112) | 16 % (6 %; 26 %) |
| Kliininen vastei | |||
| Kokonaispotilasjoukko | 35 % (N = 139) | 66 % (N = 279) | 31 % (22 %; 40 %)e |
| Ei aiempaa biologista hoitoa, JAK-estäjähoitoa tai hoitoa S1P-reseptorin muuntajallaf | 42 % (N = 79) | 71 % (N = 164) | 30 % (17 %; 43 %) |
| Aiempi biologinen hoito, JAK-estäjähoito ja/tai hoito S1P-reseptorin muuntajalla epäonnistunutg | 25 % (N = 56) | 57 % (N = 112) | 31 % (17 %; 45 %) |
| Histologis-endoskooppinen limakalvon paraneminenj | |||
| Kokonaispotilasjoukko | 11 % (N = 139) | 30 % (N = 279) | 20 % (12 %; 27 %)e |
| Ei aiempaa biologista hoitoa, JAK-estäjähoitoa tai hoitoa S1P-reseptorin muuntajallaf | 14 % (N = 79) | 38 % (N = 164) | 25 % (14 %; 35 %) |
| Aiempi biologinen hoito, JAK-estäjähoito ja/tai hoito S1P-reseptorin muuntajalla epäonnistunutg | 7 % (N = 56) | 19 % (N = 112) | 11 % (1 %; 20 %) |
a 400 mg guselkumabia ihon alle induktiohoitona viikolla 0, viikolla 4 ja viikolla 8. b Korjattu hoitojen ero ja luottamusvälit perustuvat yleiseen eroon riskissä ja on saatu käyttämällä Mantel–Haenszelin ositepainoja ja Saton varianssiestimaattoria. Osituksen muuttujina käytettiin aiemman biologisen hoidon, JAK-estäjähoidon ja/tai S1P-reseptorin muuntajalla annetun hoidon epäonnistumisstatusta (kyllä tai ei) sekä Mayon endoskopialöydöksiä koskevan osa-alueen pisteitä lähtötilanteessa (keskivaikea [2] tai vaikea [3]). c Ulostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eivätkä suurentuneet lähtötilanteesta, peräsuoliverenvuotoa koskevan osa-alueen pisteet 0 ja endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa. d Ulostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eivätkä suurentuneet induktiohoidon lähtötilanteesta sekä peräsuoliverenvuotoa koskevan osa-alueen pisteet 0. e p < 0,001 f Lisäksi 4 potilasta lumeryhmässä ja 3 potilasta guselkumabiryhmässä oli aiemmin altistunut biologiselle hoidolle, JAK-estäjälle tai S1P-reseptorin muuntajalle eikä hoito ollut epäonnistunut. g Sisältää riittämättömän vasteen haavaiseen paksusuolitulehdukseen annettuun biologiseen hoitoon (TNF-estäjät, vedolitsumabi), JAK-estäjään ja/tai S1P-reseptorin muuntajaan, vasteen häviämisen näille hoidoille tai kyvyttömyyden sietää näitä hoitoja. h Endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa. i Modifioitujen Mayo-pisteiden pieneneminen lähtötilanteesta ≥ 30 % ja ≥ 2 pistettä ja joko peräsuoliverenvuotoa koskevassa osa-alueessa ≥ 1 pisteen pieneneminen lähtötilanteesta tai peräsuoliverenvuotoa koskevan osa-alueen pisteet 0 tai 1. j Endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa ja Geboes-pisteytys ≤ 3.1 (mikä osoittaa neutrofiilien infiltraatiota < 5 %:ssa kryptoista, ei kryptojen tuhoutumista eikä eroosioita, haavaumia eikä granulaatiokudosta). | |||
| Taulukko 16. Tehon päätetapahtumat ASTRO-tutkimuksen viikon 24 aikapisteessä täyttäneiden potilaiden osuus | |||||
| Päätetapahtuma | Lumelääke % | 400 mg guselkumabia ihon alle induktio-hoitona→ 100 mg injektiona ihon alle 8 viikon välein a % | 400 mg guselkumabia ihon alle induktio-hoitona→ 200 mg injektiona ihon alle 4 viikon välein b % | Hoidon ero lumehoitoon verrattuna (95 %:n luottamusväli)c | |
| 100 mg guselkumabia | 200 mg guselkumabia | ||||
| Kliininen remissiod | |||||
| Kokonaispotilasjoukko | 9 % (N = 139) | 35 % (N = 139) | 36 % (N = 140) | 26 % (17 %; 35 %)e | 27 % (18 %; 36 %)e |
| Ei aiempaa biologista hoitoa, JAK-estäjähoitoa tai hoitoa S1P-reseptorin muuntajallaf | 13 % (N = 79) | 49 % (N = 81) | 43 % (N = 83) | 37 % (24 %; 50 %) | 31 % (18 %; 44 %) |
| Aiempi biologinen hoito, JAK-estäjähoito ja/tai hoito S1P-reseptorin muuntajalla epäonnistunutg | 5 % (N = 56) | 16 % (N = 57) | 27 % (N = 55) | 10 % (‑1 %; 21 %) | 21 % (9 %; 34 %) |
| Endoskooppinen paraneminenh | |||||
| Kokonaispotilasjoukko | 12 % (N = 139) | 40 % (N = 139) | 45 % (N = 140) | 28 % (18 %; 38 %)e | 33 % (23 %; 42 %)e |
| Ei aiempaa biologista hoitoa, JAK-estäjähoitoa tai hoitoa S1P-reseptorin muuntajallaf | 18 % (N = 79) | 54 % (N = 81) | 52 % (N = 83) | 37 % (23 %; 51 %) | 34 % (21 %; 48 %) |
| Aiempi biologinen hoito, JAK-estäjähoito ja/tai hoito S1P-reseptorin muuntajalla epäonnistunutg | 5 % (N = 56) | 19 % (N = 57) | 36 % (N = 55) | 13 % (1 %; 25 %) | 30 % (17 %; 44 %) |
a 400 mg guselkumabia ihon alle induktiohoitona viikoilla 0, 4 ja 8, minkä jälkeen 100 mg guselkumabia ihon alle ylläpitohoitona 8 viikon välein. b 400 mg guselkumabia ihon alle induktiohoitona viikoilla 0, 4 ja 8, minkä jälkeen 200 mg guselkumabia ihon alle ylläpitohoitona 4 viikon välein c Korjattu hoitojen ero ja luottamusvälit perustuvat yleiseen eroon riskissä ja on saatu käyttämällä Mantel–Haenszelin ositepainoja ja Saton varianssiestimaattoria. Osituksen muuttujina käytettiin aiemman biologisen hoidon, JAK-estäjähoidon ja/tai S1P-reseptorin muuntajalla annetun hoidon epäonnistumisstatusta (kyllä tai ei) sekä Mayon endoskopialöydöksiä koskevan osa-alueen pisteitä lähtötilanteessa (keskivaikea [2] tai vaikea [3]). d Ulostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eivätkä suurentuneet lähtötilanteesta, peräsuoliverenvuotoa koskevan osa-alueen pisteet 0 ja endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa. e p < 0,001 f Lisäksi 4 potilasta lumeryhmässä, 1 potilas 100 mg guselkumabia saaneessa ryhmässä ja 2 potilasta 200 mg guselkumabia saaneessa ryhmässä oli aiemmin altistunut biologiselle hoidolle, JAK-estäjälle tai S1P-reseptorin muuntajalle eikä hoito ollut epäonnistunut. g Sisältää riittämättömän vasteen haavaiseen paksusuolitulehdukseen annettuun biologiseen hoitoon (TNF-estäjät, vedolitsumabi), JAK-estäjään ja/tai S1P-reseptorin muuntajaan, vasteen häviämisen näille hoidoille tai kyvyttömyyden sietää näitä hoitoja. h Endoskopialöydöksiä koskevan osa-alueen pisteet 0 tai 1 eikä kosketusverenvuotoa | |||||
Oireenmukainen remissio ajan kuluessa
ASTRO-tutkimuksessa oireenmukaiseksi remissioksi määriteltiin ulostamistiheyttä koskevan osa-alueen pisteet 0 tai 1 eikä pisteiden suurenemista lähtötilanteesta ja peräsuoliverenvuotoa koskevan osa-alueen pisteet 0 viikon 12 aikapisteeseen saakka. Oireenmukainen remissio todettiin suuremmalla osalla guselkumabihoitoryhmien potilaista kuin lumeryhmän potilaista (kuva 9):
Kuva 9. Oireenmukaisessa remissiossa olevien potilaiden osuus ASTRO-tutkimuksen viikkoon 12 mennessä
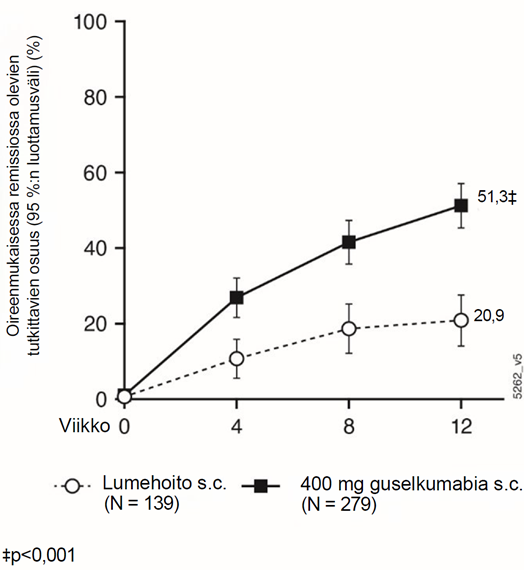
Peräsuoliverenvuotoa ja ulostamistiheyttä koskevien osa-alueiden pisteet
Peräsuoliverenvuotoa ja ulostamistiheyttä koskevien osa-alueiden pisteiden havaittiin pienentyneen guselkumabihoitoa saaneilla potilailla lumehoitoon verrattuna jo viikolla 2.
Histologinen ja endoskooppinen arviointi
Histologinen remissio todettiin viikon 12 aikapisteessä 44 %:lla potilaista, jotka olivat saaneet induktiohoitona 400 mg guselkumabia ihon alle, verrattuna 20 %:iin lumehoitoa saaneista potilaista.
Endoskooppinen normalisoituminen todettiin viikon 24 aikapisteessä 21 %:lla potilaista, jotka olivat saaneet induktiohoitona 400 mg guselkumabia ihon alle ja sen jälkeen 100 mg guselkumabia injektiona ihon alle viikolla 16 ja sitten 8 viikon välein, ja 26 %:lla potilaista, jotka olivat saaneet induktiohoitona 400 mg guselkumabia ihon alle ja sen jälkeen 200 mg guselkumabia injektiona ihon alle viikolla 12 ja sitten 4 viikon välein, verrattuna 4 %:iin lumehoitoa saaneista potilaista.
Vatsakipu ja ulostamispakko
Suuremmalla osalla 400 mg guselkumabia ihon alle induktiohoitona saaneista potilaista kuin lumehoitoa saaneista potilaista ei ollut viikon 12 aikapisteessä vatsakipua (56 % vs. 31 %) eikä ulostamispakkoa (49 % vs. 24 %).
Terveyteen liittyvä elämänlaatu
Sairausspesifistä terveyteen liittyvää elämänlaatua arvioitiin tulehduksellista suolistosairautta (IBD) koskevalla spesifisellä elämänlaatuarvioinnilla (IBDQ). IBDQ-pisteillä arvioituna remissio todettiin viikon 12 aikapisteessä suuremmalla osalla potilaista yhdistetyissä guselkumabiryhmissä (61%), jotka saivat 400 mg:n annoksia ihon alle, kuin lumeryhmässä (34 %).
Crohnin tauti
Guselkumabin tehoa ja turvallisuutta arvioitiin kolmessa vaiheen III kliinisessä tutkimuksessa aikuisilla potilailla, joilla oli keskivaikea tai vaikea aktiivinen Crohnin tauti ja joiden vaste suun kautta otettaviin kortikosteroideihin, tavanomaisiin immunomodulaattoreihin (atsatiopriini, merkaptopuriini, metotreksaatti) ja/tai biologiseen hoitoon (TNF-estäjät tai vedolitsumabi) oli ollut riittämätön, joiden vaste näihin oli hävinnyt tai jotka eivät olleet sietäneet tällaisia hoitoja. Kyseiset tutkimukset olivat kaksi 48 viikon pituista satunnaistettua, kaksoissokkoutettua, lumevalmisteella ja vaikuttavalla aineella (ustekinumabi) kontrolloitua rinnakkaisryhmillä tehtyä monikeskustutkimusta, joiden tutkimusasetelmat olivat identtiset (GALAXI 2 ja GALAXI 3), sekä yksi 24 viikon pituinen satunnaistettu, kaksoissokkoutettu, lumekontrolloitu, rinnakkaisryhmillä tehty monikeskustutkimus (GRAVITI). Kaikissa kolmessa tutkimuksessa oli treat-through-tutkimusasetelma, mikä tarkoitti, että guselkumabihoitoon (tai GALAXI 2- ja GALAXI 3 ‑tutkimuksissa ustekinumabihoitoon) satunnaistetut potilaat jatkoivat samaa heille osoitettua hoitoa koko tutkimuksen ajan.
GALAXI 2 ja GALAXI 3
Vaiheen III GALAXI 2- ja GALAXI 3 ‑tutkimuksissa keskivaikean tai vaikean aktiivisen Crohnin taudin määritelmä oli CDAI (Crohn’s Disease Activity Index) ‑pisteet ≥ 220 – ≤ 450 ja SES-CD (Simple Endoscopic Score for CD) ‑pisteet ≥ 6 (tai ≥ 4, jos potilaalla oli ileumiin rajoittunut tauti). Muita kriteereitä GALAXI 2/3 ‑tutkimuksissa olivat keskimääräistä päivittäistä ulostustiheyttä koskevat pisteet > 3 tai keskimääräiset päivittäistä vatsakipua koskevat pisteet > 1.
GALAXI 2- ja GALAXI 3 ‑tutkimuksissa potilaat satunnaistettiin suhteessa 2:2:2:1 saamaan 200 mg guselkumabia laskimoon induktiohoitona viikoilla 0, 4 ja 8 ja sen jälkeen 200 mg guselkumabia ihon alle neljän viikon välein ylläpitohoitona tai 200 mg guselkumabia laskimoon induktiohoitona viikoilla 0, 4 ja 8 ja sen jälkeen 100 mg guselkumabia ihon alle kahdeksan viikon välein ylläpitohoitona tai noin 6 mg/kg ustekinumabia laskimoon induktiohoitona viikolla 0 ja sen jälkeen 90 mg ustekinumabia ihon alle kahdeksan viikon välein ylläpitohoitona tai lumelääkettä. Tutkittavat, jotka eivät saaneet vastetta lumelääkkeeseen, saivat viikosta 12 alkaen ustekinumabia.
GALAXI 2- (n = 508) ja GALAXI 3 (n = 513) ‑tutkimuksissa arvioitiin yhteensä 1 021 potilasta. Iän mediaani oli 34 vuotta (vaihteluväli 18–83 vuotta); 57,6 % oli miehiä, ja 74,3 % identifioitui valkoihoisiksi, 21,3 % aasialaisiksi ja 1,5 % mustaihoisiksi.
GALAXI 2 ‑tutkimuksessa 52,8 %:lla potilaista vähintään yksi aiempi biologinen hoito oli epäonnistunut (50,6 % ei ollut sietänyt vähintään yhtä aiempaa TNFα-estäjähoitoa tai se oli epäonnistunut, 7,5 % ei ollut sietänyt aiempaa vedolitsumabihoitoa tai se oli epäonnistunut), 41,9 % ei ollut aiemmin saanut biologista hoitoa ja 5,3 % oli aiemmin saanut biologista hoitoa eikä hoito ollut epäonnistunut. Lähtötilanteessa 37,4 % potilaista sai suun kautta otettavia kortikosteroideja ja 29,9 % potilaista sai tavanomaisia immunomodulaattoreita.
GALAXI 3 ‑tutkimuksessa 51,9 %:lla potilaista vähintään yksi aiempi biologinen hoito oli epäonnistunut (50,3 % ei ollut sietänyt vähintään yhtä aiempaa TNFα-estäjähoitoa tai se oli epäonnistunut, 9,6 % ei ollut sietänyt aiempaa vedolitsumabihoitoa tai se oli epäonnistunut), 41,5 % ei ollut aiemmin saanut biologista hoitoa ja 6,6 % oli aiemmin saanut biologista hoitoa eikä hoito ollut epäonnistunut. Lähtötilanteessa 36,1 % potilaista sai suun kautta otettavia kortikosteroideja ja 30,2 % potilaista sai tavanomaisia immunomodulaattoreita.
Rinnakkaisten ensisijaisten ja tärkeimpien toissijaisten päätetapahtumien tulosten vertailu lumelääkkeeseen GALAXI 2- ja GALAXI 3 ‑tutkimuksissa esitetään taulukoissa 17 (viikko 12) ja 18 (viikko 48). Tärkeimpien toissijaisten päätetapahtumien viikon 48 tulosten vertailu ustekinumabiin esitetään taulukoissa 19 ja 20.
| Taulukko 17. Guselkumabihoidolla rinnakkaiset ensisijaiset päätetapahtumat ja tärkeimmät toissijaiset tehon päätetapahtumat GALAXI 2- ja GALAXI 3 ‑tutkimusten viikon 12 aikapisteessä saavuttaneiden potilaiden osuus verrattuna lumelääkkeeseen | ||||
| GALAXI 2 | GALAXI 3 | |||
Lumelääke % | Guselkumabi laskimoon induktiohoitonaa % | Lumelääke % | Guselkumabi laskimoon induktiohoitonaa % | |
| Rinnakkaiset ensisijaiset päätetapahtumat | ||||
| Kliininen remissiob viikon 12 aikapisteessä | ||||
| Kokonaispotilasjoukko | 22 % (N = 76) | 47 %i (N = 289) | 15 % (N = 72) | 47 %i (N = 293) |
| Ei aiempaa biologista hoitoac | 18 % (N = 34) | 50 % (N = 121) | 15 % (N = 27) | 50 % (N = 123) |
| Aiempi biologinen hoito epäonnistunutd | 23 % (N = 39) | 45 % (N = 150) | 15 % (N = 39) | 47 % (N = 150) |
| Endoskooppinen vastee viikon 12 aikapisteessä | ||||
| Kokonaispotilasjoukko | 11 % (N = 76) | 38 %i (N = 289) | 14 % (N = 72) | 36 %i (N = 293) |
| Ei aiempaa biologista hoitoac | 15 % (N = 34) | 51 % (N = 121) | 22 % (N = 27) | 41 % (N = 123) |
| Aiempi biologinen hoito epäonnistunutd | 5 % (N = 39) | 27 % (N = 150) | 8 % (N = 39) | 31 % (N = 150) |
| Tärkeimmät toissijaiset tehon päätetapahtumat | ||||
| PRO-2-remissiof viikon 12 aikapisteessä | ||||
| Kokonaispotilasjoukko | 21 % (N = 76) | 43 %i (N = 289) | 14 % (N = 72) | 42 %i (N = 293) |
| Ei aiempaa biologista hoitoac | 24 % (N = 34) | 43 % (N = 121) | 15 % (N = 27) | 47 % (N = 123) |
| Aiempi biologinen hoito epäonnistunutd | 13 % (N = 39) | 41 % (N = 150) | 13 % (N = 39) | 39 % (N = 150) |
| Uupumuksen suhteen todettu vasteg viikon 12 aikapisteessä | ||||
| Kokonaispotilasjoukko | 29 % (N = 76) | 45 %j (N = 289) | 18 % (N = 72) | 43 %i (N = 293) |
| Ei aiempaa biologista hoitoac | 32 % (N = 34) | 48 % (N = 121) | 19 % (N = 27) | 46 % (N = 123) |
| Aiempi biologinen hoito epäonnistunutd | 26 % (N = 39) | 41 % (N = 150) | 18 % (N = 39) | 43 % (N = 150) |
| Endoskooppinen remissioh viikon 12 aikapisteessä | ||||
| Kokonaispotilasjoukko | 1 % (N = 76) | 15 % (N = 289) | 8 % (N = 72) | 16 % (N = 293) |
| Ei aiempaa biologista hoitoac | 3 % (N = 34) | 22 % (N = 121) | 19 % (N = 27) | 25 % (N = 123) |
| Aiempi biologinen hoito epäonnistunutd | 0 % (N = 39) | 9 % (N = 150) | 0 % (N = 39) | 9 % (N = 150) |
a 200 mg guselkumabia laskimoon induktiohoitona viikolla 0, viikolla 4 ja viikolla 8 – Tässä sarakkeessa kaksi guselkumabihoitoryhmää on yhdistetty, sillä potilaat saivat samansuuruisina annoksina laskimoon annettavan induktiohoito-ohjelman ennen viikon 12 aikapistettä. b Kliiniseksi remissioksi on määritelty CDAI-pisteet < 150. c Lisäksi 9 potilasta lumeryhmässä ja 38 potilasta 200 mg guselkumabia laskimoon saaneessa ryhmässä oli aiemmin altistunut biologiselle hoidolle eikä hoito ollut epäonnistunut. d Sisältää riittämättömän vasteen Crohnin tautiin annettuun biologiseen hoitoon (TNF-estäjät, vedolitsumabi) sekä vasteen häviämisen näille hoidoille tai kyvyttömyyden sietää näitä hoitoja. e Endoskooppiseksi vasteeksi on määritelty SES-CD-pisteiden ≥ 50 %:n paraneminen lähtötilanteesta tai SES-CD-pisteet ≤ 2. f PRO-2-remissioksi on määritelty vatsakipua koskevat keskimääräiset päivittäiset pisteet enintään 1 ja ulostustiheyttä koskevat keskimääräiset päivittäiset pisteet enintään 3 eikä vatsakivun pahenemista tai ulostustiheyden lisääntymistä lähtötilanteesta. g Uupumuksen suhteen todetuksi vasteeksi on määritelty ≥ 7 pisteen paraneminen PROMIS Fatigue Short Form 7a ‑kyselyssä. h Endoskooppiseksi remissioksi on määritelty SES-CD-pisteet ≤ 2. i p < 0,001 j p < 0,05 | ||||
| Taulukko 18. Guselkumabihoidolla tärkeimmät toissijaiset tehon päätetapahtumat GALAXI 2- ja GALAXI 3 ‑tutkimusten viikon 48 aikapisteessä saavuttaneiden potilaiden osuus verrattuna lumelääkkeeseen | ||||||
| GALAXI 2 | GALAXI 3 | |||||
Lumelääke
| Guselkumabi laskimoon induktio-hoitona → 100 mg injektiona ihon alle 8 viikon väleina
| Guselkumabi laskimoon induktio-hoitona → 200 mg injektiona ihon alle 4 viikon väleinb
| Lumelääke (N = 72) | Guselkumabi laskimoon induktio-hoitona → 100 mg injektiona ihon alle 8 viikon väleina
| Guselkumabi laskimoon induktio-hoitona → 200 mg injektiona ihon alle 4 viikon väleinb
| |
| Kliininen remissio ilman kortikosteroidejac viikon 48 aikapisteessäf | ||||||
| Kokonais-potilas-joukko | 12 % (N = 76) | 45 %e (N = 143) | 51 %e (N = 146) | 14 % (N = 72) | 44 %e (N = 143) | 48 %e (N = 150) |
| Endoskooppinen vasted viikon 48 aikapisteessäf | ||||||
| Kokonais-potilas-joukko | 7 % (N = 76) | 38 %e (N = 143) | 38 %e (N = 146) | 6 % (N = 72) | 33 %e (N = 143) | 36 %e (N = 150) |
a 200 mg guselkumabia laskimoon induktiohoitona viikolla 0, viikolla 4 ja viikolla 8, jonka jälkeen 100 mg guselkumabia ihon alle 8 viikon välein enintään 48 viikon ajan. b 200 mg guselkumabia laskimoon induktiohoitona viikolla 0, viikolla 4 ja viikolla 8, jonka jälkeen 200 mg guselkumabia ihon alle 4 viikon välein enintään 48 viikon ajan. c Kliiniseksi remissioksi ilman kortikosteroideja määriteltiin CDAI-pisteet < 150 viikon 48 aikapisteessä eikä kortikosteroidihoitoa viikon 48 aikapisteessä. d Endoskooppiseksi vasteeksi on määritelty SES-CD-pisteiden ≥ 50 %:n paraneminen lähtötilanteesta tai SES-CD-pisteet ≤ 2. e p < 0,001 f Riittämättömän vasteen kriteerit viikon 12 aikapisteessä täyttäneiden tutkittavien suhteen katsottiin, ettei heillä ollut vastetta viikon 48 aikapisteessä hoitohaarasta riippumatta. | ||||||
| Taulukko 19. Guselkumabihoidolla tärkeimmät toissijaiset tehon päätetapahtumat GALAXI 2- ja GALAXI 3 ‑tutkimusten viikon 48 aikapisteessä saavuttaneiden potilaiden osuus verrattuna ustekinumabiin | ||||||
| GALAXI 2 | GALAXI 3 | |||||
| 6 mg/kg ustekinumabia laskimoon induktio-hoitona → 90 mg injektiona ihon alle 8 viikon väleina | Guselkumabi laskimoon induktio-hoitona → 100 mg injektiona ihon alle 8 viikon väleinb | Guselkumabi laskimoon induktio-hoitona → 200 mg injektiona ihon alle 4 viikon väleinc | 6 mg/kg ustekinumabia laskimoon induktio-hoitona → 90 mg injektiona ihon alle 8 viikon väleina | Guselkumabi laskimoon induktio-hoitona→ 100 mg injektiona ihon alle 8 viikon väleinb | Guselkumabi laskimoon induktio-hoitona → 200 mg injektiona ihon alle 4 viikon väleinc | |
| Kliininen remissio viikon 48 aikapisteessä ja endoskooppinen vasted viikon 48 aikapisteessä | ||||||
| Kokonais-potilas-joukko | 39 % (N = 143) | 42 % (N = 143) | 49 % (N = 146) | 28 % (N = 148) | 41 %k (N = 143) | 45 %k (N = 150) |
| Endoskooppinen vastee viikon 48 aikapisteessäl | ||||||
| Kokonais-potilas-joukko | 42 % (N = 143) | 49 % (N = 143) | 56 % (N = 146) | 32 % (N = 148) | 47 % (N = 143) | 49 % (N = 150) |
| Endoskooppinen remissiof viikon 48 aikapisteessä | ||||||
| Kokonais-potilas-joukko | 20 % (N = 143) | 27 % (N = 143) | 24 % (N = 146) | 13 % (N = 148) | 24 %k (N = 143) | 19 % (N = 150) |
| Kliininen remissiog viikon 48 aikapisteessä | ||||||
| Kokonais-potilas-joukko | 65 % (N = 143) | 64 % (N = 143) | 75 % (N = 146) | 61 % (N = 148) | 66 % (N = 143) | 66 % (N = 150) |
| Kliininen remissio ilman kortikosteroidejah viikon 48 aikapisteessäl | ||||||
| Kokonais-potilas-joukko | 61 % (N = 143) | 63 % (N = 143) | 71 % (N = 146) | 59 % (N = 148) | 64 % (N = 143) | 64 % (N = 150) |
| Pysyvä kliininen remissioi viikon 48 aikapisteessä | ||||||
| Kokonais-potilas-joukko | 45 % (N = 143) | 46 % (N = 143) | 52 % (N = 146) | 39 % (N = 148) | 50 % (N = 143) | 49 % (N = 150) |
| PRO-2-remissioj viikon 48 aikapisteessä | ||||||
| Kokonais-potilas-joukko | 59 % (N = 143) | 60 % (N = 143) | 69 % (N = 146) | 53 % (N = 148) | 58 % (N = 143) | 56 % (N = 150) |
a 6 mg/kg ustekinumabia laskimoon induktiohoitona viikolla 0, jonka jälkeen 90 mg ustekinumabia ihon alle 8 viikon välein enintään 48 viikon ajan. b 200 mg guselkumabia laskimoon induktiohoitona viikolla 0, viikolla 4 ja viikolla 8, jonka jälkeen 100 mg guselkumabia ihon alle 8 viikon välein enintään 48 viikon ajan. c 200 mg guselkumabia laskimoon induktiohoitona viikolla 0, viikolla 4 ja viikolla 8, jonka jälkeen 200 mg guselkumabia ihon alle 4 viikon välein enintään 48 viikon ajan. d Kliinisen remission ja endoskooppisen vasteen yhdistelmä, kuten määritelty jäljempänä. e Endoskooppiseksi vasteeksi on määritelty SES-CD-pisteiden ≥ 50 %:n paraneminen lähtötilanteesta tai SES-CD-pisteet ≤ 2. f Endoskooppiseksi remissioksi on määritelty SES-CD-pisteet ≤ 2. g Kliiniseksi remissioksi on määritelty CDAI-pisteet < 150. h Kliiniseksi remissioksi ilman kortikosteroideja on määritelty CDAI-pisteet < 150 viikon 48 aikapisteessä eikä kortikosteroidihoitoa viikon 48 aikapisteessä. i Pysyväksi kliiniseksi remissioksi on määritelty CDAI-pisteet < 150, jotka todetaan ≥ 80 %:lla kaikista käynneistä viikon 12 ja viikon 48 välillä (vähintään kahdeksalla käynnillä kymmenestä), jonka täytyy sisältää viikko 48. j PRO-2-remissioksi on määritelty vatsakipua koskevat keskimääräiset päivittäiset pisteet enintään 1 ja ulostustiheyttä koskevat keskimääräiset päivittäiset pisteet enintään 3 eikä vatsakivun pahenemista tai ulostustiheyden lisääntymistä lähtötilanteesta. k p < 0,05 l Vasteet arvioitiin viikon 48 aikapisteessä riippumatta kliinisestä vasteesta viikon 12 aikapisteessä. | ||||||
| Taulukko 20.Guselkumabihoidolla tehon päätetapahtumat yhdistettyjen GALAXI 2- ja GALAXI 3 ‑tutkimusten viikon 48 aikapisteessä saavuttaneiden potilaiden osuus verrattuna ustekinumabiin | |||
| 6 mg/kg ustekinumabia laskimoon induktiohoitona → 90 mg injektiona ihon alle 8 viikon väleina | Guselkumabi laskimoon induktiohoitona → 100 mg injektiona ihon alle 8 viikon väleinb | Guselkumabi laskimoon induktiohoitona → 200 mg injektiona ihon alle 4 viikon väleinc | |
| Kliininen remissio viikon 48 aikapisteessä ja endoskooppinen vasted viikon 48 aikapisteessä | |||
| Kokonais-potilasjoukko | 34 % (N = 291) | 42 % (N = 286) | 47 % (N = 296) |
| Ei aiempaa biologista hoitoae | 43 % (N = 121) | 51 % (N = 116) | 55 % (N = 128) |
| Aiempi biologinen hoito epäonnistunutf | 26 % (N = 156) | 37 % (N = 153) | 41 % (N = 147) |
| Endoskooppinen vasteg viikon 48 aikapisteessä | |||
| Kokonais-potilasjoukko | 37 % (N = 291) | 48 % (N = 286) | 53 % (N = 296) |
| Ei aiempaa biologista hoitoae | 43 % (N = 121) | 59 % (N = 116) | 59 % (N = 128) |
| Aiempi biologinen hoito epäonnistunutf | 31 % (N = 156) | 43 % (N = 153) | 47 % (N = 147) |
| Endoskooppinen remissioh viikon 48 aikapisteessä | |||
| Kokonais-potilasjoukko | 16 % (N = 291) | 25 % (N = 286) | 21 % (N = 296) |
| Ei aiempaa biologista hoitoae | 19 % (N = 121) | 34 % (N = 116) | 27 % (N = 128) |
| Aiempi biologinen hoito epäonnistunutf | 13 % (N = 156) | 21 % (N = 153) | 14 % (N = 147) |
| Kliininen remissioi viikon 48 aikapisteessä | |||
| Kokonais-potilasjoukko | 63 % (N = 291) | 65 % (N = 286) | 70 % (N = 296) |
| Ei aiempaa biologista hoitoae | 75 % (N = 121) | 73 % (N = 116) | 77 % (N = 128) |
| Aiempi biologinen hoito epäonnistunutf | 53 % (N = 156) | 61 % (N = 153) | 64 % (N = 147) |
a 6 mg/kg ustekinumabia laskimoon induktiohoitona viikolla 0, jonka jälkeen 90 mg ustekinumabia ihon alle 8 viikon välein enintään 48 viikon ajan. b 200 mg guselkumabia laskimoon induktiohoitona viikolla 0, viikolla 4 ja viikolla 8, jonka jälkeen 100 mg guselkumabia ihon alle 8 viikon välein enintään 48 viikon ajan. c 200 mg guselkumabia laskimoon induktiohoitona viikolla 0, viikolla 4 ja viikolla 8, jonka jälkeen 200 mg guselkumabia ihon alle 4 viikon välein enintään 48 viikon ajan. d Kliinisen remission ja endoskooppisen vasteen yhdistelmä, kuten määritelty jäljempänä. e Lisäksi 14 potilasta ustekinumabiryhmässä, 21 potilasta 200 mg guselkumabia ihon alle 4 viikon välein saaneessa ryhmässä ja 17 potilasta 100 mg guselkumabia ihon alle 8 viikon välein saaneessa ryhmässä oli aiemmin altistunut biologiselle hoidolle eikä hoito ollut epäonnistunut. f Sisältää riittämättömän vasteen Crohnin tautiin annettuun biologiseen hoitoon (TNF-estäjät, vedolitsumabi) sekä vasteen häviämisen näille hoidoille tai kyvyttömyyden sietää näitä hoitoja. g Endoskooppiseksi vasteeksi on määritelty SES-CD-pisteiden ≥ 50 %:n paraneminen lähtötilanteesta tai SES-CD-pisteet ≤ 2. h Endoskooppiseksi remissioksi on määritelty SES-CD-pisteet ≤ 2. i Kliiniseksi remissioksi on määritelty CDAI-pisteet < 150. | |||
GALAXI 2- ja GALAXI 3 ‑tutkimuksissa guselkumabin teho ja turvallisuus osoitettiin yhdenmukaisesti iästä, sukupuolesta, etnisestä taustasta ja painosta riippumatta.
Yhdistettyjen vaiheen III GALAXI-tutkimusten potilaiden osajoukkoanalyysissa potilaat, joilla oli suuri tulehdustaakka induktiohoidon päättymisen jälkeen, saivat lisähyötyä 200 mg:n guselkumabiannoksista ihon alle 4 viikon välein verrattuna 100 mg:n annoksina ihon alle 8 viikon välein annettuun ylläpitohoitoon. Potilailla, joiden CRP-arvo oli induktiohoidon päättymisen jälkeen > 5 mg/l, havaittiin kahden guselkumabiannosryhmän välillä kliinisesti merkittävä ero kliinistä remissiota viikon 48 aikapisteessä koskevissa päätetapahtumissa (100 mg ihon alle 8 viikon välein 54,1 % vs. 200 mg ihon alle 4 viikon välein 71,0 %), endoskooppisessa vasteessa viikon 48 aikapisteessä (100 mg ihon alle 8 viikon välein 36,5 % vs. 200 mg ihon alle 4 viikon välein 50,5 %) ja PRO-2-remissiossa viikon 48 aikapisteessä (100 mg ihon alle 8 viikon välein 51,8 % vs. 200 mg ihon alle 4 viikon välein 61,7 %).
Kliininen remissio ajan kuluessa
CDAI-pisteet kirjattiin potilaiden jokaisella käynnillä. Kliinisessä remissiossa olevien potilaiden osuus viikkoon 48 mennessä esitetään kuvassa 10.
Kuva 10. Kliinisessä remissiossa olevien potilaiden osuus yhdistettyjen GALAXI 2- ja GALAXI 3 ‑tutkimusten viikkoon 48 mennessä
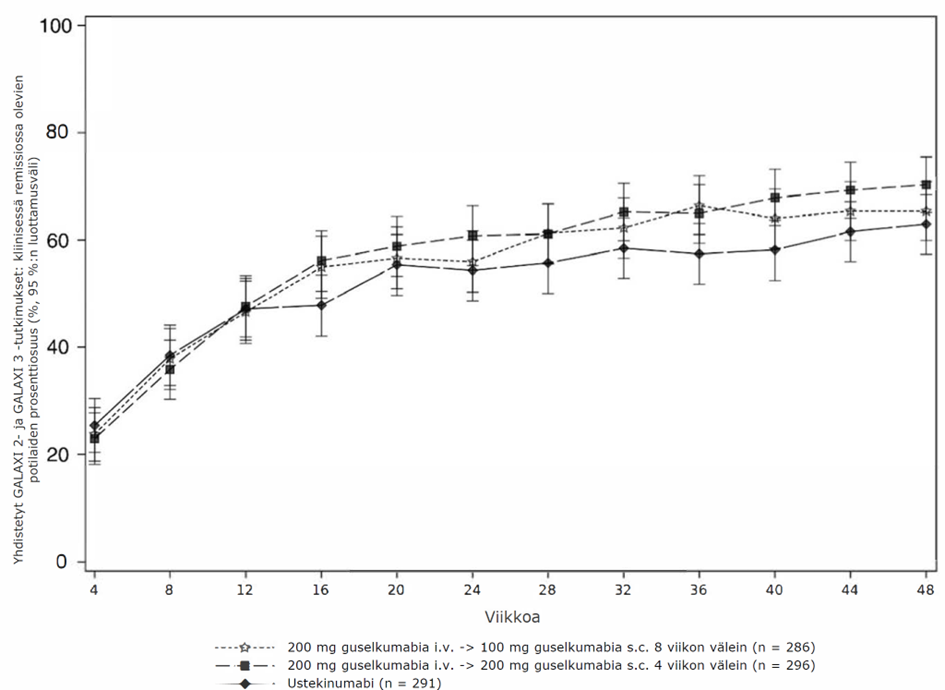
Terveyteen liittyvä elämänlaatu
Guselkumabihoitoryhmissä havaittiin viikon 12 aikapisteessä elämänlaadun paranemista lähtötilanteesta enemmän kuin lumehoidon yhteydessä, mikä todettiin tulehduksellista suolistosairautta (IBD) koskevan spesifisen elämänlaatuarvioinnin IBDQ-kokonaispisteillä arvioituna. Tämä paraneminen säilyi kummassakin tutkimuksessa viikkoon 48 saakka.
GRAVITI
Vaiheen III GRAVITI-tutkimuksessa keskivaikean tai vaikean aktiivisen Crohnin taudin määritelmä oli CDAI-pisteet ≥ 220 – ≤ 450 ja Crohnin tautia koskevat (SES-CD) pisteet ≥ 6 (tai ≥ 4, jos potilaalla oli ileumiin rajoittunut tauti) sekä keskimääräinen päivittäinen ulostamistiheys ≥ 4 tai keskimääräiset päivittäiset vatsakipua koskevat pisteet ≥ 2.
GRAVITI-tutkimuksessa potilaat satunnaistettiin suhteessa 1:1:1 saamaan 400 mg guselkumabia ihon alle induktiohoitona viikoilla 0, 4 ja 8 ja sen jälkeen 100 mg guselkumabia 8 viikon välein ihon alle ylläpitohoitona tai 400 mg guselkumabia ihon alle induktiohoitona viikoilla 0, 4 ja 8 ja sen jälkeen 200 mg guselkumabia 4 viikon välein ihon alle ylläpitohoitona tai lumelääkettä. Kaikki lumeryhmän potilaat, jotka täyttivät varalääkityksen kriteerit, saivat induktioannostuksena 400 mg guselkumabia ihon alle viikoilla 16, 20 ja 24 ja sen jälkeen 100 mg guselkumabia ihon alle 8 viikon välein.
Yhteensä 347 potilasta arvioitiin. Potilaiden iän mediaani oli 36 vuotta (vaihteluväli 18–83 vuotta); 58,5 % oli miehiä, ja 66 % identifioitui valkoihoisiksi, 21,9 % aasialaisiksi ja 2,6 % mustaihoisiksi.
GRAVITI-tutkimuksessa 46,4 %:lla potilaista vähintään yksi aiempi biologinen hoito oli epäonnistunut, 46,4 % ei ollut aiemmin saanut biologista hoitoa ja 7,2 % oli aiemmin saanut biologista hoitoa eikä hoito ollut epäonnistunut. Lähtötilanteessa 29,7 % potilaista sai suun kautta otettavia kortikosteroideja ja 28,5 % potilaista sai tavanomaisia immunomodulaattoreita.
Rinnakkaisten ensisijaisten ja tärkeimpien toissijaisten tehon päätetapahtumien viikon 12 aikapisteen tulosten vertailu lumelääkkeeseen esitetään taulukossa 21.
| Taulukko 21. Guselkumabihoidolla rinnakkaiset ensisijaiset ja tärkeimmät toissijaiset tehon päätetapahtumat GRAVITI‑tutkimuksen viikon 12 aikapisteessä saavuttaneiden potilaiden osuus verrattuna lumelääkkeeseen | |||
Lumelääke
| 400 mg guselkumabia injektiona ihon allea
| ||
| Rinnakkaiset ensisijaiset tehon päätetapahtumat | |||
| Kliininen remissiob viikon 12 aikapisteessä | |||
| Kokonaispotilasjoukko | 21 % (N = 117) | 56 %c (N = 230) | |
| Ei aiempaa biologista hoitoad | 25 % (N = 56) | 50 % (N = 105) | |
| Aiempi biologinen hoito epäonnistunute | 17 % (N = 53) | 60 % (N = 108) | |
| Endoskooppinen vastef viikon 12 aikapisteessä | |||
| Kokonaispotilasjoukko | 21 % (N = 117) | 41 %c (N = 230) | |
| Ei aiempaa biologista hoitoad | 27 % (N = 56) | 49 % (N = 105) | |
| Aiempi biologinen hoito epäonnistunute | 17 % (N = 53) | 33 % (N = 108) | |
| Tärkeimmät toissijaiset tehon päätetapahtumat | |||
| Kliininen vasteg viikon 12 aikapisteessä | |||
| Kokonaispotilasjoukko | 33 % (N = 117) | 73 %c (N = 230) | |
| Ei aiempaa biologista hoitoad | 38 % (N = 56) | 68 % (N = 105) | |
| Aiempi biologinen hoito epäonnistunute | 28 % (N = 53) | 78 % (N = 108) | |
| PRO-2-remissioh viikon 12 aikapisteessä | |||
| Kokonaispotilasjoukko | 17 % (N = 117) | 49 %c (N = 230) | |
| Ei aiempaa biologista hoitoad | 18 % (N = 56) | 44 % (N = 105) | |
| Aiempi biologinen hoito epäonnistunute | 17 % (N = 53) | 52 % (N = 108) | |
a 400 mg guselkumabia ihon alle viikolla 0, viikolla 4 ja viikolla 8 b Kliininen remissio: CDAI-pisteet < 150 c p < 0,001 d Lisäksi 8 potilasta lumeryhmässä ja 17 potilasta 400 mg guselkumabia ihon alle saaneessa ryhmässä oli aiemmin altistunut biologiselle hoidolle eikä hoito ollut epäonnistunut. e Sisältää riittämättömän vasteen Crohnin tautiin annettuun biologiseen hoitoon (TNF-estäjät, vedolitsumabi) sekä vasteen häviämisen näille hoidoille tai kyvyttömyyden sietää näitä hoitoja. f Endoskooppinen vaste: SES-CD-pisteiden paraneminen ≥ 50 % lähtötilanteesta. g Kliininen vaste: CDAI-pisteiden pieneneminen ≥ 100 pistettä lähtötilanteesta tai CDAI-pisteet < 150. h PRO-2-remissio: vatsakipua koskevat keskimääräiset päivittäiset pisteet enintään 1 ja ulostustiheyttä koskevat keskimääräiset päivittäiset pisteet enintään 3 eikä vatsakivun pahenemista tai ulostustiheyden lisääntymistä lähtötilanteesta. | |||
Merkittävästi suuremmalla osuudella potilaista, jotka saivat 400 mg guselkumabia ihon alle induktiohoitona ja sen jälkeen 100 mg guselkumabia ihon alle 8 viikon välein tai 200 mg ihon alle 4 viikon välein, todettiin kliininen remissio viikon 24 aikapisteessä verrattuna lumelääkettä saaneisiin (400 mg guselkumabia ihon alle induktiohoitona ja sen jälkeen 100 mg guselkumabia ihon alle 8 viikon välein saaneista 60,9 % ja 400 mg guselkumabia ihon alle induktiohoitona ja sen jälkeen 200 mg ihon alle 4 viikon välein saaneista 58,3 % vs. lumehoitoa saaneista 21,4 %, kummankin vertailun p-arvo < 0,001). Kliininen remissio todettiin viikon 48 aikapisteessä 60 %:lla potilaista, jotka saivat 400 mg guselkumabia ihon alle induktiohoitona ja sen jälkeen 100 mg guselkumabia ihon alle 8 viikon välein, ja 66,1 %:lla potilaista, jotka saivat 400 mg guselkumabia ihon alle induktiohoitona ja sen jälkeen 200 mg ihon alle 4 viikon välein (kummankin p-arvo < 0,001 lumelääkkeeseen verrattuna).
Endoskooppinen vaste todettiin viikon 48 aikapisteessä 44,3 %:lla potilaista, jotka saivat 400 mg guselkumabia ihon alle induktiohoitona ja sen jälkeen 100 mg guselkumabia ihon alle 8 viikon välein, ja 51,3 %:lla potilaista, jotka saivat 400 mg guselkumabia ihon alle induktiohoitona ja sen jälkeen 200 mg ihon alle 4 viikon välein (kummankin p-arvo < 0,001 lumelääkkeeseen verrattuna).
Terveyteen liittyvä elämänlaatu
GRAVITI-tutkimuksessa IBD-spesifisessä elämänlaadussa havaittiin kliinisesti merkittävää paranemista lumelääkkeeseen verrattuna arvioitaessa sitä IBDQ-kokonaispisteillä viikon 12 ja viikon 24 aikapisteissä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset guselkumabin käytöstä nivelpsoriaasin, haavaisen paksusuolitulehduksen ja Crohnin taudin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä. 100 mg:n esitäytettyjä kyniä (PushPen ja OnePress) ei ole tutkittu pediatrisilla potilailla eikä niitä suositella pediatristen potilaiden käyttöön (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Lapsuusiän läiskäpsoriaasi
Guselkumabin turvallisuutta ja tehoa arvioitiin yhdessä satunnaistetussa, lumelääkkeellä ja vaikuttavalla biologisella vertailuvalmisteella kontrolloidussa monikeskustutkimuksessa (PROTOSTAR) 120 pediatrisella potilaalla, jotka olivat iältään 6–17-vuotiaita ja joilla oli keskivaikea tai vaikea läiskäpsoriaasi, joille harkittiin valohoitoa tai systeemistä hoitoa ja joiden sairaus ei ollut valohoidon ja/tai paikallisten hoitojen avulla riittävässä hoitotasapainossa. PROTOSTAR-tutkimus tehtiin kahdessa osassa. Osa 1 käsitti 16 viikon satunnaistetun lumelääkkeellä ja vaikuttavalla vertailuvalmisteella kontrolloidun jakson, jonka jälkeen oli kontrolloimaton jakso, jolloin hoito lopetettiin ja sitä jatkettiin tai aloitettiin viikkoon 52 jatkunut guselkumabihoito. Osa 2 käsitti avoimen guselkumabiryhmän viikkoon 52 saakka.
Mukaan otettujen potilaiden IGA-pisteet olivat ≥ 3 sairauden kokonaisvaikeusastetta kuvaavalla viisiportaisella asteikolla, PASI-vaste oli ≥ 12 ja ihottumaa oli vähimmillään ≥ 10 %:ssa kehon pinta-alasta, ja heillä oli vähintään yksi seuraavista: 1) hyvin paksuja ihomuutoksia, 2) kliinisesti oleellisia psoriaasin oireita kasvoissa, sukupuolielimissä tai käsissä/jalkaterissä, 3) PASI-vaste ≥ 20, 4) ihottumaa > 20 %:ssa kehon pinta-alasta tai 5) IGA-pisteet = 4. Tutkimukseen ei otettu mukaan tutkittavia, joilla oli pisarapsoriaasi, erytroderminen psoriaasi tai märkärakkulainen psoriaasi.
Osassa 1 satunnaistettiin 92 iältään 6–17-vuotiasta potilasta saamaan injektioina ihon alle joko guselkumabia (n = 41) tai lumelääkettä (n = 25) viikoilla 0, 4 ja 12 tai vaikuttavaa biologista vertailuvalmistetta (n = 26) viikoittain. Osassa 2 mukaan otettiin vielä 28 iältään 12–17-vuotiasta nuorta potilasta, jotka saivat guselkumabia injektioina ihon alle viikoilla 0 ja 4 ja sen jälkeen 8 viikon välein. Guselkumabiryhmässä alle 70 kg:n painoiset potilaat saivat annoksen 1,3 mg/kg, joka annettiin 45 mg/0,45 ml esitäytetyllä kynällä, ja vähintään 70 kg:n painoiset potilaat saivat annoksen 100 mg, joka annettiin esitäytetyllä ruiskulla.
Yhdistettyjä ensisijaisia päätetapahtumia olivat niiden potilaiden osuus, jotka saavuttivat PASI 75 ‑vasteen, ja niiden potilaiden osuus, jotka saavuttivat IGA-pisteet 0 (”parantunut”) tai 1 (”minimaalinen”) viikolla 16. Toissijaisia päätetapahtumia olivat niiden potilaiden osuus, jotka saavuttivat PASI 90 ‑vasteen, IGA-pisteet 0 (”parantunut”) tai PASI 100 -vasteen viikolla 16, näihin kuitenkaan rajoittumatta.
Tutkimuksen kontrolloidun osan 92 potilaan lähtötilanteen demografiset ominaisuudet olivat yleisesti verrannolliset tutkimusryhmien välillä. Kaikkiaan yli 55 % oli miespuolisia, 85 % oli valkoihoisia, keskimääräinen paino oli noin 57,3 kg, keskimääräinen ikä oli 12,9 vuotta ja 33 % potilaista oli alle 12-vuotiaita.
Sairauden lähtötilanteen ominaisuudet olivat yleisesti verrannolliset hoitoryhmien välillä, ja ihottumaa oli lähtötilanteessa 20 %:ssa (mediaani) kehon pinta-alasta, lähtötilanteen PASI-pisteet olivat noin 17 (mediaani), lähtötilanteessa vaikea-asteista sairautta osoittavat IGA-pisteet todettiin 20 %:lla (lumelääke) ja 24 %:lla (guselkumabi) potilaista ja 3,3 %:lla potilaista oli anamneesissa nivelpsoriaasi.
Ihosairauden kokonaistilanne
Guselkumabihoito paransi taudin aktiivisuutta koskevaa hoitotulosta osoittavia mittareita merkittävästi lumehoitoon verrattuna viikolla 16. Tutkimuksen päätetapahtumien keskeiset tehoa koskevat tulokset esitetään jäljempänä taulukossa 22.
| Taulukko 22. Yhteenveto päätetapahtumista PROTOSTAR-tutkimuksen viikolla 16 | |||
Lumelääke (N = 25) | Guselkumabi (N = 41) | p‑arvo | |
| IGA-pisteet ”parantunut” (0) tai ”minimaalinen” (1), n (%) | 4 (16,0 %) | 27 (65,9 %) | < 0,001 |
| IGA-pisteet ”parantunut” (0), n (%) | 1 (4,0 %) | 16 (39,0 %) | 0,004 |
| PASI 75 ‑vasteen saaneet, n (%) | 5 (20,0 %) | 31 (75,6 %) | < 0,001 |
| PASI 90 ‑vasteen saaneet, n (%) | 4 (16,0 %) | 23 (56,1 %) | 0,003 |
| PASI 100 ‑vasteen saaneet, n (%) | 0 | 14 (34,1 %) | 0,002 |
| CDLQI-pisteiden muutos lähtötilanteesta, pienimmän neliösumman keskiarvo (95 %:n luottamusväli) | ‑1,88 (‑3,81; 0,05) | ‑7,28 (‑8,87; ‑5,68) | < 0,001 |
| CDLQI = ihotauteihin liittyvä elämänlaatumittarina käytettävä kysymyssarja (Children’s Dermatology Life Quality Index) | |||
PROTOSTAR-tutkimuksen osaan 1 kuuluneen 16 viikon pituisen lumekontrolloidun jakson jälkeen niiden potilaiden, jotka olivat saaneet guselkumabihoitoa ja saavuttaneet PASI 90 ‑vasteen viikolla 16, hoito lopetettiin. PASI 90 ‑vasteen todettiin hävinneen jo 12 viikon kuluttua guselkumabihoidon lopettamisesta, ja PASI 90 ‑vasteen häviämiseen kuluneen ajan mediaani oli noin 24 viikkoa. Niistä guselkumabihoitoa saaneista potilaista, jotka eivät olleet viikolla 16 saavuttaneet PASI 90 ‑vastetta, guselkumabihoitoa vielä 32 lisäviikon ajan jatkaneista potilaista 72,2 % oli saavuttanut PASI 75 ‑vasteen viikolla 52 ja 61,1 % oli saavuttanut PASI 90 ‑vasteen viikolla 52.
Lumehoitoon viikolla 0 satunnaistetut potilaat, jotka eivät olleet viikolla 16 saavuttaneet PASI 90 ‑vastetta, siirtyivät guselkumabihoitoon. Heistä 95,0 % oli saavuttanut PASI 75 ‑vasteen ja 65,0 % oli saavuttanut PASI 90 ‑vasteen viikolla 52.
Farmakokinetiikka
Imeytyminen
Terveille tutkittaville ihon alle annetun 100 mg:n kertainjektion jälkeen guselkumabin maksimipitoisuus (Cmax) seerumissa oli keskimäärin (± keskihajonta) 8,09 ± 3,68 mikrog/ml noin 5,5 vuorokauteen mennessä annoksen jälkeen. Guselkumabin absoluuttisen biologisen hyötyosuuden arvioitiin olevan terveille tutkittaville ihon alle annetun 100 mg:n kertainjektion jälkeen noin 49 %.
Läiskäpsoriaasia sairastavilla potilailla guselkumabin vakaan tilan pitoisuus seerumissa saavutettiin viikoilla 0 ja 4 ja sen jälkeen 8 viikon välein ihon alle annettujen 100 mg:n guselkumabiannosten jälkeen viikkoon 20 mennessä. Vakaan tilan pienimmät guselkumabipitoisuudet seerumissa olivat läiskäpsoriaasia sairastavilla potilailla tehdyissä kahdessa vaiheen III tutkimuksessa keskimäärin (± keskihajonta) 1,15 ± 0,73 mikrog/ml ja 1,23 ± 0,84 mikrog/ml.
Guselkumabin farmakokinetiikka oli nivelpsoriaasia sairastavilla potilailla samankaltainen kuin psoriaasia sairastavilla potilailla. Viikoilla 0 ja 4 sekä sen jälkeen 8 viikon välein ihon alle annettujen 100 mg:n guselkumabiannosten jälkeen vakaan tilan pienimmät guselkumabipitoisuudet seerumissa olivat myös keskimäärin noin 1,2 mikrog/ml. Ihon alle 4 viikon välein annettujen 100 mg:n guselkumabiannosten jälkeen vakaan tilan pienimmät guselkumabipitoisuudet seerumissa olivat keskimäärin noin 3,8 mikrog/ml.
Guselkumabin farmakokinetiikka oli haavaista paksusuolitulehdusta ja Crohnin tautia sairastavilla potilailla samankaltainen. Haavaista paksusuolitulehdusta sairastavilla potilailla guselkumabin keskimääräinen huippupitoisuus seerumissa oli 8 viikon aikapisteessä suositellun laskimoon annettavan induktiohoito-ohjelman jälkeen, eli viikoilla 0, 4 ja 8 annettujen 200 mg:n guselkumabiannosten jälkeen, 68,27 mikrog/ml, ja Crohnin tautia sairastavilla potilailla se oli 70,5 mikrog/ml.
Guselkumabin keskimääräiseksi huippupitoisuudeksi seerumissa viikon 8 aikapisteessä arvioitiin haavaista paksusuolitulehdusta sairastavilla potilailla 28,8 mikrog/ml ja Crohnin tautia sairastavilla potilailla 27,7 mikrog/ml, kun potilaat olivat saaneet suositellun induktiohoito-ohjelman 400 mg guselkumabia ihon alle viikoilla 0, 4 ja 8. Systeeminen kokonaisaltistus (AUC) oli suositellun induktiohoidon jälkeen samankaltainen annettaessa induktiohoito ihon alle tai laskimoon.
Haavaista paksusuolitulehdusta sairastavilla potilailla guselkumabin keskimääräinen vakaan tilan pienin (trough) pitoisuus seerumissa oli ihon alle annettavan ylläpitohoidon jälkeen seuraava: 8 viikon välein annettujen 100 mg:n guselkumabiannosten jälkeen 1,4 mikrog/ml ja 4 viikon välein annettujen 200 mg:n guselkumabiannosten jälkeen 10,7 mikrog/ml.
Crohnin tautia sairastavilla potilailla guselkumabin keskimääräinen vakaan tilan pienin (trough) pitoisuus seerumissa oli ihon alle annettavan ylläpitohoidon jälkeen seuraava: 8 viikon välein annettujen 100 mg:n guselkumabiannosten jälkeen 1,2 mikrog/ml ja 4 viikon välein annettujen 200 mg:n guselkumabiannosten jälkeen 10,1 mikrog/ml.
Jakautuminen
Terminaalisen vaiheen (Vz) jakautumistilavuuden keskiarvo oli terveille tutkittaville laskimoon annetun kerta-annoksen jälkeen kaikissa tutkimuksissa noin 7–10 l.
Biotransformaatio
Guselkumabin tarkkaa metaboliareittiä ei ole selvitetty. Guselkumabi on ihmisen IgG monoklonaalinen vasta-aine, joten se hajoaa oletettavasti pieniksi peptideiksi ja aminohapoiksi kataboliareittien kautta samalla tavoin kuin endogeeninen IgG.
Eliminaatio
Systeemisen puhdistuman (CL) keskiarvo oli terveille tutkittaville laskimoon annetun kerta-annoksen jälkeen kaikissa tutkimuksissa 0,288–0,479 l/vrk. Guselkumabin puoliintumisajan (T½) keskiarvo oli terveillä tutkittavilla noin 17 vuorokautta, läiskäpsoriaasia sairastavilla potilailla noin 15–18 vuorokautta kaikissa tutkimuksissa ja haavaista paksusuolitulehdusta tai Crohnin tautia sairastavilla potilailla noin 17 vuorokautta.
Populaatiofarmakokineettiset analyysit osoittivat, että tulehduskipulääkkeiden, atsatiopriinin, merkaptopuriinin, suun kautta otettavien kortikosteroidien ja csDMARD-lääkkeiden, kuten metotreksaatin, samanaikainen käyttö ei vaikuttanut guselkumabin puhdistumaan.
Lineaarisuus/ei-lineaarisuus
Terveille tutkittaville tai läiskäpsoriaasia sairastaville potilaille ihon alle annetun 10−300 mg:n kertainjektion jälkeen guselkumabin systeeminen altistus (Cmax ja AUC) suureni suunnilleen suhteessa annokseen. Haavaista paksusuolitulehdusta tai Crohnin tautia sairastavilla potilailla guselkumabipitoisuus seerumissa oli suunnilleen suhteessa annokseen, kun valmiste annettiin laskimoon.
Pediatriset potilaat
Iältään 6–17-vuotiailla pediatrisilla potilailla, joilla oli keskivaikea tai vaikea läiskäpsoriaasi ja jotka saivat ihon alle guselkumabi-injektioita 45 mg/0,45 ml esitäytetyillä kynillä tai 100 mg:n esitäytetyillä ruiskuilla (ks. kohta Annostus ja antotapa), vakaan tilan pienimmät guselkumabipitoisuudet saavutettiin seerumissa viikkoon 20 mennessä, ja pitoisuudet olivat samansuuruisia kuin aikuisilla oli havaittu.
Suositellusta hoito-ohjelmasta saadaan läiskäpsoriaasia sairastavien pediatristen potilaiden seerumiin samankaltainen ennustettu guselkumabialtistus kuin eripainoisilla aikuisilla.
Iäkkäät potilaat
Iäkkäillä potilailla ei ole tehty erityisiä tutkimuksia. Guselkumabille vaiheen III kliinisissä tutkimuksissa altistuneista ja populaatiofarmakokineettiseen analyysiin mukaan otetuista 1 384 läiskäpsoriaasia sairastavasta potilaasta 70 potilasta oli vähintään 65-vuotiaita, ja heistä 4 potilasta oli vähintään 75-vuotiaita. Guselkumabille vaiheen III kliinisissä tutkimuksissa altistuneista 746 nivelpsoriaasia sairastavasta potilaasta yhteensä 38 potilasta oli vähintään 65-vuotiaita ja yksikään potilas ei ollut 75‑vuotias tai vanhempi. Guselkumabille vaiheen II/III kliinisissä tutkimuksissa altistuneista ja populaatiofarmakokineettiseen analyysiin mukaan otetuista 859:stä haavaista paksusuolitulehdusta sairastavasta potilaasta yhteensä 52 potilasta oli vähintään 65-vuotiaita ja 9 potilasta oli vähintään 75-vuotiaita. Guselkumabille vaiheen III kliinisissä tutkimuksissa altistuneista ja populaatiofarmakokineettiseen analyysiin mukaan otetuista 1 009 Crohnin tautia sairastavasta potilaasta yhteensä 39 potilasta oli vähintään 65-vuotiaita ja 5 potilasta oli vähintään 75‑vuotiaita.
Läiskäpsoriaasia, nivelpsoriaasia, haavaista paksusuolitulehdusta ja Crohnin tautia sairastavista potilaista tehdyt populaatiofarmakokineettiset analyysit eivät osoittaneet CL/F-estimaatissa ilmeisiä muutoksia ≥ 65-vuotiailla verrattuna < 65-vuotiaisiin, mikä viittaa siihen, että iäkkäiden potilaiden annosta ei tarvitse muuttaa.
Munuaisten tai maksan vajaatoimintaa sairastavat potilaat
Munuaisten tai maksan vajaatoiminnan vaikutuksesta guselkumabin farmakokinetiikkaan ei ole tehty spesifisiä tutkimuksia. Muuttumaton guselkumabi on monoklonaalinen IgG vasta-aine, ja sen eliminaatio munuaisten kautta on oletettavasti vähäistä ja merkitys on vähäinen. Vastaavasti maksan vajaatoiminta ei oletettavasti vaikuta guselkumabin puhdistumaan, koska monoklonaaliset IgG vasta-aineet eliminoituvat pääasiassa kataboloitumalla solunsisäisesti. Populaatiofarmakokineettisten analyysien perusteella kreatiniinipuhdistumalla tai maksan toiminnalla ei ollut merkittävää vaikutusta guselkumabin puhdistumaan.
Paino
Guselkumabin puhdistuma lisääntyy ja jakautumistilavuus suurenee painon lisääntyessä, mutta kliinisiin tutkimustietoihin perustuvat havainnot osoittavat, ettei annosta tarvitse muuttaa painon perusteella.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, lisääntymistoksisuutta ja pre- ja postnataalista kehitystä koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Cynomolgus-apinoilla tehdyissä toistuvan altistuksen aiheuttamaa toksisuutta koskeneissa tutkimuksissa laskimoon ja ihon alle annettu guselkumabi oli hyvin siedetty. Viikoittain apinoille ihon alle annetuista 50 mg/kg annoksista aiheutuva altistus (AUC) oli vähintään 23-kertainen verrattuna suurimpaan kliiniseen altistukseen laskimoon annetun 200 mg:n annoksen jälkeen. Cynomolgus-apinoilla tehdyissä toistuvan altistuksen aiheuttamaa toksisuutta tai kohdennettua kardiovaskulaarista turvallisuutta koskeneissa farmakologisissa tutkimuksissa ei havaittu haitallista immunotoksisuutta tai kardiovaskulaarista turvallisuutta koskevia farmakologisia vaikutuksia.
Histopatologisissa tutkimuksissa ei havaittu preneoplastisia muutoksia eläimillä, joita hoidettiin enintään 24 viikon ajan, eikä 12 viikon toipumisjakson jälkeen, jolloin vaikuttavaa ainetta oli seerumissa havaittavissa.
Guselkumabilla ei tehty mutageenisuus- eikä karsinogeenisuustutkimuksia.
Guselkumabia ei havaittu cynomolgus-apinoiden maidossa, kun mittaus tehtiin 28 päivää synnytyksen jälkeen.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Histidiinimonohydrokloridimonohydraatti
Polysorbaatti 80 (E433)
Sakkaroosi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
2 vuotta.
Säilytys
Säilytä jääkaapissa (2 °C – 8 ºC). Ei saa jäätyä.
Pidä esitäytetty kynä ulkopakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TREMFYA ONEPRESS injektioneste, liuos, esitäytetty kynä
100 mg (L:ei) 1 kpl (1 ml (100 mg/ml), automaattinen neulanpistosuoja) (1854,18 €)
TREMFYA PUSHPEN injektioneste, liuos, esitäytetty kynä
100 mg (L:ei) 1 kpl (1 ml (100 mg/ml), automaattinen neulanpistosuoja) (1854,18 €)
PF-selosteen tieto
Tremfya 100 mg OnePress injektioneste, liuos, esitäytetty kynä
1 ml liuosta lasisessa esitäytetyssä ruiskussa esitäytetyn kynän sisällä. Esitäytetyssä ruiskussa on bromobutyylikumitulppa, ja kynässä on automaattinen neulanpistosuoja.
Tremfya-injektionestettä on saatavana yhden esitäytetyn kynän pakkauksina sekä kahden esitäytetyn kynän (kaksi yhden esitäytetyn kynän pakkausta sisältävinä) kerrannaispakkauksina.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Tremfya 100 mg PushPen injektioneste, liuos, esitäytetty kynä
1 ml liuosta lasisessa esitäytetyssä ruiskussa esitäytetyn kynän sisällä. Esitäytetyssä ruiskussa on bromobutyylikumitulppa, ja kynässä on automaattinen neulanpistosuoja.
Tremfya-injektionestettä on saatavana yhden esitäytetyn kynän pakkauksina sekä kahden esitäytetyn kynän (kaksi yhden esitäytetyn kynän pakkausta sisältävinä) kerrannaispakkauksina.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Liuos on kirkasta ja väritöntä tai vaaleankeltaista ja se saattaa sisältää joitakin pieniä valkoisia tai kirkkaita hiukkasia. Sen tavoite-pH on 5,8 ja likimääräinen osmolaarisuus on 367,5 mOsm/l.
Käyttö- ja käsittelyohjeet
Kun OnePress- esitäytetty kynä tai PushPen- esitäytetty kynä on otettu jääkaapista, anna esitäytetyn kynän olla ulkopakkauksessa ja anna sen lämmetä huoneenlämpöiseksi. Odota 30 minuuttia ennen kuin annat Tremfya‑injektion. Esitäytettyjä kyniä ei saa ravistaa.
Esitäytetyt kynät suositellaan tarkistamaan silmämääräisesti ennen käyttöä. Liuoksen pitää olla kirkasta, väritöntä tai vaaleankeltaista, ja se saattaa sisältää joitakin pieniä valkoisia tai kirkkaita hiukkasia. Jos liuos on sameaa tai värjäytynyttä tai siinä on isoja hiukkasia, Tremfya‑liuosta ei saa käyttää.
Jokainen pakkaus sisältää Käyttöohjeet, joissa on tarkat ohjeet injektion valmisteluun ja esitäytettyjen kynien käyttöön.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TREMFYA ONEPRESS injektioneste, liuos, esitäytetty kynä
100 mg 1 kpl
TREMFYA PUSHPEN injektioneste, liuos, esitäytetty kynä
100 mg 1 kpl
- Alempi erityiskorvaus (65 %). Guselkumabi, risankitsumabi, ustekinumabi ja vedolitsumabi (tulehdukselliset suolistosairaudet): Haavaisen paksusuolitulehduksen ja Crohnin taudin hoito erityisin edellytyksin (256), Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313), Adalimumabi, bimekitsumabi, brodalumabi, etanersepti, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sekukinumabi, sertolitsumabipegoli, tildrakitsumabi ja ustekinumabi (ihopsoriaasi): Vaikean kroonisen ihopsoriaasin hoito erityisin edellytyksin (319), Adalimumabi, golimumabi, guselkumabi, infliksimabi, risankitsumabi, ustekinumabi ja vedolitsumabi (tulehdukselliset suolistosairaudet): Eräiden suolistosairauksien hoito erityisin edellytyksin (326).
ATC-koodi
L04AC16
Valmisteyhteenvedon muuttamispäivämäärä
18.12.2025
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com