
BALVERSA tabletti, kalvopäällysteinen 3 mg, 4 mg, 5 mg
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Balversa 3 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 3 mg erdafitinibiä.
Balversa 4 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 4 mg erdafitinibiä.
Balversa 5 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 5 mg erdafitinibiä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
Balversa monoterapiana on tarkoitettu sellaisten aikuispotilaiden hoitoon, joilla on leikkaukseen soveltumaton tai metastasoitunut uroteelikarsinooma, jossa on tietynlaisia fibroblastikasvutekijäreseptorin 3 (FGFR3) geenimuutoksia, ja jotka ovat aiemmin saaneet vähintään yhden PD-1:n tai PD-L1:n estäjää sisältävän hoitolinjan leikkaukseen soveltumattoman tai metastasoituneen sairauden hoitoon (ks. kohta Farmakodynamiikka).
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Balversa-hoidon tulee aloittaa ja sitä tulee valvoa syöpälääkkeiden käyttöön perehtynyt lääkäri.
Lääkärillä on ennen Balversa-hoitoa oltava varmistus tietynlaisesta FGFR3-geenimuutoksesta / tietynlaisista FGFR3-geenimuutoksista (ks. kohta Farmakodynamiikka) kyseistä tarkoitusta vastaavalla CE-merkityllä in vitro ‑diagnostisella (IVD) lääkinnällisellä laitteella arvioituna. Jos CE-merkittyä in vitro ‑diagnostista laitetta ei ole saatavissa, on käytettävä vaihtoehtoista validoitua testiä.
Annostus
Suositeltu Balversa-aloitusannos on 8 mg suun kautta kerran päivässä.
Tämän annoksen käyttöä jatketaan, ja seerumin fosfaattipitoisuus arvioidaan 14–21 päivää hoidon aloittamisen jälkeen. Jos seerumin fosfaattipitoisuus on < 9,0 mg/dl (< 2,91 mmol/l) eikä lääkkeeseen liittyvää toksisuutta todeta, annos titrataan suuremmaksi tasolle 9 mg kerran päivässä. Jos fosfaattipitoisuus on 9,0 mg/dl (2,91 mmol/l) tai suurempi, noudatetaan taulukossa 2 mainittuja soveltuvia annosmuutoksia. Päivän 21 jälkeen seerumin fosfaattipitoisuutta ei pidä käyttää annoksen suuremmaksi titraamisessa ohjaavana tekijänä.
Jos potilas oksentaa milloin tahansa Balversa-valmisteen ottamisen jälkeen, seuraava annos pitää ottaa seuraavana päivänä.
Hoidon kesto
Hoitoa jatketaan, kunnes sairaus etenee tai ilmaantuu toksisuutta, joka ei ole hyväksyttävissä.
Annoksen jääminen ottamatta
Jos Balversa-annos jää ottamatta, se voidaan ottaa mahdollisimman pian. Seuraavana päivänä jatketaan säännöllisen päivittäisen Balversa-hoitoaikataulun noudattamista. Ottamatta jääneen annoksen korvaamiseksi ei pidä ottaa ylimääräisiä tabletteja.
Annoksen pienentäminen ja haittavaikutusten hoito
Ks. annoksen pienentämistä koskevat suositukset taulukoista 1–5.
Taulukko 1. Balversa-annoksen pienentäminen
| Annos | Annoksen 1. pienennys-kerta | Annoksen 2. pienennys-kerta | Annoksen 3. pienennys-kerta | Annoksen 4. pienennys-kerta | Annoksen 5. pienennys-kerta |
9 mg (esim. kolme 3 mg:n tablettia) | 8 mg (esim. kaksi 4 mg:n tablettia) | 6 mg (kaksi 3 mg:n tablettia) | 5 mg (yksi 5 mg:n tabletti) | 4 mg (yksi 4 mg:n tabletti) | Lopeta |
8 mg (esim. kaksi 4 mg:n tablettia) | 6 mg (kaksi 3 mg:n tablettia) | 5 mg (yksi 5 mg:n tabletti) | 4 mg (yksi 4 mg:n tabletti) | Lopeta | |
Hyperfosfatemian hoito
Hyperfosfatemia on FGFR:n estäjien oletettu, ohimenevä farmakodynaaminen vaikutus (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Farmakodynamiikka). Fosfaattipitoisuus pitää määrittää ennen ensimmäistä annosta ja sen jälkeen sitä seurataan kuukausittain. Jos Balversa-hoitoa saavan potilaan fosfaattipitoisuus on koholla, pitää noudattaa taulukon 2 ohjeita annoksen muuttamisesta. Jos fosfaattipitoisuus pysyy koholla pitkään, on harkittava tarpeen mukaan kalsiumia sisältämättömän fosfaatinsitojan (esim. sevelameerikarbonaatin) lisäämistä hoitoon (ks. taulukko 2).
Taulukko 2. Seerumin fosfaattipitoisuuteen perustuvat suositukset Balversa-valmisteen annosmuutoksista annoksen suuremmaksi titraamisen jälkeen
| Seerumin fosfaattipitoisuus | Balversa-hoito |
| Jos fosfaattipitoisuus on ≥ 5,5 mg/dl (≥ 1,75 mmol/l), rajoita fosfaatin saanti 600–800 mg:aan/vrk. | |
< 6,99 mg/dl (< 2,24 mmol/l) | Jatka Balversa-hoitoa parhaillaan käytössä olevalla annoksella. |
7,00–8,99 mg/dl (2,25–2,90 mmol/l) | Jatka Balversa-hoitoa. Aloita fosfaatinsitojan käyttö ruokailun yhteydessä, kunnes fosfaattipitoisuus on < 7,00 mg/dl (< 2,25 mmol/l). Annosta on pienennettävä, jos seerumin fosfaattipitoisuus on pitkäkestoisesti 2 kuukauden ajan ≥ 7,00 mg/dl (≥ 2,25 mmol/l) tai jos sen lisäksi on muita haittavaikutuksia tai muita elektrolyyttitasapainon häiriöitä, jotka liittyvät pitkittyneeseen hyperfosfatemiaan. |
9,00–10,00 mg/dl (2,91–3,20 mmol/l) | Keskeytä Balversa-hoito, kunnes seerumin fosfaattipitoisuus korjautuu tasolle < 7,00 mg/dl (< 2,25 mmol/l) (viikoittaista testausta suositellaan). Aloita fosfaatinsitojan käyttö ruokailun yhteydessä, kunnes seerumin fosfaattipitoisuus korjautuu tasolle < 7,00 mg/dl (< 2,25 mmol/l). Aloita hoito uudelleen samalla annostuksella (ks. taulukko 1). Annosta on pienennettävä, jos seerumin fosfaattipitoisuus on pitkäkestoisesti 1 kuukauden ajan ≥ 9,00 mg/dl (≥ 2,91 mmol/l) tai jos sen lisäksi on muita haittavaikutuksia tai muita elektrolyyttitasapainon häiriöitä, jotka liittyvät pitkittyneeseen hyperfosfatemiaan. |
> 10,00 mg/dl (> 3,20 mmol/l) | Keskeytä Balversa-hoito, kunnes seerumin fosfaattipitoisuus korjautuu tasolle < 7,00 mg/dl (< 2,25 mmol/l) (viikoittaista testausta suositellaan). Aloita hoito uudelleen ensimmäisen pienennyskerran annostuksella (ks. taulukko 1). Jos seerumin fosfaattipitoisuus on pitkäkestoisesti > 2 viikon ajan ≥ 10,00 mg/dl (≥ 3,20 mmol/l), Balversa-hoito pitää lopettaa pysyvästi. Oireiden kliinisesti asianmukainen lääketieteellinen hoito (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). |
| Merkittävä muutos lähtötilanteeseen verrattuna munuaisten toiminnassa tai 3. asteen hypokalsemia hyperfosfatemian seurauksena. | Balversa-hoito pitää lopettaa pysyvästi. Kliinisesti asianmukainen lääketieteellinen hoito. |
Silmähaittojen hoito
Balversa-hoito pitää lopettaa tai sitä pitää muuttaa erdafitinibiin liittyvän toksisuuden perusteella, kuten taulukossa 3 kuvataan.
Taulukko 3. Ohjeet Balversa-hoitoon liittyvien silmähaittojen hoitoon
| Vaikeusasteluokka | Balversa-annoksen muuttaminen |
| 1. aste Oireeton tai lieviä oireita; vain kliinisiä tai diagnostisia havaintoja tai poikkeava Amslerin testikarttatulos. | Tee lähete silmätutkimukseen. Jos silmätutkimusta ei voida tehdä 7 päivän kuluessa, keskeytä Balversa-hoito, kunnes silmätutkimus voidaan tehdä. Jos silmätutkimuksessa ei todeta silmätoksisuutta, jatka Balversa-hoitoa samalla annoksella. Jos silmätutkimuksessa todetaan keratiitti tai verkkokalvon poikkeavuus (esim. sentraalinen seroosi korioretinopatiaa), keskeytä Balversa-hoito, kunnes ne ovat hävinneet. Jos ne häviävät 4 viikossa silmätutkimuksesta, jatka hoitoa yhtä annoksen pienennyskertaa pienemmällä annoksella. Aloitettaessa Balversa-hoito uudelleen seuraa silmätoksisuuden uusiutumista kuukauden ajan 1–2 viikon välein ja sen jälkeen kliinisesti asianmukaisesti. Jos silmätoksisuus ei uusiudu, harkitse annoksen suurentamista uudelleen. |
| 2. aste Keskivaikea, rajoittaa iänmukaisia itsestään huolehtimiseen liittyviä päivittäisiä toimia. | Keskeytä Balversa-hoito välittömästi ja tee lähete silmätutkimukseen. Jos silmätoksisuutta ei havaita, jatka erdafitinibihoitoa oireiden hävittyä yhtä annoksen pienennyskertaa pienemmällä annoksella. Jos silmätoksisuus häviää (häviää täysin tai stabiloituu ja potilas oireeton) 4 viikon kuluessa silmätutkimuksesta, aloita Balversa-hoito uudelleen yhtä annoksen pienennyskertaa pienemmällä annoksella. Aloitettaessa Balversa-hoito uudelleen seuraa silmätoksisuuden uusiutumista kuukauden ajan 1–2 viikon välein ja sen jälkeen kliinisesti asianmukaisesti. |
| 3. aste Vaikea-asteinen tai lääketieteellisesti merkitsevä, mutta ei välittömästi näkökykyä uhkaava; rajoittaa itsestään huolehtimiseen liittyviä päivittäisiä toimia. | Keskeytä Balversa-hoito välittömästi ja tee lähete silmätutkimukseen. Jos silmätoksisuus häviää (häviää täysin tai stabiloituu ja potilas oireeton) 4 viikon kuluessa, aloita Balversa-hoito uudelleen kahta annoksen pienennyskertaa pienemmällä annoksella. Aloitettaessa Balversa-hoito uudelleen seuraa silmätoksisuuden uusiutumista kuukauden ajan 1–2 viikon välein ja sen jälkeen kliinisesti asianmukaisesti. Harkitse Balversa-hoidon lopettamista pysyvästi uusiutumisen vuoksi. |
| 4. aste Näkökykyä uhkaavia seurauksia; sokeus (20/200 tai huonompi). | Lopeta Balversa-hoito pysyvästi. Seuraa, kunnes silmätoksisuus häviää täysin tai stabiloituu. |
| a Sentraalinen seroosi korioretinopatia, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet | |
Kynsi-, iho- ja limakalvomuutokset
Balversa-hoidon yhteydessä on havaittu kynsi-, iho- ja limakalvomuutoksia. Balversa-hoito pitää lopettaa tai sitä pitää muuttaa erdafitinibiin liittyvän toksisuuden perusteella, kuten taulukossa 4 kuvataan.
Taulukko 4. Annosmuutossuositukset Balversa-hoitoon liittyvien kynsiin, ihoon ja limakalvoihin kohdistuvien haittavaikutusten vuoksi
| Haittavaikutuksen vaikeusaste | Balversa |
| Kynsimuutokset | Balversa-annoksen muuttaminen |
| 1. aste | Jatka Balversa-hoitoa parhaillaan käytössä olevalla annoksella. |
| 2. aste | Keskeytä Balversa-hoito ja arvioi tilanne uudelleen 1–2 viikon kuluessa. Jos kyseessä on ensimmäinen ilmaantumiskerta, joka lievenee ≤ 1. asteeseen tai lähtötilanteeseen 2 viikon kuluessa, aloita hoito uudelleen samalla annoksella. Jos kyseessä on uusiutunut tapahtuma tai lieveneminen ≤ 1. asteeseen tai lähtötilanteeseen kestää > 2 viikkoa, aloita hoito uudelleen yhtä annoksen pienennyskertaa pienemmällä annoksella. |
| 3. aste | Keskeytä Balversa-hoito ja arvioi tilanne uudelleen 1–2 viikon kuluessa. Kun kynsimuutokset lievenevät ≤ 1. asteeseen tai lähtötilanteeseen, aloita hoito uudelleen yhtä annoksen pienennyskertaa pienemmällä annoksella. |
| 4. aste | Lopeta Balversa-hoito. |
| Kuiva iho ja ihotoksisuus | |
| 1. aste | Jatka Balversa-hoitoa parhaillaan käytössä olevalla annoksella. |
| 2. aste | Jatka Balversa-hoitoa parhaillaan käytössä olevalla annoksella. |
| 3. aste | Keskeytä Balversa-hoito (enintään 28 päivän ajaksi) ja arvioi kliininen tilanne viikoittain uudelleen. Kun kuiva iho ja ihotoksisuus lievenevät ≤ 1. asteeseen tai lähtötilanteeseen, aloita hoito uudelleen yhtä annoksen pienennyskertaa pienemmällä annoksella. |
| 4. aste | Lopeta Balversa-hoito. |
| Suun limakalvotulehdus | |
| 1. aste | Jatka Balversa-hoitoa parhaillaan käytössä olevalla annoksella. |
| 2. aste | Keskeytä Balversa-hoito, jos potilaalla on samanaikaisesti muita erdafitinibiin liittyviä 2. asteen haittavaikutuksia. Keskeytä Balversa-hoito, jos potilaan oireita on hoidettu jo yli viikon ajan. Jos Balversa-hoito keskeytetään, arvioi tilanne uudelleen 1–2 viikon kuluessa. Jos kyseessä on toksisuuden ensimmäinen ilmaantumiskerta ja jos toksisuus lievenee ≤ 1. asteeseen tai lähtötilanteeseen 2 viikon kuluessa, aloita hoito uudelleen samalla annoksella. Jos kyseessä on uusiutunut tapahtuma tai jos lieveneminen ≤ 1. asteeseen tai lähtötilanteeseen kestää > 2 viikkoa, aloita hoito uudelleen yhtä annoksen pienennyskertaa pienemmällä annoksella. |
| 3. aste | Keskeytä Balversa-hoito ja arvioi kliininen tilanne uudelleen 1–2 viikon kuluessa. Kun toksisuus lievenee ≤ 1. asteeseen tai lähtötilanteeseen, aloita hoito uudelleen yhtä annoksen pienennyskertaa pienemmällä annoksella. |
| 4. aste | Lopeta Balversa-hoito. |
| Suun kuivuminen | |
| 1. aste | Jatka Balversa-hoitoa parhaillaan käytössä olevalla annoksella. |
| 2. aste | Jatka Balversa-hoitoa parhaillaan käytössä olevalla annoksella. |
| 3. aste | Keskeytä Balversa-hoito (enintään 28 päivän ajaksi) ja arvioi kliininen tilanne viikoittain uudelleen. Kun suun kuivuminen lievenee ≤ 1. asteeseen tai lähtötilanteeseen, aloita hoito uudelleen yhtä annoksen pienennyskertaa pienemmällä annoksella. |
Taulukko 5. Annosmuutossuositukset Balversa-hoitoon liittyvien muiden haittavaikutusten vuoksi
| Muut haittavaikutukseta | |
| 3. aste | Keskeytä Balversa-hoito, kunnes toksisuus lievenee 1. asteeseen tai lähtötilanteeseen, minkä jälkeen Balversa-hoito voidaan aloittaa uudelleen yhtä annoksen pienennyskertaa pienemmällä annoksella. |
| 4. aste | Lopeta hoito pysyvästi. |
| a Annosmuutokset luokiteltu NCI CTCAEv5.0 (National Cancer Institute Common Terminology Criteria for Adverse Events) ‑kriteerien mukaan. | |
Erityispotilasryhmät
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei populaatiofarmakokineettisten analyysien perusteella tarvitse muuttaa (ks. kohta Farmakokinetiikka). Balversa-valmisteen käytöstä vaikeaa munuaisten vajaatoimintaa sairastaville potilaille ei ole tietoja. Vaikeaa munuaisten vajaatoimintaa sairastaville potilaille pitää harkita muita hoitovaihtoehtoja (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka). Balversa-valmisteen käytöstä vaikeaa maksan vajaatoimintaa sairastaville potilaille on vähän tietoja saatavissa. Vaikeaa maksan vajaatoimintaa sairastaville potilaille pitää harkita muita hoitovaihtoehtoja (ks. kohta Farmakokinetiikka).
Iäkkäät
Erityisiä annosmuutoksia ei katsota iäkkäille potilaille tarpeellisiksi (ks. kohta Farmakokinetiikka).
Yli 85-vuotiaista potilaista on vähän tietoja saatavissa.
Pediatriset potilaat
Ei ole asianmukaista käyttää erdafitinibiä pediatrisille potilaille uroteelikarsinooman hoitoon. Erdafitinibin turvallisuutta ja tehoa pediatristen potilaiden (< 18 vuotta) hoidossa ei ole vahvistettu. Tällä hetkellä saatavilla olevat turvallisuustiedot ovat kohdassa Haittavaikutukset.
Antotapa
Balversa otetaan suun kautta. Tabletit niellään kokonaisina ruoan kanssa tai tyhjään mahaan joka päivä suunnilleen samaan aikaan päivästä.
Greippihedelmien ja pomeranssin käyttämistä tulee välttää Balversa-hoidon aikana niiden voimakkaan CYP3A4:ää estävän vaikutuksen vuoksi (ks. kohta Yhteisvaikutukset).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Silmähaitat
Ennen Balversa-hoidon aloittamista pitää tehdä lähtötilanteen silmätutkimus, mukaan lukien Amslerin testikartta, silmänpohja- ja näöntarkkuustutkimus ja jos mahdollista, valokerroskuvaus (optinen koherenssitomografia, OCT).
Balversa-valmisteesta voi aiheutua silmäsairauksia, mukaan lukien sentraalinen seroosi korioretinopatia (ryhmitelty termi, joka sisältää verkkokalvon pigmenttiepiteelin irtauman), josta aiheutuu näkökenttäpuutos (ks. kohdat Vaikutus ajokykyyn ja koneidenkäyttökykyyn ja Haittavaikutukset). Sentraalisen seroosin korioretinopatian kokonaisilmaantuvuus oli ≥ 65‑vuotiailla potilailla suurempi (33,3 %) kuin < 65‑vuotiailla potilailla (28,8 %). Verkkokalvon pigmenttiepiteelin irtaumatapahtumia raportoitiin yleisemmin ≥ 65‑vuotiailla potilailla (6,3 %) kuin < 65‑vuotiailla potilailla (2,1 %). Tarkka kliininen seuranta on suositeltavaa, jos potilas on vähintään 65-vuotias, samoin kuin jos potilaalla on kliinisesti merkittävä silmäsairaus, kuten verkkokalvon sairaus, mukaan lukien mm. sentraalinen seroosi korioretinopatia, makula- tai verkkokalvorappeuma, diabeettinen retinopatia ja aiempi verkkokalvon irtauma (ks. kohta Haittavaikutukset).
Kuivasilmäisyyttä todettiin Balversa-hoidon aikana 16,7 %:lla potilaista, ja 0,3 %:lla sen vaikeusaste oli 3. tai 4. aste (ks. kohta Haittavaikutukset). Kaikkien potilaiden pitää hereilläoloaikana käyttää vähintään joka toinen tunti kuivasilmäisyyttä estävää hoitoa tai hoitoa silmä-ärsytystä lievittävillä aineilla (esim. keinokyyneleitä, kosteuttavia ja voitelevia silmägeelejä tai ‑voiteita). Silmälääkärin pitää tutkia vaikea-asteinen hoitoon liittyvä kuivasilmäisyys.
Silmätutkimuksia, mukaan lukien Amslerin testikartta, tehdään ensimmäisten neljän hoitokuukauden aikana kuukausittain ja sen jälkeen joka 3. kuukausi sekä näköoireiden yhteydessä kiireellisesti (ks. kohta Annostus ja antotapa). Jos poikkeavuuksia havaitaan, noudatetaan taulukon 3 hoito-ohjeita. Silmätutkimukseen pitää kuulua näöntarkkuuden arviointi, rakolamppututkimus, silmänpohjatutkimus ja valokerroskuvaus. Potilaita, jotka ovat aloittaneet Balversa-hoidon uudelleen silmiin liittyneen haittavaikutuksen jälkeen, pitää seurata tarkoin, mukaan lukien tehdä kliinisiä silmätutkimuksia.
Jos sentraalinen seroosi korioretinopatia ilmaantuu, Balversa-hoito pitää keskeyttää. Hoito pitää lopettaa pysyvästi, jos sentraalinen seroosi korioretinopatia ei häviä 4 viikon kuluessa tai jos sen vaikeusaste on 4. aste. Noudata silmiin kohdistuvien haittavaikutusten yhteydessä ohjeita annoksen muuttamisesta (ks. kohta Annostus ja antotapa Silmäsairauksien hoito).
Hyperfosfatemia
Balversa voi aiheuttaa hyperfosfatemiaa. Pitkittyneestä hyperfosfatemiasta voi aiheutua pehmytkudosten mineralisaatiota, ihon kalsinoosia, ei-ureemista kalsifylaksia, hypokalsemiaa, anemiaa, sekundaarista hyperparatyreoosia, lihaskramppeja, kouristusaktiivisuutta, QT-ajan pitenemistä ja sydämen rytmihäiriöitä. Hyperfosfatemiaa raportoitiin Balversa-hoidon varhaisvaiheessa, ja valtaosa tapahtumista ilmeni ensimmäisten 3–4 kuukauden kuluessa ja 3. asteen tapahtumia ilmeni yleensä ensimmäisen kuukauden kuluessa.
Seuraa hyperfosfatemiaa koko hoidon ajan. Jos seerumin fosfaattipitoisuus on ≥ 5,5 mg/dl (≥ 1,75 mmol/l), fosfaatin saantia ruokavaliosta pitää rajoittaa (600–800 mg päivässä) ja seerumin fosfaattipitoisuutta mahdollisesti suurentavien lääkeaineiden samanaikaista käyttöä pitää välttää (ks. kohta Annostus ja antotapa). Erdafitinibiä saaville potilaille ei suositella D-vitamiinilisää, koska se voi osaltaan suurentaa seerumin fosfaatti- ja kalsiumpitoisuutta.
Jos seerumin fosfaattipitoisuus on yli 7,0 mg/dl (2,25 mmol/l), harkitse suun kautta otettavan fosfaatinsitojan lisäämistä hoitoon, kunnes seerumin fosfaattipitoisuus pienenee tasolle < 7,0 mg/dl (< 2,25 mmol/l). Harkitse Balversa-hoidon keskeyttämistä, annoksen pienentämistä tai hoidon lopettamista pysyvästi hyperfosfatemian keston ja vaikeusasteen perusteella taulukon 2 mukaisesti (ks. kohta Annostus ja antotapa).
Käyttö QT-aikaa tunnetusti pidentävien valmisteiden kanssa
Balversa-valmisteen käytössä seuraavien lääkevalmisteiden kanssa suositellaan varovaisuutta: lääkevalmisteet, joiden tiedetään pidentävän QT‑aikaa tai jotka voivat aiheuttaa kääntyvien kärkien takykardiaa, kuten ryhmän IA (esim. kinidiini, disopyramidi) tai ryhmän III (esim. amiodaroni, sotaloli, ibutilidi) rytmihäiriölääkkeet, makrolidiantibiootit, serotoniinin takaisinoton estäjät (esim. sitalopraami, essitalopraami), metadoni, moksifloksasiini ja psykoosilääkkeet (esim. haloperidoli ja tioridatsiini).
Hypofosfatemia
Balversa-hoidon aikana voi ilmetä hypofosfatemiaa. Seerumin fosfaattipitoisuutta pitää seurata erdafitinibihoidon aikana ja erdafitinibihoidon taukojen aikana. Jos seerumin fosfaattipitoisuus pienenee normaaliarvojen alle, fosfaattipitoisuutta pienentävä hoito ja ruokavalion fosfaattirajoitukset (jos soveltuvat) pitää lopettaa. Vaikea-asteinen hypofosfatemia saattaa ilmetä sekavuutena, kouristuskohtauksina, fokaalisina neurologisina löydöksinä, sydämen vajaatoimintana, hengityksen vajaatoimintana, lihasheikkoutena, rabdomyolyysinä ja hemolyyttisenä anemiana. Annosmuutokset, ks. kohta Annostus ja antotapa. Potilaista 1,0 %:lla oli 3.–4. asteen hypofosfatemiareaktioita.
Kynsien häiriöt
Kynsien häiriöitä, mukaan lukien kynnen irtoaminen, kynsien värinmuutoksia ja kynsivallitulehduksia, voi ilmetä Balversa-hoidon yhteydessä hyvin yleisesti (ks. kohta Haittavaikutukset).
Potilaita pitää seurata kynsitoksisuuden oireiden ja löydösten havaitsemiseksi. Potilaille pitää kertoa ennalta ehkäisevästä hoidosta, kuten hyvästä hygieniasta ja tarvittaessa itsehoitona käytettävästä kynsienvahvistajasta, ja potilaita on seurattava infektion oireiden havaitsemiseksi. Balversa-hoito pitää lopettaa tai sitä pitää muuttaa taulukossa 4 kuvatun erdafitinibiin liittyvän toksisuuden perusteella.
Ihohaitat
Balversa-hoidon aikana voi ilmetä hyvin yleisesti ihohaittoja, mukaan lukien kuiva iho, käsi-jalkaoireyhtymä, alopesia ja kutina (ks. kohta Haittavaikutukset). Potilaita pitää seurata ja heille pitää antaa tukihoitoa, kuten välttää tarpeetonta altistumista auringonvalolle sekä liiallista saippuan käyttöä ja kylpemistä. Potilaiden pitää käyttää säännöllisesti ihoa kosteuttavia voiteita ja välttää hajustettuja tuotteita. Balversa-hoito pitää lopettaa tai sitä pitää muuttaa erdafitinibiin liittyvän toksisuuden perusteella, kuten taulukossa 4 kuvataan.
Valoherkkyysreaktiot
Altistumista auringolle pitää varoa käyttämällä suojaavaa vaatetusta ja/tai auringonsuojavoidetta, sillä Balversa-hoitoon liittyy valoherkkyysreaktioiden mahdollinen riski.
Limakalvohaitat
Balversa-hoidon yhteydessä voi ilmetä hyvin yleisesti stomatiittia ja suun kuivumista (ks. kohta Haittavaikutukset). Potilaita pitää neuvoa hakeutumaan lääkäriin, jos oireet pahenevat. Potilaita pitää seurata ja heille pitää antaa tukihoitoa, jota ovat mm. hyvä suuhygienia, suun huuhtelu ruokasoodalla tarvittaessa 3 tai 4 kertaa päivässä sekä mausteisten ja/tai happamien ruokien välttäminen. Balversa-hoito pitää lopettaa tai sitä pitää muuttaa erdafitinibiin liittyvän toksisuuden perusteella, kuten taulukossa 4 kuvataan.
Laboratoriokokeet
Balversa-hoitoa saavilla potilailla on raportoitu kohonneita kreatiniinipitoisuuksia, hyponatremiaa, kohonneita transaminaasipitoisuuksia ja anemiaa (ks. kohta Haittavaikutukset). Täydellinen verenkuva ja seerumin kemialliset ominaisuudet pitää tutkia säännöllisin väliajoin Balversa-hoidon aikana tällaisten muutosten seuraamiseksi.
Lisääntymis- ja kehitystoksisuus
Eläimillä tehtyjen lisääntymistutkimusten havaintojen ja valmisteen vaikutusmekanismin perusteella erdafitinibi on alkiotoksinen ja teratogeeninen (ks. kohta Prekliiniset tiedot turvallisuudesta). Raskaana oleville naisille pitää kertoa sikiölle aiheutuvasta mahdollisesta riskistä. Naispotilaita, jotka voivat tulla raskaaksi, pitää kehottaa käyttämään erittäin tehokasta ehkäisyä ennen hoitoa ja hoidon aikana sekä 1 kuukauden ajan viimeisen annoksen jälkeen (ks. kohta Raskaus ja imetys). Miespotilaita pitää neuvoa käyttämään tehokasta ehkäisyä (esim. kondomia) ja heille pitää kertoa, etteivät he saa luovuttaa tai heiltä ei tule ottaa talteen siemennestettä hoidon aikana eikä 1 kuukauteen viimeisen Balversa-annoksen jälkeen (ks. kohta Raskaus ja imetys).
Naisille, jotka voivat tulla raskaaksi, suositellaan tekemään raskaustesti erittäin herkällä määritysmenetelmällä ennen Balversa-hoidon aloittamista.
Yhdistelmähoito voimakkailla tai kohtalaisilla CYP2C9:n tai CYP3A4:n estäjillä
Balversa-valmisteen samanaikainen käyttö kohtalaisten CYP2C9:n tai voimakkaiden CYP3A4:n estäjien kanssa edellyttää annosmuutosta (ks. kohta Yhteisvaikutukset).
Yhdistelmähoito voimakkailla tai kohtalaisilla CYP3A4:n indusoijilla
Balversa-valmisteen samanaikaista käyttöä voimakkaiden CYP3A4:n indusoijien kanssa ei suositella. Balversa-valmisteen samanaikainen käyttö kohtalaisten CYP3A4:n indusoijien kanssa edellyttää annosmuutosta (ks. kohta Yhteisvaikutukset).
Yhdistelmähoito hormonaalisilla ehkäisyvalmisteilla
Balversa-valmisteen samanaikainen käyttö voi heikentää hormonaalisten ehkäisyvalmisteiden tehoa. Hormonaalisia ehkäisyvalmisteita käyttäviä potilaita pitää neuvoa käyttämään jotakin vaihtoehtoista ehkäisymenetelmää, joihin entsyymien indusoijat eivät vaikuta (esim. hormoniton kierukka), tai käyttämään lisäksi jotakin ei-hormonaalista ehkäisymenetelmää (esim. kondomia) hoidon aikana ja 1 kuukauden ajan viimeisen Balversa-annoksen ottamisen jälkeen (ks. kohdat Yhteisvaikutukset ja Raskaus ja imetys).
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää alle 1 mmol natriumia (23 mg) eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus Balversa-valmisteeseen
Kohtalaiset CYP2C9:n tai voimakkaat CYP3A4:n estäjät
Kohtalaisen CYP2C9:n tai voimakkaan CYP3A4:n estäjän samanaikainen käyttö suurensi erdafitinibialtistusta ja saattaa lisätä lääkkeeseen liittyvää toksisuutta. Käytettäessä samanaikaisesti flukonatsolia, joka on kohtalainen CYP2C9:n ja CYP3A4:n estäjä, erdafitinibin Cmax -arvon keskimääräinen suhde (90 %:n luottamusväli) oli 121 % (99,9–147) ja AUC∞-arvon keskimääräinen suhde (90 %:n luottamusväli) oli 148 % (120–182) verrattuna pelkän erdafitinibin käyttöön. Käytettäessä samanaikaisesti itrakonatsolia, joka on voimakas CYP3A4:n estäjä ja P‑gp:n estäjä, erdafitinibin Cmax-arvo oli 105 % (90 %:n luottamusväli: 86,7–127) ja AUC∞-arvo oli 134 % (90 %:n luottamusväli: 109–164) verrattuna pelkän erdafitinibin käyttöön. Harkitse vaihtoehtoisia lääkeaineita, jotka eivät estä entsyymejä tai estävät niitä minimaalisesti. Jos Balversa-valmistetta käytetään samanaikaisesti kohtalaisen CYP2C9:n tai voimakkaan CYP3A4:n estäjän (esim. itrakonatsoli, ketokonatsoli, posakonatsoli, vorikonatsoli, flukonatsoli, mikonatsoli, seritinibi, klaritromysiini, telitromysiini, elvitegraviiri, ritonaviiri, paritapreviiri, sakinaviiri, nefatsodoni, nelfinaviiri, tipranaviiri, lopinaviiri, amiodaroni, piperiini) kanssa, pienennä Balversa-annos siedettävyyden perusteella seuraavaan pienempään annokseen (ks. kohta Annostus ja antotapa). Jos kohtalaisen CYP2C9:n tai voimakkaan CYP3A4:n estäjän käyttö lopetetaan, Balversa-annosta voidaan muuttaa potilaan sietokyvyn mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Greippihedelmien ja pomeranssin käyttämistä tulee välttää Balversa-hoidon aikana niiden voimakkaan CYP3A4:ää estävän vaikutuksen vuoksi (ks. kohta Annostus ja antotapa).
Voimakkaat tai kohtalaiset CYP3A4:n indusoijat
Samanaikainen käyttö karbamatsepiinin kanssa, joka on voimakas CYP3A4:n ja heikko CYP2C9:n indusoija, pienentää erdafitinibialtistusta. Käytettäessä samanaikaisesti karbamatsepiinia erdafitinibin Cmax-arvon keskimääräinen suhde oli 65,4 % (90 %:n luottamusväli: 60,8–70,5) ja AUC∞-arvon keskimääräinen suhde oli 37,7 % (90 %:n luottamusväli: 35,4–40,2) verrattuna pelkän erdafitinibin käyttöön. Vältä Balversa-valmisteen samanaikaista käyttöä voimakkaiden CYP3A4:n indusoijien kanssa (kuten apalutamidi, entsalutamidi, lumakaftori, ivosidenibi, mitotaani, rifapentiini, rifampisiini, karbamatsepiini, fenytoiini ja mäkikuisma). Jos Balversa-valmistetta käytetään samanaikaisesti kohtalaisen CYP3A4:n indusoijan (esim. dabrafenibi, bosentaani, senobamaatti, elagoliksi, efavirentsi, etraviriini, lorlatinibi, mitapivaatti, modafiniili, peksidartinibi, fenobarbitaali, primidoni, repotrektinibi, rifabutiini, sotorasibi, telotristaattietyyli) kanssa, annosta pitää suurentaa varoen 1–2 mg ja säätää sitä asteittain kahden tai kolmen viikon välein haittavaikutusten kliinisen seurannan perusteella, mutta 9 mg:n annosta ei saa ylittää. Jos kohtalaisen CYP3A4:n indusoijan käyttö lopetetaan, Balversa-annosta voidaan säätää potilaan sietokyvyn mukaan (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Balversa-valmisteen vaikutus muihin lääkevalmisteisiin
Pääasialliset CYP-isoformin substraatit (mukaan lukien hormonaaliset ehkäisyvalmisteet)
Erdafitinibin ja midatsolaamin (herkkä CYP3A4:n substraatti) samanaikaisessa käytössä midatsolaamin Cmax-arvon keskimääräinen suhde oli 86,3 % (90 %:n luottamusväli: 73,5–101) ja AUC∞-arvon keskimääräinen suhde oli 82,1 % (90 %:n luottamusväli: 70,8–95,2) verrattuna pelkän midatsolaamin käyttöön. Erdafitinibillä ei ole kliinisesti merkittävää vaikutusta midatsolaamin farmakokinetiikkaan. Ei kuitenkaan voida sulkea pois sitä, että Balversa-valmisteen yksinään antamisen jälkeinen tai muiden CYP3A4:n indusoijien ja Balversa-valmisteen samanaikaisen antamisen jälkeinen CYP3A4:n induktio voi vähentää hormonaalisten ehkäisyvalmisteiden tehoa.
Hormonaalisia ehkäisyvalmisteita käyttäviä potilaita pitää neuvoa käyttämään jotakin vaihtoehtoista ehkäisymenetelmää, johon entsyymien indusoijat eivät vaikuta (esim. hormoniton kierukka), tai käyttämään lisäksi jotakin ei-hormonaalista ehkäisymenetelmää (esim. kondomia) hoidon aikana ja 1 kuukauden ajan viimeisen Balversa-annoksen ottamisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
P-glykoproteiinin (P-gp) substraatit
Erdafitinibi on P‑gp:n estäjä. Balversa-valmisteen samanaikainen antaminen P‑gp:n substraattien kanssa voi lisätä niiden systeemistä altistusta. Suun kautta otettavat P‑gp:n substraatit, joiden terapeuttinen indeksi on kapea (esim. kolkisiini, digoksiini, dabigatraani ja apiksabaani), pitää ottaa vähintään 6 tuntia ennen erdafitinibin ottamista tai aikaisintaan 6 tuntia sen ottamisen jälkeen, jotta yhteisvaikutusten mahdollisuus minimoidaan.
Orgaanisen kationin kuljettaja 2:n (OCT2) substraatit
Erdafitinibin ja metformiinin (herkkä OCT2:n substraatti) samanaikaisessa käytössä metformiinin Cmax-arvon keskimääräinen suhde oli 109 % (90 %:n luottamusväli: 90,3–131) ja AUC∞-arvon keskimääräinen suhde oli 114 % (90 %:n luottamusväli: 93,2–139) verrattuna pelkän metformiinin käyttöön. Erdafitinibillä ei ole kliinisesti merkittävää vaikutusta metformiinin farmakokinetiikkaan.
Seerumin fosfaattipitoisuutta mahdollisesti muuttavat lääkevalmisteet
Balversa-hoitoa saavilla potilailla pitää välttää lääkevalmisteita, jotka voivat muuttaa seerumin fosfaattipitoisuutta, kunnes seerumin fosfaattipitoisuus on määritetty 14–21 päivää hoidon aloittamisen jälkeen, sillä se voi vaikuttaa päätökseen annoksen titraamisesta suuremmaksi.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehillä ja naisilla
Vaikutusmekanismin ja eläimillä tehtyjen lisääntymistutkimusten havaintojen perusteella raskaana olevalle naiselle annettu erdafitinibi voi vahingoittaa sikiötä. Naisia, jotka voivat tulla raskaaksi, pitää kehottaa käyttämään erittäin tehokasta ehkäisyä ennen hoitoa ja hoidon aikana sekä 1 kuukauden ajan viimeisen Balversa-annoksen jälkeen. Miespotilaita pitää kehottaa käyttämään tehokasta ehkäisyä (esim. kondomia) ja heille pitää kertoa, etteivät he saa luovuttaa siemennestettä tai antaa sitä talletettavaksi hoidon aikana eikä 1 kuukauteen viimeisen Balversa-annoksen jälkeen.
Balversa-valmisteen samanaikainen käyttö voi heikentää hormonaalisten ehkäisyvalmisteiden tehoa. Hormonaalisia ehkäisyvalmisteita käyttäviä potilaita pitää neuvoa käyttämään jotakin vaihtoehtoista ehkäisymenetelmää, johon entsyymien indusoijat eivät vaikuta (esim. hormoniton kierukka), tai käyttämään lisäksi jotakin ei-hormonaalista ehkäisymenetelmää (esim. kondomia) hoidon aikana ja 1 kuukauden ajan viimeisen Balversa-annoksen ottamisen jälkeen (ks. kohta Yhteisvaikutukset).
Raskaustesti
Naisille, jotka voivat tulla raskaaksi, suositellaan tekemään raskaustesti erittäin herkällä määritysmenetelmällä ennen Balversa-hoidon aloittamista.
Raskaus
Erdafitinibin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Erdafitinibin vaikutusmekanismin ja eläimillä tehtyjen lisääntymistutkimusten havaintojen perusteella Balversa-valmistetta ei pidä käyttää raskauden aikana, paitsi jos naisen kliininen tila edellyttää erdafitinibihoitoa.
Jos Balversa-valmistetta käytetään raskauden aikana tai jos potilas tulee raskaaksi Balversa-valmistetta käyttäessään, kerro potilaalle sikiölle aiheutuvasta mahdollisesta vaarasta ja kerro hänelle, mitkä ovat kliiniset ja hoidolliset vaihtoehdot. Potilasta pitää kehottaa ottamaan yhteyttä terveydenhuollon ammattilaiseen, jos hän tulee raskaaksi tai epäilee raskautta Balversa-hoidon aikana tai 1 kuukauden aikana sen jälkeen.
Imetys
Erdafitinibin erittymisestä äidinmaitoon, vaikutuksista imetettävään vauvaan vai vaikutuksista maidontuotantoon ei ole tietoja.
Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Imetys pitää lopettaa hoidon ajaksi ja 1 kuukaudeksi viimeisen Balversa-annoksen jälkeen.
Hedelmällisyys
Erdafitinibin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoja. Erdafitinibillä ei ole tehty varsinaisia hedelmällisyyttä koskevia eläinkokeita (ks. kohta Prekliiniset tiedot turvallisuudesta). Yleisissä eläinkokeissa tehtyjen hedelmällisyyttä koskevien alustavien arvioiden perusteella (ks. kohta Prekliiniset tiedot turvallisuudesta) ja erdafitinibin farmakologian perusteella miehen ja naisen hedelmällisyyden heikkenemistä ei voida sulkea pois.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Balversa-valmisteella on kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Silmähaittoja, kuten sentraalista seroosia korioretinopatiaa tai keratiittia on havaittu liittyen hoitoon FGFR:n estäjillä ja Balversa-valmisteella. Jos potilaalle ilmaantuu näkökykyyn vaikuttavia hoitoon liittyviä oireita, hänen on suositeltavaa olla ajamatta moottoriajoneuvoa ja käyttämättä koneita, kunnes tällainen vaikutus häviää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät haittavaikutukset olivat hyperfosfatemia (78,5 %), ripuli (55,5 %), stomatiitti (52,8 %), suun kuivuminen (39,9 %), heikentynyt ruokahalu (31,7 %), anemia (28,2 %), kuiva iho (28,0 %), sentraalinen seroosi korioretinopatia (28,0 %), ummetus (27,3 %), makuhäiriö (26,3 %), käsi-jalkaoireyhtymä (25,5 %), alopesia (23,2 %), astenia (23,0 %), suurentunut alaniiniaminotransferaasipitoisuus (21,7 %), kynnen irtoaminen (21,7 %), uupumus (20,3 %), pahoinvointi (18,6 %), painon lasku (18,4 %), suurentunut aspartaattiaminotransferaasipitoisuus (18,0 %), kuivat silmät (16,7 %), kynsien värimuutos (15,9 %), oksentelu (13,8 %), suurentunut veren kreatiniinipitoisuus (13,8 %), hyponatremia (13,4 %), kynnenvierustulehdus (12,5 %), kynsien surkastuminen (11,9 %), onykomadeesi (11,5 %), nenäverenvuoto (10,6 %), kynsisairaus (10,2 %) ja vatsakipu (10,0 %).
Yleisimmät 3. asteen tai vaikeampiasteiset haittavaikutukset olivat stomatiitti (10,6 %), hyponatremia (8,8 %), käsi-jalkaoireyhtymä (7,9 %), kynnen irtoaminen (4,8 %), ripuli (4,0 %), hyperfosfatemia (2,9 %), heikentynyt ruokahalu (2,5 %) ja kynsien surkastuminen (2,5 %). Hoidosta aiheutuvia 3. ja 4. asteen haittavaikutuksia raportoitiin yleisemmin vähintään 65-vuotiailla potilailla kuin < 65-vuotiailla potilailla (47,6 % vs. 43,5 %), kuten myös vakavia haittavaikutuksia (14,6 % vs. 10,5 %).
Annoksen pienentämiseen johtaneita haittavaikutuksia ilmeni 59,7 %:lle potilaista. Yleisimmät annoksen pienentämiseen johtaneet haittavaikutukset olivat stomatiitti (15,4 %), käsi-jalkaoireyhtymä (9,6 %), kynnen irtoaminen (7,3 %) ja hyperfosfatemia (5,2 %).
Hoidon lopettamiseen johtaneita haittavaikutuksia ilmeni 19,4 %:lle potilaista. Yleisimmät hoidon lopettamiseen johtaneet haittavaikutukset olivat verkkokalvon pigmenttisolukerroksen irtauma (1,7 %) ja stomatiitti (1,5 %).
Haittavaikutustaulukko
Turvallisuusprofiili perustuu kliinisissä Balversa-tutkimuksissa hoitoa saaneista 479:stä paikallisesti edennyttä leikkaukseen soveltumatonta tai metastasoitunutta uroteelikarsinoomaa sairastaneesta potilaasta saatuihin yhdistettyihin tietoihin. Potilaat saivat Balversa-hoitoa suun kautta otetulla 8/9 mg:n aloitusannoksella kerran päivässä. Hoidon keston mediaani oli 4,8 kuukautta (vaihteluväli 0,1–43,4 kuukautta).
Kliinisten tutkimusten aikana havaitut haittavaikutukset luetellaan jäljempänä taulukossa 6 yleisyysluokittain. Yleisyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 6. Kliinisissä tutkimuksissa tunnistetut haittavaikutukset
| Elinjärjestelmä | Esiintyvyys | Haittavaikutus |
| Umpieritys | yleinen | hyperparatyreoosi |
| Aineenvaihdunta ja ravitsemus | hyvin yleinen | hyperfosfatemia, hyponatremia, heikentynyt ruokahalu |
| yleinen | hyperkalsemia, hypofosfatemia | |
| Hermosto | hyvin yleinen | makuhäiriö |
| Silmät | hyvin yleinen | sentraalinen seroosi korioretinopatiaa, kuivat silmät |
| yleinen | haavainen sarveiskalvotulehdus, sarveiskalvotulehdus, sidekalvotulehdus, silmien kuivuus, kaihi, luomitulehdus, lisääntynyt kyyneleritys | |
| Verisuonisto | melko harvinainen | verisuonten kalkkiutuminen |
| Hengityselimet, rintakehä ja välikarsina | hyvin yleinen | nenäverenvuoto |
| yleinen | nenän kuivuus | |
| Ruoansulatuselimistö | hyvin yleinen | ripuli, stomatiittib, suun kuivuminen, ummetus, pahoinvointi, oksentelu, vatsakipu |
| yleinen | dyspepsia | |
| Iho ja ihonalainen kudos | hyvin yleinen | kynnenvierustulehdus, kynnen irtoaminen, onykomadeesi, kynsien surkastuminen, kynsisairaus, kynsien värimuutos, käsi-jalkaoireyhtymä, alopesia, kuiva iho |
| yleinen | kynsikipu, kynsien murtuminen, harjanteet kynsissä, ihon halkeilu, kutina, ihon kesiminen, kseroderma, hyperkeratoosi, iholeesiot, ekseema, ihottuma | |
| melko harvinainen | kynsimarron verenvuoto, epämukavat tuntemukset kynsissä, ihon atrofia, kämmenien punoitus, ihotoksisuus | |
| Munuaiset ja virtsatiet | yleinen | akuutti munuaisvaurio, munuaisten vajaatoiminta, munuaisten toiminnan häiriö |
| Maksa ja sappi | yleinen | maksasolujen hajoaminen, poikkeava maksan toiminta, hyperbilirubinemia |
| Yleisoireet ja antopaikassa todettavat haitat | hyvin yleinen | astenia, uupumus |
| melko harvinainen | limakalvojen kuivuus | |
| Veri ja imukudos | hyvin yleinen | anemia |
| Tutkimukset | hyvin yleinen | painon lasku, suurentunut veren kreatiniinipitoisuus, suurentunut alaniiniaminotransferaasipitoisuus, suurentunut aspartaattiaminotransferaasipitoisuus |
a Sentraalinen seroosi korioretinopatia sisältää seuraavat: verkkokalvon irtauma, lasiaisen irtauma, verkkokalvon turvotus, retinopatia, korioretinopatia, verkkokalvon pigmenttisolukerroksen irtauma, verkkokalvon makula-alueen pigmenttiepiteelin irtauma, makulan irtauma, verkkokalvon seroosi irtauma, verkkokalvonalainen neste, verkkokalvon paksuuntuminen, korioretiniitti, seroosi retinopatia, makulopatia, suonikalvon effuusio, näön sumeneminen, näkökyvyn heikkeneminen, heikentynyt näöntarkkuus. b Stomatiitti sisältää seuraavat: suun haavaumat. | ||
Valikoitujen haittavaikutusten kuvaus
Sentraalinen seroosi korioretinopatia
Sentraalista seroosia korioretinopatiaa raportoitiin haittavaikutuksena 31,5 %:lla potilaista, ja sen ensimmäiseen ilmaantumiskertaan (minkä tahansa vaikeusasteen tapahtuma) kuluneen ajan mediaani oli 51 päivää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Yleisimmin raportoituja tapahtumia olivat näön sumeneminen, korioretinopatia, verkkokalvon pigmenttisolukerroksen irtauma, heikentynyt näöntarkkuus, näkökyvyn heikentyminen, verkkokalvon irtauma, retinopatia ja verkkokalvonalainen neste. 3. tai 4. asteen sentraalista seroosia korioretinopatiaa raportoitiin 2,7 %:lla potilaista. Valtaosa sentraalista seroosia korioretinopatiaa koskeneista tapahtumista ilmeni hoidon ensimmäisten 90 päivän aikana. Tietojen keruun katkaisuajankohtana sentraalinen seroosi korioretinopatia oli parantunut 43,0 %:lla potilaista. Potilaista, joille sentraalinen seroosi korioretinopatia ilmaantui, 11,3 %:lla hoito keskeytettiin ja 14,6 %:lla annosta pienennettiin. 3,3 % potilaista lopetti Balversa-hoidon seuraavien vuoksi: verkkokalvon pigmenttisolukerroksen irtauma (1,7 %), korioretinopatia (0,6 %), heikentynyt näöntarkkuus (0,6 %), makulopatia (0,4 %), näön sumeneminen (0,2 %), näkökyvyn heikentyminen (0,2 %), verkkokalvon irtauma (0,2 %) ja verkkokalvonalainen neste (0,2 %).
Muut silmähaitat
Silmähaittoja (muita kuin sentraalista seroosia korioretinopatiaa) raportoitiin 36,3 %:lla potilaista. Yleisimmin raportoituja tapahtumia olivat kuivat silmät (16,7 %), sidekalvotulehdus (9,8 %) ja lisääntynyt kyynelvuoto (9,2 %). Niistä potilaista, joille tapahtumia ilmaantui, 4,8 %:lla annosta pienennettiin ja 6,7 %:lla hoito keskeytettiin. 1,3 % lopetti erdafitinibihoidon silmähaittojen vuoksi. Silmähaittojen ensimmäiseen ilmaantumiskertaan kuluneen ajan mediaani oli 53 päivää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kynsihaitat
Kynsihaittoja raportoitiin 62,6 %:lla potilaista. Yleisimmin raportoituja tapahtumia olivat kynnen irtoaminen (21,7 %), kynsien värimuutos (15,9 %), kynnenvierustulehdus (12,5 %), kynsien surkastuminen (11,9 %) ja onykomadeesi (11,5 %). Kynsihaittojen ilmaantuvuus lisääntyi ensimmäisen altistuskuukauden jälkeen. Minkä tahansa vaikeusasteen kynsihaitan ilmenemiseen kuluneen ajan mediaani oli 63 päivää.
Ihohaitat
Ihohaittoja raportoitiin 54,5 %:lla potilaista. Yleisimmin raportoituja tapahtumia olivat kuiva iho (28 %) ja käsi-jalkaoireyhtymä (25,5 %). Minkä tahansa vaikeusasteen ihohaitan ilmenemiseen kuluneen ajan mediaani oli 47 päivää.
Ruoansulatuselimistön haitat
Ruoansulatuselimistön haittoja raportoitiin 83,9 %:lla potilaista. Yleisimmin raportoituja tapahtumia olivat ripuli (55,5 %), stomatiitti (52,8 %) ja suun kuivuminen (39,9 %). Minkä tahansa vaikeusasteen ruoansulatuselimistön haitan ilmenemiseen kuluneen ajan mediaani oli 15 päivää.
Hyperfosfatemia ja pehmytkudosten mineralisaatio
Erdafitinibi voi aiheuttaa hyperfosfatemiaa. Fosfaattipitoisuuden suureneminen on oletettu ja ohimenevä farmakodynaaminen vaikutus (ks. kohta Farmakodynamiikka). Hyperfosfatemiaa raportoitiin haittavaikutuksena 78,5 %:lla Balversa-hoitoa saaneista potilaista. Hyperfosfatemiaa raportoitiin erdafitinibihoidon varhaisvaiheessa, ja 1.–2. asteen tapahtumat ilmenivät yleensä ensimmäisten 3 tai 4 kuukauden kuluessa ja 3. asteen tapahtumia ilmeni yleensä ensimmäisen kuukauden kuluessa. Minkä tahansa vaikeusasteen hyperfosfatemian ilmenemiseen kuluneen ajan mediaani oli 16 päivää. 0,2 %:lla Balversa-hoitoa saaneista potilaista on havaittu verisuonten kalkkiutumista (ks. kohta Annostus ja antotapa). Hyperkalsemiaa havaittiin 6,1 %:lla ja hyperparatyreoosia havaittiin 2,9 %:lla Balversa-hoitoa saaneista potilaista (ks. taulukko 2 kohdassa Annostus ja antotapa).
Hypofosfatemia
Erdafitinibi voi aiheuttaa hypofosfatemiaa. Hypofosfatemia ilmaantui 5,6 %:lle potilaista. Hypofosfatemiareaktiot olivat 1,0 %:lla potilaista 3.–4. asteen tapahtumia. 3. asteen reaktion ilmenemiseen kuluneen ajan mediaani oli 140 päivää. Yksikään tapahtumista ei ollut vakava eikä johtanut hoidon lopettamiseen tai annoksen pienentämiseen. Hoito keskeytettiin 0,2 %:lla potilaista.
Poikkeavuudet laboratoriokoetuloksissa
Poikkeavuuksia laboratoriokoetuloksissa (muita kuin hyperfosfatemia, joka kuvataan erikseen), ilmaantui 53,4 %:lle potilaista. Yleisimmin raportoidut laboratoriokoetulosten poikkeavuudet olivat anemia (28,2 % [135 potilasta]; sen ilmenemiseen kuluneen ajan mediaani oli 44 päivää, ja 38,5 % [52/135] tapahtumista korjautui), suurentunut alaniiniaminotransferaasipitoisuus (21,7 % [104 potilasta]; sen ilmenemiseen kuluneen ajan mediaani oli 41 päivää, ja 75 % [78/104] tapahtumista korjautui), suurentunut aspartaattiaminotransferaasipitoisuus (18 % [86 potilasta]; sen ilmenemiseen kuluneen ajan mediaani oli 37 päivää, ja 73,3 % [63/86] tapahtumista korjautui), suurentunut veren kreatiniinipitoisuus (14,2 % [68 potilasta]; sen ilmenemiseen kuluneen ajan mediaani oli 57 päivää, ja 44,1 % [30/68] tapahtumista korjautui) ja hyponatremia (13,4 % [64 potilasta]; sen ilmenemiseen kuluneen ajan mediaani oli 55 päivää, ja 51,6 % [33/64] tapahtumista korjautui).
Pediatriset potilaat
Pediatrisista potilaista (< 18-vuotiaat), jotka ovat saaneet erdafitinibiä kliinisissä tutkimuksissa hyväksytystä käyttöaiheesta poiketen ja myyntiluvasta poikkeavalla tavalla (off label -käyttö) myyntiluvan myöntämisen jälkeen, on saatu ilmoituksia kasvun kiihtymisestä ja reisiluun pään epifyseolyysistä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Balversa-yliannokseen ei tunneta spesifistä vasta-ainetta. Yliannoksen yhteydessä lopeta Balversa-hoito ja ryhdy yleisiin tukitoimenpiteisiin, kunnes kliininen toksisuus on vähentynyt tai hävinnyt.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, proteiinikinaasin estäjät. ATC-koodi: L01EN01
Vaikutusmekanismi
Erdafitinibi on fibroblastikasvutekijäreseptorin (FGFR) tyrosiinikinaasin yleisestäjä.
Farmakodynaamiset vaikutukset
Seerumin fosfaattipitoisuus
Erdafitinibi suurentaa seerumin fosfaattipitoisuutta, mikä on FGFR:n estymisestä aiheutuva vaikutus (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Kliininen teho
Balversa-valmisteen tehoa arvioitiin vaiheen 3 satunnaistetun, avoimen, BLC3001-monikeskustutkimuksen kohortissa 1, jossa verrattiin kokonaiselossaoloa erdafitinibin ja solunsalpaajahoidon (dosetakseli tai vinfluniini) välillä potilailla, joilla oli pitkälle edennyt (leikkaukseen soveltumaton tai metastasoitunut) uroteelikarsinooma, jossa oli tietynlaisia FGFR-muutoksia, kun potilaiden syöpä oli edennyt yhden tai kahden aiemman hoidon jälkeen, joista vähintään toinen sisälsi jonkin PD-1-reseptorin (PD‑1) tai PD-1-ligandin (PD-L1) estäjän, jota käytetään paikallisesti edenneen leikkaukseen soveltumattoman tai metastasoituneen taudin hoitoon.
Jos potilas oli saanut solunsalpaajahoitoa neoadjuvantti- tai adjuvanttihoitona tai immuno-onkologista hoitoa ja sairauden oli todettu edenneen 12 kuukauden kuluessa viimeisestä annoksesta, hänen katsottiin saaneen systeemistä hoitoa metastasoituneeseen sairauteen. Potilaita, joilla oli huonossa hoitotasapainossa oleva sydän- ja verisuonitauti edeltävien 3 kuukauden aikana tai 2. asteen tai vaikeampiasteinen (≥ 481 ms) QTc-ajan pidentymä ja haavojen paraneminen oli heikentynyt, ei otettu mukaan tutkimukseen. Tutkimukseen ei myöskään otettu mukaan potilaita, joilla oli minkä tahansa vaikeusasteen sentraalinen seroosi korioretinopatia tai verkkokalvon pigmenttiepiteelin irtauma.
Pääasialliset tehotiedot perustuvat 266 potilaaseen, jotka olivat saaneet aiemmin PD-(L)1:n estäjähoitoa ja jotka oli satunnaistettu saamaan erdafitinibiä (8 mg, jota suurennettiin yksilöllisesti titraamalla 9 mg:aan, jos seerumin fosfaattipitoisuus oli < 9,0 mg/dl (< 2,91 mmol/l) eikä lääkkeeseen liittyvää toksisuutta todettu) tai solunsalpaajahoitoa (75 mg/m2 dosetakselia 3 viikon välein tai 320 mg/m2 vinfluniinia 3 viikon välein).
Tutkimukseen mukaan soveltuneilla potilailla piti olla vähintään yksi seuraavista FGFR-fuusioista: FGFR2-BICC1, FGFR2-CASP7, FGFR3-TACC3, FGFR3-BAIAP2L1. Vaihtoehtoisesti heillä piti olla yksi seuraavista FGFR3-geenimutaatioista: R248C, S249C, G370C, Y373C. Molekulaarinen soveltuvuus selvitettiin keskitettyjen (74,6 %) tai paikallisten (25,4 %) FGFR-tulosten perusteella. Kasvainnäytteistä tutkittiin keskuslaboratoriossa FGFR-geenimuutokset käyttämällä Qiagen Therascreen FGFR RGQ ‑RT-PCR-testikittiä. Aiemmat paikalliset testit kasvain- tai verinäytteistä perustuivat paikalliseen uuden sukupolven sekvensointimenetelmään (NGS). Paikallisen määrityksen perusteella mukaan otetussa pienessä potilasjoukossa, josta oli kasvainnäytteet saatavissa varmistusmääritystä varten, 75,6 %:lla tuloksen havaittiin vastanneen keskitettyä määritystä.
Tutkimuskohortin potilaista 99,2 %:lla oli FGFR-geenimuutoksia (2 potilaalla ei ollut FGFR-muutoksia; 80,8 %:lla potilaista oli FGFR3-mutaatioita, 16,5 %:lla potilaista oli FGFR3-fuusioita ja 1,9 %:lla potilaista oli sekä FGFR3-mutaatioita että ‑fuusioita). Tässä tutkimuskohortissa yhdelläkään potilaalla ei havaittu FGFR2-muutoksia. Kasvaimessa, jossa katsottiin olevan altistavia FGFR3-geenimuutoksia, oli vähintään yksi seuraavista FGFR-fuusioista: FGFR3-TACC3, FGFR3-BAIAP2L1 tai vaihtoehtoisesti yksi seuraavista FGFR3-geenimutaatioista: R248C, S249C, G370C, Y373C. Kaikilla tutkimuskohortin potilailla, joilla oli FGFR-muutoksia, oli vähintään yksi FGFR3-muutos. Vallitsevin (46,6 %) muutos oli FGFR3-S249C ja sen jälkeen vallitsevimpia olivat FGFR3-Y373C (16,9 %) ja FGFR3-TACC3-fuusio (9,8 %).
Demografiset tiedot olivat tasapainossa erdafitinibiä ja solunsalpaajahoitoa saaneiden ryhmien välillä. Iän mediaani tutkimuksen täydellisen seulonnan ajankohtana oli 67 vuotta (vaihteluväli: 32–86 vuotta). Valtaosa potilaista oli 65-vuotiaita tai vanhempia: 19,9 % oli 65–69-vuotiaita, 19,9 % oli 70–74-vuotiaita ja 21,1 % oli 75‑vuotiaita tai vanhempia. Valtaosa potilaista oli miehiä (71,4 %), valkoihoisia (54,1 %) ja eurooppalaisia (60,9 %).
Kaikilla potilailla oli transitiosellulaarinen karsinooma, ja pienellä osalla (5,3 %) potilaista oli histologisten muunnosten vähäisempiä komponentteja (kaikkiaan < 50 %:lla). Kasvaimen ensisijainen sijaintipaikka oli ylemmät virtsatiet 33,5 %:lla potilaista ja alemmat virtsatiet 66,5 %:lla potilaista. Potilaiden lähtötilanteen ECOG-pisteet olivat 0 (42,9 %), 1 (47,7 %) tai 2 (9,4 %).
Kaikki potilaat olivat saaneet aiemmin vähintään yhtä syöpähoitolinjaa, johon piti kuulua jokin PD‑(L)‑1:n estäjä. Yleisimmin saatuja PD‑(L)1:n estäjähoitoja olivat pembrolitsumabi (35,3 %), avelumabi (22,2 %) ja atetsolitsumabi (19,5 %). Aiempaa solunsalpaajahoitoa ei edellytetty, mutta valtaosa potilaista (89,1 %) oli saanut aiemmin vähintään yhden hoitolinjan solunsalpaajia. Lähes kaikki potilaat olivat saaneet platinapohjaista solunsalpaajahoitoa (89,7 % erdafitinibiryhmässä, 85,4 % solunsalpaajaryhmässä); yleisimmin sisplatiinia (55,9 % erdafitinibiryhmässä, 45,4 % solunsalpaajaryhmässä) ja sen jälkeen yleisimmin karboplatiinia (27,2 % erdafitinibiryhmässä, 31,5 % solunsalpaajaryhmässä).
Ensisijainen tehon päätetapahtuma oli kokonaiselossaolo (Overall Survival, OS). Tutkijat arvioivat radiologista vastetta RECIST (Response Evaluation Criteria in Solid Tumours, Versio 1.1) ‑kriteerien mukaisesti, kunnes sairaus eteni, ilmaantui sietämätöntä toksisuutta, suostumus peruttiin, tutkija päätti lopettaa hoidon tai tutkimus päättyi sen mukaan, mikä näistä toteutui ensimmäisenä. Toissijaisia tehon päätetapahtumia olivat etenemättömyysaika (Progression‑Free Survival, PFS), objektiivinen vasteluku (Objective Response Rate, ORR) ja vasteen kesto.
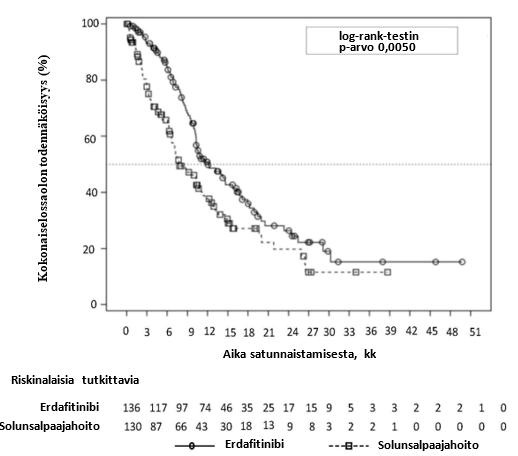
Erdafitinibihoidon osoitettiin pidentävän erdafitinibihoitoa saaneiden potilaiden kokonaiselossaoloa tilastollisesti merkitsevästi solunsalpaajahoitoon verrattuna (kokonaiselossaolon mediaani 12,1 vs 7,8 kuukautta) (ks. taulukko 7).
Yhteenveto tehoa koskevista tuloksista on taulukossa 7.
Taulukko 7. Tehoa koskevat tulokset BLC3001-tutkimuksen kohortissa 1
Erdafitinibi (n = 136) | Solunsalpaajahoito (n = 130) | |
| Kokonaiselossaolo (OS) | ||
| Tapahtumien lukumäärä (%) | 77 (56,6 %) | 78 (60,0 %) |
| Mediaani, kuukautta (95 %:n luottamusväli) | 12,06 (10,28–16,36) | 7,79 (6,54–11,07) |
| Riskitiheyksien suhde (Hazard Ratio, HR) (95 %:n luottamusväli) | 0,64 (0,44–0,93)a 0,0050 | |
| p-arvo | ||
| Etenemättömyysaika (PFS) | ||
| Tapahtumien lukumäärä (%) | 101 (74,3 %) | 90 (69,2 %) |
| Mediaani, kuukautta (95 %:n luottamusväli) | 5,55 (4,40–5,65) | 2,73 (1,81–3,68) |
| Riskitiheyksien suhde (Hazard Ratio, HR) (95 %:n luottamusväli) | 0,58 (0,41–0,82)a 0,0002 | |
| p-arvo | ||
| Objektiivinen vasteluku (ORR), vahvistettu | ||
| ORR (CR + PR) | 48 (35,3 %) | 11 (8,5 %) |
| Vasteen kesto, tutkijan arvioima, vahvistettu | ||
| Mediaani, kuukautta (95 %:n luottamusväli) | 5,55 (4,17–8,31) | 5,75 (4,86–7,16) |
Kaikki raportoidut p-arvot ovat kaksisuuntaisia. a Toistetut luottamusvälit ilmoitettu. | ||
Näiden kahden hoitoryhmän kokonaiselossaolon Kaplan-Meierin käyrä on kuvassa 1.
Kuva 1. Kokonaiselossaolon Kaplan-Meierin kuvaaja – osittamaton analyysi (BLC3001-tutkimuksen kohortti 1)
Iäkkäät potilaat
Kliinisessä Balversa-tutkimuksessa 60,9 % potilaista oli vähintään 65-vuotiaita (39,8 % oli 65- – < 75-vuotiaita ja 21,1 % potilaista oli vähintään 75-vuotiaita). Tehossa ei yleisesti ottaen havaittu eroa iäkkäiden ja nuorempien aikuisten potilaiden välillä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset erdafitinibin käytöstä uroteelikarsinooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdista Annostus ja antotapa ja Haittavaikutukset ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Kerta-annosten ja toistuvan kerran päivässä annon jälkeen erdafitinibialtistus (suurin plasmassa havaittava pitoisuus [Cmax] ja pitoisuus-aikakuvaajan pinta-ala [AUC]) suurenivat koko 0,5–12 mg:n annosvälillä suhteessa annokseen. Vakaa tila saavutettiin kerran päivässä annon yhteydessä 2 viikon jälkeen, ja syöpäpotilailla keskimääräinen kertymissuhde oli 4-kertainen. Annettaessa suositeltua aloitusannosta 8 mg kerran päivässä syöpäpotilaiden keskimääräinen (variaatiokerroin [CV%]) erdafitinibin vakaan tilan Cmax-arvo oli 1 399 ng/ml (50,8 %), AUCτ-arvo oli 29 268 ng∙h/ml (59,9 %) ja plasmassa havaittu pienin pitoisuus (Cmin) oli 936 ng/ml (64,9 %). Plasman erdafitinibipitoisuuden päivittäiset vaihtelut olivat pieniä, sillä kerran päivässä antamisen yhteydessä keskimääräinen (CV%) huippupitoisuuden ja pienimmän pitoisuuden välinen suhde vakaassa tilassa oli 1,47 (23 %).
Imeytyminen
Suun kautta otetun kerta-annoksen jälkeen huippupitoisuuden (tmax) plasmassa saavuttamiseen terveillä vapaaehtoisilla kuluneen ajan mediaani oli 2,5 tuntia (vaihteluväli: 2–6 tuntia), ja oraalinen imeytyminen oli lähes täydellistä.
Ruoan vaikutus
Annettaessa erdafitinibiä terveille vapaaehtoisille paastotilassa ja runsasrasvaisen aterian yhteydessä Cmax- ja AUC-arvoissa ei tapahtunut kliinisesti oleellisia muutoksia. Otettaessa erdafitinibi runsasrasvaisen aterian yhteydessä keskimääräinen AUC∞-arvo pieneni 6 % ja Cmax-arvo pieneni 14 %. Ruokailun yhteydessä ottaminen pidensi huippupitoisuuden saavuttamiseen kuluneen ajan (tmax) mediaania noin 1,5 tuntia (ks. kohta Annostus ja antotapa).
Jakautuminen
Erdafitinibin keskimääräinen laskennallinen jakautumistilavuus syöpäpotilailla oli 0,411 l/kg. Erdafitinibi sitoutui ihmisen plasman proteiineihin 99,7-prosenttisesti, lähinnä happamaan α1-glykoproteiiniin.
Biotransformaatio
Metabolia on erdafitinibin pääasiallinen eliminaatioreitti. Erdafitinibi metaboloituu ihmisellä pääasiassa CYP2C9:n ja CYP3A4:n välityksellä, jolloin muodostuu pääasiallinen O-demetyloitunut metaboliitti. CYP2C9:n osuudeksi erdafitinibin kokonaispuhdistumassa arvioidaan 39 % ja CYP3A4:n osuudeksi 20 %. Pääasiallinen lääkkeeseen liittyvä fraktio plasmassa oli muuttumaton erdafitinibi eikä kiertäviä metaboliitteja todettu.
Eliminaatio
Erdafitinibin laskennallisen kokonaispuhdistuman (CL/F) keskiarvo syöpäpotilailla oli 0,362 l/h.
Erdafitinibin efektiivisen puoliintumisajan keskiarvo syöpäpotilailla oli 58,9 tuntia.
Terveille vapaaehtoisille suun kautta annetun radioaktiivisesti merkityn [14C]‑erdafitinibikerta-annoksen jälkeen 16 päivän kuluessa 69 % annoksesta havaittiin ulosteessa (14–21 % muuttumattomana erdafitinibinä) ja 19 % havaittiin virtsassa (13 % muuttumattomana erdafitinibinä).
Erityispotilasryhmät
Erdafitinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja iän (21–92 vuotta), sukupuolen, etnisen taustan (valkoihoinen, latinalaisamerikkalainen tai aasialainen), painon (36–166 kg), lievän tai keskivaikean munuaisten vajaatoiminnan ja lievän tai keskivaikean maksan vajaatoiminnan perusteella.
Pediatriset potilaat
Erdafitinibin farmakokinetiikkaa ei ole tutkittu pediatrisilla potilailla.
Munuaisten vajaatoiminta
Erdafitinibin farmakokinetiikassa ei havaittu populaatiofarmakokineettisen analyysin perusteella kliinisesti merkittäviä eroja sellaisten tutkittavien välillä, joilla oli normaali munuaisten toiminta (absoluuttinen GFR-MDRD [ruokavalioon liittyvä absoluuttisen glomerulusten suodatusnopeuden muutos munuaissairauden yhteydessä] ≥ 90 ml/min), lievä (absoluuttinen GFR‑MDRD 60–89 ml/min) tai keskivaikea munuaisten vajaatoiminta (absoluuttinen GFR‑MDRD 30–59 ml/min). Vaikeaa munuaisten vajaatoimintaa (absoluuttinen GFR‑MDRD alle 30 ml/min) tai dialyysihoitoa vaativaa munuaisten vajaatoimintaa sairastavista tutkittavista ei ole tietoja farmakokineettisten tietojen vähyyden vuoksi (n = 7, 0,8 %).
Maksan vajaatoiminta
Erdafitinibin farmakokinetiikkaa selvitettiin tutkittavilla, joilla oli ennestään lievä (Child–Pugh-luokka A; n = 8) tai keskivaikea (Child–Pugh-luokka B; n = 8) maksan vajaatoiminta, sekä terveillä verrokeilla, joiden maksan toiminta oli normaali (n = 8). AUC∞-kokonaisarvo oli lievää maksan vajaatoimintaa sairastavilla 82 % ja keskivaikeaa maksan vajaatoimintaa sairastavilla 61 % verrattuna tutkittaviin, joiden maksan toiminta oli normaali. Cmax-kokonaisarvo oli lievää maksan vajaatoimintaa sairastavilla 83 % ja keskivaikeaa maksan vajaatoimintaa sairastavilla 74 % verrattuna tutkittaviin, joiden maksan toiminta oli normaali. Vapaan lääkeaineen AUC∞-arvo oli lievää maksan vajaatoimintaa sairastavilla 95 % ja keskivaikeaa maksan vajaatoimintaa sairastavilla 88 % verrattuna tutkittaviin, joiden maksan toiminta oli normaali. Vapaan lääkeaineen Cmax-arvo oli lievää maksan vajaatoimintaa sairastavilla 96 % ja keskivaikeaa maksan vajaatoimintaa sairastavilla 105 % verrattuna tutkittaviin, joiden maksan toiminta oli normaali. Erdafitinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja niiden tutkittavien välillä, joilla oli lievä (Child–Pugh A) tai keskivaikea (Child–Pugh B) maksan vajaatoiminta tai joilla maksan toiminta oli normaali. Erdafitinibin farmakokinetiikkaa tutkittavilla, joilla on vaikea maksan vajaatoiminta, ei tunneta tietojen vähyyden vuoksi.
Lääkkeiden yhteisvaikutukset
P‑gp:n estäjien vaikutus erdafitinibiin
Erdafitinibi on P‑gp:n substraatti. P‑gp:n estäjät eivät oletettavasti vaikuta kliinisesti oleellisesti erdafitinibin farmakokinetiikkaan.
Mahan happamuutta vähentävien lääkeaineiden vaikutus erdafitinibiin
Erdafitinibin liukoisuus on riittävä pH-arvoilla 1–7,4. Mahan happamuutta vähentävät lääkeaineet (esim. antasidit, H2:n estäjät tai protonipumpun estäjät) eivät oletettavasti vaikuta erdafitinibin biologiseen hyötyosuuteen.
Sevelameerin vaikutus erdafitinibiin
Sevelameeria käyttävillä potilailla ei havaittu kliinisesti merkittäviä eroja erdafitinibin farmakokinetiikassa.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttama toksisuus
Toistuvasta erdafitinibialtistuksesta aiheutuneet pääasialliset toksikologiset havainnot liittyivät sekä rotilla että koirilla erdafitinibin farmakologiseen aktiivisuuteen palautumattomana FGFR:n estäjänä. Niitä olivat mm. plasman suurentunut epäorgaanisen fosforin ja kalsiumin pitoisuus, eri elinten ja kudosten ektooppinen mineralisaatio, sekä luusto-/rustoleesiot erdafitinibialtistuksilla, jotka olivat suositellusta kliinisestä annoksesta ihmiselle aiheutuvaa altistusta pienemmät. Rotilla havaittiin sarveiskalvon atrofiaa (sarveiskalvon epiteelin ohenemista), ja rotilla ja koirilla havaittiin kyynelrauhasten atrofiaa, turkin ja kynsien muutoksia sekä hammasmuutoksia kolmen kuukauden hoidon jälkeen. Rotilla ja koirilla havaittiin fosfaattihomeostaasin häiriöitä altistuksilla, jotka olivat kaikkien tutkittujen annosten yhteydessä ihmisen altistusta pienempiä.
Rottien ja koirien pehmytkudosten mineralisaatio (lukuun ottamatta koiran aortan mineralisaatiota) ja kondroididysplasia sekä rottien nisärauhasdysplasia olivat korjautuneet osittain tai täysin 4 viikon hoidottoman jakson lopussa.
Erdafitinibin ominaisuuksiin kuuluu ihmisen ether-à-go-go-related -geenin (human ether-à-go-go-related gene, hERG) salpaaminen, joka edistää alttiutta sydämen rytmihäiriölle. Tämä ilmeni pitkittyneenä repolarisaationa (korjattu QT-aika), kun nukutetuille koirille ja marsuille oli annettu valmistetta laskimoon ja tajuissaan oleville koirille oli annettu valmistetta suun kautta. Vaikutukseton annostaso tarkoittaa turvallisuusmarginaalia 2,4 suhteessa 9 mg:n annoksesta kerran päivässä aiheutuvaan kliiniseen vakaan tilan huippupitoisuuteen plasmassa (Cmax, u).
Karsinogeenisuus ja mutageenisuus
Erdafitinibin karsinogeenisuuden arvioimiseksi ei ole tehty pitkäkestoisia eläinkokeita. Erdafitinibiä ei katsottu genotoksiseksi hyvän laboratoriokäytännön (GLP) mukaisessa tavanomaisessa genotoksisuusmäärityspaneelissa.
Lisääntymistoksikologia
Erdafitinibi oli rotilla teratogeeninen ja alkiotoksinen altistuksilla, jotka olivat ihmiselle aiheutuvaa altistusta pienempiä. Sikiötoksisuudelle tyypillistä olivat etu-/takajalkojen viat sekä epämuodostumat joissakin suurissa verisuonissa, kuten aortassa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys).
Hedelmällisyys
Erdafitinibillä ei ole tehty varsinaisia hedelmällisyyttä koskevia eläinkokeita. Kolme kuukautta kestäneessä yleistä toksisuutta koskeneessa tutkimuksessa erdafitinibilla todettiin kuitenkin vaikutuksia rottanaaraiden lisääntymiselimiin (keltarauhasen nekroosi) altistuksella, joka vastasi suunnilleen suurinta suositeltua annosta 9 mg kerran päivässä saaneilla potilailla todettua AUC-arvoa.
Farmaseuttiset tiedot
Apuaineet
Balversa 3 mg kalvopäällysteiset tabletit
Tablettiydin
Kroskarmelloosinatrium
Magnesiumstearaatti (E572)
Mannitoli (E421)
Meglumiini
Mikrokiteinen selluloosa (E460)
Kalvopäällyste (Opadry amb II)
Glyserolimonokaprylokapraatti, tyyppi I
Polyvinyylialkoholi, osittain hydrolysoitu
Natriumlauryylisulfaatti
Talkki
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Balversa 4 mg kalvopäällysteiset tabletit
Tablettiydin
Kroskarmelloosinatrium
Magnesiumstearaatti (E572)
Mannitoli (E421)
Meglumiini
Mikrokiteinen selluloosa (E460)
Kalvopäällyste (Opadry amb II)
Glyserolimonokaprylokapraatti, tyyppi I
Polyvinyylialkoholi, osittain hydrolysoitu
Natriumlauryylisulfaatti
Talkki
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Punainen rautaoksidi (E172)
Balversa 5 mg kalvopäällysteiset tabletit
Tablettiydin
Kroskarmelloosinatrium
Magnesiumstearaatti (E572)
Mannitoli (E421)
Meglumiini
Mikrokiteinen selluloosa (E460)
Kalvopäällyste (Opadry amb II)
Glyserolimonokaprylokapraatti, tyyppi I
Polyvinyylialkoholi, osittain hydrolysoitu
Natriumlauryylisulfaatti
Talkki
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Punainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Purkit
4 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BALVERSA tabletti, kalvopäällysteinen
3 mg (L:ei) 84 kpl (9820,55 €)
4 mg (L:ei) 56 kpl (9820,55 €)
5 mg (L:ei) 28 kpl (9820,55 €)
PF-selosteen tieto
Purkki
HDPE (suurtiheyspolyeteeni) ‑purkki, jossa on polypropeeninen turvasuljin ja induktiosinettitiiviste. Yksi kartonkikotelo sisältää yhden purkin, jossa on 28, 56 tai 84 kalvopäällysteistä tablettia.
3 mg:n tabletit:
- Yksi 56 kalvopäällysteisen tabletin kartonkikotelo sisältää yhden 56 tablettia sisältävän purkin.
- Yksi 84 kalvopäällysteisen tabletin kartonkikotelo sisältää yhden 84 tablettia sisältävän purkin.
4 mg:n tabletit:
- Yksi 28 kalvopäällysteisen tabletin kartonkikotelo sisältää yhden 28 tablettia sisältävän purkin.
- Yksi 56 kalvopäällysteisen tabletin kartonkikotelo sisältää yhden 56 tablettia sisältävän purkin.
5 mg:n tabletit:
- Yksi 28 kalvopäällysteisen tabletin kartonkikotelo sisältään yhden 28 tablettia sisältävän purkin.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
3 mg:n tabletit
Keltaisia, pyöreitä, kaksoiskuperia kalvopäällysteisiä tabletteja, joiden läpimitta on 7,6 mm ja joiden toiselle puolelle on kaiverrettu 3 ja vastakkaiselle puolelle EF.
4 mg:n tabletit
Oransseja, pyöreitä, kaksoiskuperia kalvopäällysteisiä tabletteja, joiden läpimitta on 8,1 mm ja joiden toiselle puolelle on kaiverrettu 4 ja vastakkaiselle puolelle EF.
5 mg:n tabletit
Ruskeita, pyöreitä, kaksoiskuperia kalvopäällysteisiä tabletteja, joiden läpimitta on 8,6 mm ja joiden toiselle puolelle on kaiverrettu 5 ja vastakkaiselle puolelle EF.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BALVERSA tabletti, kalvopäällysteinen
3 mg 84 kpl
4 mg 56 kpl
5 mg 28 kpl
- Ylempi erityiskorvaus (100 %). Erdafitinibi: Aikuisten leikkaukseen soveltumattoman tai etäpesäkkeisen uroteelisyövän hoito monoterapiana erityisin edellytyksin (1552).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Erdafitinibi: Aikuisten leikkaukseen soveltumattoman tai etäpesäkkeisen uroteelisyövän hoito monoterapiana erityisin edellytyksin (3099).
ATC-koodi
L01EN01
Valmisteyhteenvedon muuttamispäivämäärä
24.04.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com