
VYLOY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 100 mg, 300 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Vyloy 100 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä varten sisältää 100 mg tsolbetuksimabia.
Vyloy 300 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä varten sisältää 300 mg tsolbetuksimabia.
Käyttökuntoon saattamisen jälkeen yksi millilitra liuosta sisältää 20 mg tsolbetuksimabia.
Tsolbetuksimabi tuotetaan kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla.
Apuaine, jonka vaikutus tunnetaan
Yksi millilitra konsentraattia sisältää 0,21 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Vyloy yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa on tarkoitettu ensilinjan hoidoksi aikuispotilaille, joilla on paikallisesti edennyt, leikkaukseen soveltumaton tai metastaattinen HER2-negatiivinen mahalaukun tai ruokatorvi-mahalaukkurajan adenokarsinooma ja joiden kasvaimet ovat klaudiini (CLDN) 18.2 ‑positiivisia (ks. kohta Annostus ja antotapa).
Ehto
Tämä valmiste on reseptilääke, jonka määräämiseen liittyy rajoitus, ja hoidon saa aloittaa vain syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa. Valmisteen antajan tulee olla terveydenhuollon ammattilainen, jolla on valmius anafylaksian hoitoon, ja ensiapuvälineiden on oltava saatavilla.
Annostus ja antotapa
Hoidon määrää, aloittaa ja sitä valvoo syöpähoitoihin perehtynyt lääkäri. Resurssien tulee olla saatavilla yliherkkyysreaktioiden ja/tai anafylaktisten reaktioiden hoitoon.
Potilasvalinta
Hoitoon soveltuvilla potilailla on oltava CLDN18.2 -positiivinen kasvainstatus määriteltynä niin, että ≥ 75 %:ssa kasvainsoluista näkyy kohtalainen tai voimakas membranoottinen CLDN18:n immunohistokemiallinen värjäys arvioituna kyseiseen käyttötarkoitukseen suunnitellulla, CE-merkinnän saaneella IVD-testillä. Jos CE-merkinnän saanutta IVD-testiä ei ole saatavana, voidaan käyttää jotakin toista validoitua testiä.
Annostus
Ennen antoa
Jos potilaalla esiintyy pahoinvointia ja/tai oksentelua ennen tsolbetuksimabin antoa, oireiden on lievityttävä asteeseen ≤ 1 ennen ensimmäisen infuusion antamista.
Ennen jokaista tsolbetuksimabi-infuusiota potilaille on annettava esilääkityksenä pahoinvointilääkkeitä (esim. NK‑1-reseptorin salpaajia ja 5‑HT3-reseptorin salpaajia sekä muita lääkevalmisteita tarpeen mukaan).
Esilääkitys pahoinvointilääkkeillä on tärkeää, sillä pahoinvoinnin ja oksentelun hallitseminen auttaa välttämään tsolbetuksimabihoidon varhaista lopettamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Myös esilääkitystä systeemisillä kortikosteroidella voi harkita paikallisten hoito-ohjeiden mukaan, etenkin ennen ensimmäistä tsolbetuksimabi-infuusiota.
Suositeltu annos
Taulukossa 1 luetellut tsolbetuksimabin suositellut lataus- ja ylläpitoannokset tulee laskea kehon pinta-alan (body surface area, BSA) mukaan.
| Taulukko 1. Kehon pinta-alaan perustuva suositeltu tsolbetuksimabiannos | |||
| Yksi latausannos | Ylläpitoannokset | Hoidon kesto | |
Sykli 1 päivänä 1a, 800 mg/m2 laskimoon Anna tsolbetuksimabi yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa (ks. kohta Farmakodynamiikka).b | Alkaen 3 viikkoa yhden latausannoksen jälkeen 600 mg/m2 laskimoon 3 viikon välein tai Alkaen 2 viikkoa yhden latausannoksen jälkeen 400 mg/m2 laskimoon 2 viikon välein Anna tsolbetuksimabi yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa (ks. kohta Farmakodynamiikka).b | Sairauden etenemiseen tai ei-hyväksyttävään toksisuuteen asti. | |
a. Tsolbetuksimabihoitosyklin kesto määräytyy samanaikaisesti käytettävän kemoterapian mukaan (ks. kohta Farmakodynamiikka).
b. Katso kemoterapian annostusohjeet fluoropyrimidiinin tai platinaa sisältävän kemoterapian valmistetiedoista.
Annosmuutokset
Tsolbetuksimabiannoksen pienentämistä ei suositella. Tsolbetuksimabin haittavaikutukset hoidetaan pienentämällä infuusionopeutta, keskeyttämällä infuusio ja/tai lopettamalla hoito taulukon 2 ohjeistuksen mukaisesti.
Taulukko 2. Tsolbetuksimabin annosmuutokset
| Haittavaikutus | Vaikeusastea | Annosmuutos |
| Yliherkkyysreaktiot | Anafylaktinen reaktio, epäilty anafylaksia, aste 3 tai 4 | Lopeta infuusio välittömästi ja lopeta hoito pysyvästi. |
| Aste 2 | Keskeytä infuusio, kunnes haittavaikutus on lievittynyt asteeseen ≤ 1, ja jatka sitten pienemmällä infuusionopeudellab infuusion loppuun asti. Anna potilaalle esilääkityksenä antihistamiineja seuraavan infuusion yhteydessä, ja anna infuusio taulukossa 3 kuvatulla infuusionopeudella. | |
| Infuusioon liittyvä reaktio | Aste 3 tai 4 | Lopeta infuusio välittömästi ja lopeta hoito pysyvästi. |
| Aste 2 | Keskeytä infuusio, kunnes haittavaikutus on lievittynyt asteeseen ≤ 1, ja jatka sitten pienemmällä infuusionopeudellab infuusion loppuun asti. Anna potilaalle esilääkityksenä antihistamiineja seuraavan infuusion yhteydessä, ja anna infuusio taulukossa 3 kuvatulla infuusionopeudella. | |
| Pahoinvointi | Aste 2 tai 3 | Keskeytä infuusio, kunnes haittavaikutus on lievittynyt asteeseen ≤ 1, ja jatka sitten pienemmällä infuusionopeudellab infuusion loppuun asti. Anna seuraava infuusio taulukossa 3 kuvatulla infuusionopeudella. |
| Oksentelu | Aste 4 | Lopeta hoito pysyvästi. |
| Aste 2 tai 3 | Keskeytä infuusio, kunnes haittavaikutus on lievittynyt asteeseen ≤ 1, ja jatka sitten pienemmällä infuusionopeudellab infuusion loppuun asti. Anna seuraava infuusio taulukossa 3 kuvatulla infuusionopeudella. | |
a. Toksisuus luokiteltiin National Cancer Institute Common Terminology Criteria for Adverse Events -luokituksen version 4.03 (NCI-CTCAE v4.03) mukaan, missä aste 1 on lievä, aste 2 keskivaikea, aste 3 vaikea ja aste 4 henkeä uhkaava. b. Lääkäri määrittää pienemmän infuusionopeuden kliinisellä arviolla potilaan sietokyvyn, toksisuuden vakavuuden ja potilaan aiemmin sietämän infuusionopeuden perusteella (ks. potilaan seurantaa koskevat suositukset kohdasta Varoitukset ja käyttöön liittyvät varotoimet). | ||
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa ≥ 65‑vuotiaille potilaille (ks. kohta Farmakokinetiikka). Tsolbetuksimabihoitoa saaneista vähintään 75‑vuotiaista potilaista on vain vähän tietoa.
Munuaisten vajaatoiminta
Lievää (kreatiniinipuhdistuma [CrCL] ≥ 60 – < 90 ml/min) tai keskivaikeaa (CrCL ≥ 30 – < 60 ml/min) munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Vaikeaa munuaisten vajaatoimintaa (CrCL ≥ 15 – < 30 ml/min) sairastaville potilaille ei ole laadittu annossuosituksia (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa (kokonaisbilirubiini [TB] ≤ viitealueen yläraja [ULN] ja aspartaattiaminotransferaasi [ASAT] > ULN, tai TB > 1–1,5 × ULN ja ASAT mikä tahansa) sairastavien potilaiden annosta ei tarvitse muuttaa. Keskivaikeaa (TB > 1,5–3 × ULN ja ASAT mikä tahansa) tai vaikeaa (TB > 3–10 × ULN ja ASAT mikä tahansa) maksan vajaatoimintaa sairastaville potilaille ei ole laadittu annossuosituksia (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää tsolbetuksimabia pediatrisille potilaille gastrisen tai gastroesofageaalisen junktion adenokarsinooman hoitoon.
Antotapa
Tsolbetuksimabi annetaan laskimoon. Suositeltu annos annetaan vähintään 2 tuntia kestävänä laskimoinfuusiona. Lääkevalmistetta ei saa antaa nopeana laskimoinfuusiona tai bolusinjektiona.
Jos tsolbetuksimabi ja fluoropyrimidiiniä ja platinaa sisältävä kemoterapia annetaan samana päivänä, tsolbetuksimabi on annettava ensimmäisenä.
Mahdollisten haittavaikutusten vähentämiseksi on suositeltavaa aloittaa kukin infuusio pienemmällä antonopeudella ensimmäisten 30–60 minuutin ajan, minkä jälkeen nopeutta voidaan lisätä vähitellen siedettävyyden mukaan (ks. taulukko 3).
Jos infuusioaika ylittää suositellun säilytysajan huoneenlämmössä (≤ 25 °C 8 tuntia infuusioliuoksen valmistelun päättymisestä), infuusiopussi on hävitettävä ja uusi infuusiopussi on valmisteltava infuusion jatkamista varten (suositellut säilytysajat, ks. kohta Kestoaika).
Taulukko 3. Kullekin tsolbetuksimabi-infuusiolle suositellut infuusionopeudet
| Tsolbetuksimabiannos | Infuusionopeus | ||
| Ensimmäiset 30‑60 minuuttia | Jäljellä oleva infuusioaikab | ||
| Yksi latausannos (sykli 1, päivä 1)a | 800 mg/m2 | 75 mg/m2/h | 150–300 mg/m2/h |
| Ylläpitoannokset | 600 mg/m2 3 viikon välein | 75 mg/m2/h | 150–300 mg/m2/h |
| tai | tai | tai | |
| 400 mg/m2 2 viikon välein | 50 mg/m2/h | 100–200 mg/m2/h | |
a. Tsolbetuksimabihoitosyklin kesto määräytyy samanaikaisesti käytettävän kemoterapian mukaan (ks. kohta Farmakodynamiikka).
b. Jos 30–60 minuutin kuluessa ei ilmene haittavaikutuksia, infuusionopeutta voidaan suurentaa siedettävyyden mukaan.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyysreaktiot
Kliinisissä tutkimuksissa tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa saaneilla potilailla ilmeni yliherkkyysreaktioita, kuten anafylaktisia reaktioita ja lääkeyliherkkyyttä (ks. kohta Haittavaikutukset).
Potilaita on seurattava tsolbetuksimabi-infuusion aikana ja sen jälkeen (vähintään 2 tuntia tai pidempään, jos se on kliinisesti aiheellista) sellaisten yliherkkyysreaktioiden varalta, joiden merkit ja oireet viittaavat voimakkaasti anafylaksiaan (nokkosihottuma, toistuva yskä, hengityksen vinkuminen ja puristava tunne kurkussa/äänen muutokset).
Yliherkkyysreaktiot on hoidettava taulukossa 2 annettujen, annosmuutoksia koskevien suositusten mukaisesti.
Infuusioon liittyvät reaktiot
Infuusioon liittyviä reaktioita on esiintynyt kliinisissä tutkimuksissa potilailla, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa (ks. kohta Haittavaikutukset).
Potilaita on seurattava infuusioon liittyvien reaktioiden merkkien ja oireiden varalta, joita ovat esimerkiksi pahoinvointi, oksentelu, vatsakipu, syljen liikaeritys, kuume, epämukava tunne rintakehässä, vilunväristykset, selkäkipu, yskä ja hypertensio. Nämä merkit ja oireet yleensä korjaantuvat, kun infuusio keskeytetään.
Infuusioon liittyvät reaktiot on hoidettava taulukossa 2 annettujen, annosmuutoksia koskevien suositusten mukaisesti.
Pahoinvointi ja oksentelu
Kliinisissä tutkimuksissa pahoinvointi ja oksentelu olivat useimmin todettuja ruoansulatuskanavan haittavaikutuksia potilailla, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa (ks. kohta Haittavaikutukset).
Pahoinvoinnin ja oksentelun ehkäisemiseksi suositellaan pahoinvointilääkkeiden antamista esilääkityksenä ennen kutakin tsolbetuksimabi-infuusiota (ks. kohta Annostus ja antotapa).
Potilaita on seurattava infuusion aikana ja sen jälkeen, ja heille on annettava tavanomaista hoitoa, kuten pahoinvointilääkkeitä tai nesteytystä, kliinisen tarpeen mukaan.
Pahoinvointi ja oksentelu on hoidettava taulukossa 2 annettujen, annosmuutoksia koskevien suositusten mukaisesti.
Riskienhallintatoimet ennen tsolbetuksimabihoidon aloittamista
Ennen kuin tsolbetuksimabia annetaan yhdistelmähoitona fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa, hoidon määräävän lääkärin on arvioitava potilaan riskit ruoansulatuskanavaan kohdistuvan toksisuuden osalta. Tsolbetuksimabi- ja/tai kemoterapia-altistuksen heikkenemisen riskin pienentämiseksi on tärkeää pyrkiä ennaltaehkäisevästi hallitsemaan pahoinvointia ja oksentelua.
Pahoinvoinnin ja oksentelun ehkäisemiseksi suositellaan pahoinvointilääkkeiden antamista esilääkityksenä ennen kutakin tsolbetuksimabi-infuusiota. On tärkeää seurata potilaita tarkasti infuusion aikana ja hallita ruoansulatuskanavaan kohdistuvaa toksisuutta keskeyttämällä infuusio ja/tai pienentämällä infuusionopeutta vakavien haittavaikutusten ja varhaisten hoidon lopettamisten riskin minimoimiseksi. Infuusion aikana ja sen jälkeen potilaita on seurattava ja hoidettava antaen tavanomaista hoitoa, mukaan lukien antiemeettiset lääkkeet tai nesteen korvaaminen kliinisen tarpeen mukaan.
Kliinisistä tutkimuksista pois suljetut potilaat
Potilaat suljettiin pois kliinisistä tutkimuksista, jos heillä oli täydellinen tai osittainen mahalaukun ulosvirtauskanavan oireyhtymä, positiivinen ihmisen immuunikatovirustesti (HIV-testi) tai tiedossa oleva aktiivinen B- tai C‑hepatiitti, merkittävä sydän- ja verisuonitauti (esim. luokan III tai IV kongestiivinen sydämen vajaatoiminta New York Heart Association -luokituksen mukaan, aiempia merkittäviä kammioperäisiä rytmihäiriöitä, QTc-aika > 450 ms miehillä ja > 470 ms naisilla) tai aiempia keskushermoston metastaaseja.
Tietoa apuaineista
Tämä lääke sisältää 1,05 mg polysorbaatti 80:tä yhdessä 100 mg:n injektiopullossa ja 3,15 mg polysorbaatti 80:tä yhdessä 300 mg:n injektiopullossa. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Tämä lääke ei sisällä natriumia, mutta tsolbetuksimabin laimentamiseen ennen antoa käytetään natriumkloridi-infuusioliuosta, jonka pitoisuus on 9 mg/ml (0,9‑prosenttista), mikä tulee ottaa huomioon potilaan päivittäisessä natriumin saannissa.
Yhteisvaikutukset
Tsolbetuksimabista ei ole tehty virallisia farmakokineettisiä yhteisvaikutustutkimuksia. Koska tsolbetuksimabi puhdistuu verenkierrosta katabolisesti, sillä ei odoteta olevan metabolisia yhteisvaikutuksia muiden lääkkeiden kanssa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, on neuvottava käyttämään varmuuden vuoksi tehokasta ehkäisyä raskauden ehkäisemiseksi hoidon aikana.
Raskaus
Tsolbetuksimabin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä lisääntymis- ja kehitystutkimuksissa, joissa tsolbetuksimabia annettiin tiineille hiirille laskimoon organogeneesin aikana, ei todettu haittavaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Tsolbetuksimabia saa antaa raskaana oleville naisille vain, jos hoidon hyödyt ovat mahdollisia riskejä suuremmat.
Imetys
Tietoa ei ole saatavilla tsolbetuksimabin erittymisestä äidinmaitoon ihmisillä, vaikutuksista imetettävään vauvaan eikä vaikutuksista maidontuotantoon. Koska vasta-aineiden tiedetään voivan erittyä äidinmaitoon ihmisillä, ja koska imetettävään vauvaan kohdistuvat haittavaikutukset saattavat olla vakavia, imetystä ei suositella tsolbetuksimabihoidon aikana.
Hedelmällisyys
Tsolbetuksimabin vaikutusta hedelmällisyyteen arvioivia tutkimuksia ei ole tehty. Näin ollen tsolbetuksimabin vaikutusta miesten ja naisten hedelmällisyyteen ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tsolbetuksimabilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Yleisimpiä tsolbetuksimabihoidon yhteydessä todettuja haittavaikutuksia olivat pahoinvointi (77,2 %), oksentelu (66,9 %), vähentynyt ruokahalu (42 %), neutropenia (30,7 %), pienentynyt neutrofiilimäärä (28,4 %), painonlasku (21,9 %), kuume (17,4 %), hypoalbuminemia (17,1 %), perifeerinen edeema (13,9 %), hypertensio (9 %), dyspepsia (7,8 %), vilunväristykset (5,2 %), syljen liikaeritys (3,8 %), infuusioon liittyvä reaktio (3,2 %) ja lääkeyliherkkyys (1,6 %).
Vakavia haittavaikutuksia esiintyi 45 %:lla tsolbetuksimabihoitoa saaneista potilaista. Yleisimpiä vakavia haittavaikutuksia olivat oksentelu (6,8 %) pahoinvointi (4,9 %) ja vähentynyt ruokahalu (1,9 %).
20 % potilaista lopetti tsolbetuksimabihoidon pysyvästi haittavaikutusten vuoksi; yleisimpiä lääkkeenannon lopettamiseen johtaneita haittavaikutuksia olivat oksentelu (3,8 %) ja pahoinvointi (3,3 %).
Tsolbetuksimabin annon keskeyttämiseen johtaneita haittavaikutuksia esiintyi 60,9 %:lla potilaista; yleisimpiä lääkkeenannon keskeyttämiseen johtaneita haittavaikutuksia olivat oksentelu (26,6 %), pahoinvointi (25,5 %), neutropenia (9,8 %), pienentynyt neutrofiilimäärä (5,9 %), hypertensio (3,2 %), vilunväristykset (2,2 %), infuusioon liittyvä reaktio (1,6 %), vähentynyt ruokahalu (1,6 %) ja dyspepsia (1,1 %).
Taulukkomuotoinen luettelo haittavaikutuksista
Haittavaikutusten yleisyydet perustuvat kahteen vaiheen 2 tutkimukseen ja kahteen vaiheen 3 tutkimukseen 631 potilaalla, jotka saivat vähintään yhden latausannoksen 800 mg/m2 tsolbetuksimabia ja sen jälkeen ylläpitoannoksia 600 mg/m2 3 viikon välein yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa. Tsolbetuksimabialtistuksen mediaanikesto oli 174 vuorokautta (vaihteluväli: 1–1 791 vuorokautta).
Kliinisissä tutkimuksissa todetut haittavaikutukset on lueteltu tässä kohdassa yleisyyden mukaan. Yleisyysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kussakin yleisyysluokassa haittavaikutukset on esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 4. Haittavaikutukset
|
Valikoitujen haittavaikutusten kuvaukset
Yliherkkyysreaktiot
Integroidussa turvallisuusanalyysissa kaikenasteisia anafylaktisia reaktioita ja lääkeyliherkkyyttä esiintyi tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa saaneilla potilailla yleisyydellä 0,5 % ja vastaavasti 1,6 %.
Vaikeita (asteen 3) anafylaktisia reaktioita esiintyi 0,5 %:lla ja vaikeaa lääkeyliherkkyyttä 0,2 %:lla potilaista, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa.
Anafylaktinen reaktio johti tsolbetuksimabihoidon pysyvään lopettamiseen 0,3 %:lla potilaista. Lääkeyliherkkyys johti tsolbetuksimabin annon keskeyttämiseen 0,3 %:lla potilaista.
Tsolbetuksimabin tai fluoropyrimidiiniä ja platinaa sisältävän kemoterapian infuusionopeutta pienennettiin 0,2 %:lla potilaista lääkeyliherkkyyden takia.
Infuusioon liittyvä reaktio
Integroidussa turvallisuusanalyysissa kaikenasteisia infuusioon liittyviä reaktioita esiintyi 3,2 %:lla potilaista, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa.
Vaikeita (asteen 3) infuusioon liittyviä reaktioita esiintyi 0,5 %:lla potilaista, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa.
Infuusioon liittyvä reaktio johti tsolbetuksimabihoidon pysyvään lopettamiseen 0,5 %:lla potilaista ja lääkkeenannon keskeyttämiseen 1,6 %:lla potilaista. Tsolbetuksimabin tai fluoropyrimidiiniä ja platinaa sisältävän kemoterapian infuusionopeutta pienennettiin 0,3 %:lla potilaista infuusioon liittyvän reaktion takia.
Pahoinvointi ja oksentelu
Integroidussa turvallisuusanalyysissa kaikenasteista pahoinvointia esiintyi 77,2 %:lla ja kaikenasteista oksentelua 66,9 %:lla potilaista, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa. Pahoinvointia ja oksentelua esiintyi useammin ensimmäisen hoitosyklin aikana ja harvemmin myöhempien hoitosyklien aikana. Mediaaniaika sekä pahoinvoinnin että oksentelun alkamiseen oli 1 vuorokausi potilailla, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa. Pahoinvoinnin mediaanikesto oli 3 vuorokautta ja oksentelun mediaanikesto 1 vuorokausi potilailla, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa.
Vaikeaa (asteen 3) pahoinvointia esiintyi 11,6 %:lla ja vaikeaa oksentelua 13,6 %:lla potilaista, jotka saivat tsolbetuksimabia yhdessä fluoropyrimidiiniä ja platinaa sisältävän kemoterapian kanssa.
Pahoinvointi johti tsolbetuksimabihoidon pysyvään lopettamiseen 3,3 %:lla potilaista ja lääkkeenannon keskeyttämiseen 25,5 %:lla potilaista. Oksentelu johti tsolbetuksimabihoidon pysyvään lopettamiseen 3,8 %:lla potilaista ja lääkkeenannon keskeyttämiseen 26,6 %:lla potilaista. Tsolbetuksimabin tai fluoropyrimidiiniä ja platinaa sisältävän kemoterapian infuusionopeutta pienennettiin 9,7 %:lla potilaista pahoinvoinnin takia ja 7,8 %:lla potilaista oksentelun takia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksessa potilaan vointia on seurattava tiiviisti haittavaikutusten varalta, ja tukihoitoa on annettava tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset aineet, muut monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ATC-koodi: L01FX31
Vaikutusmekanismi
Tsolbetuksimabi on kimeerinen (hiiren/ihmisen IgG1) monoklonaalinen vasta-aine, jonka vaikutus kohdistuu tiiviin liitoksen molekyyliin CLDN18.2. Ei-kliiniset tiedot viittaavat siihen, että tsolbetuksimabi sitoutuu selektiivisesti solulinjoihin, jotka on transfektoitu CLDN18.2:lla tai jotka ilmentävät endogeenisesti CLDN18.2:ta. Tsolbetuksimabi näivettää CLDN18.2-positiiviset solut vasta-aineesta riippuvaisen soluvälitteisen sytotoksisuuden (ADCC) ja komplementista riippuvaisen sytotoksisuuden (CDC) kautta. Sytotoksisten lääkevalmisteiden osoitettiin lisäävän CLDN18.2:n ilmentymistä ihmisen syöpäsoluissa ja parantavan tsolbetuksimabin indusoimaa ADCC- ja CDC-aktiivisuutta.
Farmakodynaamiset vaikutukset
Tehoa ja turvallisuutta koskevien altistus-vasteanalyysien perusteella tsolbetuksimabiannosten 800/400 mg/m2 2 viikon välein ja 800/600 mg/m2 3 viikon välein tehossa ja turvallisuudessa ei odoteta olevan kliinisesti merkittäviä eroja potilailla, joilla on paikallisesti edennyt, leikkauskelvoton tai metastaattinen HER2-negatiivinen gastrinen tai gastroesofageaalisen junktion adenokarsinooma ja joiden kasvaimet ovat CLDN18.2-positiivisia.
Immunogeenisuus
Kahdesta vaiheen 3 tutkimuksesta saatujen tietojen yhdistetyn analyysin perusteella immunogeenisuuden yleinen ilmaantuvuus oli 9,5 % (46 potilasta sai positiivisen tuloksen lääkevasta-ainetestistä [ADA] yhteensä 485 potilaasta, joille annettiin 800/600 mg/m2 tsolbetuksimabia 3 viikon välein yhdessä mFOLFOX6/CAPOX-hoidon kanssa). Neutraloivien vasta-aineiden ilmaantuvuus oli 3,3 % (16 potilasta yhteensä 485 potilaasta). Koska lääkevasta-aineiden ilmaantuvuus oli niin pieni, näiden vasta-aineiden vaikutus tsolbetuksimabin farmakokinetiikkaan, turvallisuuteen ja/tai tehoon on epävarma.
Kliininen teho ja turvallisuus
Gastrinen tai gastroesofageaalisen junktion adenokarsinooma
SPOTLIGHT (8951-CL-0301) ja GLOW (8951-CL-0302)
Tsolbetuksimabin ja kemoterapian yhdistelmän turvallisuutta ja tehoa arvioitiin kahdessa vaiheen 3 kaksoissokkoutetussa, satunnaistetussa monikeskustutkimuksissa, joihin otetuilla 1 072 potilaalla oli paikallisesti edennyt, leikkauskelvoton tai metastaattinen gastrinen tai gastroesofageaalisen junktion adenokarsinooma ja joiden kasvaimet olivat CLDN18.2-positiivisia ja HER2-negatiivisia. CLDN18.2-positiivisuus (määritelmän mukaan ≥ 75 %:ssa kasvainsoluista oli nähtävissä kohtalainen tai voimakas membranoottinen CLDN18:n värjäys) määritettiin immunohistokemiallisesti mahalaukun tai gastroesofageaalisen junktion kasvainkudosnäytteestä kaikilta potilailta, ja VENTANA CLDN18 (43-14A) RxDx -määritys tehtiin keskuslaboratoriossa.
Potilaat satunnaistettiin suhteessa 1:1 saamaan joko tsolbetuksimabia yhdessä kemoterapian kanssa (n = 283 SPOTLIGHT-tutkimuksessa, n = 254 GLOW-tutkimuksessa) tai lumelääkettä yhdessä kemoterapian kanssa (n = 282 SPOTLIGHT-tutkimuksessa, n = 253 GLOW-tutkimuksessa). Tsolbetuksimabi annettiin laskimoon seuraavasti: latausannos 800 mg/m2 (syklin 1 päivänä 1) ja sen jälkeen ylläpitoannoksena 600 mg/m2 3 viikon välein yhdessä joko mFOLFOX6-hoidon (oksaliplatiini, foliinihappo ja fluorourasiili) tai CAPOX-hoidon (oksaliplatiini ja kapesitabiini) kanssa.
SPOTLIGHT-tutkimukseen osallistuneille potilaille annettiin 1–12 mFOLFOX6-hoitokertaa (85 mg/m2 oksaliplatiinia, 400 mg/m2 foliinihappoa (leukovoriinia tai paikallista vastinetta), 400 mg/m2 fluorourasiilia boluksena ja 2 400 mg/m2 fluorourasiilia jatkuvana infuusiona) 42 vuorokauden pituisen syklin päivinä 1, 15 ja 29. Kahdentoista hoitokerran jälkeen potilaat saivat jatkaa hoitoa tsolbetuksimabilla, 5‑fluorourasiililla ja foliinihapolla (leukovoriinilla tai paikallisella vastineella) tutkijan harkinnan mukaan taudin etenemiseen tai ei-hyväksyttävään toksisuuteen asti.
GLOW-tutkimukseen osallistuneille potilaille annettiin 1–8 CAPOX-hoitokertaa 21 vuorokauden pituisen syklin päivänä 1 (130 mg/m2 oksaliplatiinia) ja päivinä 1–14 (1 000 mg/m2 kapesitabiinia). Kahdeksan oksaliplatiinihoitokerran jälkeen potilaat saivat jatkaa hoitoa tsolbetuksimabilla ja kapesitabiinilla tutkijan harkinnan mukaan taudin etenemiseen tai ei-hyväksyttävään toksisuuteen asti.
Lähtötilanteen ominaisuudet olivat tutkimuksissa yleisesti ottaen samankaltaiset lukuun ottamatta aasialaisten potilaiden osuutta muihin kuin aasialaisiin potilaisiin verrattuna.
SPOTLIGHT-tutkimuksessa mediaani-ikä oli 61 vuotta (vaihteluväli: 20–86), 62 % tutkittavista oli miehiä, 53 % oli kaukaasialaisia, 38 % oli aasialaisia; 31 % oli kotoisin Aasiasta ja 69 % muualta kuin Aasiasta. Potilaiden Eastern Cooperative Oncology Group (ECOG) -suorituskyky oli lähtötilanteessa 0 (43 %) tai 1 (57 %). Potilaiden kehon pinta-ala oli keskimäärin 1,7 m2 (vaihteluväli: 1,1–2,5). Mediaaniaika diagnoosin saamisesta oli 56 vuorokautta (vaihteluväli: 2–5 366), 36 % kasvaimista oli diffuusia tyyppiä, 24 % oli suoliston kasvaimia, 76 %:lla potilaista oli gastrinen adenokarsinooma, 24 %:lla oli gastroesofageaalisen junktion adenokarsinooma, 16 %:lla oli paikallisesti edennyt tauti ja 84 %:lla oli metastaattinen tauti.
GLOW-tutkimuksessa mediaani-ikä oli 60 vuotta (vaihteluväli: 21–83), 62 % tutkittavista oli miehiä, 37 % oli kaukaasialaisia, 63 % oli aasialaisia; 62 % oli kotoisin Aasiasta ja 38 % muualta kuin Aasiasta. Potilaiden ECOG-suorituskyky oli lähtötilanteessa 0 (43 %) tai 1 (57 %). Potilaiden kehon pinta-ala oli keskimäärin 1,7 m2 (vaihteluväli: 1,1–2,3). Mediaaniaika diagnoosin saamisesta oli 44 vuorokautta (vaihteluväli: 2–6 010), 37 % kasvaimista oli diffuusia tyyppiä, 15 % oli suoliston kasvaimia, 84 %:lla potilaista oli gastrinen adenokarsinooma, 16 %:lla oli gastroesofageaalisen junktion adenokarsinooma, 12 %:lla oli paikallisesti edennyt tauti ja 88 %:lla oli metastaattinen tauti.
Ensisijainen tehotulos oli taudin etenemättömyysaika (progression-free survival, PFS), jonka riippumaton arviointilautakunta arvioi RECIST v1.1 -kriteerien mukaisesti. Tärkein toissijainen tehotulos oli kokonaiselinaika (overall survival, OS). Muita toissijaisia tehotuloksia olivat objektiivinen vasteprosentti (objective response rate, ORR) ja vasteen kesto (duration of response, DOR), jotka riippumaton arviointilautakunta arvioi RECIST v1.1 -kriteerien mukaisesti.
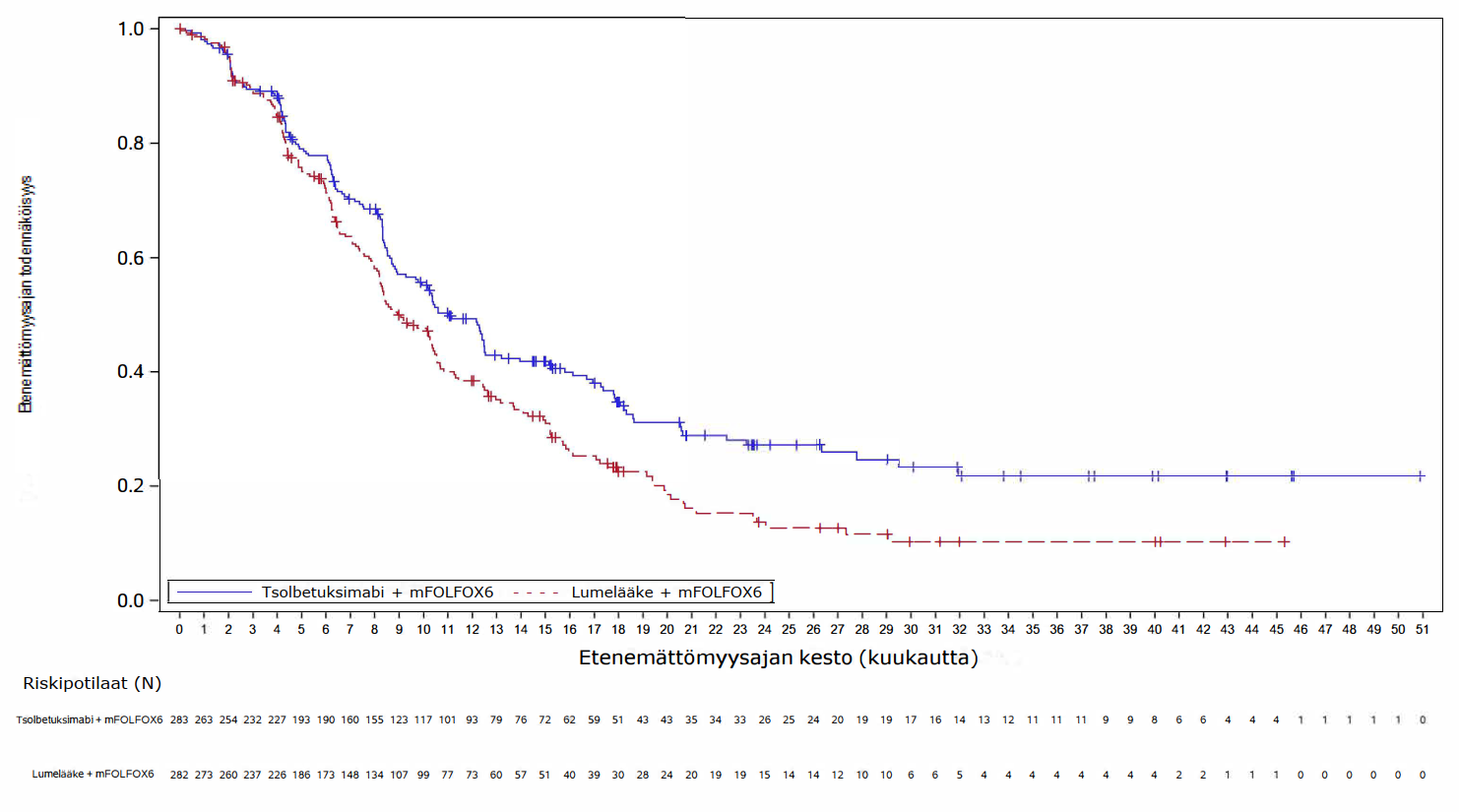
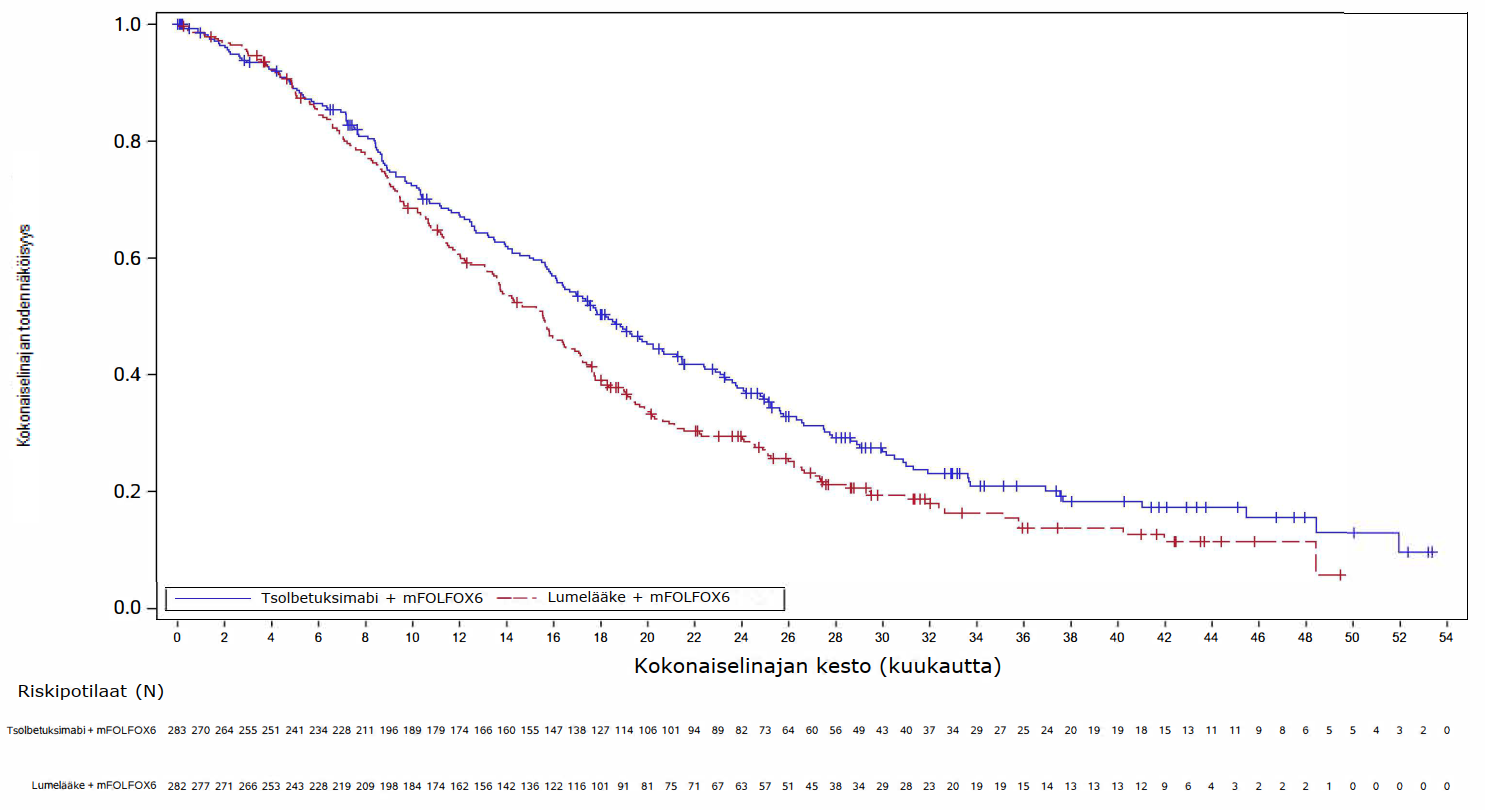
Ensisijaisessa analyysissä (lopullinen PFS-analyysi ja OS-välianalyysi) SPOTLIGHT-tutkimuksessa tsolbetuksimabin ja mFOLFOX6-hoidon yhdistelmää saaneilla potilailla todettiin tilastollisesti merkitsevää hyötyä PFS:n suhteen (riippumattoman arviointilautakunnan arvioimana) ja OS:n suhteen verrattuna potilaisiin, jotka saivat lumelääkkeen ja mFOLFOX6-hoidon yhdistelmää. PFS:n riskisuhde oli 0,751 (95 %:n luottamusväli: 0,598; 0,942; 1‑puolinen P = 0,0066) ja OS:n riskisuhde oli 0,750 (95 %:n luottamusväli: 0,601; 0,936; 1‑puolinen P = 0,0053).
Päivitetty PFS-analyysi ja lopullinen OS-analyysi SPOTLIGHT-tutkimuksesta on esitetty taulukossa 5 ja Kaplan-Meierin käyrät kuvissa 1-2.
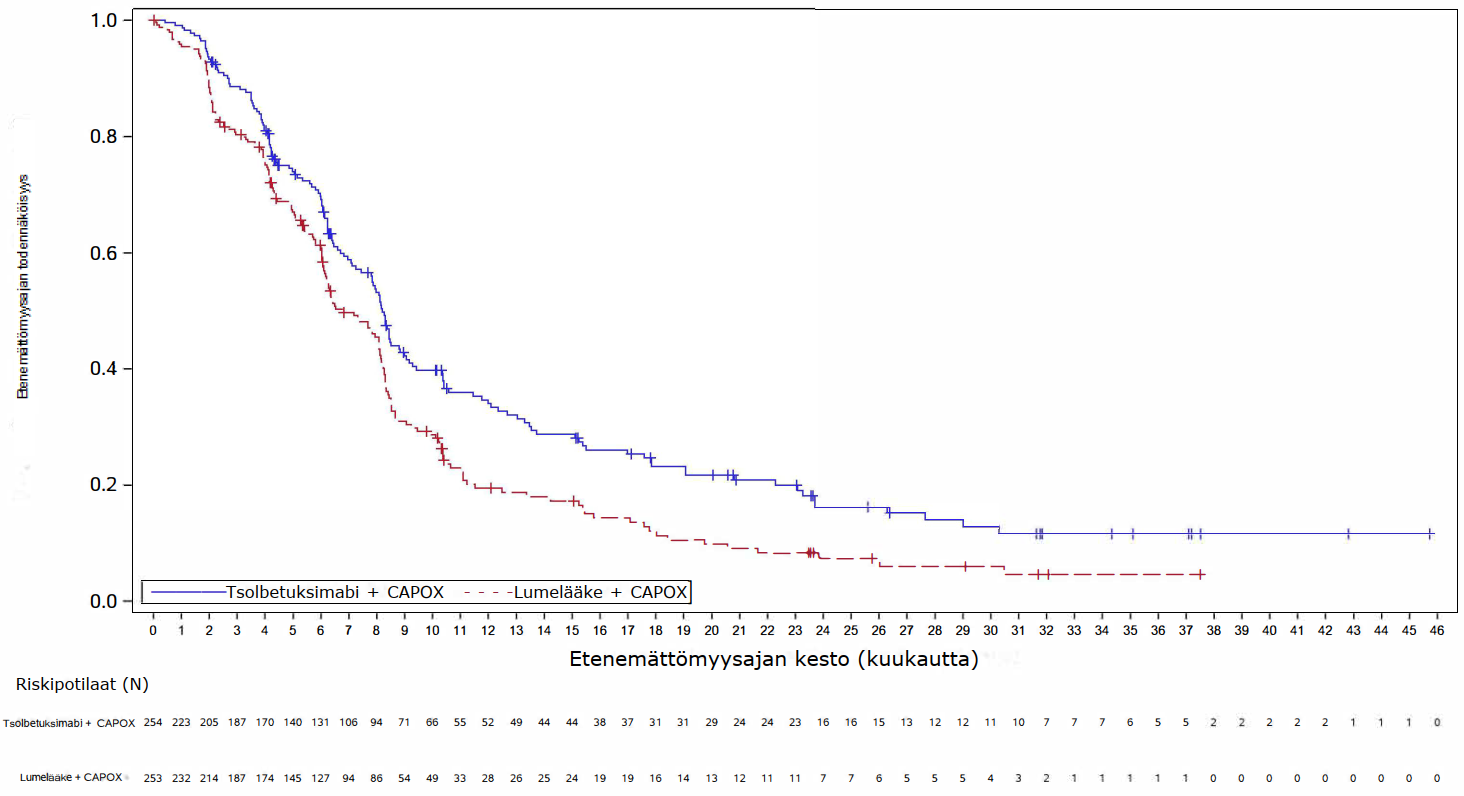
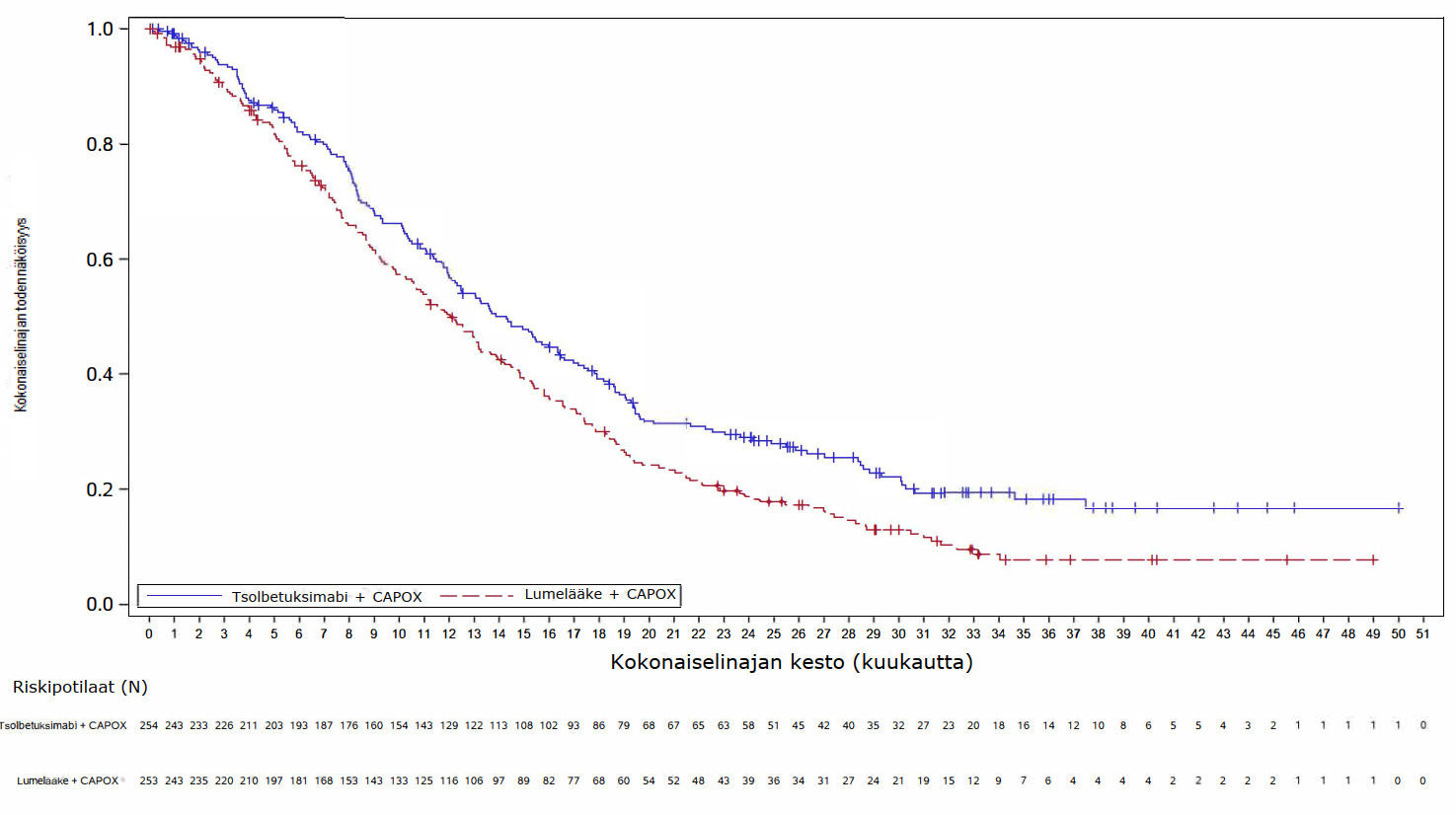
Ensisijaisessa analyysissä (lopullinen PFS-analyysi ja OS-välianalyysi) GLOW-tutkimuksessa tsolbetuksimabin ja CAPOX-hoidon yhdistelmää saaneilla potilailla todettiin tilastollisesti merkitsevää hyötyä PFS:n suhteen (riippumattoman arviointilautakunnan arvioimana) ja OS:n suhteen verrattuna potilaisiin, jotka saivat lumelääkkeen ja CAPOX-hoidon yhdistelmää. PFS:n riskisuhde oli 0,687 (95 %:n luottamusväli: 0,544; 0,866; 1‑puolinen P = 0,0007) ja OS:n riskisuhde oli 0,771 (95 %:n luottamusväli: 0,615; 0,965; 1‑puolinen P = 0,0118).
Päivitetty PFS-analyysi ja lopullinen OS-analyysi GLOW-tutkimuksesta on esitetty taulukossa 5 ja Kaplan-Meierin käyrät kuvissa 3–4.
Taulukko 5. SPOTLIGHT- ja GLOW-tutkimusten tehotulokset
| Päätetapahtuma | SPOTLIGHTa | GLOWb | ||
Tsolbetuksimabi ja mFOLFOX6 n = 283 | Lumelääke ja mFOLFOX6 n = 282 | Tsolbetuksimabi ja CAPOX n = 254 | Lumelääke ja CAPOX n = 253 | |
| Taudin etenemättömyysaika | ||||
| Niiden potilaiden lukumäärä (%), joilla esiintyi tapahtumia | 159 (56,2) | 187 (66,3) | 153 (60,2) | 182 (71,9) |
Mediaani kuukausina (95 %:n luottamusväli)c | 11,0 (9,7; 12,5) | 8,9 (8,2; 10,4) | 8,2 (7,3; 8,8) | 6,8 (6,1; 8,1) |
| Riskisuhde (95 %:n luottamusväli)d,e | 0,734 (0,591; 0,910) | 0,689 (0,552; 0,860) | ||
| Kokonaiselinaika | ||||
| Niiden potilaiden lukumäärä (%), joilla esiintyi tapahtumia | 197 (69,6) | 217 (77,0) | 180 (70,9) | 207 (81,8) |
Mediaani kuukausina (95 %:n luottamusväli)c | 18,2 (16,1; 20,6) | 15,6 (13,7; 16,9) | 14,3 (12,1; 16,4) | 12,2 (10,3; 13,7) |
| Riskisuhde (95 %:n luottamusväli)d,e | 0,784 (0,644; 0,954) | 0,763 (0,622; 0,936) | ||
| Objektiivinen vasteprosentti (ORR), Vasteen kesto (DOR) | ||||
ORR (%) (95 %:n luottamusväli) | 48,1 (42,1; 54,1) | 47,5 (41,6; 53,5) | 42,5 (36,4; 48,9) | 39,1 (33,1; 45,4) |
| DOR mediaani kuukausina (95 %:n luottamusväli) | 9,0 (7,5; 10,4) | 8,1 (6,5; 11,4) | 6,3 (5,4; 8,3) | 6,1 (4,4; 6,3) |
a. Tiedonkeruun katkaisupiste SPOTLIGHT-tutkimuksessa: 8.9.2023, tsolbetuksimabin ja mFOLFOX6-tutkimushaaran yhdistelmän seuranta-ajan mediaani 18 kuukautta. b. Tiedonkeruun katkaisupiste GLOW-tutkimuksessa: 12.1.2024, tsolbetuksimabin ja CAPOX-tutkimushaaran yhdistelmän seuranta-ajan mediaani 20,6 kuukautta. c. Perustuu Kaplan-Meierin menetelmän mukaiseen arviointiin. d. Ositustekijöitä olivat alue, metastaasikohtien lukumäärä ja aiempi gastrektomia interaktiivisesta vasteteknologiasta sekä tutkimuksen tunniste (SPOTLIGHT/GLOW). e. Perustuu Coxin suhteellisen riskin malliin, jossa selittäviä muuttujia olivat hoito, alue, niiden elinten lukumäärä, joissa oli metastaasikohtia, aiempi gastrektomia sekä tutkimuksen tunniste (SPOTLIGHT/GLOW). f. Perustuu riippumattoman arviointilautakunnan arvioon ja vahvistamattomiin vastauksiin. | ||||
SPOTLIGHT- ja GLOW-tutkimuksista yhdistetyn tehoanalyysin (lopullinen OS-analyysi ja päivitetty PFS-analyysi) tulosten mukaan PFS-mediaani (riippumattoman arviointilautakunnan arvioimana) oli tsolbetuksimabin ja mFOLFOX6/CAPOX-hoidon yhdistelmällä 9,2 kuukautta (95 %:n luottamusväli: 8,4; 10,4) ja lumelääkkeen ja mFOLFOX6/CAPOX-hoidon yhdistelmällä 8,2 kuukautta (95 %:n luottamusväli: 7,6; 8,4) [riskisuhde 0,712, 95 %:n luottamusväli: 0,610; 0,831], ja OS-mediaani oli tsolbetuksimabin ja mFOLFOX6/CAPOX-hoidon yhdistelmällä 16,4 kuukautta (95 %:n luottamusväli: 15,0; 17,9) ja lumelääkkeen ja mFOLFOX6/CAPOX-hoidon yhdistelmällä 13,7 kuukautta (95 %:n luottamusväli: 12,3; 15,3) [riskisuhde 0,774, 95 %:n luottamusväli: 0,672; 0,892].
Kuva 1. Kaplan-Meierin käyrä taudin etenemättömyysajalle, SPOTLIGHT
Kuva 2. Kaplan-Meierin käyrä kokonaiselinajalle, SPOTLIGHT
Kuva 3. Kaplan-Meierin käyrä taudin etenemättömyysajalle, GLOW
Kuva 4. Kaplan-Meierin käyrä kokonaiselinajalle, GLOW
SPOTLIGHT- ja GLOW-tutkimusten tehoa koskevat eksploratiiviset alaryhmäanalyysit osoittivat, että kaukaasialaisten ja aasialaisten potilaiden PFS- ja OS-arvoissa oli eroja.
SPOTLIGHTin osalta kaukaasialaisilla potilailla tämä johti siihen, että (riippumattoman arviointilautakunnan mukaan arvioituna) tsolbetuksimabin ja mFOLFOX6:n yhdistelmällä saavutetun PFS:n riskisuhde oli 0,872 [95 %:n luottamusväli: 0,653; 1,164] ja OS:n riskisuhde oli 0,940 [95 %:n luottamusväli: 0,718; 1,231] verrattuna lumelääkkeen ja mFOLFOX6:n yhdistelmään.
Aasialaisilla potilailla tämä johti siihen, että (riippumattoman arviointilautakunnan mukaan arvioituna) tsolbetuksimabilla yhdessä mFOLFOX6:n kanssa saavutetun PFS:n riskisuhde oli 0,526 [95 %:n luottamusväli: 0,354; 0,781] ja OS:n riskisuhde oli 0,636 [95 %:n luottamusväli: 0,450; 0,899] verrattuna lumelääkkeen ja mFOLFOX6:n yhdistelmään. GLOW-tutkimuksen osalta kaukaasialaisilla potilailla tämä johti siihen, että (riippumattoman arviointilautakunnan mukaan arvioituna) tsolbetuksimabilla yhdessä CAPOX-hoidon kanssa saavutetun PFS:n riskisuhde oli 0,891 [95 %:n luottamusväli: 0,622; 1,276] ja OS:n riskisuhde oli 0,805 [95 %:n luottamusväli: 0,579; 1,120] verrattuna lumelääkkeeseen CAPOX-hoidon kanssa. Aasialaisilla potilailla tämä johti siihen, että (riippumattoman arviointilautakunnan arvion mukaan) tsolbetuksimabilla yhdessä CAPOX-hoidon kanssa saavutetun PFS:n riskisuhde oli 0,616 [95 %:n luottamusväli: 0,467; 0,813] ja OS:n riskisuhde 0,710 [95 %:n luottamusväli: 0,549; 0,917] verrattuna lumelääkkeellä CAPOX-hoidon kanssa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset tsolbetuksimabin käytöstä gastrisen tai gastroesofageaalisen junktion adenokarsinooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Laskimoon annon jälkeen tsolbetuksimabin farmakokinetiikka oli suhteessa annokseen annosalueella 33 mg/m2 – 1 000 mg/m2. Kun valmistetta annettiin annoksena 800/600 mg/m2 3 viikon välein, vakaa tila saavutettiin populaatiofarmakokineettisen analyysin perusteella 24 viikossa, ja keskimääräinen (keskihajonta) Cmax oli 453 (82) µg/ml ja keskimääräinen AUCtau oli 4 125 (1 169) vrk•µg/ml. Kun valmistetta annetaan annoksena 800/400 mg/m2 2 viikon välein, vakaa tila odotetaan saavutettavan populaatiofarmakokineettisen analyysin perusteella 22 viikossa, ja keskimääräinen (keskihajonta) Cmax on 359 (68) µg/ml ja keskimääräinen AUCtau on 2 758 (779) vrk•µg/ml.
Jakautuminen
Tsolbetuksimabin arvioitu keskimääräinen jakautumistilavuus vakaassa tilassa oli 5,5 l.
Biotransformaatio
Tsolbetuksimabin odotetaan kataboloituvan pieniksi peptideiksi ja aminohapoiksi.
Eliminaatio
Tsolbetuksimabin puhdistuma (CL) pieneni ajan myötä. Suurin lasku lähtötilanteen arvoista oli 57,6 %, ja tällöin populaation keskimääräinen vakaan tilan puhdistuma (CLss) oli 0,0117 l/h. Tsolbetuksimabin puoliintumisaika oli 7,6–15,2 vuorokautta hoidon aikana.
Erityisryhmät
Iäkkäät
Populaatiofarmakokineettisen analyysin mukaan iällä [vaihteluväli: 22–83 vuotta; 32,2 % (230/714) oli > 65-vuotiaita, 5,0 % (36/714) oli > 75-vuotiaita] ei ollut kliinisesti merkittävää vaikutusta tsolbetuksimabin farmakokinetiikkaan.
Rotu ja sukupuoli
Populaatiofarmakokineettisen analyysin mukaan tsolbetuksimabin farmakokinetiikassa ei todettu mitään kliinisesti merkitseviä, sukupuoleen [62,3 % miehiä, 37,7 % naisia] tai rotuun [50,1 % kaukaasialaisia, 42,2 % aasialaisia, 4,2 %:lta tiedot puuttuivat, 2,7 % muita ja 0,8 % tummaihoisia] perustuvia eroja.
Munuaisten vajaatoiminta
Populaatiofarmakokineettisessä analyysissa, jossa hyödynnettiin gastrista tai gastroesofageaalisen junktion adenokarsinoomaa sairastavilla potilailla tehdyistä kliinisistä tutkimuksista saatuja tietoja, tsolbetuksimabin farmakokinetiikassa ei todettu mitään kliinisesti merkitseviä eroja potilailla, jotka sairastivat lievää (CrCL ≥ 60 – < 90 ml/min; n = 298) tai keskivaikeaa (CrCL ≥ 30 – < 60 ml/min; n = 109) munuaisten vajaatoimintaa (CrCL arvioitiin Cockcroft-Gaultin kaavalla). Tsolbetuksimabia on arvioitu vain pienellä määrällä vaikeaa munuaisten vajaatoimintaa (CrCL ≥ 15 – < 30 ml/min; n = 1) sairastavia potilaita. Vaikean munuaisten vajaatoiminnan vaikutusta tsolbetuksimabin farmakokinetiikkaan ei tunneta.
Maksan vajaatoiminta
Populaatiofarmakokineettisessä analyysissa, jossa hyödynnettiin gastrista tai gastroesofageaalisen junktion adenokarsinoomaa sairastavilla potilailla tehdyistä kliinisistä tutkimuksista saatuja tietoja, tsolbetuksimabin farmakokinetiikassa ei todettu mitään kliinisesti merkitseviä eroja potilailla, jotka sairastivat TB- ja ASAT-arvojen perusteella mitattua lievää maksan vajaatoimintaa (TB ≤ ULN ja ASAT > ULN, tai TB > 1–1,5 × ULN ja ASAT mikä tahansa; n = 108). Tsolbetuksimabia on arvioitu vain pienellä määrällä keskivaikeaa maksan vajaatoimintaa (TB > 1,5–3 × ULN ja ASAT mikä tahansa; n = 4) sairastavia potilaita, eikä sitä ole arvioitu vaikeaa maksan vajaatoimintaa (TB > 3–10 × ULN ja ASAT mikä tahansa) sairastavilla potilailla. Keskivaikean tai vaikean maksan vajaatoiminnan vaikutusta tsolbetuksimabin farmakokinetiikkaan ei tunneta.
Prekliiniset tiedot turvallisuudesta
Karsinogeenisuutta tai mutageenisuutta ei ole arvioitu eläimillä tehdyissä tutkimuksissa.
Toksisuutta tai muita tsolbetuksimabiin liittyviä, sydämeen ja verisuonistoon, hengityselimiin tai keskushermostoon kohdistuvia haittavaikutuksia ei todettu hiirillä, joille annettiin tsolbetuksimabia 13 viikon ajan systeemisinä altistuksina, jotka olivat enintään 7,0‑kertaisia verrattuna ihmisen altistukseen suositellulla annoksella 600 mg/m2 (AUC-arvon perusteella), eikä makakeilla, joille annettiin tsolbetuksimabia 4 viikon ajan systeemisinä altistuksina, jotka olivat enintään 6,1‑kertaisia verrattuna ihmisen altistukseen suositellulla annoksella 600 mg/m2 (AUC-arvon perusteella).
Tsolbetuksimabi läpäisi istukan alkion- ja sikiönkehitystä selvittäneessä toksisuustutkimuksessa, jossa tsolbetuksimabia annettiin tiineille hiirille organogeneesin aikana systeemisinä annoksina, jotka olivat enintään noin 6,2-kertaisia verrattuna ihmisen altistukseen suositellulla annoksella 600 mg/m2 (AUC-arvon perusteella). Tämän seurauksena tsolbetuksimabin pitoisuus sikiön seerumissa tiineyspäivänä 18 oli suurempi kuin pitoisuus emon seerumissa tiineyspäivänä 16. Tsolbetuksimabi ei aiheuttanut sikiöille mitään ulkoisia tai sisäelinten poikkeavuuksia (epämuodostumia tai variaatioita).
Farmaseuttiset tiedot
Apuaineet
Arginiini
Fosforihappo (E338)
Sakkaroosi
Polysorbaatti 80 (E433)
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
4 vuotta.
Käyttökuntoon saatettu liuos injektiopullossa
Käyttökuntoon saatettuja injektiopulloja voidaan säilyttää huoneenlämmössä (≤ 25 °C) enintään 6 tunnin ajan. Ne eivät saa jäätyä eivätkä altistua suoralle auringonvalolle. Käyttökuntoon saatettua liuosta sisältävät käyttämättömät injektiopullot on hävitettävä suositellun säilytysajan umpeuduttua.
Laimennettu liuos infuusiopussissa
Mikrobiologiselta kannalta infuusiopussissa oleva laimennettu liuos on annettava välittömästi. Jos sitä ei anneta välittömästi, käyttövalmista infuusiopussia voidaan säilyttää:
- kylmässä (2 °C – 8 °C) enintään 24 tunnin ajan infuusiopussin valmistelun päättymisestä, mukaan lukien infuusioon kuluva aika. Ei saa jäätyä.
- huoneenlämmössä (≤ 25 °C) enintään 8 tunnin ajan siitä, kun käyttövalmis infuusiopussi otetaan jääkaapista, mukaan lukien infuusioon kuluva aika.
Ei saa altistua suoralle auringonvalolle. Käyttämättömät, valmistellut infuusiopussit on hävitettävä suositellun säilytysajan umpeuduttua.
Säilytys
Säilytä jääkaapissa (2 ºC – 8 ºC).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VYLOY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
100 mg (L:ei) 1 kpl (20 mg/ml) (645,76 €)
300 mg (L:ei) 1 kpl (20 mg/ml) (1850,00 €)
PF-selosteen tieto
Vyloy 100 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos, injektiopullo
Tyypin I lasista valmistettu 20 ml:n injektiopullo, jossa tulppaa lukitseva sisennös, harmaa bromobutyylikumitulppa ja eteenitetrafluorieteenikalvo sekä alumiinisinetti ja vihreä korkki.
Vyloy 300 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos, injektiopullo
Tyypin I lasista valmistettu 50 ml:n injektiopullo, jossa tulppaa lukitseva sisennös, harmaa bromobutyylikumitulppa ja eteenitetrafluorieteenikalvo sekä alumiinisinetti ja violetti korkki.
Pakkauskoot 100 mg: yksi kotelo sisältää 1 injektiopullon tai 3 injektiopulloa.
Pakkauskoko 300 mg: yksi kotelo sisältää 1 injektiopullon.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen kylmäkuivattu jauhe.
Käyttö- ja käsittelyohjeet
Valmistelu- ja anto-ohjeet
Käyttökuntoon saattaminen kerta-annosinjektiopullossa
- Syöpälääkkeiden asianmukaista käsittelyä ja hävittämistä koskevia toimenpiteitä on noudatettava.
- Käytä asianmukaista aseptista tekniikkaa liuosten käyttökuntoon saattamiseen ja valmisteluun.
- Laske potilaan kehon pinta-alaan perustuva suositeltu annos ja määritä tarvittavien injektiopullojen lukumäärä.
-
Saata jokainen injektiopullo käyttökuntoon seuraavasti. Mikäli mahdollista, suuntaa injektionesteisiin käytettävä vesi injektiopullon seinämään eikä suoraan kylmäkuivattuun jauheeseen:
- 100 mg:n injektiopullo: lisää hitaasti 5 ml steriiliä injektionesteisiin käytettävää vettä, jolloin tsolbetuksimabin vahvuudeksi saadaan 20 mg/ml.
- 300 mg:n injektiopullo: lisää hitaasti 15 ml steriiliä injektionesteisiin käytettävää vettä, jolloin tsolbetuksimabin vahvuudeksi saadaan 20 mg/ml.
- Pyörittele kutakin injektiopulloa hitaasti, kunnes sisältö on liuennut kokonaan. Anna käyttökuntoon saatetun/saatettujen injektiopullon/injektiopullojen asettua. Tarkkaile liuosta silmämääräisesti, kunnes ilmakuplat ovat hävinneet. Älä ravista injektiopulloa.
- Tarkista liuos silmämääräisesti hiukkasten ja värimuutosten varalta. Käyttökuntoon saatetun liuoksen pitäisi olla kirkasta tai hieman opalisoivaa, väritöntä tai vaaleankeltaista, eikä siinä saa olla näkyviä hiukkasia. Jos injektiopullossa näkyy hiukkasia tai värimuutoksia, hävitä se.
- Injektiopullon/-pullojen sisältämä käyttökuntoon saatettu liuos on lisättävä infuusiopussiin välittömästi lasketun annoksen mukaisesti. Tämä valmiste ei sisällä säilytysaineita. Jos sitä ei käytetä välittömästi, katso käyttökuntoon saatettujen injektiopullojen säilytysohjeet kohdasta Kestoaika.
Infuusiopussiin laimentaminen
- Vedä laskettu annos käyttökuntoon saatettua liuosta injektiopullosta/-pulloista ja siirrä se infuusiopussiin.
- Laimenna käyttämällä natriumkloridi-infuusionestettä, jonka pitoisuus on 9 mg/ml (0,9‑prosenttista). Infuusiopussiin pitää mahtua riittävästi laimenninta, jotta lopulliseksi pitoisuudeksi saadaan 2 mg/ml tsolbetuksimabia.
Laimennettu, potilaalle annettava tsolbetuksimabiliuos on yhteensopiva seuraavien kanssa: infuusiopussit, jotka on valmistettu polyeteenistä (PE), polypropeenista (PP), polyvinyylikloridista (PVC), jossa on pehmitintä [di-2-etyyliheksyyli)ftalaattia (DEHP) tai trioktyylitrimellitaattia (TOTM)], eteenipropeenikopolymeeristä, eteenivinyyliasetaatti (EVA) -kopolymeeristä, PP- ja styreenieteenibutyleenistyreenikopolymeerista, sekä lasi (lääkkeen antoon tarkoitettu pullo) ja sellaiset infuusioletkut, jotka on valmistettu PE:stä, polyuretaanista (PU), PVC:stä, jossa on pehmitintä [DEHP, TOTM tai di(2‑etyyliheksyyli)tereftalaatti], polybutadieenista (PB) tai elastomeerillä modifioidusta PP:stä ja joissa on polyeetterisulfonista (PES) tai polysulfonista valmistettu letkun sisäinen suodatinkalvo (huokoskoko 0,2 μm).
- Sekoita laimennettu liuos varovasti kääntelemällä. Älä ravista pussia.
- Tarkista infuusiopussi silmämääräisesti hiukkasten varalta ennen käyttöä. Laimennetussa liuoksessa ei saa olla näkyviä hiukkasia. Älä käytä infuusiopussia, jos näet siinä hiukkasia.
- Hävitä kerta-annosinjektiopulloihin jäänyt käyttämätön liuos.
Anto
- Saman infuusioletkuston kautta ei saa antaa samanaikaisesti muita lääkevalmisteita.
- Anna lääke välittömästi infuusioletkun kautta vähintään 2 tuntia kestävänä infuusiona. Ei saa antaa nopeana infuusiona eikä boluksena laskimoon.
Yhteensopimattomuuksia ei ole havaittu sellaisten suljetun järjestelmän siirtolaitteistojen kanssa, jotka on valmistettu PP:stä, PE:stä, ruostumattomasta teräksestä, silikonista (kumi/öljy/hartsi), polyisopreenistä, PVC:stä, jossa on pehmitintä [TOTM], akrylonitriilibutadieenistyreeni (ABS) -kopolymeeristä, metyylimetakrylaatti-ABS-kopolymeeristä, termoplastisesta elastomeeristä, polytetrafluorieteenistä, polykarbonaatista, PES:stä, akryylikopolymeeristä, polybutyleenitereftalaatista, PB:stä tai EVA-kopolymeeristä.
Yhteensopimattomuuksia ei ole todettu sellaisten keskusporttien kanssa, jotka on valmistettu silikonikumista, titaaniseoksesta tai PVC:stä, jossa on pehmitintä [TOTM].
- Annon aikana suositellaan käyttämään letkun sisäisiä suodattimia (huokoskoko 0,2 μm, yllä luetellut materiaalit).
- Jos lääkettä ei anneta välittömästi, katso käyttövalmiin infuusiopussin säilytysohjeet kohdasta Kestoaika.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VYLOY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
100 mg 1 kpl
300 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FX31
Valmisteyhteenvedon muuttamispäivämäärä
22.01.2026
Yhteystiedot
Hatsinanpuisto 8
02600 Espoo
09 8560 6000
www.astellas.fi