
BEYFORTUS injektioneste, liuos, esitäytetty ruisku 50 mg, 100 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Beyfortus 50 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 50 mg nirsevimabia 0,5 ml:ssa liuosta (100 mg/ml).
Beyfortus 100 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 100 mg nirsevimabia 1 ml:ssa liuosta (100 mg/ml).
Nirsevimabi (nirsevimab) on humaani monoklonaalinen vasta-aine (immunoglobuliini G1 kappa (IgG1κ)) , joka on tuotettu kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 0,1 mg polysorbaatti 80:aa (E433) per 50 mg:n (0,5 ml:n) annos ja 0,2 mg per 100 mg:n (1 ml:n) annos (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste).
Kliiniset tiedot
Käyttöaiheet
Beyfortus on tarkoitettu estämään RS-viruksen (RSV) aiheuttamia alahengitystiesairauksia
- vastasyntyneillä ja imeväisillä lapsen ensimmäisen RSV-kauden aikana
- enintään 24 kuukauden ikäisillä lapsilla, jotka ovat edelleen alttiita vaikealle RSV-taudille toisella RSV-kaudellaan (ks. kohta Farmakodynamiikka).
Beyfortus-valmisteen käytössä on noudatettava virallisia suosituksia.
Annostus ja antotapa
Annostus
Imeväiset ensimmäisen RSV-kautensa aikana
Suositeltu annos on yksi 50 mg:n annos lihakseen alle 5 kg painaville imeväisille ja yksi 100 mg:n annos lihakseen vähintään 5 kg painaville imeväisille.
RSV-kaudella syntyneille imeväisille Beyfortus annetaan syntymän jälkeen. Muille kuin RSV-kauden aikana syntyneille Beyfortus on annettava mieluiten ennen RSV-kautta.
1,0 – < 1,6 kg painavilla imeväisillä annostus perustuu ekstrapolointiin. Kliinisiä tietoja ei ole saatavilla. Altistuksen odotetaan olevan suurempi alle 1 kg painavilla imeväisillä kuin imeväisillä, jotka painavat tätä enemmän. Nirsevimabin käytön hyödyt ja riskit alle 1 kg painavilla imeväisillä on arvioitava huolellisesti.
Alle 8 viikon ikäisistä erittäin ennenaikaisina syntyneistä imeväisistä (gestaatioikä < 29 viikkoa) on saatavilla vain vähän tietoja. Kliinisiä tietoja ei ole saatavilla imeväisistä, joiden postmenstruaalinen ikä (gestaatioikä syntymähetkellä + kronologinen ikä) on alle 32 viikkoa (ks. kohta Farmakodynamiikka).
Lapset, jotka ovat edelleen alttiita vaikealle RSV‑taudille toisella RSV‑kaudellaan
Suositeltu annos on yksi 200 mg:n annos kahtena injektiona lihakseen (2 x 100 mg). Beyfortus on annettava mieluiten ennen toisen RSV‑kauden alkamista.
Lapsille, joille tehdään sydän-keuhkokoneen käyttöä vaativa sydänleikkaus, voidaan antaa lisäannos heti, kun lapsen tila on vakaa leikkauksen jälkeen, jotta varmistetaan riittävä nirsevimabin pitoisuus seerumissa. Jos lisäannos annetaan 90 päivän kuluessa ensimmäisestä Beyfortus-annoksesta, lisäannoksen suuruus on ensimmäisen RSV-kauden aikana 50 mg tai 100 mg riippuen lapsen painosta tai toisen RSV-kauden aikana 200 mg. Jos ensimmäisestä annoksesta on kulunut yli 90 päivää, voidaan antaa lapsen painosta riippumatta yksi 50 mg:n lisäannos ensimmäisen RSV-kauden aikana tai 100 mg:n lisäannos toisen RSV-kauden aikana jäljellä olevan RSV-kauden kattamiseksi.
Nirsevimabin turvallisuutta ja tehoa 2–18-vuotiaiden lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Beyfortus on tarkoitettu annettavaksi vain injektiona lihakseen.
Se annetaan lihakseen, mieluiten reiteen anterolateraalisesti. Pakaralihasta ei pidä käyttää rutiininomaisesti injektiokohtana iskiashermon vaurioitumisriskin takia. Jos injektioita tarvitaan kaksi, on käytettävä eri injektiokohtia.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet käsittelyä koskevista erityisvaatimuksista.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyys (myös anafylaksia)
Beyfortus‑valmisteen annon jälkeen on ilmoitettu vakavia yliherkkyysreaktioita. Humaanien monoklonaalisten immunoglobuliini G1 ‑vasta-aineiden (IgG1‑vasta-aineiden) käytön yhteydessä on havaittu anafylaksiaa. Jos anafylaksian tai muun kliinisesti merkittävän yliherkkyysreaktion merkkejä ja oireita ilmenee, on välittömästi keskeytettävä valmisteen anto ja aloitettava asianmukaiset lääkehoidot ja/tai tukihoidot.
Kliinisesti merkittävät verenvuotohäiriöt
Kuten muitakin lihakseen annettavia injektioita annettaessa, nirsevimabin antamisessa on noudatettava varovaisuutta lapsilla, joilla on trombosytopenia tai mikä tahansa hyytymishäiriö.
Immuunipuutteiset lapset
Kliinisissä tutkimuksissa on havaittu nirsevimabin suuri puhdistuma joillakin immuunipuutteisilla lapsilla, joilla on proteiinihukkaa aiheuttava tila (ks. kohta Farmakokinetiikka). Nirsevimabi ei välttämättä tuota tällaisille henkilöille samantasoista suojaa.
Polysorbaatti 80 (E433)
Tämä lääkevalmiste sisältää 0,1 mg polysorbaatti 80:aa per 50 mg:n (0,5 ml:n) annos ja 0,2 mg per 100 mg:n (1 ml:n) annos. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Monoklonaalisilla vasta-aineilla yhteisvaikutusten todennäköisyys ei yleensä ole merkittävä, sillä ne eivät suoraan vaikuta sytokromi P450 ‑entsyymeihin eivätkä ole maksan tai munuaisten kuljettajaproteiinien substraatteja. Epäsuorat vaikutukset sytokromi P450 -entsyymeihin ovat epätodennäköisiä, sillä nirsevimabin vaikutus kohdistuu eksogeeniseen virukseen.
Nirsevimabi ei häiritse käänteiskopiointipolymeraasiketjureaktiotekniikalla (RT‑PCR) tai antigeenipikatestillä toteutettavia RSV:n diagnostisia määritysmenetelmiä, joissa hyödynnetään kaupallisesti saatavilla olevia, RSV:n fuusioproteiinin (F‑proteiinin) antigeenikohtaan I, II tai IV kohdentuvia vasta‑aineita.
Samanaikainen anto rokotteiden kanssa
Koska nirsevimabi on passiiviseen immunisaatioon tarkoitettu RSV-spesifinen monoklonaalinen vasta-aine, sen ei odoteta häiritsevän aktiivista immuunivastetta samanaikaisesti annetuille rokotteille.
Samanaikaisesta annosta rokotteiden kanssa on vain vähän kokemusta. Kliinisissä tutkimuksissa, joissa nirsevimabia annettiin tavanomaisten lapsuusiän rokotteiden kanssa, samanaikaisesti annetun ohjelman turvallisuus- ja reaktogeenisuusprofiilit olivat samankaltaiset kuin yksinään annetuilla lapsuusiän rokotteilla. Nirsevimabi voidaan antaa samanaikaisesti lapsuusiän rokotteiden kanssa.
Nirsevimabia ei saa sekoittaa minkään rokotteen kanssa samaan ruiskuun tai injektiopulloon (ks. kohta Yhteensopimattomuudet). Kun nirsevimabia annetaan samanaikaisesti injisoitavien rokotteiden kanssa, ne on annettava omilla ruiskuillaan eri injektiokohtiin.
Raskaus ja imetys
Ei merkityksellinen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ei merkityksellinen.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisin haittavaikutus oli ihottuma (0,7 %), joka ilmeni 14 päivän kuluessa annoksen saamisesta. Suurin osa tapauksista oli vaikeusasteeltaan lieviä tai kohtalaisia. Lisäksi kuumetta ilmoitettiin 0,5 %:lla ja injektiokohdan reaktioita 0,3 %:lla 7 päivän kuluessa annoksen saamisesta. Injektiokohdan reaktiot eivät olleet vakavia.
Haittavaikutustaulukko
Taulukossa 1 on esitetty haittavaikutukset, joita ilmoitettiin 2 966:lla täysiaikaisena ja ennenaikaisena syntyneellä imeväisellä (gestaatioikä ≥ 29 viikkoa), jotka saivat nirsevimabia kliinisissä tutkimuksissa, sekä haittavaikutukset, joita on ilmoitettu markkinoilletulon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kontrolloiduissa kliinisissä tutkimuksissa ilmoitetut haittavaikutukset on luokiteltu MedDRA:n elinjärjestelmäluokituksen mukaisesti. Suositellut termit on esitetty kussakin elinjärjestelmässä ensin esiintymistiheyden mukaan alenevassa järjestyksessä ja sitten vakavuuden mukaan alenevassa järjestyksessä. Haittavaikutusten esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1 / 1 000, < 1/100); harvinainen (≥ 1 / 10 000, < 1 / 1 000); hyvin harvinainen (< 1 / 10 000); ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1: Haittavaikutukset
| MedDRA-elinjärjestelmä | MedDRA:n suositeltu termi | Esiintymistiheys |
| Immuunijärjestelmä | Yliherkkyysa | Tuntematon |
| Iho ja ihonalainen kudos | Ihottumab | Melko harvinainen |
| Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan reaktioc | Melko harvinainen |
| Kuume | Melko harvinainen |
a Spontaanisti ilmoitettu haittavaikutus.
b Ihottuma määriteltiin seuraavilla suositelluilla termeillä: ihottuma, makulopapulaarinen ihottuma, makulaarinen ihottuma.
c Injektiokohdan reaktio määriteltiin seuraavilla suositelluilla termeillä: injektiokohdan reaktio, injektiokohdan kipu, injektiokohdan kovettuma, injektiokohdan edeema, injektiokohdan turvotus.
Imeväiset, joilla on suurentunut riski saada vaikea RSV-tauti ensimmäisellä RSV-kaudellaan
Turvallisuutta arvioitiin MEDLEY-tutkimuksessa 918 imeväisellä, joilla oli suurentunut riski saada vaikea RSV-tauti, mukaan lukien 196 erittäin ennenaikaisena syntynyttä imeväistä (gestaatioikä < 29 viikkoa) ja 306 imeväistä, joilla oli keskosen krooninen keuhkosairaus tai hemodynaamisesti merkittävä synnynnäinen sydänvika. Tutkittavien ensimmäinen RSV-kausi oli alkamassa, ja heille annettiin nirsevimabia (n = 614) tai palivitsumabia (n = 304). Nirsevimabin turvallisuusprofiili imeväisillä, jotka saivat nirsevimabia ensimmäisellä RSV-kaudellaan, oli verrannollinen vertailuvalmiste palivitsumabin kanssa ja vastasi täysiaikaisina ja ennenaikaisina syntyneillä imeväisillä (gestaatioikä ≥ 29 viikkoa) todettua nirsevimabin turvallisuusprofiilia (D5290C00003 ja MELODY).
Lapset, jotka ovat toisella RSV-kaudellaan edelleen alttiita vaikealle RSV-taudille
Turvallisuutta arvioitiin MEDLEY‑tutkimuksessa 220 lapsella, joilla oli keskosen krooninen keuhkosairaus tai hemodynaamisesti merkittävä synnynnäinen sydänvika. Lapset saivat nirsevimabia tai palivitsumabia ensimmäisellä RSV‑kaudellaan ja nirsevimabia silloin, kun heidän toinen RSV‑kautensa oli alkamassa (180 tutkittavaa sai nirsevimabia sekä kaudella 1 että kaudella 2, ja 40 tutkittavaa sai palivitsumabia kaudella 1 ja nirsevimabia kaudella 2). Nirsevimabin turvallisuusprofiili lapsilla, jotka saivat nirsevimabia toisella RSV‑kaudellaan, vastasi nirsevimabin turvallisuusprofiilia täysiaikaisina ja ennenaikaisina syntyneillä (gestaatioikä ≥ 29 viikkoa) imeväisillä (D5290C00003 ja MELODY).
Turvallisuutta arvioitiin myös avoimessa, kontrolloimattomassa MUSIC‑kerta‑annostutkimuksessa 100 immuunipuutteisella imeväisellä ja lapsella (ikä ≤ 24 kuukautta), jotka saivat nirsevimabia ensimmäisellä tai toisella RSV‑kaudellaan. Tutkimukseen osallistui tutkittavia, joilla oli vähintään yksi seuraavista tiloista: immuunivajavuus (sekamuotoinen immuunivajavuus, vasta‑aineiden vajavuuksina ilmenevä immuunivajavuus tai etiologialtaan muunlainen immuunivajavuus) (n = 33), systeeminen suuriannoksinen kortikosteroidihoito (n = 29), elimen tai luuytimen siirto (n = 16), meneillään oleva immunosuppressiivinen solunsalpaajahoito (n = 20), muu immunosuppressiivinen hoito (n = 15) ja HIV‑infektio (n = 8). Nirsevimabin turvallisuusprofiili vastasi immuunipuutteisten lasten populaatiossa odotettavissa olevaa turvallisuusprofiilia ja nirsevimabin turvallisuusprofiilia täysiaikaisina ja ennenaikaisina syntyneillä (gestaatioikä ≥ 29 viikkoa) imeväisillä (D5290C00003 ja MELODY).
Nirsevimabin turvallisuusprofiili lapsilla heidän toisen RSV-kautensa aikana vastasi heidän ensimmäisellä RSV-kaudellaan havaittua nirsevimabin turvallisuusprofiilia.
Täysiaikaisina tai ennenaikaisina syntyneet imeväiset, joiden ensimmäinen RSV-kausi on alkamassa
Nirsevimabin turvallisuutta arvioitiin myös satunnaistetussa, avoimessa HARMONIE-monikeskustutkimuksessa 8 034:llä täysiaikaisena tai ennenaikaisena syntyneellä imeväisellä (gestaatioikä ≥ 29 viikkoa), joiden ensimmäinen RSV-kausi oli alkamassa (ja jotka eivät soveltuneet saamaan palivitsumabia). Tutkittaville joko annettiin nirsevimabia (n = 4 016) tai ei tehty mitään interventiota (n = 4 018) sairaalahoitoon johtavan RSV-alahengitystieinfektion ehkäisemiseksi. Ensimmäisellä RSV-kaudella annetun nirsevimabin turvallisuusprofiili vastasi nirsevimabin turvallisuusprofiilia lumekontrolloiduissa tutkimuksissa (D5290C00003 ja MELODY).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Nirsevimabin yliannostukseen ei ole spesifistä hoitoa. Yliannostustapauksessa henkilön tilaa on seurattava haittavaikutusten varalta ja häntä on hoidettava oireenmukaisesti tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunoseerumit ja immunoglobuliinit, antiviraaliset monoklonaaliset vasta-aineet, ATC-koodi: J06BD08
Vaikutusmekanismi
Nirsevimabi on rekombinantti neutraloiva humaani pitkävaikutteinen monoklonaalinen vasta-aine (IgG1ĸ) RSV:n F-proteiinin fuusiotumista edeltävälle pre-F-konformaatiolle. Sen puoliintumisaikaa seerumissa on pidennetty kolmella Fc-alueeseen kohdennetulla aminohapposubstituutiolla (YTE). Nirsevimabi sitoutuu proteiinin pre-F-konformaation antigeenikohdan Ø hyvin konservoituneeseen epitooppiin. Dissosiaatiovakio KD on 0,12 nM RS-viruksen alatyypissä A ja 1,22 nM alatyypissä B. Nirsevimabi estää viruksen soluunpääsyprosessissa keskeisen solukalvojen fuusioitumisvaiheen. Tämä neutraloi viruksen ja estää solujen fuusioitumisen.
Farmakodynaamiset vaikutukset
Antiviraalinen vaikutus
Nirsevimabin RS-virukseen kohdistuvaa neutralointivaikutusta soluviljelmässä mitattiin annos-vastemallissa, jossa käytettiin viljeltyjä Hep‑2-soluja. Nirsevimabi neutraloi RSV A- ja RSV B -isolaatteja. EC50-arvon mediaanit 3,2 ng/ml (vaihteluväli 0,48–15 ng/ml) ja 2,9 ng/ml (vaihteluväli 0,3–59,7 ng/ml) , vastaavasti. Kliiniset RSV-isolaatit (70 RSV A -isolaattia ja 49 RSV B -isolaattia) kerättiin vuosina 2003–2017 tutkittavilta eri puolilta Yhdysvaltoja, Australiaa, Alankomaita, Italiaa, Kiinaa ja Israelia, ja niihin sisältyivät yleisimmät RS-viruksen kiertävissä kannoissa esiintyvät F-sekvenssin polymorfismit.
Nirsevimabin osoitettiin sitoutuvan immobilisoituihin ihmisen Fcγ-reseptoreihin (FcγRI, FcγRIIA, FcγRIIB ja FcγRIII) in vitro, ja sillä todettiin vastaava neutralointivaikutus kuin alkuperäisillä monoklonaalisilla vasta-aineilla IG7:llä ja IG7‑TM:llä (Fc-aluetta muokattu vähentämään sitoutumista Fc-reseptoriin ja efektorivaikutusta). Pumpulirotilla tehdyssä RSV-infektiomallissa IG7:n ja IG7‑TM:n todettiin vähentävän vastaavalla tavalla annosriippuvaisesti RS-viruksen replikoitumista keuhkoissa ja nenäkuorikoissa, mikä viittaa vahvasti siihen, että suoja RSV-infektiota vastaan perustuu nirsevimabin neutralointivaikutukseen eikä Fc‑välitteiseen efektorivaikutukseen.
Viruslääkeresistenssi
Soluviljelmässä
Neutraloitumista väistäviä variantteja valikoitui kolmen siirrostuksen jälkeen RSV A2 - ja B9320-kantojen soluviljelmässä, jossa oli nirsevimabia. Rekombinantteihin RSV A -variantteihin, joiden herkkyys nirsevimabille oli heikentynyt, sisältyi sellaisia, joissa oli tunnistetut substituutiot N67I + N208Y (103‑kertainen vertailuvalmisteeseen nähden). Rekombinantteihin RSV B -variantteihin, joiden herkkyys nirsevimabille oli heikentynyt, sisältyi sellaisia, joissa oli tunnistetut substituutiot N208D (> 90 000‑kertainen), N208S (> 24 000‑kertainen), K68N + N201S (> 13 000‑kertainen) tai K68N + N208S (> 90 000‑kertainen). Kaikki neutraloitumista väistävistä varianteista tunnistetut resistenssiin liittyvät substituutiot sijaitsivat nirsevimabin sitoutumiskohdassa (aminohapot 62–69 ja 196–212), ja niiden osoitettiin vähentävän sitoutumisaffiniteettia RSV F‑proteiiniin.
Kliinisissä tutkimuksissa
MELODY‑, MEDLEY- ja MUSIC-tutkimuksissa yhdelläkään tutkittavalla, jolla oli lääkärissäkäyntiin johtanut RSV-alahengitystieinfektio, ei missään hoitoryhmässä ollut nirsevimabiresistenssiin liittyviä substituutioita sisältävää RSV-isolaattia.
D5290C00003-tutkimuksessa (tutkittavat, jotka saivat 50 mg:n kerta-annoksen nirsevimabia riippumatta painosta antohetkellä) kahdella 40:stä nirsevimabiryhmän tutkittavasta, joilla oli lääkärissäkäyntiin johtanut RSV-alahengitystieinfektio, oli nirsevimabiresistenssiin liittyviä substituutioita sisältävä RSV-isolaatti. Yhdelläkään lumeryhmän tutkittavalla ei ollut nirsevimabiresistenssiin liittyviä substituutioita sisältävää RSV-isolaattia. Rekombinantit RSV B -variantit, joissa oli tunnistetut F-proteiinia koodaavan sekvenssin variaatiot I64T + K68E + I206M + Q209R (> 447,1‑kertainen) tai N208S (> 386,6‑kertainen) nirsevimabin sitoutumiskohdassa, heikensivät herkkyyttä nirsevimabin neutraloivalle vaikutukselle.
Nirsevimabin vaikutus säilyi sellaisia rekombinantteja RSV-variantteja vastaan, joissa oli molekyyliepidemiologisissa tutkimuksissa ja palivitsumabin neutraloivaa vaikutusta väistävistä varianteista tunnistettuja, palivitsumabiresistenssiin liittyviä substituutioita. On mahdollista, että nirsevimabille resistenteillä varianteilla saattaa olla ristiresistenssiä muille monoklonaalisille vasta-aineille, joiden vaikutus kohdentuu RSV:n F‑proteiiniin.
Immunogeenisuus
Lääkevasta-aineita havaittiin yleisesti.
Käytetyllä immunogeenisuusmäärityksellä ei pystytä varmuudella toteamaan lääkevasta‑aineita varhaisessa vaiheessa (ennen päivää 361), kun lääkepitoisuus on suuri, joten lääkevasta‑aineiden ilmaantuvuutta ei välttämättä ole määritetty kattavasti. Vaikutus nirsevimabin puhdistumaan on epävarma. Tutkittavilla, jotka olivat lääkevasta‑ainepositiivisia päivänä 361, nirsevimabipitoisuudet olivat pienemmät päivänä 361 verrattuna nirsevimabia saaneisiin tutkittaviin, jotka olivat lääkevasta‑ainenegatiivisia.
Lääkevasta-aineiden vaikutusta nirsevimabin tehoon ei ole määritetty. Lääkevasta-aineiden vaikutuksesta turvallisuuteen ei havaittu näyttöä.
Kliininen teho
Nirsevimabin tehoa ja turvallisuutta lääkärikäyntiin johtavan RSV-alahengitystieinfektion ehkäisyssä täysiaikaisina syntyneillä ja ennenaikaisina syntyneillä (gestaatioikä ≥ 29 viikkoa) imeväisillä, joiden ensimmäinen RSV-kausi oli alkamassa, arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (D5290C00003 [vaihe IIb] ja MELODY [vaihe III]). Nirsevimabin turvallisuutta ja farmakokinetiikkaa arvioitiin myös satunnaistetussa, kaksoissokkoutetussa, palivitsumabikontrolloidussa monikeskustutkimuksessa (MEDLEY [vaihe II/III]). Tutkimukseen otettiin imeväisiä, joiden ensimmäinen RSV-kausi oli alkamassa, joiden gestaatioikä oli alle 35 viikkoa ja joilla oli suurentunut riski saada vaikea RSV-tauti, mukaan lukien erittäin ennenaikaisina syntyneitä imeväisiä (gestaatioikä < 29 viikkoa) ja imeväisiä, joilla oli keskosen krooninen keuhkosairaus tai hemodynaamisesti merkittävä synnynnäinen sydänvika. Tutkimukseen otettiin myös lapsia, joilla oli keskosen krooninen keuhkosairaus tai hemodynaamisesti merkittävä synnynnäinen sydänvika ja joiden toinen RSV‑kausi oli alkamassa.
Nirsevimabin turvallisuutta ja farmakokinetiikkaa arvioitiin myös avoimessa, kontrolloimattomassa, kerta‑annos‑ ja monikeskustutkimuksessa (MUSIC [vaihe II]) immuunipuutteisilla imeväisillä ja ≤ 24 kuukauden ikäisillä lapsilla.
Nirsevimabin tehoa ja turvallisuutta ei interventioon verrattuna sairaalahoitoon johtavan RSV-alahengitystieinfektion ehkäisemisessä arvioitiin myös yhdessä satunnaistetussa avoimessa monikeskustutkimuksessa (HARMONIE, vaihe IIIb) täysiaikaisina tai ennenaikaisina syntyneillä imeväisillä (gestaatioikä ≥ 29 viikkoa), joiden ensimmäinen RSV-kausi oli alkamassa tai jotka syntyivät kauden aikana (ja jotka eivät soveltuneet saamaan palivitsumabia).
Teho lääkärikäyntiin johtaneen RSV-alahengitystieinfektion, lääkärikäyntiin ja sairaalahoitoon johtaneen RSV-alahengitystieinfektion sekä hyvin vaikean lääkärikäyntiin johtaneen RSV-alahengitystieinfektion ehkäisemisessä täysiaikaisina ja ennenaikaisina syntyneillä imeväisillä (D5290C00003 ja MELODY)
D5290C00003-tutkimukseen otettiin yhteensä 1 453 hyvin tai kohtalaisen ennenaikaisina syntynyttä imeväistä (gestaatioikä ≥ 29 – < 35 viikkoa), joiden ensimmäinen RSV-kausi oli alkamassa. Tutkittavat satunnaistettiin (2:1) saamaan 50 mg nirsevimabia tai lumelääkettä kerta-annoksena lihakseen. Satunnaistamishetkellä gestaatioikä oli 20,3 %:lla imeväisistä ≥ 29 – < 32 viikkoa ja 79,7 %:lla ≥ 32 – < 35 viikkoa; 52,4 % oli poikia; 72,2 % oli valkoihoisia; 17,6 % oli afrikkalaistaustaisia; 1,0 % oli aasialaisia; 59,5 % painoi < 5 kg (17,0 % painoi < 2,5 kg); 17,3 % imeväisistä oli ≤ 1,0 kuukauden ikäisiä, 35,9 % oli > 1,0 – ≤3,0 kuukauden ikäisiä, 32,6 % oli > 3,0 – ≤ 6,0 kuukauden ikäisiä ja 14,2 % oli > 6,0 kuukauden ikäisiä.
MELODY-tutkimuksessa (ensisijaisessa kohortissa) yhteensä 1 490 täysiaikaisina tai hieman ennenaikaisina syntynyttä imeväistä (gestaatioikä ≥ 35 viikkoa), joiden ensimmäinen RSV-kausi oli alkamassa, satunnaistettiin (2:1) saamaan kerta-annos nirsevimabia (50 mg nirsevimabia, jos paino antohetkellä < 5 kg, tai 100 mg nirsevimabia, jos paino antohetkellä ≥ 5 kg) tai lumelääkettä lihakseen. Satunnaistamishetkellä gestaatioikä oli 14,0 %:lla imeväisistä 35 – < 37 viikkoa ja 86,0 %:lla ≥ 37 viikkoa; 51,6 % oli poikia; 53,5 % oli valkoihoisia; 28,4 % oli afrikkalaistaustaisia; 3,6 % oli aasialaisia; 40,0 % painoi < 5 kg (2,5 % painoi < 2,5 kg); 24,5 % imeväisistä oli ≤ 1,0 kuukauden ikäisiä, 33,4 % oli > 1,0 – ≤ 3,0 kuukauden ikäisiä, 32,1 % oli > 3,0 – ≤ 6,0 kuukauden ikäisiä ja 10,0 % oli > 6,0 kuukauden ikäisiä.
Tutkimuksista suljettiin pois imeväiset, joilla oli anamneesissa keskosen krooninen keuhkosairaus / bronkopulmonaalinen dysplasia tai hemodynaamisesti merkittävä synnynnäinen sydänvika (lukuun ottamatta imeväisiä, joilla oli komplisoitumaton synnynnäinen sydänvika). Demografiset tiedot ja lähtötilanteen tiedot olivat molemmissa tutkimuksissa verrannolliset nirsevimabi- ja lumeryhmien välillä.
D5290C00003-tutkimuksessa ja MELODY-tutkimuksen ensisijaisessa kohortissa ensisijainen päätetapahtuma oli lääkärikäyntiin johtaneiden (MA; medically attended, mukaan lukien sairaalahoitoa vaatineet) RTPCR-tutkimuksella vahvistettujen RSV:n aiheuttamien alahengitystieinfektioiden (LRTI, lower respiratory track infection) eli RSV MA LRTI:n, tyypillisesti bronkioliitin tai keuhkokuumeen, ilmaantuvuus 150 vuorokauden kuluessa annoksen saamisesta. Alahengitystieinfektion merkkien määritelmä oli, että lääkärintarkastuksessa todettiin vähintään yksi alahengitystieinfektioon viittaava löydös (esimerkiksi rohina, rahina, ritinä tai vinkuna) ja vähintään yksi kliinisen tilan vakavuuden merkki (suurentunut hengitystiheys, hypoksemia, akuutti hypokseeminen hengitysvajaus tai vajaatuuletus, uutena oireena ilmenevät hengityskatkokset, nenänsiipihengitys, kylkiluuvälien sisään vetäytyminen, valittelu tai hengitysvaikeudesta johtuva kuivuminen). Toissijainen päätetapahtuma oli sairaalahoidon ilmaantuvuus imeväisillä, joilla oli lääkärikäyntiin johtanut RSV-alahengitystieinfektio (sairaalahoitoon johtanut RSV MA LRTI). RSV sairaalahoidon määritelmänä oli sairaalahoitoa vaativa alahengitystieinfektio, johon liittyi positiivinen RSV-testitulos, tai jo sairaalahoidossa olevan potilaan hengitysstatuksen paheneminen ja positiivinen RSV-testitulos. Myös hyvin vaikeaa lääkärikäyntiin johtanutta RSV-alahengitystieinfektiota arvioitiin (hyvin vaikea RSV MA LRTI). Se määriteltiin lääkärikäyntiin johtaneeksi RSV-alahengitystieinfektioksi, joka johti sairaalahoitoon ja jonka hoidossa tarvittiin lisähappea tai laskimonsisäistä nesteytystä.
Nirsevimabin teho RSV MA LRTI:a, sairaalahoitoon johtanutta RSV MA LRTI:a sekä hyvin vaikeaa RSV MA LRTI:a vastaan täysiaikaisina ja ennenaikaisina syntyneillä (gestaatioikä ≥ 29 viikkoa) imeväisillä, joiden ensimmäinen RSV-kausi oli alkamassa, on esitetty taulukossa 2.
Taulukko 2: Teho täysiaikaisina ja ennenaikaisina syntyneillä imeväisillä RSV MA LRTI:a, sairaalahoitoon johtanutta RSV MA LRTI:a sekä hyvin vaikeaa RSV MA LRTI:a vastaan 150 vuorokauden ajanjaksolla annoksen jälkeen (D5290C00003 ja MELODY [ensisijainen kohortti])
| Ryhmä | Hoito | N | Esiintyvyys, % (n) | Tehoa (95 %:n luottamusväli) |
| Teho imeväisillä RSV MA LRTI:a vastaan 150 vuorokauden ajanjaksolla annoksen jälkeen | ||||
| Hyvin tai kohtalaisen ennenaikaisina syntyneet, gestaatioikä ≥ 29 – < 35 viikkoa (D5290C00003)b | Nirsevimabi | 969 | 2,6 (25) | 70,1 % (52,3; 81,2)c |
| Lumelääke | 484 | 9,5 (46) | ||
| Täysiaikaisina tai hieman ennenaikaisina syntyneet, gestaatioikä ≥ 35 viikkoa (MELODY, ensisijainen kohortti) | Nirsevimabi | 994 | 1,2 (12) | 74,5 % (49,6; 87,1)c |
| Lumelääke | 496 | 5,0 (25) | ||
| Teho imeväisillä sairaalahoitoon johtanutta RSV MA LRTI:a vastaan 150 vuorokauden ajanjaksolla annoksen jälkeen | ||||
| Hyvin tai kohtalaisen ennenaikaisina syntyneet, gestaatioikä ≥ 29 – < 35 viikkoa (D5290C00003)b | Nirsevimabi | 969 | 0,8 (8) | 78,4 % (51,9; 90,3)c |
| Lumelääke | 484 | 4,1 (20) | ||
| Täysiaikaisina tai hieman ennenaikaisina syntyneet, gestaatioikä ≥ 35 viikkoa (MELODY, ensisijainen kohortti) | Nirsevimabi | 994 | 0,6 (6) | 62,1 % (-8,6; 86,8) |
| Lumelääke | 496 | 1,6 (8) | ||
| Teho imeväisillä hyvin vaikeaa RSV MA LRTI:a vastaan 150 vuorokauden ajanjaksolla annoksen jälkeen | ||||
| Hyvin tai kohtalaisen ennenaikaisina syntyneet, gestaatioikä ≥ 29 – < 35 viikkoa (D5290C00003)b | Nirsevimabi | 969 | 0,4 (4) | 87,5 % (62,9; 95,8)d |
| Lumelääke | 484 | 3,3 (16) | ||
| Täysiaikaisina tai hieman ennenaikaisina syntyneet, gestaatioikä ≥ 35 viikkoa (MELODY, ensisijainen kohortti) | Nirsevimabi | 994 | 0,5 (5) | 64,2 % (-12,1; 88,6)d |
| Lumelääke | 496 | 1,4 (7) | ||
a Perustuu suhteellisen riskin vähenemään lumelääkkeeseen verrattuna.
b Kaikki tutkittavat, jotka saivat 50 mg:n annoksen riippumatta painosta antohetkellä.
c Ennalta määritelty, monivertailukorjattu; p-arvo =< 0,001.
d Ei monivertailukorjausta.
Ensisijaisen tehoa koskevan päätemuuttujan alaryhmäanalyysit gestaatioiän, sukupuolen, etnisen taustan ja alueen mukaan osoittivat, että tulokset vastasivat koko populaation tuloksia.
Tutkittavien, joilla oli sairaalahoitoon johtanut RSV MA LRTI, läpimurtoinfektioiden vaikeusastetta arvioitiin. Lisähappea tarvinneiden tutkittavien osuus oli 44,4 % (4/9) ja 81,0 % (17/21), CPAP-hoitoa tai suurivirtauksista nenäkanyyliä (HFNC) tarvinneiden osuus oli 11,1 % (1/9) ja 23,8 % (5/21), ja teho-osastolle otettujen osuus oli 0 % (0/9) ja 28,6 % (6/21) nirsevimabiryhmässä ja lumeryhmässä, vastaavasti.
Imeväisten ottamista MELODY-tutkimukseen jatkettiin primaarianalyysin jälkeen, ja yhteensä 3 012 imeväistä satunnaistettiin saamaan Beyfortus-valmistetta (n = 2 009) tai lumelääkettä (n = 1 003). 150 päivän kuluttua annoksen antamisesta nirsevimabin teho suhteellisen riskin vähenemän perusteella oli lääkärikäyntiin johtanutta RSV-alahengitystieinfektiota vastaan 76,4 % (95 %:n luottamusväli 62,3; 85,2), lääkärikäyntiin ja sairaalahoitoon johtanutta RSV-alahengitystieinfektiota vastaan 76,8 % (95 %:n luottamusväli 49,4; 89,4) ja hyvin vaikeaa lääkärikäyntiin johtanutta RSV-alahengitystieinfektiota vastaan 78,6 % (95 %:n luottamusväli 48,8; 91,0).
Lääkärikäyntiin johtaneiden RSV‑alahengitystieinfektiotapahtumien osuudet toisen RSV‑kauden aikana (päivä 361 – päivä 510 annoksen saamisen jälkeen) olivat hoitoryhmissä samankaltaiset (19 [1,0 %] nirsevimabia saaneilla ja 10 [1,0 %] lumelääkettä saaneilla).
Teho lääkärikäyntiin johtanutta RSV-alahengitystieinfektiota vastaan imeväisillä, joilla on suurentunut riski saada vaikea RSV-tauti, ja toisella RSV-kaudellaan vaikealle RSV-taudille edelleen alttiilla lapsilla (MEDLEY ja MUSIC)
MEDLEY-tutkimuksessa satunnaistettiin yhteensä 925 imeväistä, joilla oli suurentunut riski saada vaikea RSV-tauti ja joiden ensimmäinen RSV-kausi oli alkamassa. Tutkittavien joukossa oli imeväisiä, joilla oli keskosen krooninen keuhkosairaus tai hemodynaamisesti merkittävä synnynnäinen sydänvika, sekä ennenaikaisina syntyneitä imeväisiä, joiden gestaatioikä oli alle 35 viikkoa. Imeväiset satunnaistettiin (2:1) saamaan joko kerta-annos nirsevimabia lihakseen (50 mg nirsevimabia, jos paino antohetkellä oli < 5 kg, tai 100 mg nirsevimabia, jos paino antohetkellä oli ≥ 5 kg) ja sen jälkeen lumelääkettä lihakseen kerran kuukaudessa 4 kuukauden ajan tai 15 mg/kg palivitsumabia lihakseen kerran kuukaudessa 5 kuukauden ajan. Satunnaistamishetkellä gestaatioikä oli 21,6 %:lla imeväisistä < 29 viikkoa, 21,5 %:lla ≥ 29 – < 32 viikkoa, 41,9 %:lla ≥ 32 – < 35 viikkoa ja 14,9 %:lla ≥ 35 viikkoa. Näistä imeväisistä 23,5 %:lla oli keskosen krooninen keuhkosairaus; 11,2 %:lla oli hemodynaamisesti merkittävä synnynnäinen sydänvika; 53,5 % oli poikia; 79,2 % oli valkoihoisia; 9,5 % oli afrikkalaistaustaisia; 5,4 % oli aasialaisia; 56,5 % painoi < 5 kg (9,7 % painoi < 2,5 kg); 11,4 % imeväisistä oli ≤ 1,0 kuukauden ikäisiä, 33,8 % oli > 1,0 – ≤3,0 kuukauden ikäisiä, 33,6 % oli > 3,0 – ≤ 6,0 kuukauden ikäisiä ja 21,2 % oli > 6,0 kuukauden ikäisiä.
Korkeintaan 24 kuukauden ikäiset lapset, joilla oli suurentunut vaikean RSV‑taudin riski keskosen kroonisen keuhkosairauden tai hemodynaamisesti merkittävän synnynnäisen sydänvian vuoksi ja jotka olivat edelleen alttiita vaikealle taudille, jatkoivat tutkimuksessa toisen RSV‑kautensa ajan. Tutkittavat, jotka saivat nirsevimabia ensimmäisen RSV‑kautensa aikana, saivat toisen 200 mg:n kerta‑annoksen nirsevimabia, kun heidän toinen RSV‑kautensa oli alkamassa (n = 180). Tämän jälkeen tutkittaville annettiin lumelääkettä lihakseen kerran kuukaudessa 4 kuukauden ajan. Palivitsumabia ensimmäisen RSV‑kautensa aikana saaneet tutkittavat satunnaistettiin uudelleen suhteessa 1:1 joko nirsevimabi‑ tai palivitsumabiryhmään, kun heidän toinen RSV‑kautensa oli alkamassa. Nirsevimabiryhmän tutkittaville (n = 40) annettiin 200 mg:n kiinteä kerta‑annos ja sen jälkeen lumelääkettä lihakseen kerran kuukaudessa 4 kuukauden ajan. Palivitsumabiryhmän tutkittaville (n = 42) annettiin 15 mg/kg palivitsumabia lihakseen kerran kuukaudessa 5 kuukauden ajan. Näistä lapsista 72,1 %:lla oli keskosen krooninen keuhkosairaus, 30,9 %:lla oli hemodynaamisesti merkittävä synnynnäinen sydänvika, 57,6 % oli poikia, 85,9 % oli valkoihoisia, 4,6 % oli afrikkalaistaustaisia, 5,7 % oli aasialaisia ja 2,3 % painoi < 7 kg. Demografiset tiedot ja lähtötilanteen tiedot olivat verrannolliset nirsevimabi‑nirsevimabi‑, palivitsumabi‑nirsevimabi‑ ja palivitsumabi‑palivitsumabiryhmien välillä.
Nirsevimabin teho imeväisillä, joilla oli suurentunut riski saada vaikea RSV-tauti, mukaan lukien erittäin ennenaikaisina syntyneet imeväiset (gestaatioikä < 29 viikkoa), joiden ensimmäinen RSV‑kausi oli alkamassa, ja ≤ 24 kuukauden ikäiset lapset, joilla oli keskosen krooninen keuhkosairaus tai hemodynaamisesti merkittävä synnynnäinen sydänvika ja joiden ensimmäinen tai toinen RSV‑kausi oli alkamassa, määritettiin ekstrapoloinnilla D5290C00003-tutkimuksessa ja MELODY-tutkimuksen ensisijaisessa kohortissa todetun tehon pohjalta, ja se perustuu farmakokineettiseen altistukseen (ks. kohta Farmakokinetiikka). MEDLEY-tutkimuksessa lääkärikäyntiin johtaneen RSV-alahengitystieinfektion (RSV MA LRTI) ilmaantuvuus 150 vuorokauden ajanjaksolla annoksen jälkeen oli 0,6 % (4/616) nirsevimabiryhmässä ja 1,0 % (3/309) palivitsumabiryhmässä ensimmäisellä RSV-kaudella. Toisella RSV-kaudella ei ilmennyt lääkärikäyntiin johtaneita RSV-alahengitystieinfektiotapauksia 150 päivän kuluessa annoksen saamisesta.
MUSIC‑tutkimuksessa nirsevimabin teho 100 immuunipuutteisella imeväisellä ja ≤ 24 kuukauden ikäisellä lapsella, jotka saivat nirsevimabia suositellulla annoksella, määritettiin ekstrapoloinnilla D5290C00003‑tutkimuksessa ja MELODY‑tutkimuksen ensisijaisessa kohortissa todetun tehon pohjalta, ja se perustuu farmakokineettiseen altistukseen (ks. kohta Farmakokinetiikka). Lääkärikäyntiin johtaneita RSV‑alahengitystieinfektiotapauksia ei ilmennyt 150 vuorokauden kuluessa annoksen saamisesta.
Teho sairaalahoitoon johtavan RSV-alahengitystieinfektion ehkäisemisessä täysiaikaisina tai ennenaikaisina syntyneillä imeväisillä (HARMONIE)
HARMONIE-tutkimuksessa satunnaistettiin yhteensä 8 058 täysiaikaisena tai ennenaikaisena syntynyttä imeväistä (gestaatioikä ≥ 29 viikkoa), joiden ensimmäinen RSV-kausi oli alkamassa tai jotka syntyivät kauden aikana. Imeväisille joko annettiin kerta-annos nirsevimabia lihakseen (50 mg, jos paino antohetkellä oli < 5 kg, tai 100 mg, jos paino antohetkellä oli ≥ 5 kg) tai ei tehty mitään interventiota. Mediaani-ikä oli satunnaistamishetkellä 4 kuukautta (vaihteluväli 0–12 kuukautta). 48,6 % imeväisistä oli ≤ 3 kuukauden ikäisiä, 23,7 % oli > 3 – ≤ 6 kuukauden ikäisiä ja 27,7 % oli > 6 kuukauden ikäisiä. Näistä imeväisistä 52,1 % oli poikia ja 47,9 % tyttöjä. Puolet näistä imeväisistä oli syntynyt RSV-kauden aikana. Suurin osa (85,2 %) tutkittavista oli täysiaikaisina syntyneitä imeväisiä, joiden gestaatioikä syntymähetkellä oli ≥ 37 viikkoa.
HARMONIE-tutkimuksen ensisijainen päätetapahtuma oli sairaalahoitoon johtaneiden, vahvistetusti RSV:n aiheuttamien alahengitystieinfektioiden kokonaisilmaantuvuus RSV-kauden aikana täysiaikaisina tai ennenaikaisina syntyneillä imeväisillä. Tosielämän olosuhteiden jäljittelemiseksi seuranta-aika otettiin huomioon, kun arvioitiin nirsevimabin tehoa sairaalahoitoon johtavien RSV-alahengitystieinfektioiden ehkäisemisessä verrattuna siihen, että mitään interventiota ei tehty. Tutkittavien seuranta-ajan mediaani oli 2,3 kuukautta (vaihteluväli 0–7,0 kuukautta) nirsevimabiryhmässä ja 2,0 kuukautta (vaihteluväli 0–6,8 kuukautta) ryhmässä, jossa ei tehty mitään interventiota.
Sairaalahoitoon johtaneita RSV-alahengitystieinfektioita ilmeni 11:llä imeväisellä 4 037:stä nirsevimabiryhmässä (ilmaantuvuus = 0,001) ja 60:llä imeväisellä 4 021:stä ryhmässä, jossa ei tehty mitään interventiota (ilmaantuvuus = 0,006). Tämä vastasi 83,2 %:n tehoa (95 %:n luottamusväli 67,8; 92,0) sairaalahoitoon johtavien RSV-alahengitystieinfektioiden ehkäisemisessä RSV-kauden aikana, ja teho säilyi 180 päivän ajan antohetkestä/satunnaistamisesta (82,7 %; 95 %:n luottamusväli 67,8; 91,5).
Suojan kesto
Kliinisten ja farmakokineettisten tietojen perusteella nirsevimabin antaman suojan kesto on vähintään 5–6 kuukautta.
Farmakokinetiikka
Nirsevimabin farmakokineettiset ominaisuudet perustuvat yksittäisistä tutkimuksista ja populaatiofarmakokineettisistä analyyseistä saatuihin tietoihin. Nirsevimabin farmakokinetiikka oli annoksesta riippuvaista lapsilla ja aikuisilla, joille annettiin kliinisesti relevantteja 25 mg:n – 300 mg:n nirsevimabiannoksia lihakseen.
Imeytyminen
Lihakseen annon jälkeen huippupitoisuus saavutettiin 6 vuorokauden kuluessa (vaihteluväli 1–28 vuorokautta) ja arvioitu absoluuttinen hyötyosuus oli 84 %.
Jakautuminen
Nirsevimabin arvioitu sentraalinen jakautumistilavuus 5 kg painavalla imeväisellä oli 216 ml ja perifeerinen jakautumistilavuus 261 ml. Jakautumistilavuus suurenee painon kasvaessa.
Biotransformaatio
Nirsevimabi on humaani monoklonaalinen IgG1κ-vasta-aine, jota hajottavat laajalti elimistössä esiintyvät proteolyyttiset entsyymit. Nirsevimabi ei metaboloidu maksaentsyymien välityksellä.
Eliminaatio
Monoklonaalisille vasta-aineille tyypilliseen tapaan nirsevimabi eliminoituu intrasellulaarisen katabolian välityksellä, eikä kohdevälitteisestä puhdistumasta ole näyttöä kliinisissä tutkimuksissa käytetyillä annoksilla.
Nirsevimabin arvioitu puhdistuma 5 kg:n painoisella imeväisellä oli 3,42 ml/vrk, ja terminaalinen puoliintumisaika oli noin 71 vuorokautta. Nirsevimabin puhdistuma suurenee painon kasvaessa.
Erityisryhmät
Etninen tausta
Etnisellä taustalla ei ollut kliinisesti merkittävää vaikutusta.
Munuaisten vajaatoiminta
Monoklonaalisille IgG-vasta-aineille tyypilliseen tapaan nirsevimabi ei suuren molekyylipainonsa takia poistu munuaisten kautta. Munuaistoiminnan muutosten ei odoteta vaikuttavan nirsevimabin puhdistumaan. Kliinisissä tutkimuksissa yhdellä nefroottista oireyhtymää sairastavalla potilaalla todettiin kuitenkin suurentunut nirsevimabin puhdistuma.
Maksan vajaatoiminta
Monoklonaaliset IgG‑vasta-aineet eivät ensisijaisesti poistu maksan kautta. Kliinisissä tutkimuksissa todettiin kuitenkin suurentunut nirsevimabin puhdistuma joillakin henkilöillä, joilla oli krooninen maksasairaus, johon saattoi liittyä proteiinihukka.
Imeväiset, joilla on suurentunut riski saada vaikea RSV-tauti, ja toisella RSV-kaudellaan vaikealle RSV-taudille edelleen alttiit lapset
Keskosen kroonisella keuhkosairaudella tai hemodynaamisesti merkittävällä synnynnäisellä sydänvialla ei ollut merkittävää vaikutusta nirsevimabin farmakokinetiikkaan. Pitoisuudet seerumissa päivänä 151 MEDLEY‑tutkimuksessa olivat verrannolliset MELODY‑tutkimuksessa todettujen pitoisuuksien kanssa.
Toisella RSV‑kaudellaan 200 mg:n nirsevimabiannoksen lihakseen saaneilla lapsilla, joilla oli keskosen krooninen keuhkosairaus tai hemodynaamisesti merkittävä synnynnäinen sydänvika (MEDLEY) tai jotka olivat immuunipuutteisia (MUSIC), nirsevimabialtistukset seerumissa olivat hieman suuremmat ja suurelta osin samalla alueella verrattuna MELODY‑tutkimuksen tutkittavilla todettuihin altistuksiin (ks. taulukko 3).
Taulukko 3: Altistukset lihakseen annettujen nirsevimabiannosten käytössä: keskiarvo (keskihajonta) [vaihteluväli]; johdettu yksilöllisten populaatiofarmakokineettisten parametrien perusteella
| Tutkimus/kausi | N (AUC) | AUC0–365 mg*vrk/ml | AUCbaseline CL mg*vrk/ml | N (pitoisuus seerumissa päivänä 151) | Pitoisuus seerumissa päivänä 151 µg/ml |
| MELODY (ensisijainen kohortti) | 954 | 12,2 (3,5) [3,3–24,9] | 21,3 (6,5) [5,2–48,7] | 636 | 26,6 (11,1) [2,1–76,6] |
| MEDLEY/kausi 1 | 591 | 12,3 (3,3) [4,1–23,4] | 22,6 (6,2) [7–43,8] | 457 | 27,8 (11,1) [2,1–66,2] |
| MEDLEY/kausi 2 | 189 | 21,5 (5,5) [7,5–41,9] | 23,6 (7,8) [8,2–56,4] | 163 | 55,6 (22,8) [11,2–189,3] |
| MUSIC/kausi 1 | 46 | 11,2 (4,3) [1,2–24,6] | 16,7 (7,3) [3,1–43,4] | 37 | 25,6 (13,4) [5,1–67,4] |
| MUSIC/kausi 2 | 50 | 16 (6,3) [2,2–25,5] | 21 (8,4) [5,6–35,5] | 42 | 33,2 (19,3) [0,9–68,5] |
AUC0–365 = pitoisuus‑aikakäyrän alle jäävä pinta‑ala 0–365 päivän kuluttua annoksen saamisesta, AUCbaseline CL = seerumin pitoisuus‑aikakäyrän alle jäävä pinta‑ala, joka on johdettu post hoc ‑puhdistuma‑arvoista annon yhteydessä, pitoisuus seerumissa päivänä 151 = pitoisuus käyntipäivänä 151 ± 14 päivää.
Farmakokineettiset/farmakodynaamiset suhteet
D5290C00003-tutkimuksessa ja MELODY-tutkimuksen ensisijaisessa kohortissa havaittiin positiivinen korrelaatio arvon 12,8 mg*vrk/ml ylittävän seerumin AUC:n (pitoisuus‑aikakäyrän alle jäävän pinta‑alan) ja lääkärikäyntiin johtaneiden RSV-alahengitystieinfektioiden matalamman ilmaantuvuuden välillä. AUC perustui lähtötilanteen puhdistumaan. Suositeltu annostusohjelma, jossa imeväisille annetaan ensimmäisellä RSV-kaudella 50 mg:n tai 100 mg:n annos lihakseen ja lapsille annetaan 200 mg:n annos lihakseen, kun toinen RSV-kausi on alkamassa, valittiin näiden tulosten perusteella.
MEDLEY-tutkimuksessa yli 80 %:lla imeväisistä, joilla oli suurentunut riski saada vaikea RSV-tauti, mukaan lukien erittäin ennenaikaisena syntyneet imeväiset (gestaatioikä < 29 viikkoa), ja joiden ensimmäinen RSV-kausi oli alkamassa, sekä imeväiset/lapset, joilla oli keskosen krooninen keuhkosairaus tai hemodynaamisesti merkittävä synnynnäinen sydänvika ja joiden ensimmäinen tai toinen RSV-kausi oli alkamassa, saavutettiin yhden annoksen jälkeen RSV:ltä suojaavaan vaikutukseen liittyvä nirsevimabialtistus (seerumin AUC yli 12,8 mg*vrk/ml) (ks. kohta Farmakodynamiikka).
MUSIC‑tutkimuksessa 75 %:lla (72/96) immuunipuutteisista imeväisistä/lapsista, joiden ensimmäinen tai toinen RSV‑kausi oli alkamassa, saavutettiin RSV:ltä suojaavaan vaikutukseen liittyvä nirsevimabialtistus. Kun tutkittavista suljettiin pois 14 lasta, joilla nirsevimabin puhdistuma oli suurentunut, 87 %:lla (71/82) saavutettiin RSV:ltä suojaavaan vaikutukseen liittyvä nirsevimabialtistus.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta ja kudosten ristireaktiivisuutta koskevien tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
L‑histidiini
L‑histidiinihydrokloridi
L‑arginiinihydrokloridi
Sakkaroosi
Polysorbaatti 80 (E433)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
4 vuotta
Beyfortus-valmistetta voidaan säilyttää huoneenlämmössä (20 °C –25 °C) valolta suojattuna enintään 48 tunnin ajan. Tämän jälkeen ruisku on hävitettävä.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Älä ravista äläkä altista suoralle lämmönlähteelle.
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BEYFORTUS injektioneste, liuos, esitäytetty ruisku
50 mg (L:ei) 0,5 ml (100 mg/ml) (471,71 €), 5 x 0,5 ml (2196,87 €)
100 mg (L:ei) 1 ml (100 mg/ml) (471,71 €), 5 x 1 ml (2196,87 €)
PF-selosteen tieto
Silikonoitu tyypin I lasista valmistettu esitäytetty Luer lock ‑ruisku, jossa on FluroTec-materiaalilla päällystetty männän pysäytin.
Yksi esitäytetty ruisku sisältää 0,5 ml tai 1 ml liuosta.
Pakkauskoot:
- 1 tai 5 esitäytettyä ruiskua ilman neulaa
- 1 esitäytetty ruisku ja kaksi erillistä, erikokoista neulaa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai keltainen liuos, jonka pH on 6,0.
Käyttö- ja käsittelyohjeet
Tämän lääkevalmisteen saa antaa vain koulutettu terveydenhuollon ammattilainen, ja antamisen yhteydessä on noudatettava aseptista tekniikkaa steriiliyden varmistamiseksi.
Tarkista ennen lääkevalmisteen antamista, näkyykö valmisteessa hiukkasia tai värimuutoksia. Lääkevalmiste on kirkas tai opaalinhohtoinen, väritön tai keltainen liuos. Älä anna injektiota, jos neste on sameaa, värjäytynyttä tai sisältää suuria hiukkasia tai vierasaineita.
Älä käytä, jos esitäytetty ruisku on pudonnut tai vahingoittunut tai jos pakkauksen turvasinetti on rikkoutunut.
Anto-ohjeet
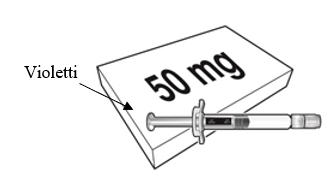
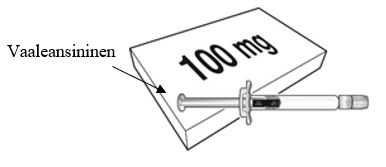
Beyfortus on saatavilla 50 mg:n ja 100 mg:n esitäytetyssä ruiskussa. Tarkista pakkauksen ja esitäytetyn ruiskun etiketeistä, että olet valinnut oikean vahvuuden, joko 50 mg tai 100 mg.
| Beyfortus 50 mg (50 mg / 0,5 ml) esitäytetty ruisku, jossa on violetti männänvarsi. | Beyfortus 100 mg (100 mg / 1 ml) esitäytetty ruisku, jossa on vaaleansininen männänvarsi. |
 |  |
Esitäytetyn ruiskun osat on esitetty kuvassa 1.
Kuva 1: Luer lock -ruiskun osat
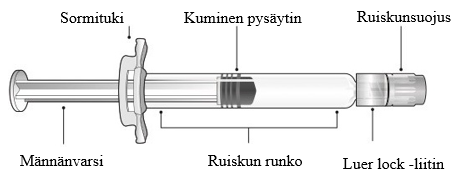
Vaihe 1: Pidä toisella kädellä kiinni Luer lock -liittimestä (älä pidä kiinni männänvarresta tai ruiskun rungosta) ja irrota ruiskunsuojus kiertämällä sitä vastapäivään toisella kädellä.
Vaihe 2: Kiinnitä Luer lock ‑neula esitäytettyyn ruiskuun kiertämällä neulaa varovasti myötäpäivään kiinni ruiskuun, kunnes tunnet kevyen vastuksen.
Vaihe 3: Pidä kiinni ruiskun rungosta toisella kädellä ja poista toisella kädellä neulansuojus vetämällä sitä varovasti suoraan ruiskusta poispäin. Älä pidä kiinni männänvarresta poistaessasi neulansuojusta, ettei kuminen pysäytin liiku. Älä koske neulaan äläkä anna sen koskea mihinkään. Älä laita neulansuojusta takaisin paikalleen äläkä irrota neulaa ruiskusta.
Vaihe 4: Anna esitäytetyn ruiskun koko sisältö lihakseen, mieluiten reiteen anterolateraalisesti. Pakaralihasta ei pidä käyttää rutiininomaisesti injektiokohtana iskiashermon vaurioitumisriskin takia.
Vaihe 5: Pane käytetty ruisku ja neula välittömästi terävälle jätteelle tarkoitettuun astiaan tai hävitä ne paikallisten vaatimusten mukaisesti.
Jos injektioita tarvitaan kaksi, toista vaiheet 1–5 käyttämällä eri injektiokohtaa.
Hävittäminen
Jokainen esitäytetty ruisku on tarkoitettu vain kertakäyttöön. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BEYFORTUS injektioneste, liuos, esitäytetty ruisku
50 mg 0,5 ml, 5 x 0,5 ml
100 mg 1 ml, 5 x 1 ml
- Ei korvausta.
ATC-koodi
J06BD08
Valmisteyhteenvedon muuttamispäivämäärä
12.03.2026
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi