
AQUIPTA tabletti 10 mg, 60 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
AQUIPTA 10 mg tabletit
Yksi tabletti sisältää 10 mg atogepanttia.
AQUIPTA 60 mg tabletit
Yksi tabletti sisältää 60 mg atogepanttia.
Apuaine, jonka vaikutus tunnetaan
Yksi 60 mg:n tabletti sisältää 31,5 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti
Kliiniset tiedot
Käyttöaiheet
AQUIPTA on tarkoitettu:
- aurallisen tai aurattoman migreenin kohtaushoitoon aikuisille
- migreenin estohoitoon aikuisille, joilla on vähintään 4 migreenipäivää kuukaudessa.
Annostus ja antotapa
Annostus
Enimmäisvuorokausiannos on 60 mg atogepanttia.
Migreenikohtauksen hoitoon tarvittaessa otettuna, suositeltu annos on 60 mg atogepanttia.
Migreenin estohoitoon suositeltu annos on 60 mg atogepanttia kerran päivässä.
Tabletit voidaan ottaa ruoan kanssa tai ilman ruokaa.
Annoksen unohtuminen
Migreenin estohoidossa unohtunut annos on otettava heti muistettaessa. Jos potilas ei muista ottaa annosta koko päivänä, unohtunut annos on jätettävä väliin ja seuraava annos otettava aikataulun mukaisesti.
Annosmuutokset
Annosmuutokset, jotka ovat tarpeen samanaikaisessa käytössä tiettyjen lääkevalmisteiden kanssa, on annettu taulukossa 1 (ks. kohta Yhteisvaikutukset).
Taulukko 1: Annosmuutokset yhteisvaikutusten vuoksi
Annosmuutokset | Suositeltu kerran päivässä otettava annos |
Voimakkaat CYP3A4:n estäjät | 10 mg |
Voimakkaat OATP:n estäjät | 10 mg |
Erityisryhmät
Iäkkäät
Populaatiofarmakokineettinen mallinnus ei viittaa kliinisesti merkittäviin farmakokineettisiin eroihin iäkkäiden ja nuorempien tutkittavien välillä. Annosta ei tarvitse muuttaa iäkkäille potilaille.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma [CLcr] 15–29 ml/min) ja loppuvaiheen munuaissairautta (ESRD) (kreatiniinipuhdistuma < 15 ml/min) sairastaville potilaille suositeltu annos on 10 mg kerran päivässä. Jaksoittaista dialyysihoitoa saavien ESRD-potilaiden on suositeltavaa ottaa AQUIPTA-valmiste dialyysin jälkeen.
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa lievää tai keskivaikeaa maksan vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka). Atogepantin käyttöä on vältettävä potilaille, joilla on vaikea maksan vajaatoiminta.
Pediatriset potilaat
Atogepantin turvallisuutta ja tehoa (alle 18-vuotiaiden) lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
AQUIPTA otetaan suun kautta. Tabletit on nieltävä kokonaisina, eikä niitä saa halkaista, murskata tai pureskella.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Vakavat yliherkkyysreaktiot
Vakavia yliherkkyysreaktioita, mukaan lukien anafylaksiaa, hengenahdistusta, ihottumaa, kutinaa, nokkosihottumaa ja kasvojen turvotusta, on raportoitu AQUIPTA-valmisteen käytön yhteydessä (ks. kohta Haittavaikutukset). Useimmat vakavat reaktiot ovat ilmenneet 24 tunnin kuluessa ensimmäisestä käytöstä, mutta joitain yliherkkyysreaktioita voi esiintyä päiviä annostelun jälkeen. Potilaita on varoitettava yliherkkyyteen liittyvistä oireista. Jos yliherkkyysreaktio ilmenee, keskeytä AQUIPTA ja aloita asianmukainen hoito.
Maksan vajaatoiminta
Atogepanttia ei suositella potilaille, joilla on vaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
Apuaineet, joiden vaikutus tunnetaan
AQUIPTA 10 mg tabletit sisältävät alle 1 mmol natriumia (23 mg) per tabletti eli niiden voidaan sanoa olevan ”natriumittomia”.
AQUIPTA 60 mg tabletit sisältävät 31,5 mg natriumia per tabletti, joka vastaa 1,6 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Yhteisvaikutukset
CYP3A4:n estäjät
Voimakkaat CYP3A4:n estäjät (esim. ketokonatsoli, itrakonatsoli, klaritromysiini ja ritonaviiri) voivat lisätä merkittävästi atogepantin systeemistä altistusta. Atogepantin samanaikainen anto itrakonatsolin kanssa lisäsi atogepanttialtistusta terveillä tutkittavilla (Cmax suurentui 2,15-kertaiseksi ja AUC 5,5-kertaiseksi) (ks. kohta Annostus ja antotapa). Atogepanttialtistuksen ei odoteta muuttuvan kliinisesti merkittävästi samanaikaisessa käytössä heikkojen tai kohtalaisten CYP3A4:n estäjien kanssa.
Kuljettajaproteiinien estäjät
Orgaanisten anionien kuljettajapolypeptidien (OATP) estäjät (esim. rifampisiini, siklosporiini ja ritonaviiri) voivat lisätä merkittävästi atogepantin systeemistä altistusta. Atogepantin samanaikainen anto rifampisiinin kerta-annoksen kanssa lisäsi atogepanttialtistusta terveillä tutkittavilla (Cmax suurentui 2,23-kertaiseksi ja AUC 2,85-kertaiseksi) (ks. kohta Annostus ja antotapa).
Usein samanaikaisesti käytettävät lääkevalmisteet
Atogepantin samanaikainen anto suun kautta otettavien ehkäisyvalmisteiden komponenttien etinyyliestradiolin ja levonorgestreelin, parasetamolin, naprokseenin, sumatriptaanin tai ubrogepantin kanssa ei vaikuttanut merkittävästi atogepantin eikä samanaikaisesti annettujen lääkevalmisteiden farmakokinetiikkaan. Famotidiinin tai esomepratsolin samanaikainen anto ei muuttanut atogepanttialtistusta kliinisesti merkittävällä tavalla.
Raskaus ja imetys
Raskaus
On vain vähän tietoja atogepantin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Atogepantin käyttöä ei suositella raskauden aikana eikä hedelmällisessä iässä oleville naisille, jotka eivät käytä ehkäisyä.
Imetys
Farmakokineettiset tiedot osoittivat, että yksittäisen annoksen annon jälkeen atogepantin erittyminen rintamaitoon oli vähäistä (ks. kohta Farmakokinetiikka).
Atogepantin vaikutuksista imetettävään lapseen tai rintamaidontuotantoon ei ole tietoja.
Imetyksen kehitykseen ja terveyteen liittyvät hyödyt on arvioitava suhteessa äidin kliiniseen atogepantin käytön tarpeeseen sekä mahdollisiin haittavaikutuksiin, joita äidin atogepantin käytöstä tai äidillä olevasta sairaudesta aiheutuu imetettävälle lapselle.
Hedelmällisyys
Tietoja atogepantin vaikutuksesta ihmisen hedelmällisyyteen ei ole saatavilla. Eläinkokeissa atogepanttihoidon ei havaittu vaikuttavan naaraiden ja urosten hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Atogepantilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Se voi kuitenkin aiheuttaa joillekin henkilöille uneliaisuutta. Potilaiden on oltava varovaisia ennen ajamista tai koneiden käyttöä, kunnes ovat varmistuneet, että atogepantti ei vaikuta haitallisesti suorituskykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Turvallisuutta arvioitiin 3 852 migreenipotilaalla, jotka saivat vähintään yhden annoksen atogepanttia kliinisissä tutkimuksissa. Näistä potilaista 1 225 potilaan altistus päivittäisessä estohoidossa kesti vähintään 6 kuukautta ja 826 potilaan altistus 12 kuukautta. Migreenikohtausten hoidossa (lääkkeen otto tarpeen mukaan) 895 potilasta altistettiin 24 viikon ajan.
Lumekontrolloiduissa, 12 viikon pituisissa kliinisissä estohoitotutkimuksissa 678 potilasta sai vähintään yhden 60 mg:n atogepanttiannoksen kerran päivässä, ja 663 potilasta sai lumelääkettä. Lumekontrolloidussa kliinisessä tutkimuksessa, jossa tutkittiin migreenin kohtaushoitoa, 1 195 potilasta sai vähintään yhden 60 mg:n atogepanttiannoksen ja 1 177 potilasta sai lumelääkettä; potilaat saivat sekä atogepanttia että lumelääkettä määritelmän täyttävien migreenikohtausten hoitoon.
Lumekontrolloiduissa estohoitotutkimuksissa yleisimmin raportoidut haittavaikutukset olivat pahoinvointi (9 %), ummetus (8 %) ja väsymys/uneliaisuus (5 %). Useimmat reaktiot olivat vakavuudeltaan lieviä tai kohtalaisia. Estohoidon lopettamiseen yleisimmin johtanut haittavaikutus oli pahoinvointi (0,4 %). Pahoinvointi (1,3 %) oli yleisin kohtaushoidossa raportoitu haittavaikutus.
Haittavaikutustaulukko
Kliinisissä tutkimuksissa ja markkinoille tulon jälkeen raportoidut haittavaikutukset on lueteltu alla elinjärjestelmän ja esiintymistiheyden mukaan. Yleisimmin esiintyneet haittavaikutukset on lueteltu ensin. Esiintymistiheysluokat ovat seuraavat: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on kussakin esiintymistiheysluokassa esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2. Atogepanttiin liittyvät haittavaikutukset
| Elinjärjestelmäluokka | Esiintymisti-heys | Haittavaikutus |
| Migreenin kohtaushoito | ||
| Ruoansulatuselimistö | Yleinen | Pahoinvointi |
| Tutkimukset | Melko harvinainen | ALAT-/ASAT-arvojen kohoaminen** |
| Migreenin estohoito | ||
| Immuunijärjestelmä | Tuntematon | Yliherkkyys (esim. anafylaksia, hengenahdistus, ihottuma, kutina, nokkosihottuma, kasvojen turvotus) |
| Aineenvaihdunta ja ravitsemus | Yleinen | Ruokahalun heikentyminen |
| Ruoansulatuselimistö | Yleinen | Pahoinvointi Ummetus |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Väsymys/uneliaisuus |
| Tutkimukset | Yleinen | Painonlasku* |
| Melko harvinainen | ALAT-/ASAT-arvojen kohoaminen** | |
* Määriteltiin kliinisissä tutkimuksissa vähintään 7 %:n painonlaskuna missä tahansa vaiheessa.
** Kliinisissä tutkimuksissa havaittiin, että atogepantilla oli ajallinen yhteys ALAT-/ASAT-arvojen kohoamisiin (kohonneiksi määriteltiin vähintään 3 kertaa viitealueen ylärajaa suuremmat arvot). Mukana olivat tapaukset, joissa haittavaikutukset olivat hävinneet 8 viikon kuluessa hoidon keskeyttämisestä, kun altistus lääkkeelle lopetettiin. Maksaentsyymiarvojen kokonaisesiintymistiheys oli kuitenkin samankaltainen atogepantti- ja lumelääkeryhmissä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa atogepanttia annettiin enintään 300 mg:n kerta-annoksena ja enintään 170 mg:n toistuvina annoksina kerran päivässä. Haittavaikutukset vastasivat pienemmillä annoksilla raportoituja haittavaikutuksia, eikä erityisiä toksisuuksia havaittu. Atogepantille ei ole tunnettua vastalääkettä. Yliannostusta on hoidettava yleisillä tukitoimenpiteillä mukaan lukien elintoimintojen tarkkailu ja potilaan kliinisen tilan seuranta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Analgeetit, kalsitoniinigeeniin liittyvän peptidin (CGRP) antagonistit, ATC-koodi: N02CD07
Vaikutusmekanismi
Ei-kliiniset tutkimukset reseptoreihin sitoutumisesta ja toiminnalliset in vitro -tutkimukset viittaavat siihen, että useampi kuin yksi reseptorityyppi on yhteydessä atogepantin farmakologisiin vaikutuksiin. Atogepantilla on havaittu affiniteettia useisiin kalsitoniinin/kalsitoniinigeeniin liittyvän peptidin (CGRP) reseptoriperheen reseptoreihin. Kun otetaan huomioon atogepantin kliinisesti merkitykselliset vapaat plasmapitoisuudet (60 mg:n annoksella Cmax > 20 nM) ja se, että CGRP:n ja amyliini-1:n reseptorien katsotaan olevan yhteydessä migreenin patogysiologiaan, atogepantin inhiboivilla vaikutuksilla näihin reseptoreihin (CGRP:n Ki-arvo on 26 pM ja amyliini-1:n 2,4 nM) voi olla kliinistä merkitystä. Atogepantin tarkkaa vaikutusmekanismia migreenin estohoidossa tai kohtaushoidossa ei kuitenkaan ole vielä selvitetty.
Kliininen teho ja turvallisuus
Migreenin kohtaushoito
AQUIPTA-valmisteen tehoa aurallisen tai aurattoman migreenin kohtaushoidossa aikuisilla tutkittiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (ECLIPSE). 16 viikon sokkoutetun hoitojakson aikana 1 328 tutkittavaa satunnaistettiin yhteen neljästä sekvenssiryhmästä hoitamaan 4 määritelmän täyttävää migreenikohtausta, joissa päänsäryn voimakkuus oli kohtalainen tai vaikea, joko 60 mg:lla atogepanttia (3 kohtausta) tai lumelääkkeellä (1 kohtaus) ennalta määritetyssä järjestyksessä. Tutkittavat, jotka suorittivat tutkimuksen sokkoutetun jakson, hoitivat myöhemmät kohtaukset avoimessa vaiheessa 60 mg:n atogepantilla viikon 24 loppuun asti. Noin 22 prosenttia tutkittavista käytti samanaikaisesti migreenin estohoitolääkitystä (esim. topiramaatti, amitriptyliini, propranololi). Varalääkitys sallittiin alkaen 2 tuntia tutkimuslääkkeen oton jälkeen. CGRP-reittiin vaikuttavien lääkevalmisteiden samanaikainen käyttö migreenin kohtaus- tai estohoitoon ei ollut sallittua.
Kaksoissokkoutetun tutkimusjakson aikana 1 037 tutkittavaa hoiti 4 määritelmän täyttävää migreenikohtausta. Migreenin keskimääräinen esiintymistiheys oli noin 4 kohtalaista tai vaikeaa migreenikohtausta kuukaudessa tutkimuksen mukaanottoa edeltävien 3 kuukauden aikana.
Ensisijainen tehon päätetapahtuma oli kivuttomuus (määritelty päänsäryn vaikeusasteen vähenemisenä kohtalaisesta/vaikeasta kivuttomuuteen) 2 tunnin kohdalla kohtauksen 1 aikana (taulukko 3). Toissijaisiin päätetapahtumiin kuuluivat potilaan määrittämän häiritsevimmän oireen poissaolo 2 tunnin kohdalla, kivunlievitys (määritelty päänsäryn vaikeusasteen vähenemisenä kohtalaisesta/vaikeasta lievään/kivuttomuuteen) 2 tunnin kohdalla sekä yhtämittainen kivuttomuus 2 tunnista 48 tuntiin. AQUIPTA-valmiste osoitti tilastollisesti merkitsevää ja kliinisesti merkityksellistä paranemista näiden päätetapahtumien osalta lumelääkkeeseen verrattuna. Lisäksi tilastollisesti merkitseviä vaikutuksia osoitettiin toissijaisten päätetapahtumien osalta, jotka koskivat varalääkitystä 24 tunnin sisällä ja kykyä toimia normaalisti 2 tunnin kohdalla.
Taulukko 3: Tehon päätetapahtumat kohtauksen 1 aikana ECLIPSE-tutkimuksessa
AQUIPTA 60 mg N = 602 | Lumelääke N = 612 | |
| Kivuttomuus 2 tunnin kohdalla | ||
| Vasteen saavuttaneiden osuus (%) | 24,3 | 13,1 |
| Kerroinsuhde (95 %:n luottamusväli) | 2,36 (1,76; 3,15) | |
| p-arvo | < 0,0001a | |
| Vapaus häiritsevimmästä oireestab 2 tunnin kohdalla | ||
| Vasteen saavuttaneiden osuus (%) | 43,7 | 32,7 |
| Kerroinsuhde (95 %:n luottamusväli) | 1,77 (1,41; 2,24) | |
| p-arvo | < 0,0001a | |
| Kivunlievitys 2 tunnin kohdalla | ||
| Vasteen saavuttaneiden osuus (%) | 72,1 | 54,4 |
| Kerroinsuhde (95 %:n luottamusväli) | 2,35 (1,85; 2,98) | |
| p-arvo | < 0,0001a | |
| Yhtämittainen kivuttomuus 2 tunnista 48 tuntiin | ||
| Vasteen saavuttaneiden osuus (%) | 16,6 | 5,7 |
| Kerroinsuhde (95 %:n luottamusväli) | 3,62 (2,43; 5,39) | |
| p-arvo | < 0,0001a | |
ap-arvoja muokattu useita vertailuja varten b Potilaan määrittäminä pahoinvointi (31 %), valonarkuus (44 %) tai fonofobia (25 %) | ||
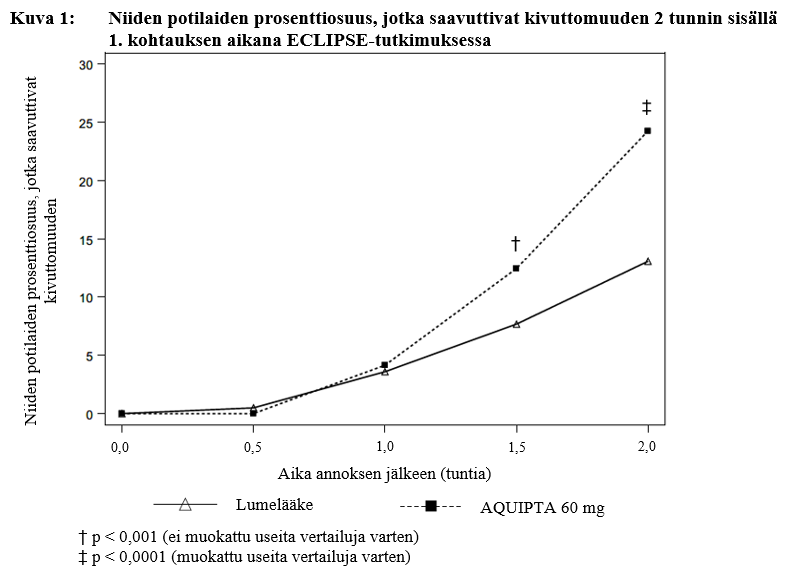
Suurempi prosenttiosuus AQUIPTA 60 mg-valmisteella hoidetuista potilaista saavutti kivuttomuuden 1,5 tunnista alkaen kohtauksen 1 aikana verrattuna lumelääkettä saaneisiin potilaisiin (kuva 1). Atogepantti osoitti myös kliinisesti merkittävän ja johdonmukaisen vaikutuksen, kun 26,3 prosenttia potilaista saavutti kivuttomuuden 2 tunnin kohdalla vähintään kahdessa kolmesta migreenikohtauksesta.
Migreenin estohoito
Atogepanttia migreenin estohoidossa arvioitiin kahdessa kroonista ja episodista migreenityyppiä koskevassa pivotaalitutkimuksessa. Episodista migreeniä koskevaan tutkimukseen (ADVANCE) otettiin potilaita, jotka täyttivät aurallisen tai aurattoman migreenin ICHD (International Classification of Headache Disorders) -diagnoosikriteerit. Kroonista migreeniä koskevaan tutkimukseen (PROGRESS) otettiin potilaita, jotka täyttivät lisäksi kroonisen migreenin ICHD-kriteerit. Kumpaankaan tutkimukseen ei otettu potilaita, joilla oli seulontaa edeltävien 6 kuukauden aikana ollut sydäninfarkti, aivohalvaus tai TIA-kohtaus.
Episodinen migreeni
Atogepanttia episodisen migreenin (4–14 migreenipäivää kuukaudessa) estohoidossa arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (ADVANCE). Potilaat satunnaistettiin saamaan AQUIPTA 60 mg -valmistetta (N = 235) tai lumelääkettä (N = 223) kerran päivässä 12 viikon ajan. Potilaiden sallittiin käyttää päänsäryn kohtaushoitoja (eli triptaaneja, ergotamiinin johdoksia, NSAID-lääkkeitä, parasetamolia ja opioideja) tarpeen mukaan. CGRP-reittiin vaikuttavien kohtaus- tai estolääkevalmisteiden samanaikaista käyttöä ei sallittu.
Potilaista 88 % oli mukana 12 viikon pituisen kaksoissokkoutetun tutkimusjakson loppuun asti. Potilaiden keski-ikä oli 42 vuotta (vaihteluväli: 18–73 vuotta). Potilaista 4 % oli vähintään 65 vuoden ikäisiä, 89 % naisia ja 83 % valkoihoisia. Kuukausittaisten migreenipäivien keskilukumäärä lähtötilanteessa oli noin 8, ja tämä lukumäärä oli samankaltainen hoitoryhmien välillä.
Ensisijainen tehon päätetapahtuma oli kuukausittaisten migreenipäivien keskilukumäärän muutos lähtötilanteesta 12 viikon pituisen hoitojakson aikana. Kerrannaisuuden suhteen kontrolloituja toissijaisia päätetapahtumia olivat kuukausittaisten päänsärkypäivien keskilukumäärän muutos lähtötilanteesta, kuukausittaisten kohtauslääkityksen käyttöpäivien keskilukumäärän muutos lähtötilanteesta, niiden potilaiden osuus, joiden kuukausittaisten migreenipäivien keskilukumäärä pieneni vähintään 50 % lähtötilanteesta (3 kuukauden keskiarvo), ja useat potilaiden raportoimat, toimintakykyä arvioivat elämänlaatumittareihin (elämänlaatuun) liittyvät tulosmittaukset.
ADVANCE-tutkimuksessa saatiin tilastollisesti merkitsevää näyttöä AQUIPTA-valmisteen tehosta lumelääkkeeseen verrattuna ensisijaisissa ja toissijaisissa tehon päätetapahtumissa. Yhteenveto tästä on esitetty taulukossa 4.
Taulukko 4: Tehon päätetapahtumat ADVANCE-tutkimuksessa
AQUIPTA 60 mg N = 226 | Lumelääke N = 216 | |
| Kuukausittaiset migreenipäivät 12 viikon aikana | ||
| Lähtötilanne | 7,8 | 7,5 |
| Keskimääräinen muutos lähtötilanteesta | -4,1 | -2,5 |
| Ero lumelääkkeeseen | -1,7 | |
| p-arvo | < 0,001 | |
| Kuukausittaiset päänsärkypäivät 12 viikon aikana | ||
| Lähtötilanne | 9,0 | 8,5 |
| Keskimääräinen muutos lähtötilanteesta | -4,2 | -2,5 |
| Ero lumelääkkeeseen | -1,7 | |
| p-arvo | < 0,001 | |
| Kuukausittaiset kohtauslääkityksen käyttöpäivät12 viikon aikana | ||
| Lähtötilanne | 6,9 | 6,5 |
| Keskimääräinen muutos lähtötilanteesta | -3,8 | -2,3 |
| Ero lumelääkkeeseen | -1,4 | |
| p-arvo | < 0,001 | |
| Kuukausittaisten migreenipäivien vähentyminen ≥ 50 %:lla 12 viikon aikana | ||
| Vasteen saavuttaneiden osuus (%) | 59 | 29 |
| Kerroinsuhde (95 %:n luottamusväli) | 3,55 (2,39, 5,28) | |
| p-arvo | < 0,001 | |
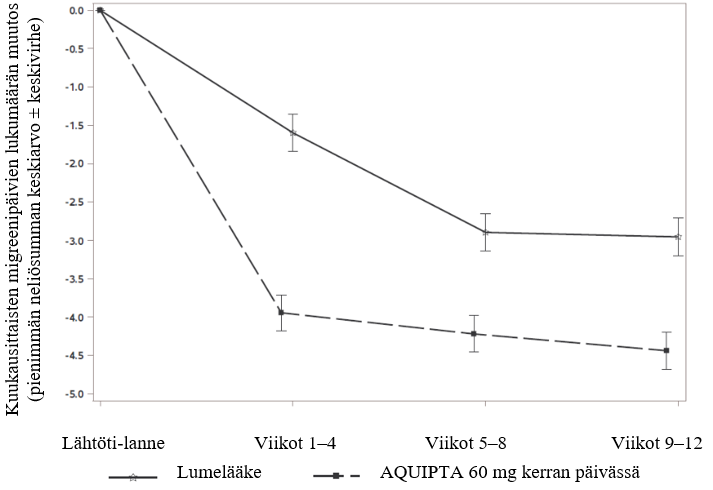
Kuvassa 2 on esitetty kuukausittaisten migreenipäivien keskimääräisen lukumäärän muutos lähtötilanteesta ADVANCE-tutkimuksessa. Kuukausittaisten migreenipäivien keskimäärä väheni lähtötilanteesta 12 viikon pituisen hoitojakson aikana enemmän potilailla, jotka saivat AQUIPTA 60 mg -valmistetta kerran päivässä, verrattuna lumelääkettä saaneisiin potilaisiin. Ensimmäisten 4 hoitoviikon aikana AQUIPTA 60 mg kerran päivässä pienensi kuukausittaisten migreenipäivien keskilukumäärää lähtötilanteeseen nähden merkitsevästi verrattuna lumelääkettä saaneisiin potilaisiin.
Kuva 2: Kuukausittaisten migreenipäivien lukumäärän muutos lähtötilanteesta ADVANCE-tutkimuksessa
Pitkäaikainen teho
Teho säilyi jopa yhden vuoden ajan avoimessa tutkimuksessa, jossa 546 episodista migreeniä sairastavaa potilasta satunnaistettiin saamaan AQUIPTA 60 mg -valmistetta kerran päivässä. Potilaista 68 % (373/546) oli mukana koko hoitojakson ajan. Kuukausittaisten migreenipäivien vähenemän pienimmän neliösumman keskiarvo oli ensimmäisenä kuukautena (viikot 1–4) -3,8 päivää, ja se parani viimeisenä kuukautena (viikot 49–52) -5,2 päivään. Viimeisenä kuukautena (viikot 49–52) n. 84 % potilaista raportoi kuukausittaisten migreenipäiviensä vähentyneen ≥ 50 %:lla, n. 70 % raportoi niiden vähentyneen ≥ 75 %:lla ja n. 48 % raportoi niiden vähentyneen 100 %:lla.
Potilaat, joilla 2–4 eri luokan suun kautta otettava estohoito on aiemmin epäonnistunut
ELEVATE-tutkimuksessa 315 episodista migreeniä sairastavaa aikuispotilasta, joilla oli 2–4 aiempaa tehon ja/tai siedettävyyden perusteella epäonnistunutta suun kautta otettua estohoitoa (esim. topiramaatti, trisykliset masennuslääkkeet, beetasalpaajat), satunnaistettiin 1:1 saamaan joko 60 mg atogepanttia (N = 157) tai lumelääkettä (N = 158) 12 viikon ajan. Tämän tutkimuksen tulokset olivat yhdenmukaisia aiempien episodisen migreenin tehoa koskevien tutkimusten päähavaintojen kanssa ja tilastollisesti merkitseviä primaaristen ja sekundaaristen tehon päätetapahtumien osalta, mukaan lukien useat potilaiden ilmoittamat toimintakykyä arvioivat tulosmittaukset. Atogepanttihoito vähensi kuukausittaisten migreenipäivien keskilukumäärää 4,2 vuorokautta verrattuna lumeryhmän 1,9 vuorokauden vähenemään (p <0,001). 50,6 % (78/154) atogepanttiryhmän potilaista saavutti kuukausittaisten migreenipäivien keskilukumäärän vähenemisen vähintään 50 %:lla lähtötilanteesta verrattuna lumelääkeryhmän 18,1 %:n vähenemään (28/155) (kerroinsuhde [95 %:n luottamusväli]: 4,82 [2,85; 8,14], p <0,001).
Krooninen migreeni
Atogepanttia kroonisen migreenin (kuukaudessa vähintään 15 päänsärkypäivää, joista vähintään 8 migreenipäivää) estohoidossa arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (PROGRESS). Potilaat satunnaistettiin saamaan AQUIPTA 60 mg -valmistetta (N = 262) tai lumelääkettä (N = 259) kerran päivässä 12 viikon ajan. Osan potilaista (11 %) sallittiin käyttää samanaikaisesti yhtä migreenin estohoitoon tarkoitettua lääkevalmistetta (esim. amitriptyliinia, propranololia tai topiramaattia). Kaikkien potilaiden sallittiin käyttää päänsäryn kohtaushoitoja (eli triptaaneja, ergotamiinin johdoksia, NSAID-lääkkeitä, parasetamolia ja opioideja) tarpeen mukaan. Tutkimukseen otettiin myös potilaita, joilla oli kohtauslääkkeiden ylikäyttöä ja lääkepäänsärkyä. CGRP-reittiin vaikuttavien kohtaus- tai estolääkevalmisteiden samanaikaista käyttöä ei sallittu.
Yhteensä 463 potilasta (89 %) oli mukana 12 viikon pituisen kaksoissokkoutetun tutkimuksen loppuun asti. Potilaiden keski-ikä oli 42 vuotta (vaihteluväli: 18–74 vuotta). Potilaista 3 % oli vähintään 65 vuoden ikäisiä, 87 % naisia ja 59 % valkoihoisia. Kuukausittaisten migreenipäivien keskilukumäärä lähtötilanteessa oli noin 19, ja tämä lukumäärä oli samankaltainen hoitoryhmien välillä.
Ensisijainen tehon päätetapahtuma oli kuukausittaisten migreenipäivien keskilukumäärän muutos lähtötilanteesta 12 viikon pituisen hoitojakson aikana. Kerrannaisuuden suhteen kontrolloituja toissijaisia päätetapahtumia olivat kuukausittaisten päänsärkypäivien keskilukumäärän muutos lähtötilanteesta, kuukausittaisten kohtauslääkityksen käyttöpäivien keskilukumäärän muutos lähtötilanteesta, niiden potilaiden osuus, joiden kuukausittaisten migreenipäivien keskilukumäärä pieneni vähintään 50 % lähtötilanteesta (3 kuukauden keskiarvo), ja useat potilaiden raportoimat, toimintakykyä arvioivat elämänlaatumittareihin (elämänlaatuun) liittyvät tulosmittaukset. PROGRESS-tutkimuksessa saatiin tilastollisesti merkitsevää näyttöä AQUIPTA-valmisteen tehosta lumelääkkeeseen verrattuna ensisijaisissa ja toissijaisissa tehon päätetapahtumissa. Yhteenveto näistä tuloksista on esitetty taulukossa 5.
Taulukko 5: Tehon päätetapahtumat PROGRESS-tutkimuksessa
AQUIPTA 60 mg N = 257 | Lumelääke N = 249 | |
| Kuukausittaiset migreenipäivät 12 viikon aikana | ||
| Lähtötilanne | 19,2 | 19,0 |
| Keskimääräinen muutos lähtötilanteesta | -6,8 | -5,1 |
| Ero lumelääkkeeseen | -1,7 | |
| p-arvo | 0,002 | |
| Kuukausittaiset päänsärkypäivät 12 viikon aikana | ||
| Lähtötilanne | 21,5 | 21,4 |
| Keskimääräinen muutos lähtötilanteesta | -6,9 | -5,2 |
| Ero lumelääkkeeseen | -1,7 | |
| p-arvo | 0,002 | |
| Kuukausittaiset kohtauslääkityksen käyttöpäivät12 viikon aikana | ||
| Lähtötilanne | 15,5 | 15,3 |
| Keskimääräinen muutos lähtötilanteesta | -6,2 | -4,1 |
| Ero lumelääkkeeseen | -2,1 | |
| p-arvo | 0,002 | |
| Kuukausittaisten migreenipäivien vähentyminen ≥ 50 %:lla 12 viikon aikana | ||
| Vasteen saavuttaneiden osuus (%) | 40 | 27 |
| Kerroinsuhde (95 %:n luottamusväli) | 1,90 (1,29, 2,79) | |
| p-arvo | 0,002 | |
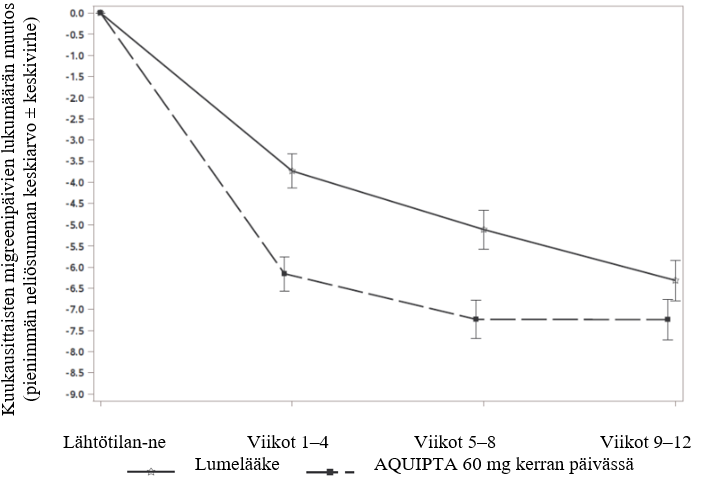
Kuvassa 3 on esitetty kuukausittaisten migreenipäivien keskimäärän muutos lähtötilanteesta PROGRESS-tutkimuksessa. Kuukausittaisten migreenipäivien keskimäärä väheni lähtötilanteesta 12 viikon pituisen hoitojakson aikana enemmän potilailla, jotka saivat AQUIPTA 60 mg -valmistetta kerran päivässä, verrattuna lumelääkettä saaneisiin potilaisiin.
Kuva 3: Kuukausittaisten migreenipäivien lukumäärän muutos lähtötilanteesta PROGRESS-tutkimuksessa
Siedettävyys ja teho verrattuna topiramaattiin
TEMPLE-tutkimuksessa 540 aikuispotilasta, joilla oli episodinen tai krooninen migreeni, satunnaistettiin saamaan joko 60 mg atogepanttia kerran vuorokaudessa (N = 273) tai 50–100 mg topiramaattia vuorokaudessa (N = 267) 24 viikon ajan kaksoissokkoutetun vaiheen aikana siedettävyyden, turvallisuuden ja tehon arvioimiseksi. Atogepantti osoitti parempaa siedettävyyttä verrattuna topiramaattiin perustuen haittatapahtumien vuoksi hoidon keskeyttäneiden osuuteen (atogepantti: 12,1 %, topiramaatti: 29,6 %; p < 0,0001; ensisijainen päätetapahtuma). Atogepanttiryhmän potilaista 64,1 % saavutti vähintään 50 %:n vähenemän kuukausittaisten migreenipäivien määrässä tutkimuksen kuukausien 4–6 aikana verrattuna topiramaattia saaneen ryhmän 39,3 %:iin (p < 0,0001).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset AQUIPTA-valmisteen käytöstä migreenin kohtaushoidossa ja estohoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Suun kautta annetun atogepantin huippupitoisuus plasmassa saavutetaan noin 1–2 tunnissa. Kerran päivässä annossa atogepantin farmakokinetiikka on annosriippuvaista 170 mg:n annokseen saakka (noin 3-kertainen suurin suositeltu annos), eikä kumuloitumista tapahdu.
Ruoan vaikutus
Atogepantin anto runsasrasvaisen aterian kanssa pienensi AUC-arvoa noin 18 % ja Cmax-arvoa noin 22 %. Vaikutusta atogepantin huippupitoisuutta plasmassa edeltävään mediaaniaikaan ei havaittu. Tehoa koskevissa kliinisissä tutkimuksissa atogepanttia annettiin ilman ruokaan liittyvää ohjeistusta.
Jakautuminen
Atogepantin sitoutuminen plasman proteiineihin ei ollut pitoisuusriippuvaista välillä 0,1–10 µM. Atogepantin vapaa fraktio ihmisen plasmassa oli noin 4,7 %. Suun kautta annetun atogepantin keskimääräinen näennäinen jakautumistilavuus (Vz/F) oli noin 292 l.
Biotransformaatio
Atogepantti eliminoituu pääasiassa metaboloitumalla, ensisijaisesti CYP3A4:n välityksellä. Pääasialliset kiertävät komponentit ihmisen plasmassa olivat kanta-aine (atogepantti) ja glukuronidikonjugaattimetaboliitti (M23).
CYP3A4:n indusoijat
Atogepantin samanaikainen anto voimakkaan CYP3A4:n indusoija rifampisiinin kanssa rifampisiinin vakaan tilan pitoisuuden saavuttamisen jälkeen vähensi merkittävästi atogepanttialtistusta terveillä tutkittavilla (Cmax pieneni 30 % ja AUC 60 %).
Atogepantin samanaikainen anto heikon CYP3A4:n indusoija topiramaatin kanssa topiramaatin vakaan tilan pitoisuuden saavuttamisen jälkeen vähensi atogepanttialtistusta (Cmax pieneni 24 % ja AUC 25 %).
In vitro -tutkimusten perusteella atogepantti ei ole CYP3A4:n, CYP1A2:n, CYP2B6:n, CYP2C8:n, CYP2C9:n, CYP2C19:n, CYP2D6, MAO-A:n tai UGT1A1:n estäjä kliinisesti oleellisilla pitoisuuksilla. Atogepantti ei myöskään ole CYP1A2:n, CYP2B6:n tai CYP3A4:n indusoija kliinisesti oleellisilla pitoisuuksilla.
Eliminaatio
Atogepantin eliminaation puoliintumisaika on noin 11 tuntia. Suun kautta annetun atogepantin näennäinen puhdistuma (CL/F) on keskimäärin noin 19 l/h. Terveille miestutkittaville suun kautta annetusta 14C‑atogepantin 50 mg:n kerta-annoksesta 42 % erittyi ulosteeseen ja 5 % virtsaan muuttumattomana atogepanttina.
Kuljettajaproteiinit
Atogepantti on P‑gp:n, BCRP:n, OATP1B1:n, OATP1B3:n ja OAT1:n substraatti. Voimakkaalla OATP:n estäjällä tehdyn kliinisen yhteisvaikutustutkimuksen perusteella atogepantin annosta on suositeltavaa muuttaa samanaikaisessa käytössä voimakkaiden OATP:n estäjien kanssa. Atogepantti ei ole OAT3:n, OCT2:n tai MATE1:n substraatti.
Atogepantti ei ole P‑gp:n, BCRP:n, OAT1:n, OAT3:n, NTCP:n, BSEP:n, MRP3:n tai MRP4:n estäjä kliinisesti oleellisilla pitoisuuksilla. Atogepantti on heikko OATP1B1:n, OATP1B3:n, OCT1:n ja MATE1:n estäjä, mutta kliinisesti merkittäviä yhteisvaikutuksia ei odoteta muodostuvan.
Erityisryhmät
Munuaisten vajaatoiminta
Munuaisten kautta eliminoitumisella on vähäinen rooli atogepantin puhdistumassa. Populaatiofarmakokineettisen analyysin perusteella atogepantin farmakokinetiikka lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (kreatiniinipuhdistuma 30–89 ml/min) ei merkittävästi eroa potilaista, joiden munuaistoiminta on normaali (kreatiniinipuhdistuma > 90 ml/min). Koska potilaita, joilla on vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaissairaus (kreatiniinipuhdistuma < 30 ml/min), ei ole tutkittu, näille potilaille on suositeltavaa käyttää 10 mg:n atogepanttiannosta.
Maksan vajaatoiminta
Kokonaisaltistus atogepantille oli 24 % suurempi lievää (Child-Pugh-luokka A), 15 % suurempi keskivaikeaa (Child-Pugh-luokka B) ja 38 % suurempi vaikeaa (Child-Pugh-luokka C) maksan vajaatoimintaa sairastavilla potilailla. Vaikeaa maksan vajaatoimintaa sairastavien potilaiden altistus vapaalle atogepantille oli kuitenkin noin 3-kertainen. AQUIPTA-valmisteen käyttöä on vältettävä vaikeaa maksan vajaatoimintaa sairastaville potilaille.
Erittyminen rintamaitoon
Tutkimuksessa, jossa 12 tervettä imettävää naista sai 60 mg atogepanttia kerta-annoksena suun kautta 1–6 kuukautta synnytyksen jälkeen, havaittiin atogepantin huippupitoisuuksien esiintyvän rintamaidossa 1–3 tuntia annon jälkeen. Atogepantin Cmax ja AUC rintamaidossa olivat merkittävästi, noin 93 %, alhaisemmat verrattuna pitoisuuksiin naisten plasmassa. Imetettävän lapsen keskimääräinen suhteellinen annos oli noin 0,19 % (vaihteluväli 0,06–0,33 %) äidin painoon suhteutettuun annokseen nähden, ja keskimääräinen maito–plasma-suhde oli 0,08 (vaihteluväli 0,02–0,10). Rintamaitoon 24 tunnin aikana erittynyt atogepantin kumulatiivinen määrä oli vähäinen, alle 0,01 mg.
Muut erityisryhmät
Populaatiofarmakokineettisen analyysin perusteella sukupuolella, rodulla ja painolla ei ollut merkittävää vaikutusta atogepantin farmakokinetiikkaan (Cmax ja AUC). Annosta ei siksi tarvitse muuttaa näiden tekijöiden perusteella.
Prekliiniset tiedot turvallisuudesta
Huolimatta merkittävistä lajien välisistä eroista atogepantin CGRP-reseptoriaffiniteetissa, farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, fototoksisuutta tai karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Hedelmällisyyden heikentyminen
Atogepantin anto suun kautta uros- ja naarasrotille ennen pariutumista ja sen aikana sekä naaraille myös parittelun jälkeen 7. tiineyspäivään asti ei aiheuttanut hedelmällisyyteen tai lisääntymiskykyyn kohdistuvia haittavaikutuksia. Plasmapitoisuus (AUC) vastasi enintään noin 15-kertaista ihmisen altistusta suurimmalla ihmisille suositellulla annoksella (MRHD).
Lisääntymis- ja kehitystoksisuus
Tiineille rotille ja kaneille organogeneesin aikana suun kautta annettu atogepantti aiheutti rottien sikiöiden painon laskua ja sikiöiden sisäelin- ja luustomuutoksien ilmaantuvuuden lisääntymistä annoksilla, jotka olivat emolle vähäisesti toksisisia. Plasmapitoisuus (AUC) suurimmalla annoksella, joka ei aiheuttanut alkion ja sikiön kehitykseen kohdistuvia haittavaikutuksia, oli rotilla noin 4-kertaista ja kaneilla noin 3-kertaista verrattuna ihmisen altistukseen suurimmalla ihmisille suositellulla annoksella 60 mg/vrk.
Atogepantin anto suun kautta rotille koko tiineyden ja imetyksen ajan aiheutti poikasten painonkehityksen ei-haitallista, mutta merkittävää hidastumista, joka säilyi täysikasvuisuuteen asti. Pitoisuus plasmassa (AUC) suurimmalla annoksella, joka ei aiheuttanut sikiöiden ja poikasten kehitykseen kohdistuvia haittavaikutuksia, vastasi noin 5-kertaista ihmisen altistusta suurimmalle ihmisille suositellulle annokselle. Atogepanttia suun kautta saavien imettävien rottien maidon atogepanttipitoisuus oli noin 2 kertaa suurempi kuin emojen plasmassa.
Farmaseuttiset tiedot
Apuaineet
Polyvinyylipyrrolidoni/vinyyliasetaattikopolymeeri
E‑vitamiinin polyeteeniglykolisuksinaatti
Mannitoli
Mikrokiteinen selluloosa
Natriumkloridi
Kroskarmelloosinatrium
Kolloidinen piidioksidi
Natriumstearyylifumaraatti
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
AQUIPTA tabletti
10 mg (L:ei) 28 fol (418,72 €)
60 mg (L:ei) 28 fol (418,72 €)
PF-selosteen tieto
AQUIPTA 10 mg tabletit
Alumiinifolio-PVC/PE/PCTFE-läpipainopakkaukset, kussakin 7 tablettia.
Pakkaus sisältää 7, 28 tai 98 tablettia.
AQUIPTA 60 mg tabletit
Alumiinifolio-PVC/PE/PCTFE-läpipainopakkaukset, kussakin 2 tablettia.
Pakkaus sisältää 2 tablettia.
Alumiinifolio-PVC/PE/PCTFE-läpipainopakkaukset, kussakin 7 tablettia.
Pakkaus sisältää 7, 28 tai 98 tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
AQUIPTA 10 mg tabletit
Valkoinen tai luonnonvalkoinen, pyöreä, kaksoiskupera, halkaisijaltaan 6 mm:n tabletti, jonka toiselle puolelle on kohokuvioitu ”A” ja ”10”.
AQUIPTA 60 mg tabletit
Valkoinen tai luonnonvalkoinen, soikea, kaksoiskupera, 16 mm × 9 mm:n tabletti, jonka toiselle puolelle on kohokuvioitu ”A60”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
AQUIPTA tabletti
10 mg 28 fol
60 mg 28 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Atogepantti, eptinetsumabi, erenumabi, fremanetsumabi, galkanetsumabi ja rimegepantti (migreenin estohoito): Aikuisten migreenin estohoito erityisin edellytyksin (3007).
ATC-koodi
N02CD07
Valmisteyhteenvedon muuttamispäivämäärä
29.05.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi