
LYFNUA tabletti, kalvopäällysteinen 45 mg
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää gefapiksanttisitraattia määrän, joka vastaa 45 mg:aa gefapiksanttia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
Lyfnua on tarkoitettu aikuisille refraktorisen pitkäkestoisen yskän tai selittämättömän pitkäkestoisen yskän hoitoon.
Annostus ja antotapa
Annostus
Suositeltu gefapiksanttiannos on yksi 45 mg:n tabletti suun kautta kahdesti päivässä ruoan kanssa tai tyhjään mahaan.
Unohtunut annos
Potilaille on neuvottava, että jos annos unohtuu, kyseinen annos jätetään väliin ja hoitoa jatketaan tavanomaisen aikataulun mukaisesti. Seuraavaa annosta ei pidä ottaa kaksinkertaisena eikä määrättyä annosta suurempana.
Erityisryhmät
Iäkkäät potilaat (≥ 65-vuotiaat)
Annoksen muuttaminen ei ole tarpeen iäkkäillä potilailla (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Gefapiksantin tiedetään erittyvän huomattavissa määrin munuaisten kautta. Iäkkäillä potilailla munuaisten toiminnan heikentyminen on todennäköisempää, joten gefapiksantin aiheuttamien haittavaikutusten riski saattaa olla näillä potilailla suurentunut. Alkuvaiheessa lääkkeen antotiheyden suhteen on noudatettava varovaisuutta.
Munuaisten vajaatoiminta
Annoksen muuttaminen on tarpeen potilailla, joilla on vaikea munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus [eGFR] < 30 ml/min/1,73 m2) ja jotka eivät tarvitse dialyysihoitoa. Annosta on pienennettävä yhteen 45 mg:n tablettiin, joka otetaan kerran vuorokaudessa.
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta (eGFR ≥ 30 ml/min/1,73 m2). Potilaista, joilla on dialyysihoitoa vaativa loppuvaiheen munuaissairaus, on saatavilla liian vähän tietoa annostussuositusten tekemiseksi (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastavia potilaita ei ole tutkittu. Annoksen muuttamista ei kuitenkaan suositella, koska gefapiksantti eliminoituu vain vähäisessä määrin metaboloitumalla maksassa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää Lyfnua-valmistetta pediatrisille potilaille (alle 18-vuotiaille) refraktorisen tai selittämättömän, pitkäkestoisen yskän hoitoon.
Antotapa
Suun kautta.
Tabletit niellään kokonaisina, ja ne voidaan ottaa joko ruoan kanssa tai tyhjään mahaan. Potilaita on neuvottava, ettei tabletteja saa rikkoa, murskata eikä pureskella.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Obstruktiivinen uniapnea
Potilailla, joilla oli keskivaikea tai vaikea obstruktiivinen uniapnea (n = 19) ja jotka eivät käyttäneet PAP-hoitoa (ylipainehengityslaitetta), joka ilta nukkumaan mennessä otettuun 180 mg:n gefapiksanttiannokseen liittyi lumehoitoon verrattuna pienempi SaO2-keskiarvo ja suurempi sen ajan keskiarvo, jona SaO2 oli alle 90 % kaikkien univaiheiden aikana. Näiden löydösten kliinistä merkitystä gefapiksantin (45 mg kahdesti päivässä) käytölle sellaisten potilaiden hoidossa, joilla on refraktorinen tai selittämätön pitkäkestoinen yskä ja samanaikainen obstruktiivinen uniapnea, ei tunneta. Obstruktiivista uniapneaa sairastaville potilaille on harkittava uniapnean asianmukaista hoitoa ennen gefapiksanttihoidon aloittamista.
Yliherkkyys
Gefapiksantti sisältää sulfonamidiosan, mutta lääkeaineen katsotaan olevan ei-sulfonyyliaryyliamiini. Gefapiksanttia ei ole tutkittu potilailla, joilla on anamneesissa yliherkkyyttä sulfonamideille. Siksi ristiyliherkkyyttä sulfonamidiyliherkkyyden kanssa ei voida sulkea pois. Gefapiksanttia on käytettävä varoen potilailla, joiden tiedetään olevan yliherkkiä sulfonamideille.
Akuutti alahengitystieinfektio
Gefapiksanttihoito on arvioitava ja sitä on muutettava yksilöllisesti potilailla, joille kehittyy akuutti alahengitystieinfektio (ks. kohta Farmakodynamiikka).
Makuaistiin liittyvät haittavaikutukset
Makuaistiin liittyviä haittavaikutuksia ilmoitettiin kliinisissä tutkimuksissa hyvin yleisesti. Useimmilla potilailla nämä haittavaikutukset korjaantuivat pian gefapiksanttihoidon lopettamisen jälkeen (mediaaniaika 5 päivää). Muutamilla potilailla nämä reaktiot jatkuivat yli vuoden ajan hoidon lopettamisen jälkeen (ks. kohta Haittavaikutukset).
Apuaineet
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
In vitro -tutkimusten perusteella (ks. kohta Farmakokinetiikka) tehtiin asiaankuuluvia kliinisiä yhteisvaikutustutkimuksia. Kliinisesti merkittäviä yhteisvaikutuksia ei ole tunnistettu.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Gefapiksantin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Lyfnua-valmisteen käyttöä on suositeltavaa välttää raskauden aikana ja naisilla, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä.
Imetys
Olemassa olevat farmakokineettiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet gefapiksantin erittyvän maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois.
On päätettävä, lopetetaanko imetys vai pidättäydytäänkö Lyfnua-hoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Gefapiksantin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Gefapiksanttihoidolla ei ole todettu olevan vaikutuksia rottien paritteluun tai hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Gefapiksantilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Yksittäistapauksissa gefapiksantin annon jälkeen saattaa ilmetä huimausta, joka saattaa vaikuttaa ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettuja haittavaikutuksia ovat olleet dysgeusia (41 %), ageusia (15 %) ja hypogeusia (11 %).
Haittavaikutustaulukko
Gefapiksantin turvallisuutta arvioitiin kahdessa vaiheen 3 kliinisessä tutkimuksessa (COUGH‑1 ja COUGH‑2), jotka kestivät 52 viikkoa. Niihin osallistui yhteensä 1 369 potilasta, joilla oli refraktorinen tai selittämätön pitkäkestoinen yskä ja jotka saivat gefapiksanttia (15 mg tai 45 mg kahdesti päivässä) (ks. kohta Farmakodynamiikka). Kahdesta 12 viikon pituisesta vaiheen 3b kliinisestä tutkimuksesta saatiin valmisteen turvallisuutta puoltavia tietoja. Näihin tutkimuksiin osallistui lisäksi 391 potilasta, joilla oli refraktorinen tai selittämätön pitkäkestoinen yskä ja jotka saivat gefapiksanttia (45 mg kahdesti päivässä). Mukana oli 185 naispotilasta, joilla oli yskään liittyvää ponnistusinkontinenssia.
Seuraavassa taulukossa luetellaan kliinisissä tutkimuksissa gefapiksanttihoidon yhteydessä ilmoitetut haittavaikutukset MedDRA-elinjärjestelmäluokituksen ja esiintymistiheyden mukaan. Esiintymistiheydet määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000) ja hyvin harvinainen (< 1 / 10 000).
Taulukko 1: Haittavaikutukset
| Elinjärjestelmä | Haittavaikutukset |
| Infektiot | |
| Yleinen | Ylähengitystieinfektio |
| Aineenvaihdunta ja ravitsemus | |
| Yleinen | Ruokahalun heikentyminen |
| Hermosto | |
| Hyvin yleinen | Dysgeusia*, ageusia, hypogeusia |
| Yleinen | Makuaistin häiriö, huimaus, päänsärky† |
| Hengityselimet, rintakehä ja välikarsina | |
| Yleinen | Yskä‡, kipu suun ja nielun alueella |
| Ruoansulatuselimistö | |
| Yleinen | Pahoinvointi, ripuli, suun kuivuminen, syljen liikaeritys, ylävatsakipu, dyspepsia, suun hypoestesia, suun parestesia |
| Psyykkiset häiriöt | |
| Yleinen | Unettomuus |
| Munuaiset ja virtsatiet | |
| Melko harvinainen | Virtsatiekivi, nefrolitiaasi, virtsarakkokivi |
*Dysgeusiaa ilmoitettiin yleisesti seuraavasti: karvas maku, metallinen maku tai suolainen maku.
†Päänsärkyä ilmoitettiin vaiheen 3b kliinisessä tutkimuksessa naispotilailla, joilla oli yskään liittyvää ponnistusinkontinenssia.
‡Yskä-haittavaikutus sisältää seuraavat ilmoitukset: ”yskän paheneminen”, ”yskän pahenemisvaiheet”, ”yskän lisääntyminen” ja ”lisääntynyt yskä”.
Valikoitujen haittavaikutusten kuvaus
Makuaistiin liittyvät haittavaikutukset
Suurimmalla osalla potilaista, joilla oli makuaistiin liittyviä haittavaikutuksia (dysgeusia, ageusia, hypogeusia tai makuaistin häiriö), haittavaikutus ilmeni 9 päivän kuluessa gefapiksanttihoidon aloittamisesta; valtaosa haittavaikutuksista oli vaikeusasteeltaan lieviä (65 %) tai keskivaikeita (32 %). Makuaistiin liittyvät haittavaikutukset korjaantuivat 96 %:lla potilaista, ja 25 % ilmoitti haitan korjaantuneen viimeisen gefapiksanttiannoksen saamisen aikaan tai ennen sitä. Makuaistiin liittyvät haittavaikutukset kestivät yli vuoden ajan hoidon lopettamisen jälkeen 1,6 %:lla potilaista (7/447) gefapiksanttiryhmässä ja 12,8 %:lla potilaista (6/47) lumelääkeryhmässä. Gefapiksanttihoitoa saaneista potilaista 22 %:lla ilmeni haittavaikutuksia, jotka johtivat hoidon lopettamiseen. Yleisimmin ilmoitetut hoidon lopettamiseen johtaneet haittavaikutukset olivat dysgeusia (9 %) ja ageusia (4 %).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisessä tutkimuksessa, jossa 8 terveelle tutkittavalle annettiin gefapiksanttia 1 800 mg kahdesti päivässä (40‑kertainen annos ihmiselle suositeltuun annokseen nähden) enintään 14 päivän ajan, tutkittavien virtsassa havaittiin gefapiksantista muodostuneita kiteitä. Munuaisiin tai virtsateihin kohdistuvista vaurioista ei havaittu näyttöä.
Vaiheen 3 tutkimusten aikana ilmoitettujen yliannostustapausten yhteydessä ei ilmoitettu haittatapahtumia.
Yliannostustapauksessa potilasta on tarkkailtava haittavaikutusten varalta, ja asianmukaiset tukitoimet on aloitettava. Gefapiksantti poistuu osittain hemodialyysilla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: muut yskänhillitsijät, ATC-koodi: R05DB29
Vaikutusmekanismi
Gefapiksantti on selektiivinen P2X3-reseptorin antagonisti. Gefapiksantti vaikuttaa myös P2X2/3-reseptorialatyyppiä vastaan. P2X3-reseptorit ovat ATP:n säätelemiä ionikanavia, joita on hengitysteissä kiertäjähermon sensorisissa C-hermosyissä. C-hermosyyt aktivoituvat reaktiona tulehdukseen tai ärsyttäviin kemikaaleihin. ATP:ta vapautuu hengitysteiden limakalvojen soluista tulehdusolosuhteissa. C-hermosyyt tulkitsevat solunulkoisen ATP:n sitoutumisen P2X3-reseptoreihin vaurion merkiksi. Potilas aistii C-hermosyiden aktivaation tarpeena yskiä, ja yskänrefleksi aktivoituu. P2X3-reseptorivälitteisen ATP-signaalinvälityksen salpaus vähentää solunulkoisen ATP:n indusoimaa liiallista tuntohermojen aktivaatiota ja liiallista yskää.
Kliininen teho ja turvallisuus
Refraktorista tai selittämätöntä pitkäkestoista yskää koskeneet tutkimukset, joissa arvioitiin objektiivista yskimistiheyttä
Lyfnua-valmisteen tehoa refraktorisen tai selittämättömän pitkäkestoisen yskän hoidossa arvioitiin kahdessa 52 viikon pituisessa, satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa, johon osallistuneilla aikuisilla oli joko refraktorinen tai selittämätön pitkäkestoinen yskä. Refraktorisen pitkäkestoisen yskän määritelmänä oli samanaikaiseen sairauteen (esim. astmaan, ruokatorven refluksitautiin tai ylähengitysteiden yskäoireyhtymään) liittyvä yskä, joka jatkui samanaikaisen sairauden asianmukaisesta hoidosta huolimatta. Selittämättömän pitkäkestoisen yskän määritelmänä oli yskä, johon ei havaittu liittyvän mitään samanaikaista sairautta huolellisesta kliinisestä arvioinnista huolimatta.
Kummankin vaiheen 3 tutkimuksen ensisijaisena tavoitteena oli arvioida Lyfnua-hoidon tehoa 24 tunnin yskimistiheyden pienentämisessä lumelääkkeeseen verrattuna. Toissijaisia tavoitteita olivat pienentynyt yskimistiheys valveillaolon aikana ja yskäspesifinen elämänlaatu. Kummassakin tutkimuksessa potilaat satunnaistettiin saamaan Lyfnua-valmistetta kahdesti päivässä annoksilla 45 mg tai 15 mg tai lumelääkettä. COUGH-1-tutkimuksen (NCT03449134) ensisijainen tehon arviointijakso kesti 12 viikkoa, minkä jälkeen oli 40 viikon pituinen sokkoutettu jatkovaihe. COUGH-2-tutkimuksen (NCT03449147) ensisijainen tehon arviointijakso kesti 24 viikkoa, minkä jälkeen oli 28 viikon pituinen sokkoutettu jatkovaihe.
COUGH-1- ja COUGH-2-tutkimuksiin otetut potilaat olivat sillä hetkellä tupakoimattomia, eivät käyttäneet angiotensiinikonvertaasin (ACE:n) estäjiä, heillä oli todettu refraktorinen tai selittämätön pitkäkestoinen yskä, ja yskä oli jatkunut yli 1 vuoden ajan. Useimmat potilaat olivat naispuolisia (75 %), valkoihoisia (80 %) ja eurooppalaisia (53 %); ikäkeskiarvo oli 58 vuotta (vaihteluväli 19–89 vuotta), ja 7 % potilaista oli yli 75‑vuotiaita. Yhteensä 61,5 %:lla potilaista oli todettu refraktorinen pitkäkestoinen yskä ja 38,5 %:lla selittämätön pitkäkestoinen yskä. Pitkäkestoisen yskän keskimääräinen kesto oli 11 vuotta.
Yskimistiheys
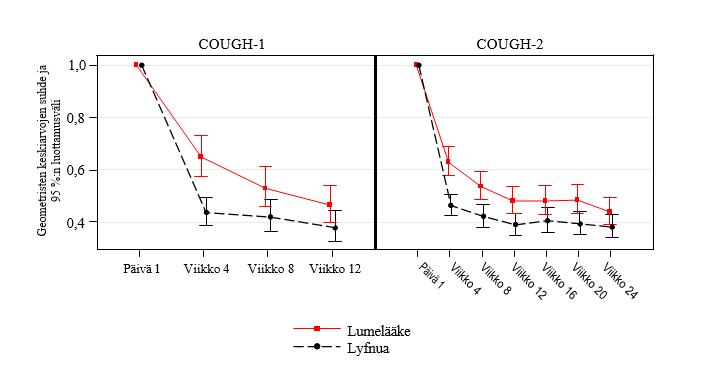
COUGH-1- ja COUGH-2-tutkimuksissa potilailla, jotka saivat Lyfnua-hoitoa 45 mg kahdesti päivässä, todettiin 24 tunnin yskimistiheyden pienentyneen merkitsevästi verrattuna lumehoitoon (taulukko 2). 24 tunnin yskimistiheyden pieneneminen havaittiin viikkoon 4 mennessä, ja vaikutus säilyi koko ensisijaisen tehojakson ajan (12 viikkoa COUGH-1-tutkimuksessa ja 24 viikkoa COUGH-2-tutkimuksessa; kuva 1).
Gefapiksanttia 15 mg kahdesti päivässä saaneiden tutkittavien ryhmässä ei havaittu merkitsevää 24 tunnin yskimistiheyden pienenemistä kummassakaan tutkimuksessa.
Taulukko 2: 24 tunnin yskimistiheys, Lyfnua 45 mg kahdesti päivässä -hoidon tulokset (COUGH-1 ja COUGH-2)
| COUGH‑1 | COUGH‑2 | |||
| Lyfnua | Lumelääke | Lyfnua | Lumelääke | |
| N | 243 | 243 | 439 | 435 |
| Ensisijainen tehoa koskeva päätemuuttuja | ||||
| 24 tunnin yskimistiheys (yskäisyjen määrä tunnissa) | ||||
| Lähtötilanne (geometrinen keskiarvo) | 18,24 | 22,83 | 18,55 | 19,48 |
| Viikko 12 (COUGH‑1) tai viikko 24 (COUGH‑2) (geometrinen keskiarvo) | 7,05 | 10,33 | 6,83 | 8,34 |
| Viikko 12 (COUGH-1) tai viikko 24 (COUGH-2) (prosentuaalinen pieneneminen lähtötilanteesta) | ‑61,35 | ‑54,77 | ‑63,17 | ‑57,19 |
| Väheneminen suhteessa lumelääkkeeseen (prosentuaalinen pieneneminen ja 95 %:n luottamusväli)† | ‑18,52 (‑32,76, ‑1,28) | ‑13,29 (‑24,74, ‑0,10) | ||
| p-arvo | 0,036 | 0,048 | ||
| N = Analyysiin sisältyneiden tutkittavien kokonaismäärä. †Puuttuvat lähtötilanteen arvot imputoitiin sukupuolen ja maantieteellisen alueen perusteella, minkä jälkeen tehtiin puuttuvien tietojen moni-imputointi (m = 50 imputoitua tietoaineistoa) kaikkien seurantakäyntien osalta käyttäen kovariaatteina hoitoa, sukupuolta, maantieteellistä aluetta ja muita seurantakäyntejä. Imputoinnin jälkeen suoritettiin kovarianssianalyysi (ANCOVA) tarkasteltuna ajankohtana, ja tulokset korjattiin hoidon, lähtötilanteen, sukupuolen ja maantieteellisen alueen kovariaattien suhteen. | ||||
Kuva 1: 24 tunnin yskimistiheyden analyysi ajan myötä Lyfnua 45 mg kahdesti päivässä -hoidolla (COUGH-1 ja COUGH-2)
Yskäspesifinen elämänlaatu
COUGH-2-tutkimus suunniteltiin nimenomaan arvioimaan Lyfnua-hoidon vaikutusta yskäspesifiseen elämänlaatuun verrattuna lumehoitoon ja mitattuna LCQ-kyselylomakkeella (Leicester Cough Questionnaire; mahdollisten pistemäärien vaihteluväli on 3–21, ja suurempi pistemäärä tarkoittaa parempaa elämänlaatua). LCQ-kokonaispistemäärän suureneminen lähtötilanteesta vähintään 1,3 pisteellä määriteltiin kliinisesti merkittäväksi. COUGH-2-tutkimuksessa yskäspesifisen elämänlaadun kliinisesti merkittävä paraneminen oli merkitsevästi todennäköisempää Lyfnua 45 mg -hoitoryhmässä kuin lumelääkeryhmässä viikon 24 kohdalla mitattuna (ks. taulukko 3).
Taulukko 3: Yskäspesifinen elämänlaatu Lyfnua 45 mg kahdesti päivässä -hoitoa saaneilla (COUGH-2): niiden potilaiden osuus, joilla LCQ-kokonaispistemäärä viikolla 24 oli suurentunut lähtötilanteesta vähintään 1,3 pisteellä
| Lyfnua | Lumelääke | |
| N | 439 | 435 |
| Vasteen saaneita* (%) | 75,7 | 68,1 |
| Arvioitu kerroinsuhde vs. lumelääke (95 %:n luottamusväli)† | 1,46 (1,07, 1,99) | |
| Arvioitu ero† vs. lumelääke (95 %:n luottamusväli)†† | 7,63 (1,34, 13,76) | |
| p-arvo† | 0,016 | |
| N = Sellaisten tutkittavien määrä, joilta oli tietoja saatavilla viikon 24 kohdalla. * Vasteen saaneiden prosenttiosuus viikon 24 kohdalla. Vasteen saaneiden lukumäärä on ilmoitettu usean imputoinnin keskiarvona. Lyfnua-valmistetta saaneessa ryhmässä oli noin 332 vasteen saanutta ja lumeryhmässä noin 296 vasteen saanutta. LCQ = Leicester Cough Questionnaire. † Puuttuvat lähtötilanteen arvot imputoitiin sukupuolen ja maantieteellisen alueen perusteella, minkä jälkeen tehtiin puuttuvien tietojen moni-imputointi (m = 50 imputoitua tietoaineistoa) kaikkien seurantakäyntien osalta käyttäen kovariaatteina hoitoa, sukupuolta, maantieteellistä aluetta ja muita seurantakäyntejä. Imputoinnin jälkeen suoritettiin logistinen regressioanalyysi tarkastellun ajankohdan dikotomisoiduista pistemääristä, ja tulokset korjattiin hoidon, lähtötilanteen (jatkuvan) LCQ-kokonaispistemäärän, sukupuolen ja maantieteellisen alueen kovariaattien suhteen. †† Perustuu bootstrap-menetelmään. | ||
Hiljattain ilmaantunutta refraktorista tai selittämätöntä pitkäkestoista yskää koskenut tutkimus, jossa arvioitiin potilaiden ilmoittamia hoitotuloksia
Lyfnua-valmisteen tehoa hiljattain ilmaantuneen refraktorisen tai selittämättömän pitkäkestoisen yskän hoidossa aikuisilla arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (NCT04193202). Hiljattain ilmaantuneeksi määriteltiin refraktorinen tai selittämätön pitkäkestoinen yskä, joka oli kestänyt yli 8 viikkoa mutta alle 12 kuukautta.
Tutkimuksen ensisijaisena tavoitteena oli osoittaa, että Lyfnua parantaa tehokkaasti yskäspesifistä terveyteen liittyvää elämänlaatua. Mittarina käytettiin LCQ-kokonaispistemäärän muutosta lähtötilanteesta 12 viikon kohdalla. Potilaat satunnaistettiin saamaan Lyfnua-valmistetta kahdesti päivässä annoksella 45 mg tai lumelääkettä.
Tutkimukseen otetut potilaat olivat sillä hetkellä tupakoimattomia, eivät käyttäneet angiotensiinikonvertaasin estäjiä, heillä oli todettu refraktorinen tai selittämätön pitkäkestoinen yskä, yskän vaikeusastetta kuvaavalla VAS-asteikolla (visual analogue scale) lukema oli vähintään 40 mm, ja pitkäkestoinen yskä oli jatkunut alle 12 kuukauden ajan. Useimmat potilaat olivat naispuolisia (65 %), valkoihoisia (72 %) ja eurooppalaisia (59 %), ja ikäkeskiarvo oli 53 vuotta (vaihteluväli 18–83 vuotta). Yhteensä 70,8 %:lla potilaista oli todettu refraktorinen pitkäkestoinen yskä ja 29,2 %:lla selittämätön pitkäkestoinen yskä. Pitkäkestoisen yskän keskimääräinen kesto oli 7,2 kuukautta.
Yskäspesifinen elämänlaatu
Potilailla, jotka saivat Lyfnua-valmistetta 45 mg kahdesti päivässä, LCQ-kokonaispistemäärä oli parantunut viikon 12 kohdalla lähtötilanteesta merkitsevästi enemmän kuin lumelääkettä saaneilla (taulukko 4).
Taulukko 4: LCQ-kokonaispistemäärän analyysi Lyfnua-valmistetta 45 mg kahdesti päivässä saaneilla
| Hoito | N | Lähtötilanne, keskiarvo (keskihajonta) | Viikko 12, keskiarvo (keskihajonta) | Muutos lähtötilanteesta, pienimmän neliösumman keskiarvo (95 %:n luottamusväli)* |
| Lumelääke | 199 | 11,30 (2,80) | 14,73 (3,48) | 3,59 (3,09, 4,09) |
| Lyfnua | 199 | 10,82 (3,08) | 15,32 (3,91) | 4,34 (3,84, 4,83) |
| Hoitojen välinen ero | Arvioitu ero (95 %:n luottamusväli) | p‑arvo | ||
| Lyfnua vs. lumelääke | 0,75 (0,06, 1,44) | 0,034 | ||
| N = Analyysissa mukana olleiden tutkittavien määrä. LCQ = Leicester Cough Questionnaire. *Laskettiin kaavalla (viikon 12 arvo – lähtötilanteen arvo) / lähtötilanteen arvo ja kovarianssimallin pitkittäisanalyysin perusteella, kun mallin vasteena oli LCQ-kokonaispistemäärän muutos lähtötilanteeseen nähden kullakin lähtötilanteen jälkeisellä käynnillä (viikkoon 12 asti). Mallissa olivat termeinä hoito, käynti, hoidon ja käynnin yhdysvaikutus, sukupuoli ja lähtötilanteen LCQ-kokonaispistemäärä. | ||||
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Lyfnua-valmisteen käytöstä selittämättömän tai pitkäkestoisen refraktorisen yskän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Gefapiksantin farmakokinetiikkaa tutkittiin terveillä aikuisilla ja aikuisilla, joilla oli refraktorinen tai selittämätön pitkäkestoinen yskä, ja farmakokinetiikka oli näissä populaatioissa samankaltainen. Kun gefapiksanttia annetaan 45 mg kaksi kertaa vuorokaudessa, vakaan tilan keskimääräinen AUC plasmassa on 4 144 ng h/ml ja huippupitoisuus (Cmax) 531 ng/ml. Vakaa tila saavutetaan 2 vuorokauden kuluessa, ja kumuloitumissuhde on 1,4–1,5-kertainen.
Imeytyminen
Kun gefapiksanttia annettiin suun kautta, aika huippupitoisuuden saavuttamiseen plasmassa (tmax) oli 1–4 tuntia. Toistuvilla annoksilla altistus suurenee suhteessa annokseen, kun annokset ovat enintään 300 mg kaksi kertaa vuorokaudessa. Gefapiksantin imeytyvä osuus on vähintään 78 %.
Ruoan vaikutus
Gefapiksantin 50 mg:n kerta-annoksen anto suun kautta tavanomaisen runsasrasvaisen ja runsaskalorisen aterian yhteydessä ei paastotilaan verrattuna vaikuttanut gefapiksantin AUC- tai Cmax-arvoihin.
Jakautuminen
Populaatiofarmakokineettisten analyysien perusteella keskimääräinen vakaan tilan näennäinen jakautumistilavuus on suun kautta otetun 45 mg:n annoksen jälkeen arviolta 138 litraa.
In vitro gefapiksantin sitoutuminen proteiineihin on vähäistä (55 %), ja veren ja plasman gefapiksanttipitoisuuksien suhde on 1,1. Prekliinisten tutkimusten perusteella gefapiksantin pääsy keskushermostoon on vähäistä.
Biotransformaatio
Maksametabolian merkitys gefapiksantin eliminaatiossa on pieni, ja siihen kuuluvat hapettuminen ja glukuronidaatio. Kun [14C]-gefapiksanttia annettiin suun kautta, 14 % annetusta annoksesta havaittiin metaboliitteina virtsassa ja ulosteessa. Muuttumaton gefapiksantti on pääasiallinen lääkeaineeseen liittyvä komponentti plasmassa (87 %), ja kunkin verenkierrossa havaitun metaboliitin osuus oli alle 10 %:sta havaitusta kokonaisradioaktiivisuudesta.
Eliminaatio
Gefapiksantin pääasiallinen eliminaatioreitti on erittyminen munuaisten kautta, ja siihen kuuluu sekä passiivisen suodatuksen että aktiivisen kuljetuksen mekanismeja. Gefapiksanttia havaitaan virtsassa kanta-aineena (~ 64 %) tai metaboliitteina (~ 12 %), ja loppuosa on ulosteessa kanta-aineena (~ 20 %) tai metaboliitteina (~ 2 %). Aktiivisen munuaisten kautta tapahtuvan erityksen osuudeksi arvioitiin ≤ 50 % kokonaiseliminaatiosta. In vitro gefapiksantti on MATE1-, MATE2K-, P-gp- ja BCRP-kuljettajaproteiinien substraatti. Gefapiksantin terminaalinen puoliintumisaika (t½) on 6–10 tuntia.
Erityisryhmät
Munuaisten vajaatoiminta
Gefapiksantin pääasiallinen eliminaatioreitti on erittyminen munuaisten kautta. Lievä tai kohtalainen munuaisten vajaatoiminta (eGFR≥30 ml/min/1,73 m2) ei vaikuta gefapiksanttialtistukseen kliinisesti merkittävästi.
Populaatiofarmakokineettisessa analyysissa arvioitiin potilaita, joilla oli refraktorinen tai selittämätön pitkäkestoinen yskä. Keskimääräisen AUC-arvon ennustettiin suurenevan 89 % ja keskimääräisen Cmax-arvon 54 % potilailla, joilla oli vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min/1,73 m2), verrattuna potilaisiin, joilla munuaiset toimivat normaalisti. Jotta systeeminen altistus pysyisi samankaltaisena kuin potilailla, joilla munuaiset toimivat normaalisti, suositellaan annoksen muuttamista (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Maksametabolian merkitys lääkeaineen eliminaatiossa on pieni. Suurin osa suun kautta otetusta annoksesta todettiin muuttumattomana kanta-aineena virtsassa (64 %) tai ulosteessa (20 %). Nimenomaan maksan vajaatoimintaa sairastavilla potilailla ei ole tehty erillistä tutkimusta, koska maksan vajaatoiminta ei todennäköisesti vaikuta altistukseen kliinisesti merkittävästi (ks. kohta Annostus ja antotapa).
Iän, painon, sukupuolen, etnisen taustan ja rodun vaikutukset
Populaatiofarmakokineettisen analyysin perusteella iällä, painolla, sukupuolella, etnisellä taustalla ja rodulla ei ole kliinisesti merkittäviä vaikutuksia gefapiksantin farmakokinetiikkaan.
Lääkkeiden yhteisvaikutukset
Muiden lääkkeiden vaikutukset gefapiksantin farmakokinetiikkaan
Maksametabolian merkitys gefapiksantin eliminaatiossa on vähäinen, ja kliinisesti merkittävien yhteisvaikutusten todennäköisyys on pieni, kun gefapiksanttia annetaan samanaikaisesti sytokromi P450:n (CYP:n) tai uridiini-5'-difosfaattiglukuronyylitransferaasin (UGT:n) estäjien tai indusorien kanssa.
Protonipumpun estäjä omepratsolin samanaikainen käyttö ei vaikuttanut kliinisesti merkittävästi gefapiksantin farmakokinetiikkaan.
In vitro -tutkimusten perusteella gefapiksantti on substraatti effluksikuljettajaproteiineille MATE1 (multidrug and toxin extrusion 1), MATE2K, P-glykoproteiini (P-gp) ja rintasyövän resistenssiproteiini (BCRP). Vaiheen 1 kliinisessä tutkimuksessa kerta-annos pyrimetamiinia, joka on MATE1:n/MATE2K:n estäjä, suurensi gefapiksantin AUC-arvoa 24 %, mikä ei ole kliinisesti merkittävä suureneminen eikä vaikuttanut gefapiksantin Cmax-arvoon.
Gefapiksantin vaikutukset muiden lääkkeiden farmakokinetiikkaan
In vitro -tutkimusten perusteella gefapiksantin kyky estää tai indusoida CYP-entsyymejä on pieni, joten on epätodennäköistä, että gefapiksantti vaikuttaisi muiden lääkkeiden CYP-välitteiseen metaboliaan.
Gefapiksantti on MATE1:n, MATE2K:n sekä orgaanisia anioneja kuljettavien polypeptidien 1B1 (OATP1B1) ja 1B3 (OATP1B3) estäjä in vitro. Näiden kuljettajaproteiinien estymisen vaikutuksesta ilmenevien kliinisesti merkittävien yhteisvaikutusten riski on kuitenkin pieni, kun gefapiksanttia annetaan 45 mg kaksi kertaa vuorokaudessa. Gefapiksantti estää orgaanisten kationien kuljettajaproteiini 1:n (OCT1:n) toimintaa in vitro, mutta tämän seikan kliinistä merkitystä ei ole varmistettu. Vaiheen 1 kliinisessä tutkimuksessa gefapiksantin anto toistuvina 45 mg:n annoksina ei vaikuttanut altistukseen pitavastatiinille, OATP1B:n substraatille.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttama toksisuus
Gefapiksanttia saaneilla laboratorioeläimillä ilmeni kristalluriaa, ja suurimman osan virtsan kiteistä vahvistettiin koostuvan gefapiksantista.
Kuuden kuukauden pituisessa toistuvan altistuksen aiheuttamaa toksisuutta arvioineessa tutkimuksessa rotilla havaittiin mikroskooppisia muutoksia munuaisissa (kidemateriaalin takia pingottuneet tubulukset, tubulusten sisäpinnan epiteelisolujen rappeutuminen ja interstitiumin tulehdusreaktio), virtsanjohtimissa (laajentuminen ja tulehdusreaktio) sekä virtsarakossa (transitiosellulaarinen hyperplasia), kun altistus oli 9-kertainen ihmiselle suositellun enimmäisannoksen aiheuttamaan altistukseen nähden.
Yhdeksän kuukauden pituisessa toistuvien oraalisten annosten aiheuttamaa toksisuutta arvioineessa tutkimuksessa koirilla havaittiin kiteitä virtsassa, ja mikroskoopilla havaittiin fokaalista, minimaalista tubulusten rappeumaa, myös joissakin kortikaalisissa tubuluksissa, yhdellä uroskoiralla, jonka altistus oli 35-kertainen ihmiselle suositellun enimmäisannoksen aiheuttamaan altistukseen nähden.
Karsinogeenisuus
Gefapiksantin karsinogeenisuutta koskeneissa tutkimuksissa rotilla (kesto 2 vuotta) ja rasH2-siirtogeenisillä hiirillä (kesto 6 kuukautta) ei havaittu näyttöä karsinogeenisuudesta (ei hoitoon liittyviä kasvaimia), kun altistukset olivat ihmiselle suositellun enimmäisannoksen aiheuttamaan altistukseen verrattuna enintään 9-kertaisia (rotilla) ja 4-kertaisia (hiirillä).
Mutageneesi
Gefapiksantti ei ollut genotoksinen in vitro- ja in vivo -määritysten sarjassa, jossa arvioitiin mutageneesiä mikrobeilla sekä kromosomipoikkeavuuksia ihmisen perifeerisen veren lymfosyyteissä ja in vivo mikrotumatestissä rotilla.
Lisääntymistoksisuus
Eläimillä tehdyissä lisääntymistutkimuksissa tiineille rotille ja kaneille annettiin organogeneesivaiheen aikana suun kautta gefapiksanttia. Tutkimuksissa ei havaittu näyttöä teratogeenisuudesta tai alkio-/sikiöletaliteetista, kun altistukset (AUC) olivat ihmiselle suositellun enimmäisannoksen aiheuttamaan altistukseen nähden 6-kertaisia (rotilla) ja 34-kertaisia (kaneilla). Rotilla havaittiin sikiöiden painon vähäinen pieneneminen ja sen yhteydessä toksisuus emoille altistuksella, joka oli ihmiselle suositellun enimmäisannoksen aiheuttamaan altistukseen nähden noin 11-kertainen.
Tiineillä rotilla ja kaneilla tehdyt tutkimukset osoittivat, että gefapiksantti läpäisee istukan ja kulkeutuu sikiöön. Pitoisuus sikiön plasmassa oli enintään 21 % (rotilla) ja 25 % (kaneilla) emoilla tiineyspäivänä 20 havaituista pitoisuuksista.
Imetystä koskeneessa tutkimuksessa gefapiksantti erittyi imettävien rottien maitoon, kun se annettiin suun kautta imetyspäivänä 10 (jolloin altistus oli enintään 9-kertainen verrattuna ihmiselle suositeltua enimmäisannosta vastaavaan altistukseen). Pitoisuus maidossa oli 4-kertainen verrattuna pitoisuuksiin, jotka todettiin emon plasmassa 1 tunnin kuluttua annoksen antamisen jälkeen imetyspäivänä 10.
Kun gefapiksanttia annettiin naaras- ja urosrotille enintään 9-kertaisilla altistuksilla ihmiselle suositellun enimmäisannoksen aiheuttamaan altistukseen nähden, ei havaittu hedelmällisyyteen, parittelukykyyn eikä alkioiden varhaiskehitykseen kohdistuvia vaikutuksia.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Vedetön kolloidinen piidioksidi (E551)
Krospovidoni (E1202)
Hypromelloosi (E464)
Magnesiumstearaatti (E470b)
Mannitoli (E421)
Mikrokiteinen selluloosa (E460)
Natriumstearyylifumaraatti
Kalvopäällyste
Hypromelloosi (E464)
Titaanidioksidi (E171)
Triasetiini (E1518)
Punainen rautaoksidi (E172)
Karnaubavaha (E903)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LYFNUA tabletti, kalvopäällysteinen
45 mg (L:ei) 56 fol (138,21 €)
PF-selosteen tieto
Läpikuultamattomat valkoiset PVC/PE/PVdC-läpipainopakkaukset, joissa on läpipainettava alumiinifoliokuori.
Pakkauksissa on 28, 56 tai 98 kalvopäällysteistä tablettia läpipainopakkauksissa, joissa ei ole repäisyviivoja (14 tablettia levyssä), ja monipakkauksissa on 196 (2 pakkausta, joissa on 98) kalvopäällysteistä tablettia läpipainopakkauksissa, joissa ei ole repäisyviivoja.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Vaaleanpunainen, 10 mm:n kokoinen, pyöreä ja kupera tabletti, jossa toisella puolella on merkintä ”777” ja toisella puolella ei ole merkintöjä.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LYFNUA tabletti, kalvopäällysteinen
45 mg 56 fol
- Ei korvausta.
ATC-koodi
R05DB29
Valmisteyhteenvedon muuttamispäivämäärä
03.04.2025
Yhteystiedot
 MSD FINLAND OY
MSD FINLAND OY Keilaniementie 1, PL 46
02151 Espoo
09 804 650
www.msd.fi
info@msd.fi