
SPEXOTRAS jauhe oraaliliuosta varten 0,05 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi pullo sisältää trametinibidimetyylisulfoksidia määrän, joka vastaa 4,7 mg trametinibia.
Yksi millilitra käyttökuntoon saatettua liuosta sisältää 0,05 mg trametinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi millilitra käyttökuntoon saatettua liuosta sisältää 100 mg sulfobutyylibeetadeksinatriumia, 0,8 mg metyyliparahydroksibentsoaattia ja 1,98 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Jauhe oraaliliuosta varten.
Kliiniset tiedot
Käyttöaiheet
Matala-asteinen gliooma
Spexotras yhdistelmänä dabrafenibin kanssa on tarkoitettu sellaisten vähintään 1 vuoden ikäisten pediatristen potilaiden hoitoon, joilla on BRAF V600E -mutaatiopositiivinen matala-asteinen gliooma ja jotka tarvitsevat systeemistä hoitoa.
Korkea-asteinen gliooma
Spexotras yhdistelmänä dabrafenibin kanssa on tarkoitettu sellaisten vähintään 1 vuoden ikäisten pediatristen potilaiden hoitoon, joilla on BRAF V600E -mutaatiopositiivinen korkea-asteinen gliooma ja jotka ovat aiemmin saaneet vähintään yhtä säde- ja/tai solunsalpaajahoitoa.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Syövän hoitoon perehtyneen lääkärin on aloitettava Spexotras-hoito ja valvottava sen toteuttamista.
Ennen Spexotras-hoidon aloittamista BRAF V600E -mutaatio on vahvistettava kyseiseen käyttötarkoitukseen soveltuvalla CE-merkityllä in vitro -diagnostisella (IVD) lääkinnällisellä laitteella. Jos CE-merkittyä IVD-laitetta ei ole saatavilla, BRAF V600E -mutaatio on arvioitava vaihtoehtoisella validoidulla testillä.
Spexotras-valmistetta käytetään yhdessä dispergoituvien dabrafenibitablettien kanssa. Dispergoituvien dabrafenibitablettien annostus, ks. kyseisen valmisteen valmisteyhteenveto.
Annostus
Suositeltu kerran vuorokaudessa otettava Spexotras-annos määräytyy potilaan painon perusteella (taulukko 1).
Taulukko 1 Painon mukainen annostus
| Paino* | Suositeltu annos | |
| Oraaliliuoksen määrä (ml) kerran vuorokaudessa | Vastaava trametinibin mg-määrä | |
| 8 kg | 6 ml | 0,30 mg |
| 9–10 kg | 7 ml | 0,35 mg |
| 11 kg | 8 ml | 0,40 mg |
| 12–13 kg | 9 ml | 0,45 mg |
| 14–17 kg | 11 ml | 0,55 mg |
| 18–21 kg | 14 ml | 0,70 mg |
| 22–25 kg | 17 ml | 0,85 mg |
| 26–29 kg | 18 ml | 0,90 mg |
| 30–33 kg | 20 ml | 1 mg |
| 34–37 kg | 23 ml | 1,15 mg |
| 38–41 kg | 25 ml | 1,25 mg |
| 42–45 kg | 28 ml | 1,40 mg |
| 46–50 kg | 32 ml | 1,60 mg |
| ≥ 51 kg | 40 ml | 2 mg |
*Paino pyöristetään tarvittaessa lähimpään kilogrammaan. Alle 8 kg painavien potilaiden suositusannosta ei ole määritetty. Spexotras-valmisteen kanssa yhdessä käytettävän dabrafenibihoidon annostusohjeet, ks. dispergoituvien dabrafenibitablettien valmisteyhteenvedon kohdat ”Annostus” ja ”Antotapa”. | ||
Hoidon kesto
Spexotras-hoitoa jatketaan, kunnes tauti etenee tai potilaalla ilmenee liiallista toksisuutta. Yli 18-vuotiaita glioomapotilaita koskevia tietoja on vain vähän, minkä vuoksi hoidon jatkamisen aikuisiällä on perustuttava lääkärin arvioon potilaan yksilöllisistä hyödyistä ja riskeistä.
Väliin jääneet tai myöhästyneet annokset
Jos Spexotras-annos jää väliin, se otetaan vain, jos seuraavan annoksen suunniteltuun ajankohtaan on yli 12 tuntia aikaa. Jos potilas oksentaa Spexotras-valmisteen ottamisen jälkeen, lisäannosta ei pidä ottaa ja seuraava annos otetaan seuraavana suunniteltuna ajankohtana.
Annoksen sovittaminen
Haittavaikutukset saattavat vaatia annoksen pienentämistä, hoidon keskeyttämistä tai hoidon lopettamista (ks. taulukot 2 ja 3).
Jos hoitoon liittyviä haittoja esiintyy, sekä trametinibin että dabrafenibin annosta on pienennettävä samanaikaisesti tai hoidot on keskeytettävä tai lopetettava. Poikkeustapaukset, joissa vain toisen valmisteen annoksen muuttaminen on tarpeen, on esitetty jäljempänä seuraavien haittojen yhteydessä: uveiitti, muut RAS-mutaatiopositiiviset syövät kuin ihosyöpä (liittyy ensisijaisesti dabrafenibiin), vasemman kammion ejektiofraktion pieneneminen, verkkokalvon laskimotukos, verkkokalvon pigmenttiepiteelin irtauma ja interstitiaalinen keuhkosairaus/pneumoniitti (liittyy ensisijaisesti trametinibiin).
Annoksen sovittamista tai hoidon keskeyttämistä ei suositella, jos potilaalle ilmaantuu haittavaikutuksena ihomaligniteetti (lisätiedot, ks. dispergoituvien dabrafenibitablettien valmisteyhteenveto).
Taulukko 2 Annoksen sovittaminen haittavaikutuksen vaikeusasteen mukaan (kuumetta lukuun ottamatta)
| Vaikeusaste (CTCAE)* | Suositeltu trametinibiannoksen sovittaminen |
| 1. tai 2. aste (siedettävissä) | Hoitoa jatketaan ja potilaan tilaa seurataan kliinisen tilanteen mukaan. |
| 2. aste (kestämätön) tai 3. aste | Hoito keskeytetään, kunnes vaikeusaste on laskenut tasolle 0–1. Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella. Annostasosuositukset, ks. taulukko 3. |
| 4. aste | Hoito lopetetaan pysyvästi tai keskeytetään, kunnes vaikeusaste on laskenut tasolle 0–1. Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella. Annostasosuositukset, ks. taulukko 3. |
| *Kliinisten haittavaikutusten vaikeusaste on määritetty CTCAE-kriteerien (Common Terminology Criteria for Adverse Events) mukaan. | |
Suositukset annoksen pienentämisestä noin 75 %:iin suositusannoksesta (annostaso ensimmäisen pienennyksen jälkeen) ja noin 50 %:iin suositusannoksesta (annostaso toisen pienennyksen jälkeen) on esitetty taulukossa 3.
Taulukko 3 Annostasosuositukset annoksen pienentämiseen haittavaikutuksen yhteydessä
| Paino | Suositeltu annos | Pienennetty annos | |
ml liuosta (mg trametinibia) (kerran vuorokaudessa) | Annos ensimmäisen pienennyksen jälkeen (kerran vuorokaudessa) | Annos toisen pienennyksen jälkeen (kerran vuorokaudessa) | |
| 8 kg | 6 ml (0,30 mg) | 5 ml | 3 ml |
| 9–10 kg | 7 ml (0,35 mg) | 5 ml | 4 ml |
| 11 kg | 8 ml (0,40 mg) | 6 ml | 4 ml |
| 12–13 kg | 9 ml (0,45 mg) | 7 ml | 5 ml |
| 14–17 kg | 11 ml (0,55 mg) | 8 ml | 6 ml |
| 18–21 kg | 14 ml (0,70 mg) | 11 ml | 7 ml |
| 22–25 kg | 17 ml (0,85 mg) | 13 ml | 9 ml |
| 26–29 kg | 18 ml (0,90 mg) | 14 ml | 9 ml |
| 30–33 kg | 20 ml (1 mg) | 15 ml | 10 ml |
| 34–37 kg | 23 ml (1,15 mg) | 17 ml | 12 ml |
| 38–41 kg | 25 ml (1,25 mg) | 19 ml | 13 ml |
| 42–45 kg | 28 ml (1,40 mg) | 21 ml | 14 ml |
| 46–50 kg | 32 ml (1,60 mg) | 24 ml | 16 ml |
| ≥ 51 kg | 40 ml (2 mg) | 30 ml | 20 ml |
| Spexotras-annoksen pienentämistä alle 50 %:iin suositusannoksesta ei suositella. | |||
Kun potilaan haittavaikutukset on saatu tehokkaasti hallintaan, voidaan harkita annoksen nostamista uudelleen samojen annostasojen kautta kuin annosta pienennettäessä. Trametinibiannos ei saa ylittää taulukossa 1 esitettyä suositeltua annosta.
Annosmuutokset valikoitujen haittavaikutusten kohdalla
Kuume
Trametinibi- ja dabrafenibihoito on keskeytettävä, jos potilaan ruumiinlämpö on ≥ 38 °C. Kuumeilun toistuessa hoito voidaan keskeyttää myös ensimmäisten kuumeen oireiden ilmaannuttua. Hoito kuumelääkkeillä, kuten ibuprofeenilla tai parasetamolilla on aloitettava. Oraalisten kortikosteroidien käyttöä on harkittava tapauksissa, joissa kuumelääkkeet ovat riittämättömiä. Potilaan infektio-oireet ja -löydökset on arvioitava ja tarvittaessa hoidettava paikallisten hoitokäytäntöjen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hoito on aloitettava uudelleen, kun potilas on ollut oireeton vähintään 24 tunnin ajan joko (1) samalla annoksella tai (2) yhtä pienemmällä annoksella, jos kuume oli toistuvaa ja/tai siihen liittyi muita vaikeita oireita, kuten nestehukka, verenpaineen lasku tai munuaisten vajaatoiminta.
Annosmuutoksia koskevat poikkeukset (kun vain toisen valmisteen annosta pienennetään) valikoitujen haittavaikutusten osalta
Vasemman kammion ejektiofraktion (LVEF) pieneneminen / vasemman kammion toimintahäiriö
Trametinibihoito on keskeytettävä, jos potilaalla todetaan oireeton, > 10 %:n absoluuttinen vasemman kammion ejektiofraktion lasku lähtötasoon verrattuna ja ejektiofraktio alittaa hoitolaitoskohtaisen viitealueen alarajan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Dabrafenibin annosta ei tarvitse muuttaa yhdistelmähoidossa trametinibin kanssa. Jos vasemman kammion ejektiofraktio korjaantuu, trametinibihoito voidaan aloittaa uudelleen, mutta annosta on pienennettävä yhden annostason verran ja potilaan vointia on seurattava tarkoin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Trametinibihoito on lopetettava pysyvästi, jos vasemman kammion toimintahäiriön vaikeusaste on 3 tai 4 tai jos vasemman kammion ejektiofraktio on pienentynyt kliinisesti merkittävästi eikä korjaannu 4 viikon kuluessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verkkokalvon laskimotukos ja verkkokalvon pigmenttiepiteelin irtauma
Jos potilas ilmoittaa uusista näköhäiriöistä, kuten keskeisen näön heikkenemisestä, näön hämärtymisestä tai näön menetyksestä, milloin tahansa trametinibin ja dabrafenibin yhdistelmähoidon aikana, on suositeltavaa tehdä oftalmologinen tutkimus viipymättä. Jos verkkokalvon laskimotukos diagnosoidaan, trametinibi on lopetettava pysyvästi. Jos potilaalla todetaan verkkokalvon pigmenttiepiteelin irtauma, trametinibiannosta muutetaan alla taulukossa 4 esitetyn ohjelman mukaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Dabrafenibin annosta ei tarvitse muuttaa yhdistelmähoidossa trametinibin kanssa vahvistetun verkkokalvon laskimotukoksen tai verkkokalvon pigmenttiepiteelin irtauman takia.
Taulukko 4 Suositukset trametinibiannoksen muuttamisesta verkkokalvon pigmenttiepiteelin irtauman vuoksi
Verkkokalvon pigmenttiepiteelin 1. asteen irtauma | Jatka hoitoa ja tarkasta verkkokalvo kuukausittain, kunnes irtauma korjaantuu. Jos irtauma pahenee, noudata alla olevia ohjeita, ja keskeytä trametinibihoito enintään 3 viikon ajaksi. |
Verkkokalvon pigmenttiepiteelin 2. tai 3. asteen irtauma | Keskeytä trametinibihoito enintään 3 viikon ajaksi. |
Verkkokalvon pigmenttiepiteelin 2. tai 3. asteen irtauma, joka lievittyy 0. tai 1. asteen tasolle kolmessa viikossa | Aloita trametinibi uudelleen pienemmällä annoksella (ks. taulukko 3) tai lopeta trametinibi, jos potilaan trametinibiannos on jo pienimmällä annostasolla. |
Verkkokalvon pigmenttiepiteelin 2. tai 3. asteen irtauma, joka ei lievity vähintään 1. asteen tasolle kolmessa viikossa | Lopeta trametinibihoito pysyvästi. |
Interstitiaalinen keuhkosairaus/pneumoniitti
Jos potilaalla epäillään interstitiaalista keuhkosairautta tai pneumoniittia ja myös jos potilaalla on uusia tai eteneviä keuhko-oireita ja -löydöksiä, kuten yskää, hengenahdistusta, hypoksiaa, nestettä keuhkopussissa tai infiltraatteja, trametinibihoito on keskeytettävä, kunnes kliiniset tutkimukset on saatu päätökseen. Trametinibi on lopettava pysyvästi, jos potilaalla diagnosoidaan hoitoon liittyvä interstitiaalinen keuhkosairaus tai pneumoniitti. Dabrafenibin annosta ei tarvitse muuttaa yhdistelmähoidossa trametinibin kanssa interstitiaalisen keuhkosairauden tai pneumoniitin takia.
Uveiitti
Annosta ei tarvitse muuttaa uveiitin takia, jos silmätulehdus saadaan pidettyä hallinnassa tehokkailla paikallishoidoilla. Jos uveiitti ei reagoi silmän paikalliseen hoitoon, dabrafenibihoito on tauotettava, kunnes silmätulehdus on parantunut, ja aloitettava uudelleen yhtä annostasoa pienemmällä annoksella. Trametinibin annosta ei tarvitse muuttaa yhdistelmähoidossa dabrafenibin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muut RAS-mutaatiopositiiviset syövät kuin ihosyöpä
Hyötyjä ja riskejä on punnittava ennen dabrafenibihoidon jatkamista, jos potilaalla on muu RAS-mutaatiopositiivinen syöpä kuin ihosyöpä. Trametinibin annosta ei ole tarpeen muuttaa yhdistelmähoidossa dabrafenibin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erityisryhmät
Maksan vajaatoiminta
Annosta ei tarvitse sovittaa lievässä maksan vajaatoiminnassa. Kliinisen farmakologian tutkimus on osoittanut, että keskivaikealla tai vaikealla maksan vajaatoiminnalla on rajallinen vaikutus trametinibialtistukseen (ks. kohta Farmakokinetiikka). Trametinibin käytössä on noudatettava varovaisuutta keskivaikeassa tai vaikeassa maksan vajaatoiminnassa
Munuaisten vajaatoiminta
Annosta ei tarvitse sovittaa lievässä tai keskivaikeassa munuaisten vajaatoiminnassa. Trametinibin käytöstä vaikeassa munuaisten vajaatoiminnassa ei ole tietoa, joten annoksen sovittamisen tarvetta ei voida arvioida (ks. kohta Farmakokinetiikka). Trametinibin käytössä on noudatettava varovaisuutta vaikeassa munuaisten vajaatoiminnassa.
Pediatriset potilaat
Trametinibi-dabrafenibiyhdistelmähoidon turvallisuutta ja tehoa alle 1 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla. Eläinkokeissa, joissa trametinibia on annettu nuorille eläimille, on tullut esiin vaikutuksia, joita ei havaittu täysikasvuisilla eläimillä (ks. kohta Prekliiniset tiedot turvallisuudesta). Pediatrisia potilaita koskevia pitkän aikavälin tietoja on toistaiseksi vain vähän.
Antotapa
Spexotras on tarkoitettu otettavaksi suun kautta.
Apteekkihenkilökunnan on saatettava Spexotras-jauhe käyttökuntoon oraaliliuokseksi ennen valmisteen toimittamista. Lisäksi on suositeltavaa, että terveydenhuollon ammattilainen keskustelee ennen ensimmäisen annoksen antoa potilaan tai häntä avustavan henkilön kanssa siitä, miten lääkärin määräämä vuorokausiannos oraaliliuosta annetaan.
Ruoka ei vaikuta Spexotras-altistukseen (ks. kohta Farmakokinetiikka). Spexotras on otettava samanaikaisesti dispergoituvan dabrafenibitabletin kanssa, jonka altistus ruoan kanssa annettuna pienenee. Siksi Spexotras otetaan ilman ruokaa vähintään 1 tunti ennen ateriaa tai 2 tuntia aterian jälkeen (ks. kohta Farmakokinetiikka). Rintamaitoa ja/tai äidinmaidonkorviketta voidaan antaa tarpeen mukaan, jos potilas ei siedä paasto-olosuhteita.
On suositeltavaa ottaa Spexotras-annos joka päivä samaan aikaan käyttäen pakkauksessa olevaa uudelleenkäytettävää mittaruiskua. Spexotras-valmisteen kerran vuorokaudessa otettava annos on otettava samaan aikaan joka päivä joko dabrafenibin aamuannoksen tai ilta-annoksen yhteydessä.
Jos potilas ei kykene nielemään ja hänellä on nenä-mahaletku, Spexotras-oraaliliuos voidaan antaa letkun kautta.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet oraaliliuoksen valmistuksesta.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Spexotras on tarkoitettu käytettäväksi yhdessä dispergoituvien dabrafenibitablettien kanssa, koska trametinibi- ja dabrafenibimonoterapian tehoa koskevat tiedot ovat rajalliset BRAF V600 -mutaatiopositiivisen gliooman hoidossa. Dispergoituvien dabrafenibitablettien valmisteyhteenvetoon on perehdyttävä ennen hoidon aloittamista. Lisätiedot dabrafenibihoitoon liittyvistä varoituksista ja varotoimista, ks. dispergoituvien dabrafenibitablettien valmisteyhteenveto.
BRAF V600E -testaus
Trametinibin tehoa ja turvallisuutta yhdessä dabrafenibin kanssa ei ole arvioitu niiden potilaiden hoidossa, joiden gliooma on BRAF V600E -mutaation suhteen negatiivinen.
Uudet maligniteetit
Uusia maligniteetteja (ihomaligniteetteja ja muita maligniteetteja) saattaa esiintyä, kun trametinibia käytetään yhdessä dabrafenibin kanssa.
Ihomaligniteetit
Trametinibin ja dabrafenibin yhdistelmällä hoidetuilla aikuispotilailla on todettu ihomaligniteetteja, kuten ihon okasolusyöpää (mukaan lukien keratoakantooma) ja uusia primaarimelanoomia (ks. kohta Haittavaikutukset). Dermatologinen tutkimus tulisi tehdä ennen trametinibihoidon aloittamista ja kuukausittain hoidon aikana sekä kuuden kuukauden ajan hoidon jälkeen. Seurantaa on jatkettava kuuden kuukauden ajan trametinibihoidon lopettamisen jälkeen tai kunnes aloitetaan jokin muu syöpälääkitys.
Epäilyttävät ihomuutokset on poistettava kirurgisesti. Hoitoa ei tarvitse muuttaa. Potilaita on kehotettava ottamaan heti yhteys lääkäriin, jos uusia ihomuutoksia ilmaantuu.
Muut syöpäsairaudet kuin ihosyöpä
Vaikutusmekanisminsa perusteella dabrafenibi saattaa suurentaa muiden syöpäsairauksien kuin ihosyöpien riskiä, kun potilaalla on RAS-mutaatio. Ks. dispergoituvien dabrafenibitablettien valmisteyhteenveto (kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos potilaalla on RAS-mutaatiopositiivinen maligniteetti, trametinibin annosta ei tarvitse muuttaa yhdistelmähoidossa dabrafenibin kanssa.
Verenvuoto
Trametinibin ja dabrafenibin yhdistelmää käyttävillä aikuisilla ja pediatrisilla potilailla on raportoitu verenvuototapahtumia (ks. kohta Haittavaikutukset). Trametinibin ja dabrafenibin yhdistelmää käyttävillä aikuispotilailla on esiintynyt merkittäviä verenvuototapahtumia ja kuolemaan johtaneita verenvuotoja. Näiden tapahtumien mahdollisuutta ei ole varmistettu potilailla, joilla on alhainen trombosyyttiarvo (< 75 E9/l), sillä tällaiset potilaat suljettiin pois kliinisistä tutkimuksista. Samanaikainen antitromboottinen lääkitys tai antikoagulanttihoito saattaa suurentaa verenvuotoriskiä. Jos verenvuotoa esiintyy, potilasta on hoidettava kliinisen tilanteen mukaisesti.
Vasemman kammion ejektiofraktion (LVEF) pieneneminen / vasemman kammion toimintahäiriö
Trametinibin ja dabrafenibin yhdistelmähoidon on todettu pienentävän vasemman kammion ejektiofraktiota sekä aikuisilla että pediatrisilla potilailla (ks. kohta Haittavaikutukset). Pediatrisilla potilailla toteutetuissa kliinisissä tutkimuksissa mediaaniaika vasemman kammion ejektiofraktion ensimmäiseen pienenemiseen oli noin 1 kuukausi. Aikuispotilailla toteutetuissa kliinisissä tutkimuksissa mediaaniaika vasemman kammion toimintahäiriön ja sydämen vajaatoiminnan ensimmäiseen ilmaantumiseen ja vasemman kammion ejektiofraktion ensimmäiseen pienenemiseen oli 2–5 kuukautta.
Trametinibia on käyttävä varoen, jos potilaan vasemman kammion toiminta on heikentynyt. Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli vasemman kammion toimintahäiriö, NYHA (New York Heart Association) -luokan II, III tai IV sydämen vajaatoiminta, äkillinen sepelvaltimo-oireyhtymä kuuden edellisen kuukauden aikana, kliinisesti merkittäviä hallitsemattomia rytmihäiriöitä ja hallitsematon hypertensio. Siksi lääkkeen käytön turvallisuutta tämän potilasryhmän hoidossa ei tunneta. Kaikkien potilaiden vasemman kammion ejektiofraktio on tarkistettava ennen trametinibihoidon aloittamista, kuukauden kuluttua hoidon aloittamisesta ja sen jälkeen noin 3 kuukauden välein hoidon aikana (ks. annoksen muuttamista koskevat ohjeet kohdasta Annostus ja antotapa).
Potilailla, jotka käyttävät trametinibia yhdessä dabrafenibin kanssa, on raportoitu yksittäisiä myokardiitista johtuvia akuutteja vaikeita vasemman kammion toimintahäiriöitä. Potilaat toipuivat täysin, kun hoito lopetettiin. Lääkäreiden pitää ottaa huomioon myokardiitin mahdollisuus potilailla, joille ilmaantuu uusia tai pahenevia sydämeen liittyviä löydöksiä tai oireita.
Kuume
Kliinisissä trametinibitutkimuksissa aikuispotilailla ja pediatrisilla potilailla on ilmoitettu kuumetta (ks. kohta Haittavaikutukset). Kuumeen ilmaantuvuus ja vaikeusaste ovat suurempia yhdistelmähoidossa (ks. dispergoituvien dabrafenibitablettien valmisteyhteenveto, kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos potilas käyttää trametinibia yhdessä dabrafenibin kanssa, kuumeeseen saattaa liittyä vaikeaa vilutusta, nestehukkaa ja hypotensiota. Tämä voi johtaa joissakin tapauksissa akuuttiin munuaisten vajaatoimintaan. Trametinibin ja dabrafenibin yhdistelmää saaneilla pediatrisilla potilailla mediaaniaika kuumeen ensimmäiseen ilmaantumiseen oli 1,5 kk.
Trametinibi- ja dabrafenibihoito on keskeytettävä, jos potilaan ruumiinlämpö on ≥ 38 °C (ks. kohta Farmakodynamiikka). Toistuvassa kuumeilussa hoito voidaan keskeyttää myös ensimmäisten kuumeen oireiden ilmaannuttua. Hoito kuumelääkkeillä, kuten ibuprofeenilla tai parasetamolilla, on aloitettava. Oraalisten kortikosteroidien käyttöä on harkittava tapauksissa, joissa kuumelääkkeet ovat riittämättömiä. Potilaan infektio-oireet ja -löydökset on arvioitava. Hoito voidaan aloittaa uudelleen kuumeen laskettua. Jos kuumeeseen liittyy muita vaikeita oireita tai löydöksiä, hoitoa on jatkettava pienemmällä annoksella kuumeen laskettua ja kun se on kliinisesti tarkoituksenmukaista (ks. kohta Annostus ja antotapa).
Verenpaineen muutokset
Kliinisissä tutkimuksissa trametinibin ja dabrafenibin yhdistelmää käyttäneillä potilailla on raportoitu sekä hypertensiota että hypotensiota (ks. kohta Haittavaikutukset). Verenpaine on mitattava lähtötilanteessa, ja sitä on seurattava hoidon aikana. Hypertensiota on tarvittaessa hoidettava tavanomaisella hoidolla.
Interstitiaalinen keuhkosairaus/pneumoniitti
Aikuispotilailla toteutetussa vaiheen III tutkimuksessa 2,4 %:lle (5/211) pelkkää trametinibihoitoa saaneista potilaista kehittyi interstitiaalinen keuhkosairaus tai pneumoniitti, ja kaikki viisi potilasta tarvitsivat sairaalahoitoa. Mediaaniaika interstitiaalisen keuhkosairauden tai pneumoniitin ensimmäiseen ilmaantumiseen oli 160 päivää (vaihteluväli: 60–172 päivää). Kun aikuispotilaita hoidettiin trametinibin ja dabrafenibin yhdistelmällä kahdessa tutkimuksessa, 1 %:lle potilaista kehittyi pneumoniitti tai interstitiaalinen keuhkosairaus (ks. kohta Haittavaikutukset).
Jos potilaalla epäillään interstitiaalista keuhkosairautta tai pneumoniittia ja myös jos potilaalla on uusia tai eteneviä keuhko-oireita ja -löydöksiä, kuten yskää, hengenahdistusta, hypoksiaa, nestettä keuhkopussissa tai infiltraatteja, trametinibihoito on keskeytettävä, kunnes kliiniset tutkimukset on saatu päätökseen. Trametinibi on lopettava pysyvästi, jos potilaalla diagnosoidaan hoitoon liittyvä interstitiaalinen keuhkosairaus tai pneumoniitti (ks. kohta Annostus ja antotapa). Dabrafenibihoitoa voidaan jatkaa samalla annoksella.
Näön heikkeneminen
Näköhäiriöitä aiheuttavia sairauksia, kuten verkkokalvon pigmenttiepiteelin irtaumia ja verkkokalvon laskimotukoksia saattaa esiintyä trametinibin käytön yhteydessä. Joissakin tapauksissa niiden alkamiseen on kulunut useita kuukausia. Oireita, kuten näön hämärtymistä, näöntarkkuuden heikkenemistä ja muita näköoireita on raportoitu trametinibin aikuisilla toteutetuissa kliinisissä tutkimuksissa. Kliinisissä tutkimuksissa on ilmoitettu myös uveiittia ja iridosykliittiä, kun aikuisille ja pediatrisille potilaille on annettu trametinibin ja dabrafenibin yhdistelmää.
Trametinibia ei suositella potilaille, joilla on ollut verkkokalvon laskimotukos. Trametinibin turvallisuutta ei ole vahvistettu potilailla, joilla on verkkokalvon laskimotukokselle altistavia tekijöitä, kuten huonossa hoitotasapainossa oleva glaukooma tai kohonnut silmänpaine, huonossa hoitotasapainossa oleva hypertensio, huonossa hoitotasapainossa oleva diabetes mellitus tai aiemmin todettu hyperviskositeettioireyhtymä tai veren liiallinen hyytymistaipumus.
Jos potilas ilmoittaa uusista näköhäiriöistä, kuten keskeisen näön heikkenemisestä, näön hämärtymisestä tai näön menetyksestä, milloin tahansa trametinibihoidon aikana, oftalmologinen tutkimus on suositeltavaa tehdä viipymättä. Jos potilaalla todetaan verkkokalvon pigmenttiepiteelin irtauma, annosta muutetaan taulukossa 4 esitetyn ohjelman mukaisesti (ks. kohta Annostus ja antotapa). Jos potilaalla todetaan uveiitti, ks. dispergoituvien dabrafenibitablettien valmisteyhteenveto (kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos verkkokalvon laskimotukos diagnosoidaan, trametinibihoito on lopetettava pysyvästi.
Dabrafenibin annosta ei tarvitse muuttaa yhdistelmähoidossa trametinibin kanssa verkkokalvon laskimotukos- tai verkkokalvon pigmenttiepiteelin irtaumadiagnoosin jälkeen. Trametinibin annosta ei tarvitse muuttaa yhdistelmähoidossa dabrafenibin kanssa uveiittidiagnoosin jälkeen.
Ihottuma
Kliinisissä tutkimuksissa on havaittu ihottumaa 49 %:lla pediatrisista potilaista, kun trametinibia on käytetty yhdessä dabrafenibin kanssa (ks. kohta Haittavaikutukset). Suurimmassa osassa näistä tapauksista ihottuman vaikeusaste oli 1 tai 2, eikä se vaatinut hoidon keskeyttämistä eikä annoksen pienentämistä.
Vaikeat ihoon kohdistuvat haittavaikutukset
Trametinibin ja dabrafenibin yhdistelmähoidon aikana aikuispotilailla on ilmoitettu vaikeita ihoon kohdistuneita haittavaikutuksia, mukaan lukien Stevens–Johnsonin oireyhtymää ja DRESS-reaktioita (lääkereaktio, johon liittyy eosinofiliaa ja systeemisiä oireita), jotka voivat olla henkeä uhkaavia tai johtaa kuolemaan. Ennen hoidon aloitusta potilaille on kerrottava ihoreaktioiden oireista ja löydöksistä, ja heitä on seurattava niiden varalta tarkoin. Jos vaikeisiin ihoon kohdistuviin haittavaikutuksiin viittaavia oireita ja löydöksiä esiintyy, trametinibin ja dabrafenibin käyttö on lopetettava.
Rabdomyolyysi
Rabdomyolyysia on raportoitu aikuispotilailla, jotka ovat käyttäneet trametinibia. Joissakin tapauksissa potilaat pystyivät jatkamaan trametinibihoitoa. Vaikeammat tapaukset vaativat sairaalahoitoa tai hoidon keskeyttämistä tai pysyvää lopettamista. Rabdomyolyysin oireet ja löydökset vaativat asianmukaista kliinistä selvittelyä ja tarvittaessa hoitoa.
Haimatulehdus
Kliinisissä tutkimuksissa trametinibin ja dabrafenibin yhdistelmällä hoidetuilla aikuisilla ja pediatrisilla potilailla on raportoitu haimatulehdusta (ks. kohta Haittavaikutukset). Selittämätön vatsakipu on tutkittava heti, ja seerumin amylaasi- ja lipaasiarvot on tarkistettava. Potilaan tilaa on seurattava tarkoin, kun hoito aloitetaan uudelleen haimatulehdusepisodin jälkeen.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa on todettu ≤ 1 %:lla aikuispotilaista, joita on hoidettu trametinibilla yhdessä dabrafenibin kanssa. Aikuispotilailla todettuihin tapauksiin liittyi yleensä kuumetta ja nestehukkaa ja potilaat reagoivat hyvin hoidon keskeyttämiseen ja tavanomaisiin tukitoimenpiteisiin. Myös granulomatoottista nefriittiä on raportoitu aikuispotilailla. Potilaan seerumin kreatiniinia on säännöllisesti seurattava hoidon aikana. Jos kreatiniiniarvo nousee, potilaan kliininen tila saattaa vaatia hoidon keskeyttämistä. Trametinibia ei ole tutkittu munuaisten vajaatoiminnassa (kreatiniini > 1,5 x ULN), joten varovaisuutta on noudatettava näillä potilailla (ks. kohta Farmakokinetiikka).
Maksaan liittyvät tapahtumat
Kliinisissä tutkimuksissa aikuisilla ja pediatrisilla potilailla on raportoitu maksaan kohdistuvia haittavaikutuksia, kun trametinibia on käytetty yhdessä dabrafenibin kanssa (ks. kohta Haittavaikutukset). Potilaiden maksan toimintaa suositellaan seurattavan neljän viikon välein kuuden kuukauden ajan hoidon aloittamisen jälkeen. Maksa-arvojen seurantaa voidaan jatkaa myös tämän jälkeen kliinisen tarpeen mukaan.
Maksan vajaatoiminta
Trametinibi eliminoituu ensisijaisesti metaboloitumalla ja erittymällä sappeen, ja siksi trametinibin käytössä on noudatettava varovaisuutta, jos potilaalla on keskivaikea tai vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Syvä laskimotukos/keuhkoembolia
Keuhkoembolioita tai syviä laskimotukoksia saattaa esiintyä. Jos potilaalla ilmenee keuhkoembolian tai syvän laskimotukoksen oireita, kuten hengenahdistusta, rintakipua tai käsivarren tai jalan turvotusta, hänen on hakeuduttava välittömästi lääkärin hoitoon. Hoito on lopetettava pysyvästi, jos potilaalla on henkeä uhkaava keuhkoembolia.
Ruoansulatuselimistö
Trametinibin ja dabrafenibin yhdistelmällä hoidetuilla pediatrisilla potilailla on raportoitu koliittia ja enterokoliittia (ks. kohta Haittavaikutukset). Koliittia ja ruoansulatuskanavan perforaatioita (myös kuolemaan johtaneita tapauksia) on raportoitu aikuispotilailla. Trametinibin käytössä on noudatettava varovaisuutta hoidettaessa potilaita, joilla on ruoansulatuskanavan perforaation riskitekijöitä, kuten aikaisempi divertikuliitti, ruoansulatuskanavan metastaasit tai sellaisten lääkevalmisteiden samanaikainen käyttö, joihin tiedetään liittyvän ruoansulatuskanavan perforaation riski.
Sarkoidoosi
Trametinibin ja dabrafenibin yhdistelmällä hoidetuilla aikuispotilailla on ilmoitettu sarkoidoositapauksia. Haittavaikutukset ovat kohdistuneet pääasiassa ihoon, keuhkoihin, silmiin ja imusolmukkeisiin. Suurimmassa osassa tapauksia trametinibi- ja dabrafenibihoitoa jatkettiin. Sarkoidoosidiagnoosin yhteydessä on harkittava asianmukaista hoitoa.
Naiset, jotka voivat tulla raskaaksi / Miesten hedelmällisyys
Naisille, jotka voivat tulla raskaaksi, on annettava asianmukaista neuvontaa tehokkaista ehkäisymenetelmistä ennen hoidon aloittamista. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja 16 viikon ajan viimeisen Spexotras-annoksen jälkeen. Trametinibia yhdessä dabrafenibin kanssa käyttäville miespotilaille on kerrottava mahdollisesta siittiötuotannon vähenemisestä, joka voi olla pysyvää (ks. kohta Raskaus ja imetys).
Hemofagosyyttinen lymfohistiosytoosi
Myyntiluvan myöntämisen jälkeen trametinibia ja dabrafenibia yhdistelmähoitona saavilla aikuispotilailla on havaittu hemofagosyyttista lymfohistiosytoosia (HLH). Käytettäessä trametinibia yhdessä dabrafenibin kanssa on noudatettava varovaisuutta. Jos potilaalla on vahvistettu HLH, on lopetettava trametinibin ja dabrafenibin yhdistelmähoidon antaminen ja aloitettava HLH:n hoito.
Tuumorilyysioireyhtymä
Trametinibin ja dabrafenibin yhdistelmähoidon käyttöön on liittynyt tuumorilyysioireyhtymää, joka voi johtaa kuolemaan (ks. kohta Haittavaikutukset). Tuumorilyysioireyhtymän riskitekijöitä ovat suuri kasvaintaakka, olemassa oleva krooninen munuaisten vajaatoiminta, oliguria, nestehukka, hypotensio ja hapan virtsa. Potilaita, joilla on tuumorilyysioireyhtymän riskitekijöitä, on seurattava huolellisesti ja profylaktista nesteytystä on harkittava. Tuumorilyysioireyhtymä on hoidettava viipymättä kliinisen tarpeen mukaan.
Apuaineet
Sulfobutyylibeetadeksinatrium
Spexotras-oraaliliuos sisältää syklodekstriini sulfobutyylibeetadeksinatriumia (100 mg/ml). Syklodekstriinit ovat apuaineita, jotka voivat vaikuttaa vaikuttavan aineen ja muiden lääkkeiden ominaisuuksiin. Prekliinisissä tutkimuksissa eläimillä, joille annettiin syklodekstriineja laskimoon, havaittiin munuaistoksisuutta ja ototoksisuutta. Syklodekstriinien turvallisuusnäkökohdat on huomioitava lääkevalmisteen tuotekehityksen ja turvallisuusarvioinnin aikana. Turvallisuutta koskevaa tietoa syklodekstriinien vaikutuksista alle 2-vuotiaisiin lapsiin on vain vähän.
Metyyliparahydroksibentsoaatti
Tämä lääkevalmiste sisältää metyyliparahydroksibentsoaattia, joka saattaa aiheuttaa allergisia reaktioita (mahdollisesti viivästyneitä).
Natrium
Tämä lääkevalmiste sisältää 1,98 mg natriumia per ml Spexotras-oraaliliuosta, joka vastaa 4 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuiselle käytettäessä trametinibin enimmäisvuorokausiannosta 2 mg (40 ml).
Kalium
Tämä lääkevalmiste sisältää kaliumia alle 1 mmol (39 mg) per enimmäisvuorokausiannos, eli sen voidaan sanoa olevan ”kaliumiton”.
Yhteisvaikutukset
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Muiden lääkkeiden vaikutus trametinibiin
Trametinibin metaboloituminen tapahtuu pääasiassa hydrolyyttisten entsyymien (esim. karboksyyliesteraasien) välittämän deasetylaation kautta, joten muut lääkeaineet eivät todennäköisesti vaikuta sen farmakokinetiikkaan metabolisten yhteisvaikutusten kautta (ks. kohta Farmakokinetiikka). Näiden hydrolyyttisten entsyymien välittämiä lääkeaineiden yhteisvaikutuksia ei voida sulkea pois, ja ne voivat vaikuttaa trametinibialtistukseen.
Trametinibi on P-gp-kuljetusproteiinin substraatti in vitro. Koska maksan P-gp:n voimakkaan eston aiheuttamaa trametinibitason nousua ei voida sulkea pois, on varovaisuutta noudatettava annettaessa trametinibia yhdessä sellaisten lääkevalmisteiden kanssa, jotka ovat voimakkaita P-gp-estäjiä (esim. verapamiili, siklosporiini, ritonaviiri, kinidiini, itrakonatsoli).
Trametinibin vaikutus muihin lääkkeisiin
In vitro ja in vivo -tutkimusten perusteella trametinibi ei todennäköisesti vaikuta merkittävästi muiden lääkevalmisteiden farmakokinetiikkaan CYP-entsyymien tai kuljettajaproteiinien välityksellä (ks. kohta Farmakokinetiikka). Trametinibi saattaa estää tilapäisesti BCRP:n substraatteja (esim. pitavastatiinia) suolistossa. Tätä vaikutusta voidaan vähentää porrastamalla näiden lääkeaineiden ja trametinibin antaminen (lääkkeiden välillä on oltava 2 tuntia).
Kliinisten tietojen perusteella hormonaalisten ehkäisyvalmisteiden tehon ei odoteta heikkenevän, kun niitä annetaan samanaikaisesti trametinibimonoterapian kanssa (ks. kohta Farmakokinetiikka). Yhteiskäyttö dabrafenibin kanssa voi kuitenkin heikentää hormonaalisten ehkäisyvalmisteiden tehoa.
Apuaine sulfobutyylibeetadeksinatriumin vaikutus muihin suun kautta otettaviin lääkevalmisteisiin, joiden biologinen hyötyosuus on pieni ja terapeuttinen leveys kapea
Trametinibioraaliliuos sisältää 100 mg/ml sulfobutyylibeetadeksinatriumia, joka saattaa vaikuttaa muiden suun kautta otettavien lääkkeiden liukoisuuteen ja biologiseen hyötyosuuteen. Varovaisuutta on noudatettava, kun trametinibioraaliliuosta annetaan samanaikaisesti sellaisten suun kautta otettavien lääkevalmisteiden kanssa, joiden biologinen hyötyosuus on pieni ja terapeuttinen leveys kapea (esim. imipramiini, desipramiini).
Ks. lisäksi ohjeet dabrafenibin yhteisvaikutuksista muiden lääkevalmisteiden kanssa dispergoituvien dabrafenibitablettien valmisteyhteenvedon kohdista Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Raskauden ehkäisy naisilla
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä trametinibihoidon aikana ja 16 viikon ajan hoidon päättymisen jälkeen.
Yhteiskäyttö dabrafenibin kanssa saattaa heikentää suun kautta tai systeemisesti otettujen hormonaalisten ehkäisyvalmisteiden tehoa, joten tehokasta vaihtoehtoista ehkäisymenetelmää, kuten estemenetelmää, on käytettävä trametinibin ja dabrafenibin yhdistelmähoidon aikana. Lisätiedot, ks. dispergoituvien dabrafenibitablettien valmisteyhteenveto.
Raskaus
Ei ole olemassa tietoja trametinibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Trametinibia ei pidä antaa raskaana oleville naisille, paitsi jos hoidon odotettu hyöty äidille on suurempi kuin mahdollinen sikiölle aiheutuva riski. Jos trametinibia käytetään raskauden aikana tai jos potilas tulee raskaaksi trametinibihoidon aikana, hänelle on kerrottava mahdollisesta sikiöön kohdistuvasta vaarasta.
Imetys
Ei tiedetä, erittyykö trametinibi ihmisen rintamaitoon. Imetettävään lapseen kohdistuvia riskejä ei voida poissulkea. Trametinibia ei pidä antaa imettäville äideille. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko trametinibihoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Ihmistä koskevia tietoja ei ole trametinibin käytöstä. Eläimillä ei ole tehty hedelmällisyystutkimuksia, mutta naaraiden lisääntymiselimiin kohdistuvia vaikutuksia on havaittu (ks. kohta Prekliiniset tiedot turvallisuudesta). Trametinibi saattaa heikentää ihmisen hedelmällisyyttä.
Miehet, jotka käyttävät trametinibia yhdessä dabrafenibin kanssa
Dabrafenibia saaneilla eläimillä on havaittu spermatogeneesiin kohdistuvia vaikutuksia. Trametinibia yhdessä dabrafenibin kanssa käyttäville miespotilaille on kerrottava mahdollisesta siittiötuotannon vähenemisestä, joka voi olla pysyvää. Lisätiedot, ks. dispergoituvien dabrafenibitablettien valmisteyhteenveto.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Trametinibilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaan kliininen tila ja trametinibin haittavaikutusprofiili on otettava huomioon, kun arvioidaan potilaan kykyä selviytyä harkintakykyä ja motorisia tai kognitiivisia taitoja vaativista tehtävistä. Potilaille on kerrottava, että mahdollinen väsymys, huimaus ja silmäoireet voivat vaikuttaa näihin toimintoihin.
Haittavaikutukset
Yhteenveto turvallisuustiedoista
Kliinisissä tutkimuksissa trametinibin ja dabrafenibin yhdistelmällä hoidettujen pediatristen potilaiden yleisimmät haittavaikutukset (raportoitu ≥ 20 %:lla) olivat kuume (70 %), ihottuma (49 %), päänsärky (47 %), oksentelu (40 %), väsymys (36 %), ihon kuivuus (35 %), ripuli (34 %), verenvuoto (34 %), pahoinvointi (29 %), aknetyyppinen dermatiitti (29 %), vatsakipu (28 %), neutropenia (26 %), yskä (24 %) ja transaminaasiarvojen suureneminen (22 %). Yleisimmin raportoidut vaikeat (3. tai 4. asteen) haittavaikutukset olivat neutropenia (15 %), kuume (11 %), transaminaasiarvojen suureneminen (6 %) ja painonnousu (5 %). Pitkäaikaistietoja pediatristen potilaiden kasvusta ja luuston kypsymisestä on tällä hetkellä vain vähän (ks. kohta Prekliiniset tiedot turvallisuudesta).
Turvallisuusprofiili pediatrisilla potilailla oli laajalti yhdenmukainen aikuispotilailla aiemmin todetun turvallisuusprofiilin kanssa. Seuraavia haittavaikutuksia on toistaiseksi raportoitu vain aikuispotilailla, joita on hoidettu trametinibitableteilla ja dabrafenibikapseleilla: ihon okasolusyöpä, seborrooinen keratoosi, perifeerinen neuropatia (mukaan lukien sensorinen ja motorinen neuropatia), lymfedeema, suun kuivuus, aktiininen keratoosi, munuaisten vajaatoiminta, sädehoidon toksisuuden vahvistuminen (yleisiä); melanooma, ihopolyypit, sarkoidoosi, korioretinopatia, pneumoniitti, akuutti munuaisten vajaatoiminta, munuaistulehdus, sydämen vajaatoiminta, vasemman kammion toimintahäiriö, interstitiaalinen keuhkosairaus, rabdomyolyysi (melko harvinaisia); ruoansulatuskanavan perforaatio, hemofagosyyttinen lymfohistiosytoosi (harvinaisia); tuumorilyysioireyhtymä, myokardiitti, Stevens–Johnsonin oireyhtymä, DRESS-reaktio (lääkereaktio, johon liittyy eosinofiliaa ja systeemisiä oireita), tatuointeihin liittyvät ihoreaktiot (yleisyys tuntematon).
Haittavaikutustaulukko
Trametinibin ja dabrafenibin yhdistelmän turvallisuutta on arvioitu 171 pediatrisen potilaan yhdistetyssä turvallisuuspopulaatiossa, joka sisälsi potilaita kahdesta tutkimuksesta. Potilailla oli BRAF V600 -mutaatiopositiivisia pitkälle edenneitä kiinteitä kasvaimia. Tutkimukseenottohetkellä potilaista neljä (2,3 %) oli 1 – < 2-vuotiaita, 39 (22,8 %) oli 2 – < 6-vuotiaita, 54 (31,6 %) oli 6 – < 12-vuotiaita ja 74 (43,3 %) oli 12 – < 18-vuotiaita. Hoidon keskikesto oli 2,3 vuotta.
Haittavaikutukset (taulukko 5) on lueteltu alla MedDRA-elinjärjestelmäluokittain ja yleisyyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja yleisyys tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 5Trametinibin ja dabrafenibin yhdistelmähoitoon liittyvät haittavaikutukset
| Infektiot | |
| Hyvin yleinen | Kynnenvierustulehdus, nenänielutulehdus*1 |
| Yleinen | Virtsatieinfektio, selluliitti |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | |
| Hyvin yleinen | Ihon papillooma |
| Veri ja imukudos | |
| Hyvin yleinen | Neutropenia*2, anemia, leukopenia* |
| Yleinen | Trombosytopenia* |
| Immuunijärjestelmä | |
| Yleinen | Yliherkkyys |
| Aineenvaihdunta ja ravitsemus | |
| Yleinen | Nestehukka, ruokahalun heikkeneminen |
| Hermosto | |
| Hyvin yleinen | Päänsärky, huimaus*3 |
| Silmät | |
| Yleinen | Näön hämärtyminen, näköhäiriöt, uveiitti*4 |
| Melko harvinainen | Verkkokalvon irtauma, periorbitaalinen turvotus |
| Sydän | |
| Yleinen | Ejektiofraktion pieneneminen, bradykardia* |
| Melko harvinainen | Eteis-kammiokatkos5 |
| Verisuonisto | |
| Hyvin yleinen | Verenvuoto*6 |
| Yleinen | Hypertensio, hypotensio |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | Yskä* |
| Yleinen | Hengenahdistus |
| Ruoansulatuselimistö | |
| Hyvin yleinen | Vatsakipu*, ummetus, ripuli, pahoinvointi, oksentelu |
| Yleinen | Haimatulehdus, suutulehdus |
| Melko harvinainen | Koliitti* |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | Aknetyyppinen dermatiitti*7, ihon kuivuus*8, kutina, ihottuma*9, punoitus |
| Yleinen | Yleistynyt eksfoliatiivinen dermatiitti*10, hiustenlähtö, käsi-jalkaoireyhtymä, follikuliitti, ihomuutokset, pannikuliitti, hyperkeratoosi, valoherkkyys*11 |
| Melko harvinainen | Akuutti kuumeinen neutrofiilinen dermatoosi, ihon fissuurat, yöhikoilu, voimakas hikoilu |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleinen | Nivelkipu, raajakipu |
| Yleinen | Lihaskipu*, lihasspasmit*12 |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | Kuume*, väsymys*13, painonnousu |
| Yleinen | Limakalvotulehdus, kasvojen turvotus*, vilunväristykset, perifeerinen edeema, influenssankaltainen sairaus |
| Tutkimukset | |
| Hyvin yleinen | Transaminaasiarvojen suureneminen*14 |
| Yleinen | Hyponatremia, hypofosfatemia, hyperglykemia, veren AFOS-pitoisuuden suureneminen, GGT-arvon suureneminen, veren kreatiinikinaasipitoisuuden suureneminen |
*Kuvaa yläkäsitettä, joka kattaa vähintään kaksi kliinisesti samankaltaisiksi katsottua MedDRA-haittavaikutustermiä. 1 nenänielutulehdus sisältää nielutulehduksen 2 neutropenia sisältää neutrofiiliarvon pienenemisen ja kuumeisen neutropenian 3 huimaus sisältää kiertohuimauksen 4 uveiitti sisältää iridosykliitin 5 eteis-kammiokatkos sisältää ensimmäisen asteen eteis-kammiokatkoksen 6 verenvuoto sisältää nenäverenvuodon, hematurian, kontuusion, hematooman, INR-arvon suurenemisen, peräaukon verenvuodon, kanyylikohdan verenvuodon, aivoverenvuodon, ekkymoosin, ekstraduraalihematooman, ruoansulatuskanavan verenvuodon, hematoketsian, petekiat, toimenpiteen jälkeisen verenvuodon, peräsuolen verenvuodon, veren punasoluarvon pienenemisen, ruoansulatuskanavan yläosan verenvuodon, kohtuverenvuodon, runsaan kuukautisvuodon sekä purppuran 7 aknetyyppinen dermatiitti sisältää aknen ja pustulaarisen aknen 8 ihon kuivuus sisältää kseroosin ja kseroderman 9 ihottuma sisältää makulopapulaarisen ihottuman, pustulaarisen ihottuman, punoittavan ihottuman, papulaarisen ihottuman ja makulaarisen ihottuman 10 yleistynyt eksfoliatiivinen dermatiitti sisältää ihon kesimisen ja eksfoliatiivisen dermatiitin 11 valoherkkyys sisältää valoherkkyysreaktion ja auringonpolttaman 12 lihasspasmit sisältää tuki- ja liikuntaelimistön jäykkyyden 13 väsymys sisältää huonovointisuuden ja astenian 14 transaminaasiarvojen suureneminen sisältää ASAT-arvon suurenemisen, ALAT-arvon suurenemisen ja hypertransaminasemian | |
Valikoitujen haittavaikutusten kuvaus
Painonnousu
Painonnousua on raportoitu ainoastaan pediatrisilla potilailla. Painonnousua raportoitiin haittavaikutuksena 16 %:lla pediatrisista potilaista; 3. asteen tapauksia raportoitiin 5 %:lla potilaista, ja painonnousu johti hoidon lopettamiseen 0,6 %:lla potilaista. Mediaaniaika ensimmäiseen painonnousun raportointiin oli trametinibin ja dabrafenibin yhdistelmää saaneilla pediatrisilla potilailla 3,5 kk. Potilaista 36 %:lla havaittiin lähtötilanteeseen verrattuna painonnousu, joka vastasi ≥ 2:n iänmukaisen BMI (body mass index, painoindeksi) -persentiililuokan nousua.
Verenvuoto
Verenvuototapahtumia havaittiin 34 %:lla pediatrisista potilaista ja 3. asteen tapahtumia 1,2 %:lla potilaista. Yleisintä verenvuototapahtumaa (nenäverenvuoto) raportoitiin 18 %:lla pediatrisista potilaista. Mediaaniaika ensimmäisen verenvuototapahtuman ilmaantumiseen pediatrisilla potilailla oli 2,6 kk. Trametinibia yhdessä dabrafenibin kanssa saaneilla aikuispotilailla esiintyi verenvuototapahtumia, joihin kuului myös merkittäviä verenvuototapahtumia ja kuolemaan johtaneita verenvuotoja.
Samanaikainen antitromboottinen lääkitys tai antikoagulanttihoito saattaa suurentaa verenvuotoriskiä. Jos verenvuotoa esiintyy, potilasta hoidetaan kliinisen tilanteen mukaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vasemman kammion ejektiofraktion (LVEF) pieneneminen / vasemman kammion toimintahäiriö
Vasemman kammion ejektiofraktion pienenemistä on raportoitu 5,3 %:lla pediatrisista potilaista ja 3. asteen tapahtumia < 1 %:lla potilaista. Mediaaniaika vasemman kammion ejektiofraktion pienenemisen ensimmäiseen ilmaantumiseen oli noin 1 kk. Aikuispotilailla toteutetuissa kliinisissä tutkimuksissa aika vasemman kammion toimintahäiriön ja sydämen vajaatoiminnan ensimmäiseen ilmaantumiseen ja ejektiofraktion ensimmäiseen pienenemiseen oli 2–5 kuukautta (mediaani).
Trametinibin kliinisiin tutkimuksiin ei otettu mukaan potilaita, joiden LVEF oli tutkimuskeskuksen oman viitealueen alarajan alapuolella. Trametinibia on käytettävä dabrafenibin kanssa varoen potilailla, joilla on sairauksia, jotka voivat heikentää vasemman kammion toimintaa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kuume
Kliinisissä tutkimuksissa on ilmoitettu kuumetta, kun trametinibia on käytetty ainoana lääkkeenä tai yhdessä dabrafenibin kanssa. Kuumeen ilmaantuvuus ja vaikeusaste ovat kuitenkin suurempia yhdistelmähoidossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kuumetta ilmoitettiin 70 %:lla pediatrisista potilaista ja 3. asteen tapahtumia 11 %:lla potilaista. Ks. dispergoituvien dabrafenibitablettien valmisteyhteenveto.
Maksaan liittyvät tapahtumat
Aikuispotilailla ja pediatrisilla potilailla toteutetuissa kliinisissä tutkimuksissa on raportoitu maksaan liittyviä haittavaikutuksia, kun trametinibia on käytetty yhdessä dabrafenibin kanssa. Pediatrisessa turvallisuuspopulaatiossa ALAT-arvon suureneminen (raportoitiin 13 %:lla potilaista) ja ASAT-arvon suureneminen (raportoitiin 16 %:lla potilaista) olivat hyvin yleisiä tapahtumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Aikuispotilailla ALAT-arvon kohoaminen ja ASAT-arvon kohoaminen olivat yleisimmät maksaan liittyneet haittavaikutukset, ja suurimmassa osassa näistä tapahtumista vaikeusasteluokka oli 1 tai 2. Pelkkää trametinibia saaneilla potilailla yli 90 % näistä maksatapahtumista ilmaantui kuuden ensimmäisen hoitokuukauden aikana. Maksatapahtumat todettiin kliinisissä tutkimuksissa, joissa arvoja seurattiin neljän viikon välein. Trametinibia saavien potilaiden maksatoimintaa on suositeltavaa seurata neljän viikon välein kuuden kuukauden ajan. Maksa-arvojen seurantaa voidaan jatkaa myös tämän jälkeen, jos se on kliinisesti aiheellista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verenpaineen muutokset
Hypertensiota raportoitiin 2,3 %:lla pediatrisista potilaista ja 3. asteen tapahtumia 1,2 %:lla potilaista. Mediaaniaika ensimmäisen hypertensiotapahtuman ilmaantumiseen pediatrisilla potilailla oli 5,4 kk.
Hypotensiota raportoitiin 4,1 %:lla pediatrisista potilaista ja 3. asteen tapahtumia 2,3 %:lla potilaista. Mediaaniaika ensimmäisen hypotensiotapahtuman ilmaantumiseen pediatrisilla potilailla oli 2,2 kk.
Verenpaine on mitattava hoitoa aloitettaessa, ja sitä on seurattava hoidon aikana, ja tarvittaessa hypertensio on pidettävä hallinnassa tavanomaisella hoidolla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Interstitiaalinen keuhkosairaus/pneumoniitti
Trametinibia saaville potilaille saattaa kehittyä interstitiaalinen keuhkosairaus tai pneumoniitti. Jos potilaalla epäillään interstitiaalista keuhkosairautta tai pneumoniittia ja myös jos potilaalla on uusia tai eteneviä keuhko-oireita ja -löydöksiä, kuten yskää, hengenahdistusta, hypoksiaa, nestettä keuhkopussissa tai infiltraatteja, trametinibihoito on keskeytettävä, kunnes kliiniset tutkimukset on saatu päätökseen. Trametinibi on lopettava pysyvästi, jos potilaalla diagnosoidaan hoitoon liittyvä interstitiaalinen keuhkosairaus tai pneumoniitti (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Näön heikkeneminen
Trametinibin ja dabrafenibin yhdistelmällä hoidetuilla pediatrisilla potilailla on raportoitu silmäreaktioita, kuten uveiittia (3,5 %) ja iridosykliittiä (1,8 %). 3. asteen uveiitti todettiin 1,8 %:lla pediatrisista potilaista. Verkkokalvon pigmenttiepiteelin irtauma todettiin < 1 %:lla pediatrisista potilaista. Näköhäiriöitä aiheuttavia sairauksia, kuten verkkokalvon pigmenttiepiteelin irtaumia ja verkkokalvon laskimotukoksia, on havaittu myös aikuispotilailla trametinibihoidon yhteydessä. Trametinibin aikuisilla toteutetuissa kliinisissä tutkimuksissa on raportoitu oireita, kuten näön hämärtymistä, näöntarkkuuden heikkenemistä ja muita näköhäiriöitä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Ihottuma
Ihottumaa on havaittu 49 %:lla pediatrisista potilaista trametinibin ja dabrafenibin yhdistelmää koskevissa tutkimuksissa yhdistetyssä turvallisuuspopulaatiossa. Suurimmassa osassa näistä tapauksista vaikeusaste oli 1 tai 2, eikä se vaatinut hoidon keskeyttämistä eikä annoksen pienentämistä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Rabdomyolyysi
Rabdomyolyysia on raportoitu trametinibia käyttävillä aikuispotilailla. Rabdomyolyysin oireet ja löydökset vaativat asianmukaista kliinistä selvittelyä ja tarvittaessa hoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haimatulehdus
Haimatulehdusta raportoitiin 1,2 %:lla pediatrisista potilaista; potilaista < 1 %:lla haimatulehdus oli vaikeudeltaan 3. astetta. Selittämätön vatsakipu on tutkittava heti, ja seerumin amylaasi- ja lipaasiarvot on tarkistettava. Potilaan tilaa on seurattava tarkoin, kun hoito aloitetaan uudelleen haimatulehdusepisodin jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa on ilmoitettu trametinibin ja dabrafenibin yhdistelmähoidossa. Kuumeeseen liittyvän prerenaalisen atsotemian aiheuttamaa munuaisten vajaatoimintaa tai granulomatoottista nefriittiä esiintyi aikuispotilailla melko harvoin. Trametinibia ei kuitenkaan ole tutkittu potilailla, joilla oli munuaisten vajaatoimintaa (kreatiniini > 1,5 x viitearvojen yläraja). Tällaisissa tapauksissa on noudatettava varovaisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa ei ole raportoitu akuutteja yliannostusoireita pediatrisilla potilailla, jotka saivat trametinibia yhdessä dabrafenibin kanssa. Trametinibin jatkuva yliannostus voi aiheuttaa ihottuman lisääntymistä, vasemman kammion ejektiofraktion pienenemistä tai verkkokalvon poikkeavuuksia. Yliannostukseen ei ole spesifistä hoitoa. Yliannostustapauksissa on annettava asianmukaista tukihoitoa ja potilaan tilaa on seurattava tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, proteiinikinaasin estäjät, mitogeenin aktivoiman proteiinikinaasin (MEK) estäjät, ATC-koodi: L01EE01
Vaikutusmekanismi
Trametinibi on reversiibeli, erittäin selektiivinen, mitogeenin aktivoiman solunulkoisen signaalin säätelykinaasi 1:n (MEK1:n) ja MEK2:n aktivaation ja kinaasiaktiivisuuden allosteerinen estäjä. MEK-proteiinit ovat ERK-kinaasin signalointireitin osia. Ihmisen syövissä tämän reitin aktivoivat usein BRAF:n mutatoituneet muodot, mikä puolestaan aktivoi MEK:n. Trametinibi estää BRAF:n aiheuttaman MEK:n aktivoitumisen ja estää MEK:n kinaasiaktiivisuutta.
Käyttö yhdessä dabrafenibin kanssa
Dabrafenibi on RAF-kinaasien estäjä. BRAF:n onkogeeniset mutaatiot johtavat RAS/RAF/MEK/ERK-reitin konstitutiiviseen aktivaatioon.
Trametinibi ja dabrafenibi estävät siis kahta tämän reitin kinaasia (MEK ja RAF), ja tästä syystä yhdistelmä estää reittiä samanaikaisesti. Trametinibin ja dabrafenibin yhdistelmän on osoitettu rajoittavan kasvua BRAF V600 -mutaatiopositiivisissa syöpäsolulinjoissa in vitro ja viivästyttävän resistenssin kehittymistä in vivo BRAF V600 -mutaatiopositiivisissa ksenografteissa.
Kliininen teho ja turvallisuus
Pediatriset potilaat
Dabrafenibi-trametinibiyhdistelmähoidon kliinistä tehoa ja turvallisuutta BRAF V600 -mutaatiopositiivista glioomaa sairastavilla 1 – < 18-vuotiailla pediatrisilla potilailla arvioitiin avoimessa, kliinisessä, vaiheen II monikeskustutkimuksessa (EudraCT 2015-004015-20). Potilaat, joilla oli matala-asteinen gliooma (WHO:n vuoden 2016 luokituksen asteet 1 ja 2) ja jotka tarvitsivat systeemistä hoitoa, satunnaistettiin suhteessa 2:1 saamaan dabrafenibi-trametinibihoitoa tai karboplatiini-vinkristiinihoitoa. Potilaat, joilla oli uusiutunut tai hoitoon reagoimaton korkea-asteinen gliooma (WHO:n vuoden 2016 luokituksen asteet 3 ja 4) otettiin yksihaaraiseen dabrafenibi-trametinibikohorttiin.
BRAF-mutaatiostatus todettiin kasvainkudoksesta prospektiivisesti paikallisella testillä; tai jos paikallista testausta ei ollut saatavilla, käytettiin keskuslaboratorion bioMérieux THxID-BRAF-testiä. Lisäksi BRAF V600E -mutaatio vahvistettiin tutkimalla saatavilla olevat kasvainnäytteet retrospektiivisesti keskuslaboratoriossa.
Dabrafenibin ja trametinibin annostus kliinisessä tutkimuksessa riippui potilaan iästä ja painosta: suun kautta annetun dabrafenibin annostus oli < 12-vuotialle 2,625 mg/kg kahdesti vuorokaudessa ja 12 vuotta täyttäneille 2,25 mg/kg kahdesti vuorokaudessa, ja suun kautta annetun trametinibin annostus oli < 6-vuotiaille 0,032 mg/kg kerran vuorokaudessa ja 6 vuotta täyttäneille 0,025 mg/kg kerran vuorokaudessa. Dabrafenibin enimmäisannokseksi asetettiin 150 mg kahdesti vuorokaudessa ja trametinibin enimmäisannokseksi 2 mg kerran vuorokaudessa. Karboplatiinin ja vinkristiinin annostus perustui ikään ja kehon pinta-alaan: karboplatiinin annostus oli 175 mg/m2 ja vinkristiinin annostus 1,5 mg/m2; molemmat valmisteet annettiin infuusioina kerran viikossa. Karboplatiinilla ja vinkristiinillä toteutettiin ensin yksi 10 viikon induktiohoitojakso ja sen jälkeen kahdeksan 6 viikon ylläpitohoitojaksoa.
Ensisijainen tehon päätetapahtuma oli molemmissa kohorteissa kokonaisvasteprosentti (ORR; vahvistettujen täydellisten vasteiden [CR] ja osittaisten vasteiden [PR] summa). Vaste arvioitiin riippumattomasti, matala-asteisen gliooman kohortissa vuoden 2017 RANO-kriteerien perusteella ja korkea-asteisen gliooman kohortissa vuoden 2010 RANO-kriteerien perusteella. Ensisijainen analyysi toteutettiin, kun kummankin kohortin kaikki potilaat olivat saaneet hoitoa vähintään 32 viikon ajan. Lopullinen analyysi toteutettiin 2 vuotta tutkimukseenoton päättymisen jälkeen molemmissa kohorteissa.
Pediatristen potilaiden BRAF-mutaatiopositiivinen matala-asteinen gliooma (WHO:n luokituksen asteet 1 ja 2)
Matala-asteisen gliooman kohortissa 110 potilasta satunnaistettiin saamaan dabrafenibi-trametinibihoitoa (n = 73) tai karboplatiini-vinkristiinihoitoa (n = 37). Iän mediaani oli 9,5 vuotta; potilaista 34 (30,9 %) oli iältään 12 kk – < 6 vuotta, 36 (32,7 %) oli 6 – < 12 vuotta ja 40 (36,4 %) oli 12 – < 18 vuotta. 60 % potilaista oli naispuolisia. Suurimmalla osalla potilaista (80 %) oli ensidiagnoosin hetkellä 1. asteen gliooma. Yleisimmät kasvaintyypit olivat pilosyyttinen astrosytooma (30,9 %), gangliogliooma (27,3 %) ja tarkemmin määrittelemätön matala-asteinen gliooma (18,2 %). Etäpesäkealueita oli 9 potilaalla (8,2 %). Aiempia leikkauksia raportoitiin 91 potilaalla (82,7 %); näistä potilaista 28:lla (25,5 %) viimeisin kirurginen toimenpide oli ollut resektio. Systeemisten kortikosteroidien käyttöä raportoitiin 44 potilaalla (41,5 %).
Ensisijaisen analyysin ajankohtana dabrafenibi-trametinibiryhmän kokonaisvasteprosentti parani tilastollisesti merkitsevästi verrattuna karboplatiini-vinkristiiniryhmään. Lisäksi myöhempi hierarkkinen testaus osoitti tilastollisesti merkitsevän paranemisen etenemisvapaan elossaolon (PFS) suhteen solunsalpaajahoitoon verrattuna (taulukko 6).
Ensisijaisen analyysin ajankohtana (kun kaikki potilaat olivat saaneet hoitoa vähintään 32 viikon ajan tai keskeyttäneet tutkimukseen osallistumisen) kokonaiselossaoloa (OS) koskevat tiedot olivat vielä epäkypsät (karboplatiini-vinkristiiniryhmässä raportoitiin yksi kuolema).
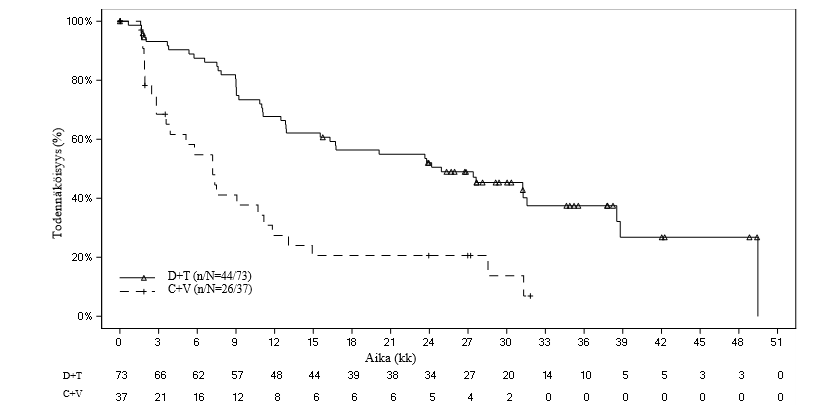
Taulukko 6 Riippumattomaan arviointiin perustuva vaste ja etenemisvapaa elossaolo G2201-avaintutkimuksessa (matala-asteisen gliooman kohortti, ensisijainen analyysi)
Dabrafenibi + trametinibi (D + T) N = 73 | Karboplatiini + vinkristiini (C + V) N = 37 | |
| Paras kokonaisvaste | ||
| Täydellinen vaste (CR), n (%) | 2 (2,7) | 1 (2,7) |
| Osittainen vaste (PR), n (%) | 32 (43,8) | 3 (8,1) |
| Vakaa tauti (SD), n (%) | 30 (41,1) | 15 (40,5) |
| Etenevä tauti (PD), n (%) | 8 (11,0) | 12 (32,4) |
| Ei tiedossa, n (%) | 1 (1,4) | 6 (16,2)1 |
| Kokonaisvasteprosentti (ORR) | ||
| ORR (CR + PR), (95 %:n lv) | 46,6 % (34,8-58,6 %) | 10,8 % (3,0-25,4 %) |
| Vetosuhde (OR)2, p-arvo | 7,19 (2,3-22,4), p < 0,001 | |
| Riskiero | 35,8 % (20,6-51,0) | |
| Etenemisvapaa elossaolo (PFS) | ||
| Mediaani (kk), (95 %:n lv) | 20,1 (12,8 - ei arv.) | 7,4 (3,6-11,8) |
| Riskitiheyssuhde (95 %:n lv), p-arvo | 0,31 (0,17-0,55), p < 0,001 | |
ei arv. = ei arvioitavissa. 1 Neljä C + V -hoitoon satunnaistettua potilasta keskeytti tutkimukseen osallistumisen ennen hoidon aloittamista. 2 Vetosuhde (D + T vs. C + V) ja 95 %:n lv on laskettu logistisella regressiolla hoidon ollessa ainoa kovariaatti (ts. vasteen havaitsemisen todennäköisyys D + T -ryhmässä vs. vasteen havaitsemisen todennäköisyys C + V -ryhmässä). Vetosuhde > 1 on D + T -hoidolle suotuisa. | ||
Lopullisen analyysin ajankohtana (seurannan keston mediaani: 39,0 kk) riippumattomaan arviointiin perustuva kokonaisvasteprosentti oli 54,8 % D+T-ryhmässä ja 16,2 % C+V-ryhmässä vetosuhteen ollessa 6,26. Analyysi vahvisti myös, että etenemisvapaa elossaolo (PFS) parani solunsalpaajahoitoon verrattuna riippumattoman arvioinnin perusteella, ja taudin etenemisen / kuoleman riski pieneni arviolta 64 % (riskitiheyssuhde 0,36). Etenemisvapaan elossaolon (PFS) mediaani oli D+T-ryhmässä 24,9 kk ja C+V-ryhmässä 7,2 kk. Kummassakaan ryhmässä ei raportoitu uusia kuolemantapauksia lopullisen analyysin ajankohtana.
Kuva 1 Riippumattomaan arviointiin perustuvaa etenemisvapaata elossaoloa G2201-avaintutkimuksessa kuvaavat Kaplan–Meier-käyrät (matala-asteisen gliooman kohortti, lopullinen analyysi)
Pediatristen potilaiden BRAF-mutaatiopositiivinen korkea-asteinen gliooma (WHO:n luokituksen asteet 3 ja 4)
Yksihaaraiseen korkea-asteisen gliooman kohorttiin otettiin 41 potilasta, joilla oli uusiutunut tai hoitoon reagoimaton korkea-asteinen gliooma. Potilaat saivat dabrafenibi-trametinibihoitoa. Iän mediaani oli 13,0 vuotta; potilaista 5 (12,2 %) oli iältään 12 kk – < 6 vuotta, 10 (24,4 %) oli 6 – < 12 vuotta ja 26 (63,4 %) oli 12 – < 18 vuotta. 56 % potilaista oli naispuolisia. Ensidiagnoosin hetkellä gliooma oli 20 potilaalla (48,8 %) histologista astetta 4; 13 potilaalla (31,7 %) astetta 3; neljällä potilaalla (9,8 %) astetta 2; kolmella potilaalla (7,3 %) astetta 1; yhdeltä potilaalta (2,4 %) puuttui tieto histologisesta asteesta. Yleisimmät kasvaintyypit olivat glioblastoma multiforme (31,7 %), anaplastinen pleomorfinen ksantoastrosytooma (14,6 %), tarkemmin määrittelemätön korkea-asteinen gliooma (9,8 %) ja pleomorfinen ksantoastrosytooma (9,8 %). Aiempia leikkauksia raportoitiin 40 potilaalla (97,6 %); näistä potilaista 24:llä (58,5 %) viimeisin kirurginen toimenpide oli ollut resektio. Aiempi syövän solunsalpaajahoito raportoitiin 33 potilaalla (80,5 %). Aiempi sädehoito raportoitiin 37 potilaalla (90,2 %). Systeemisten kortikosteroidien käyttöä tutkimushoidon aikana raportoitiin 24 potilaalla (58,5 %).
Lopullisen analyysin ajankohtana (seuranta-ajan mediaani: 45,2 kk) riippumattomaan arviointiin perustuva kokonaisvasteprosentti oli 56,1 % (23/41; 95 %:n lv 39,7–71,5): täydellinen vaste saavutettiin 14 potilaalla (34,1 %) ja osittainen vaste 9 potilaalla (22,0 %). Vasteen keston mediaani oli 27,4 kk (95 %:n lv 9,2 – ei arvioitavissa).
Farmakokinetiikka
Trametinibin farmakokineettisiä ominaisuuksia on määritetty pääasiassa aikuispotilailla, jotka käyttivät kiinteää valmistemuotoa (tabletteja). Trametinibin farmakokinetiikkaa painon mukaan määritetyn kerta-annoksen tai toistuvien annosten jälkeen arvioitiin myös 244 pediatrisella potilaalla. Trametinibin farmakokineettiset ominaisuudet (imeytymisnopeus ja puhdistuma) olivat pediatrisilla potilailla verrannollisia aikuisten farmakokineettisiin ominaisuuksiin. Painon todettiin vaikuttavan suun kautta otetun trametinibin puhdistumaan, mutta iän ei. Pediatristen potilaiden farmakokineettiset trametinibialtistukset suositelluilla painon mukaisilla annoksilla asettuivat aikuisilla havaittujen altistusten vaihteluvälille.
Imeytyminen
Trametinibioraaliliuos imeytyi nopeasti: mediaaniaika plasman huippupitoisuuteen (Tmax) oli 1 tunti annoksesta. Trametinibitablettien keskimääräinen absoluuttinen oraalinen biologinen hyötyosuus oli 72 %. Kun oraaliliuoksen ja tablettimuotoisen valmisteen suhteellista biologista hyötyosuutta verrattiin tutkimuksessa aikuisille paastotilassa annetun kerta-annoksen jälkeen, AUC(0-inf) oli oraaliliuoksen annon jälkeen 12 % suurempi, AUC(0-last) 10 % suurempi ja Cmax 71 % suurempi kuin tablettimuotoisen valmisteen annon jälkeen.
Trametinibialtistus suureni suhteessa annokseen 0,125 mg:n ja 4 mg:n välillä kerran vuorokaudessa annetun toistuvan annostelun jälkeen.
Pediatrisessa avaintutkimuksessa vakaan tilan Cmax-arvon geometrinen keskiarvo (%CV) oli matala-asteisen gliooman kohortissa 22,7 ng/ml (41,1 %) ja korkea-asteisen gliooman kohortissa 21,3 ng/ml (36,3 %). Vakaan tilan AUCtau-arvon geometrinen keskiarvo (%CV) oli matala-asteisen gliooman kohortissa 339 ng*h/ml (22,2 %) ja korkea-asteisen gliooman kohortissa 307 ng*h/ml (22,8 %).
Trametinibi kumuloituu toistuvan päivittäisen annon aikana. Kumuloitumissuhteen todettiin olevan 6,0 (keskiarvo) kerran vuorokaudessa otettavalla 2 mg:n tablettiannoksella. Vakaa tila saavutettiin 15. päivään mennessä.
Ruoan vaikutus
Trametinibioraaliliuoksen (2 mg kerta-annos) annnostelu vähärasvaisen ja vähäkalorisen aterian kanssa pienensi trametinibin Cmax-arvoa 12 %verrattuna annosteluun paasto-olosuhteissa, mitä ei pidetä kliinisesti merkittävänä. AUClast-arvo pysyi muuttumattomana.
Jakautuminen
Trametinibi sitoutuu ihmisen plasman proteiineihin 97,4-prosenttisesti. Trametinibin jakautumistilavuus on noin 1 200 litraa määritettynä 5 µg:n laskimoon annetun mikroannoksen jälkeen
Biotransformaatio
In vitro- ja in vivo -tutkimukset osoittivat, että trametinibi metaboloituu pääasiassa deasetylaation kautta joko pelkästään tai yhdessä mono-oksygenaation kanssa. Deasetyloitunut metaboliitti metaboloitui edelleen glukuronidaation kautta. CYP3A4-oksidaatiota pidetään vähäisempänä metaboloitumisreittinä. Deasetylaatio tapahtuu karboksyyliesteraasien 1b, 1c ja 2 välityksellä, ja siihen osallistuu mahdollisesti myös muita hydrolyyttisia entsyymejä.
Trametinibin kerta-annoksen ja toistuvien annosten jälkeen kanta-aine trametinibi on pääasiallinen plasmassa tavattava aineosa.
Eliminaatio
Terminaalisen puoliintumisajan keskiarvo on 127 tuntia (5,3 vuorokautta) kerta-annoksen jälkeen. Trametinibin näennäinen puhdistuma oli pediatrisilla potilailla (mediaanipaino 32,85 kg) 3,44 l/h (CV 20 %).
Kun radioaktiivisesti merkittyä trametinibiliuosta annettiin kerta-annoksena, koko erittynyt lääkemäärä 10 vuorokauden keräämisajan jälkeen oli pieni (< 50 %), mikä johtuu lääkeaineen pitkästä eliminoitumisen puoliintumisajasta. Trametinibiin liittyvä aines erittyi lähinnä ulosteeseen (osuus kerätystä radioaktiivisuudesta oli > 80 %) ja vähäisemmässä määrin virtsaan (≤ 19 %). Alle 0,1 % erittyneestä annoksesta erittyi kanta-aineena virtsaan.
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Trametinibin vaikutukset lääkeaineita metaboloiviin entsyymeihin ja kuljetusproteiineihin
In vitro ja in vivo -tutkimusten tulokset viittaavat siihen, ettei trametinibi todennäköisesti vaikuta muiden lääkevalmisteiden farmakokinetiikkaan. In vitro -tutkimusten perusteella trametinibi ei ole CYP1A2-, CYP2A6-, CYP2B6-, CYP2D6- eikä CYP3A4-entsyymien estäjä. In vitro -tutkimusten perusteella trametinibi estää CYP2C8-, CYP2C9- ja CYP2C19-entsyymejä ja indusoi CYP3A4-entsyymiä sekä estää kuljetusproteiineja OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, P-gp ja BCRP. Annos ja systeeminen altistus ovat kuitenkin pieniä verrattuna esto- tai induktiovaikutuksen aiheuttaneisiin arvoihin in vitro, joten trametinibin ei katsota estävän tai indusoivan näitä entsyymejä tai kuljetusproteiineja in vivo, vaikka BCRP:n substraattien ohimenevä estyminen suolistossa on mahdollinen (ks. kohta Yhteisvaikutukset).
Muiden lääkevalmisteiden vaikutukset trametinibiin
In vitro ja in vivo -tutkimusten perusteella muut lääkevalmisteet eivät todennäköisesti vaikuta trametinibin farmakokinetiikkaan. Trametinibi ei ole CYP-entsyymien eikä BCRP-, OATP1B1-, OATP1B3-, OATP2B1-, OCT1-, MRP2- tai MATE1-kuljetusproteiinien substraatti. Trametinibi on BSEP:n ja P-gp-kuljetusproteiinin substraatti in vitro. Vaikka BSEP:n esto ei todennäköisesti vaikuta trametinibialtistukseen, maksan P-gp:n voimakkaan eston aiheuttamaa trametinibitason nousua ei voida sulkea pois (ks. kohta Yhteisvaikutukset).
Trametinibin vaikutus muihin lääkevalmisteisiin
Toistuvien trametinibiannosten vaikutusta noretisteronia ja etinyyliestradiolia sisältävien yhdistelmäehkäisytablettien vakaan tilan farmakokinetiikkaan arvioitiin kliinisessä tutkimuksessa 19 naispotilaalla, joilla oli kiinteitä kasvaimia. Noretisteronialtistus suureni 20 % ja etinyyliestradiolialtistus oli samaa luokkaa, kun näitä lääkeaineita annettiin yhtä aikaa trametinibin kanssa. Näiden tulosten pohjalta hormonaalisten ehkäisyvalmisteiden tehon ei oleteta heikkenevän, kun niitä käytetään samanaikaisesti trametinibin kanssa.
Erityisryhmät
Maksan vajaatoiminta
Populaatiofarmakokineettinen analyysi ja kliinisen farmakologian tutkimuksesta saadut tiedot aikuispotilailta, joilla on normaali maksan toiminta tai lievästi, keskivaikeasti tai vaikeasti koholla olevat bilirubiini- ja/tai ASAT-arvot (NCI [National Cancer Institute] -luokituksen mukaan), osoittavat, ettei maksan toiminta vaikuta merkittävästi suun kautta annetun trametinibin puhdistumaan.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminta ei todennäköisesti vaikuta kliinisesti merkittävästi trametinibin farmakokinetiikkaan, koska trametinibi erittyy vain vähäisessä määrin munuaisten kautta. Trametinibin farmakokinetiikkaa tarkasteltiin populaatiofarmakokineettisen analyysin avulla kliinisiin trametinibitutkimuksiin osallistuneilla 223 aikuispotilaalla, joilla oli lievä munuaisten vajaatoiminta, ja 35 aikuispotilaalta, joilla oli keskivaikea munuaisten vajaatoiminta. Lievällä ja keskivaikealla munuaisten vajaatoiminnalla ei ollut vaikutusta trametinibialtistukseen (< 6 % kummassakin ryhmässä). Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista ei ole tutkimustietoa (ks. kohta Annostus ja antotapa).
Etninen tausta
Tietoja ei ole riittävästi, jotta etnisen taustan mahdollista vaikutusta trametinibin farmakokinetiikkaan voitaisiin arvioida, sillä kliinisiä kokemuksia on ainoastaan valkoihoisten potilaiden hoidosta.
Sukupuoli
Aikuisia ja pediatrisia potilaita koskeneiden populaatiofarmakokineettisten analyysien perusteella sukupuoli vaikutti suun kautta annetun trametinibin puhdistumaan. Vaikka naispotilaiden lääkeainealtistuksen ennustetaan olevan suurempi kuin miespotilaiden, erot eivät todennäköisesti ole kliinisesti merkityksellisiä, eikä annoksen muuttaminen ole tarpeen.
Prekliiniset tiedot turvallisuudesta
Trametinibilla ei ole tehty karsinogeenisuustutkimuksia. Trametinibi ei ollut genotoksinen bakteereilla tehdyissä takaisinmutaatiokokeissa, nisäkässoluissa tehdyissä kromosomipoikkeavuuskokeissa eikä rottien luuytimen mikrotumatestissä.
Trametinibi saattaa heikentää naisten hedelmällisyyttä, sillä toistuvilla annoksilla tehdyissä eläinkokeissa havaittiin kystisten follikkeleiden lisääntymistä ja keltarauhasen pienenemistä naarasrotilla, kun altistukset olivat pienempiä kuin ihmisen kliininen altistus AUC-arvon perusteella.
Lisäksi nuorilla, trametinibia saaneilla rotilla havaittiin munasarjojen painon laskua, vähäistä viivästymistä naaraiden sukukypsyyden tunnusmerkeissä (viivästynyt vaginan avautuminen ja lisääntynyt rintatiehyiden kasvusolukeskusten määrä) sekä vähäistä kohdun pintaepiteelin hypertrofiaa. Nämä vaikutukset korjaantuivat lääkkeettömän jakson jälkeen ja johtuivat farmakologiasta. Rotilla ja koirilla tehdyissä toksisuustutkimuksissa, joiden kesto oli enintään 13 viikkoa, hoidon ei kuitenkaan havaittu vaikuttavan urosten lisääntymiskudoksiin.
Rotilla ja kaniineilla tehdyissä alkion ja sikiön kehitystä koskeneissa toksisuustutkimuksissa trametinibilla oli emoon ja alkion- ja sikiönkehitykseen kohdistuvia haittavaikutuksia. Rotilla havaittiin sikiöiden painon laskua ja implantaation jälkeisten keskenmenojen lisääntymistä, kun altistukset olivat pienempiä tai vain vähän suurempia kuin ihmisen kliiniset altistukset AUC-arvon perusteella. Alkion ja sikiön kehitystä koskeneessa toksisuustutkimuksessa kaniineilla havaittiin sikiöiden painon laskua, keskenmenojen lisääntymistä, epätäydellisen luutumisen ilmaantuvuuden suurenemista ja luuston epämuodostumia AUC-arvon perusteella subkliinisillä altistustasoilla.
Toistuvilla annoksilla tehdyissä tutkimuksissa trametinibialtistuksen jälkeen vaikutuksia havaittiin pääasiassa ihossa, ruoansulatuselimistössä, hematologisessa järjestelmässä, luustossa ja maksassa. Suurin osa löydöksistä korjaantuu lääkkeettömän jakson jälkeen. Maksasolunekroosia ja kohonneita aminotransferaasiarvoja havaittiin rotilla, jotka olivat saaneet 8 viikon ajan ≥ 0,062 mg/kg/vrk trametinibia (noin 0,8-kertainen altistus verrattuna ihmisen kliiniseen altistukseen AUC-arvon perusteella).
Hiirillä esiintyi sydämen sykkeen hidastumista, sydämen painon laskua ja vasemman kammion toiminnan heikkenemistä mutta ei sydämen histopatologisia muutoksia 3 viikon kuluttua, kun trametinibia annettiin ≥ 0,25 mg/kg/vrk (noin 3-kertainen altistus verrattuna ihmisen kliiniseen altistukseen AUC-arvon perusteella) enintään 3 viikon ajan. Täysikasvuisilla rotilla seerumin fosforipitoisuuden suurenemiseen liittyi useiden elinten mineralisaatiota, ja se oli läheisessä yhteydessä sydämen, maksan ja munuaisten nekroosiin ja keuhkoverenvuotoon altistustasoilla, jotka vastasivat ihmisen kliinistä altistusta. Rotilla havaittiin kasvulevyn hypertrofiaa ja luun aineenvaihdunnan kiihtymistä. Rotilla ja koirilla, joille annettiin trametinibia ihmisen kliinistä altistusta vastaavina tai sen alittavina annoksina, havaittiin luuytimen nekroosia, imukudoksen atrofiaa kateenkorvassa ja suolistoon liittyvässä imukudoksessa sekä imukudoksen nekroosia imusolmukkeissa, pernassa ja kateenkorvassa. Nämä ilmiöt voivat heikentää immuunijärjestelmän toimintaa. Nuorilla rotilla havaittiin sydämen painon nousua, mutta ei histopatologisia muutoksia annoksella 0,35 mg/kg/vrk (noin kaksinkertainen altistus verrattuna ihmisen kliiniseen altistukseen AUC-arvon perusteella).
Trametinibi oli fototoksinen in vitro hiiren 3T3-fibroblastien NRU-testissä (Neutral Red Uptake), kun pitoisuudet olivat huomattavasti korkeammat kuin kliinisessä altistuksessa (IC50-pitoisuuden ollessa 2,92 mikrog/ml, ≥ 130 kertaa korkeampi kuin ihmisen kliininen altistus laskettuna Cmax-arvosta). Trametinibia saavilla potilailla fototoksisuuden riski näyttäisi näin ollen olevan matala.
Käyttö yhdessä dabrafenibin kanssa
Kun koirille annettiin tutkimuksessa trametinibin ja dabrafenibin yhdistelmää 4 viikon ajan, niillä havaittiin maha-suolikanavan haittojen ja kateenkorvan imukudoksen solukkuuden vähenemisen merkkejä, kun altistukset olivat pienempiä kuin pelkkää trametinibia saaneilla koirilla. Muutoin havaittiin samankaltaisia haittoja kuin vertailukelpoisissa monoterapiatutkimuksissa.
Farmaseuttiset tiedot
Apuaineet
Sulfobutyylibeetadeksinatrium
Sukraloosi (E955)
Sitruunahappomonohydraatti (E330)
Dinatriumfosfaatti (E339)
Kaliumsorbaatti (E202)
Metyyliparahydroksibentsoaatti (E218)
Mansikka-aromi (akaasiakumi, triasetiini, keinotekoiset aromit)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Jauhe oraaliliuosta varten
3 vuotta.
Käyttökuntoon saatettu oraaliliuos
Säilytä alle 25 ºC.
Ei saa jäätyä.
Hävitä käyttämättä jäänyt liuos 35 vuorokauden kuluttua käyttökuntoon saattamisesta.
Säilytys
Säilytä jääkaapissa (2 ºC – 8 ºC).
Säilytä alkuperäispakkauksessa. Herkkä valolle. Herkkä kosteudelle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SPEXOTRAS jauhe oraaliliuosta varten
0,05 mg/ml (L:ei) 1 kpl (4,7 mg, 1 pullon sovitinosa+mittaruisku) (457,95 €)
PF-selosteen tieto
Kullanruskea 180 ml:n lasipullo, jossa on turvasuljin (kierrekorkki) ja joka sisältää 12 g jauhetta.
Yksi kotelo sisältää yhden pullon, yhden pulloon painettavan sovitinosan ja yhden 20 ml:n uudelleenkäytettävän mittaruiskun, jossa on mitta-asteikko 0,5 ml:n välein.
Valmisteen kuvaus:
Valkoinen tai lähes valkoinen jauhe.
Käyttö- ja käsittelyohjeet
Apteekkihenkilökunnan on liuotettava Spexotras-jauhe valmiiksi oraaliliuokseksi ennen valmisteen toimittamista potilaalle.
Ohjeet käyttökuntoon saattamiseen (vain apteekkihenkilökunnalle):
1. Pese ja kuivaa kädet.
2. Tarkasta jauheen viimeinen käyttöpäivämäärä pullosta.
3. Napauta pulloa, jotta jauhe muuttuu irtonaiseksi.
4. Irrota korkki ja lisää pullossa olevaan jauheeseen 90 ml tislattua tai puhdistettua vettä.
5. Laita korkki takaisin paikoilleen ja kääntele pulloa ylösalaisin enintään 5 minuutin ajan, kunnes jauhe on liuennut täydellisesti. Pulloa voi myös ravistaa varovasti.
Huom. Valmisteelle ominaisia valkoisia kelluvia hiukkasia voi näkyä lopullisessa käyttökuntoon saatetussa liuoksessa.
6. Irrota pullon sovitinosa mittaruiskusta. Irrota pullon korkki ja aseta pullon sovitinosa pullon suuhun ja paina voimakkaasti, kunnes pullon sovitinosa on työntynyt kokonaan pulloon. Pullon sovitinosan yläreunan pitää olla samalla tasolla pullon suun yläreunan kanssa.
7. Merkitse koteloon liuoksen valmistuspäivämäärä. Liuos on käytettävä 35 vuorokauden kuluessa valmistamisesta.
8. Kerro lääkkeen vastaanottajalle annos ja liuoksen valmistuspäivämäärä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SPEXOTRAS jauhe oraaliliuosta varten
0,05 mg/ml 1 kpl
- Ylempi erityiskorvaus (100 %). Dabrafenibi ja trametinibi: Melanooman, gliooman ja keuhkosyövän hoito erityisin edellytyksin (1509).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Dabrafenibi ja trametinibi: Dabrafenibin ja trametinibin yhdistelmähoito aikuisille, kun kyseessä BRAF V600 - mutaatiopositiivisen melanooman hoito erityisin edellytyksin (3024).
ATC-koodi
L01EE01
Valmisteyhteenvedon muuttamispäivämäärä
19.05.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com