
VEYVONDI injektiokuiva-aine ja liuotin, liuosta varten 650 IU, 1300 IU
Vaikuttavat aineet ja niiden määrät
VEYVONDI 650 IU injektiokuiva‑aine ja liuotin, liuosta varten
Yksi injektiokuiva‑ainepullo sisältää nimellisesti 650 kansainvälistä yksikköä (IU) vonikogi alfaa.
Kun VEYVONDI on saatettu käyttökuntoon käyttämällä 5 ml pakkauksessa olevaa liuotinta, se sisältää noin 130 IU/ml vonikogi alfaa.
VEYVONDI 1 300 IU injektiokuiva‑aine ja liuotin, liuosta varten
Yksi injektiokuiva‑ainepullo sisältää nimellisesti 1 300 kansainvälistä yksikköä (IU) vonikogi alfaa.
Kun VEYVONDI on saatettu käyttökuntoon käyttämällä 10 ml pakkauksessa olevaa liuotinta, se sisältää 130 IU/ml vonikogi alfaa.
VEYVONDI‑valmisteen spesifinen aktiivisuus on noin 110 IU VWF:RCo/mg proteiinia.
Von Willebrand ‑tekijän (WWF) teho (IU) mitataan Euroopan farmakopean ristosetiinikofaktorin aktiivisuusmäärityksen (VWF:RCo) mukaan. Rekombinantin ihmisen von Willebrand ‑tekijän ristosetiinikofaktorin aktiivisuus määritettiin kansainvälisen von Willebrand ‑tekijän konsentraattistandardin (WHO) mukaan.
Vonikogi alfa on puhdistettu, rekombinantti ihmisen von Willebrand ‑tekijä (rVWF). Sitä tuotetaan yhdistelmä‑DNA‑tekniikalla (rDNA) kiinanhamsterin munasarjan (CHO) solulinjassa lisäämättä ulkoista ihmis‑ tai eläinperäistä proteiinia soluviljelyprosessin, puhdistuksen tai lopullisen koostumuksen muodostuksen aikana.
Valmiste sisältää vain hyvin pieniä määriä ihmisen rekombinanttia hyytymistekijää VIII (≤ 0,01 IU FVIII / IU VWF:RCo), mikä on määritetty Euroopan farmakopean tekijä VIII (FVIII) ‑kromogeenisuusmäärityksellä.
Apuaineet, joiden vaikutus tunnetaan
Yksi 650 IU:n injektiokuiva-ainepullo sisältää 5,2 mg natriumia ja 0,5 mg polysorbaattia 80.
Yksi 1 300 IU:n injektiokuiva-ainepullo sisältää 10,4 mg natriumia ja 1,0 mg polysorbaattia 80.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva‑aine ja liuotin, liuosta varten.
Kuiva‑aine on valkoinen tai luonnonvalkoinen kylmäkuivattu jauhe.
Liuotin on kirkas ja väritön liuos.
Kliiniset tiedot
Käyttöaiheet
Verenvuotojen tai kirurgisiin toimenpiteisiin liittyvien verenvuotojen ehkäisy ja hoito von Willebrandin tautia (VWD) sairastavilla aikuisilla (vähintään 18‑vuotiailla), kun desmopressiinihoito (DDAVP) ei yksinään ole tehonnut tai on vasta-aiheista.
Verenvuotojen hoito von Willebrandin tautia (VWD) sairastavilla lapsilla (alle 18‑vuotiailla), kun desmopressiinihoito (DDAVP) ei yksinään ole tehonnut tai on vasta-aiheista.
VEYVONDI‑valmistetta ei saa käyttää A‑hemofilian hoitoon.
Ehto
Hoito tulee aloittaa hyytymishäiriöihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hemostaasin häiriöiden hoitoon perehtyneen lääkärin on valvottava von Willebrandin taudin (VWD) hoitoa.
Annostus
Annos ja antoväli määritetään yksilöllisesti lääkärin harkinnan perusteella, ja seuraavat asiat on otettava huomioon: potilaan paino, verenvuototapahtumien/leikkauksen tyyppi ja vaikeusaste sekä asianmukaisten kliinisten arvojen ja laboratorioarvojen seuranta. Painoon perustuvaa annosta saatetaan joutua säätämään ali‑ ja ylipainoisille potilaille.
1 IU/kg (VWF:RCo/VEYVONDI/vonikogi alfa) suurentaa plasman VWF:RCo‑pitoisuutta yleensä 0,02 IU/ml (2 %).
Hemostaasin saavuttaminen edellyttää vähintään tekijän VIII koagulaatioaktiivisuutta (FVIII:C) 0,4 IU/ml (≥ 40 % normaalista aktiivisuudesta). Potilaan FVIII:C‑perustasosta riippuen yksi rVWF‑infuusio suurentaa endogeenisen FVIII:C‑aktiivisuuden useimmilla potilailla yli 40 %:iin 6 tunnin kuluessa ja pitää sen tällä tasolla jopa 72 tunnin ajan infuusion jälkeen. Annos ja hoidon kesto riippuvat potilaan kliinisestä tilasta, verenvuodon tyypistä ja vaikeusasteesta ja sekä VWF:RCo‑ että FVIII:C‑pitoisuuksista. Jos potilaan plasman FVIII:C‑perustaso on < 40 % tai se ei ole tiedossa, ja kaikissa tilanteissa, joissa hemostaasi on korjattava nopeasti (kuten akuutin verenvuodon hoito, vaikea trauma tai hätäleikkaus), ensimmäisen VEYVONDI‑infuusion yhteydessä on annettava rekombinanttia hyytymistekijä VIII ‑valmistetta plasman FVIII:C‑pitoisuuden suurentamiseksi hemostaattiselle tasolle.
Jos FVIII:C‑pitoisuuden välitön nostaminen ei kuitenkaan ole tarpeen tai jos FVIII:C‑perustaso riittää varmistamaan hemostaasin, lääkäri voi harkintansa mukaan olla antamatta rFVIII‑valmistetta ensimmäisen VEYVONDI‑infuusion yhteydessä.
Usein toistettuja infuusioita vaativien merkittävien verenvuototapahtumien tai suurten leikkausten yhteydessä suositellaan FVIII:C‑pitoisuuksien seurantaa, jotta voidaan päättää, onko myöhempien infuusioiden yhteydessä annettava rFVIII‑valmistetta ja jotta vältytään FVIII:C‑pitoisuuden liialliselta nousulta.
Verenvuototapahtumien hoito (tarvittaessa annettava hoito) aikuisilla ja lapsilla
Hoidon aloitus
Ensimmäisen VEYVONDI‑annoksen pitää olla 40–80 IU/kg. VWF:RCo‑tavoitetaso on > 0,6 IU/ml (60 %) ja FVIII:C‑tavoitetaso > 0,4 IU/ml (40 %). Annostusohjeet lievien ja merkittävien verenvuotojen hoitoon on annettu taulukossa 1.
VEYVONDI‑valmistetta pitää antaa yhdessä rekombinantin tekijä VIII:n kanssa verenvuodon hallitsemiseksi, jos FVIII:C‑pitoisuudet ovat alle 40 % tai jos ne eivät ole tiedossa. Potilaan rFVIII‑annos lasketaan plasman FVIII:C‑perustason ja halutun FVIII:C‑huippupitoisuuden välisen eron perusteella, jotta saavutetaan keskimääräiseen saantoon 0,02 (IU/ml)/(IU/kg) perustuva asianmukainen plasman FVIII:C‑pitoisuus. rFVIII‑valmistetta pitää antaa 10 minuutin sisällä siitä, kun VEYVONDI‑annos on kokonaisuudessaan annettu.
Annoksen laskeminen
VEYVONDI‑annos [IU] = annos [IU/kg] x kehon paino [kg]
Seuraavat infuusiot
Seuraavat annokset VEYVONDI‑valmistetta 40–60 IU/kg annetaan infuusiona 8–24 tunnin välein taulukossa 1 annettujen annostusohjeiden mukaisesti tai niin kauan kuin se on kliinisesti tarpeellista. Merkittävien verenvuotojen kohdalla VWF:RCo‑pitoisuus on pidettävä vähintään 50 %:ssa niin kauan kuin se katsotaan tarpeelliseksi.
Kliinisistä tutkimuksista kertyneen kokemuksen perusteella endogeeniset FVIII‑pitoisuudet pysyvät VWF‑korvaushoidon jälkeen normaaleina tai lähes normaaleina niin kauan kuin VEYVONDI‑valmisteen antamista jatketaan.
| Taulukko 1. Annossuositukset lievien ja merkittävien verenvuotojen hoitoon | |||
| Verenvuoto | Aloitusannosa (IU VWF:RCo / kg) | Seuraava annos | |
Lievä (esim. verenvuoto nenästä tai suusta, runsaat kuukautiset) | 40–50 IU/kg | 40–50 IU/kg 8–24 tunnin välein (tai kliinisen tarpeen mukaan) | |
Merkittäväb (esim. vaikea tai sitkeä nenäverenvuoto, runsaat kuukautiset, ruoansulatuskanavan verenvuoto, keskushermoston vaurio, verinivel tai vamman aiheuttama verenvuoto) | 50–80 IU/kg | 40–60 IU/kg 8–24 tunnin välein noin 2–3 päivän ajan (tai kliinisen tarpeen mukaan) | |
a Jos potilaalle annetaan rFVIII‑valmistetta, tarkista ohjeet rFVIII‑valmisteen käyttökuntoon saattamisesta ja antamisesta kyseisen valmisteen pakkausselosteesta. b Verenvuoto voidaan katsoa merkittäväksi, jos potilas tarvitsee välttämättä tai mahdollisesti punasolusiirron tai jos verenvuoto sijaitsee jossakin kriittisen tärkeässä paikassa (esim. kallonsisäinen tai ruoansulatuskanavan verenvuoto). | |||
Verenvuodon ennaltaehkäisy ja hoito elektiivisen leikkauksen yhteydessä aikuisilla
Ennen leikkausta
Potilaille, joiden FVIII‑tasot ovat riittämättömät, pitää antaa 40–60 IU/kg VEYVONDI‑valmistetta 12–24 tuntia ennen elektiivistä leikkausta (leikkausta edeltävä annos) sen varmistamiseksi, että elimistön FVIII‑pitoisuus on vähintään 0,4 IU/ml ennen pientä ja vähintään 0,8 IU/ml ennen suurta leikkausta.
Runsaan verenvuodon estämiseksi elektiivisen leikkauksen yhteydessä FVIII:C‑pitoisuus pitää määrittää 3 tunnin sisällä ennen kirurgista toimenpidettä. Jos FVIII:C‑pitoisuus on suositellulla tavoitetasolla
- vähintään 0,4 IU/ml pienten kirurgisten toimenpiteiden ja suun kirurgisten toimenpiteiden yhteydessä ja
- vähintään 0,8 IU/ml suurten leikkausten yhteydessä,
potilaalle pitää antaa annos pelkkää VEYVONDI‑valmistetta enintään 1 tunti ennen toimenpidettä.
Jos FVIII:C‑pitoisuudet eivät ole suositellulla tavoitetasolla, potilaalle pitää antaa vonikogi alfan lisäksi tunnin sisällä ennen toimenpidettä rFVIII‑valmistetta VWF:RCo‑ ja FVIII:C‑pitoisuuksien suurentamiseksi. Tarkista taulukosta 2 suositellut FVIII:C‑tavoitearvot. Annos riippuu potilaan VWF‑ ja FVIII‑tasoista sekä mahdollisen vuodon tyypistä ja vakavuusasteesta.
| Taulukko 2. Suositetut VWF:RCo:n ja FVIII:C:n huipputavoitetasot plasmassa ennen leikkausta runsaan verenvuodon ehkäisemiseksi leikkauksen aikana ja sen jälkeen | |||
| Leikkauksen tyyppi | Haluttu plasman VWF:RCo‑ huippupitoisuus | Haluttu plasman FVIII:C‑ huippupitoisuusa | rVWF‑annoksen (annetaan enintään 1 tunti ennen leikkausta) laskeminen (tarvittava IU VWF:RCo) |
| Lievä | 0,50–0,60 IU/ml | 0,40–0,50 IU/ml | ∆b VWF:RCo x paino (kg) /IRc |
| Merkittävä | 1 IU/ml | 0,80–1 IU/ml | ∆b VWF:RCo x paino (kg) /IRc |
| a rFVIII‑lisäannos voi olla tarpeen plasman suositellun FVIII:C‑huippupitoisuuden saavuttamiseksi. Annostusohjeet pitää antaa IR‑arvon perusteella. | |||
| b ∆ = plasman VWF:RCo‑huippupitoisuus – plasman VWF:RCo‑perustaso | |||
| c IR = potilaasta mitattu inkrementaalinen saanto (incremental recovery). Jos IR‑arvoa ei ole käytettävissä, sen oletetaan olevan 0,02 IU/ml per IU/kg. | |||
Leikkauksen aikana ja leikkauksen jälkeen
Kun kirurginen toimenpide on aloitettu, plasman VWF:RCo‑ ja FVIII:C‑pitoisuuksia on seurattava, ja leikkauksen aikana ja sen jälkeen annettava korvaushoito on räätälöitävä yksilöllisesti farmakokineettisten tulosten, verenvuodon voimakkuuden ja keston sekä laitoksen omien hoitokäytäntöjen perusteella. Leikkauksen jälkeen annettavassa korvaushoidossa VEYVONDI‑valmisteen annosväli on 12–48 tuntia. Katso myöhempiä ylläpitoannoksia koskevat hoitosuositukset taulukosta 3.
| Taulukko 3. Suositetut VWF:RCo:n ja FVIII:C:n vähimmäistavoitetasot ennen leikkausta ja hoidon vähimmäiskestot annettaessa myöhempiä ylläpitoannoksia runsaan verenvuodon ennaltaehkäisyyn leikkauksen jälkeen | ||||||
| Leikkauksen tyyppi | VWF:RCo Haluttu plasman vähimmäispitoisuus | FVIII:C Haluttu plasman vähimmäispitoisuus | Hoidon vähimmäiskesto | Antoväli | ||
| Enintään 72 tuntia leikkauksen jälkeen | Yli 72 tuntia leikkauksen jälkeen | Enintään 72 tuntia leikkauksen jälkeen | Yli 72 tuntia leikkauksen jälkeen | |||
| Lievä | ≥ 0,30 IU/ml | ‑ | > 0,40 IU/ml | ‑ | 48 tuntia | 12–24 tunnin välein / joka toinen päivä |
| Merkittävä | > 0,50 IU/ml | > 0,30 IU/ml | > 0,50 IU/ml | > 0,40 IU/ml | 72 tuntia | 12–24 tunnin välein / joka toinen päivä |
Estohoito aikuisilla
Verenvuotojen pitkäaikaisen estohoidon aloitukseen VWD-potilailla on harkittava 40–60 IU:n/kg:n VEYVONDI-annosta kahdesti viikossa. Potilaan tila ja kliininen vaste, välivuodot mukaan luettuina, voivat edellyttää suurempia annoksia (enintään 80 IU/kg) ja/tai tiheämpää antoväliä (enintään kolme kertaa viikossa).
Pediatriset potilaat
VEYVONDI‑valmisteen turvallisuus ja teho alle 18 vuoden ikäisten lasten verenvuotojen hoidossa on varmistettu. Annostus perustuu samoihin ohjeisiin kuin aikuisilla, ja se on mukautettava potilaan kliiniseen tilaan sekä plasman VWF:RCo- ja FVIII:C-pitoisuuksiin. Nuorilla potilailla lyhyemmät antovälit tai suuremmat annokset voivat olla tarpeen (ks. kohta Farmakokinetiikka). VEYVONDI-valmisteen turvallisuutta ja tehoa verenvuotojen estohoidossa tai kirurgisiin toimenpiteisiin liittyvien verenvuotojen ehkäisyssä tai hoidossa ei ole vielä varmistettu alle 18‑vuotiailla lapsilla.
Antotapa
VEYVONDI‑valmiste annetaan laskimoon. Käyttökuntoon saatettu lääkevalmiste on tarkistettava silmämääräisesti ennen käyttöä.
Antonopeus on määritettävä potilaan mukavuus huomioiden, ja se saa olla enintään 4 ml/min. Potilasta on tarkkailtava välittömien reaktioiden varalta. Jos potilaalle ilmaantuu valmisteen antoon mahdollisesti liittyvä reaktio, kuten takykardia, infuusionopeutta on hidastettava tai valmisteen anto on keskeytettävä potilaan kliinisen tilan mukaan.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Tiedossa oleva hiiren tai hamsterin proteiinien aiheuttama allerginen reaktio.
Varoitukset ja käyttöön liittyvät varotoimet
Jos potilaalla on aktiivinen verenvuoto, ensilinjan hoidoksi suositellaan FVIII‑valmisteen ja VEYVONDI‑valmisteen yhteiskäyttöä FVIII‑aktiivisuustasoista riippuen (ks. kohta Annostus ja antotapa).
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyysreaktiot
Yliherkkyysreaktioita (mukaan lukien anafylaksia) on esiintynyt. Potilaille ja/tai heitä hoitaville henkilöille on kerrottava yliherkkyysreaktioiden varhaisista merkeistä, joita voivat olla esimerkiksi takykardia, puristava tunne rinnassa, hengityksen vinkuminen ja/tai akuutti hengitysvaikeus, hypotensio, yleistynyt nokkosihottuma, kutina, rinokonjunktiviitti, angioedeema, letargia, pahoinvointi, oksentelu, parestesia ja levottomuus. Oireet voivat edetä anafylaktiseksi sokiksi. Sokin ilmetessä on noudatettava tavanomaista sokin hoitokäytäntöä.
Potilasta on seurattava tiiviisti ja huolellisesti mahdollisten oireiden varalta koko infuusion ajan. Jos potilaalle kehittyy vaikean allergisen reaktion merkkejä ja oireita, VEYVONDI‑valmisteen anto on keskeytettävä välittömästi ja potilaalle on annettava asianmukaista tukihoitoa.
Käytettävissä on oltava riittävä hoitovalmius ja välineet, jotta mahdollinen anafylaktinen reaktio voidaan hoitaa välittömästi. Tämä koskee etenkin potilaita, joilla on aiemmin esiintynyt allergisia reaktioita.
VEYVONDI sisältää hyvin pieniä määriä hiiren immunoglobuliinia G ja hamsterin proteiineja (enintään 2 ng/IU VEYVONDI‑valmistetta). Tällä valmisteella hoidetuille potilaille voi kehittyä näiden ei‑ihmisperäisten nisäkäsproteiinien aiheuttamia yliherkkyysreaktioita. VEYVONDI sisältää hyvin pieniä määriä rekombinanttia hyytymistekijä VIII:aa.
Tromboosi ja embolia
Tromboottisten tapahtumien riski on olemassa etenkin, jos potilaalla tiedetään olevan kliinisiä tai laboratoriokokein todettuja riskitekijöitä, vähäinen ADAMTS13‑aktiivisuus mukaan lukien. Riskiryhmään kuuluvia potilaita on siksi seurattava tromboosin varhaisten merkkien varalta, ja tromboembolioiden estohoito on aloitettava voimassa olevien suositusten ja hoitokäytäntöjen mukaisesti.
Jos potilas tarvitsee usein toistuvia VEYVONDI‑annoksia yhdessä rekombinantin tekijä VIII:n_ kanssa, plasman FVIII:C‑aktiivisuutta on seurattava, jotta vältetään plasman FVIII:C‑pitoisuuden liiallinen ja pitkäkestoinen nousu, sillä se voi suurentaa tromboottisten tapahtumien riskiä.
VEYVONDI‑valmisteen lisäksi annettavien FVIII‑valmisteiden on oltava puhtaita FVIII‑valmisteita. VWF:ää sisältävän FVIII‑valmisteen samanaikainen käyttö suurentaisi tromboottisten tapahtumien riskiä.
Neutraloivien vasta‑aineiden (inhibiittorien) kehittyminen
Von Willebrandin tautia (etenkin tyypin 3 tautimuotoa) sairastaville potilaille saattaa kehittyä von Willebrand ‑tekijää neutraloivia vasta‑aineita (inhibiittoreita). Jos odotettua VWF:RCo‑pitoisuutta ei saavuteta plasmassa tai jos verenvuotoa ei saada hallintaan asianmukaisella annoksella, potilaalta on määritettävä von Willebrand ‑tekijän inhibiittorien mahdollinen esiintyminen asianmukaisella menetelmällä. Jos potilaan neutraloivien VWF‑vasta‑aineiden pitoisuus on suuri, hoito von Willebrand ‑tekijällä ei ehkä ole tehokasta, joten muita hoitovaihtoehtoja on harkittava hemostaasin saavuttamiseksi.
Hoidettaessa VWD‑potilaita, joilla sitovia vasta‑aineita esiintyy korkeina pitoisuuksina aiemman plasmaperäisen VWF‑valmistehoidon (pdVWF) seurauksena, sitovien vasta‑aineiden vaikutuksen kumoaminen voi vaatia suurempaa annosta. Tällaisia potilaita voidaan hoitaa antamalla suurempia vonikogi alfa ‑annoksia kunkin yksittäisen potilaan farmakokineettisten tulosten perusteella.
Apuaineisiin liittyvät huomioon otettavat asiat
Natriumsisältö
Tämä lääkevalmiste sisältää 5,2 mg natriumia per 650 IU:n injektiopullo tai 10,4 mg natriumia per 1 300 IU:n injektiopullo, mikä vastaa 2,2 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille, kun henkilön oletettu paino on 70 kg ja annos on 80 IU/kg. Potilaiden, joilla on ruokavalion natriumrajoitus, on otettava tämä huomioon.
Polysorbaattisisältö
Tämä lääkevalmiste sisältää 0,5 mg polysorbaattia 80 per 650 IU:n injektiopullo tai 1,0 mg polysorbaattia 80 per 1 300 IU:n injektiopullo, mikä vastaa 0,1 mg:aa/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Ihmisen von Willebrand ‑tekijää sisältävien valmisteiden yhteisvaikutuksia muiden lääkevalmisteiden kanssa ei tunneta.
Raskaus ja imetys
VEYVONDI‑valmisteella ei ole tehty lisääntymistä koskevia eläinkokeita.
Raskaus
Raskaana olevien tai imettävien naisten hoidosta ei ole kokemusta. VEYVONDI‑valmistetta saa antaa raskaana oleville naisille vain, jos se on selvästi aiheellista, kun otetaan huomioon, että tällä potilasryhmällä on synnytyksen yhteydessä suurentunut verenvuotoriski.
Imetys
Ei tiedetä, erittyykö VEYVONDI ihmisillä äidinmaitoon. Siksi VEYVONDI‑valmistetta saa antaa imettäville, von Willebrand ‑tekijän puutoksesta kärsiville naisille vain, jos se on selvästi aiheellista. Terveydenhuollon ammattilaisten on punnittava mahdollisia riskejä ja määrättävä VEYVONDI‑valmistetta vain, jos se on tarpeellista.
Hedelmällisyys
VEYVONDI‑valmisteen vaikutusta hedelmällisyyteen ei ole vahvistettu.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
VEYVONDI‑valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Hoidettaessa potilaita VEYVONIDI‑valmisteella seuraavat haittatapahtumat ovat mahdollisia: yliherkkyys tai allergiset reaktiot, tromboemboliat, inhibiittorien syntyminen VWF:ia vastaan.
Haittatapahtumataulukko
Taulukossa 4 on luettelo haittavaikutuksista, joita on raportoitu kliinisissä tutkimuksissa, myyntiluvan myöntämisen jälkeisissä turvallisuustutkimuksissa ja myyntiin tulon jälkeisessä raportoinnissa. Haittavaikutusten yleisyydet on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 4. Yhteenveto haittavaikutuksista, joita raportoitiin VEYVONDI‑valmisteella tehdyissä kliinisissä tutkimuksissa,myyntiluvan myöntämisen jälkeisissä turvallisuustutkimuksissa ja myyntiin tulon jälkeen von Willebrandin tautia sairastavilla potilailla | ||
| MedDRA‑ elinjärjestelmäluokitus | Haittavaikutus (suositellut termit) | Yleisyysluokka tutkittavien mukaan |
| Immuunijärjestelmä | Anafylaktinen reaktio* | Tuntematon |
| Hermosto | Päänsärky | Hyvin yleinen |
| Heitehuimaus | Yleinen | |
| Kiertohuimaus | Yleinen | |
| Makuhäiriö | Melko harvinainen | |
| Vapina | Melko harvinainen | |
| Sydän | Takykardia | Melko harvinainen |
| Verisuonisto | Hypertensio | Yleinen |
| Syvä laskimotukos | Melko harvinainen | |
| Kuumat aallot | Melko harvinainen | |
| Ruoansulatuselimistö | Oksentelu | Yleinen |
| Pahoinvointi | Yleinen | |
| Iho ja ihonalainen kudos | Yleistynyt kutina | Yleinen |
| Yleisoireet ja antopaikassa todettavat haitat | Epämiellyttävät rintatuntemukset | Melko harvinainen |
| Infuusiokohdan parestesia | Melko harvinainen | |
| Infuusioon liittyvä reaktio (mukaan lukien takykardia, kuumat aallot, ihottuma, hengenahdistus, näön sumeneminen)* | Tuntematon | |
| Tutkimukset | T‑aallon inversio sydänsähkökäyrässä | Melko harvinainen |
| Syketiheyden nousu | Melko harvinainen | |
* Haittavaikutukset tunnistettu myyntiluvan myöntämisen jälkeisessä valvonnassa.
Kuvaus valikoiduista haittavaikutuksista
Yliherkkyys
Yliherkkyysreaktiot tai allergiset reaktiot (joiden oireita voivat olla esimerkiksi angioedeema, infuusiokohdan kuumotus ja kirvely, vilunväristykset, punastelu, rinokonjunktiviitti, yleistynyt nokkosihottuma, päänsärky, nokkosihottuma, hypotensio, letargia, pahoinvointi, levottomuus, takykardia, puristava tunne rinnassa, kihelmöinti, oksentelu, hengityksen vinkuminen) ovat mahdollisia, ja ne saattavat joissakin tapauksissa edetä anafylaksiaksi (sokki mukaan lukien).
Von Willebrandin tautia (etenkin tyypin 3 tautimuotoa) sairastaville potilaille voi hyvin harvoin kehittyä von Willebrand ‑tekijää neutraloivia vasta‑aineita (inhibiittoreita). Tällaisten inhibiittorien kehittyminen saattaa ilmetä riittämättömänä kliinisenä vasteena hoidolle. Näillä vasta‑aineilla voi olla läheinen yhteys yliherkkyys‑ tai anafylaktisiin reaktioihin. Siksi kaikki yliherkkyysreaktioita tai anafylaktisia reaktioita saavat potilaat on tutkittava ja arvioitava inhibiittorien varalta.
Tällaisissa tapauksissa on suositeltavaa ottaa yhteyttä hemofilian hoitoon erikoistuneeseen hoitokeskukseen.
Trombogeenisuus
Tromboottisten tapahtumien riski on olemassa etenkin, jos potilaalla tiedetään olevan kliinisiä tai laboratoriokokein todettuja riskitekijöitä, vähäinen ADAMTS13‑aktiivisuus mukaan lukien. Riskiryhmään kuuluvia potilaita on siksi seurattava tromboosin varhaisten merkkien varalta, ja tromboembolioiden estohoito on aloitettava voimassa olevien suositusten ja hoitokäytäntöjen mukaisesti.
Immunogeenisuus
VEYVONDI‑valmisteen immunogeenisuutta arvioitiin kliinisissä tutkimuksissa seuraamalla VWF:ää ja FVIII:aa neutraloivien vasta‑aineiden kehittymistä sekä VWF:ää, furiinia, kiinanhamsterin munasarjan (CHO) proteiineja ja hiiren IgG:tä sitovia vasta‑aineita. Ihmisen VWF:ää tai ihmisen rFVIII:aa neutraloivien vasta‑aineiden kehittymistä hoidon aikana ei todettu. Kliinisissä tutkimuksissa perioperatiivista VEYVONDI‑hoitoa saaneista 132 tutkittavasta yhdelle kehittyi hoidon aikana VWF:ää sitovia vasta‑aineita leikkauksen jälkeen. Kyseisellä potilaalla ei raportoitu haittatapahtumia tai hemostaattisen tehon puutetta. Epäpuhtauksia, kuten rekombinanttia furiinia, CHO‑proteiinia tai hiiren IgG:tä sitovia vasta‑aineita ei todettu VEYVONDI‑hoidon jälkeen.
Pediatriset potilaat
Lapsilla, jotka saavat VEYVONDI-valmistetta verenvuotojen hoitoon, haittavaikutusten esiintymistiheyden, tyypin ja vaikeusasteen odotetaan olevan samat kuin aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista liitteessä V luetellun kansallisen ilmoitusjärjestelmän kautta.
Yliannostus
Von Willebrand ‑tekijän yliannostusoireita ei ole raportoitu. Tromboembolisia tapahtumia saattaa esiintyä suurissa yliannostuksissa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: hemostaatit, veren hyytymistekijät, ATC‑koodi: B02BD10
Vaikutusmekanismi
VEYVONDI on rekombinantti ihmisen von Willebrand ‑tekijä (rVWF). VEYVONDI toimii samalla tavalla kuin endogeeninen von Willebrand ‑tekijä.
VEYVONDI‑valmisteella voidaan korjata hemostaasin häiriöitä potilailla, joilla on von Willebrand ‑tekijän puutos (von Willebrandin tauti). Vaikutus tapahtuu kahdella tasolla:
- VEYVONDI palauttaa verihiutaleiden kiinnittymiskyvyn verisuonen vauriokohdan subendoteeliin (se sitoutuu sekä verisuonen subendoteelin matriksiin, esim. kollageeniin, että verihiutaleen solukalvoon) ja mahdollistaa primaarin hemostaasin. Tämä näkyy vuotoajan lyhentymisenä. Tämä vaikutus on välitön, ja sen tiedetään riippuvan pitkälti proteiinin polymerisaatioasteesta.
- VEYVONDI korjaa viiveellä tapahtumaan liittyvän tekijä VIII ‑vajauksen. Laskimoon annettuna VEYVONDI sitoutuu endogeeniseen tekijä VIII:aan (jota potilaan elimistö tuottaa normaalisti) ja vakauttamalla tämän tekijän estää sen nopean hajoamisen. Tämän vuoksi VEYVONDI‑valmisteen antaminen palauttaa toissijaisena vaikutuksena FVIII:C‑pitoisuuden normaaliksi. Ensimmäisen infuusion annon jälkeen FVIII:C‑pitoisuuden odotetaan nousevan yli 40 %:iin 6 tunnin kuluessa, ja huippupitoisuus saavutetaan useimmilla potilailla 24 tunnin kuluessa FVIII:C‑perustasosta riippuen.
VEYVONDI on rVWF‑valmiste, joka sisältää erittäin suurikokoisia multimeereja kaikkien plasman multimeerien lisäksi, koska se ei altistu ADAMTS13:n aiheuttamalle proteolyysille valmistusprosessin aikana.
Kliininen teho ja turvallisuus
Kliinistä turvallisuutta, tehoa ja farmakokinetiikkaa arvioitiin viidessä loppuun suoritetussa tutkimuksessa von Willebrandin tautia sairastavilla aikuisilla ja lapsilla (070701, 071001, 071101, 071301 ja SHP677‑304) sekä yhdessä edelleen käynnissä olevassa tutkimuksessa von Willebrandin tautia sairastavilla lapsilla (071102). Yhteensä VEYVONDI‑valmistetta annettiin 144 yksittäiselle tutkittavalle (103 von Willebrandin tautia sairastavalle aikuiselle tutkittavalle, 29 von Willebrandin tautia sairastavalle pediatriselle tutkittavalle sekä 12 A‑hemofiliaa sairastavalle tutkittavalle tutkimuksessa 071104) kliinisen kehitysohjelman aikana.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset VEYVONDI‑valmisteen käytöstä von Willebrandin taudin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
VEYVONDI‑valmisteen farmakokinetiikkaa (PK) selvitettiin aikuisilla kolmessa kliinisessä tutkimuksessa arvioimalla plasman VWF:RCo‑pitoisuuksia, von Willebrand ‑tekijän antigeenin (VWF:Ag) pitoisuuksia ja von Willebrand ‑tekijän kollageeniin sitoutumista (VWF:CB). Kaikissa kolmessa tutkimuksessa tutkittavat arvioitiin tilanteessa, jossa heillä ei ollut verenvuotoja. FVIII:C‑pitoisuuden pitkäkestoinen nousu oli todettavissa kuuden tunnin kuluessa VEYVONDI‑kertainfuusiosta.
Taulukossa 5 on yhteenveto VEYVONDI‑valmisteen farmakokineettisistä ominaisuuksista aikuisilla 50 IU:n/kg VWF:Rco (PK50)- tai 80 IU:n/kg VWF:Rco (PK80) ‑infuusioiden jälkeen. Infuusion keskimääräinen kesto oli 16,5 minuuttia (keskihajonta ± 3,51 minuuttia), kun VWF:Rco‑annos oli 50 IU/kg (PK50), ja 11,8 minuuttia (± 2,86 minuuttia), kun VWF:Rco‑annos oli 80 IU/kg (PK80).
| Taulukko 5. VWF:RCo‑hoidon farmakokineettinen arviointia aikuisilla | ||||
| Parametri | Vaihe 1, PK50 VEYVONDI yhdessä oktokogi alfan kanssag(tutkimus 070701)Keskiarvo (95 % CI)Keskihajonta | Vaihe 3, PK50 VEYVONDI(tutkimus 071001)Keskiarvo (95 % CI)Keskihajonta | Vaihe 3, PK80 VEYVONDI(tutkimus 071001)Keskiarvo (95 % CI)Keskihajonta | Leikkaus, PK50 VEYVONDI(tutkimus 071101)Keskiarvo (95 % CI)Keskihajonta |
| T1/2b | 19,3 (14,3; 24,3) 10,99 | 22,6 (19,5; 25,7) 5,34 | 19,1 (16,7; 21,5) 4,32 | 17,8 (12,9; 22,8) 7,34 |
| Clc | 0,04 (0,03; 0,05) 0,028 | 0,02 (0,02; 0,03) 0,005 | 0,03 (0,02; 0,03) 0,009 | 0,03 (0,02; 0,04) 0,011 |
| IR, Cmaxd | 1,7 (1,4; 2,0) 0,62 | 1,9 (1,6; 2,1) 0,41 | 2,0 (1,7; 2,2) 0,39 | 2,0 (1,7; 2,3) 0,45 |
| AUC0‑infe | 1 541,4 (1 295,7; 1 787,2) 554,31 | 2 105,4 (1 858,6; 2 352,3) 427,51 | 2 939,0 (2 533,2; 3 344,8) 732,72 | 1 834,4 (1 259,0; 2 409,7) 856,45 |
| AUC0‑inf/annosf | 33,4 (27,2; 39,5) 13,87 | 42,1 (37,3; 46,9) 8,31 | 36,8 (31,8; 41,8) 8,97 | 37,5 (25,3; 49,7) 18,14 |
a [Käytössä oli VWF:RCo‑määrityksiä, joiden herkkyys ja tunnistusalueet vaihtelivat: vaihe 1: automatisoitu määritys 0,08–1,50 IU/ml ja herkkä manuaalinen määritys 0,01–0,08 IU/ml; vaihe 3: automatisoitu määritys 0,08–1,50 IU/ml b [tuntia] c [dl/kg/h] d [(IU/dl)/(IU VWF:RCo/kg)] e [(h*IU/dl)] f [(h*IU/dl)/(IU VWF:RCo/kg)] g Tämä tutkimus tehtiin ADVATE‑valmisteella, joka on rekombinantti tekijä VIII | ||||
Tutkimusten 070701 ja 071001 yhdistettyjen tietojen eksploratiivinen analyysi viittasi siihen, että VWF:RCo:n keskimääräinen pysyvyys oli tilastollisesti merkitsevästi (5 %:n tasolla) suurentunut, terminaalinen puoliintumisaikaa oli tilastollisesti merkitsevästi (5 %:n tasolle) pidentynyt ja AUC0inf‑arvo oli tilastollisesti merkitsevästi (5 %:n tasolla) suurentunut VEYVONDI‑valmisteen annon (50 IU/kg VWF:RCo) ja VEYVONDI‑valmisteen ja oktokogi alfan samanaikaisen annon (50 IU/kg VWF:RCo ja 38,5 IU/kg rFVIII) jälkeen verrattuna pdVWF:n ja plasmaperäisen hyytymistekijä VIII:n (pdFVIII) antoon (50 IU/kg pdVWF:RCo ja 38,5 IU/kg pdFVIII).
Lisäksi VEYVONDI-valmisteesta tehtiin täydelliset farmakokineettiset arvioinnit kerta-annon ja toistuvan annon jälkeen tutkimuksessa 071301, jossa tutkittiin pitkäaikaista estohoitoa yhteensä 23 aikuisella tutkittavalla, joilla oli vaikea VWD (N = 3, tyyppi 1; N = 1, tyyppi 2A; N = 1, tyyppi 2B; N = 18, tyyppi 3). Näistä arvioinneista saadut farmakokineettiset parametrit vahvistivat aikaisempien tutkimusten tulokset (ks. taulukko 5 edellä), eikä VWF:n keskeisten farmakokineettisten parametrien tilastollinen vertailu osoittanut merkitseviä eroja estohoidon aloituksen ja kuukauden 12 välillä.
Kuudesta tutkimuksesta saatuja VWF:n (N = 134) farmakokineettisiä tietoja arvioitiin käyttämällä populaatiofarmakokineettistä mallinnusta ja simulaatioon perustuvaa lähestymistapaa. Nämä tulokset vahvistivat, että VWF:RCo:n farmakokinetiikka on riippumaton sekä annoksesta (vaihteluväli: 2,0–80 IU/kg) että ajasta (enintään 4,3 vuotta). Kovariaattien arvioinnit eivät osoittaneet sukupuolella, rodulla tai VWF:n tyypillä olevan kliinisesti merkityksellistä vaikutusta VWF:RCo:n farmakokinetiikkaan; kehonpaino ja ikä tunnistettiin merkittäviksi kovariaateiksi.
Pediatriset potilaat
VWF:n farmakokinetiikka arvioitiin 24:llä von Willebrandin tautia (VWD) sairastavalla pediatrisella potilaalla potilasryhmän PK-mallinnuksen perusteella käyttäen harvalukuisia PK‑näytteitä, jotka otettiin pediatrisen tutkimuksen aikana kolmessa ikäryhmässä (alle 6‑vuotiaat [N = 5], 6 –< 12‑vuotiaat [N = 10] ja 12 –< 18‑vuotiaat [N = 9]) sen jälkeen, kun tutkittavat olivat saaneet kertainfuusiona 50 ± 5 IU/kg rVWF:RCo:ta (ks. taulukko 6).
Taulukko 6. VWF:RCo-hoidon farmakokineettinen arviointi pediatrisilla tutkittavillaa
| Parametri | PK50 VEYVONDI (tutkimus 071102) Keskiarvo (95 % CI) Keskihajonta | |||
| Ikäryhmä | < 6 vuotta (n = 5) | 6 – < 12 vuotta (n = 10) | 12 – < 18 vuotta (n = 9) | Yhteensä (n = 24) |
| T1/2b | 12,4 (9,91; 15,0) 2,90 | 14,5 (13,6; 15,4) 1,47 | 15,1 (14,2; 16,1) 1,50 | 14,3 (13,5; 15,1) 2,03 |
| CLc | 0,082 (0,047; 0,118) 0,041 | 0,051 (0,041; 0,061) 0,016 | 0,043 (0,038; 0,048) 0,007 | 0,055 (0,045; 0,065) 0,025 |
| IR, Cmaxd | 1,25 (0,92; 1,58) 0,378 | 1,54 (1,30; 1,77) 0,378 | 1,58 (1,43; 1,72) 0,225 | 1,49 (1,36; 1,63) 0,339 |
| AUC0-infe | 1 260 (690; 1 840) 654 | 1 630 (1 080; 2 170) 882 | 1 600 (1 140; 2 060) 704 | 1 540 (1 240; 1 840) 757 |
| AUC0-inf/annosf | 25,6 (14,4; 36,9) 12,8 | 32,5 (21,3; 43,7) 18,1 | 32,6 (23,2; 41,9) 14,3 | 31,1 (25,0; 37,2) 15,3 |
a Tiedot edustavat neljää harvalukuista PK-näytettä, jotka otettiin kultakin tutkittavalta pediatrisen tutkimuksen 071102 aikana, sekä potilasryhmän PK-mallinnuksen tuloksia.
b [tuntia]
c [dl/kg/h]
d [(IU/dl)/(IU VWF:RCo/kg)]]
e [(h*IU/dl)]
f [(h*IU/dl)/(IU VWF:RCo/kg)]
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, geenitoksisuutta, karsinogeenisuutta sekä lisääntymis‑ ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Tutkimuksia karsinogeenisuudesta, hedelmällisyyden heikkenemisestä tai sikiönkehityksestä ei ole tehty. Ihmisen istukan ex vivo ‑perfuusiomallissa on osoitettu, että VEYVONDI ei läpäise istukkaa.
Farmaseuttiset tiedot
Apuaineet
Kuiva‑aine
Natriumsitraatti (E 331)
Glysiini (E 640)
Trehaloosidihydraatti
Mannitoli (E 421)
Polysorbaatti 80 (E 433)
Liuotin
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Kestoaika käyttökuntoon saattamisen jälkeen:
Valmisteen käytönaikaisen kemiallisen ja fysikaalisen säilyvyyden on osoitettu olevan 3 tuntia 25 °C:ssa.
Mikrobiologiselta kannalta valmiste on käytettävä heti. Jos valmistetta ei käytetä heti, käytönaikaiset säilytysajat ja ‑olosuhteet ovat käyttäjän vastuulla.
Säilytys
Kuiva‑aine
Säilytä alle 30 °C.
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saattamisen jälkeen
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VEYVONDI injektiokuiva-aine ja liuotin, liuosta varten
650 IU (L:ei) 1 kpl (Mix2Vial, liuotin 5 ml (130 IU/ml)) (639,25 €)
1300 IU (L:ei) 1 kpl (Mix2Vial, liuotin 10 ml (130 IU/ml)) (1235,72 €)
PF-selosteen tieto
VEYVONDI 650 IU injektiokuiva‑aine ja liuotin, liuosta varten
Kukin pakkaus sisältää:
- kuiva‑aine injektiopullossa (tyypin I lasia), jossa on butyylikumitulppa
- 5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on kumitulppa (klooributyyliä tai bromobutyyliä)
- yhden laitteen valmisteen käyttökuntoon saattamista varten (Mix2Vial)
VEYVONDI 1 300 IU injektiokuiva‑aine ja liuotin, liuosta varten
Kukin pakkaus sisältää:
- kuiva‑aine injektiopullossa (tyypin I lasia), jossa on butyylikumitulppa.
- 10 ml liuotinta injektiopullossa (tyypin I lasia), jossa on kumitulppa (bromobutyyliä)
- yhden laitteen valmisteen käyttökuntoon saattamista varten (Mix2Vial)
Valmisteen kuvaus:
Kuiva‑aine on valkoinen tai luonnonvalkoinen kylmäkuivattu jauhe
Liuotin on kirkas ja väritön liuos.
Käyttö- ja käsittelyohjeet
Yleiset ohjeet
- Tarkista viimeinen käyttöpäivämäärä ja varmista, että VEYVONDI‑kuiva‑aine ja injektionesteisiin käytettävä vesi (liuotin) ovat huoneenlämpöisiä ennen valmisteen käyttökuntoon saattamista. Älä käytä etiketeissä ja kotelossa mainitun viimeisen käyttöpäivämäärän jälkeen.
- Käytä käyttökuntoon saattamisen aikana aseptista (puhdasta ja vähäbakteerista) tekniikkaa ja tasaista työskentelyalustaa. Pese kädet ja pue puhtaat tutkimuskäsineet (käsineiden käyttäminen on vapaaehtoista).
- Käyttökuntoon saatettu valmiste tulee käyttää (kun kuiva‑aine on sekoitettu mukana toimitettuun veteen) mahdollisimman pian kolmen tunnin kuluessa. Käyttökuntoon saatettua valmistetta voi säilyttää huoneenlämmössä enintään 25 °C:ssa enintään kolmen tunnin ajan.
- Varmista, että VEYVONDI‑kuiva‑aine ja injektionesteisiin käytettävä vesi (liuotin) ovat huoneenlämpöisiä ennen valmisteen käyttökuntoon saattamista.
- Käytä muoviruiskuja tämän valmisteen antamiseen, sillä valmisteen sisältämillä proteiineilla on taipumus tarttua lasisten ruiskujen pintaan.
-
Älä sekoita VEYVONDI-valmistetta muiden lääkevalmisteiden kuin oktokogi alfan (ADVATE) kanssa.
Ohjeet käyttökuntoon saattamiseksi ja lääkkeen antamiseksi
| Vaiheet | Kuvaesimerkki | |
| 1 | Poista VEYVONDI‑kuiva‑aineen ja liuottimen sisältävistä injektiopulloista korkit ja paljasta kumitulppien keskustat. | 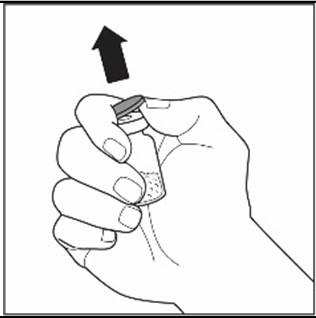 |
| 2 | Desinfioi jokainen tulppa erillisellä steriilillä desinfiointipyyhkeellä (tai muulla sopivalla, lääkärin tai hemofilian hoitoon erikoistuneen keskuksen suosittelemalla steriilillä liuoksella) pyyhkimällä tulppaa useiden sekuntien ajan. Anna kumitulpan kuivua. Aseta injektiopullot tasaiselle alustalle. | 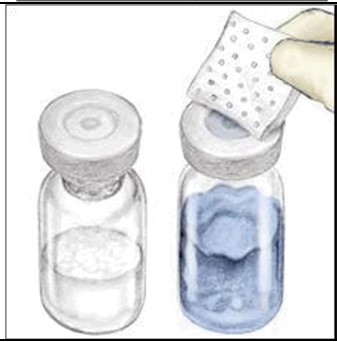 |
| 3 | Avaa Mix2Vial‑laitteen pakkaus vetämällä suojakansi kokonaan pois ilman, että kosketat pakkauksen sisäpuolta. Älä poista Mix2Vial‑laitetta pakkauksesta. | - |
| 4 | Käännä Mix2Vial‑laitteen pakkaus ylösalaisin ja aseta se liuottimen sisältävän injektiopullon päälle. Työnnä laitteen sininen muovipiikki kohtisuoraan liuotinpullon tulpan keskustan läpi. Tartu pakkaukseen sen reunasta ja vedä pakkaus irti Mix2Vial‑laitteesta. Älä koske läpinäkyvään muovipiikkiin. Liuottimen sisältävä injektiopullo on nyt kiinnitetty Mix2Vial‑laitteeseen ja valmis kiinnitettäväksi VEYVONDI‑injektiopulloon. | 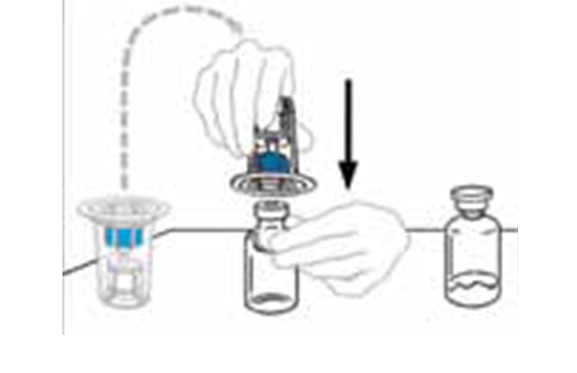 |
| 5 | Kiinnitä liuottimen sisältävä injektiopullo VEYVONDI‑injektiopulloon kääntämällä liuotinpullo ylösalaisin ja asettamalla se VEYVONDI‑kuiva‑aineen sisältävän injektiopullon päälle. Työnnä läpinäkyvä muovipiikki kohtisuoraan VEYVONDI‑injektiopullon tulpan läpi. Tämä on tehtävä heti, jotta nesteeseen ei pääse taudinaiheuttajia. Tyhjiö vetää liuottimen VEYVONDI‑injektiopulloon. Tarkista, että liuotin on kokonaan siirtynyt. Älä käytä, jos tyhjiö ei ole vetänyt liuotinta VEYVONDI‑injektiopulloon. | 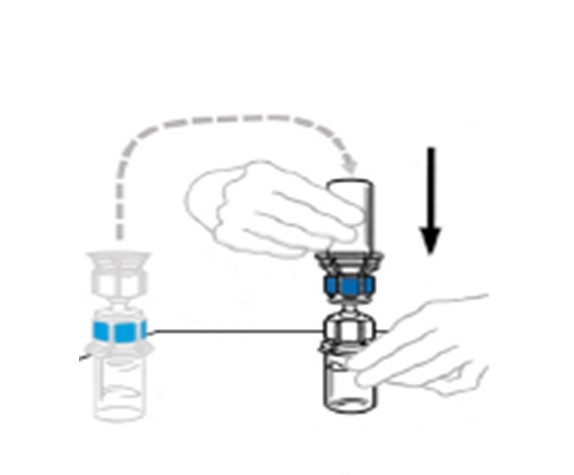 |
| 6 | Pyörittele toisiinsa liitettyjä injektiopulloja varovasti ja jatkuvasti, tai anna käyttökuntoon saatetun valmisteen tasaantua 5 minuutin ajan ja pyörittele sitä sitten varovasti, kunnes kuiva‑aine on liuennut täysin. Älä ravista. Ravistaminen huonontaa valmisteen laatua. Älä säilytä kylmässä käyttökuntoon saattamisen jälkeen. | 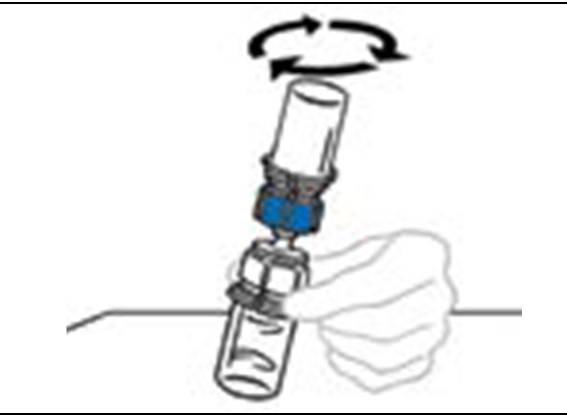 |
| 7 | Irrota Mix2Vial‑laitteen puoliskot toisistaan tarttumalla toisella kädellä VEYVONDI‑injektiopulloon kiinnitetyn Mix2Vial‑laitteen läpinäkyvään muoviosaan ja toisella kädellä liuotinpulloon kiinnitetyn Mix2Vial‑laitteen siniseen muoviosaan. Irrota injektiopullot varovasti toisistaan kiertämällä sinistä muoviosaa vastapäivään. Älä koske liuotetun valmisteen sisältävään VEYVONDI‑injektiopulloon kiinnitetyn muovisen liitoskappaleen päähän. Aseta VEYVONDI‑injektiopullo tasaiselle työtasolle. Hävitä tyhjä liuotinpullo. | 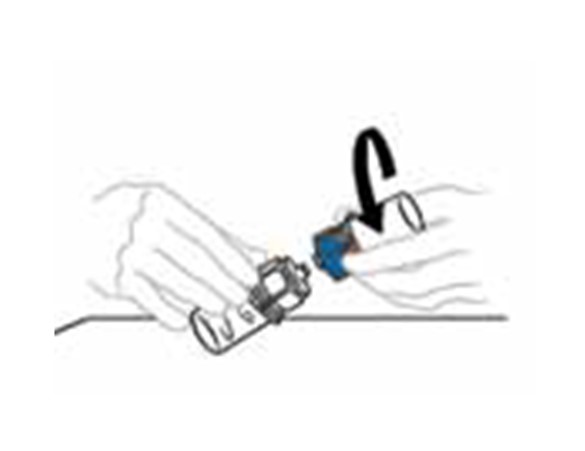 |
| 8 | Vedä tyhjään, steriiliin, kertakäyttöiseen muoviruiskuun ilmaa vetämällä mäntää taaksepäin. Ruiskuun vedettävän ilman määrän tulisi olla yhtä suuri kuin injektiopullosta vedettävän, käyttökuntoon saatetun VEYVONDI‑valmisteen määrä. | 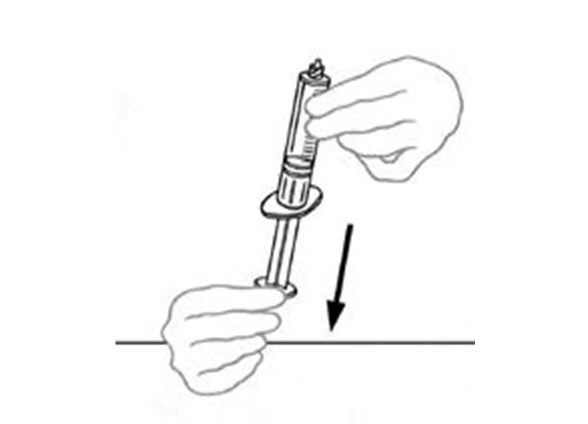 |
| 9 | Laita VEYVONDI‑injektiopullo (joka sisältää käyttökuntoon saatetun valmisteen) tasaiselle työtasolle, kiinnitä ruisku läpinäkyvään muoviseen liitoskappaleeseen ja kierrä ruiskua myötäpäivään | 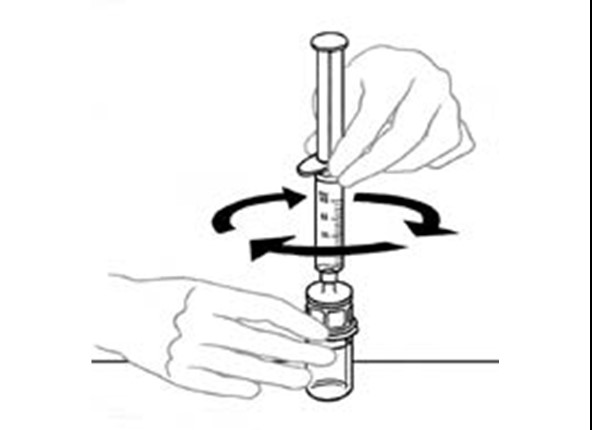 |
| 10 | Pidä injektiopulloa toisessa kädessä ja paina toisella kädellä kaikki ilma ruiskusta injektiopulloon. | 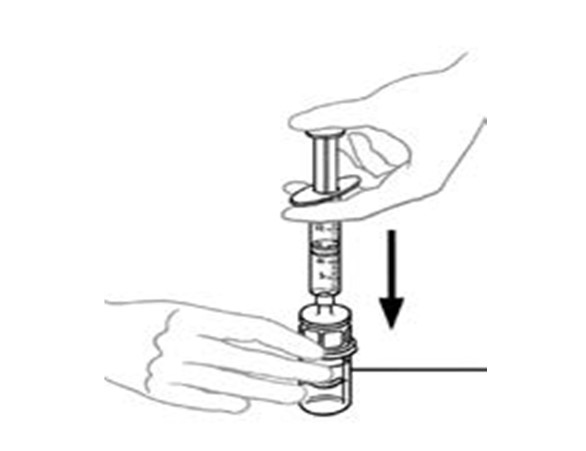 |
| 11 | Käännä toisiinsa liitetyt ruisku ja VEYVONDI‑injektiopullo ylösalaisin niin, että injektiopullo on päällimmäisenä. Varmista, että ruiskun mäntä on painettu pohjaan. Vedä VEYVONDI‑valmiste ruiskuun vetämällä mäntää hitaasti taaksepäin. | 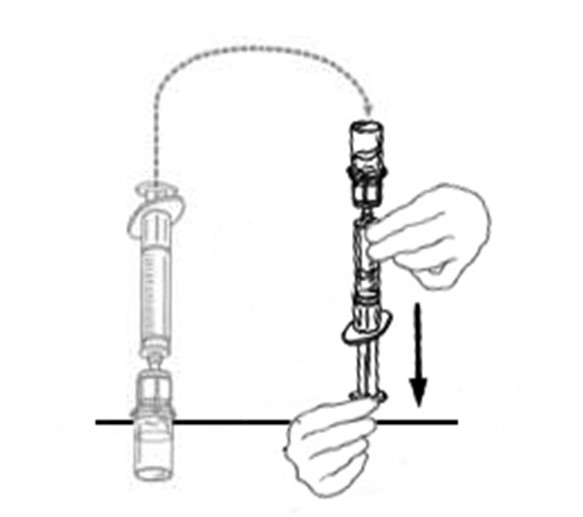 |
| 12 | Älä siirtele liuosta ruiskusta injektiopulloon ja takaisin. Se voi heikentää lääkkeen laatua. Kun olet valmis antamaan infuusion, irrota ruisku kiertämällä sitä vastapäivään. Tarkista ruisku silmämääräisesti hiukkasten varalta. Liuoksen tulee olla kirkasta ja väritöntä. Jos liuoksessa on saostumia tai hiukkasia, sitä ei saa käyttää. Ota yhteys lääkäriin. | 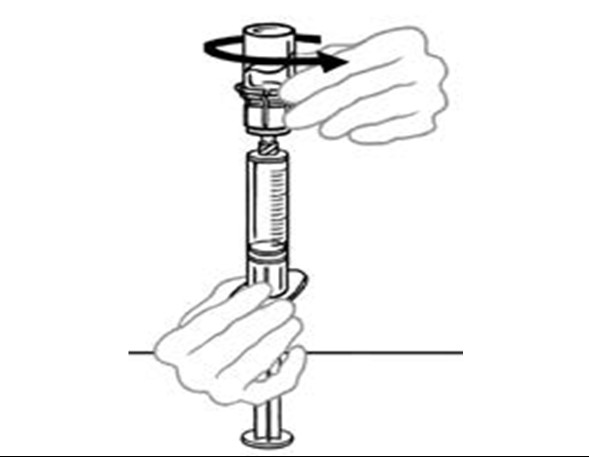 |
| 13 | Jos annokseen tarvitaan enemmän kuin yksi VEYVONDI‑injektiopullo: Jätä ruisku kiinni injektiopulloon, kunnes seuraava injektiopullo on valmis käytettäväksi. Valmistele seuraava VEYVONDI‑injektiopullo käyttökuntoon uutta Mix2Vial‑laitetta käyttäen ja yllä kuvattuja vaiheita (2–8) noudattaen. | |
| 14 | Yhteen ruiskuun voi vetää kahden injektiopullon sisällön. HUOMAUTUS: kun painat ilmaa toiseen VEYVONDI‑injektiopulloon, jonka sisältö on tarkoitus vetää samaan ruiskuun, varmista, että ruiskuun liitetty injektiopullo on ylösalaisin (injektiopullo ruiskun yläpuolella). |
Ohjeet lääkkeen antamiseksi
Tarkista ruiskussa oleva, käyttökuntoon saatettu liuos hiukkasten ja värimuutosten varalta ennen antoa (liuoksen tulee olla kirkasta ja väritöntä, eikä siinä saa olla hiukkasia). Injektiopulloon jää usein hiukan saostumia tai hiukkasiavalmisteen käyttökuntoon saattamisen jälkeen. Mix2Vial‑laitteen suodatin poistaa nämä hiukkaset täysin. Suodatus ei vaikuta annoksen laskentaan. Ruiskussa olevaa liuosta ei saa käyttää, jos se on sameaa tai sisältää suodatuksen jälkeen saostumia tai hiukkasia.
1. Kiinnitä infuusioneula VEYVONDI‑liuoksen sisältävään ruiskuun. Potilasmukavuuden vuoksi on suositeltavaa käyttää siipineulalla varustettua infuusiolaitteistoa. Aseta neulan kärki ylöspäin ja poista kaikki ilmakuplat naputtamalla ruiskua kevyesti sormellasi. Poista ilma ruiskusta ja neulasta painamalla mäntää hitaasti ja varovasti.
2. Tee kiristysside ja valmistele infuusiokohta puhdistamalla iho perusteellisesti steriilillä desinfiointipyyhkeellä (tai muulla sopivalla, lääkärin tai hemofilian hoitoon erikoistuneen keskuksen suosittelemalla steriilillä liuoksella).
3. Vie neula laskimoon ja avaa kiristysside. Anna VEYVONDI hitaana infuusiona. Infuusionopeus saa olla enintään 4 ml minuutissa. Irrota tyhjä ruisku. Jos annokseen tarvitaan useita ruiskuja, kiinnitä ja annostele seuraavat VEYVONDI‑valmistetta sisältävät ruiskut yksi kerrallaan.
Huomautus:
älä poista siipineulaa äläkä koske ruiskun liittimenä toimivaan Luer‑porttiin, ennen kuin kaikkien ruiskujen sisältö on annettu.
Jos lääkäri on määrännyt rekombinanttia tekijää VIII, anna rekombinantti tekijä VIII 10 minuutin kuluessa siitä, kun koko VEYVONDI‑infuusio on annettu.
4. Poista neula laskimosta ja paina infuusiokohtaa steriilillä harsotaitoksella useiden minuuttien ajan.
Jos VEYVONDI‑valmistetta on tarpeen antaa suurina tilavuuksina, kahden VEYVONDI‑injektiopullon sisältö voidaan yhdistää. Käyttökuntoon saatettujen VEYVONDI‑injektiopullojen sisällön voi vetää samaan ruiskuun. Tällöin käyttökuntoon saatettua VEYVONDI‑liuosta ei saa kuitenkaan laimentaa edelleen.
Liuos on annettava hitaasti laskimoon (ks. kohta Annostus ja antotapa) enintään nopeudella 4 ml/min.
Älä laita neulansuojusta takaisin paikoilleen. Laita neula, ruisku ja tyhjä(t) VEYVONDI‑valmisteen ja liuottimen injektiopullo(t) terävälle jätteelle tarkoitettuun kovaseinäiseen astiaan odottamaan asianmukaista hävittämistä. Älä hävitä näitä tarvikkeita talousjätteen mukana.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VEYVONDI injektiokuiva-aine ja liuotin, liuosta varten
650 IU 1 kpl
1300 IU 1 kpl
- Ylempi erityiskorvaus (100 %). Von Willebrand -tekijävalmiste ja vonikogi alfa: Kroonisten hyytymishäiriöiden hoito erityisin edellytyksin (161).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Von Willebrand -tekijävalmiste ja vonikogi alfa: von Willebrandin taudin hoito erityisin edellytyksin (357).
ATC-koodi
B02BD10
Valmisteyhteenvedon muuttamispäivämäärä
18.12.2025
Yhteystiedot
 TAKEDA OY
TAKEDA OY PL 1406, Ilmalankuja 3
00101 Helsinki
0800 774 051
www.takeda.fi
etunimi.sukunimi@takeda.com