
SPEXOTRAS pulver till oral lösning 0,05 mg/ml
Kvalitativ och kvantitativ sammansättning
En flaska innehåller trametinibdimetylsulfoxid motsvarande 4,7 mg trametinib.
Varje ml av den färdigberedda lösningen innehåller 0,05 mg trametinib.
Hjälpämnen med känd effekt
Varje ml av den färdigberedda lösningen innehåller 100 mg sulfobutylbetadexnatrium, 0,8 mg metylparahydroxibensoat och 1,98 mg natrium.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Pulver till oral lösning.
Kliniska uppgifter
Terapeutiska indikationer
Låggradigt gliom
Spexotras i kombination med dabrafenib är avsett för behandling av pediatriska patienter från 1 års ålder med låggradigt gliom (LGG) med BRAF V600E-mutation, som kräver systemisk behandling.
Höggradigt gliom
Spexotras i kombination med dabrafenib är avsett för behandling av pediatriska patienter från 1 års ålder med höggradigt gliom (HGG) med BRAF V600E-mutation, som tidigare har fått minst en behandling med radioterapi och/eller kemoterapi.
Villkor
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Dosering och administreringssätt
Behandling med Spexotras ska inledas och övervakas av kvalificerad läkare med erfarenhet av användning av cancerläkemedel.
Innan patienterna tar Spexotras måste de ha en bekräftelse på BRAF V600E-mutation bedömd med en CE-märkt medicinsk utrustning för in vitro-diagnostik (IVD) med motsvarande avsedda syfte. Om den CE-märkta IVD:n inte är tillgänglig, bör bekräftelsen på BRAF V600E bedömas med ett alternativt validerat test.
Spexotras används i kombination med dabrafenib dispergerbara tabletter. Se produktresumén angående dosering av dabrafenib dispergerbara tabletter.
Dosering
Rekommenderad dos Spexotras en gång dagligen är beroende av kroppsvikten (tabell 1).
Tabell 1 Dosregim baserad på kroppsvikt
| Kroppsvikt* | Rekommenderad dos | |
| Volym oral lösning (ml) en gång dagligen | Motsvarande mg av trametinib | |
| 8 kg | 6 ml | 0,30 mg |
| 9-10 kg | 7 ml | 0,35 mg |
| 11 kg | 8 ml | 0,40 mg |
| 12-13 kg | 9 ml | 0,45 mg |
| 14-17 kg | 11 ml | 0,55 mg |
| 18-21 kg | 14 ml | 0,70 mg |
| 22-25 kg | 17 ml | 0,85 mg |
| 26-29 kg | 18 ml | 0,90 mg |
| 30-33 kg | 20 ml | 1 mg |
| 34-37 kg | 23 ml | 1,15 mg |
| 38-41 kg | 25 ml | 1,25 mg |
| 42-45 kg | 28 ml | 1,40 mg |
| 46-50 kg | 32 ml | 1,60 mg |
| ≥ 51 kg | 40 ml | 2 mg |
*Kroppsvikten avrundas vid behov till närmaste kg. Rekommenderad dos till patienter som väger under 8 kg har inte fastställts. Doseringsanvisningar för behandling med dabrafenib i kombination med Spexotras finns i produktresumén för dabrafenib dispergerbara tabletter, avsnitten ”Dosering” och ”Administreringssätt”. | ||
Behandlingstid
Behandling med Spexotras ska pågå fram till sjukdomsprogression eller tills oacceptabel toxicitet uppträder. Data hos patienter över 18 år med gliom är begränsade. Fortsatt behandling i vuxen ålder ska därför baseras på en av läkaren utförd nytta-riskbedömning för varje enskild patient.
Missade eller försenade doser
Om en dos Spexotras missas ska den inte tas om det är mindre än 12 timmar till nästa planerade dos. Om patienten kräks efter att ha tagit Spexotras ska inte någon extra dos tas utan nästa dos ska tas vid den planerade tiden.
Dosjustering
För att hantera biverkningar kan dossänkning, behandlingsuppehåll eller utsättning av behandlingen bli nödvändigt (se tabell 2 och 3).
Om behandlingsrelaterade toxiciteter uppträder ska både trametinib och dabrafenib dossänkas, avbrytas eller sättas ut samtidigt. Undantag där dosjusteringar är nödvändiga för endast en av de två behandlingarna beskrivs nedan för uveit, RAS-mutationspositiva icke-kutana maligniteter (främst relaterat till dabrafenib), reducerad vänsterkammarejektionsfraktion (LVEF), retinalvensocklusion (RVO), näthinneavlossning (RPED) och interstitiell lungsjukdom (ILD)/pneumonit (främst relaterat till trametinib).
Dosjusteringar eller behandlingsuppehåll rekommenderas inte vid biverkningar i form av kutana maligniteter (se produktresumén för dabrafenib dispergerbara tabletter för ytterligare information).
Tabell 2 Dosjusteringsschema baserat på graden av eventuella biverkningar (exklusive feber)
| Grad (CTCAE)* | Rekommenderade justeringar av trametinibdosen |
| Grad 1 eller grad 2 (tolererbara) | Fortsätt behandlingen och följ upp efter kliniskt behov. |
| Grad 2 (ej tolererbara) eller grad 3 | Avbryt behandlingen tills biverkningarna avtagit till grad 0 till 1 och sänk dosen med ett steg när behandlingen återupptas. Doseringsriktlinjer finns i tabell 3. |
| Grad 4 | Avbryt behandlingen permanent, eller avbryt tills biverkningarna avtagit till grad 0 till 1 och sänk dosen med ett steg när behandlingen återupptas. Doseringsriktlinjer finns i tabell 3. |
| * Intensitet hos kliniska biverkningar graderade efter Common Terminology Criteria for Adverse Events (CTCAE) | |
Rekommenderade dossänkningar till cirka 75 % av den rekommenderade dosen (första dossänkningen) och till cirka 50 % av den rekommenderade dosen (andra dossänkningen) visas i tabell 3.
Tabell 3 Rekommenderade dossänkningar på grund av biverkningar
| Kroppsvikt | Rekommenderad dos | Reducerad dos | |
ml lösning (mg trametinib) (en gång dagligen) | Dos efter första sänkningen (en gång dagligen) | Dos efter andra sänkningen (en gång dagligen) | |
| 8 kg | 6 ml (0,30 mg) | 5 ml | 3 ml |
| 9-10 kg | 7 ml (0,35 mg) | 5 ml | 4 ml |
| 11 kg | 8 ml (0,40 mg) | 6 ml | 4 ml |
| 12-13 kg | 9 ml (0,45 mg) | 7 ml | 5 ml |
| 14-17 kg | 11 ml (0,55 mg) | 8 ml | 6 ml |
| 18-21 kg | 14 ml (0,70 mg) | 11 ml | 7 ml |
| 22-25 kg | 17 ml (0,85 mg) | 13 ml | 9 ml |
| 26-29 kg | 18 ml (0,90 mg) | 14 ml | 9 ml |
| 30-33 kg | 20 ml (1 mg) | 15 ml | 10 ml |
| 34-37 kg | 23 ml (1,15 mg) | 17 ml | 12 ml |
| 38-41 kg | 25 ml (1,25 mg) | 19 ml | 13 ml |
| 42-45 kg | 28 ml (1,40 mg) | 21 ml | 14 ml |
| 46-50 kg | 32 ml (1,60 mg) | 24 ml | 16 ml |
| ≥ 51 kg | 40 ml (2 mg) | 30 ml | 20 ml |
| Dosjusteringar av Spexotras till under 50 % av den rekommenderade dosen rekommenderas inte. | |||
När patientens biverkningar kan hanteras effektivt kan man överväga att åter öka dosen och därvid följa samma doseringssteg som användes vid dossänkningen. Trametinibdosen ska inte överstiga den rekommenderade dosen i tabell 1.
Dosjusteringar vid utvalda biverkningar
Feber
Om en patients kroppstemperatur är ≥ 38 °C ska behandlingen med trametinib och dabrafenib avbrytas. Vid återfall kan behandlingen också avbrytas vid det första symtomet på feber. Behandling med antipyretika såsom ibuprofen eller paracetamol ska inledas. Användning av orala kortikosteroider ska övervägas i de fall där antipyretika är otillräckliga. Patienterna ska utvärderas för tecken och symtom på infektion och vid behov behandlas i linje med lokal praxis (se avsnitt Varningar och försiktighet). Behandlingen ska återinsättas om patienten är symtomfri i minst 24 timmar antingen 1) på samma dosnivå eller 2) reducerad med en dosnivå om febern är återkommande och/eller åtföljs av andra allvarliga symtom såsom uttorkning, hypotoni eller njursvikt.
Undantag från dosjustering (där endast en av två behandlingar är dossänkt) för utvalda biverkningar
Reducerad vänsterkammarejektionsfraktion (LVEF)/vänsterkammardysfunktion
Behandlingen med trametinib ska avbrytas om patienterna får en asymtomatisk, absolut minskning av LVEF på > 10 % jämfört med vid baslinjen och om ejektionsfraktionen ligger under institutionens lägsta normalvärden (LLN) (se avsnitt Varningar och försiktighet). Ingen dosjustering av dabrafenib krävs när det tas i kombination med trametinib. Om LVEF återgår till det normala kan behandling med trametinib återupptas, dock med dosen sänkt ett steg och under noggrann uppföljning (se avsnitt Varningar och försiktighet).
Trametinib ska sättas ut permanent hos patienter med vänsterkammardysfunktion grad 3 eller 4, eller kliniskt signifikant LVEF-minskning, som inte återgår till det normala inom 4 veckor (se avsnitt Varningar och försiktighet).
Retinalvensocklusion (RVO) och näthinneavlossning (RPED)
Om patienterna rapporterar nya synstörningar, såsom centralt skotom, dimsyn eller synförlust någon gång under kombinationsbehandling med trametinib och dabrafenib, rekommenderas omedelbar undersökning av oftalmolog. Behandling med trametinib ska sättas ut permanent hos patienter som diagnostiseras med RVO. Vid diagnostiserad RPED, följ dosändringarna i tabell 4 nedan för trametinib (se avsnitt Varningar och försiktighet). Ingen dosjutering av dabrafenib krävs när det tas i kombination med trametinib vid bekräftade fall av RVO eller RPED.
Tabell 4 Rekommenderade dosändringar för trametinib vid RPED
| RPED grad 1 | Fortsätt behandlingen med månatliga undersökningar av näthinnan tills problemet försvunnit. Om RPED förvärras, följ anvisningarna nedan och gör uppehåll i trametinibbehandlingen i upp till 3 veckor. |
| RPED grad 2 eller 3 | Gör uppehåll i trametinibbehandlingen i upp till 3 veckor. |
| RPED grad 2 eller 3 som återgår till grad 0 eller 1 inom 3 veckor | Återuppta trametinibbehandlingen med lägre dos (se tabell 3) eller sätt ut trametinibbehandlingen hos patienter som står på den lägsta dosen. |
| RPED grad 2 eller 3 som inte återgår till åtminstone grad 1 inom 3 veckor | Sätt ut trametinib permanent. |
Interstitiell lungsjukdom (ILD)/pneumonit
Ett uppehåll måste göras i trametinibbehandlingen hos patienter med misstänkt ILD eller pneumonit, inklusive patienter med nya eller progressiva pulmonella symtom och symtom som hosta, dyspné, hypoxi, pleurautgjutning eller -infiltrat, i avvaktan på kliniska undersökningar. Behandlingen med trametinib måste sättas ut permanent hos patienter med behandlingsrelaterad ILD eller pneumonit. Ingen dosjustering av dabrafenib krävs när det tas i kombination med trametinib vid fall av ILD eller pneumonit.
Uveit
Inga dosjusteringar krävs för uveit så länge effektiva lokala behandlingar kan kontrollera inflammation i ögonen. Vid uteblivet svar på lokal ögonbehandling ska uppehåll med dabrafenib göras tills ögoninflammationen läkt. Därefter kan dabrafenib återinsättas, reducerat med en dosnivå. Ingen dosjustering av trametinib krävs när det tas i kombination med dabrafenib (se avsnitt Varningar och försiktighet).
RAS-mutationspositiva icke-kutana maligniteter
Nyttan och riskerna måste övervägas innan behandling med dabrafenib fortsätter hos patienter med icke-kutan malignitet som har en RAS-mutation. Ingen dosjustering av trametinib krävs när det tas i kombination med dabrafenib (se avsnitt Varningar och försiktighet).
Särskilda populationer
Nedsatt leverfunktion
Ingen dosjustering krävs för patienter med lätt nedsatt leverfunktion. Tillgängliga data från en klinisk farmakologisk studie indikerar en begränsad påverkan av måttligt till kraftigt nedsatt leverfunktion på exponeringen av trametinib (se avsnitt Farmakokinetiska egenskaper). Trametinib ska användas med försiktighet till patienter med måttligt eller kraftigt nedsatt leverfunktion.
Nedsatt njurfunktion
Ingen dosjustering krävs för patienter med lätt eller måttligt nedsatt njurfunktion. Det finns inga data om trametinib hos patienter med kraftig njurfunktionsnedsättning och eventuellt behov av dosjustering kan därför inte fastställas (se avsnitt Farmakokinetiska egenskaper). Trametinib ska användas med försiktighet till patienter med kraftig njurfunktionsnedsättning.
Pediatrisk population
Säkerhet och effekt för kombinationsbehandling med trametinib och dabrafenib för barn under 1 år har inte fastställts. Inga data finns tillgängliga. Studier på unga djur har visat på påverkan av trametinib som inte sågs hos vuxna djur (se avsnitt Prekliniska säkerhetsuppgifter). Data om långtidssäkerhet hos pediatriska patienter är för närvarande begränsade.
Administreringssätt
Spexotras är avsedd för oral användning.
Spexotras pulver måste beredas till oral lösning av apotekspersonal innan den dispenseras. Det rekommenderas att hälso- och sjukvårdspersonalen informerar patient eller vårdare om hur den ordinerade dagliga dosen oral lösning ska administreras innan den första dosen tas.
Exponeringen för Spexotras påverkas inte av föda (se avsnitt Farmakokinetiska egenskaper). Spexotras ska tas samtidigt som dabrafenib dispergerbara tabletter, vars exponering minskar vid samtidigt intag av föda. Spexotras ska därför tas utan föda, minst en timme före eller två timmar efter måltid (se avsnitt Farmakokinetiska egenskaper). Bröstmjölk och/eller modersmjölksersättning kan ges vid behov om en patient inte tolererar fasta.
Det rekommenderas att Spexotras-dosen tas vid ungefär samma tidpunkt varje dag med den återanvändbara orala spruta som ingår i förpackningen. Den dagliga engångsdosen av Spexotras ska tas tillsammans med antingen morgon- eller kvällsdosen av dabrafenib vid samma tidpunkt varje dag.
Om patienten inte kan svälja och har nasogastrisk sond kan Spexotras oral lösning ges via sonden.
Anvisningar för beredning finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spexotras är avsedd att användas i kombination med dabrafenib dispergerbara tabletter då det finns begränsade effektdata för trametinib i monoterapi och för dabrafenib i monoterapi vid BRAF V600-mutationspositivt gliom. Produktresumén för dabrafenib dispergerbara tabletter måste läsas innan behandling påbörjas. För ytterligare information om varningar och försiktighet associerat med behandling med dabrafenib, se produktresumén för dabrafenib dispergerbara tabletter.
BRAF V600E-test
Effekt och säkerhet för trametinib i kombination med dabrafenib har inte undersökts hos patienter vars gliom testats negativt med avseende på BRAF V600E-mutationen.
Nya maligniteter
Nya maligniteter, kutana och icke-kutana, kan uppkomma när trametinib används i kombination med dabrafenib.
Kutana maligniteter
Kutana maligniteter som kutant skivepitelkarcinom (cuSCC), inklusive keratoakantom och nytt primärt melanom, har observerats hos vuxna patienter som behandlats med trametinib i kombination med dabrafenib (se avsnitt Biverkningar). Undersökning av huden rekommenderas innan behandling med trametinib sätts in och varje månad under behandlingsperioden och i upp till sex månader efter behandling. Övervakningen ska fortsätta i 6 månader efter utsättning av trametinib eller fram tills annan cancerbehandling sätts in.
Misstänkta hudlesioner ska behandlas genom dermatologisk excision och ingen justering av behandlingen krävs. Patienterna ska instrueras att informera läkaren omedelbart om nya hudlesioner utvecklas.
Icke-kutana maligniteter
Baserat på dess verkningsmekanism kan dabrafenib öka risken för icke-kutana maligniteter när RAS-mutationer finns närvarande. Se produktresumén för dabrafenib dispergerbara tabletter (avsnitt Varningar och försiktighet). Ingen dosjustering av trametinib krävs för RAS-mutationspositiva maligniteter när det tas i kombination med dabrafenib.
Blödning
Blödningar har rapporterats hos vuxna och pediatriska patienter som tagit trametinib i kombination med dabrafenib (se avsnitt Biverkningar). Större blödningar och blödningar med dödlig utgång har förekommit hos vuxna patienter som behandlas med trametinib i kombination med dabrafenib. Risken för dessa händelser hos patienter med lågt antal trombocyter (< 75 000/mm3) har inte fastställts eftersom sådana patienter exkluderades från kliniska studier. Blödningsrisken kan öka vid samtidig användning av trombocythämmare eller vid antikoagulantiabehandling. Om blödningar uppstår ska patienten behandlas efter kliniskt behov.
Reducerad vänsterkammarejektionsfraktion LVEF/vänsterkammardysfunktion
Trametinib i kombination med dabrafenib har rapporterats reducera LVEF hos både vuxna och pediatriska patienter (se avsnitt Biverkningar). I kliniska studier på pediatriska patienter var mediantiden till debut av första förekomsten av LVEF-minskning cirka en månad. I kliniska studier på vuxna patienter var mediantiden till debut av första förekomsten av vänsterkammardysfunktion, hjärtsvikt och reducerad LVEF mellan 2 och 5 månader.
Trametinib ska användas med försiktighet till patienter med nedsatt vänsterkammarfunktion. Patienter med vänsterkammardysfunktion, hjärtsvikt av New York Heart Association-klass II, III eller IV, akut koronarsyndrom under de senaste 6 månaderna, kliniskt signifikanta okontrollerade arytmier eller okontrollerad hypertoni, uteslöts från de kliniska studierna. Säkerheten vid användning i dessa patientgrupper är därför okänd. LVEF ska bedömas hos alla patienter innan behandling med trametinib sätts in, en månad efter insättningen och därefter med cirka 3 månaders intervall under hela behandlingstiden (se avsnitt Dosering och administreringssätt avseende dosjusteringar).
Hos patienter som behandlas med trametinib i kombination med dabrafenib har det förekommit enstaka rapporter om akut, svår vänsterkammardysfunktion på grund av myokardit. Fullständig återhämtning observerades vid avbrytande av behandlingen. Läkarna ska vara uppmärksamma på risken för myokardit hos patienter som utvecklar nya eller förvärrade tecken eller symtom på hjärtbesvär.
Feber
Feber har rapporterats i kliniska studier med trametinib hos vuxna och pediatriska patienter (se avsnitt Biverkningar). Förekomst och allvarlighetsgrad av feber ökar vid kombinationsbehandling (se produktresumén för dabrafenib dispergerbara tabletter, avsnitt Varningar och försiktighet). För patienter som får trametinib i kombination med dabrafenib kan feber åtföljas av svåra frossbrytningar, dehydrering och hypotoni som i vissa fall kan leda till akut njurinsufficiens. Hos pediatriska patienter som fick trametinib i kombination med dabrafenib var mediantiden till feberdebut 1,5 månader.
Behandling med trametinib och dabrafenib ska avbrytas om patientens kroppstemperatur är ≥ 38 ºC (se avsnitt Farmakodynamiska egenskaper). Vid återfall kan behandlingen också avbrytas vid det första symtomet på feber. Behandling med antipyretika såsom ibuprofen eller paracetamol ska inledas. Användning av orala kortikosteroider ska övervägas i de fall där antipyretika är otillräckliga. Patienterna ska utvärderas för tecken och symtom på infektion. Behandlingen kan återinsättas när febern försvinner. Om febern är förknippad med andra allvarliga symtom ska behandlingen återinsättas med reducerad dos när febern väl upphört och enligt vad som är kliniskt lämpligt (se avsnitt Dosering och administreringssätt).
Förändrat blodtryck
Såväl hypertoni som hypotoni har rapporterats hos patienter i kliniska studier med trametinib i kombination med dabrafenib (se avsnitt Biverkningar). Blodtrycket ska mätas vid behandlingsstarten och övervakas under behandlingen och hypertoni åtgärdas vid behov med gängse behandling.
Interstitiell lungsjukdom (ILD)/pneumonit
I en fas III-studie på vuxna patienter utvecklade 2,4 % (5/211) av patienterna som behandlades med trametinib monoterapi ILD eller pneumonit, sjukhusvistelse krävdes för samtliga fem patienter. Mediantiden till debut av den första presentationen på ILD eller pneumonit var 160 dagar (mellan 60 och 172 dagar). I två studier på vuxna patienter som behandlades med trametinib i kombination med dabrafenib utvecklade 1 % av patienterna pneumonit eller ILD (se avsnitt Biverkningar).
Behandlingen med trametinib måste avbrytas hos patienter med misstänkt interstitiell lungsjukdom eller pneumonit, inklusive patienter med nya eller progressiva lungsymtom och symtom som hosta, dyspné, hypoxi, pleurautgjutning eller infiltrat, i avvaktan på kliniska undersökningar. Behandlingen med trametinib ska sättas ut permanent hos patienter med behandlingsrelaterad ILD eller pneumonit (se avsnitt Dosering och administreringssätt). Behandling med dabrafenib kan fortsätta med samma dosering.
Synnedsättning
Sjukdomar förknippade med synstörningar, t.ex. RPED och RVO kan förekomma med trametinib, som i vissa fall debuterade efter flera månader. Symtom som dimsyn, nedsatt synskärpa och andra synfenomen har rapporterats hos vuxna i kliniska studier med trametinib. I kliniska studier har även uveit och iridocyklit rapporterats hos vuxna och pediatriska patienter som behandlas med trametinib i kombination med dabrafenib.
Trametinib rekommenderas inte till patienter med RVO i anamnesen. Säkerheten för trametinib hos patienter med predisponerande faktorer för RVO, inklusive okontrollerat glaukom eller okulär hypertension, okontrollerad hypertoni, okontrollerad diabetes mellitus eller hyperviskositets- eller hyperkoagulationssyndrom i anamnesen, har inte fastställts.
Om patienterna rapporterar nya synstörningar, såsom centralt skotom, dimsyn eller synförlust någon gång under trametinibbehandlingen, rekommenderas omedelbar undersökning av oftalmolog. Vid diagnostiserad RPED ska doseringsschemat i tabell 4 följas (se avsnitt Dosering och administreringssätt), vid diagnostiserad uveit se produktresumén för dabrafenib dispergerbara tabletter (avsnitt Varningar och försiktighet). Patienter som fått diagnosen RVO ska omedelbart avsluta behandlingen med trametinib.
Vid diagnostiserad RVO eller PRED krävs ingen dosjustering av dabrafenib när det tas i kombination med trametinib. Vid diagnostiserad uveit krävs ingen dosjustering av trametinib när det tas i kombination med dabrafenib.
Hudutslag
Hudutslag har observerats hos 49 % av pediatriska patienter i kliniska studier när trametinib användes i kombination med dabrafenib (se avsnitt Biverkningar). Majoriteten av dessa fall var av grad 1 eller 2 och krävde inte behandlingsavbrott eller dossänkning.
Allvarliga hudbiverkningar
Fall av allvarliga hudbiverkningar (SCAR), inklusive Stevens-Johnsons syndrom och läkemedelsreaktion med eosinofili och systemiska symtom (DRESS), som kan vara livshotande eller ha dödlig utgång, har rapporterats hos vuxna patienter vid kombinationsbehandling med trametinib och dabrafenib. Innan behandling påbörjas ska patienterna informeras om tecken och symtom och övervakas noga för hudreaktioner. Om tecken och symtom som tyder på SCAR uppträder ska behandlingen sättas ut.
Rabdomyolys
Rabdomyolys har rapporterats hos vuxna patienter som tar trametinib. I vissa fall kunde patienterna fortsätta behandlingen med trametinib. I mer allvarliga fall krävdes sjukhusvistelse, behandlingsavbrott eller permanent utsättning av behandlingen. Tecken och symtom på rabdomyolys ska utgöra grund för lämplig klinisk utredning och behandling enligt indikation.
Pankreatit
Pankreatit har rapporterats hos vuxna och pediatriska patienter som behandlats med trametinib i kombination med dabrafenib i kliniska studier (se avsnitt Biverkningar). Oförklarliga buksmärtor ska undersökas skyndsamt och inkludera mätning av serumamylas och -lipas. Patienterna ska följas noga när behandlingen återinsätts efter en pankreatitepisod.
Njursvikt
Njursvikt har identifierats hos ≤ 1 % av de vuxna patienterna som behandlades med trametinib i kombination med dabrafenib. Observerade fall hos vuxna patienter förknippades i allmänhet med feber och dehydrering och svarade väl på doseringsavbrott och allmän stödjande behandling. Granulomatös nefrit har också rapporterats hos vuxna patienter. Patienternas serumkreatinin ska rutinmässigt övervakas då de är under behandling. Om kreatininet ökar kan det vara kliniskt lämpligt att avbryta behandlingen. Trametinib har inte studerats hos patienter med njurinsufficiens (definierat som kreatinin > 1,5 x ULN) och ska därför hanteras med stor försiktighet under dessa omständigheter (se avsnitt Farmakokinetiska egenskaper).
Leverpåverkan
Leverbiverkningar har rapporterats hos vuxna och pediatriska patienter i kliniska studier med trametinib i kombination med dabrafenib (se avsnitt Biverkningar). Kontroll av leverfunktionen rekommenderas var fjärde vecka under 6 månader från behandlingsstarten. Därefter kan leverkontroller göras efter kliniskt behov.
Nedsatt leverfunktion
Eftersom metabolism och biliär utsöndring är de viktigaste elimineringsvägarna för trametinib ska administrering av trametinib ske med försiktighet till patienter med måttligt till kraftigt nedsatt leverfunktion (se avsnitt Dosering och administreringssätt och Farmakokinetiska egenskaper).
Djup ventrombos/lungemboli
Lungemboli eller djup ventrombos kan förekomma. Om patienterna utvecklar symtom på djup ventrombos eller lungemboli såsom andnöd, bröstsmärta eller arm- eller bensvullnad ska de omedelbart söka sjukvård. Behandlingen ska sättas ut permanent vid livshotande lungemboli.
Gastrointestinala störningar
Kolit och enterokolit har rapporterats hos pediatriska patienter som behandlats med trametinib i kombination med dabrafenib (se avsnitt Biverkningar). Kolit och gastrointestinal perforation, inklusive fall med dödlig utgång, har rapporterats hos vuxna patienter. Trametinib ska användas med försiktighet hos patienter med riskfaktorer för gastrointestinal perforation, som omfattar anamnes på divertikulit, metastaser till magtarmkanalen och samtidig användning av läkemedel med en erkänd risk för gastrointestinal perforation.
Sarkoidos
Fall av sarkoidos har rapporterats hos vuxna patienter som behandlats med trametinib i kombination med dabrafenib, oftast i hud, lungor, ögon och lymfkörtlar. I de flesta fallen fortsatte behandlingen med trametinib och dabrafenib. Om sarkoidos fastställs ska lämplig behandling övervägas.
Fertila kvinnor/fertilitet hos män
Innan behandling sätts in hos fertila kvinnor ska lämplig rådgivning om effektiva preventivmetoder ges. Fertila kvinnor måste använda effektiva preventivmetoder under behandlingen och i 16 veckor efter den sista dosen av Spexotras. Manliga patienter som tar trametinib i kombination med dabrafenib ska informeras om den potentiella risken för försämrad spermatogenes, som kan vara irreversibel (se avsnitt Fertilitet, graviditet och amning).
Hemofagocyterande lymfohistiocytos
Efter godkännandet för försäljning har hemofagocyterande lymfohistiocytos (HLH) observerats hos vuxna patienter som behandlats med trametinib i kombination med dabrafenib. Försiktighet ska iakttas när trametinib administreras i kombination med dabrafenib. Om HLH bekräftas ska administreringen av trametinib och dabrafenib avbrytas och behandling av HLH inledas.
Tumörlyssyndrom (TLS)
Förekomsten av TLS, som kan vara dödlig, har associerats med användning av trametinib i kombination med dabrafenib (se avsnitt Biverkningar). Riskfaktorer för TLS inkluderar hög tumörbörda, redan existerande kronisk njurinsufficiens, oliguri, uttorkning, hypotoni och sur urin. Patienter med riskfaktorer för TLS ska övervakas noggrant och profylaktisk hydrering ska övervägas. TLS ska behandlas omedelbart, enligt klinisk indikation.
Hjälpämnen
Sulfobutylbetadexnatrium
Spexotras oral lösning innehåller cyklodextrinet sulfobutylbetadexnatrium (100 mg/ml). Cyklodextriner (CD) är hjälpämnen som kan påverka egenskaperna (så som toxicitet eller hudpenetration) av aktiv substans eller andra läkemedel. I prekliniska studier på djur som gavs CD intravenöst, observerades njurtoxicitet och ototoxicitet. Säkerhetsaspekter för CD har övervägts under utveckling och utredning av säkerhet för läkemedlet. Det finns begränsade säkerhetsdata om effekterna av CD hos barn < 2 år.
Metylparahydroxibensoat
Detta läkemedel innehåller metylparahydroxibensoat, som kan ge allergisk reaktion (eventuellt fördröjd).
Natrium
Detta läkemedel innehåller 1,98 mg natrium per ml Spexotras oral lösning. Detta motsvarar 4 % av WHO:s högsta rekommenderat dagligt intag av 2 g natrium för vuxna vid den högsta dagliga dosen trametinib på 2 mg (40 ml).
Kalium
Detta läkemedel innehåller mindre än 1 mmol (39 mg) kalium per högsta dagliga dos, d.v.s. är näst intill ”kaliumfritt”.
Interaktioner
Interaktionsstudier har endast utförts på vuxna.
Andra läkemedels effekt på trametinib
Eftersom trametinib främst metaboliseras genom deacetylering som medieras av hydrolytiska enzymer (t.ex. karboxylesteraser) påverkas dess farmakokinetik sannolikt inte av andra substanser genom metabola interaktioner (se avsnitt Farmakokinetiska egenskaper). Interaktioner mellan läkemedel via dessa hydrolytiska enzymer kan inte uteslutas och kan påverka exponeringen för trametinib.
Trametinib är ett in vitro-substrat för effluxtransportören P-gp. Eftersom det inte kan uteslutas att en stark hämning av lever-P-gp kan leda till ökade nivåer av trametinib, rekommenderas försiktighet vid samtidig användning av trametinib med läkemedel som är starka hämmare av P-gp (t.ex. verapamil, ciklosporin, ritonavir, kinidin, itrakonazol).
Trametinibs effekt på andra läkemedel
Baserat på in vitro- och in vivo-data är det osannolikt att trametinib skulle ha någon signifikant inverkan på farmakokinetiken hos andra läkemedel genom interaktion med CYP-enzymer eller transportenzymer (se avsnitt Farmakokinetiska egenskaper). Trametinib kan orsaka en övergående hämning av BCRP-substrat (t.ex. pitavastatin) i tarmen, vilken kan minimeras genom att dosera dessa ämnen och trametinib med 2 timmars mellanrum.
Baserat på kliniska data förväntas ingen förlust av effekten av hormonella preventivmedel vid samtidig administrering med trametinib som monoterapi (se avsnitt Farmakokinetiska egenskaper). Dock kan användning i samtidigt med dabrafenib göra hormonella preventivmedel mindre effektiva.
Effekt av hjälpämnet sulfobutylbetadexnatrium på andra orala läkemedel med låg biotillgänglighet och smalt terapeutiskt index
Trametinib oral lösning innehåller 100 mg/ml sulfobutylbetadexnatrium som kan ha potential att påverka lösligheten och biotillgängligheten hos andra orala läkemedel. Försiktighet bör iakttas när trametinib oral lösning ges med orala läkemedel som har låg biotillgänglighet och ett smalt terapeutiskt index (t.ex. imipramin, desipramin).
Se även riktlinjerna för läkemedelsinteraktioner vid användning av dabrafenib i avsnitt Varningar och försiktighet och Interaktioner i produktresumén för dabrafenib dispergerbara tabletter.
Fertilitet, graviditet och amning
Fertila kvinnor/preventivmedel för kvinnor
Fertila kvinnor måste använda effektiva preventivmetoder under behandlingen med trametinib och i 16 veckor efter avslutad behandling.
Användning tillsammans med dabrafenib kan minska effekten av orala eller andra systemiska, hormonella preventivmedel och en effektiv, alternativ preventivmetod, t.ex. en barriärmetod, ska användas under kombinationsbehandling med trametinib/dabrafenib. Se produktresumén för dabrafenib dispergerbara tabletter för ytterligare information.
Graviditet
Det finns inga data gällande användning av trametinib till gravida kvinnor. Djurstudier har visat reproduktionstoxikologiska effekter (se avsnitt Prekliniska säkerhetsuppgifter). Trametinib ska inte ges till gravida kvinnor om inte den potentiella nyttan för modern överstiger den eventuella risken för fostret. Om trametinib används under graviditet, eller om patienten blir gravid under behandlingen med trametinib, ska patienten informeras om den eventuella risken för fostret.
Amning
Det är okänt om trametinib utsöndras i bröstmjölk. En risk för det ammade barnet kan inte uteslutas. Trametinib ska inte ges till kvinnor som ammar. Ett beslut måste fattas om man ska avbryta amningen eller avbryta behandlingen med trametinib efter att man tagit hänsyn till fördelen med amning för barnet och fördelen med behandling för kvinnan.
Fertilitet
Det finns inga data som gäller människa för trametinib. Inga fertilitetsstudier har utförts på djur, men påverkan sågs på reproduktionsorganen hos hondjur (se avsnitt Prekliniska säkerhetsuppgifter). Trametinib kan försämra fertiliteten hos människa.
Män som tar trametinib i kombination med dabrafenib
Påverkan på spermatogenesen har observerats hos djur som fått dabrafenib. Manliga patienter som tar trametinib i kombination med dabrafenib ska informeras om den potentiella risken för försämrad spermatogenes, som kan vara irreversibel. Se produktresumén för dabrafenib dispergerbara tabletter för ytterligare information.
Effekter på förmågan att framföra fordon och använda maskiner
Trametinib har mindre effekt på förmågan att framföra fordon och använda maskiner. Patientens kliniska status och trametinibs biverkningsprofil ska beaktas vid bedömning av patientens förmåga att utföra uppgifter som kräver omdöme, motorisk eller kognitiv förmåga. Patienterna ska uppmärksammas på risken för att trötthet, yrsel och ögonproblem kan inverka på dessa aktiviteter.
Biverkningar
Sammanfattning av säkerhetsprofilen
I kliniska studier med pediatriska patienter som behandlades med trametinib i kombination med dabrafenib var de vanligaste biverkningarna (rapporterade med en frekvens på ≥ 20 %) feber (70 %), hudutslag (49 %), huvudvärk (47 %), kräkningar (40 %), trötthet (36 %), torr hud (35 %), diarré (34 %), blödningar (34 %), illamående (29 %), akneiform dermatit (29 %), buksmärtor (28 %), neutropeni (26 %), hosta (24 %) och förhöjda transaminaser (22 %). De vanligaste allvarliga biverkningarna (grad 3/4) var neutropeni (15 %), feber (11 %), förhöjda transaminaser (6 %) och viktökning (5 %). Långtidsdata vad gäller tillväxt och benmognad hos pediatriska patienter är för närvarande begränsade (se avsnitt Prekliniska säkerhetsuppgifter).
Säkerhetsprofilen för pediatriska patienter var i stort sett densamma som den som tidigare fastställts för vuxna patienter. Följande ytterligare biverkningar har hittills endast rapporterats hos vuxna patienter som behandlats med trametinibtabletter och dabrafenibkapslar: kutant skivepitelkarcinom, seborroisk keratos, perifer neuropati (inklusive sensorisk och motorisk neuropati), lymfödem, muntorrhet, aktinisk keratos, njursvikt, potentiering av strålningstoxicitet (vanliga), melanom, akrokordon, sarkoidos, korioretinopati, pneumonit, akut njursvikt, nefrit, hjärtsvikt, vänsterkammardysfunktion, interstitiell lungsjukdom, rabdomyolys (mindre vanliga), gastrointestinal perforation, hemofagocytisk lymfohistiocytos (sällsynta), tumörlyssyndrom, myokardit, Stevens-Johnsons syndrom, läkemedelsreaktion med eosinofili och systemiska symtom, tatueringsrelaterade hudreaktioner (ingen känd frekvens).
Tabell över biverkningar
Säkerheten för trametinib i kombination med dabrafenib har utvärderats i en poolad säkerhetspopulation bestående av 171 pediatriska patienter i två studier av patienter med avancerade solida tumörer med BRAF V600-mutation. Fyra patienter (2,3 %) var 1 till < 2 år gamla, 39 patienter (22,8 %) var 2 till < 6 år gamla, 54 patienter (31,6 %) var 6 till < 12 år gamla och 74 patienter (43,3 %) var 12 till < 18 år gamla vid rekryteringen. Genomsnittlig varaktighet var 2,3 år.
Biverkningar (tabell 5) redovisas nedan indelade efter MedDRA:s organsystem och efter frekvens enligt följande princip: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), sällsynta (≥ 1/10 000, < 1/1 000), mycket sällsynta (< 1/10 000) och ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom varje frekvensgrupp presenteras biverkningarna i fallande allvarlighetsgrad.
Tabell 5Biverkningar med trametinib i kombination med dabrafenib
| Infektioner och infestationer | |
| Mycket vanliga | Paronyki, nasofaryngit*1 |
| Vanliga | Urinvägsinfektion, cellulit |
| Neoplasier; benigna, maligna och ospecificerade (inkl. cystor och polyper) | |
| Mycket vanliga | Hudpapillom |
| Blodet och lymfsystemet | |
| Mycket vanliga | Neutropeni*2, anemi, leukopeni* |
| Vanliga | Trombocytopeni* |
| Immunsystemet | |
| Vanliga | Överkänslighet |
| Metabolism och nutrition | |
| Vanliga | Dehydrering, minskad aptit |
| Centrala och perifera nervsystemet | |
| Mycket vanliga | Huvudvärk, yrsel*3 |
| Ögon | |
| Vanliga | Dimsyn, synnedsättning, uveit*4 |
| Mindre vanliga | Näthinneavlossning, periorbitalt ödem |
| Hjärtat | |
| Vanliga | Minskad ejektionsfraktion, bradykardi* |
| Mindre vanliga | Atrioventrikulärt block5 |
| Blodkärl | |
| Mycket vanliga | Blödning*6 |
| Vanliga | Hypertoni, hypotoni |
| Andningsvägar, bröstkorg och mediastinum | |
| Mycket vanliga | Hosta* |
| Vanliga | Dyspné |
| Magtarmkanalen | |
| Mycket vanliga | Buksmärta*, förstoppning, diarré, illamående, kräkningar |
| Vanliga | Pankreatit, stomatit |
| Mindre vanliga | Kolit* |
| Hud och subkutan vävnad | |
| Mycket vanliga | Akneiform dermatit*7, torr hud*8, klåda, utslag*9, erytem |
| Vanliga | Generaliserad exfoliativ dermatit*10, alopeci, palmoplantar erytrodysestesi, follikulit, hudlesion, pannikulit, hyperkeratos, ljuskänslighet*11 |
| Mindre vanliga | Akut febril neutrofil dermatos, hudfissurer, nattliga svettningar, hyperhidros |
| Muskuloskeletala systemet och bindväv | |
| Mycket vanliga | Artralgi, smärta i extremiteter |
| Vanliga | Myalgi*, muskelspasmer*12 |
| Allmänna symtom och/eller symtom vid administreringsstället | |
| Mycket vanliga | Feber*, trötthet*13, viktökning |
| Vanliga | Slemhinneinflammation, ansiktsödem*, frossa, perifert ödem, influensaliknande sjukdom |
| Undersökningar | |
| Mycket vanliga | Förhöjda transaminaser*14 |
| Vanliga | Hyponatremi, hypofosfatemi, hyperglykemi, förhöjt alkaliskt fosfatas i blodet, förhöjt gammaglutamyltransferas, förhöjt kreatinfosfokinas i blodet |
*Indikerar sammanslagning av två eller fler MedDRA-termer som bedömts som kliniskt likartade. 1 nasofaryngit inkluderar faryngit 2 neutropeni inkluderar minskat antal neutrofiler samt febril neutropen 3 yrsel inkluderar vertigo 4 uveit inkluderar iridocyklit 5 atrioventrikulärt block inkluderar atrioventrikulärt block, första graden 6 blödning inkluderar epistaxis, hematuri, kontusion, hematom, förhöjt INR (international normalised ratio), analblödning, blödning vid kateterställe, cerebral blödning, ekkymos, extraduralt hematom, gastrointestinal blödning, hematochezi, petekier, blödning efter ingrepp, rektalblödning, minskat antal röda blodkroppar, blödning i övre magtarmkanalen, uterin blödning, kraftiga menstruationsblödningar samt purpura 7 akneiform dermatit inkluderar akne och pustulös akne 8 torr hud inkluderar xeros och xeroderma 9 utslag inkluderar makulopapulöst, pustulöst, erytematöst, papulöst och makulärt utslag 10 generaliserad exfoliativ dermatit inkluderar hudexfoliering och exfoliativ dermatit 11 ljuskänslighet inkluderar ljuskänslighetsreaktion och solbränna 12 muskelspasmer inkluderar muskuloskeletal stelhet 13 trötthet inkluderar sjukdomskänsla och asteni 14 förhöjda transaminaser inkluderar förhöjt aspartataminotransferas (ASAT), förhöjt alaninaminotransferas (ALAT) och hypertransaminasemi | |
Beskrivning av utvalda biverkningar
Viktökning
Viktökning har endast rapporterats i den pediatriska populationen. Den rapporterades som en biverkning hos 16 % av de pediatriska patienterna, med svårighetsgrad 3 hos 5 % av patienterna, och med en utsättningsfrekvens på 0,6 % av patienterna. Mediantid till den första rapporterade viktökningen hos pediatriska patienter som fick trametinib i kombination med dabrafenib var 3,5 månader. Viktökning från baslinjen på ≥ 2 BMI (kroppsmasseindex) för ålderspercentilen observerades hos 36 % av patienterna.
Blödning
Blödningar har observerats hos 34 % av de pediatriska patienterna, med svårighetsgrad 3 hos 1,2 % av patienterna. Den vanligaste typen av blödning (epistaxis) rapporterades hos 18 % av de pediatriska patienterna. Mediantid till första blödningen hos pediatriska patienter var 2,6 månader. Blödningar, inklusive större blödningar och blödningar med dödlig utgång, har förekommit hos vuxna patienter som tar trametinib i kombination med dabrafenib.
Blödningsrisken kan vara förhöjd vid samtidig användning av trombocythämmare eller antikoagulantia. Vid blödning ska patienten behandlas enligt kliniska indikationer (se avsnitt Varningar och försiktighet).
Reducerad vänsterkammarejektionsfraktion (LVEF)/vänsterkammardysfunktion
Reducerad LVEF har rapporterats hos 5,3 % av de pediatriska patienterna, med svårighetsgrad 3 hos < 1 % av patienterna. Mediantiden till första LVEF-reduceringen var cirka en månad. I kliniska studier på vuxna patienter var mediantiden till debut av första förekomsten av vänsterkammardysfunktion, hjärtsvikt och reducerad LVEF mellan 2 och 5 månader.
Patienter med LVEF som understeg institutionellt lägsta normalvärde inkluderades inte i kliniska studier med trametinib. Trametinib i kombination med dabrafenib ska användas med försiktighet till patienter med tillstånd som kan försämra vänsterkammarfunktionen (se avsnitt Dosering och administreringssätt och Varningar och försiktighet).
Feber
Feber har rapporterats i kliniska studier med trametinib som monoterapi och i kombination med dabrafenib; förekomsten och allvarlighetsgraden av feber ökade emellertid med kombinationsbehandlingen (se avsnitt Varningar och försiktighet). Feber rapporterades hos 70 % av de pediatriska patienterna, med svårighetsgrad 3 hos 11 % av patienterna. Se avsnitt Biverkningar i produktresumén för dabrafenib dispergerbara tabletter.
Leverpåverkan
Leverbiverkningar har rapporterats hos vuxna och pediatriska patienter i kliniska studier med trametinib i kombination med dabrafenib. I den pediatriska säkerhetspopulationen var förhöjt ALAT och ASAT mycket vanligt, rapporterat hos 13 respektive 16 % av patienterna (se avsnitt Varningar och försiktighet). Ökat ALAT och ASAT var de vanligaste leverbiverkningarna hos vuxna patienter och majoriteten av dessa var antingen av grad 1 eller 2. Vid monoterapi med trametinib inträffade mer än 90 % av dessa fall av leverpåverkan inom de första 6 månaderna av behandlingen. Leverpåverkan upptäcktes i kliniska studier där provtagning gjordes var fjärde vecka. Vid behandling med trametinib som monoterapi eller i kombination med dabrafenib rekommenderas det att kontrollera patientens leverfunktion var fjärde vecka under 6 månader. Därefter kan provtagning av leverfunktion fortgå om detta är kliniskt indicerat (se avsnitt Varningar och försiktighet).
Förändrat blodtryck
Hypertoni har rapporterats hos 2,3 % av pediatriska patienter, med svårighetsgrad 3 hos 1,2 % av patienterna. Mediantiden till första tillfället av hypertoni hos pediatriska patienter var 5,4 månader.
Hypotoni har rapporterats hos 4,1 % av de pediatriska patienterna, med svårighetsgrad ≥ 3 hos 2,3 % av patienterna. Mediantiden till första tillfället av hypotoni hos pediatriska patienter var 2,2 månader.
Blodtrycket ska mätas vid baslinjen och kontrolleras under behandling, med standardbehandling för att hålla hypertonin under kontroll efter behov (se avsnitt Varningar och försiktighet).
Interstitiell lungsjukdom (ILD)/pneumonit
Patienter behandlade med trametinib kan utveckla ILD eller pneumonit. Behandlingen ska avbrytas hos patienter med misstänkt ILD eller pneumonit, inklusive patienter med nya eller progressiva pulmonella symtom och symtom som hosta, dyspné, hypoxi, pleurautgjutning eller infiltrat, i avvaktan på kliniska undersökningar. Behandlingen med trametinib ska sättas ut permanent för patienter med behandlingsrelaterad ILD eller pneumonit (se avsnitt Dosering och administreringssätt och Varningar och försiktighet).
Synnedsättning
Oftalmologiska reaktioner, inklusive uveit hos 3,5 % och iridocyklit hos 1,8 %, har rapporterats hos pediatriska patienter som behandlats med trametinib i kombination med dabrafenib. Uveit grad 3 förekom hos 1,8 % av de pediatriska patienterna. Näthinneavlossning (RPED) förekom hos < 1 % av de pediatriska patienterna. Sjukdomar förknippade med synnedsättning, inklusive RPED och RVO, har även observerats med trametinib hos vuxna patienter. Symtom som dimsyn, nedsatt synskärpa och andra synstörningar har rapporterats i kliniska studier på vuxna som fått trametinib (se avsnitt Dosering och administreringssätt och Varningar och försiktighet).
Hudutslag
Hudutslag har observerats hos 49 % av de pediatriska patienterna i studier av kombinationsbehandling med trametinib och dabrafenib i den integrerade säkerhetspopulationen. Majoriteten av dessa fall var av grad 1 eller 2 och krävde inte behandlingsavbrott eller dossänkning (se avsnitt Dosering och administreringssätt och Varningar och försiktighet).
Rabdomyolys
Rabdomyolys har rapporterats hos vuxna patienter som tar trametinib. Tecken eller symtom på rabdomyolys ska utgöra grund för lämplig klinisk bedömning och behandling vid behov (se avsnitt Varningar och försiktighet).
Pankreatit
Pankreatit rapporterades hos 1,2 % av de pediatriska patienterna, med svårighetsgrad 3 hos < 1 % av patienterna. Oförklarliga buksmärtor ska undersökas skyndsamt och omfatta mätning av serumamylas och -lipas. Patienterna ska följas upp noga när behandlingen återinsätts efter en pankreatitepisod (se avsnitt Varningar och försiktighet).
Njursvikt
Njursvikt har rapporterats med trametinib i kombination med dabrafenib. Njursvikt orsakad av feberorsakad prerenal azotemi eller granulomatös nefrit var mindre vanligt hos vuxna patienter. Trametinib har dock inte studerats hos patienter med nedsatt njurfunktion (definierad som kreatinin > 1,5 x ULN). Försiktighet ska iakttas under dessa förutsättningar (se avsnitt Varningar och försiktighet).
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Inga akuta symtom relaterade till överdosering har rapporterats hos pediatriska patienter som fått trametinib i kombination med dabrafenib i kliniska studier. Ihållande överdosering av trametinib kan resultera i ökade hudutslag, minskad LVEF eller retinala abnormiteter. Det finns ingen specifik behandling av överdosering. Vid överdosering ska patienten ges understödjande vård med lämplig övervakning efter behov.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Antineoplastiska medel, proteinkinashämmare, mitogen-aktiverade proteinkinas (MEK) hämmare, ATC-kod: L01EE01
Verkningsmekanism
Trametinib är en reversibel, starkt selektiv, alloster hämmare av aktiveringen av mitogenaktiverat extracellulärt signalreglerat kinas 1 (MEK1) och MEK2 och kinasaktivitet. MEK-proteiner är komponenter i signalvägen för extracellulärt signalreglerat kinas (ERK). Vid cancer hos människa är denna signalväg ofta aktiverad av muterade BRAF-former som aktiverar MEK. Trametinib hämmar BRAF:s aktivering av MEK och hämmar aktiviteten hos MEK-kinas.
Kombination med dabrafenib
Dabrafenib är en RAF-kinashämmare. Onkogena mutationer i BRAF leder till konstitutiv aktivering av RAS-/RAF-/MEK-/ERK-signaleringsvägen.
Sålunda inhiberar trametinib och dabrafenib två kinaser i denna väg, MEK och RAF och därför ger kombinationen samtidig hämning av signalvägen. Kombinationen av trametinib och dabrafenib har visat antitumöraktivitet i BRAF V600-mutationspositiva cancercellinjer in vitro och försenar uppkomsten av resistens in vivo i BRAF V600-mutationspositiva xenografter.
Klinisk effekt och säkerhet
Pediatrisk population
Klinisk effekt och säkerhet för kombinationsbehandling med dabrafenib plus trametinib hos pediatriska patienter i åldern 1 till < 18 år med BRAF V600-mutationspositivt gliom har utvärderats i en öppen, klinisk multicenterstudie i fas 2 (EudraCT 2015-004015-20). Patienter med låggradigt gliom (grad 1 eller 2 enligt WHO 2016) som behövde en första systemisk behandling randomiserades i förhållandet 2:1 till dabrafenib plus trametinib eller karboplatin plus vinkristin, medan patienter med recidiverat eller refraktärt höggradigt gliom (grad 3 eller 4 enligt WHO 2016) placerades i en kohort med en singelarm som fick dabrafenib plus trametinib.
BRAF-mutationsstatus fastställdes prospektivt i tumörvävnad genom ett lokalt test eller om ett lokalt test inte var möjligt, med hjälp av bioMérieux THxID-BRAF-test utfört av centrallaboratorium. Dessutom utfördes retrospektiva tester av tillgängliga tumörprover av centrallaboratorium för att bekräfta BRAF V600E-mutation.
Dosen av dabrafenib och trametinib i den kliniska studien var beroende av ålder och vikt. Dabrafenib gavs peroralt med en dos på 2,625 mg/kg två gånger dagligen till åldersgruppen < 12 år och med 2,25 mg/kg två gånger dagligen till åldersgruppen 12 år och äldre. Trametinib gavs peroralt med dosen 0,032 mg/kg en gång dagligen till åldersgruppen < 6 år och med 0,025 mg/kg en gång dagligen till åldersgruppen 6 år och äldre. Dabrafenibdoserna begränsades till 150 mg två gånger dagligen och trametinibdoserna till 2 mg en gång dagligen. Karboplatin och vinkristin doserades baserat på ålder och kroppsyta och gavs som veckovis infusion med dosen 175 mg/m2 respektive 1,5 mg/m2. Karboplatin och vinkristin administrerades som en 10-veckors induktionsbehandling med efterföljande underhållsbehandling i åtta 6-veckorscykler.
Primärt effektmått i båda kohorterna var total responsfrekvens (ORR, summan av bekräftad komplett respons [CR] och partiell respons [PR]), bedömt genom oberoende granskning baserat på RANO-kriterierna (2017) för LGG-kohorten och RANO-kriterierna (2010) för HGG-kohorten. Den primära analysen utfördes när samtliga patienter i båda kohorterna hade fått minst 32 veckors behandling. Den slutliga analysen utfördes 2 år efter avslutad registrering av deltagande i båda kohorterna.
BRAF-mutationspositivt pediatriskt låggradigt gliom (WHO grad 1 och 2)
I kohorten med låggradigt gliom randomiserades 110 patienter till dabrafenib plus trametinib (n = 73) eller karboplatin plus vinkristin (n = 37). Medianåldern var 9,5 år, med 34 patienter (30,9 %) i åldern 12 månader till < 6 år, 36 patienter (32,7 %) i åldern 6 till < 12 år och 40 patienter (36,4 %) i åldern 12 till < 18 år. 60 % var flickor. Majoriteten (80 %) hade gliom av grad 1 när första diagnosen ställdes. De vanligaste patologierna var astrocytom (30,9 %), gangliogliom (27,3 %) och LGG utan närmare specifikation (NOS) (18,2 %). Metastaser fanns hos 9 patienter (8,2 %). 91 patienter (82,7 %) hade tidigare opererats, hos 28 av dessa patienter (25,5 %) var den senaste operationen resektion. Systemiska kortikosteroider användes av 44 patienter (41,5 %).
Vid tidpunkten för den primära analysen, visade ORR i dabrafenib plus trametinib-armen en statistiskt signifikant förbättring som översteg karboplatin plus vinkristin. Efterföljande hierarkisk testning visade också på en statistiskt signifikant förbättring vad gällde progressionsfri överlevnad (PFS) jämfört med kemoterapi (tabell 6).
Vid tidpunkten för den primära analysen, som utfördes när samtliga patienter hade genomgått minst 32 veckors behandling eller hade avbrutit tidigare, var data för total överlevnad (OS) fortfarande inte mogna (ett dödsfall hade rapporterats i karboplatin- plus vinkristin-armen [C+V]).
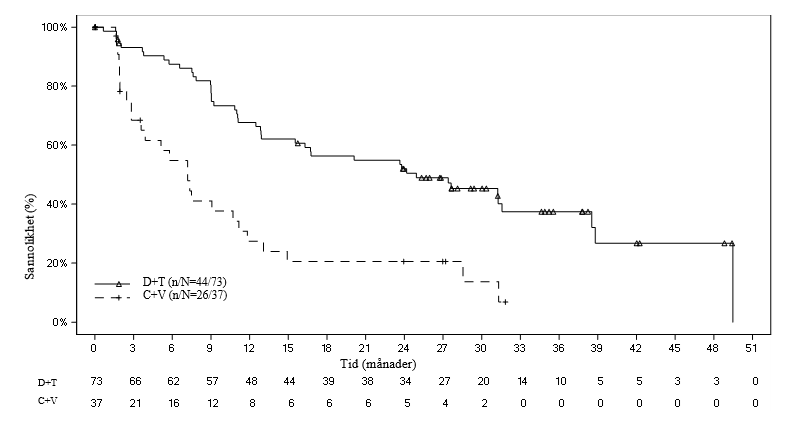
Tabell 6 Respons och progressionsfri överlevnad baserat på oberoende granskning i den pivotala studien G2201 (LGG-kohort, primär analys)
Dabrafenib + trametinib (D+T) N = 73 | Karboplatin + vinkristin (C+V) N = 37 | |
| Bästa totala respons | ||
| Komplett respons (CR), n (%) | 2 (2,7) | 1 (2,7) |
| Partiell respons (PR), n (%) | 32 (43,8) | 3 (8,1) |
| Stabil sjukdom (SD), n (%) | 30 (41,1) | 15 (40,5) |
| Progressiv sjukdom (PD), n (%) | 8 (11,0) | 12 (32,4) |
| Okänd, n (%) | 1 (1,4) | 6 (16,2)1 |
| Total responsfrekvens | ||
| ORR (CR+PR), (95 % CI) | 46,6 % (34,8-58,6 %) | 10,8 % (3,0-25,4 %) |
| Oddskvot2, p-värde | 7,19 (2,3-22,4); p < 0,001 | |
| Riskskillnad | 35,8 % (20,6-51,0) | |
| Progressionsfri överlevnad (PFS) | ||
| Median (månader), (95 % CI) | 20,1 (12,8-NE) | 7,4 (3,6-11,8) |
| Riskkvot (95 % CI), p-värde | 0,31 (0,17-0,55); p < 0,001 | |
NE = kan ej uppskattas 1 4 patienter randomiserade till C+V avbröt innan de fått behandling. 2 Oddskvot (D+T vs C+V) och 95 % CI beräknad genom logistisk regression med behandling som enda kovariat, dvs. oddsen för observation av respons i D+T-armen jämfört med oddsen för observation av respons i C+V-armen. Oddskvot > 1 innebär fördel för D+T. | ||
Vid tidpunkten för den slutliga analysen (medianlängd för uppföljning: 39,0 månader) var ORR baserad på oberoende granskning 54,8 % i D+T-armen och 16,2 % i C+V-armen med en oddskvot på 6,26. Analysen bekräftade också förbättrad PFS jämfört med kemoterapi baserat på oberoende granskning med en uppskattad 64 % riskreduktion i progression/död (riskkvot 0,36). Median PFS var 24,9 månader i D+T-armen och 7,2 månader i C+V-armen. Inga ytterligare dödsfall rapporterades i någon av armarna vid tidpunkten för den slutliga analysen.
Figur 1 Kaplan–Meier-kurvor för progressionsfri överlevnad baserade på oberoende granskning i den pivotala studien G2201 (LGG-kohort, slutlig analys)
BRAF-mutationspositivt pediatriskt höggradigt gliom (WHO grad 3 och 4)
Till kohorten med en singelarm med höggradigt gliom rekryterades 41 patienter med recidiverat eller refraktärt HGG och behandlades med dabrafenib plus trametinib. Medianåldern var 13,0 år, med 5 patienter (12,2 %) i åldern 12 månader till < 6 år, 10 patienter (24,4 %) i åldern 6 till < 12 år och 26 patienter (63,4 %) i åldern 12 till < 18 år. 56 % var flickor. Histologisk grad när diagnosen först ställdes var grad 4 hos 20 patienter (48,8 %), grad 3 hos 13 patienter (31,7 %), grad 2 hos 4 patienter (9,8 %), grad 1 hos 3 patienter (7,3 %) och saknades hos 1 patient (2,4 %). De vanligaste patologierna var glioblastoma multiforme (31, 7%), anaplastiskt pleomorft xantoastrocytom (14,6 %), HGG NOS (9,8 %) och pleomorft xantoastrocytom (9,8 %). Tidigare kirurgi rapporterades hos 40 patienter (97,6 %), hos 24 av dessa (58,5 %) var den senaste operationen resektion. 33 patienter (80,5 %) hade tidigare fått antineoplastisk kemoterapi och 37 patienter (90,2 %) hade tidigare strålbehandlats. Systemiska kortikosteroider under studiebehandlingen rapporterades för 24 patienter (58,5 %).
Vid tidpunkten för den slutliga analysen (medianlängd för uppföljning: 45,2 månader) var ORR baserat på oberoende granskning 56,1 % (23/41), (95 %CI: 39,7; 71,5): CR hos 14 patienter (34,1 %) och PR hos 9 patienter (22,0 %). Medianvärde för responsduration (DOR) var 27,4 månader (95 % CI: 9,2; NE).
Farmakokinetiska egenskaper
Trametinibs farmakokinetiska egenskaper har till största delen fastställts för vuxna patienter med användning av läkemedlet i fast form (tablett). Trametinibs farmakokinetik efter viktanpassad engångs- eller upprepad dosering har dessutom utvärderats hos 244 pediatriska patienter. Farmakokinetiska egenskaper (läkemedelsabsorption och -clearance) för trametinib hos pediatriska patienter var jämförbara med dem hos vuxna. Kroppsvikten befanns påverka oral clearance av trametinib, medan ålder inte hade någon effekt. Farmakokinetisk exponering för trametinib vid den rekommenderade viktanpassade dosen till pediatriska patienter låg inom samma intervall som hos vuxna.
Absorption
Trametinib oral lösning absorberades snabbt med en mediantid till högsta plasmakoncentration (Tmax) på 1 timme efter dosen. Genomsnittlig absolut oral biotillgänglighet för trametinibtabletter var 72 %. I en studie av relativ biotillgänglighet där man jämförde oral lösning med tabletter efter en engångsdos till fastande vuxna, resulterade administrering av oral lösning i 12 % högre AUC(0-inf), 10 % högre AUC(0-last) och 71 % högre Cmax än tabletterna.
Exponeringen för trametinib ökade på ett dosproportionellt sätt mellan 0,125 mg och 4 mg efter upprepad dosering en gång dagligen.
I den pivotala pediatriska studien var geometriskt medelvärde (%CV) för Cmax och AUCtau vid steady state 22,7 ng/ml (41,1 %) respektive 339 ng*h/ml (22,2 %) i LGG-kohorten och 21,3 ng/ml (36,3 %) respektive 307 ng*h/ml (22,8 %) i HGG-kohorten.
Trametinib ackumuleras vid upprepad daglig dosering. En genomsnittlig ackumuleringskvot på 6,0 observerades för tablettformuleringen vid doseringen 2 mg en gång dagligen. Steady state uppnåddes dag 15.
Effekt av föda
Administrering av en engångsdos på 2 mg av trametinib oral lösning tillsammans med en måltid med låg fetthalt och lågt kaloriinnehåll resulterade i en 12 % minskning av Cmax jämfört med fastande förhållanden, vilket inte anses vara kliniskt signifikant. AUClast förblev oförändrad.
Distribution
Trametinibs plasmaproteinbindningsgrad hos människa är 97,4 %. Trametinib har en distributionsvolym på cirka 1 200 l, fastställt efter administrering av 5 mikrogram som intravenös mikrodos.
Metabolism
In vitro- och in vivo-studier har visat att trametinib främst metaboliseras genom deacetylering, enbart eller med samtidig mono-oxygenering. Den deacetylerade metaboliten metaboliseras ytterligare genom glukuronidering. CYP3A4-oxidation bedöms vara en mindre viktig metaboliseringsväg. Deacetyleringen medieras av karboxylesterasen 1b, 1c och 2 och med eventuella bidrag av andra hydrolytiska enzymer.
Efter engångs- och upprepade doser trametinib är trametinib som modersubstans den huvudsakliga cirkulerande komponenten i plasma.
Eliminering
Genomsnittlig terminal halveringstid för trametinib är 127 timmar (5,3 dagar) efter administrering av en engångsdos. Skenbar clearance av trametinib hos pediatriska patienter (mediankroppsvikt: 32,85 kg) var 3,44 l/h (CV 20 %).
På grund av den långa halveringstiden var den totala dos som återfanns efter 10 dagars uppsamling låg (< 50 %) efter administrering av en peroral engångsdos av radiomärkt trametinib i lösning. Trametinibrelaterat material utsöndrades huvudsakligen i feces (> 80 % av den återfunna radioaktiviteten) och i mindre utsträckning i urinen (< 19 %). Mindre än 0,1 % av den utsöndrade dosen återfanns som modersubstans i urinen.
Läkemedelsinteraktioner
Trametinibs effekt på enzymer som bryter ner läkemedel och transportproteiner
Data in vitro och in vivo tyder på att trametinib sannolikt inte påverkar farmakokinetiken för andra läkemedel. Baserat på in vitro-studier är trametinib inte en hämmare av CYP1A2, CYP2A6, CYP2B6, CYP2D6 eller CYP3A4. Baserat på in vitro-studier är trametinib en hämmare av CYP2C8, CYP2C9 och CYP2C19, en inducerare av CYP3A4 samt en hämmare av transportproteinerna OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, P-gp och BCRP. Baserat på den låga dosen och den låga kliniska systemiska exponeringen i jämförelse med värdena för in vitro-hämning eller -induktion, anses trametinib inte hämma dessa enzymer eller transportproteiner in vivo, även om en tillfällig hämning av BCRP-substrat i tarmen kan förekomma (se avsnitt Interaktioner).
Andra läkemedels effekt på trametinib
Data in vitro och in vivo tyder på att trametinibs farmakokinetik sannolikt inte påverkas av andra läkemedel. Trametinib är inte ett substrat för CYP-enzymer eller transportörerna P-gp eller BCRP, OATP1B1, OATP1B3, OATP2B1, OCT1, MRP2 och MATE1. Trametinib är ett in vitro-substrat för BSEP och effluxtransportören P-gp. Även om trametinibexponering är osannolikt att påverkas av hämning av BSEP, kan ökade nivåer av trametinib inte uteslutas vid stark hämning av lever-P-gp (se avsnitt Interaktioner).
Trametinibs effekt på andra läkemedel:
Effekten av upprepad dosering med trametinib på farmakokinetiken vid steady state av kombinerade p-piller, noretindron och etinylestradiol, utvärderades i en klinisk studie som bestod av 19 kvinnliga patienter med solida tumörer. Exponeringen av noretindron ökade med 20 % och exponeringen för etinylestradiol var liknande vid samtidig administrering med trametinib. Baserat på dessa resultat förväntas ingen förlust av effekten av hormonella preventivmedel vid samtidig administrering med trametinib.
Särskilda patientpopulationer
Nedsatt leverfunktion
Populationsfarmakokinetiska analyser och data från en klinisk farmakologisk studie på vuxna patienter med normal leverfunktion eller med milda, måttliga eller kraftiga bilirubin- och/eller ASAT-förhöjningar (baserat på National Cancer Institute [NCI]-klassificering) visar att leverfunktionen inte har någon signifikant inverkan på oral clearance av trametinib.
Nedsatt njurfunktion
Nedsatt njurfunktion har sannolikt ingen kliniskt relevant effekt på farmakokinetiken för trametinib, med tanke på den låga renala utsöndringen av trametinib. Trametinibs farmakokinetik har beskrivits i kliniska studier där populationsfarmakokinetiska analyser utfördes på 223 vuxna patienter med lätt nedsatt njurfunktion och 35 vuxna patienter med måttligt nedsatt njurfunktion. Lätt och måttligt nedsatt njurfunktion hade ingen effekt på trametinibexponeringen (< 6 % för båda grupperna). Det finns inga data för patienter med kraftigt nedsatt njurfunktion (se avsnitt Dosering och administreringssätt).
Etnisk tillhörighet
Data är otillräckliga för bedömning av om etnisk tillhörighet har någon inverkan på trametinibs farmakokinetik. De kliniska erfarenheterna gäller endast kaukasier.
Kön
I populationsfarmakokinetiska analyser av vuxna och pediatriska patienter fann man att kön påverkar oral clearance av trametinib. Även om kvinnliga patienter förmodas exponeras i högre omfattning än manliga patienter har dessa skillnader sannolikt ingen klinisk relevans och utgör inte grund för någon dosjustering.
Prekliniska säkerhetsuppgifter
Trametinib har inte studerats med avseende på karcinogenicitet. Trametinib var inte gentoxiskt i studier där man undersökte bakteriell återmutation, kromosomavvikelser hos däggdjursceller och mikronuklei i benmärgen hos råtta.
Trametinib kan försämra fertiliteten hos kvinnor. I studier med upprepad dosering sågs ökat antal cystiska folliklar och färre gulkroppar hos honråttor vid exponeringar som låg under den kliniska exponeringen hos människa, baserat på AUC.
Dessutom, hos unga råttor som gavs trametinib, har minskad äggstocksvikt, mindre förseningar i kännetecken på könsmognad hos hondjur (slidöppning och ökad förekomst av framträdande slutanlag i bröstkörteln) och lätt hypertrofi av ytepitelet i livmodern observerats. Alla dessa effekter var reversibla efter en period utan behandling och kan tillskrivas farmakologi. I toxikologiska studier på råtta och hund, som varade i upp till 13 veckor, sågs dock inga behandlingseffekter på reproduktionsvävnad hos handjur.
I studier av embryo-fosterutveckling på råtta och kanin inducerade trametinib toxiska effekter på moderdjur och fosterutveckling. Hos råtta sågs lägre fostervikt och ökade förluster efter implantation vid exponeringar under eller något över kliniska exponeringar hos människa, baserat på AUC. I en toxicitetsstudie med embryo-fosterutveckling på kaniner observerades minskad fostervikt, ökat antal aborter, ökad frekvens ofullständig benbildning samt skelettmissbildningar vid subkliniska exponeringar, baserat på AUC.
I studier med upprepad dosering ses effekter av trametinibexponeringen främst i huden, magtarmkanalen, hematologiska systemet, skelett och lever. De flesta av dessa är reversibla och går tillbaka efter en behandlingsfri period. Hos råtta sågs hepatocellulär nekros och förhöjda transaminaser efter 8 veckor med dosen ≥ 0,062 mg/kg/dag (omkring 0,8 gånger den kliniska exponeringen hos människa, baserat på AUC).
Hos möss observerades lägre hjärtfrekvens, lägre hjärtvikt och försämrad vänsterkammarfunktion, utan kardiell histopatologi, efter 3 veckor med trametinib ≥ 0,25 mg/kg/dag (omkring 3 gånger den kliniska exponeringen hos människa, baserat på AUC), i upp till 3 veckor. Hos vuxna råttor var mineralisering av flera organ associerad med förhöjt serumfosfat och nära relaterat till nekros i hjärta, lever och njurar samt blödning i lungorna, vid exponeringar som är jämförbara med den kliniska exponeringen hos människa. Hos råtta sågs hypertrofi av epifysplattan och ökad benbildning. Hos råtta och hund som fick trametinib vid eller under klinisk exponering hos människa observerades benmärgsnekros, atrofi av lymfatisk vävnad i tymus och GALT (gut associated lymphatic tissue) samt nekros av lymfatisk vävnad i lymfkörtlar, mjälte och tymus, vilket skulle kunna försvaga immunsystemet. Hos unga råttor, observerades ökad hjärtvikt utan histopatologi vid 0,35 mg/kg/dag (ungefär två gånger den kliniska exponeringen hos människa, baserat på AUC).
I en 3T3 Neutral Red Uptake (NRU) analys på musfibroblast in vitro uppvisade trametinib fototoxicitet vid signifikant högre koncentrationer än vad som används vid klinisk exponering hos människa (IC50 på 2,92 mikrogram/ml, ≥ 130 gånger den kliniska exponeringen hos människa baserat på Cmax). Detta indikerar att risken för fototoxicitet för patienter som tar trametinib är låg.
Kombination med dabrafenib
I en studie på hundar där trametinib och dabrafenib gavs i kombination under 4 veckor observerades tecken på gastrointestinal toxicitet samt minskad lymfoid cellularitet i tymus vid lägre exponeringsgrad än hos hundar som fått trametinib ensamt. I övrigt har liknande toxicitet observerats som i jämförbara monoterapi-studier.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Sulfobutylbetadexnatrium
Sukralos (E 955)
Citronsyramonohydrat (E 330)
Dinatriumfosfat (E 339)
Kaliumsorbat (E 202)
Metylparahydroxibensoat (E 218)
Jordgubbssmak (akaciagummi, triacetin, artificiella smakämnen)
Inkompatibiliteter
Ej relevant.
Hållbarhet
Pulver till oral lösning
3 år.
Färdigberedd oral lösning
Förvaras vid högst 25 °C.
Får ej frysas.
Ej använd lösning ska kasseras 35 dagar efter beredning.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C-8 °C).
Förvaras i originalförpackningen. Ljus- och fuktkänsligt.
Förvaringsanvisningar för läkemedlet efter beredning finns i avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
SPEXOTRAS jauhe oraaliliuosta varten
0,05 mg/ml (L:ei) 1 kpl (4,7 mg, 1 pullon sovitinosa+mittaruisku) (457,95 €)
PF-selosteen tieto
Ljusbrun glasflaska om 180 ml med ett barnskyddande skruvlock, innehållande 12 g pulver.
En kartong innehåller en flaska, en flaskadapter och en 20 ml återanvändbar oral doseringsspruta med 0,5 ml graderingsmärken.
Läkemedlets utseende:
Vitt eller nästan vitt pulver.
Särskilda anvisningar för destruktion och övrig hantering
Spexotras pulver måste beredas till oral lösning av apotekspersonalen innan läkemedlet lämnas ut.
Anvisningar för beredning (endast avsedda för apotekspersonal):
1. Tvätta och torka händerna.
2. Kontrollera pulvrets utgångsdatum på flaskan.
3. Knacka på flaskan så att pulvret luckas upp.
4. Ta av locket och tillsätt 90 ml destillerat eller renat vatten till pulvret i flaskan.
5. Sätt på locket och vänd flaskan upp och ner flera gånger i upp till 5 minuter, tills allt pulver har lösts upp helt. Du kan även skaka flaskan försiktigt.
Notera: Vita flytande partiklar som är naturligt förekommande i produkten kan vara synliga i den färdigberedda lösningen.
6. Ta loss flaskadaptern från den orala sprutan. Ta bort flasklocket och tryck in flaskadaptern i flaskhalsen. Tryck hårt tills flaskadaptern förts ner i flaskhalsen. Flaskadaptern ska sitta jäms med kanten på flaskhalsen.
7. Notera beredningsdatum på kartongen. Utgångsdatum för lösningen är 35 dagar efter beredning.
8. Informera patienten om dosen och om datum för beredning av lösningen.
Ej använt läkemedel eller avfall ska kasseras enligt gällande anvisningar.
Ersättning
SPEXOTRAS jauhe oraaliliuosta varten
0,05 mg/ml 1 kpl
- Ylempi erityiskorvaus (100 %). Dabrafenibi ja trametinibi: Melanooman, gliooman ja keuhkosyövän hoito erityisin edellytyksin (1509).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Dabrafenibi ja trametinibi: Dabrafenibin ja trametinibin yhdistelmähoito aikuisille, kun kyseessä BRAF V600 - mutaatiopositiivisen melanooman hoito erityisin edellytyksin (3024).
Atc-kod
L01EE01
Datum för översyn av produktresumén
19.05.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com