
EYLEA injektioneste, liuos 114,3 mg/ml, injektioneste, liuos, esitäytetty ruisku 114,3 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Potilasoppaan äänitiedosto
Terveydenhuollon ammattilainen
EYLEA - lasiaisinjektion antaminen
EYLEA Lääkkeen määrääjän opas
Vaikuttavat aineet ja niiden määrät
1 ml injektionestettä, liuosta, sisältää 114,3 mg afliberseptiä*.
Eylea 114,3 mg/ml injektioneste, liuos
Yksi injektiopullo sisältää 30,1 mg afliberseptiä 0,263 ml:ssä injektionestettä. Tästä saadaan annettavaksi yksi 0,07 ml:n annos, jossa on 8 mg afliberseptiä.
Eylea 114,3 mg/ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 21 mg afliberseptiä 0,184 ml:ssä injektionestettä. Tästä saadaan annettavaksi yksi 0,07 ml:n annos, jossa on 8 mg afliberseptiä.
* Aflibersepti on fuusioproteiini, joka sisältää osia ihmisen verisuonten endoteelin kasvutekijän (VEGF) reseptoreiden 1 ja 2 solunulkoisista domeeneista yhdistettynä ihmisen IgG1:n Fc-osaan. Se on tuotettu kiinanhamsterin munasarja (CHO) K1-soluissa yhdistelmä-DNA-tekniikalla.
Apuaine, jonka vaikutus tunnetaan
1 ml injektionestettä, liuosta, sisältää 0,3 mg polysorbaatti 20:tä (E 432).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektio)
Kliiniset tiedot
Käyttöaiheet
Eylea on tarkoitettu aikuisille
- neovaskulaarisen (kostean) silmänpohjan ikärappeuman (AMD) hoitoon (ks. kohta Farmakodynamiikka)
- diabeettisesta makulaturvotuksesta (DME) johtuvan näön heikkenemisen hoitoon (ks. kohta Farmakodynamiikka)
- verkkokalvon laskimotukoksesta (verkkokalvon haaralaskimotukos (BRVO), keskuslaskimotukos (CRVO) ja hemiretinaalinen laskimotukos (HRVO)) johtuvan makulaturvotuksen aiheuttaman näön heikkenemisen hoitoon (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista.
Annostus ja antotapa
Eylea-valmistetta saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista.
Annostus
Kostea silmänpohjan ikärappeuma ja diabeettinen makulaturvotus
Suositeltu annos on 8 mg afliberseptiä, joka vastaa 0,07 ml:aa liuosta. Annostus on samanlainen kostean silmänpohjan ikärappeuman ja diabeettisen makulaturvotuksen hoidossa. 8 mg:n annos edellyttää Eylea 114,3 mg/ml ‑valmisteen käyttämistä.
Eylea-hoidon aloittaville potilaille annetaan yksi injektio kuukaudessa kolmen kuukauden ajan. Tämän jälkeen injektioväliä voidaan pidentää neljään kuukauteen lääkärin arvion ja näkö- ja/tai anatomiavasteiden perusteella. Hoitoväliä voidaan pidentää edelleen enintään kuuteen kuukauteen, kuten treat-and-extend-hoito-ohjelmassa, näkö- ja/tai anatomiavasteiden ollessa vakaita (ks. kohta Farmakodynamiikka).
Jos potilas on saanut aiemmin Eylea 40 mg/ml -hoitoa tai muita anti-VEGF-lääkevalmisteita ja siirtyy Eylea 114,3 mg/ml -hoitoon, hoito-ohjelma voi olla erilainen kuin aiemmin hoitamattomilla potilailla. Hoitoväli määritetään näkö- ja/tai anatomiavasteiden perusteella (ks. kohta Farmakodynamiikka).
- Potilailla, joiden näkö- ja anatomiavasteet ovat vakaat, aiemmat hoitovälit voidaan säilyttää tai niitä voidaan pidentää ensimmäisen Eylea 114,3 mg/ml -injektion jälkeen kuten treat-and-extend-hoito-ohjelmassa.
- Potilailla, joiden näkö- ja/tai anatomiavasteet ovat suboptimaaliset, Eylea 114,3 mg/ml -hoito voidaan aloittaa antamalla yksi injektio kuukaudessa enintään kolmen peräkkäisen annoksen verran, ja tämän jälkeen injektioväliä säädetään kuten treat-and-extend-hoito-ohjelmassa.
Jos näkö- ja/tai anatomiavasteet huonontuvat, tulee hoitovälejä vastaavasti lyhentää lääkärin arvion mukaisesti. Hoitovälin pitää olla vähintään 1 kuukausi.
Jos näkö- ja/tai anatomiavasteet viittaavat siihen, että potilas ei hyödy hoidon jatkamisesta, Eylea 114,3 mg/ml -hoito tulisi lopettaa.
Eylea-valmisteen kuukausittaisia 8 mg:n annoksia ei ole tutkittu kolmea peräkkäistä annosta pidempään PULSAR (kostea silmänpohjan ikärappeuma) ja PHOTON (diabeettinen makulaturvotus) ‑tutkimuksissa. Saatavilla olevat tiedot tukevat yli kolmen peräkkäisen kuukausiannoksen antamista tietyille potilaille, mutta tiedot ovat tällä hetkellä rajoitetut.
Seurantakäyntien tiheyden on perustuttava potilaan tilaan ja lääkärin arvioon. Katso kohdasta Varoitukset ja käyttöön liittyvät varotoimet tapahtumat, joiden perusteella hoito on syytä keskeyttää.
Verkkokalvon laskimotukos
Suositeltu annos on 8 mg afliberseptiä, joka vastaa 0,07 ml:aa liuosta. 8 mg:n annos edellyttää Eylea 114,3 mg/ml -valmisteen käyttämistä.
Eylea-hoidon aloittaville potilaille annetaan yksi injektio kuukaudessa kolmen kuukauden ajan. Tämän jälkeen injektioväliä voidaan pidentää lääkärin arvion ja näkö- ja/tai anatomiavasteiden perusteella (ks. kohta Farmakodynamiikka).
Jos potilas on saanut aiemmin Eylea 40 mg/ml -hoitoa tai muita anti-VEGF-lääkevalmisteita ja siirtyy Eylea 114,3 mg/ml -hoitoon, hoito-ohjelma voi olla erilainen kuin aiemmin hoitamattomilla potilailla. Hoitoväli määritetään näkö- ja/tai anatomiavasteiden perusteella (ks. kohta Farmakodynamiikka).
- Potilailla, joiden näkö- ja anatomiavasteet ovat vakaat, aiemmat hoitovälit voidaan säilyttää tai niitä voidaan pidentää ensimmäisen Eylea 114,3 mg/ml -injektion jälkeen kuten treat-and-extend-hoito-ohjelmassa.
- Potilailla, joiden näkö- ja/tai anatomiavasteet ovat suboptimaaliset, Eylea 114,3 mg/ml -hoito voidaan aloittaa antamalla yksi injektio kuukaudessa enintään kolmen peräkkäisen annoksen verran, ja tämän jälkeen injektioväliä säädetään kuten treat-and-extend-hoito-ohjelmassa.
Jos näkö- ja/tai anatomiavasteet huonontuvat, tulee hoitovälejä vastaavasti lyhentää lääkärin arvion mukaisesti (ks. kohta Farmakodynamiikka). Injektiovälin pitää ollan vähintään 1 kuukausi.
Jos näkö- ja/tai anatomiavasteet viittaavat siihen, että potilas ei hyödy hoidon jatkamisesta, Eylea 114,3 mg/ml -hoito tulisi lopettaa.
Seurantakäyntien tiheyden on perustuttava potilaan tilaan ja lääkärin arvioon. Katso kohdasta Varoitukset ja käyttöön liittyvät varotoimet tapahtumat, joiden perusteella hoito on syytä keskeyttää.
Erityisryhmät
Munuaisten tai maksan vajaatoiminta
Erityisiä tutkimuksia ei ole tehty potilaille, joilla on munuaisten tai maksan vajaatoiminta.
Saatavilla olevat tiedot eivät viittaa tarpeeseen muuttaa näiden potilaiden Eylea-annosta (ks. kohta Farmakokinetiikka).
Iäkkäät
Saatavilla olevat tiedot eivät viittaa tarpeeseen muuttaa näiden potilaiden Eylea-annosta.
Pediatriset potilaat
Eylea 114,3 mg/ml -valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Ei ole asianmukaista käyttää Eylea 114,3 mg/ml -valmistetta pediatrisille potilaille neovaskulaarisen (kostean) silmänpohjan ikärappeuman, diabeettisen makulaturvotuksen ja verkkokalvon laskimotukoksen hoitoon.
Antotapa
Eylea on tarkoitettu vain silmän lasiaiseen injektoitavaksi.
Lasiaiseen annettavat injektiot on annettava lääketieteellisten standardien sekä soveltuvien ohjeiden mukaisesti. Injektiot saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista. Yleisesti ottaen on varmistettava riittävä puudutus ja aseptiikka, mukaan lukien topikaalinen laajakirjoinen mikrobisidi (esim. jodattu povidoni silmää ympäröivälle iholle, silmäluomelle ja silmän pinnalle). On suositeltavaa käyttää kirurgian edellyttämää käsien desinfiointia, steriilejä käsineitä, steriiliä liinaa ja steriiliä luomenlevitintä (tai vastaavaa).
Injektioneula pistetään 3,5–4,0 mm limbuksesta posteriorisesti lasiaiseen, vältetään horisontaalista meridiaania ja tähdätään silmämunan keskikohtaan ja annettava 0,07 ml:n määrä injisoidaan. Kovakalvon pistoskohtaa vaihdetaan seuraavissa injektioissa.
Potilaita on seurattava silmänpaineen kohoamisen varalta välittömästi lasiaisinjektion jälkeen. Sopiva seuranta voi sisältää näköhermon pään perfuusion tarkistuksen tai tonometrian. Steriilejä parasenteesivälineitä on tarvittaessa oltava saatavilla.
Lasiaisinjektion jälkeen potilaita on neuvottava viipymättä ilmoittamaan kaikista endoftalmiittiin viittaavista oireista (esim. silmäkipu, silmän punoitus, valoherkkyys, näön sumeneminen).
Yhtä injektiopulloa tai esitäytettyä ruiskua saa käyttää vain yhden silmän hoitamiseen.
Injektion jälkeen hävitä käyttämätön valmiste tai jäte paikallisten vaatimusten mukaisesti.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen käsittelystä ennen lääkkeen antoa.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Silmän tai silmänympärysalueen infektio.
- Aktiivinen vakava silmänsisäinen inflammaatio.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Lasiaisinjektioihin liittyvät reaktiot
Lasiaisinjektioihin, Eylea-injektiot mukaan lukien, on liittynyt endoftalmiittia, silmänsisäisiä tulehdusreaktioita, verkkokalvon irtaumia, verkkokalvon repeämiä ja traumaperäistä kaihia (ks. kohta Haittavaikutukset). Oikeanlaisia aseptisia injektointitekniikoita on aina käytettävä injektoitaessa Eylea-valmistetta. Potilaita on kehotettava ilmoittamaan viipymättä kaikista endoftalmiittiin tai yllä mainittuihin tapahtumiin viittaavista oireista.
Silmänpaineen kohoaminen
Ohimenevää silmänpaineen kohoamista on havaittu 60 minuutin sisällä lasiaiseen annetusta injektiosta, myös Eylea-valmisteen yhteydessä (ks. kohta Haittavaikutukset). Siksi sekä silmänpainetta että näköhermon pään perfuusiota on seurattava ja hoidettava asianmukaisesti. Erityistä varovaisuutta tarvitaan sellaisten potilaiden kohdalla, joilla on huonosti hallinnassa oleva glaukooma (Eylea-valmistetta ei saa injektoida, kun silmänpaine on ≥ 30 mmHg).
Immunogeenisuus
Afliberseptin käyttöön voi liittyä immunogeenisuutta, koska kyseessä on terapeuttinen proteiini (ks. kohta Farmakodynamiikka). Potilaita on kehotettava ilmoittamaan, mikäli heillä ilmenee silmänsisäisen tulehduksen oireita kuten kipua, valonarkuutta tai punoitusta, mikä voi olla yliherkkyydestä johtuva kliininen oire.
Systeemiset vaikutukset
Systeemisiä haittatapahtumia, mukaan lukien silmän ulkopuolisia verenvuotoja ja valtimoiden tromboemboliatapahtumia, on raportoitu verisuonikasvutekijän (VEGF) estäjien lasiaisinjektioiden jälkeen, ja on teoreettinen mahdollisuus, että ne liittyvät VEGF-estoon (ks. kohta Haittavaikutukset).
Sellaisten potilaiden hoidon turvallisuudesta, jotka sairastavat kosteaa silmänpohjan ikärappeumaa, diabeettista makulaturvotusta tai verkkokalvon laskimotukosta, joilla lisäksi on aiemmin ollut aivohalvaus tai ohimeneviä iskeemisiä kohtauksia tai sydäninfarkti edeltävien kuuden kuukauden aikana, on vain vähän tietoa. Näiden potilaiden hoidossa on noudatettava varovaisuutta.
Molemminpuolinen hoito
Molempiin silmiin samanaikaisesti annettavan Eylea 114,3 mg/ml -hoidon turvallisuutta ja tehoa ei ole tutkittu (ks. kohta Farmakodynamiikka). Jos molemminpuolinen hoito toteutetaan samanaikaisesti, systeeminen altistus voi kasvaa, mikä voi lisätä systeemisten haittavaikutusten riskiä.
Muiden anti-VEGF-hoitojen samanaikainen käyttö
Saatavilla on vain rajallisesti tietoa Eylea-valmisteen käytöstä muiden anti-VEGF-valmisteiden kanssa (systeeminen tai paikallinen).
Hoidon antamatta jättäminen
Hoitoa ei saa antaa seuraavissa tapauksissa:
- paras korjattu näöntarkkuus (BCVA) on laskenut ≥ 30 kirjainta verrattuna edelliseen näöntarkkuuden mittaamiseen
- repeytymisestä johtuva verkkokalvon irtauma tai 3. tai 4. asteen makulareikiä
- verkkokalvon repeytyminen
- verkkokalvon alainen verenvuoto keskeisen näön alueella tai jos verenvuodon laajuus on ≥ 50 % leesion kokonaispinta-alasta
- silmäleikkaus suoritettu edellisten 28 vuorokauden sisällä tai suunnitteilla seuraavien 28 vuorokauden sisällä.
Verkkokalvon pigmenttiepiteelin repeytymä
Kostean silmänpohjan ikärappeuman anti-VEGF-hoidon jälkeen verkkokalvon pigmenttiepiteelin repeytymän kehittymiseen liittyviä riskitekijöitä ovat laaja ja/tai korkea verkkokalvon pigmenttiepiteelin irtoaminen. Aloitettaessa afliberseptihoitoa on noudatettava varovaisuutta, jos potilaalla on näitä verkkokalvon pigmenttiepiteelin repeytymien riskitekijöitä.
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 4 kuukautta viimeisen lasiaiseen annettavan Eylea 114,3 mg/ml ‑injektion jälkeen (ks. kohta Raskaus ja imetys).
Potilasryhmät, joista on rajallisesti tietoa
Eylea-hoidosta diabeetikoilla, joiden HbA1c on yli 12 % tai jotka sairastavat proliferatiivista diabeettista retinopatiaa, on vain rajallisesti tietoa.
Eylea-valmistetta ei ole tutkittu potilailla, joilla on aktiivisia systeemisiä tulehduksia, eikä potilailla, joilla on samanaikaisesti silmäsairauksia, kuten verkkokalvon irtauma tai makulareikä. Eylea-hoidosta ei myöskään ole kokemusta diabeetikoilla, joiden verenpaine ei ole hoitotasapainossa. Tällaisten potilaiden hoidossa lääkärin on otettava huomioon tietojen puutteellisuus.
Tietoa apuaineista
Tämä lääkevalmiste sisältää 0,021 mg polysorbaatti 20:tä per 0,07 ml:n annos, joka vastaa 0,3 mg/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 4 kuukautta viimeisen lasiaiseen annettavan Eylea 114,3 mg/ml -injektion jälkeen.
Raskaus
Tietoja Eylea 114,3 mg/ml -valmisteen käytöstä raskaana oleville naisille on rajoitetusti.
Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Eylea 114,3 mg/ml -valmistetta ei pidä käyttää raskauden aikana, ellei mahdollinen hyöty ole sikiölle aiheutuvaa mahdollista riskiä suurempi.
Imetys
Hyvin niukkojen ihmisistä saatujen tietojen perusteella aflibersepti saattaa erittyä ihmisen äidinmaitoon vähäisissä määrin. Aflibersepti on suuri proteiinimolekyyli, ja vauvaan imetyksen yhteydessä imeytyvän lääkkeen määrän odotetaan olevan vähäinen. Afliberseptin vaikutuksia imetettävään vauvaan ei tunneta.
Varotoimena imetystä ei suositella Eylea 114,3 mg/ml -valmisteen käytön aikana.
Hedelmällisyys
Ihmisen hedelmällisyyttä koskevia tietoja ei ole saatavilla. Eläinkokeet suurella systeemisellä altistuksella viittaavat siihen, että aflibersepti saattaa heikentää miesten ja naisten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Eylea-injektiolla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn johtuen lasiaiseen annetun injektion tai injektioon liittyvien silmätutkimusten aiheuttamista väliaikaisista näköhäiriöistä. Potilaat eivät saa ajaa tai käyttää koneita, ennen kuin heidän näkökykynsä on palautunut riittävästi.
Haittavaikutukset
Yhteenveto turvallisuudesta
Vakavia haittavaikutuksia olivat kaihi (7,1 %), kohonnut silmänpaine (3,8 %), verkkokalvoverenvuoto (2,8%), lasiaisverenvuoto (1,1 %), subkapsulaarinen kaihi (0,6 %), verkkokalvon repeämä (0,5 %), tumakaihi (0,4 %) ja verkkokalvon irtauma (0,4 %).
Yleisimpiä haittavaikutuksia (Eylea 114,3 mg/ml -valmisteella hoidetuista potilaista) olivat kaihi (7,1 %), näöntarkkuuden heikkeneminen (4,3 %), sidekalvon verenvuoto (4,0 %), kohonnut silmänpaine (3,8 %), lasiaisirtauma (3,5 %), lasiaiskellujat (3,2 %) ja verkkokalvon verenvuoto (2,8 %).
Neljässä kliinisessä tutkimuksessa todettu turvallisuusprofiili oli samankaltainen Eylea 114,3 mg/ml ‑hoitoa saaneilla (N = 1 808) ja Eylea 40 mg/ml -hoitoa saaneilla potilailla (N = 857) sekä kosteaa silmänpohjan ikärappeumaa, diabeettista makulaturvotusta ja verkkokalvon laskimotukosta sairastavilla potilailla.
Haittavaikutustaulukko
Neljässä vaiheen II/III kliinisessä tutkimuksessa (CANDELA, PULSAR, PHOTON, QUASAR) turvallisuusryhmän muodosti 1 808 potilasta, jotka saivat Eylea 114,3 mg/ml -hoitoa enintään 96 viikon ajan.
Alla kuvatut turvallisuustiedot sisältävät kaikki sellaiset haittavaikutukset, joilla oli kohtuullinen mahdollisuus syy-seuraus-suhteesta injektointitoimenpiteeseen tai lääkevalmisteen käyttöön.
Haittavaikutukset on lueteltu elinjärjestelmäluokan ja esiintymistiheyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Todetut haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Kaikki vaiheen II/III tutkimuksissa tai markkinoille tulon jälkeisessä valvonnassa kosteaa silmänpohjan ikärappeumaa, diabeettista makulaturvotusta tai verkkokalvon laskimotukosta sairastavilla, Eylea 114,3 mg/ml -hoitoa saaneilla potilailla raportoidut hoidosta johtuvat haittavaikutukset
| Elinjärjestelmäluokka | Esiintymistiheys | Haittavaikutus |
| Immuunijärjestelmä | Yleinen | Yliherkkyys* |
| Silmät | Yleinen | Kaihi, kohonnut silmänpaine, lasiaiskellujat, lasiaisen irtauma, lasiaisen verenvuoto, verkkokalvon verenvuoto, näöntarkkuuden heikkeneminen, silmäkipu, sidekalvon verenvuoto, pisteinen sarveiskalvotulehdus, sarveiskalvon naarmu |
| Melko harvinainen | Verkkokalvon irtauma, verkkokalvon repeämä, verkkokalvon pigmenttiepiteelin repeämä, verkkokalvon pigmenttiepiteelin irtauma, uveiitti, iriitti, iridosykliitti, lasiaistulehdus, kortikaalinen kaihi, tumakaihi, subkapsulaarinen kaihi, sarveiskalvon eroosio, näön hämärtyminen, injektiokohdan kipu, vierasainetuntemus silmissä, lisääntynyt kyynelnesteen eritys, injektiokohdan verenvuoto, sidekalvon verekkyys, mykiön samentuma, silmäluomen turvotus, silmän verekkyys, injektiokohdan ärsytys, verkkokalvorappeuma, sarveiskalvoturvotus | |
| Harvinainen | Sokeutuminen, endoftalmiitti, silmäluomen ärsytys | |
| Tuntematon | Skleriitti** |
* Raportoituun yliherkkyyteen liittyi ihottumaa, kutinaa ja nokkosihottumaa.
** Markkinoille tulon jälkeinen raportointi.
Seuraavia Eylea 40 mg/ml -valmisteen käytön yhteydessä esiintyneitä haittavaikutuksia odotetaan esiintyvän myös Eylea 114,3 mg/ml -valmisteen käytön yhteydessä: epänormaali tunne silmässä, sarveiskalvon epiteelin vaurio, etukammion punoitus, traumaperäinen kaihi, silmän etukammion märkäsakka, vaikeat anafylaktiset/anafylaktoidiset reaktiot.
Kuvaus valikoiduista haittavaikutuksista
Valmisteluokkaan liittyvät haittavaikutukset
Valtimon tromboemboliatapahtumat ovat haittavaikutuksia, jotka mahdollisesti liittyvät systeemiseen verisuonikasvutekijän (VEGF) estoon. Verisuonikasvutekijän estäjien lasiaisen sisäisen käytön jälkeen on olemassa teoreettinen riski valtimon tromboemboliatapahtumiin, mukaan lukien aivohalvaus ja sydäninfarkti. Afliberseptin kliinisissä tutkimuksissa todettiin matala insidenssi valtimoiden tromboembolisten tapahtumien suhteen kosteaa silmänpohjan ikärappeumaa, diabeettista makulaturvotusta ja verkkokalvon laskimotukosta sairastavilla potilailla. Eri käyttöaiheissa ei todettu merkittävää eroa Eylea 114,3 mg/ml -valmistetta saaneiden ryhmien ja Eylea 40 mg/ml -valmistetta saaneiden vertailuryhmien välillä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostus ja suuri injektiomäärä voivat nostaa silmänpainetta. Siksi yliannostustapauksissa silmänpainetta on seurattava ja hoitavan lääkärin suosituksen mukaan aloitettava sopiva hoito (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Käyttö- ja käsittelyohjeet).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: silmälääkkeet/antineovaskularisaatioaineet, ATC-koodi: S01LA05
Aflibersepti on rekombinantti fuusioproteiini, joka sisältää osia ihmisen VEGF-reseptorien 1 ja 2 solunulkoisista domeeneista yhdistettynä ihmisen IgG1:n Fc-osaan.
Aflibersepti on tuotettu kiinanhamsterin munasarja (CHO) K1-soluissa yhdistelmä-DNA-tekniikalla.
Vaikutusmekanismi
Verisuonikasvutekijä A (VEGF-A) ja plasentaalinen kasvutekijä (PlGF) kuuluvat angiogeenisiin kasvutekijöihin (VEGF), jotka toimivat endoteelisolujen potentteina mitogeenisinä, kemotaktisina ja vaskulaarista permeabiliteettia lisäävinä tekijöinä. VEGF toimii kahden endoteelisolujen pinnalla esiintyvän reseptorityrosiinikinaasin, VEGFR-1:n ja VEGFR-2:n, välityksellä. PlGF sitoutuu vain VEGFR-1:een, jota esiintyy myös leukosyyttien pinnalla. Jos VEGF-A aktivoi näitä reseptoreita liikaa, se voi aiheuttaa patologista neovaskularisaatiota ja liiallista vaskulaarista permeabiliteettia. PlGF voi aktivoida VEGFR‑1:tä itsenäisesti ja aiheuttaa verkkokalvolla tulehdusvasteen, ja sen määrän tiedetään lisääntyvän patologisissa tiloissa, kuten kosteassa silmänpohjan ikärappeumassa, diabeettisessa retinopatiassa, diabeettisessa makulaturvotuksessa ja verkkokalvon keskuslaskimotukoksessa.
Farmakodynaamiset vaikutukset
Aflibersepti toimii liukoisena syöttireseptorina, joka sitoo VEGF-A:ta ja PlGF:ä suuremmalla affiniteetilla kuin näiden luonnolliset reseptorit, ja siten se estää näihin kuuluvien VEGF-reseptoreiden sitoutumista ja aktivaatiota.
Eläintutkimusten perusteella aflibersepti saattaa estää patologista verisuonten uudismuodostusta ja uudissuonivuotoa monissa eri silmätautimalleissa.
Kostea silmänpohjan ikärappeuma
Kosteaan silmänpohjan ikärappeumaan liittyy patologinen suonikalvon verisuonten uudismuodostus (CNV). Suonikalvon verisuonten uudismuodostuksen (CNV) aiheuttama veren ja nesteen vuoto voi aiheuttaa verkkokalvon turvotusta ja/tai verkkokalvon alaista/sisäistä verenvuotoa, johtaen näöntarkkuuden heikkenemiseen.
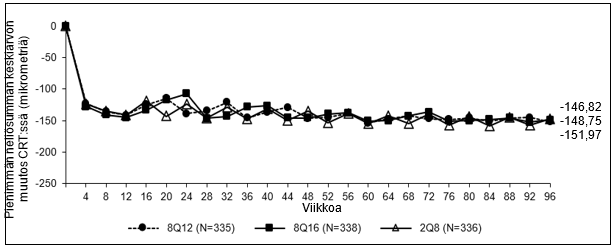
12 viikon välein (8Q12) ja 16 viikon välein (8Q16) annetun aflibersepti 114,3 mg/ml ‑valmisteen farmakodynaamiset vaikutukset kuvataan verrattuna 8 viikon välein (2Q8) annettuun aflibersepti 40 mg/ml ‑valmisteeseen kostean silmänpohjan ikärappeuman hoidossa. Nämä vaikutukset esitetään suonikalvon verisuonten uudismuodostuksen (CNV) koon muutoksena lähtötilanteesta viikolle 12; leesion kokonaispinta-alan muutoksena lähtötilanteesta viikoille 48, 60 ja 96; ja keskeisen verkkokalvon paksuuden (CRT) muutoksena lähtötilanteesta.
Yhdistetyssä 8Q12- tai 8Q16-hoitoa saaneiden potilaiden ryhmässä CNV:n muutos [LS (pienimpien neliösummien keskiarvo), perustuu toistomittausten sekamalliin (MMRM)] viikolle 12 oli -1,63 mm2, kun taas 2Q8-hoitoa saaneiden potilaiden ryhmässä vastaava muutos oli -1,17 mm2.
Farmakodynaamiset vaikutukset säilyivät yleisesti ottaen viikolle 156.
Taulukko 2: Farmakodynaamiset muuttujat (täysi analyysijoukko) PULSAR-tutkimuksessa
| Tehoa kuvaava tulos | Viikko | Eylea 8Q12 (N = 335) | Eylea 8Q16 (N = 338) | Eylea 2Q8 (N = 336) |
| Leesion kokonaispinta-alan muutos lähtötilanteesta [mm2] | ||||
| Keskimääräinen LS A | 12 | -0,55 | -0,30 | |
| Keskiarvo (SD), havaittu | 48 | ‑0,4 (2,9) | ‑0,2 (3,1) | 0,1 (3,6) |
| LS-keskiarvo (SE) A | ‑0,46 (0,19) | ‑0,35 (0,20) | 0,09 (0,22) | |
Ero keskimääräisenä muutoksena, LS (95% CI) A,B | ‑0,55 (‑1,04; ‑0,06) | ‑0,44 (‑0,94; ‑0,06) | ||
| Keskiarvo (SD), havaittu | 60 | ‑0,5 (2,8) | ‑0,4 (3,2) | ‑0,3 (3,2) |
| LS- keskiarvo (SE) A | ‑0,48 (0,20) | ‑0,54 (0,21) | ‑0,24 (0,20) | |
Ero keskimääräisenä muutoksena, LS (95% CI) A,B | ‑0,24 (‑0,72; 0,24) | ‑0,29 (‑0,79; 0,20) | ||
| Keskiarvo (SD), havaittu | 96 | -0,3 (3,3) | -0,3 (3,2) | -0,2 (3,4) |
| LS-keskiarvo (SE) A | -0,43 (0,20) | -0,42 (0,20) | -0,18 (0,20) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) A,B | -0,25 (-0,72; 0,21) | -0,24 (-0,71; 0,22) | ||
A Keskimääräinen LS, CI ja p-arvo perustuen toistomittausten sekamalliin (MMRM), jossa lähtötilanteen mittaus on kovariaattina ja hoitoryhmä tekijänä. Käyntiä ja ositusmuuttujia (maantieteellinen alue, lähtötilanteen BCVA-luokka) käytettiin sekä satunnaistamisen kiinteinä tekijöinä että lähtötilanteen mittauksen ja käynnin sekä hoidon ja käynnin välisinä vuorovaikutusehtoina (interaction term).
B Absoluuttinen ero on Eylea 8Q12‑ tai 8Q16‑ryhmän arvo miinus 2Q8‑ryhmän arvo.
CI: luottamusväli
LS: pienin neliösumma
SD: keskihajonta
SE: keskivirhe
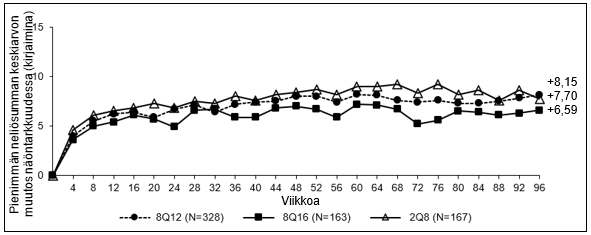
Kuva 1: Pienimmän neliösumman keskiarvon muutos keskeisen verkkokalvon paksuudessa (CRT) lähtötilanteesta viikolle 96 (täysi analyysijoukko) PULSAR-tutkimuksessa
Diabeettinen makulaturvotus
Diabeettisen makulaturvotuksen merkkejä ovat lisääntynyt verisuonien läpäisevyys ja verkkokalvon hiussuonien vaurioituminen, mikä voi johtaa näöntarkkuuden heikkenemiseen.
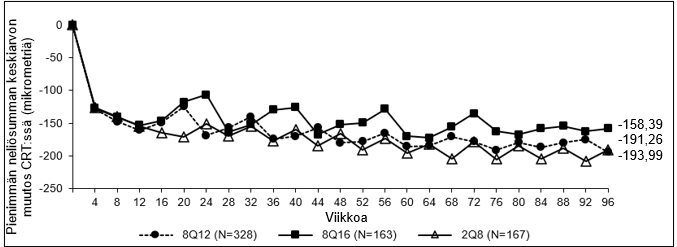
12 viikon välein (8Q12) ja 16 viikon välein (8Q16) annetun aflibersepti 114,3 mg/ml ‑valmisteen farmakodynaamiset vaikutukset kuvataan verrattuna 8 viikon välein (2Q8) annettuun aflibersepti 40 mg/ml ‑valmisteeseen diabeettisen makulaturvotuksen hoidossa. Nämä vaikutukset esitetään vuotoalueen pinta-alan muutoksena lähtötilanteesta viikoille 48, 60 ja 96.
Farmakodynaamiset vaikutukset säilyivät yleisesti ottaen viikolle 156.
Taulukko 3: Farmakodynaamiset muuttujat (täysi analyysijoukko) PHOTON-tutkimuksessa
| Tehoa kuvaava tulos | Viikko | Eylea 8Q12 (N = 328) | Eylea 8Q16 (N = 163) | Eylea 2Q8 (N = 167) |
| Vuotoalueen pinta-alan muutosA lähtötilanteesta [mm2] | ||||
| Keskiarvo (SD), havaittu | 48 | ‑13,9 (13,91) | ‑9,4 (11,50) | ‑9,2 (12,11) |
| 60 | ‑13,9 (13,54) | ‑12,0 (13,26) | ‑14,4 (12,89) | |
| 96 | -12,8 (10,98) | -9,4 (10,61) | -11,9 (11,26) | |
A perustuu fluoreseiiniangiografiamittaukseen
SD: keskihajonta
Kuva 2: Pienimmän neliösumman keskiarvon muutos keskeisen verkkokalvon paksuudessa (CRT) lähtötilanteesta viikolle 96 (täysi analyysijoukko) PHOTON-tutkimuksessa
Immunogeenisuus
Enintään 96 viikkoa kestäneen Eylea 114,3 mg/ml ‑valmisteen annostelun jälkeen hoidosta johtuvia vasta-aineita Eylea 114,3 mg/ml ‑valmisteelle havaittiin 2,5–4,4 prosentilla potilaista, joita hoidettiin diabeettisen makulaturvotuksen ja kostean silmänpohjan ikärappeuman vuoksi. Lääkevasta-aineilla ei havaittu olevan vaikutusta farmakokinetiikkaan, tehokkuuteen tai turvallisuuteen.
Verkkokalvon laskimotukos
Verkkokalvon laskimotukoksen yhteydessä ilmenevä verkkokalvon iskemia johtaa verisuonikasvutekijän (VEGF) vapautumiseen. VEGF:n vaikutuksesta tiiviit liitokset heikentyvät ja endoteelisolujen proliferaatio stimuloituu. Verisuonikasvutekijän vaikutuksen tehostumiseen (up-regulation) liittyy veri-verkkokalvoesteen hajoamista, mikä lisää verisuonten läpäisevyyttä, ja johtaa verkkokalvon edeemaan, endoteelisolujen kasvun stimulaatioon ja verisuonten uudismuodostukseen.
Taulukko 4: Farmakodynaamiset muuttujat (täysi analyysijoukko) QUASAR-tutkimuksessa
| Tehoa kuvaava tulos | Viikko | Eylea 8Q8/3 (N = 293) | Eylea 2Q4 (N = 301) |
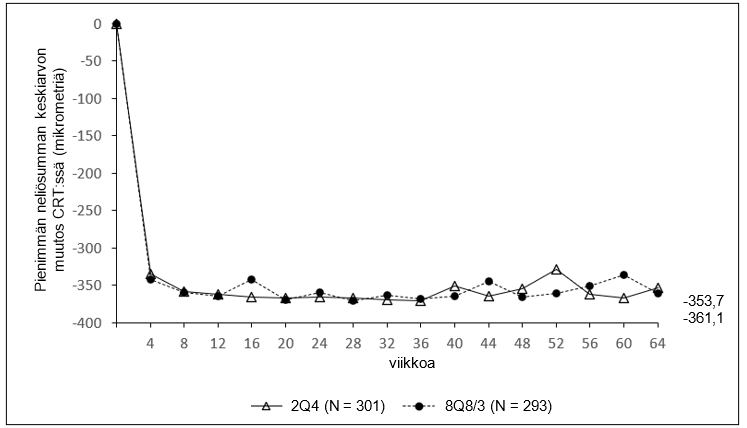
| Muutos keskeisen verkkokalvon paksuudessa (CRT) [mikrometriä] | |||
| Keskiarvo (SD), havaittu | 36 | -365,9 (239,9) | -397,3 (257,7) |
| LS-keskiarvo (SE) A | -370,9 (3,1) | -370,8 (3,9) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) A,B | -0,1 (-10,0; 9,8) | ||
| Keskiarvo (SD), havaittu | 64 | -355,5 (239,5) | -373,0 (252,1) |
| LS-keskiarvo (SE) A | -361,1 (4,3) | -353,7 (5,2) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) A,B | -7,4 (-20,7; 5,9) | ||
A Keskimääräinen LS, CI ja p-arvo perustuen toistomittausten sekamalliin (MMRM), jossa lähtötilanteen CRT-mittaus on kovariaattina ja hoitoryhmä tekijänä. Käyntiä ja ositusmuuttujia (maantieteellinen alue, lähtötilanteen BCVA-luokka ja verkkokalvon laskimotukoksen tyyppi) käytettiin sekä satunnaistamisen kiinteinä tekijöinä että lähtötilanteen CRT-mittauksen ja käynnin sekä hoidon ja käynnin välisinä vuorovaikutusehtoina (interaction term).
B Absoluuttinen ero on Eylea 8Q8/3 ryhmän arvo miinus 2Q4 ryhmän arvo.
CI: luottamusväli
CRT: keskeisen verkkokalvon paksuus
LS: pienin neliösumma
SD: keskihajonta
SE: keskivirhe
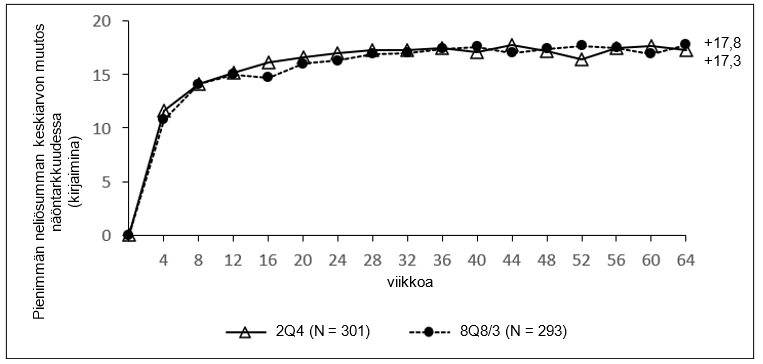
Kuva 3: Pienimmän neliösumman keskiarvon muutos keskeisen verkkokalvon paksuudessa (CRT) lähtötilanteesta viikolle 64 (täysi analyysijoukko) QUASAR-tutkimuksessa
Kliininen teho ja turvallisuus
Kostea silmänpohjan ikärappeuma
Tutkimuksen tavoitteet
Eylea 114,3 mg/ml -valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa monikeskustutkimuksessa (PULSAR) potilailla, jotka eivät olleet saaneet aiempaa hoitoa kosteaan silmänpohjan ikärappeumaan.
Ensisijainen tavoite oli määrittää, onko 12 viikon (8Q12) tai 16 viikon välein (8Q16) annettava Eylea 114,3 mg/ml ‑hoito vertailukelpoinen (non-inferior) parhaan korjatun näöntarkkuuden (BCVA) muutoksena mitattuna kahdeksan viikon välein annettuun Eylea 40 mg/ml ‑hoitoon verrattuna kosteaa silmänpohjan ikärappeumaa sairastavilla potilailla.
Toissijaisina tavoitteina oli verrata Eylea 114,3 mg/ml- ja Eylea 40 mg/ml ‑hoitojen tehoa mitattavien anatomia- ja muiden näkövasteiden perusteella sekä arvioida afliberseptin turvallisuutta, immunogeenisuutta ja farmakokinetiikkaa.
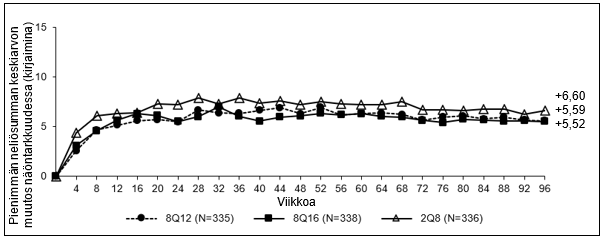
Ensisijainen tehoa koskeva päätetapahtuma oli parhaan korjatun näöntarkkuuden (BCVA) muutos ETDRS (Early Treatment Diabetic Retinopathy Study) -kirjainpisteillä mitattuna lähtötilanteesta viikolle 48.
Tärkeimmät toissijaiset päätetapahtumat olivat parhaan korjatun näöntarkkuuden (BCVA) muutos lähtötilanteesta viikolle 60 sekä niiden potilaiden osuus, joilla ei ollut verkkokalvonsisäistä tai -alaista nestettä verkkokalvon keskikuopassa viikolla 16.
Muita toissijaisia päätetapahtumia olivat mm. niiden potilaiden osuus, joiden paras korjattu näöntarkkuus (BCVA) parani vähintään 15 kirjainta lähtötilanteesta viikolle 48, sekä niiden potilaiden osuus, joiden ETDRS-kirjainpisteet olivat vähintään 69 (noin 20/40, Snellen-ekvivalentti) viikolla 48, sekä muutos National Eye Institute Visual Functioning Questionnaire-25 (NEI-VFQ-25) -kyselyn kokonaispisteissä lähtötilanteesta viikolle 48.
PULSAR-tutkimuksessa hoidettiin yhteensä 1 009 potilasta. Potilaat määrättiin suhteessa 1:1:1 yhteen kolmesta rinnakkaisesta hoitoryhmästä:
- Eylea 114,3 mg/ml -valmistetta annettiin 12 viikon välein (8Q12)
- Eylea 114,3 mg/ml -valmistetta annettiin 16 viikon välein (8Q16)
- Eylea 40 mg/ml -valmistetta annettiin 8 viikon välein (2Q8)
Kaikki potilaat saivat kolme ensimmäistä injektiota määrätystä annoksesta 4 viikon välein. Tutkimussuunnitelman mukaisesti 8Q12- ja 8Q16-ryhmien annosteluväliä lyhennettiin, jos molemmat seuraavista kriteereistä täyttyivät:
- parhaan korjatun näöntarkkuuden (BCVA) lasku yli viidellä kirjaimella viikosta 12,
- keskeisen verkkokalvon paksuuden (CRT) > 25 mikrometrin kasvu viikosta 12 tai uusi verenvuoto tai uudissuonittuminen verkkokalvon keskikuopassa.
Riippumatta siitä, pysyikö annosväli samana vai lyhenikö se ensimmäisen vuoden aikana, tutkimussuunnitelman mukaisesti kaikki 8Q12- ja 8Q16-ryhmien potilaat soveltuivat annosvälin pidennykseen (4 viikon lisäyksin) viikosta 52 alkaen seuraavien kriteerien täyttyessä:
- parhaan korjatun näöntarkkuuden (BCVA) lasku vähemmän kuin viisi kirjainta viikosta 12,
- ei nestettä keskeisessä verkkokalvossa valokerroskuvauksella (OCT) mitattuna,
- ei uutta verenvuotoa tai verisuonten uudismuodostusta verkkokalvon keskikuopassa.
Potilaat, jotka eivät täyttäneet kriteereitä annosteluvälin lyhentämiseksi tai pidentämiseksi, jatkoivat entisellä annosteluvälillä. Lyhyin mahdollinen aika injektioiden välillä oli 8 viikkoa kaikissa ryhmissä.
Potilaat, joilla oli molemminpuolinen sairaus, soveltuivat saamaan Eylea 40 mg/ml -hoitoa tai muuta anti-VEGF-valmistetta toiseen silmään.
Potilaiden ominaisuudet lähtötilanteessa
Potilaat olivat 50–96-vuotiaita, keskimääräinen ikä oli 74,5 vuotta.
Noin 92 % (309/335) 8Q12-ryhmään ja 87 % (295/338) 8Q16-ryhmään satunnaistetuista potilaista oli vähintään 65-vuotiaita. Noin 51 % (172/335) 8Q12-ryhmään ja noin 51 % (171/338) 8Q16-ryhmään satunnaistetuista oli vähintään 75-vuotiaita.
Tulokset
48 viikkoa tutkimuksessa jatkaneiden 8Q12-ryhmän potilaiden saamien injektioiden mediaani (keskiarvo) oli 6,0 (6,1), 8Q16-ryhmän 5,0 (5,2) ja 2Q8-ryhmän 7,0 (6,9).
Viikolla 48 8Q12-ryhmässä 79,4 % potilaista jatkoi Q12-hoitoväleillä ja 8Q16-ryhmässä 76,6 % potilaista jatkoi Q16-hoitoväleillä.
60 viikkoa tutkimuksessa jatkaneiden 8Q12-ryhmän potilaiden saamien injektioiden mediaani (keskiarvo) oli 7,0 (7,1), 8Q16-ryhmän 6,0 (6,2) ja 2Q8-ryhmän 9,0 (8,8).Viikolla 60 43,1 % 8Q12-ryhmän potilaista siirtyi 16 viikon hoitoväleihin ja 38,5 % 8Q16-ryhmän potilaista siirtyi 20 viikon hoitoväleihin.
96 viikkoa tutkimuksessa jatkaneiden 8Q12-ryhmän potilaiden saamien injektioiden mediaani (keskiarvo) oli 9,0 (9,7), 8Q16-ryhmän 8,0 (8,2) ja 2Q8-ryhmän 13,0 (12,8) injektiota.
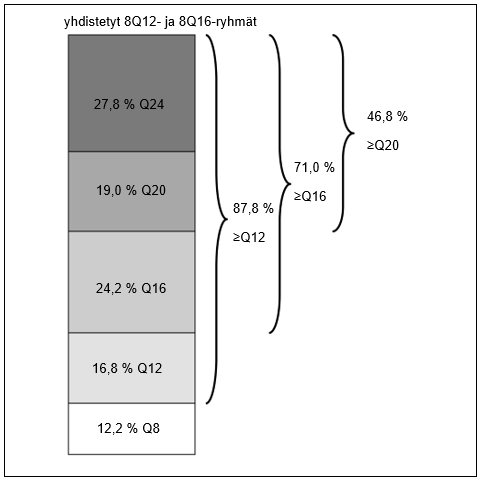
Viikolla 96 yhdistetyssä 8Q12- ja 8Q16-ryhmässä 71,0 % potilaista oli saavuttanut ≥ 16 viikon hoitovälin, 46,8 % ≥ 20 viikon hoitovälin ja 27,8 % ≥ 24 viikon hoitovälin niin, että näkö- ja anatomiatulokset säilyivät.
8Q12- ja 8Q16-hoidot osoitettiin vertailukelpoisiksi (non-inferior) 2Q8-hoidon kanssa ja niiden todettiin vastaavan kliinisesti 2Q8-hoitoa ensisijaisen tehoa koskevan päätetapahtuman ”parhaan korjatun näöntarkkuuden (BCVA) muutos lähtötilanteesta viikolle 48” sekä toissijaisen tehoa koskevan päätetapahtuman ”parhaan korjatun näöntarkkuuden (BCVA) keskimääräinen muutos viikolle 60” perusteella. Eylea 114,3 mg/ml -hoidon vaikutus parhaalla korjatulla näöntarkkuudella (BCVA) mitattuna jatkui viikolle 96.
Lisäksi Eylea-hoidon (yhdistetyt 8Q12- ja 8Q16-ryhmät) paremmuus verrattuna 2Q8-hoitoon pystyttiin osoittamaan toissijaisen tehoa koskevan päätetapahtuman ”niiden potilaiden osuus, joilla ei ollut verkkokalvonsisäistä tai -alaista nestettä verkkokalvon keskikuopassa viikolla 16” perusteella (ks. taulukko 5).
Taulukko 5: Tehoa koskevat hoitotulokset PULSAR-tutkimuksessa
| Tehoa kuvaava tulos | Viikko | Eylea 8Q12 (N = 335) | Eylea 8Q16 (N = 338) | Eylea 2Q8 (N = 336) |
| BCVA:n muutos lähtötilanteesta mitattuna ETDRS-kirjainpisteillä D | ||||
| Keskiarvo (SD), havaittu | 48 | 6,7 (12,6) | 6,2 (11,7) | 7,6 (12,2) |
| Keskimääräinen LS (SE) A | 6,06 (0,77) | 5,89 (0,72) | 7,03 (0,74) | |
Ero keskimääräisenä muutoksena, LS (95% CI) A,B | ‑0,97 (‑2,87; 0,92) | ‑1,14 (‑2,97; 0,69) | ||
| p‑arvo (yksisuuntainen non‑inferiority-testi, marginaali 4 kirjainta) A,B | 0,0009 | 0,0011 | ||
| Keskiarvo (SD), havaittu | 60 | 6,6 (13,6) | 6,6 (11,7) | 7,8 (12,6) |
| Keskimääräinen LS (SE) A | 6,37 (0,74) | 6,31 (0,66) | 7,23 (0,68) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) A,B | ‑0,86 (‑2,57; 0,84) | ‑0,92 (‑2,51; 0,66) | ||
| p‑arvo (yksisuuntainen non‑inferiority-testi, marginaali 4 kirjainta) A,B | 0,0002 | <0,0001 | ||
| Keskiarvo (SD), havaittu | 96 | 5,9 (14,2) | 5,6 (13,7) | 7,4 (13,8) |
| Keskimääräinen LS (SE) A | 5,59 (0,77) | 5,52 (0,75) | 6,60 (0,73) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) A,B | -1,01 (-2,82; 0,80) | -1,08 (-2,87; 0,71) | ||
| Potilaat, joilla ei ollut verkkokalvonsisäistä tai -alaista nestettä verkkokalvon keskikuopassaD | ||||
| Osuus (LOCF) | 16 | 63,3 % | 51,6 % | |
Osuuksien suhteellinen ero (95% CI) B,C | 11,7 % (5,3 %; 18,2 %) | |||
| p‑arvo (yksisuuntainen superiority-testi) B, C | 0,0002 | |||
| Osuus (LOCF) | 48 | 71,1 % | 66,8 % | 59,4 % |
Osuuksien suhteellinen ero (95% CI) B,C | 11,7 % (4,5 %; 18,9 %) | 7,5 % (0,1 %; 14,8 %) | ||
| Osuus (LOCF) | 60 | 74,6 % | 72,2 % | 74,6 % |
Osuuksien suhteellinen ero (95% CI) B,C | 0,0 % (‑6,6 %; 6,7 %) | ‑2,2 % (‑8,9 %; 4,4 %) | ||
| Osuus (LOCF) | 96 | 69,6 % | 63,6 % | 66,5 % |
| Osuuksien suhteellinen ero (95% CI) B,C | 3,0 % (-4,1 %; 10,1 %) | -3,0 % (-10,2 %; 4,2 %) | ||
| Potilaat, joiden ETDRS-kirjainpisteet olivat vähintään 69 (noin 20/40, Snellen-ekvivalentti) D | ||||
| Osuus (LOCF) | 48 | 56,9 % | 54,3 % | 57,9 % |
Osuuksien suhteellinen ero (95% CI) B,C | ‑0,2 % (‑6,6 %; 6,2 %) | ‑2,2 % (‑8,4 %; 4,0 %) | ||
| Osuus (LOCF) | 60 | 56,3 % | 54,6 % | 58,2 % |
Osuuksien suhteellinen ero (95% CI) B,C | ‑1,1 % (‑7,5 %; 5,3 %) | ‑2,3 % (‑8,7 %; 4,1 %) | ||
| Osuus (LOCF) | 96 | 53,3 % | 53,1 % | 56,7 % |
| Osuuksien suhteellinen ero (95% CI) B,C | -2,7 % (-9,4 %; 4,0 %) | -2,4 % (-9,1 %; 4,2 %) | ||
| Potilaat, joiden BCVA parani vähintään 15 kirjainta lähtötilanteestaD | ||||
| Osuus (LOCF) | 48 | 20,7 % | 21,7 % | 22,1 % |
Osuuksien suhteellinen ero (95% CI) B,C | ‑1,7 % (‑7,8 %; 4,3 %) | ‑0,9 % (‑7,0 %; 5,1 %) | ||
| Osuus (LOCF) | 60 | 23,7 % | 23,1 % | 23,3 % |
Osuuksien suhteellinen ero (95% CI) B,C | 0,1 % (‑6,2 %; 6,3 %) | ‑0,7 % (‑6,9 %; 5,5 %) | ||
| Osuus (LOCF) | 96 | 22,2 % | 22,8 % | 24,2 % |
Osuuksien suhteellinen ero (95% CI) B,C | -2,4 % (-8,4 %; 3,6 %) | -2,0 % (-8,0 %; 4,1 %) | ||
| Viimeisin suunniteltu hoitoväli | ||||
| Potilaat, joiden hoitoväli oli ≥ Q12 E | ||||
| Osuus (yhdistetyt 8Q12- ja 8Q16-ryhmät) | 96 | 87,8 % | - | |
| Osuus | 86,6 % | 89,0 % | - | |
| Potilaat, joiden hoitoväli oli ≥ Q16 E | ||||
| Osuus (yhdistetyt 8Q12- ja 8Q16-ryhmät) | 96 | 71,0 % | - | |
| Osuus | 63,6 % | 78,4 % | - | |
| Potilaat, joiden hoitoväli oli ≥ Q20 E | ||||
| Osuus (yhdistetyt 8Q12- ja 8Q16-ryhmät) | 96 | 46,8 % | - | |
| Osuus | 40,5 % | 53,1 % | - | |
| Potilaat, joiden hoitoväli oli Q24 E | ||||
| Osuus (yhdistetyt 8Q12- ja 8Q16-ryhmät) | 96 | 27,8 % | - | |
| Osuus | 24,7 % | 30,8 % | - | |
A Keskimääräinen LS, CI ja p-arvo perustuen toistomittausten sekamalliin (MMRM), jossa lähtötilanteen paras korjattu näöntarkkuus (BCVA) on kovariaattina ja hoitoryhmä tekijänä. Käyntiä ja ositusmuuttujia (maantieteellinen alue, lähtötilanteen BCVA-luokka) käytettiin sekä satunnaistamisen kiinteinä tekijöinä että lähtötilanteen BCVA:n ja käynnin sekä hoidon ja käynnin välisinä vuorovaikutusehtoina (interaction term).
B Absoluuttinen ero on Eylea 8Q12‑ tai 8Q16‑ryhmän arvo miinus 2Q8‑ryhmän arvo.
C Mantel‑Haenszel-painotettu hoitoero, jossa satunnaistamiseen käytettiin ositusmuuttujia (maantieteellinen alue, BCVA-luokka lähtötilanteessa), ja CI laskettiin normaalilla approksimaatiolla
D täysi analyysijoukko
E turvallisuusanalyysijoukko: potilaat, jotka olivat mukana tutkimuksessa tiettyyn aikapisteeseen asti
CI: luottamusväli
LOCF: viimeinen havainto eteenpäin
LS: pienin neliösumma
SD: keskihajonta
SE: keskivirhe
Hoitovälit analysoitiin etukäteen määrätyllä eksploratiivisella tavalla.
Kuva 4: Pienimmän neliösumman keskiarvon muutos BCVA:ssa mitattuna ETDRS-kirjainpisteinä lähtötilanteesta viikolle 96 (täysi analyysijoukko) PULSAR-tutkimuksessa
Kuva 5: Viimeisin suunniteltu hoitoväli viikolla 96
Afliberseptin kaikilla annostuksilla (8Q12, 8Q16, 2Q8) osoitettiin merkittävää nousua ennalta määritellyssä toissijaisessa päätetapahtumassa, National Eye Institute Visual Function Questionnaire (NEI VFQ-25) -kyselyn pisteissä.
8Q12-, 8Q16- ja 2Q8-ryhmien välillä ei havaittu kliinisesti merkitseviä eroja NEI VFQ‑25 -kokonaispisteiden muutoksessa lähtötilanteesta viikolle 48 ja viikolle 96.
Kaikkien ikää, sukupuolta, maantieteellistä aluetta, etnisyyttä, rotua, lähtötilanteen BVCA:ta ja leesiotyyppiä koskevien arvioitavissa olevien alaryhmien tehoa koskevat tulokset vastasivat kokonaispopulaation tuloksia.
Teho säilyi yleisesti ottaen viikolle 96.
Tulokset – PULSAR-tutkimuksen jatkovaihe
Tutkimuksen päävaiheen lopussa viikolla 96 potilaiden oli mahdollista osallistua 60 viikon pituiseen avoimeen jatkovaiheeseen. 417 potilasta, jotka oli alun perin määrätty 8Q12- ja 8Q16-ryhmiin, jatkoivat Eylea 114,3 mg/ml -hoitoa viimeisimmillä hoitoväleillään. 208 potilasta, jotka oli alun perin määrätty 2Q8-ryhmään tutkimuksen alussa, siirrettiin Eylea 114,3 mg/ml -hoitoon, joka aloitettiin 12 viikon hoitovälillä. Hoitovälejä voitiin mukauttaa edelleen lääkärin arvioimien näkö- ja/tai anatomiavasteiden perusteella.
Potilailla, jotka oli alun perin määrätty 8Q12- ja 8Q16-ryhmiin, Eylea 114,3 mg/ml -hoitovaikutus säilyi yleisesti ottaen 3 vuoden ajan (viikolle 156). Yhdistetyissä 8Q12- ja 8Q16-ryhmissä pienimmän neliösumman (LS) keskiarvon muutos lähtötilanteesta oli parhaassa korjatussa näöntarkkuudessa (BCVA) +3,41 kirjainta ja verkkokalvon keskeisen alueen paksuudessa (CRT) -148,05 mikrometriä viikolla 156.
Potilailla, jotka oli alun perin määrätty 2Q8-ryhmään, Eylea 114,3 mg/ml -hoitovaikutus oli samankaltainen. LS keskiarvon muutos lähtötilanteesta oli BCVA:ssa +4,58 kirjainta ja CRT:ssä -145,21 mikrometriä viikolla 156.
8Q12- ja 8Q16-ryhmien potilailla, jotka olivat mukana tutkimuksessa viikon 156 loppuun asti, potilaiden saamien injektioiden mediaani (keskiarvo) oli 8Q12-ryhmässä 13,0 (13,5) ja 8Q16-ryhmässä 11,0 (12,2).
Eylea 114,3 mg/ml -hoitoon siirtyneillä potilailla, jotka olivat mukana tutkimuksessa viikon 156 loppuun asti, potilaiden saamien injektioiden mediaani (keskiarvo) oli 18,0 (17,7), ja näistä injektioista 5,0 (4,9) annettiin Eylea 114,3 mg/ml -hoitoon siirtymisen jälkeen tutkimuksen jatkovaiheen 60 viikon aikana.
Yleinen turvallisuusprofiili oli jatkovaiheessa samankaltainen kuin tutkimuksen päävaiheessa.
Taulukko 6: Tehoa koskevat hoitotulokset PULSAR-tutkimuksen jatkovaiheessa viikolla 156
| Tehoa kuvaava tulos | 8Q12, jatkoi Eylea 114,3 mg/ml ‑hoitoa (N = 185) | 8Q16, jatkoi Eylea 114,3 mg/ml ‑hoitoa (N = 190) | 2Q8, siirtyi Eylea 114,3 mg/ml ‑hoitoon (N = 208) |
| BCVA:n muutos lähtötilanteesta (LS-keskiarvo) | +3,57 kirjainta | +3,23 kirjainta | +4,58 kirjainta |
| CRT:n muutos lähtötilanteesta (LS keskiarvo) | -148,42 mikrometriä | -147,54 mikrometriä | -145,21 mikrometriä |
| Viimeinen suunniteltu hoitoväli A | |||
| ≥ 12 viikkoa | 76,2 % | 78,4 % | 78,5 % |
| ≥ 16 viikkoa | 53,5 % | 62,1 % | 42,5 % |
| ≥ 20 viikkoa | 37,8 % | 42,6 % | 16,1 % |
| 24 viikkoa | 23,8 % | 24,2 % | – B |
A perustuu potilaisiin, jotka olivat mukana tutkimuksessa viikon 156 loppuun asti
B Ei sovelleta alun perin 2Q8 ryhmään satunnaistettuihin potilaisiin tutkimusasetelman / tutkimuksen keston takia
Diabeettinen makulaturvotus
Tutkimuksen tavoitteet
Eylea 114,3 mg/ml -valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa monikeskustutkimuksessa (PHOTON) potilailla, joilla oli diabeettinen makulaturvotus.
Ensisijainen tavoite oli määrittää, onko 12 viikon (8Q12) tai 16 viikon välein (8Q16) annettava Eylea 114,3 mg/ml ‑hoito vertailukelpoinen (non-inferior) parhaan korjatun näöntarkkuuden (BCVA) muutoksena mitattuna kahdeksan viikon välein annettuun Eylea 40 mg/ml -hoitoon verrattuna.
Toissijaisina tavoitteina oli verrata Eylea 114,3 mg/ml- ja Eylea 40 mg/ml ‑hoitojen tehoa mitattavien anatomia- ja muiden näkövasteiden perusteella sekä arvioida afliberseptin turvallisuutta, immunogeenisuutta ja farmakokinetiikkaa.
Ensisijainen tehoa koskeva päätetapahtuma oli BCVA:n muutos ETDRS (Early Treatment Diabetic Retinopathy Study) -kirjainpisteillä mitattuna lähtötilanteesta viikolle 48.
Yksi tärkeä toissijainen päätetapahtuma oli parhaan korjatun näöntarkkuuden (BCVA) muutos lähtötilanteesta viikolle 60.
Muita toissijaisia päätetapahtumia olivat mm. niiden potilaiden osuus, joiden paras korjattu näöntarkkuus (BCVA) parani vähintään 15 kirjainta lähtötilanteesta viikolle 48, sekä niiden potilaiden osuus, joiden ETDRS-kirjainpisteet olivat vähintään 69 (noin 20/40, Snellen-ekvivalentti) viikolla 48, sekä muutos National Eye Institute Visual Functioning Questionnaire-25 (NEI-VFQ-25) -kyselyn kokonaispisteissä lähtötilanteesta viikolle 48.
PHOTON-tutkimuksessa hoidettiin yhteensä 658 potilasta. Potilaat määrättiin suhteessa 2:1:1 yhteen kolmesta rinnakkaisesta hoitoryhmästä:
- Eylea 114,3 mg/ml -valmistetta annettiin 12 viikon välein (8Q12)
- Eylea 114,3 mg/ml -valmistetta annettiin 16 viikon välein (8Q16)
- Eylea 40 mg/ml -valmistetta annettiin 8 viikon välein (2Q8)
Potilaat, jotka siirtyivät muista anti-VEGF-lääkevalmisteista Eylea 114,3 mg/ml -hoitoon, saivat aiemman hoidon viimeisen injektion vähintään 12 viikkoa ennen Eylea 114,3 mg/ml -hoidon aloittamista.
Kaikki 8Q12- ja 8Q16-ryhmän potilaat saivat ensimmäiset kolme injektiota ja kaikki 2Q8-ryhmän potilaat saivat ensimmäiset viisi injektiota 4 viikon välein. Tutkimussuunnitelman mukaisesti 8Q12- ja 8Q16-ryhmien annosteluväliä lyhennettiin, jos molemmat seuraavista kriteereistä täyttyivät:
- parhaan korjatun näöntarkkuuden (BCVA) yli kymmenen kirjaimen lasku viikosta 12 jatkuvan tai pahenevan diabeettisen makulaturvotuksen yhteydessä ja
- keskeisen verkkokalvon paksuuden (CRT) > 50 mikrometrin kasvu viikosta 12.
Riippumatta siitä, pysyikö annosväli samana vai lyhenikö se ensimmäisen vuoden aikana, tutkimussuunnitelman mukaisesti kaikki 8Q12- ja 8Q16-ryhmien potilaat soveltuivat annosvälin pidennykseen (4 viikon lisäyksin) viikosta 52 alkaen seuraavien kriteerien täyttyessä:
- parhaan korjatun näöntarkkuuden (BCVA) lasku alle viidellä kirjaimella viikosta 12,
- keskeisen verkkokalvon paksuus < 300 mikrometriä SD-OTC:llä mitattuna (tai < 320 mikrometriä, jos mittaus sisältää RPE:n).
Potilaat, jotka eivät täyttäneet kriteereitä annosteluvälin lyhentämiseksi tai pidentämiseksi, jatkoivat entisellä annosteluvälillä. Lyhyin mahdollinen aika injektioiden välillä oli 8 viikkoa kaikissa ryhmissä.
Potilaat, joilla oli molemminpuolinen sairaus, soveltuivat saamaan Eylea 40 mg/ml -hoitoa toiseen silmään.
Potilaiden ominaisuudet lähtötilanteessa
Potilaat olivat 24–90-vuotiaita, keskimääräinen ikä oli 62,3 vuotta.
Noin 44 % (143/328) 8Q12-ryhmään ja 44 % (71/163) 8Q16-ryhmään satunnaistetuista potilaista oli vähintään 65-vuotiaita. Noin 11 % (36/328) ja 8Q12-ryhmään ja 14 % (14/163) 8Q16-ryhmään satunnaistetuista oli vähintään 75-vuotiaita.
Aiemmin diabeettiseen makulaturvotukseen hoitoa saaneiden potilaiden osuus tasapainotettiin hoitoryhmien välille (43,6 % 8Q12-ryhmässä; 43,6 % 8Q16-ryhmässä; 44,3 % 2Q8-ryhmässä).
Tulokset
48 viikkoa tutkimuksessa jatkaneiden 8Q12-ryhmän potilaiden saamien injektioiden mediaani (keskiarvo) oli 6,0 (6,0), 8Q16-ryhmän 5,0 (5,0) ja 2Q8-ryhmän 8,0 (7,9).
Viikolla 48 91,0 % 8Q12-ryhmän potilaista jatkoi Q12-hoitoväleillä, ja 89,1 % 8Q16-ryhmän potilaista jatkoi Q16-hoitoväleillä.
60 viikkoa tutkimuksessa jatkaneiden 8Q12-ryhmän potilaiden saamien injektioiden mediaani (keskiarvo) oli 7,0 (7,0), 8Q16-ryhmän 6,0 (6,0) ja 2Q8-ryhmän 10,0 (9,8). Viikolla 60 42,6 % 8Q12-ryhmän potilaista siirtyi 16 viikon hoitoväleihin ja 34,2 % 8Q16-ryhmän potilaista siirtyi 20 viikon hoitoväleihin.
96 viikkoa tutkimuksessa jatkaneiden 8Q12-ryhmän potilaiden saamien injektioiden mediaani (keskiarvo) oli 9,0 (9,5), 8Q16-ryhmän 8,0 (7,8) ja 2Q8-ryhmän 14,0 (13,8) injektiota.
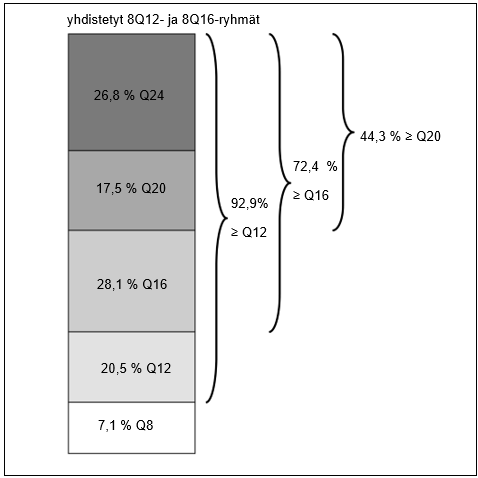
Viikolla 96 yhdistetyssä 8Q12- ja 8Q16-ryhmässä 72,4 % potilaista oli saavuttanut ≥ 16 viikon hoitovälin, 44,3 % ≥ 20 viikon hoitovälin ja 26,8 % 24 viikon hoitovälin niin, että näkö- ja anatomiatulokset säilyivät.
8Q12- ja 8Q16-hoidot osoitettiin vertailukelpoisiksi (non-inferior) 2Q8-hoidon kanssa ja niiden todettiin vastaavan kliinisesti 2Q8-hoitoa ensisijaisen tehoa koskevan päätetapahtuman ”parhaan korjatun näöntarkkuuden (BCVA) muutos lähtötilanteesta viikolle 48” sekä toissijaisen tehoa koskevan päätetapahtuman ”parhaan korjatun näöntarkkuuden (BCVA) keskimääräinen muutos viikolle 60” perusteella. Eylea 114,3 mg/ml -hoidon vaikutus parhaalla korjatulla näöntarkkuudella (BCVA) mitattuna jatkui viikolle 96.
Taulukko 7: Tehoa koskevat tulokset PHOTON-tutkimuksessa
| Tehoa kuvaava tulos | Viikko | Eylea 8Q12 (N = 328) | Eylea 8Q16 (N = 163) | Eylea 2Q8 (N = 167) |
| BCVA:n muutos lähtötilanteesta mitattuna ETDRS-kirjainpisteillä D | ||||
| Keskiarvo (SD), havaittu | 48 | 8,77 (8,95) | 7,86 (8,38) | 9,21 (8,99) |
| Keskimääräinen LS (SE) A | 8,10 (0,61) | 7,23 (0,71) | 8,67 (0,73) | |
Ero keskimääräisenä muutoksena, LS (95% CI) A,B | ‑0,57 (‑2,26; 1,13) | ‑1,44 (‑3,27; 0,39) | ||
| p‑arvo (yksisuuntainen non‑inferiority-testi, marginaali 4 kirjainta) A,B | <0,0001 | 0,0031 | ||
| Keskiarvo (SD), havaittu | 60 | 9,05 (9,27) | 7,96 (9,14) | 9,62 (9,58) |
| Keskimääräinen LS (SE) A | 8,52 (0,63) | 7,64 (0,75) | 9,40 (0,77) | |
Ero keskimääräisenä muutoksena, LS (95% CI) A,B | ‑0,88 (‑2,67; 0,91) | ‑1,76 (‑3,71; 0,19) | ||
| p‑arvo (yksisuuntainen non‑inferiority-testi, marginaali 4 kirjainta) A,B | 0,0003 | 0,0122 | ||
| Keskiarvo (SD), havaittu | 96 | 8,82 (9,93) | 7,50 (9,86) | 8,41 (11,10) |
| Keskimääräinen LS (SE) A | 8,15 (0,63) | 6,59 (0,77) | 7,70 (0,89) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) A,B | 0,45 (-1,55; 2,45) | -1,11 (-3,27; 1,05) | ||
| Potilaat, joiden ETDRS-kirjainpisteet olivat vähintään 69 (noin 20/40, Snellen-ekvivalentti) D | ||||
| Osuus (LOCF) | 48 | 65,3 % | 62,6 % | 63,0 % |
Osuuksien suhteellinen ero (95% CI) B,C | 2,45 % (‑6,47 %; 11,36 %) | ‑0,67 % (‑11,16 %; 9,82 %) | ||
| Osuus (LOCF) | 60 | 64,7 % | 62,0% | 60,6 % |
Osuuksien suhteellinen ero (95% CI) B,C | 4,34 % (‑4,72 %; 13,40 %) | 1,63 % (‑8,91 %; 12,17 %) | ||
| Osuus (LOCF) | 96 | 66,9 % | 61,3 % | 63,0 % |
| Osuuksien suhteellinen ero (95% CI) B,C | 4,01 % (-4.99 %, 13,01 %) | -1,51 % (-11,91 %, 8,89 %) | ||
| Potilaat, joiden BCVA parani vähintään 15 kirjainta lähtötilanteestaD | ||||
| Osuus (LOCF) | 48 | 18,7 % | 16,6 % | 23,0 % |
Osuuksien suhteellinen ero (95% CI) B,C | ‑4,64 % (‑12,30 %; 3,02 %) | ‑7,14 % (‑15,45 %; 1,17 %) | ||
| Osuus (LOCF) | 60 | 21,5 % | 16,0 % | 26,1 % |
Osuuksien suhteellinen ero (95% CI) B,C | ‑5,01 % (‑13,04 %; 3,02 %) | ‑10,78 % (‑19,27 %; ‑2,29 %) | ||
| Osuus (LOCF) | 96 | 24,5 % | 19,6 % | 26,1 % |
| Osuuksien suhteellinen ero (95% CI) B,C | -1,88 % (-10,03 %, 6,28 %) | -7,07 % (-15,94 %, 1,80 %) | ||
| Viimeisin suunniteltu hoitoväli | ||||
| Potilaat, joiden hoitoväli oli ≥ Q12 E | ||||
| Osuus (yhdistetyt 8Q12- ja 8Q16-ryhmät) | 96 | 92,9 % | - | |
| Osuus | 91,8 % | 95,0 % | - | |
| Potilaat, joiden hoitoväli oli ≥ Q16 E | ||||
| Osuus (yhdistetyt 8Q12- ja 8Q16-ryhmät) | 96 | 72,4 % | - | |
| Osuus | 64,1 % | 87,8 % | - | |
| Potilaat, joiden hoitoväli oli ≥ Q20 E | ||||
| Osuus (yhdistetyt 8Q12- ja 8Q16-ryhmät) | 96 | 44,3 % | - | |
| Osuus | 43,0 % | 46,8 % | - | |
| Potilaat, joiden hoitoväli oli Q24 E | ||||
| Osuus (yhdistetyt 8Q12- ja 8Q16-ryhmät) | 96 | 26,8 % | - | |
| Osuus | 23,8 % | 32,4 % | - | |
A Keskimääräinen LS, CI ja p-arvo perustuen toistomittausten sekamalliin (MMRM), jossa lähtötilanteen paras korjattu näöntarkkuus (BCVA) on kovariaattina ja hoitoryhmä tekijänä. Käyntiä ja ositusmuuttujia (maantieteellinen alue, lähtötilanteen BCVA-luokka) käytettiin sekä satunnaistamisen kiinteinä tekijöinä että lähtötilanteen BCVA:n ja käynnin sekä hoidon ja käynnin välisinä vuorovaikutusehtoina (interaction term).
B Absoluuttinen ero on Eylea 8Q12‑ tai 8Q16‑ryhmän arvo miinus 2Q8‑ryhmän arvo.
C Mantel‑Haenszel-painotettu hoitoero, jossa satunnaistamiseen käytettiin ositusmuuttujia (maantieteellinen alue, BCVA-luokka lähtötilanteessa), ja CI laskettiin normaalilla approksimaatiolla
D täysi analyysijoukko
E turvallisuusanalyysijoukko: potilaat, jotka olivat mukana tutkimuksessa tiettyyn aikapisteeseen asti
CI: luottamusväli
LOCF: viimeinen havainto eteenpäin
LS: pienin neliösumma
SD: keskihajonta
SE: keskivirhe
Hoitovälit analysoitiin etukäteen määrätyllä eksploratiivisella tavalla.
Kuva 6: Pienimmän neliösumman keskiarvon muutos BCVA:ssa mitattuna ETDRS-kirjainpisteinä lähtötilanteesta viikolle 96 (täysi analyysijoukko) PHOTON-tutkimuksessa
Kuva 7: Viimeisin suunniteltu hoitoväli viikolla 96
Eylea-valmisteen kaikilla annostuksilla (8Q12, 8Q16, 2Q8) osoitettiin merkittävää nousua ennalta määritellyssä toissijaisessa päätetapahtumassa, National Eye Institute Visual Function Questionnaire (NEI VFQ-25) -kyselyn pisteissä.
8Q12-, 8Q16- ja 2Q8-ryhmien välillä ei havaittu kliinisesti merkitseviä eroja NEI VFQ-25-kokonaispisteiden muutoksessa lähtötilanteesta viikolle 48 ja viikolle 96.
Kaikkien ikää, sukupuolta, maantieteellistä aluetta, etnisyyttä, rotua, lähtötilanteen BVCA:ta ja diabeettisen makulaturvotuksen aiempaa hoitoa koskevien arvioitavissa olevien alaryhmien tehoa koskevat tulokset vastasivat kokonaispopulaation tuloksia.
Teho säilyi yleisesti ottaen viikolle 96.
Aiemmin hoitoa saaneiden potilaiden alaryhmässä havaitut hoitotulokset vastasivat aiemmin hoitoa saamattomien potilaiden tuloksia.
Tulokset – PHOTON-tutkimuksen jatkovaihe
Tutkimuksen päävaiheen lopussa viikolla 96 potilaiden oli mahdollista osallistua 60 viikon pituiseen avoimeen jatkovaiheeseen. 195 potilasta, jotka oli alun perin määrätty 8Q12- ja 8Q16-ryhmiin, jatkoivat Eylea 114,3 mg/ml -hoitoa viimeisimmillä hoitoväleillään. 70 potilasta, jotka oli alun perin määrätty 2Q8-ryhmään tutkimuksen alussa, siirrettiin Eylea 114,3 mg/ml -hoitoon, joka aloitettiin 12 viikon hoitovälillä. Hoitovälejä voitiin mukauttaa edelleen lääkärin arvioimien näkö- ja/tai anatomiavasteiden perusteella.
Potilailla, jotka oli alun perin määrätty 8Q12- ja 8Q16-ryhmiin, Eylea 114,3 mg/ml -hoitovaikutus säilyi yleisesti ottaen 3 vuoden ajan (viikolle 156). Yhdistetyissä 8Q12- ja 8Q16-ryhmissä pienimmän neliösumman (LS) keskiarvon muutos lähtötilanteesta oli parhaassa korjatussa näöntarkkuudessa (BCVA) +7,2 kirjainta ja verkkokalvon keskeisen alueen paksuudessa (CRT) -192,4 mikrometriä viikolla 156.
Potilailla, jotka oli alun perin määrätty 2Q8-ryhmään, Eylea 114,3 mg/ml -hoitovaikutus oli samankaltainen. LS keskiarvon muutos lähtötilanteesta oli BCVA:ssa +6,5 kirjainta ja CRT:ssä ‑197,4 mikrometriä viikolla 156.
8Q12- ja 8Q16-ryhmien potilailla, jotka olivat mukana tutkimuksessa viikon 156 loppuun asti, potilaiden saamien injektioiden mediaani (keskiarvo) oli 8Q12-ryhmässä 13,0 (13,2) ja 8Q16-ryhmässä 11,0 (11,4).
Eylea 114,3 mg/ml -hoitoon siirtyneillä potilailla, jotka olivat mukana tutkimuksessa viikon 156 loppuun asti, potilaiden saamien injektioiden mediaani (keskiarvo) oli 19,0 (18,6), ja näistä injektioista 5,0 (4,8) annettiin Eylea 114,3 mg/ml -hoitoon siirtymisen jälkeen tutkimuksen jatkovaiheen 60 viikon aikana.
Yleinen turvallisuusprofiili oli jatkovaiheessa samankaltainen kuin tutkimuksen päävaiheessa.
Taulukko 8: Tehoa koskevat hoitotulokset PHOTON-tutkimuksen jatkovaiheessa viikolla 156
| Tehoa kuvaava tulos | 8Q12, jatkoi Eylea 114,3 mg/ml ‑hoitoa (N = 103) | 8Q16, jatkoi Eylea 114,3 mg/ml ‑hoitoa (N = 49) | 2Q8, siirtyi Eylea 114,3 mg/ml ‑hoitoon (N = 70) |
| BCVA:n muutos lähtötilanteesta (LS keskiarvo) | +6,8 kirjainta | +8,1 kirjainta | +6,5 kirjainta |
| CRT:n muutos lähtötilanteesta (LS keskiarvo) | -190,3 mikrometriä | -198,1 mikrometriä | -197,4 mikrometriä |
| Viimeinen suunniteltu hoitoväli A | |||
| ≥ 12 viikkoa | 85,4 % | 91,8 % | 82,8 % |
| ≥ 16 viikkoa | 62,1 % | 81,6 % | 50,0 % |
| ≥ 20 viikkoa | 40,8 % | 63,3 % | 19,0 % |
| 24 viikkoa | 20,4 % | 42,9 % | – B |
A perustuu potilaisiin, jotka olivat mukana tutkimuksessa viikon 156 loppuun asti
B Ei sovelleta alun perin 2Q8 ryhmään satunnaistettuihin potilaisiin tutkimusasetelman / tutkimuksen keston takia
Verkkokalvon laskimotukos
Tutkimuksen tavoitteet
Eylea 114,3 mg/ml -valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa monikeskustutkimuksessa (QUASAR) potilailla, joilla oli verkkokalvon laskimotukoksesta johtuva, aiemmin hoitamaton makulaturvotus.
Ensisijainen tavoite oli määrittää, onko 8 viikon välein (8Q8) annettava Eylea 114,3 mg/ml -hoito vertailukelpoinen (non-inferior) parhaan korjatun näöntarkkuuden (BCVA) muutoksena mitattuna 4 viikon välein (2Q4) annettun Eylea 40 mg/ml -hoidon kanssa.
Toissijaisina tavoitteina oli selvittää, vaatiiko 8Q8-hoito vähemmän injektioita kuin 2Q4-hoito, verrata Eylea 114,3 mg/ml- ja Eylea 40 mg/ml -hoitojen vaikutusta vastetta mitattaviin anatomia- ja muihin näkövasteisiin sekä arvioida afliberseptin turvallisuutta ja farmakokinetiikkaa.
Ensisijainen tehoa koskeva päätetapahtuma oli BCVA:n muutos ETDRS (Early Treatment Diabetic Retinopathy Study) -kirjainpisteillä mitattuna lähtötilanteesta viikolle 36.
Tärkein toissijainen päätetapahtuma oli aktiivisten eli vaikuttavien injektioiden lukumäärä lähtötilanteesta viikolle 64.
Muita toissijaisia päätetapahtumia olivat mm. aktiivisten injektioiden lukumäärä lähtötilanteesta viikolle 36, niiden potilaiden osuus, joiden paras korjattu näöntarkkuus (BCVA) parani vähintään 15 kirjainta lähtötilanteesta viikolle 36, sekä niiden potilaiden osuus, joiden ETDRS-kirjainpisteet olivat vähintään 69 (noin 20/40, Snellen-ekvivalentti) viikolla 36, sekä muutos National Eye Institute Visual Functioning Questionnaire-25 (NEI-VFQ-25) -kyselyn kokonaispisteissä lähtötilanteesta viikolle 36.
QUASAR-tutkimuksessa hoidettiin yhteensä 892 potilasta. Potilaat määrättiin suhteessa 1:1:1 yhteen kolmesta rinnakkaisesta hoitoryhmästä:
- Eylea 114,3 mg/ml -valmistetta annettiin ensin 3 injektiota 4 viikon välein ja sen jälkeen 8 viikon välein (8Q8/3)
- Eylea 114,3 mg/ml -valmistetta annettiin ensin 5 injektiota 4 viikon välein ja sen jälkeen 8 viikon välein (8Q8/5)
- Eylea 40 mg/ml -valmistetta annettiin 4 viikon välein (2Q4)
Viikosta 16 (8Q8/3), viikosta 24 (8Q8/5) ja viikosta 40 (2Q4, jos pidennetty aikaisemmin Q8:aan) alkaen annosteluväliä voitiin lyhentää 4 viikolla, jos molemmat seuraavista kriteereistä täyttyivät annostelukäynnillä:
- yli viiden kirjaimen lasku parhaassa korjatussa näöntarkkuudessa (BCVA) viitekäynnin jälkeen ja
- > 50 mikrometrin kasvu keskeisen verkkokalvon paksuudessa (CRT) viitekäynnin jälkeen.
Annosteluvälin pidentäminen sallittiin viikosta 32 (2Q4 ja 8Q8/3) tai viikosta 40 (8Q8/5) alkaen 4 viikon lisäyksin, jos molemmat seuraavista kriteereistä täyttyivät annostelukäynnillä:
- parhaan korjatun näöntarkkuuden (BCVA) lasku alle viidellä kirjaimella viitekäynnin jälkeen, ja
- keskeisen verkkokalvon paksuus < 320 mikrometriä SD-OTC:llä mitattuna (tai < 300 mikrometriä, jos mittaus ei sisältänyt RPE:tä).
Viitekäyntejä olivat viikon 12 käynti 8Q8/3-ryhmässä ja viikon 20 käynti 8Q8/5- ja 2Q4-ryhmissä.
Potilaat, jotka eivät täyttäneet kriteereitä annosteluvälin lyhentämiseksi tai pidentämiseksi, jatkoivat entisellä annosteluvälillä. Lyhyin mahdollinen aika injektioiden välillä oli 4 viikkoa kaikissa ryhmissä.
Potilaat, joilla oli molemminpuolinen sairaus, soveltuivat saamaan Eylea 40 mg/ml -hoitoa tai muuta anti-VEGF-valmistetta toiseen silmään.
Potilaiden ominaisuudet lähtötilanteessa
Potilaat olivat 23–95-vuotiaita, keskimääräinen ikä oli 65,9 vuotta.
Noin 57 % (168/293) 8Q8/3-ryhmään satunnaistetuista potilaista ja 57 % (170/298) 8Q8/5-ryhmään satunnaistetuista potilaista oli vähintään 65‑vuotiaita, ja noin 26 % (76/293) 8Q8/3-ryhmään satunnaistetuista potilaista ja 25 % (74/298) 8Q8/5-ryhmään satunnaistetuista potilaista oli vähintään 75‑vuotiaita.
Tutkimukseen otetuista potilaista 425:llä (48 %) oli verkkokalvon keskuslaskimotukos/ hemiretinaalinen laskimotukos ja 467:llä (52 %) oli verkkokalvon haaralaskimotukos. Eri alatyyppejä sairastavien potilaiden osuudet olivat hoitoryhmissä samankaltaiset.
Tulokset
Eylea 114,3 mg/ml -hoidon osoitettiin olevan vertailukelpoinen (non-inferior) 2Q4-hoidon kanssa ja sen todettiin vastaavan kliinisesti 2Q4-hoitoa ensisijaisen tehoa koskevan päätetapahtuman ”parhaan korjatun näöntarkkuuden (BCVA) muutos ETDRS-kirjainpisteinä mitattuna lähtötilanteesta viikolle 36” perusteella.
Lisäksi Eylea 114,3 mg/ml -hoidon paremmuus 2Q4-hoitoon verrattuna osoitettiin tärkeimmän toissijaisen tehoa koskevan päätetapahtuman ”aktiivisten injektioiden lukumäärä lähtötilanteesta viikolle 64” perusteella. Eylea 8Q8/3-ryhmän potilaat saivat 3,2 injektiota vähemmän kuin 2Q4-ryhmän potilaat.
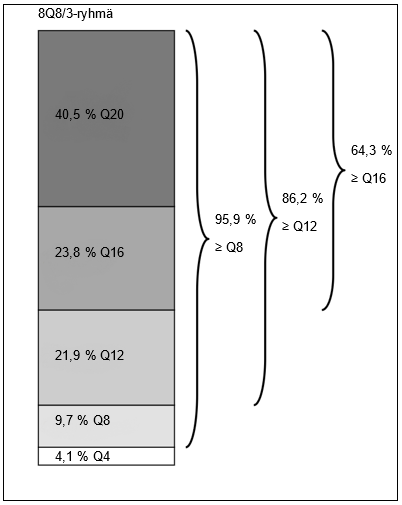
Viikolla 36 kaikkiaan 93,9 % 8Q8/3-ryhmän potilaista oli saavuttanut ≥ 8 viikon hoitovälin niin, että näkö- ja anatomiatulokset säilyivät.
Viikolla 64 kaikkiaan 56,1 % 8Q8/3-ryhmän potilaista oli saavuttanut 16 viikon hoitovälin niin, että näkö- ja anatomiatulokset säilyivät.
Viikolla 64 kaikkiaan 40,5 %:lla 8Q8/3-ryhmän potilaista viimeisin suunniteltu hoitoväli oli 20 viikkoa niin, että näkö- ja anatomiatulokset säilyivät.
Taulukko 9: Tehoa koskevat tulokset QUASAR-tutkimuksessa
| Tehoa kuvaava tulos | Viikko | Eylea 8Q8/3 (N = 293) | Eylea 2Q4 (N = 301) |
| BCVA:n muutos lähtötilanteesta mitattuna ETDRS-kirjainpisteillä A | |||
| Keskiarvo (SD), havaittu | 36 | 17,0 (11,8) | 17,8 (13,1) |
| Keskimääräinen LS (SE) B | 17,4 (0,7) | 17,5 (0,7) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) B, C | -0,1 (-2,0; 1,9) | ||
| p-arvo (yksisuuntainen non-inferiority-testi, marginaali 4 kirjainta) B, C | < 0,0001 | ||
| Keskiarvo (SD), havaittu | 64 | 17,3 (12,7) | 17,4 (14,6) |
| Keskimääräinen LS (SE) B | 17,8 (0,7) | 17,3 (0,8) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) B, C | 0,5 (-1,6; 2,7) | ||
| Verkkokalvon keskuslaskimotukos / hemiretinaalinen laskimotukosD | |||
| Keskiarvo (SD), havaittu | 36 | 16,5 (12,7) | 16,2 (14,7) |
| Keskimääräinen LS (SE) B | 16,6 (1,1) | 15,9 (1,2) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) B, C | 0,6 (-2,6; 3,9) | ||
| Keskiarvo (SD), havaittu | 64 | 16,5 (13,8) | 14,8 (16,8) |
| Keskimääräinen LS (SE) B | 17,2 (1,2) | 15,2 (1,3) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) B, C | 2,0 (-1,5; 5,6) | ||
| Verkkokalvon haaralaskimotukos D | |||
| Keskiarvo (SD), havaittu | 36 | 17,4 (10,9) | 19,4 (11,0) |
| Keskimääräinen LS (SE) B | 18,3 (0,8) | 19,0 (0,8) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) B, C | 0,8 (-2,9; 1,4) | ||
| Keskiarvo (SD), havaittu | 64 | 18,1 (11,8) | 20,1 (11,4) |
| Keskimääräinen LS (SE) B | 18,4 (0,9) | 19,6 (0,8) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) B, C | -1,1 (-3,5; 1,2) | ||
| Potilaat, joiden ETDRS-kirjainpisteet olivat vähintään 69 (noin 20/40, Snellen-ekvivalentti) A | |||
| Osuus (OC) | 36 | 72,7 % | 67,8 % |
| 64 | 70,4 % | 70,2 % | |
| Potilaat, joiden BCVA parani vähintään 15 kirjainta lähtötilanteesta A | |||
| Osuus (OC) | 36 | 58,8 % | 59,8 % |
| 64 | 61,7 % | 60,4 % | |
| Potilaat, joiden BCVA huononi vähintään 15 kirjainta lähtötilanteesta A | |||
| Osuus (OC) | 36 | 1,2 % | 1,5 % |
| 64 | 1,2 % | 2,4 % | |
| Potilaat, joilla ei ollut keskeisellä alueella verkkokalvon sisäistä eikä verkkokalvon alaista nestettä A | |||
| Osuus (OC) | 36 | 81,2 % | 83,7 % |
| 64 | 76,3 % | 66,0 % | |
| Vaikuttavien injektioiden lukumäärä | |||
| Keskiarvo (SD), havaittu E | 36 | 6,1 (0,6) | 8,8 (0,8) |
| Keskimääräinen LS (SE) F | 6,1 (0,0) | 8,8 (0,0) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) F, C | 2,7 (-2,8; -2,6) | ||
| Keskiarvo (SD), havaittu E | 64 | 8,4 (1,2) | 11,7 (1,6) |
| Keskimääräinen LS (SE) F | 8,5 (0,1) | 11,7 (0,1) | |
| Ero keskimääräisenä muutoksena, LS (95% CI) F, C | -3,2 (-3,5; -3,0) | ||
| p arvo (kaksisuuntainen paremmuustesti) G, C | < 0,0001 | ||
| Hoitovälien säilyminen | |||
| Potilaat, joiden hoitoväli säilyi tasolla ≥ Q8 E | |||
| Osuus | 36 | 88,5 % | – |
| 64 | 88,1 % | 70,0 % H | |
| Viimeinen käytetty hoitoväli | |||
| Potilaat, joiden hoitoväli oli Q4E | |||
| Osuus | 64 | 4,8 % | 13,0 % |
| Potilaat, joiden hoitoväli oli ≥ Q8 E | |||
| Osuus | 64 | 95,2 % | 87,0 % |
| Potilaat, joiden hoitoväli oli ≥ Q12E | |||
| Osuus | 64 | 81,4 % | 67,8 % |
| Potilaat, joiden hoitoväli oli Q16E | |||
| Osuus | 64 | 56,1 % | – |
| Viimeinen suunniteltu hoitoväli | |||
| Potilaat, joiden hoitoväli oli ≥ Q8E | |||
| Osuus | 36 | 93,9 % | 75,6 % |
| 64 | 95,9 % | 92,2 % | |
| Potilaat, joiden hoitoväli oli Q12E | |||
| Osuus | 36 | 69,1 % | – |
| 64 | 21,9 % | 27,8 % | |
| Potilaat, joiden hoitoväli oli ≥ Q12E | |||
| Osuus | 64 | 86,2 % | 77,8 % |
| Potilaat, joiden hoitoväli oli ≥ Q16E | |||
| Osuus | 64 | 64,3 % | 50,0 % |
| Potilaat, joiden hoitoväli oli Q20E | |||
| Osuus | 64 | 40,5 % | – |
A täysi analyysijoukko
B Keskimääräinen LS, CI ja p-arvo perustuen toistomittausten sekamalliin (MMRM), jossa lähtötilanteen parhaan korjatun näöntarkkuuden (BCVA) mittaus on kovariaattina ja hoitoryhmä tekijänä. Käyntiä ja ositusmuuttujia (maantieteellinen alue, lähtötilanteen BCVA-luokka ja verkkokalvon laskimotukoksen tyyppi) käytettiin sekä satunnaistamisen kiinteinä tekijöinä että lähtötilanteen BCVA-mittauksen ja käynnin sekä hoidon ja käynnin välisinä vuorovaikutusehtoina (interaction term).
C Absoluuttinen ero on Eylea 8Q8/3-ryhmän arvo miinus 2Q4-ryhmän arvo.
D Verkkokalvon keskuslaskimotukosta / hemiretinaalista laskimotukosta sairastavien potilaiden lukumäärä oli 8Q8/3-hoitoryhmässä 134 ja 2Q4-hoitoryhmässä 152. Verkkokalvon haaralaskimotukosta sairastavien potilaiden lukumäärä oli 8Q8/3-hoitoryhmässä 159 ja 2Q4-hoitoryhmässä 149.
E Turvallisuusanalyysijoukko: potilaat, jotka olivat mukana tutkimuksessa tiettyyn aikapisteeseen asti
F Keskimääräinen LS ja CI perustuen moni-imputointimenettelyyn, johon sovellettua lineaarista regressiomallia oli mukautettu lähtötilanteen BCVA:n ja lähtötilanteen keskeisen verkkokalvon paksuuden (CST) suhteen. Satunnaistamisen ositusmuuttujia (maantieteellinen alue, lähtötilanteen BCVA-luokka ja verkkokalvon laskimotukoksen tyyppi) sovellettiin kuhunkin imputoituun tietojoukkoon ja yhdistettyihin tuloksiin Rubinin säännön mukaisesti.
G p‑arvo perustuen moni-imputointimenettelyyn, johon sovellettua ei-parametrista kovarianssianalyysia oli mukautettu lähtötilanteen BCVA:n ja lähtötilanteen keskeisen verkkokalvon paksuuden (CST) suhteen. Satunnaistamisen ositusmuuttujia (maantieteellinen alue, lähtötilanteen BCVA-luokka ja verkkokalvon laskimotukoksen tyyppi) sovellettiin kuhunkin imputoituun tietojoukkoon ja yhdistettyihin tuloksiin Rubinin säännön mukaisesti.
H 2Q4-hoitoryhmän potilaat, joiden hoitoväliä pidennettiin viikolla 32 ja joiden hoitoväli säilyi tasolla ≥ Q8 viikolle 64.
CI: luottamusväli
ETDRS: Early Treatment Diabetic Retinopathy Study
OC: havaitut tapaukset; samanaikaisen tapauksen esiintymisen jälkeiset tiedot suljettiin pois ensisijaisen arviointistrategian mukaisesti.
LS: pienin neliösumma
MMRM: toistomittausten sekamalli
SD: keskihajonta
SE: keskivirhe
Kuva 8: Pienimmän neliösumman keskiarvon muutos BCVA:ssa mitattuna ETDRS-kirjainpisteinä lähtötilanteesta viikolle 64 (täysi analyysijoukko) QUASAR-tutkimuksessa
Eylea-valmisteen kaikilla annostuksilla (8Q8/3, 2Q4) osoitettiin merkittävää nousua lähtötilanteesta ennalta määritellyssä toissijaisessa tehoa koskevassa päätetapahtumassa, National Eye Institute Visual Function Questionnaire (NEI VFQ-25) -kyselyn pisteissä. Näiden muutosten suuruus oli samanlainen kuin julkaistuissa tutkimuksissa ja näkyi näköön liittyvän elämänlaadun paranemisena.
8Q8/3- ja 2Q4-ryhmien välillä ei havaittu kliinisesti merkittäviä eroja NEI VFQ-25 -kokonaispisteiden muutoksessa lähtötilanteesta viikolle 36 ja viikolle 64.
Verkkokalvon laskimotukoksen alatyyppiä, ikää, sukupuolta, maantieteellistä aluetta, etnisyyttä, rotua, lähtötilanteen BVCA:ta ja lähtötilanteen CRT:tä koskevien ennalta määriteltyjen ja arvioitavissa olevien alaryhmien tehoa kuvaavat tulokset vastasivat kokonaispopulaation tuloksia.
Teho säilyi yleisesti ottaen viikolle 64.
Ennalta määritellyssä toissijaisessa päätetapahtumassa ”osallistujat, joiden annosteluväli oli ainoastaan 8Q8 viikolle 36 asti Eylea 114,3 mg/ml -ryhmässä” 88,5 %:lla 8Q8/3-ryhmän potilaista hoitoväli säilyi 8 viikossa eli alkuperäisenä satunnaistamisvaiheen hoitovälinä niin, että näkö- ja anatomiavasteet säilyivät.
Kuva 9: Viimeisin suunniteltu hoitoväli viikolla 64
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset afliberseptin käytöstä kostean silmänpohjan ikärappeuman, diabeettisen makulaturvotuksen ja verkkokalvon laskimotukoksen hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen/jakautuminen
Aflibersepti imeytyy hitaasti silmästä systeemiseen verenkiertoon lasiaiseen antamisen jälkeen, ja sitä havaitaan systeemisessä verenkierrossa lähinnä inaktiivisena, stabiilina kompleksina VEGF:n kanssa. Kuitenkin vain ”vapaa aflibersepti” voi sitoa endogeenistä VEGF:ää.
Toisen silmän lasiaiseen annetun 8 mg:n afliberseptiannoksen jälkeen vapaan afliberseptin keskimääräinen (keskihajonta) Cmax plasmassa oli 0,25 (0,21) mg/l ja plasman enimmäispitoisuuden saavuttamiseen kuluneen ajan mediaani oli 1 vuorokausi kosteaa silmänpohjan ikärappeumaa ja diabeettista makulaturvotusta sairastavien potilaiden yhdistetyn populaation farmakokineettisen analyysin perusteella. Vapaan afliberseptin kertyminen plasmaan ensimmäisten kolmen kuukausittaisen annoksen jälkeen oli minimaalista. Tämän jälkeen kertymistä ei havaittu. Näitä tietoja tukevat myös populaatiofarmakokineettiset analyysit.
Nämä farmakokineettiset tulokset olivat yhdenmukaisia verkkokalvon laskimotukosta sairastavista potilaista saatujen tietojen kanssa.
Eliminaatio
Aflibersepti on proteiinipohjainen lääkevalmiste, eikä metaboliaa koskevia tutkimuksia ole tehty.
Afliberseptin odotetaan eliminoituvan sekä kohdevälitteisen jakautumisen (target-mediated disposition) kautta vapaaseen endogeeniseen VEGF:ään sitoutumalla sekä proteolyysin kautta metaboloitumalla. Lasiaiseen annetun 8 mg:n vapaan afliberseptiannoksen laskun mediaanikesto viimeiseen mitattavissa olevaan pitoisuuteen oli 3 viikkoa.
Munuaisten tai maksan vajaatoiminta
Eylea 114,3 mg/ml -valmistetta ei ole tutkittu erikseen munuaisten tai maksan vajaatoimintaa sairastavilla potilailla.
Lievää tai vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden systeeminen altistus afliberseptille vastasi niiden potilaiden altistusta, joiden munuaistoiminta oli normaalia. Rajoitetut saatavilla olevat tiedot potilaista, joilla on lievä maksan vajaatoiminta, eivät osoita vaikutusta systeemiseen afliberseptialtistukseen verrattuna potilaisiin, joiden maksan toiminta oli normaalia.
Prekliiniset tiedot turvallisuudesta
Lasiaiseen annetulla afliberseptillä hoidettujen apinoiden nenän kuorikoissa havaittiin hengitysepiteelin eroosiota ja haavaumia systeemisillä altistuksilla, jotka olivat ihmisen enimmäisaltistusta suurempia. Systeeminen altistus vapaalle afliberseptille, joka perustui Cmax- ja AUC-arvoihin, oli noin 26 ja 33 kertaa korkeampi verrattuna aikuispotilaiden vastaaviin arvoihin lasiaiseen annetun 8 mg:n annoksen jälkeen. NOAEL-pitoisuudella (No Observed Adverse Effect Level) 0,5 mg / silmä apinoiden systeeminen altistus oli 3,2 kertaa suurempi Cmax-arvon perusteella ja 3,8 kertaa suurempi AUC-arvon perusteella verrattuna aikuispotilaiden vastaaviin arvoihin.
Afliberseptin mutageenisistä tai karsinogeenisistä ominaisuuksista ei ole tehty tutkimuksia.
Afliberseptin osoitettiin vaikuttavan kohdunsisäiseen kehitykseen kaniinien alkio-sikiö-kehitystutkimuksessa, kun ainetta annettiin laskimoon (3–60 mg/kg) ja ihon alle (0,1–1 mg/kg). Emon NOAEL-tasot olivat 3 mg/kg laskimoon annettuna ja 1 mg/kg ihon alle annettuna. Kehitykseen vaikuttavaa NOAEL-tasoa ei määritetty. Annoksella 0,1 mg/kg systeeminen altistus vapaalle afliberseptille oli noin 1,0 kertaa korkeampi Cmax-arvon perusteella ja 1,0 kertaa korkeampi kumulatiivisen AUC-arvon perusteella verrattuna aikuispotilaiden vastaaviin arvoihin lasiaiseen annetun 8 mg:n annoksen jälkeen.
Vaikutukset miesten ja naisten hedelmällisyyteen arvioitiin osana 6 kuukauden tutkimusta apinoilla, joille annettiin laskimoon afliberseptiannos 3–30 mg/kg. Puuttuvat tai epäsäännölliset kuukautiset liittyivät naisten lisääntymishormonipitoisuuksien muutoksiin, ja siemennesteen morfologian ja siittiöiden liikkuvuuden muutoksia havaittiin kaikilla annostasoilla. 3 mg:n/kg laskimoon annetun annoksen kohdalla vapaan afliberseptin systeeminen altistus oli noin 377 kertaa suurempi Cmax-arvon perusteella ja 104 kertaa suurempi AUC-arvon perusteella kuin ihmisen altistus lasiaiseen annetun 8 mg:n annoksen jälkeen. Kaikki muutokset olivat palautuvia.
Farmaseuttiset tiedot
Apuaineet
Sakkaroosi
Arginiinihydrokloridi
Histidiinihydrokloridimonohydraatti
Histidiini
Polysorbaatti 20
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Eylea 114,3 mg/ml injektioneste, liuos
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Ennen käyttöä avaamaton injektiopullo voidaan säilyttää poissa jääkaapista alle 25 °C:ssa enintään 24 tunnin ajan.
Eylea 114,3 mg/ml injektioneste, liuos, esitäytetyssä ruiskussa
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä esitäytetty ruisku läpipainopakkauksessa ja ulkopakkauksessa. Herkkä valolle.
Ennen käyttöä läpipainopakkaus voidaan säilyttää poissa jääkaapista alle 25 °C:ssa enintään 24 tunnin ajan.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
EYLEA injektioneste, liuos
114,3 mg/ml (L:ei) 1 kpl (0,263 ml) (748,01 €)
EYLEA injektioneste, liuos, esitäytetty ruisku
114,3 mg/ml (L:ei) 1 kpl (0,184 ml) (748,01 €)
PF-selosteen tieto
Eylea 114,3 mg/ml injektioneste, liuos
Injektiopullo (tyypin I lasia), jossa on harmaa kumitulppa (klorobutyyli) sekä alumiinikorkki, jossa on valkoinen kansi, ja 18 G:n, 5 mikrometrin suodatinneula.
Yksi injektiopullo sisältää 0,263 ml liuosta.
Pakkauskoko: 1 injektiopullo ja 1 suodatinneula.
Eylea 114,3 mg/ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin I lasia), jossa on harmaa tulppa (elastomeerikumia), valkoinen Luer-lock-adapteri ja harmaa kärkikorkki (elastomeerikumia) ja sininen OcuClick-annostelujärjestelmä (PC/ABS-muovia).
Yksi esitäytetty ruisku sisältää 0,184 ml liuosta.
Pakkauskoko: 1 esitäytetty ruisku
Valmisteen kuvaus:
Kirkas tai hieman opalisoiva, väritön tai vaaleankeltainen iso-osmoottinen liuos, pH 5,8.
Käyttö- ja käsittelyohjeet
Eylea 114,3 mg/ml injektioneste, liuos
Injektiopullo on kertakäyttöinen ja yhden silmän hoitoon. Useiden annosten ottaminen yhdestä injektiopullosta voi lisätä kontaminaation riskiä ja sen seurauksena infektion riskiä.
Älä käytä, jos pakkaus tai sen osat ovat vanhentuneet tai vahingoittuneet tai niihin on kajottu.
Tarkista injektiopullon etiketistä, että käytössäsi on oikean vahvuinen Eylea-valmiste. 8 mg:n annos edellyttää Eylea 114,3 mg/ml -injektiopullon käyttöä.
18 G:n, 5 mikrometrin suodatinneula:
- Blunt fill -suodatinneula, ei pistämiseen ihon läpi.
- Blunt fill -suodatinneulaa ei saa autoklavoida.
- Suodatinneula on pyrogeeniton. Älä käytä sitä, jos sen pakkaus on vahingoittunut.
- Hävitä blunt fill -suodatinneula terävälle lääkejätteelle tarkoitettuun keräysastiaan.
- Varoitus: Suodatinneulan uudeelleenkäyttäminen voi aiheuttaa infektion tai muun sairauden/vamman.
Lasiaiseen annettavaan injektioon on käytettävä 30 G:n × ½ tuuman kokoista injektioneulaa (ei sisälly pakkaukseen). Suositeltua 30 G:n × ½ tuuman injektioneulaa pienemmän neulakoon (suurempi gauge) käyttö voi aiheuttaa suuremman injektiovoiman.
| 1. | Tarkista injektioneste silmämääräisesti ennen antoa. Älä käytä injektiopulloa, jos siinä on näkyviä hiukkasia, sameutta tai värimuutoksia. | |
| 2. | Poista muovikorkki ja desinfioi injektiopullon kumitulpan ulkopuoli. | 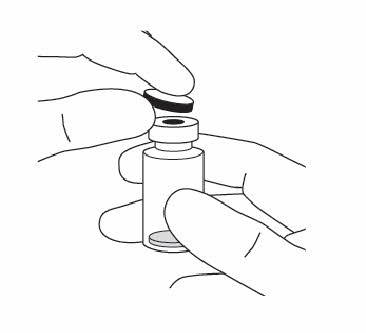 |
| 3. | Tee vaiheet 3–10 aseptista tekniikkaa käyttäen. Liitä pakkauksessa tuleva suodatinneula 1 ml:n steriiliin Luer-lock-ruiskuun. | 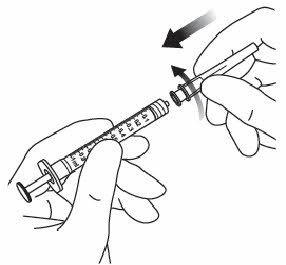 |
| 4. | Työnnä suodatinneulaa injektiopullon tulpan keskiosaan, kunnes neula on kokonaan työntynyt injektiopulloon ja kärki koskee injektiopullon pohjaan tai pohjan reunaan. | |
| 5. | Vedä Eylea-injektiopullon koko sisältö ruiskuun samalla, kun pidät injektiopulloa pystysuorassa ja hieman kallistetussa asennossa. Tämä asento helpottaa koko sisällön vetämistä ruiskuun. Estääksesi ilman pääsyn ruiskuun varmista, että suodatinneulan kärki on nesteen pinnan alla. Pidä pullo kallellaan, kun vedät nestettä ruiskuun, niin että suodatinneulan kärki on nesteen pinnan alapuolella. | |
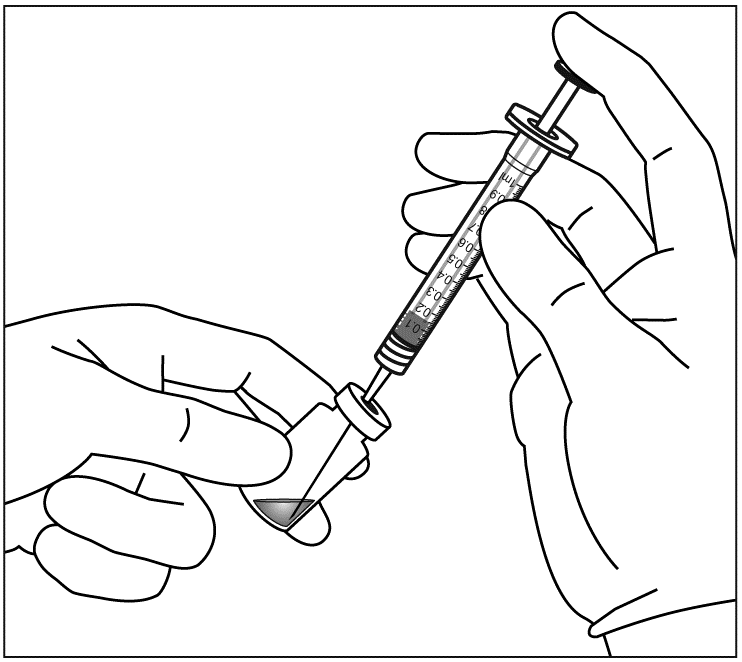 | 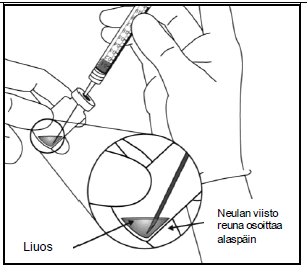 | |
| 6. | Varmista, että mäntä on vedetty tarpeeksi taakse, kun tyhjennät injektiopulloa. Näin suodatinneula tyhjenee varmasti kokonaan. Injektion jälkeen käyttämätön valmiste on hävitettävä. | |
| 7. | Poista suodatinneula ja hävitä se asianmukaisesti. Huomautus: suodatinneulaa ei saa käyttää lasiaisinjektioon. | |
| 8. | Kiinnitä 30 G:n × ½ tuuman injektioneula tiukasti Luer-lock-ruiskun kärkeen. | 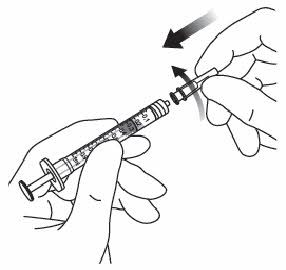 |
| 9. | Pidä ruiskua niin, että neula osoittaa ylöspäin, ja tarkista ruisku kuplien varalta. Jos ruiskussa näkyy kuplia, naputa ruiskua varovasti sormella, kunnes kuplat nousevat pinnalle. |  |
| 10. | Poista kaikki kuplat ja ylimääräinen lääkevalmiste painamalla mäntää hitaasti niin, että männän litteä reuna kohdistuu ruiskussa olevaan 0,07 ml:n annosviivaan. | |
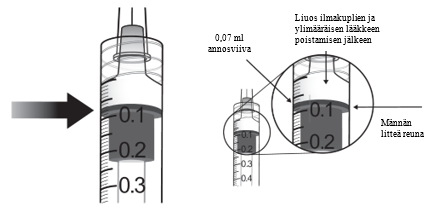 | ||
Eylea 114,3 mg/ml injektioneste, liuos, esitäytetyssä ruiskussa
OcuClick-annostelujärjestelmällä varustettu Esitäytetty ruisku on kertakäyttöinen ja yhden silmän hoitoon. Useiden annosten ottaminen yhdestä OcuClick-annostelujärjestelmällä varustetusta esitäytetystä ruiskusta voi lisätä kontaminaation riskiä ja sen seurauksena infektion riskiä.
Älä käytä, jos pakkaus tai sen osat ovat vanhentuneet tai vahingoittuneet tai niihin on kajottu. Tarkista OcuClick-annostelujärjestelmällä varustetun esitäytetyn ruiskun etiketistä, että käytössäsi on oikean vahvuinen Eylea-valmiste. 8 mg:n annos edellyttää Eylea 114,3 mg/ml esitäytetyn ruiskun käyttöä.
Lasiaiseen annettavaan injektioon on käytettävä 30 G:n × ½ tuuman injektioneulaa (ei sisälly pakkaukseen). Suositeltua 30 G:n × ½ tuuman injektioneulaa pienemmän neulakoon (suurempi gauge) käyttö voi aiheuttaa suuremman injektiovoiman.
| OcuClick-annostelujärjestelmällä varustetun esitäytetyn ruiskun kuvaus | ||
 | ||
| 1. | Valmistele Kun olet valmis antamaan Eylea 114,3 mg/ml -valmisteen, avaa pahvipakkaus ja ota steriili läpipainopakkaus. Tee vaiheet 2–9 aseptista tekniikkaa käyttäen. | |
| 2. | Ota ruisku Ota ruisku steriloidusta läpipainopakkauksesta. | |
| 3. | Tarkista ruisku ja injektioneste Älä käytä esitäytettyä ruiskua, jos | |
| 4. | Napsauta ruiskun korkki irti Napsauta (älä kierrä) ruiskun korkki irti pitämällä ruiskua yhdellä kädellä ja ruiskun korkkia toisen käden peukalolla ja etusormella. Huomautus: älä vedä mäntää taaksepäin. |  |
| 5. | Kiinnitä neula Kiinnitä 30 G:n × ½ tuuman injektioneula tiukasti Luer-lock-ruiskun kärkeen. |  |
| 6. | Naputa ilmakuplat pintaan Pidä ruiskua neula ylöspäin ja tarkista näkyykö ruiskussa kuplia. Jos ruiskussa on kuplia, naputa ruiskua varovasti sormella, kunnes kuplat nousevat pinnalle. |  |
| 7. | Poista ilmakuplat ja ylimääräinen lääkevalmiste Ruiskussa ei ole annosviivaa, koska se on suunniteltu asettamaan annos mekaanisesti kuten alla olevissa vaiheissa on selitetty. Valmistelu ja annoksen asettaminen on tehtävä noudattamalla seuraavia vaiheita. | |
 | 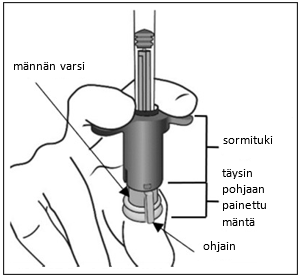 | |
| 8. | Aseta annos Käännä männän varren päätä 90 astetta myötä- tai vastapäivään, kunnes männän varren ohjain kohdistuu uran kanssa. Saatat kuulla naksahduksen. |  |
| 9. | Anna injektio Pistä neula silmän injektiokohtaan. Injisoi liuos painamalla mäntää, kunnes se pysähtyy, eli kunnes ohjain on täysin uran sisällä. Älä käytä ylimääräistä voimaa, kun ohjain on uran sisällä. On normaalia, että ruiskuun jää pieni määrä liuosta. |  |
| 10. | Esitäytetty ruisku on tarkoitettu yhden annoksen antoon, ja se on kertakäyttöinen. Hävitä käytetty ruisku injektion jälkeen terävälle jätteelle tarkoitettuun astiaan. | |
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
EYLEA injektioneste, liuos
114,3 mg/ml 1 kpl
EYLEA injektioneste, liuos, esitäytetty ruisku
114,3 mg/ml 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Aflibersepti ja farisimabi: Aikuisten neovaskulaarisen (kostean) silmänpohjan ikärappeuman (AMD) hoito (3094).
ATC-koodi
S01LA05
Valmisteyhteenvedon muuttamispäivämäärä
15.01.2026
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi