
LITFULO kapseli, kova 50 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Lääkkeen määrääjän opas
Potilas
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää ritlesitinibitosylaattia määrän, joka vastaa 50 mg:aa ritlesitinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kova kapseli sisältää 21,27 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova (kapseli)
Kliiniset tiedot
Käyttöaiheet
Litfulo on tarkoitettu vaikean pälvikaljun (alopecia areata) hoitoon aikuisille ja vähintään 12-vuotiaille nuorille (ks. kohta Farmakodynamiikka).
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Hoidon saa aloittaa vain lääkäri, jolla on kokemusta pälvikaljun diagnosoinnista ja hoidosta, ja hänen on valvottava hoitoa.
Annostus
Suositeltu annos on 50 mg kerran vuorokaudessa.
Hoidon hyötyjä ja haittoja pitää arvioida yksilöllisesti säännöllisin väliajoin.
Hoidon lopettamista on harkittava, jos potilaalla ei todeta näyttöä hoidon hyödystä 36 viikon jälkeen.
Laboratorioseuranta
Taulukko 1. Laboratorioarvot ja seurantaohjeet
Laboratorioarvot | Seurantaohje | Toiminta |
Verihiutalemäärä | Ennen hoidon aloitusta, 4 viikkoa aloituksen jälkeen ja sen jälkeen tavanomaisen hoitokäytännön mukaisesti. | Hoito pitää lopettaa, jos verihiutalemäärä on < 50 × 109/l. |
Lymfosyytit | Hoito pitää keskeyttää, jos absoluuttinen lymfosyyttimäärä on < 0,5 × 109/l, ja se voidaan aloittaa uudelleen, kun absoluuttinen lymfosyyttimäärä on palautunut tätä arvoa suuremmaksi. |
Hoidon aloitus
Ritlesitinibihoitoa ei pidä aloittaa potilaille, joiden absoluuttinen lymfosyyttimäärä on < 0,5 × 109/l tai verihiutalemäärä on < 100 × 109/l (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoidon keskeyttäminen tai lopettaminen
Jos potilaalle ilmaantuu vakava infektio tai opportunistinen infektio, ritlesitinibihoito pitää keskeyttää, kunnes infektio on saatu hallintaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoito voi olla tarpeen keskeyttää tai lopettaa hematologisten poikkeavuuksien hoitamiseksi taulukossa 1 kuvatun mukaisesti.
Jos hoito on tarpeen keskeyttää, merkittävän hiustenlähdön riski alueilla, joille hiukset ovat kasvaneet takaisin, on alle 6 viikon mittaisen tauon jälkeen pieni.
Väliin jääneet annokset
Jos annos jää väliin, potilasta pitää neuvoa ottamaan annos mahdollisimman pian, paitsi jos seuraavaan annokseen on alle 8 tuntia; tällöin potilaan ei pidä ottaa väliin jäänyttä annosta. Tämän jälkeen annostelua jatketaan tavanomaiseen aikaan.
Erityisryhmät
Munuaisten vajaatoiminta
Lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Ritlesitinibia ei ole tutkittu potilailla, joilla on loppuvaiheen munuaissairaus (ESRD) tai munuaissiirre, ja siksi sitä ei suositella näille potilaille.
Maksan vajaatoiminta
Lievää (Child–Pugh A) tai keskivaikeaa (Child–Pugh B) maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka). Ritlesitinibi on vasta-aiheista potilaille, joilla on vaikea (Child–Pugh C) maksan vajaatoiminta (ks. kohta Vasta-aiheet).
Iäkkäät
Iäkkäiden ≥ 65-vuotiaiden potilaiden annosta ei tarvitse muuttaa. ≥ 65-vuotiaista potilaista on vähän tietoja.
Pediatriset potilaat
12 – < 18-vuotiaiden potilaiden annosta ei tarvitse muuttaa.
Litfulo-valmisteen turvallisuutta ja tehoa alle 12 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Litfulo otetaan kerran päivässä ruokailun yhteydessä tai tyhjään mahaan.
Kapselit niellään kokonaisina eikä niitä pidä murskata, jakaa eikä pureskella, sillä näitä antotapoja ei ole tutkittu kliinisissä tutkimuksissa.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Vakavat aktiiviset infektiot, tuberkuloosi mukaan lukien (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Vaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
- Raskaus ja imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Vakavat infektiot
Ritlesitinibia saavilla potilailla on ilmoitettu vakavia infektioita. Yleisimpiä vakavia infektioita ovat olleet umpilisäketulehdus, COVID-19-infektio (mukaan lukien keuhkokuume) ja sepsis. Ritlesitinibihoitoa ei saa aloittaa potilaille, joilla on aktiivinen, vakava infektio (ks. kohta Vasta-aiheet).
Hoidon hyötyjä ja riskejä on arvioitava potilailla,
- joilla on pitkäkestoinen tai toistuva infektio
- jotka ovat altistuneet tuberkuloosille
- joilla on aiemmin ollut jokin vakava tai opportunistinen infektio
- jotka ovat asuneet tai matkustaneet alueilla, joilla esiintyy endeemistä tuberkuloosia tai sienitauteja, tai
- joilla on jokin perussairaus, joka voi altistaa infektiolle.
Potilaita pitää seurata tarkkaan ritlesitinibihoidon aikana ja sen jälkeen infektioiden merkkien ja oireiden havaitsemiseksi. Hoito on keskeytettävä, jos potilaalle ilmaantuu vakava tai opportunistinen infektio. Jos potilaalle ilmaantuu uusi infektio hoidon aikana, hänelle on tehtävä viipymättä kattavat immuunipuutteisiin potilaisiin sovellettavat diagnostiset kokeet, aloitettava asianmukainen mikrobilääkehoito ja potilaan tilaa on seurattava tarkkaan. Keskeytetty ritlesitinibihoito voidaan aloittaa uudelleen, kun infektio on saatu hallintaan.
Infektioiden ilmaantuvuus on iäkkäillä ja diabetesta sairastavilla yleisesti tavanomaista suurempi, joten iäkkäiden ja diabetesta sairastavien potilaiden hoidossa on noudatettava varovaisuutta ja infektioiden ilmaantumiseen on kiinnitettävä erityistä huomiota.
Tuberkuloosi
Potilaat on seulottava tuberkuloosin varalta ennen ritlesitinibihoidon aloittamista. Ritlesitinibia ei saa antaa potilaille, joilla on aktiivinen tuberkuloosi (ks. kohta Vasta-aiheet). Tuberkuloosilääkitys on aloitettava ennen ritlesitinibihoidon aloittamista, jos potilaalla on hiljattain todettu piilevä tuberkuloosi tai aiemmin hoitamaton piilevä tuberkuloosi. Tuberkuloosilääkitystä on myös harkittava ennen ritlesitinibihoidon aloitusta potilaille, joilla on suuri riski ja piilevän tuberkuloosin toteamiseksi tehdyn testin tulos on negatiivinen. Lisäksi seulontaa on harkittava potilaille, joilla on suuri tuberkuloosiriski ritlesitinibihoidon aikana.
Virusten uudelleenaktivoituminen
Virusten uudelleenaktivoitumista, mm. herpesviruksen uudelleenaktivoitumista (esim. Herpes zoster), on raportoitu (ks. kohta Haittavaikutukset). Jos potilaalle ilmaantuu vyöruusu (Herpes zoster), on harkittava hoidon keskeyttämistä, kunnes vyöruusu on parantunut.
Virushepatiitin seulonta pitää tehdä kliinisten suositusten mukaisesti ennen ritlesitinibihoidon aloittamista. Ritlesitinibilla tehtyihin tutkimuksiin ei otettu mukaan potilaita, joiden osalta oli näyttöä B- tai C-hepatiitti-infektiosta. Kliinisten suositusten mukaista seurantaa virushepatiitin uudelleenaktivoitumisen toteamiseksi suositellaan ritlesitinibihoidon aikana. Jos uudelleenaktivoitumisesta on näyttöä, on konsultoitava maksatauteihin erikoistunutta lääkäriä.
Syöpä (mukaan lukien ei-melanoottiset ihosyövät)
Ritlesitinibia saaneilla potilailla on raportoitu syöpiä, myös ei-melanoottista ihosyöpää.
Ei tiedetä, voiko selektiiviseen JAK3:n estoon liittyä sellaisia Janus-kinaasien (JAK) estoon liittyviä haittavaikutuksia, joissa pääasiassa JAK1 ja JAK2 ovat osallisina. Laajassa, satunnaistetussa, aktiivikontrolloidussa tutkimuksessa tofasitinibilla (eräs JAK-estäjä) oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä havaittiin, että tuumorinekroositekijän (TNF:n) estäjiin verrattuna tofasitinibia saaneilla potilailla syöpien, erityisesti keuhkosyövän, lymfooman ja ei-melanoottisen ihosyövän, ilmaantuvuus oli suurempi.
Kliinisistä tutkimuksista on vähän tietoa ritlesitinibille altistumisen ja syövän kehittymisen välisen mahdollisen yhteyden arvioimiseksi. Pitkäaikaiset turvallisuusarvioinnit ovat meneillään. Ritlesitinibihoidon riskejä ja hyötyjä on arvioitava ennen kuin hoito aloitetaan tai sitä jatketaan potilaalle, jolla tiedetään olevan syöpä, lukuun ottamatta onnistuneesti hoidettua ei-melanoottista ihosyöpää tai kohdunkaulan syöpää.
Jos potilaalla on tavanomaista suurempi ihosyöpäriski, säännöllistä ihon tutkimista suositellaan.
Merkittävät sydän- ja verisuonitapahtumat (MACE), syvä laskimotukos (SLT) ja keuhkoembolia
Ritlesitinibia saaneilla potilailla on raportoitu tromboembolisia laskimo- ja valtimotapahtumia, mukaan lukien merkittäviä sydän- ja verisuonitapahtumia.
Ei tiedetä, voiko selektiiviseen JAK3:n estoon liittyä sellaisia Janus-kinaasien (JAK) estoon liittyviä haittavaikutuksia, joissa pääasiassa JAK1 ja JAK2 ovat osallisina.. Laajassa, satunnaistetussa, aktiivikontrolloidussa tutkimuksessa tofasitinibilla (eräs JAK-estäjä) oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä havaittiin, että TNF:n estäjiin verrattuna tofasitinibia saaneilla potilailla merkittävien sydän- ja verisuonitapahtumien, jotka määriteltiin sydän- ja verisuonitapahtumiin liittyviksi kuolemantapauksiksi, kuolemaan johtamattomiksi sydäninfarkteiksi, ja kuolemaan johtamattomiksi aivohalvauksiksi, ilmaantuvuus oli suurempi. Myös tromboembolisten laskimotapahtumien (syvä laskimotukos ja keuhkoembolia mukaan lukien) ilmaantuvuus oli suurempi ja riippuvainen annoksesta tofasitinibihoitoa saaneilla potilailla verrattuna potilaisiin, jotka saivat hoitoa TNF:n estäjillä.
Ritlesitinibin pitkäaikaiset turvallisuusarvioinnit ovat meneillään. Ritlesitinibin käytössä on noudatettava varovaisuutta, jos potilaalla on tromboembolioiden riskitekijöitä. Jos epäillään tromboembolista tapahtumaa, suositellaan ritlesitinibihoidon lopettamista ja potilaan tilan välitöntä uudelleenarviointia. Ritlesitinibihoidon hyötyjä ja haittoja on harkittava ennen potilaan hoidon aloittamista.
Neurologiset tapahtumat
Beagleille tehdyissä toksisuustutkimuksissa on todettu ritlesitinibiin liittyvää aksonaalista dystrofiaa (ks. kohta Prekliiniset tiedot turvallisuudesta). Ritlesitinibihoito pitää lopettaa, jos esiintyy selittämättömiä neurologisia oireita.
Hematologiset poikkeavuudet
Ritlesitinibihoitoon liittyi lymfosyyttimäärän ja verihiutalemäärän pieneneminen (ks. kohta Haittavaikutukset). Ennen ritlesitinibihoidon aloitusta on tehtävä lymfosyytti- ja verihiutalemäärien määritys.
Ritlesitinibihoitoa ei pidä aloittaa potilaille, joiden absoluuttinen lymfosyyttimäärä on < 0,5 × 109/l tai verihiutalemäärä on < 100 × 109/l. Ritlesitinibihoidon aloituksen jälkeen suositellaan hoidon keskeyttämistä tai lopettamista lymfosyytti- ja verihiutalemäärien poikkeavuuksien perusteella (ks. kohta Annostus ja antotapa). Lymfosyytti- ja verihiutalemäärien määritystä suositellaan 4 viikkoa ritlesitinibihoidon aloituksen jälkeen ja sen jälkeen tavanomaisen hoitokäytännön mukaisesti.
Rokotukset
Ritlesitinibihoitoa saaneiden potilaiden rokotevasteesta ei ole tietoa. Eläviä heikennettyjä taudinaiheuttajia sisältävien rokotteiden antoa ritlesitinibihoidon aikana tai juuri ennen hoitoa pitää välttää. Kaikki rokotukset, mukaan lukien profylaktinen Herpes zoster ‑rokotus, suositellaan päivittämään ajan tasalle voimassa olevien rokotussuositusten mukaisesti ennen ritlesitinibihoidon aloittamista.
Iäkkäät potilaat
≥ 65-vuotiaista potilaista on vähän tietoja. ≥ 65-vuotiailla potilailla ikä vaikuttaa olevan pienen absoluuttisen verihiutalemäärän riskitekijä.
Apuaine, jonka vaikutus tunnetaan
Laktoosi
Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Yhteisvaikutukset
Muiden lääkevalmisteiden mahdollinen vaikutus ritlesitinibin farmakokinetiikkaan
Itrakonatsolin, voimakkaan CYP3A-estäjän, anto toistuvina 200 mg:n annoksina suurensi ritlesitinibin käyrän alle jäävää pinta-alaa (AUC)inf noin 15 %. Tätä ei pidetä kliinisesti merkityksellisenä, joten annoksen muuttaminen ei ole tarpeen annettaessa ritlesitinibia samanaikaisesti CYP3A-estäjien kanssa.
Rifampisiinin, voimakkaan CYP-entsyymien induktorin, anto toistuvina 600 mg:n annoksina pienensi ritlesitinibin (AUC)inf-arvoa noin 44 %. Tätä ei pidetä kliinisesti merkityksellisenä, joten annoksen muuttaminen ei ole tarpeen annettaessa ritlesitinibia samanaikaisesti CYP-entsyymien induktorien kanssa.
Ritlesitinibin mahdollinen vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
Ritlesitinibin anto kerran päivässä toistuvina 200 mg:n annoksina suurensi midatsolaamin (CYP3A4:n substraatti) AUCinf-arvon noin 2,7‑kertaiseksi ja Cmax-arvon noin 1,8‑kertaiseksi. Ritlesitinibi on keskivahva CYP3A:n estäjä; varovaisuutta on noudatettava käytettäessä samanaikaisesti ritlesitinibia ja CYP3A:n substraatteja (esim. kinidiini, siklosporiini, dihydroergotamiini, ergotamiini, pimotsidi), jolloin kohtalaiset muutokset pitoisuuksissa voivat johtaa vakaviin haittavaikutuksiin. CYP3A:n substraatin (esim. kolkisiini, everolimuusi, takrolimuusi, sirolimuusi) annoksen muuttamista koskevat suositukset on otettava huomioon.
Ritlesitinibin anto kerran päivässä toistuvina 200 mg:n annoksina suurensi kofeiinin (CYP1A2:n substraatti) AUCinf-arvon noin 2,7‑kertaiseksi ja Cmax-arvon noin 1,1‑kertaiseksi. Ritlesitinibi on keskivahva CYP1A2:n estäjä; varovaisuutta on noudatettava käytettäessä samanaikaisesti ritlesitinibia ja muita CYP1A2:n substraatteja (esim. titsanidiini), jolloin kohtalaiset muutokset pitoisuuksissa voivat johtaa vakaviin haittavaikutuksiin. CYP1A2:n substraatin (esim. teofylliini, pirfenidoni) annoksen muuttamista koskevat suositukset on otettava huomioon.
Ritlesitinibin kerta-annoksen (400 mg) samanaikainen anto suurensi sumatriptaanin (orgaanisten kationien kuljettajaproteiinin [OCT]1 substraatti) AUCinf-arvon noin 1,3–1,5-kertaiseksi yksinään annettuun sumatriptaaniannokseen verrattuna. Sumatriptaanialtistuksen suurenemista ei pidetä kliinisesti merkityksellisenä. Varovaisuutta on noudatettava käytettäessä samanaikaisesti ritlesitinibia ja OCT1:n substraatteja, jolloin pienet muutokset pitoisuuksissa voivat johtaa vakaviin haittavaikutuksiin.
Ritlesitinibi ei aiheuttanut kliinisesti merkityksellisiä muutoksia ehkäisytablettien (esim. etinyyliestradioli tai levonorgestreeli), CYP2B6:n substraattien (esim. efavirentsi), CYP2C:n substraattien (esim. tolbutamidi) tai orgaanisten anionien kuljettajaproteiini (OAT)P1B1:n, rintasyövän resistenssiproteiinin (BCRP) ja OAT3:n (esim. rosuvastatiini) substraattien aiheuttamaan altistukseen.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Ritlesitinibia ei suositella naisille, jotka voivat tulla raskaaksi eivätkä käytä ehkäisyä. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja yhden kuukauden ajan viimeisen Litfulo-annoksen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja ritlesitinibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Ritlesitinibi oli suurina annoksina teratogeeninen rotalla ja kaniinilla (ks. kohta Prekliiniset tiedot turvallisuudesta). Litfulo on vasta-aiheista raskauden aikana (ks. kohta Vasta-aiheet).
Imetys
Olemassa olevat farmakodynaamiset /toksikologiset tiedot koe-eläimistä ovat osoittaneet ritlesitinibin erittyvän rintamaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Litfulo on vasta-aiheista rintaruokinnan aikana (ks. kohta Vasta-aiheet).
Hedelmällisyys
Ritlesitinibin vaikutusta ihmisen hedelmällisyyteen ei ole tutkittu. Kliinisesti merkityksellisillä altistuksilla ei ollut vaikutusta rotan hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Litfulo-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoituja haittavaikutuksia ovat ripuli (9,2 %), akne (6,2 %), ylähengitystieinfektiot (6,2 %), nokkosihottuma (4,6 %), ihottuma (3,8 %), karvatuppitulehdus (3,1 %) ja huimaus (2,3 %).
Haittavaikutustaulukko
Ritlesitinibia on annettu yhteensä 1 630 potilaalle 3 751 potilasvuoden altistusta vastaava määrä. Kolme lumekontrolloitua tutkimusta yhdistettiin (130 tutkittavaa sai 50 mg/vrk ja 213 tutkittavaa sai lumelääkettä) ritlesitinibin turvallisuuden arvioimiseksi lumelääkkeeseen verrattuna enintään 24 viikon ajan hoidon aloituksen jälkeen.
Taulukossa 2 luetellaan kaikki haittavaikutukset, joita havaittiin pälvikaljua koskevissa lumekontrolloiduissa tutkimuksissa, elinjärjestelmäluokan ja esiintyvyyden mukaan. Esiintyvyydet on määritetty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutusten vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2. Haittavaikutukset
| Elinjärjestelmäluokka | Yleinen | Melko harvinainen |
| Infektiot | Vyöruusu (Herpes zoster) Karvatuppitulehdus Ylähengitystieinfektio | |
| Hermosto | Huimaus | |
| Ruoansulatuselimistö | Ripuli | |
| Iho ja ihonalainen kudos | Akne Nokkosihottuma Ihottuma | |
| Tutkimukset | Suurentunut veren kreatiinikinaasipitoisuus | Pienentynyt verihiutalemäärä Pienentynyt lymfosyyttimäärä Alaniiniaminotransferaasin suureneminen tasolle > 3 x ULNa Aspartaattiaminotransferaasin suureneminen tasolle > 3 x ULNa |
a Sisältää laboratorioseurannassa havaitut muutokset
Valikoitujen haittavaikutusten kuvaus
Infektiot
Enintään 24 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa infektioita raportoitiin 31 %:lla lumelääkettä saaneista potilaista (80,35 tapausta 100 potilasvuotta kohden) ja 33 %:lla ritlesitinibia 50 mg saaneista potilaista (74,53 tapausta 100 potilasvuotta kohden). Tutkimuksessa AA-I, jossa hoitoa annettiin enintään 48 viikon ajan, infektioita raportoitiin 51 %:lla ritlesitinibia 50 mg tai enemmän saaneista potilaista (89,32 tapausta 100 potilasvuotta kohden).
Kaikkien ritlesitinibia saaneiden potilaiden tiedoista tehdyssä yhdistetyssä turvallisuusanalyysissa, johon sisältyi pitkäaikaistutkimus sekä vitiligoa koskeva tutkimus, infektioita raportoitiin 56,3 %:lla ritlesitinibia 50 mg tai enemmän saaneista potilaista (45,3 tapausta 100 potilasvuotta kohden). Infektiot olivat vaikeusasteeltaan useimmiten lieviä tai keskivaikeita.
Lumekontrolloiduissa tutkimuksissa niiden potilaiden osuus, jotka raportoivat infektioihin liittyvänä haittavaikutuksena vyöruusua (Herpes zoster), oli ritlesitinibia 50 mg saaneessa ryhmässä 1,5 % ja lumeryhmässä 0 %. Yksikään vyöruusutapaus ei ollut vakava. Yhdelle potilaalle, joka sai ritlesitinibia 200/50 mg (200 mg kerran päivässä 4 viikon ajan ja sen jälkeen 50 mg kerran päivässä), ilmaantunut Varicella zoster ‑virusinfektio täytti opportunistisen infektion (useita dermatomeja sisältävä Herpes zoster) kriteerit. Tutkimuksessa AA-I, jossa hoitoa annettiin enintään 48 viikon ajan, vyöruusua raportoitiin 2,3 %:lla ritlesitinibia 50 mg tai enemmän saaneista potilaista (2,61 tapausta 100 potilasvuotta kohden). Kaikista ritlesitinibilla hoidetuista potilaista tehdyssä yhdistetyssä turvallisuusanalyysissa, jossa oli mukana pitkäaikaistutkimus ja vitiligoa koskeva tutkimus, vyöruusun ilmaantuvuus ritlesitinibia 50 mg tai enemmän saaneilla potilailla oli 1,05 tapausta 100 potilasvuotta kohden.
Enintään 24 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa ei raportoitu vakavia infektioita lumelääkettä tai ritlesitinibia 50 mg saaneilla potilailla. Vakavien infektioiden ilmaantuvuus ritlesitinibia 200/50 mg saaneilla potilailla oli 0,9 % (2,66 tapausta 100 potilasvuotta kohden). Tutkimuksessa AA-I, jossa hoitoa annettiin enintään 48 viikon ajan, vakavia infektioita raportoitiin 0,8 %:lla ritlesitinibia 50 mg tai enemmän saaneista potilaista (0,86 tapausta 100 potilasvuotta kohden). Kaikkien ritlesitinibia saaneiden potilaiden tiedoista tehdyssä yhdistetyssä turvallisuusanalyysissa, johon sisältyi pitkäaikaistutkimus sekä vitiligoa koskeva tutkimus, vakavien infektioiden ilmaantuvuus ritlesitinibia 50 mg tai enemmän saaneilla potilailla oli 1,3 % (0,57 sataa potilasvuotta kohden).
Opportunistiset infektiot
Useita dermatomeja sisältäviä opportunistisia Herpes zoster ‑infektioita raportoitiin lumekontrolloiduissa tutkimuksissa yhdellä ritlesitinibia 200/50 mg saaneella potilaalla (0,50 tapausta 100 potilasvuotta kohden), ei yhdelläkään potilaalla tutkimuksessa AA-I, jossa hoitoa annettiin enintään 48 viikon ajan, ja neljällä ritlesitinibia 50 mg tai enemmän saaneella potilaalla (0,12 tapausta 100 potilasvuotta kohden) kaikkien ritlesitinibia saaneiden potilaiden tiedoista tehdyssä yhdistetyssä turvallisuusanalyysissa, johon sisältyi pitkäaikaistutkimus sekä vitiligoa koskeva tutkimus. Opportunistiset Herpes zoster ‑infektiot olivat vaikeusasteeltaan lieviä tai kohtalaisia.
Pienentynyt lymfosyyttimäärä
Enintään 24 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa ja tutkimuksessa AA-I, jossa hoitoa annettiin enintään 48 viikon ajan, ritlesitinibihoitoon liittyi lymfosyyttimäärän pieneneminen. Suurimmat vaikutukset lymfosyytteihin todettiin 4 viikon kuluessa. Hoidon jatkuessa lymfosyyttimäärä pysyi vakaana alentuneella tasolla. Kaikkien ritlesitinibia saaneiden potilaiden tiedoista tehdyssä yhdistetyssä turvallisuusanalyysissa, johon sisältyi pitkäaikaistutkimus sekä vitiligoa koskeva tutkimus, kolmella ritlesitinibia 50 mg saaneella tutkittavalla (0,2 %) absoluuttinen lymfosyyttimäärä oli vahvistetusti < 0,5 × 109/l.
Pienentynyt verihiutalemäärä
Enintään 24 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa ja tutkimuksessa AA-I, jossa hoitoa annettiin enintään 48 viikon ajan, ritlesitinibihoitoon liittyi verihiutalemäärän pieneneminen. Suurimmat vaikutukset verihiutaleisiin todettiin 4 viikon kuluessa. Hoidon jatkuessa verihiutalemäärä pysyi vakaana alentuneella tasolla. Kaikkien ritlesitinibia saaneiden potilaiden tiedoista tehdyssä yhdistetyssä turvallisuusanalyysissa, johon sisältyi pitkäaikaistutkimus sekä vitiligoa koskeva tutkimus, kahdella ritlesitinibia 50 mg tai enemmän saaneella tutkittavalla (0,1 %) verihiutalemäärä oli vahvistetusti < 100 × 109/l.
Kreatiinikinaasin suureneminen
Enintään 24 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa veren kreatiinikinaasin suurenemista raportoitiin kahdella ritlesitinibia 50 mg saaneella potilaalla (1,5 %). Tutkimuksessa AA-I, jossa hoitoa annettiin enintään 48 viikon ajan, veren kreatiinikinaasin suurenemista raportoitiin 3,8 %:lla ritlesitinibia 50 mg tai enemmän saaneista potilaista. Kreatiinikinaasin suurenemista > 5 kertaa viitevälin ylärajan (ULN) suuruiseksi raportoitiin lumelääkettä saaneiden ryhmässä kahdella potilaalla (0,9 %) ja ritlesitinibia 50 mg saaneiden ryhmässä 5 potilaalla (3,9 %). Tutkimuksessa AA-I, jossa hoitoa annettiin enintään 48 viikon ajan, kreatiinikinaasin suurenemista > 5 kertaa viitevälin ylärajan (ULN) suuruiseksi raportoitiin 6,6 %:lla ritlesitinibia 50 mg tai enemmän saaneista potilaista. Arvojen suureneminen oli useimmiten ohimenevää, eikä yksikään tapaus johtanut hoidon lopettamiseen.
Transaminaasipitoisuuksien suureneminen
Enintään 24 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa ritlesitinibia 50 mg tai enemmän saaneessa ryhmässä raportoitiin ALAT-arvojen suurenemista (> 3 x ULN) kolmella potilaalla (0,9 %) ja ASAT-arvojen suurenemista (> 3 x ULN) kahdella potilaalla (0,6 %). Arvojen suureneminen oli useimmiten ohimenevää, eikä yksikään tapaus johtanut hoidon lopettamiseen.
Pediatriset potilaat
Yhteensä 181 nuorta (12 – < 18-vuotiasta) otettiin mukaan ritlesitinibilla tehtäviin pälvikaljua koskeviin tutkimuksiin.
Nuorilla todettu haittaprofiili oli samankaltainen kuin aikuispotilailla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Lumekontrolloiduissa tutkimuksissa on annettu enintään 800 mg:n kerta-annos suun kautta ja toistuvina annoksina enintään 400 mg:n annoksia suun kautta päivittäin 14 päivän ajan. Spesifistä toksisuutta ei todettu.
Yliannostustapauksessa suositellaan potilaan seurantaa haittavaikutusten merkkien ja oireiden varalta (ks. kohta Haittavaikutukset). Ritlesitinibin yliannoksen hoitoon ei ole spesifistä vasta-ainetta. Hoidon on oltava oireenmukaista ja elintoimintoja tukevaa.
Terveistä vapaaehtoisista aikuisista tutkittavista saadut farmakokineettiset tiedot suun kautta otetusta kerta-annoksesta aina 800 mg:aan saakka viittaavat siihen, että yli 90 % otetusta annoksesta oletettavasti eliminoituu 48 tunnin kuluessa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, Janus-kinaasin (JAK) estäjät, ATC-koodi: L04AF08
Vaikutusmekanismi
Ritlesitinibi estää palautumattomasti ja selektiivisesti Janus-kinaasi (JAK) 3:a ja maksasolukarsinoomassa ilmentyviä tyrosiinikinaaseja (TEC) estämällä adenosiinitrifosfaatin sitoutumiskohtaa. Solutasolla ritlesitinibi estää spesifisesti JAK3-välitteisten γ-ketjun sisältävien reseptorien välityksellä γ-tyypin sytokiinien (IL-2, IL-4, IL-7, IL-15 ja IL-21) signalointia.
Ritlesitinibi estää myös TEC-kinaaseja, mikä johtaa NK-solujen ja CD8+ T‑solujen sytolyyttisen aktiivisuuden vähenemiseen.
Sekä JAK3- että TEC-kinaasivälitteiset signaalireitit ovat osallisina pälvikaljun patogeneesissä, mutta pälvikaljun patofysiologisaa ei vielä tunneta täysin.
Farmakodynaamiset vaikutukset
Lymfosyyttien alaryhmät
Pälvikaljua sairastavilla potilailla ritlesitinibihoitoon liittyi absoluuttisen lymfosyyttimäärän, T-lymfosyyttien (CD3) määrän ja T-lymfosyyttien alaryhmien (CD4 ja CD8) solumäärien annosriippuvaista pienenemistä hoidon varhaisvaiheessa. Alkuvaiheen pienenemisen jälkeen solumäärät palautuivat osittain ja pysyivät vakaina pisimmillään 48 viikon ajan. B-lymfosyyttien (CD19) määrässä ei todettu muutoksia missään hoitoryhmässä. NK-solujen (CD16/56) määrä pieneni hoidon varhaisvaiheessa annosriippuvaisesti, ja se pysyi vakaana alentuneella tasolla pisimmillään viikolle 48 asti.
Immunoglobuliinit
Pälvikaljua sairastavilla potilailla ritlesitinibihoitoon ei liittynyt kliinisesti merkityksellisiä muutoksia IgG-, IgM- tai IgA-immunoglobuliinien pitoisuuksissa viikkoon 48 mennessä, mikä osoittaa, ettei humoraalista immunosuppressiota tapahdu.
Kliininen teho ja turvallisuus
Ritlesitinibin tehoa ja turvallisuutta arvioitiin pälvikaljua koskevassa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa avaintutkimuksessa (AA-I), johon osallistuneilla vähintään 12-vuotiailla tutkittavilla pälvikalju kattoi ≥ 50 % päänahasta, täyskaljuus (alopecia totalis) ja kaikkien ihokarvojen puuttuminen (alopecia universalis) mukaan lukien. Tutkimuksessa arvioitiin myös ritlesitinibihoidon annosvastetta. Tutkimusjakso koostui 24 viikkoa kestäneestä lumekontrolloidusta vaiheesta ja 24 viikon jatkovaiheesta. Tutkimuksessa AA‑I arvioitiin yhteensä 718:aa potilasta, jotka satunnaistettiin johonkin seuraavista hoito-ohjelmista 48 viikon ajaksi: 1) 200 mg kerran päivässä 4 viikon ajan ja sen jälkeen 50 mg kerran päivässä 44 viikon ajan, 2) 200 mg kerran päivässä 4 viikon ajan ja sen jälkeen 30 mg kerran päivässä 44 viikon ajan, 3) 50 mg kerran päivässä 48 viikon ajan, 4) 30 mg kerran päivässä 48 viikon ajan, 5) 10 mg kerran päivässä 48 viikon ajan, 6) lumelääkettä 24 viikon ajan ja sen jälkeen 200 mg kerran päivässä 4 viikon ajan ja 50 mg kerran päivässä 20 viikon ajan, tai 7) lumelääkettä 24 viikon ajan ja sen jälkeen 50 mg 24 viikon ajan.
Tutkimuksessa arviointiin ensisijaisena päätetapahtumana niiden tutkittavien osuutta, jotka saavuttivat SALT (Severity of Alopecia Tool) -asteikolla pistemäärän ≤ 10 (päänahasta vähintään 90 % hiusten peittämää) viikolla 24. Lisäksi keskeisenä toissijaisena päätetapahtumana arvioitiin PGI-C (Patient’s Global Impression of Change) -vastetta viikolla 24 ja toissijaisina päätetapahtumina SALT-pistemäärää ≤ 20 (päänahasta vähintään 80 % hiusten peittämää) viikolla 24 sekä paranemista kulmakarvojen ja/tai silmäripsien takaisinkasvussa viikolla 24.
Lähtötilanteen tiedot
Tutkimuksessa AA‑I arviointiin vähintään 12-vuotiaita mies- ja naispuolisia potilaita. Kaikilla potilailla oli pälvikalju, joka kattoi ≥ 50 % päänahasta (SALT-pistemäärä ≥ 50) ilman näyttöä terminaalisesten hiusten takaisinkasvusta 6 edellisen kuukauden aikana, meneillään oleva pälvikaljujakso oli kestänyt ≤ 10 vuotta eikä hiustenlähdölle ollut muuta tiedossa olevaa syytä (esim. miestyyppinen kaljuus).
Kaikkien hoitoryhmien yhdistetyissä tiedoissa 62,1 % tutkittavista oli naisia, 68,0 % oli valkoihoisia, 25,9 % oli aasialaista syntyperää ja 3,8 % oli tummaihoisia tai afroamerikkalaisia. Potilaiden keski-ikä oli 33,7 vuotta, ja suurin osa potilaista (85,4 %) oli aikuisia (ikä ≥ 18 vuotta). Potilaista 105 (14,6 %) oli iältään 12 – < 18-vuotiaita ja 20 potilasta (2,8 %) vähintään 65-vuotiaita. Hoitoryhmien yhdistetyissä tiedoissa lähtötilanteen SALT-pistemäärien keskiarvot vaihtelivat 88,3:sta (keskihajonta 16,87) 93,0:aan (keskihajonta 11,50); potilailla, joilla ei ollut lähtötilanteessa alopecia totalista tai alopecia universalista, SALT-pisteiden keskiarvot olivat 78,3–87,0. Suurimmalla osalla kaikkien hoitoryhmien potilaista oli lähtötilanteessa kulmakarvojen (83,0 %) ja silmäripsien (74,7 %) poikkeavuuksia. Pälvikaljun toteamisesta kulunut aika oli 6,9 vuotta (mediaani), ja meneillään olevan pälvikaljujakson kesto oli 2,5 vuotta (mediaani). Satunnaistaminen stratifioitiin alopecia totalis- / alopecia universalis ‑statuksen mukaan. 46 %:lla potilaista status oli alopecia totalis / alopecia universalis, mikä perustui lähtötilanteen SALT-pistemäärään 100.
Kliininen vaste
Lumelääkettä saaneisiin verrattuna merkitsevästi suurempi osa ritlesitinibia 50 mg saaneista potilaista saavutti SALT ≤ 10 ‑vasteen viikolla 24 (taulukko 3). Ritlesitinibi 50 mg ‑ryhmässä SALT ≤ 10 ‑vasteen saavuttaneiden osuus suureni edelleen viikkoon 48 mennessä (kuva 1).
Lumeryhmään verrattuna merkitsevästi suurempi osa ritlesitinibia 50 mg saaneista potilaista saavutti PGI-C (Patient’s Global Impression of Change) ‑vasteen viikolla 24 (taulukko 3), ja vasteluvut suurenivat edelleen viikolle 48 asti (kuva 1).
Lumelääkettä saaneisiin verrattuna merkitsevästi suurempi osa ritlesitinibia 50 mg saaneista potilaista saavutti SALT ≤ 20 ‑vasteen viikolla 24 (taulukko 3). SALT ≤ 20 ‑vasteen saavuttaneiden osuus suureni edelleen viikkoon 48 mennessä.
Ritlesitinibia 50 mg saaneessa ryhmässä kulmakarvojen ja/tai silmäripsien takaisinkasvussa todettiin paranemista viikolla 24 (taulukko 3) potilailla, joilla oli lähtötilanteessa kulmakarvojen ja/tai silmäripsien poikkeavuuksia, ja lisäkasvua todettiin viikolla 48.
Hoidon vaikutukset viikolla 24 eri alaryhmissä (ikä, sukupuoli, etninen tausta, alue, paino, aika taudin toteamisesta, meneillään olevan jakson pituus, aiempi lääkehoito) olivat yhdenmukaiset koko tutkimusjoukkoa koskevien tulosten kanssa. Alopecia totalista / alopecia universalista sairastavien alaryhmässä hoitovaikutukset viikolla 24 olivat vähäisemmät kuin alaryhmässä, johon kuuluvilla ei ollut alopecia totalista / alopecia universalista. 12 – < 18-vuotiailla nuorilla hoidon vaikutukset viikolla 24 olivat yhdenmukaiset koko tutkimusjoukkoa koskevien tulosten kanssa.
Taulukko 3.Ritlesitinibin tehoa koskevat tulokset viikolla 24
Päätetapahtuma | Ritlesitinibi 50 mg kerran päivässä (N = 130) Vasteen saavuttaneet, % | Lumelääke (N = 131) Vasteen saavuttaneet, % | Ero lumelääkkeeseen (95 %:n luottamusväli) | |
SALT ≤ 10 ‑vastea,b | 13,4 | 1,5 | 11,9 (5,4, 18,3) | |
PGI‑C-vasteb,c | 49,2 | 9,2 | 40,0 (28,9, 51,1) | |
SALT ≤ 20 ‑vasted,e | 23,0 | 1,6 | 21,4 (13,4, 29,5) | |
EBA-vastef | 29,0 | 4,7 | 24,3 (14,8, 34,5) | |
ELA-vasteg | 28,9 | 5,2 | 23,7 (13,6, 34,5) | |
Lyhenteet: EBA = kulmakarvoja koskeva arvio, ELA = silmäripsiä koskeva arvio, N = potilaiden kokonaislukumäärä, PGI‑C = Patient’s Global Impression of Change; SALT = Severity of Alopecia Tool
- SALT ≤ 10 ‑vasteen saaneita olivat potilaat, joilla hiustenlähtö kattoi ≤ 10 % päänahasta. SALT-pistemäärät olivat 0–100; 0 = ei hiustenlähtöä ja 100 = hiusten täydellinen puuttuminen.
- Tilastollisesti merkitsevä, korjattu monivertailun suhteen.
- PGI-C-vasteen saaneita olivat potilaat, joiden tulos oli ”kohtalaisesti parantunut” tai ”huomattavasti parantunut” 7‑pisteisellä asteikolla, jonka ääripäät ovat ”huomattavasti parantunut” ja ”huomattavasti pahentunut”.
- SALT ≤ 20 ‑vasteen saaneita olivat potilaat, joilla hiustenlähtö kattoi ≤ 20 % päänahasta. SALT-pistemäärät olivat 0–100; 0 = ei hiustenlähtöä ja 100 = hiusten täydellinen puuttuminen.
- Tilastollisesti merkitsevä.
- Potilailla, joilla on lähtötilanteessa kulmakarvojen poikkeavuuksia, EBA-vaste määritellään vähintään 2 asteen paranemiseksi lähtötilanteesta tai normaaliksi EBA-pistemääräksi 4‑pisteisellä asteikolla.
- Potilailla, joilla on lähtötilanteessa silmäripsien poikkeavuuksia, ELA-vaste määritellään vähintään 2 asteen paranemiseksi lähtötilanteesta tai normaaliksi ELA-pistemääräksi 4‑pisteisellä asteikolla.
Kuva 1.SALT ≤ 10 ‑vaste ja PGI-C-vaste viikkoon 48 saakka
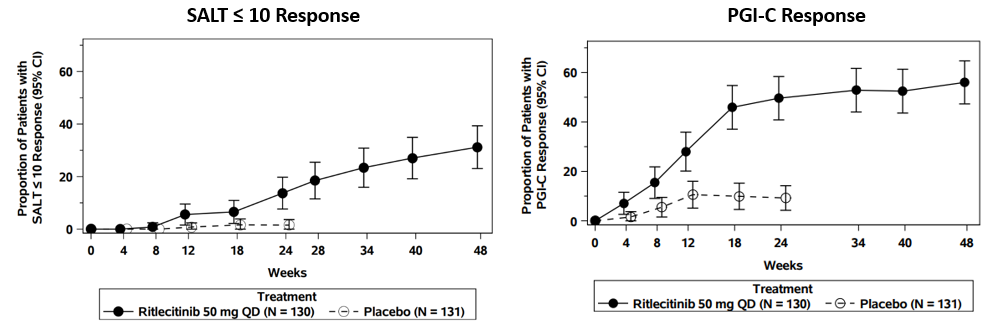
Lyhenteet: N = potilaiden kokonaismäärä, PGI-C = Patient Global Impression of Change, SALT = Severity of Alopecia Tool
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset ritlesitinibin käytöstä pälvikaljun hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Ritlesitinibin absoluuttinen biologinen hyötyosuus on noin 64 %. Leimatun vaikuttavan aineen suun kautta tai laskimoon antoa koskevien tietojen perusteella merkittyjen yhdisteiden suhteellinen (suun kautta / laskimoon) erittyminen virtsaan on noin 89 %, mikä viittaa siihen, että imeytyneen lääkeaineen osuus on suuri (fa). Annettaessa valmistetta toistuvina annoksina suun kautta huippupitoisuudet plasmassa saavutetaan 1 tunnissa. Ruoalla ei ole kliinisesti merkityksellistä vaikutusta ritlesitinibin imeytymiseen; runsasrasvainen ateria pienensi ritlesitinibin Cmax-arvoa ~ 32 % ja suurensi AUCinf-arvoa ~11 %. Lumekontrolloiduissa tutkimuksissa ritlesitinibia annettiin aterioista riippumatta (ks. kohta Annostus ja antotapa).
Ritlesitinibi on P‑glykoproteiinin (P‑gp) ja BCRP:n substraatti in vitro. Kuitenkin koska suuri osa ritlesitinibistä imeytyy (fa) ja sekä Cmax että AUC suurenevat annosriippuvaisesti (kerta-annos 20–200 mg), P‑gp:llä ja BCRP:llä ei odoteta olevan merkityksellistä vaikutusta ritlesitinibin imeytymiseen.
Jakautuminen
Laskimoon annetun ritlesitinibin jakautumistilavuus on noin 74 l. Noin 14 % verenkierrossa olevasta ritlesitinibistä sitoutuu plasman proteiineihin, pääasiassa albumiiniin. Ritlesitinibin veri/plasma-jakautumissuhde on 1,62. Ritlesitinibi on kovalenttisesti sitoutuva inhibiittori, jonka on osoitettu sitoutuvan muihin kuin kohdeproteiineihin, mukaan lukien MAP2K7, DOCK10, albumiini, CYP1A2, CYP3A, UGT1A1, ja UGT1A4; joillakin näistä voi olla kliinistä merkitystä yhteisvaikutusten suhteen (ks. kohta Yhteisvaikutukset).
Biotransformaatio
Ritlesitinibi metaboloituu useiden glutationi-S‑transferaasientsyymien (GST: sytosolinen GST A1/3, M1/3/5, P1, S1, T2, Z1 sekä mikrosomaaliset MAPEG [Membrane Associated Proteins involved in Eicosanoid and Glutathione metabolism] ‑proteiinit1/2/3) ja CYP-entsyymien (CYP3A, CYP2C8, CYP1A2 ja CYP2C9) välityksellä. Kunkin yksittäisen puhdistumareitin osuus on korkeintaan 25 %. Kutakin yksittäistä metaboliareittiä selektiivisesti estävä lääkevalmiste ei näin ollen todennäköisesti vaikuta ritlesitinibin aiheuttamaan systeemiseen altistukseen. Kuljettajaproteiinien spesifiset estäjät eivät todennäköisesti aiheuta kliinisesti merkityksellisiä muutoksia ritlesitinibin biologiseen hyötyosuuteen.
Ihmisellä radiomerkityllä lääkeaineella tehdyssä tutkimuksessa ritlesitinibi oli suun kautta annon jälkeen vallitseva verenkierrosta todettava aine (30,4 % verenkierrossa olevasta kokonaisradioaktiivisuudesta). Pääasiallinen kysteiinikonjugaattimetaboliitti M2 (16,5 %) on farmakologisesti inaktiivinen.
Eliminaatio
Ritlesitinibi eliminoituu pääasiassa metabolisen puhdistuman mekanismien kautta. Noin 4 % annoksesta erittyy virtsaan muuttumattomana vaikuttavana aineena. Noin 66 % radioleimatusta ritlesitinibiannoksesta erittyy virtsaan ja 20 % ulosteisiin. Toistuvien suun kautta annettujen annosten jälkeen vakaa tila saavutettiin arviolta päivänä 4, sillä farmakokinetiikka on epävakaata. Vakaan tilan farmakokineettiset parametrit AUCtau ja Cmax vaikuttavat suurenevan jokseenkin annosriippuvaisesti aina 200 mg:aan asti. Terminaalisen puoliintumisajan keskiarvot vaihtelevat 1,3 tunnista 2,1 tuntiin.
Erityisryhmät
Paino, sukupuoli, genotyyppi, etninen tausta ja ikä
Painolla, sukupuolella, GST P1-, M1- ja T1-genotyypillä, etnisellä taustalla ja iällä ei ollut kliinisesti merkityksellistä vaikutusta ritlesitinibin aiheuttamaan altistukseen.
Nuoret (≥ 12 – < 18 vuotta)
Populaatiofarmakokineettisen analyysin perusteella ritlesitinibin aiheuttamassa altistuksessa ei ole kliinisesti merkityksellisiä eroja nuorten ja aikuisten välillä.
Pediatriset potilaat (< 12 vuotta)
Ritlesitinibin farmakokinetiikkaa alle 12 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu.
Munuaisten vajaatoiminta
Potilailla, joilla on vaikea munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus [eGFR] < 30 ml/min), AUC24 oli noin 55 % suurempi ja Cmax noin 44 % suurempi kuin kaltaistetuilla tutkittavilla, joiden munuaisten toiminta oli normaali. Tämä vahvistettiin populaatiofarmakokineettisessa analyysissa. Eroja ei pidetä kliinisesti merkityksellisinä. Ritlesitinibia ei ole tutkittu potilailla, joilla on lievä (eGFR 60 – < 90 ml/min) tai keskivaikea (eGFR 30 – < 60 ml/min) munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista saatujen tulosten perustella ritlesitinibin aiheuttama altistus ei kuitenkaan oletettavasti suurene näillä potilailla kliinisesti merkitsevästi. Tutkittavien eGFR ja munuaisten toimintaluokka määritettiin Modification of Diet in Renal Disease (MDRD) ‑kaavalla.
Edellä mainittujen seikkojen perusteella lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Ritlesitinibia ei ole tutkittu potilailla, joilla on loppuvaiheen munuaissairaus tai jotka ovat saaneet munuaissiirteen (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Keskivaikeaa (Child–Pugh B) maksan vajaatoimintaa sairastavilla potilailla ritlesitinibin AUC24 suureni 18,5 % verrattuna tutkittaviin, joiden maksan toiminta oli normaali. Ritlesitinibia ei ole tutkittu potilailla, joilla on lievä maksan vajaatoiminta (Child–Pugh A). Keskivaikeaa maksan vajaatoimintaa sairastavista potilaista saatujen tulosten perustella ritlesitinibin aiheuttama altistus ei kuitenkaan oletettavasti suurene näillä potilailla kliinisesti merkityksellisesti. Lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Annostus ja antotapa). Ritlesitinibia ei ole tutkittu potilailla, joilla on vaikea (Child–Pugh C) maksan vajaatoiminta (ks. kohta Vasta-aiheet).
Prekliiniset tiedot turvallisuudesta
Yleinen toksisuus
Prekliinisissä tutkimuksissa havaittiin alentuneita lymfosyyttiarvoja ja lymfoidisten solujen määrän vähentymistä immuuni- ja hematopoieettisten järjestelmien elimissä/kudoksissa. Näiden katsottiin liittyvän ritlesitinibin farmakologisiin ominaisuuksiin (JAK3:n/TEC:n esto).
Ritlesitinibin pitkäaikainen anto beagleille aiheutti aksonaalista dystrofiaa systeemisillä altistuksilla, jotka olivat vähintään 7,4-kertaisia verrattuna ritlesitinibia 50 mg/vrk saaneiden potilaiden oletettuun altistukseen (sitoutumattoman lääkeaineen AUC24-arvon perusteella). Aksonaalinen dystrofia liittyy luultavasti sitoutumiseen muihin neuronaalisiin proteiineihin kuin kohdeproteiiniin. Aksonaalisen dystrofian ilmenemisestä koirilla systeemisen altistuksen ollessa edellä mainittua pienempi ei ole tietoa. Aksonaaliseen dystrofiaan liittyi neurologista kuulonmenetystä, kun systeeminen altistus oli 33-kertainen verrattuna ritlesitinibia 50 mg/vrk saaneiden potilaiden oletettuun altistukseen (sitoutumattoman lääkeaineen AUC24-arvon perusteella). Vaikka nämä löydökset korjautuivat, kun ritlesitinibin anto koirille lopetettiin, potilaaseen kohdistuvaa riskiä pitkäaikaishoidossa ei voida täysin sulkea pois (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Genotoksisuus
Ritlesitinibi ei ollut mutageeninen bakteerimutageenisuusmäärityksessä (Amesin testi). Rotan luuytimen in vivo ‑mikrotumamäärityksen tulosten perusteella ritlesitinibi ei ole aneugeeninen tai klastogeeninen altistuksella, joka vastaa 130 kertaa suurimman ihmiselle suositellun annoksen (MRHD) aiheuttamaa altistusta sitoutumattoman lääkeaineen AUC-arvon perusteella.
Karsinogeenisuus
Tuumorigeenisuudesta ei havaittu näyttöä 6 kuukauden ikäisillä Tg.ras H2 ‑hiirillä ritlesitinibialtistuksilla, jotka ovat 11-kertaisia suurimpaan ihmiselle suositeltuun annokseen nähden sitoutumattoman AUC:n perusteella. Rotalla tehdyssä kahden vuoden pituisessa karsinogeenisuustutkimuksessa naarasrotilla todettiin enemmän hyvänlaatuisia kateenkorvakasvaimia ja urosrotilla enemmän hyvänlaatuisia kilpirauhasen follikulaarisia adenoomia, kun ritlesitinibialtistus vastasi sitoutumattoman lääkeaineen AUC-arvon perusteella 29 kertaa MRHD-annoksen aiheuttamaa altistusta. Pahanlaatuisten kateenkorvakasvainten yleisyyden suurenemista naaraspuolisilla rotilla tällä altistuksella ei voida sulkea pois. Ritlesitinibiin liittyviä kateenkorvakasvaimia ja kilpirauhasen follikulaarisia adenoomia ei todettu altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 6,3 kertaa MRHD-annoksen aiheuttamaa altistusta.
Lisääntymis- ja kehitystoksisuus
Ritlesitinibilla ei ollut vaikutusta naarasrottien hedelmällisyyteen altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 55 kertaa MRHD-annoksen aiheuttamaa altistusta. Urosrottien hedelmällisyyteen kohdistuvia vaikutuksia (ennen implantaatiota kuolleiden alkioiden määrän suureneminen, mikä johti implantaatiokohtien vähenemiseen ja sen myötä pienempiin poikueisiin naarailla, jotka eivät saaneet ritlesitinibia ja jotka parittelivat ritlesitinibia saaneiden urosten kanssa) todettiin altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 55 kertaa MRHD-annoksen aiheuttamaa altistusta. Ritlesitinibilla ei ollut vaikutusta urosrottien hedelmällisyyteen altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 14 kertaa MRHD-annoksen aiheuttamaa altistusta. Rotalla tehdyssä hedelmällisyystutkimuksessa ei havaittu vaikutuksia spermatogeneesiin (siittiöiden määrä, siittiöiden muodostumisnopeus, liikkuvuus ja morfologia).
Tiineille rotille tehdyssä embryofetaalista kehitystä selvittäneessä tutkimuksessa ritlesitinibin anto suun kautta tiineyden 6.–17. päivänä aiheutti sikiöille luuston epämuodostumia ja muutoksia sekä pienensi sikiöiden painoa altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat vähintään 49 kertaa MRHD-annoksen aiheuttamaa altistusta (ks. kohta Vasta-aiheet). Ritlesitinibilla ei ollut vaikutuksia embryofetaaliseen kehitykseen altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 16 kertaa MRHD-annoksen aiheuttamaa altistusta.
Tiineille kaniineille tehdyssä embryofetaalista kehitystä selvittäneessä tutkimuksessa ritlesitinibin anto suun kautta tiineyden 7.–19. päivänä pienesi sikiöiden keskimääräistä painoa ja lisäsi sisäelinten epämuodostumien sekä luuston epämuodostumien ja luustomuutosten ilmaantuvuutta altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat vähintään 55 kertaa MRHD-annoksen aiheuttamaa altistusta (ks. kohta Vasta-aiheet). Ritlesitinibilla ei ollut vaikutuksia embryofetaaliseen kehitykseen altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 12 kertaa MRHD-annoksen aiheuttamaa altistusta.
Rotilla tehdyssä pre- ja postnataaalikehitystä selvittäneessä tutkimuksessa ritlesitinibin anto suun kautta tiineyden 6. päivästä 20. imetyspäivään asti aiheutti kehitystoksisuutta (kuten vähentynyttä syntymän jälkeistä eloonjääntiä, poikasten painon pienentymistä ja sekundaarisen kehityksen viivästymistä) altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 41 kertaa MRHD-annoksen aiheuttamaa altistusta (ks. kohta Vasta-aiheet). F1-sukupolven naarailla keltarauhasten keskimääräinen lukumäärä väheni altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 41 kertaa MRHD-annoksen aiheuttamaa altistusta. Ritlesitinibilla ei ollut vaikutuksia pre- ja postnataaliseen kehitykseen altistuksilla, jotka sitoutumattoman lääkeaineen AUC-arvon perusteella vastasivat 14 kertaa MRHD-annoksen aiheuttamaa altistusta.
Nuorilla rotilla tehdyssä toksisuustutkimuksessa ritlesitinibin suun kautta antoon 10.–60. päivänä syntymän jälkeen (vastaa ihmisen ikää vauvaiästä nuoruuteen) ei liittynyt hermostoon tai luustoon kohdistuvia vaikutuksia.
Laktaatio
Annettaessa ritlesitinibia imettäville rotille ritlesitinibipitoisuus suureni ajan myötä maidossa suuremmaksi kuin plasmassa. Maidon ja plasman AUC-arvojen suhde oli 2,2 (ks. kohta Vasta-aiheet).
Farmaseuttiset tiedot
Apuaineet
Kovan kapselin sisältö
Mikrokiteinen selluloosa
Laktoosimonohydraatti
Krospovidoni
Glyserolidibehenaatti
Kovan kapselin kuori
Hypromelloosi (E464)
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Briljanttisininen FCF (E133)
Painomuste
Shellakka
Propyleeniglykoli
Ammoniakkiliuos, väkevä
Musta rautaoksidi (E172)
Kaliumhydroksidi
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita. Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LITFULO kapseli, kova
50 mg (L:kyllä) 30 fol (958,76 €)
PF-selosteen tieto
Suurtiheyspolyeteenistä (HDPE) valmistettu purkki, jossa on kuivatusaineena piidioksidigeeliä sekä polypropeenisuljin. Purkissa on 28 kovaa kapselia.
OPA/Al/PVC/Al-läpipainopakkaus, jossa on 10 kovaa kapselia. Pakkauksessa on 30 tai 90 kovaa kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Läpinäkymättömät kovat kapselit, keltainen runko-osa ja sininen kansiosa, pituus noin 16 mm ja leveys 6 mm; runko-osaan on painettu mustalla merkintä ”RCB 50” ja kansiosaan merkintä ”Pfizer”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LITFULO kapseli, kova
50 mg 30 fol
- Ei korvausta.
ATC-koodi
L04AF08
Valmisteyhteenvedon muuttamispäivämäärä
26.02.2025
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com