
EYLEA injektionsvätska, lösning 114,3 mg/ml, injektionsvätska, lösning i förfylld spruta 114,3 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Potilasoppaan äänitiedosto
Terveydenhuollon ammattilainen
EYLEA - lasiaisinjektion antaminen
EYLEA Lääkkeen määrääjän opas
Kvalitativ och kvantitativ sammansättning
1 ml injektionsvätska, lösning innehåller 114,3 mg aflibercept*.
Eylea 114,3 mg/ml injektionsvätska, lösning
Varje injektionsflaska innehåller 30,1 mg aflibercept i 0,263 ml lösning. Denna mängd räcker till en enkeldos på 0,07 ml innehållande 8 mg aflibercept.
Eylea 114,3 mg/ml injektionsvätska, lösning i förfylld spruta
Varje förfylld spruta innehåller 21 mg aflibercept i 0,184 ml lösning. Denna mängd räcker till en enkeldos på 0,07 ml innehållande 8 mg aflibercept.
*Aflibercept är ett fusionsprotein som består av delar av extracellulära domäner av human VEGF (vaskulär endotel tillväxtfaktor)-receptorer 1 och 2 kopplade till Fc‑delen av humant IgG1 och som framställs i ovarialceller från kinesisk hamster (CHO‑K1) med rekombinant DNA‑teknologi.
Hjälpämne med känd effekt
Varje ml injektionsvätska, lösning innehåller 0,3 mg polysorbat 20 (E 432).
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Injektionsvätska, lösning (injektion)
Kliniska uppgifter
Terapeutiska indikationer
Eylea är avsett för vuxna för behandling av
- neovaskulär (våt) åldersrelaterad makuladegeneration (nAMD) (se avsnitt Farmakodynamiska egenskaper)
- nedsatt syn till följd av diabetiska makulaödem (DME) (se avsnitt Farmakodynamiska egenskaper)
- nedsatt syn till följd av makulaödem sekundärt till retinal venocklusion (RVO) (grenvensocklusion, centralvensocklusion, hemiretinal venocklusion) (se avsnitt Farmakodynamiska egenskaper).
Villkor
Valmistetta saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista.
Dosering och administreringssätt
Eylea får bara administreras av en kvalificerad läkare med erfarenhet av intravitreala injektioner.
Dosering
nAMD och DME
Den rekommenderade dosen är 8 mg aflibercept, motsvarande 0,07 ml lösning. Doseringen är densamma för indikationerna nAMD och DME. För 8 mg-dosen måste Eylea 114,3 mg/ml användas.
För patienter som påbörjar behandling administreras Eylea med 1 injektion per månad i 3 på varandra efterföljande doser. Injektionsintervallet kan sedan förlängas upp till var 4:e månad baserat på läkarens bedömning av synskärpa och/eller anatomiska resultat. Därefter kan behandlingsintervallen förlängas ytterligare upp till 6 månader, exempelvis med en ”treat-and-extend”-modell för att bibehålla stabil synskärpa och/eller anatomiska resultat (se avsnitt Farmakodynamiska egenskaper).
För patienter som tidigare har behandlats med Eylea 40 mg/ml eller andra anti-VEGF-läkemedel och som byter till Eylea 114,3 mg/ml kan behandlingsregimen skilja sig från den som används för behandlingsnaiva patienter. Behandlingsintervallerna ska bestämmas baserat på synskärpa och/eller anatomiska resultat (se avsnitt Farmakodynamiska egenskaper).
- Hos patienter med stabil synskärpa och anatomiskt resultat kan tidigare behandlingsintervall bibehållas eller förlängas efter den första injektionen med Eylea 114,3 mg/ml, exempelvis med en ”treat‑and‑extend”-modell.
- Hos patienter med suboptimal synskärpa och/eller anatomiskt resultat kan behandling med Eylea 114,3 mg/ml starta med 1 injektion per månad i upp till 3 på varandra efterföljande doser, varefter injektionsintervallerna justeras, exempelvis med en ”treat‑and‑extend”-modell.
Om synskärpa och/eller anatomiska resultat försämras ska behandlingsintervallet förkortas i motsvarande grad baserat på läkerens bedömning. Intervallet mellan 2 injektioner ska inte vara kortare än 1 månad.
Om synskärpa och/eller anatomiska resultat tyder på att patienten inte har någon nytta av fortsatt behandling ska Eylea 114,3 mg/ml sättas ut.
Eylea i månatliga doser på 8 mg har inte studerats för mer än 3 på varandra efterföljande doser i studierna PULSAR (nAMD) och PHOTON (DME). Tillgängliga data stöder administreringen av fler än 3 på varandra följande månatliga doser för vissa patienter. Det finns dock begränsat med data för närvarande.
Kontrollbesökens frekvens ska baseras på patientens tillstånd och efter läkarens bedömning. För händelser då behandling ska avbrytas, se avsnitt Varningar och försiktighet.
RVO
Den rekommenderade dosen är 8 mg aflibercept, motsvarande 0,07 ml lösning. För 8 mg-dosen måste Eylea 114,3 mg/ml användas.
För patienter som påbörjar behandling administreras Eylea med 1 injektion per månad i 3 på varandra efterföljande doser. Injektionsintervallerna kan sedan förlängas baserat på läkarens bedömning av synskärpa och/eller anatomiska resultat (se avsnitt Farmakodynamiska egenskaper).
För patienter som tidigare har behandlats med Eylea 40 mg/ml eller andra anti-VEGF-läkemedel och som byter till Eylea 114,3 mg/ml kan behandlingsregimen skilja sig från den som används för behandlingsnaiva patienter. Behandlingsintervallerna ska bestämmas baserat på synskärpa och/eller anatomiska resultat (se avsnitt Farmakodynamiska egenskaper).
- Hos patienter med stabil synskärpa och anatomiskt resultat kan tidigare behandlingsintervall bibehållas eller förlängas efter den första injektionen med Eylea 114,3 mg/ml, exempelvis med en ”treat and extend”-modell.
- Hos patienter med suboptimal synskärpa och/eller anatomiskt resultat kan behandling med Eylea 114,3 mg/ml starta med 1 injektion per månad i upp till 3 på varandra efterföljande doser, varefter injektionsintervallerna justeras, exempelvis med en ”treat and extend”-modell.
Om synskärpa och/eller anatomiska resultat försämras ska behandlingsintervallet förkortas i motsvarande grad baserat på läkarens bedömning (se avsnitt Farmakodynamiska egenskaper). Intervallet mellan 2 injektioner ska inte vara kortare än 1 månad.
Om synskärpa och/eller anatomiska resultat tyder på att patienten inte har någon nytta av fortsatt behandling ska Eylea 114,3 mg/ml sättas ut.
Kontrollbesökens frekvens ska baseras på patientens tillstånd och efter läkarens bedömning. För händelser då behandling ska avbrytas, se avsnitt Varningar och försiktighet.
Speciella populationer
Nedsatt njur- eller leverfunktion
Inga specifika studier hos patienter med nedsatt njur- eller leverfunktion har utförts.
Tillgängliga data tyder inte på något behov av dosjustering av Eylea hos dessa patienter (se avsnitt Farmakokinetiska egenskaper).
Äldre
Tillgängliga data tyder inte på något behov av dosjustering av Eylea hos dessa patienter.
Pediatrisk population
Säkerhet och effekt för Eylea 114,3 mg/ml för barn och ungdomar i åldern under 18 år har inte fastställts. Det finns ingen relevant användning av Eylea 114,3 mg/ml för en pediatrisk population för indikationerna nAMD, DME och RVO.
Administreringssätt
Eylea är endast avsett för intravitreal injektion.
Intravitreala injektioner måste utföras i enlighet med medicinska standarder och tillämpliga riktlinjer av en kvalificerad läkare med erfarenhet av administrering av intravitreala injektioner. I allmänhet måste man försäkra sig om adekvat bedövning och aseptiska förhållanden, inklusive lokal bredspektrummikrobicid (t.ex. applicering av povidonjodid på periokulär hud, ögonlock och okulär yta). Kirurgisk handdesinfektion, sterila handskar, en steril duk och ett sterilt ögonlocksspekulum (eller motsvarande) rekommenderas.
Injektionsnålen bör föras in 3,5 till 4,0 mm posteriort om limbus in i glaskroppsrummet samtidigt som man undviker den horisontella meridianen och riktar nålen mot ögonglobens centrum. Injektionsmängden på 0,07 ml injiceras därefter. Ett annat skleralt område bör användas vid efterföljande injektioner.
Omedelbart efter den intravitreala injektionen ska patienten kontrolleras för ökning av det intraokulära trycket. Lämplig metod kan bestå av en kontroll av perfusion av synnerven eller tonometri. Vid behov ska steril utrustning för paracentes finnas tillgänglig.
Efter intravitreal injektion ska patienterna instrueras att omedelbart rapportera alla symtom som tyder på endoftalmit (t.ex. ögonsmärta, ögonrodnad, fotofobi, dimsyn).
Varje injektionsflaska eller förfylld spruta får bara användas för behandling av ett öga.
Efter injektion ska allt ej använt läkemedel eller avfall kasseras i enlighet med gällande anvisningar.
Anvisningar om hantering av läkemedlet före administrering finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Kontraindikationer
- Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
- Okulär eller periokulär infektion.
- Aktiv allvarlig intraokulär inflammation.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Reaktioner relaterade till intravitreal injektion
Intravitreala injektioner, inklusive injektioner med Eylea, har förknippats med endoftalmit, intraokulär inflammation, näthinneavlossning, näthinneruptur och traumatisk katarakt (se avsnitt Biverkningar). Korrekta aseptiska injektionstekniker måste alltid användas vid administrering av Eylea. Patienter ska instrueras att omedelbart rapportera alla symtom som tyder på endoftalmit eller någon av ovanstående händelser och dessa ska behandlas på lämpligt sätt.
Ökat intraokulärt tryck
Övergående ökningar av intraokulärt tryck har setts inom 60 minuter efter en intravitreal injektion, t.ex. med Eylea (se avsnitt Biverkningar). Både det intraokulära trycket och perfusion av synnerven måste därför kontrolleras och behandlas på lämpligt sätt. Särskild försiktighet krävs hos patienter med dåligt kontrollerat glaukom (injicera inte Eylea när det intraokulära trycket är ≥ 30 mmHg).
Immunogenitet
Eftersom detta är ett terapeutiskt protein finns potential för immunogenitet med aflibercept (se avsnitt Farmakodynamiska egenskaper). Patienter bör instrueras att rapportera alla tecken eller symtom som tyder på intraokulär inflammation, t.ex. ögonsmärta, fotofobi eller rodnad i ögat, som kan vara kliniska tecken på överkänslighet.
Systemisk effekt
Systemiska biverkningar som t.ex. icke-okulära blödningar och arteriella tromboemboliska händelser har rapporterats till följd av intravitreal användning av VEGF-hämmare, och det finns en teoretisk risk att dessa kan vara relaterade till VEGF-hämmare (se avsnitt Biverkningar).
Det finns begränsade data om säkerhet vid behandling av patienter med nAMD, DME och RVO med en anamnes på stroke eller transitoriska ischemiska attacker eller myokardinfarkt de senaste 6 månaderna. Försiktighet ska iakttas vid behandling av sådana patienter.
Bilateral behandling
Säkerhet och effekt för bilateral behandling med Eylea 114,3 mg/ml per öga har inte studerats (se avsnitt Farmakodynamiska egenskaper). Om bilateral behandling utförs samtidigt kan det leda till en ökad systemisk exponering, vilket skulle kunna öka risken för systemiska biverkningar.
Samtidig användning av annat anti‑VEGF
Det finns begränsade data på samtidig användning av Eylea med andra anti‑VEGF-läkemedel (systemiska eller okulära).
Uppskjuten behandling
Behandlingen ska skjutas upp i händelse av:
- en försämring av synskärpan med bästa korrektion (BCVA) med ≥ 30 bokstäver jämfört med den senaste bedömningen av synskärpan
- en regmatogen näthinneavlossning eller makulahål i stadium 3 eller stadium 4
- näthinneruptur
- subretinal blödning som innefattar foveas centrum eller om blödningens storlek är ≥ 50 % av det totala lesionsområdet
- en utförd eller planerad intraokulär kirurgi inom föregående eller kommande 28 dagar.
Ruptur på det retinala pigmentepitelet
Riskfaktorer som förknippas med utveckling av en ruptur på det retinala pigmentepitelet efter anti‑VEGF‑behandling för nAMD innefattar en uttalad och/eller hög avlossning av retinala pigmentepitelet. När behandling med aflibercept sätts in ska försiktighet iakttas hos patienter med riskfaktorer för ruptur på det retinala pigmentepitelet.
Fertila kvinnor
Fertila kvinnor ska använda effektiv preventivmetod under behandling och minst 4 månader efter den sista intravitreala injektionen av Eylea 114,3 mg/ml (se avsnitt Fertilitet, graviditet och amning).
Populationer med begränsade data
Det finns endast begränsad erfarenhet av behandling med Eylea hos diabetespatienter med ett HbA1c över 12 % eller med proliferativ diabetesretinopati.
Eylea har inte studerats på patienter med aktiva systemiska infektioner eller patienter med samtidiga ögonsjukdomar som t.ex. näthinneavlossning eller makulahål. Det finns inte heller någon erfarenhet av behandling med Eylea hos diabetespatienter med okontrollerad hypertoni. Denna brist på information bör beaktas av läkaren vid behandling av sådana patienter.
Information om hjälpämnen
Detta läkemedel innehåller 0,021 mg polysorbat 20 per 0,07 ml dos, motsvarande 0,3 mg/ml. Polysorbater kan orsaka allergiska reaktioner.
Interaktioner
Inga interaktionsstudier har utförts.
Fertilitet, graviditet och amning
Fertila kvinnor
Fertila kvinnor ska använda effektiv preventivmetod under behandling och under minst 4 månader efter den sista intravitreala injektionen med Eylea 114,3 mg/ml.
Graviditet
Det finns begränsade data från användningen av aflibercept hos gravida kvinnor.
Djurstudier har visat reproduktionstoxicitet (se avsnitt Prekliniska säkerhetsuppgifter).
Eylea 114,3 mg/ml ska inte användas under graviditet om inte de eventuella fördelarna överväger de eventuella riskerna för fostret.
Amning
Baserat på mycket begränsade humana data kan aflibercept utsöndras i bröstmjölk i låga nivåer. Aflibercept är en stor proteinmolekyl och mängden läkemedel som absorberas av spädbarn förväntas vara minimal. Effekterna av aflibercept på ammade nyfödda barn/spädbarn är okända.
Som en försiktighetsåtgärd rekommenderas inte amning under användning av Eylea 114,3 mg/ml.
Fertilitet
Det finns inga fertilitetsdata från människa. Resultat från djurstudier med hög systematisk exponering indikerar att aflibercept kan ha en negativ effekt på manlig och kvinnlig fertilitet (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
Injektion med Eylea har mindre effekt på förmågan att framföra fordon och använda maskiner på grund av möjlig tillfällig synstörning som kan kopplas antingen till injektionen eller till ögonundersökningen. Patienten ska inte köra bil eller använda maskiner förrän synen är tillfredsställande återställd.
Biverkningar
Sammanfattning av säkerhetsprofilen
Allvarliga biverkningar var katarakt (7,1 %), ökat intraokulärt tryck (3,8 %), retinal blödning (2,8 %), blödning i glaskroppen (1,1 %), subkapsulär katarakt (0,6 %), näthinneruptur (0,5 %), nukleär katarakt (0,4 %) och näthinneavlossning (0,4 %).
De vanligaste biverkningarna hos patienter behandlade med Eylea 114,3 mg/ml var katarakt (7,1 %), nedsatt synskärpa (4,3 %), konjunktival blödning (4,0 %), ökat intraokulärt tryck (3,8 %), glaskroppsavlossning (3,5 %), fläckar i synfältet (3,2 %) och retinal blödning (2,8 %).
Säkerhetsprofilen som observerades i de 4 kliniska studierna var likartad hos patienter behandlade med Eylea 114,3 mg/ml (N = 1 808) respektive Eylea 40 mg/ml (N = 857) och hos patienter med nAMD, DME respektive RVO.
Tabell med biverkningar
Totalt 1 808 patienter behandlade med Eylea 114,3 mg/ml i upp till 96 veckor utgjorde säkerhetspopulationen i 4 kliniska fas II/III studier (CANDELA, PULSAR, PHOTON, QUASAR).
De säkerhetsdata som beskrivs nedan omfattar alla biverkningar med en rimlig möjlighet för orsakssamband med injektionsproceduren eller med läkemedlet.
Biverkningarna listas efter organsystem och frekvens enligt följande konvention: Mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100) och sällsynta (≥ 1/10 000, < 1/1 000), ingen känd frekvens (kan inte beräknas från tillgängliga data).
Biverkningar presenteras inom varje frekvensområde efter fallande allvarlighetsgrad.
Tabell 1: Alla biverkningar relaterade till behandlingen rapporterade hos patienter med nAMD, DME eller RVO behandlade med Eylea 114,3 mg/ml i fas II/III studierna eller efter marknadsintroduktionen
| Klassificering av organsystem | Frekvens | Biverkning |
| Immunsystemet | Vanliga | Överkänslighet* |
| Ögon | Vanliga | Katarakt, ökat intraokulärt tryck, fläckar i synfältet, glaskroppsavlossning, blödning i glaskroppen, retinal blödning, nedsatt synskärpa, ögonsmärta, konjunktival blödning, punktuell keratit, skrubbsår på hornhinnan |
| Mindre vanliga | Näthinneavlossning, näthinneruptur, ruptur på retinalt pigmentepitel, avlossning av retinalt pigmentepitel, uveit, irit, iridocyklit, vitrit, kortikal katarakt, nukleär katarakt, subkapsulär katarakt, korneal erosion, dimsyn, smärta vid injektionsstället, känsla av främmande kropp i ögat, ökat tårflöde, blödning vid injektionsstället, konjunktival hyperemi, linsgrumlingar, ögonlocksödem, okulär hyperemi, irritation vid injektionsstället, näthinnedegenerering, hornhinneödem | |
| Sällsynta | Blindhet, endoftalmit, ögonlocksirritation | |
| Ingen känd frekvens | Sklerit** |
* Rapporter om överkänslighet inkluderade utslag, klåda och urtikaria.
** Från rapportering efter marknadsintroduktionen.
Följande biverkningar av Eylea 40 mg/ml anses också förväntas med Eylea 114,3 mg/ml: onormal känsla i ögat, korneal epiteldefekt, ljusväg i främre kammaren, traumatisk katarakt, hypopyon och allvarliga anafylaktiska/anafylaktoida reaktioner.
Beskrivning av utvalda biverkningar
Biverkningar relaterade till läkemedelsklassen
Arteriella tromboemboliska händelser (ATE) är biverkningar som eventuellt har ett samband med systemisk VEGF‑hämning. Det finns en teoretisk risk för ATE, inklusive stroke och hjärtinfarkt, efter intravitreal användning av VEGF‑hämmare. Ett lågt incidenstal för ATE observerades i de kliniska studierna med aflibercept hos patienter med nAMD, DME och RVO. Inga märkbara skillnader sågs mellan grupperna behandlade med Eylea 114,3 mg och jämförelsegrupperna behandlade med Eylea 40 mg/ml, oavsett indikation.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Överdosering med ökad injektionsvolym kan öka det intraokulära trycket. Vid en överdosering ska därför det intraokulära trycket kontrolleras och om behandlande läkare anser det nödvändigt ska adekvat behandling sättas in (se avsnitt Varningar och försiktighet och Särskilda anvisningar för destruktion och övrig hantering).
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Oftalmologiska preparat/antineovaskulariseringspreparat, ATC-kod: S01LA05
Aflibercept är ett rekombinant fusionsprotein som består av delar av extracellulära domäner av humana VEGF‑receptorer 1 och 2 kopplade till Fc‑delen av humant IgG1.
Aflibercept framställs i ovarialceller från kinesisk hamster (CHO‑K1) med rekombinant DNA‑teknologi.
Verkningsmekanism
Vaskulär endotel tillväxtfaktor A (VEGF‑A) och placentatillväxtfaktor (PlGF) tillhör VEGF‑familjen av angiogena faktorer som kan verka som potenta mitogena, kemotaktiska och vaskulära permeabilitetsfaktorer för endotelceller. VEGF verkar via två tyrosinkinasreceptorer, VEGFR‑1 och VEGFR‑2, som finns på endotelcellernas yta. PlGF binder bara till VEGFR‑1 som också finns på leukocyternas yta. Kraftig aktivering av dessa receptorer av VEGF‑A kan leda till patologisk neovaskularisering och omfattande vaskulär permeabilitet. PlGF kan verka fristående för att aktivera VEGFR‑1 och därmed främja ett inflammatoriskt svar i näthinnan och är känt för att öka patologiska tillstånd som nAMD, diabetisk retinopati (DR), DME och retinalvensocklusion (RVO).
Farmakodynamisk effekt
Aflibercept fungerar som en löslig, falsk receptor som binder VEGF‑A och PlGF med högre affinitet än deras naturliga receptorer och som därmed kan hämma bindningen och aktiveringen av dessa besläktade VEGF‑receptorer.
I djurstudier kan aflibercept motverka patologisk neovaskularisering och vaskulärt läckage i ett antal modeller av okulära sjukdomar.
nAMD
nAMD kännetecknas av patologisk koroidal neovaskularisering (CNV). Läckage av blod och vätska från CNV kan leda till näthinneödem och/eller sub-/intraretinal blödning som leder till nedsatt synskärpa.
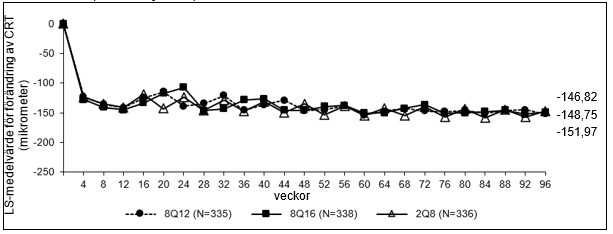
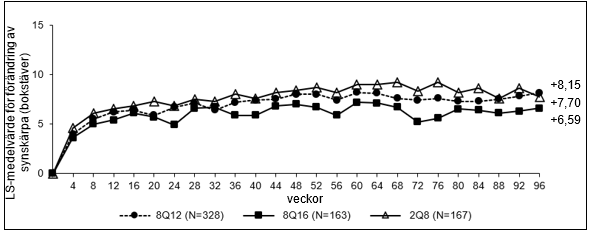
De farmakodynamiska effekterna av aflibercept 114,3 mg/ml administrerat var 12:e (8Q12) och var 16:e (8Q16) vecka beskrivs i jämförelse med aflibercept 40 mg/ml administrerat var 8 vecka (2Q8) för nAMD-indikationen. Dessa effekter visas som förändring av CNV-storlek från baslinjen till vecka 12; förändring i totalt lesionsområde från baslinjen till vecka 48, 60 och 96 samt förändring från baslinjen av central retinal tjocklek (CRT).
I den sammanslagna gruppen av patienter behandlade med 8Q12 eller 8Q16 var minskningar i CNV-storlek (LS-medelvärde, baserat på en blandad modell för upprepade mätningar [MMRM]) vecka 12 ‑1,63 mm2 jämfört med ‑1,17 mm2 för patienter behandlade med 2Q8.
Farmakodynamiska effekter bibehölls generellt till och med vecka 156.
Tabell 2: Farmakodynamiska parametrar (hela analyssetet) i PULSAR-studien
| Effektresultat | Vecka | Eylea 8Q12 (N = 335) | Eylea 8Q16 (N = 338) | Eylea 2Q8 (N = 336) |
| Förändring i totalt lesionsområde från baslinjen (mm2) | ||||
| LS meanA | 12 | -0,55 | -0,30 | |
| Aritmetiskt medelvärdet (SD), observerat | 48 | ‑0,4 (2.9) | ‑0,2 (3,1) | 0,1 (3,6) |
| LS mean (SE) A | -0,46 (0,19) | -0,35 (0,20) | 0,09 (0,22) | |
Skillnad i LS means (95 % KI) A,B | -0,55 (‑1,04; ‑0,06) | -0,44 (‑0,94; ‑0,06) | ||
| Aritmetiskt medelvärdet (SD), observerat | 60 | ‑0,5 (2,8) | ‑0,4 (3,2) | ‑0,3 (3,2) |
| LS mean (SE) A | -0,48 (0,20) | -0,54 (0,21) | ‑0,24 (0,20) | |
Skillnad i LS means (95 % KI) A,B | -0,24 (‑0,72; 0,24) | -0,29 (‑0,79; 0,20) | ||
| Aritmetiskt medelvärde (SD), observerat | 96 | -0,3 (3,3) | -0,3 (3,2) | -0,2 (3,4) |
| LS mean (SE) A | -0,43 (0,20) | -0,42 (0,20) | -0,18 (0,20) | |
| Skillnad i LS means (95 % KI) A,B | -0,25 (-0,72; 0,21) | -0,24 (-0,71; 0,22) | ||
A LS mean (minsta kvadratmedelvärde), KI och p-värde baserade på en MMRM med mätningar vid baslinjen som kovariat, behandlingsgrupp som faktor, besök och stratifieringsvariabler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen) som fastafaktorer samt villkor för interaktion mellan baslinjemätning och besök och för interaktionen mellan behandling och besök.
B Absoluta skillnader är Eylea 8Q12‑ respektive 8Q16‑grupper minus 2Q8‑grupper.
KI: Konfidensintervall
LS: Minsta kvadrat (Least square)
SD: Standardavvikelse
SE: Standardfel
Figur 1: LS-medelvärde för förändring av central retinal tjocklek (CRT) från baslinjen till och med vecka 96 (hela analyssetet) i studien PULSAR
DME
Diabetiskt makulaödem kännetecknas av ökad vasopermeabilitet och skada på retinala kapillärer som kan leda till nedsatt synskärpa.
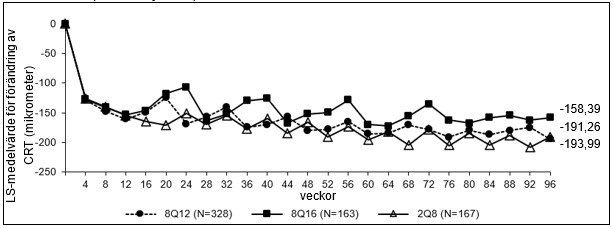
De farmakodynamiska effekterna av aflibercept 114,3 mg/ml administrerat var 12:e (8Q12) och var 16:e (8Q16) vecka beskrivs i jämförelse med aflibercept 40 mg/ml administrerat var 8:e vecka (2Q8) för DME-indikationen. Dessa effekter visas som förändring i läckageområdet från baslinjen till vecka 48, 60 och 96.
Farmakodynamiska effekter bibehölls generellt till och med vecka 156.
Tabell 3: Farmakodynamiska parametrar (hela analyssetet) i PHOTON-studien
| Effektresultat | Vecka | Eylea 8Q12 (N = 328) | Eylea 8Q16 (N = 163) | Eylea 2Q8 (N = 167) |
| Förändring i läckageområdeA från baslinjen (mm2) | ||||
| Aritmetiskt medelvärdet (SD), observerat | 48 | ‑13,9 (13,91) | ‑9,4 (11,50) | ‑9,2 (12,11) |
| 60 | ‑13,9 (13,54) | ‑12,0 (13,26) | ‑14,4 (12,89) | |
| 96 | -12,8 (10,98) | -9,4 (10,61) | -11,9 (11,26) | |
A baserat på mätning med fluoresceinangiografi
SD: Standardavvikelse
Figur 2: LS-medelvärde för förändring av central retinal tjocklek (CRT) från baslinjen till och med vecka 96 (hela analyssetet) i studien PHOTON
Immunogenicitet
Efter dosering av Eylea 114,3 mg/ml i upp till 96 veckor detekterades behandlingsrelaterade antikroppar mot Eylea 114,3 mg/ml hos 2,5 % till 4,4 % av patienterna behandlade för DME och nAMD. Inga tecken på antikroppar mot läkemedlet som påverkade farmakokinetik, effekt eller säkerhet observerades.
RVO
Vid RVO inträffar retinal ischemi som signalerar frisättning av VEGF, som i sin tur destabiliserar de täta förbindelserna och främjar endotelcellsproliferation. Uppreglering av VEGF förknippas med nedbrytningen av blod retinabarriären och denna ökade vaskulära permeabilitet leder till retinalt ödem, stimulering av endotelcellstillväxt och neovaskularisering.
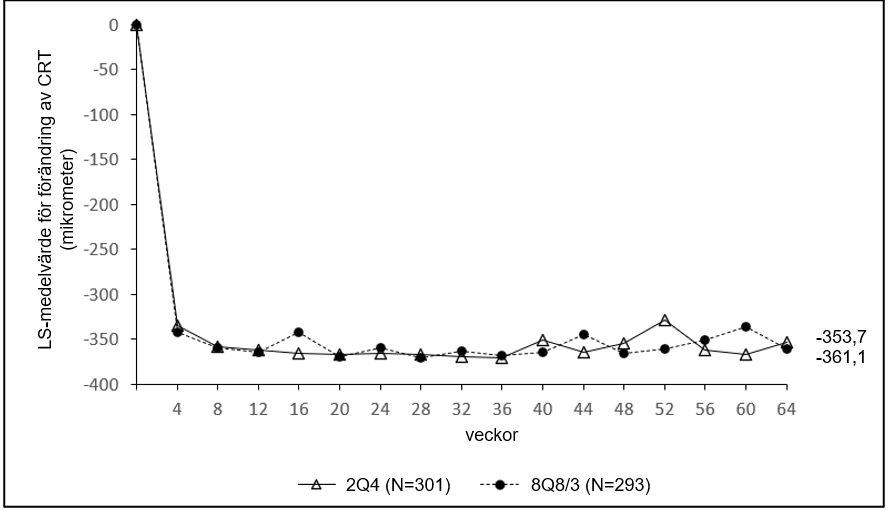
Tabell 4: Farmakodynamiska parametrar (hela analyssetet) i studien QUASAR
| Effektresultat | Vecka | Eylea 8Q8/3 (N = 293) | Eylea 2Q4 (N = 301) |
| Förändring i CRT från baslinjen [mikrometer] | |||
| Aritmetiskt medelvärdet (SD), observerat | 36 | ‑365,9 (239,9) | ‑397,3 (257,7) |
| LS mean (SE) A | -370,9 (3,1) | ‑370,8 (3,9) | |
| Skillnad i LS means (95 % KI) A,B | -0,1 (‑10,0; 9,8) | ||
| Aritmetiskt medelvärdet (SD), observerat | 64 | ‑355,5 (239,5) | ‑373,0 (252,1) |
| LS mean (SE) A | -361,1 (4,3) | ‑353,7 (5,2) | |
| Skillnad i LS means (95 % KI) A,B | -7,4 (‑20,7; 5,9) | ||
A LS mean, KI och p-värde baserade på en MMRM med CRT mätt vid baslinjen som kovariat, behandlingsgrupp som faktor, besök och stratifieringsvariabler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen och RVO-typ) som fasta faktorer samt villkor för interaktion mellan baslinje-CRT och besök och för interaktionen mellan behandling och besök.
B Absolut skillnad är Eylea 8Q8/3‑grupp minus 2Q4‑grupp.
KI: Konfidensintervall
CRT: Central retinal tjocklek
LS: Minsta kvadrat (Least square)
SD: Standardavvikelse
SE: Standardfel
Figur 3: LS-medelvärde för förändring av central retinal tjocklek (CRT) från baslinjen till och med vecka 64 (hela analyssetet) i studien QUASAR
Klinisk effekt och säkerhet
nAMD
Studiens syften
Säkerhet och effekt för Eylea 114,3 mg/ml bedömdes i en randomiserad, dubbelmaskerad, aktivt kontrollerad multicenterstudie (PULSAR) på patienter med behandlingsnaiv nAMD.
Det primära syftet var att bestämma om behandling med Eylea 114,3 mg/ml med 12 (8Q12) eller 16 veckors (8Q16) intervall ger non-inferior bästa korrigerad synskärpa (BCVA)-förändringar jämfört med Eylea 40 mg/ml var 8:e vecka hos patienter med nAMD.
Sekundära syften var att bestämma effekten av Eylea 114,3 mg/ml jämfört med Eylea 40 mg/ml på anatomiska och andra visuella responsmått och att utvärdera säkerhet, immunogenitet och farmakokinetik för aflibercept.
Det primära effektmåttet var förändring av BCVA från studiestart mätt som Early Treatment Diabetic Retinopathy Study (ETDRS)-bokstavspoäng vecka 48.
De viktiga sekundära effektmåtten var förändring i BCVA från baslinjen vecka 60 och andelen patienter utan intraretinal vätska (IRL) och subretinal vätska (SRF) i centrala näthinnan vecka 16.
Ytterligare sekundära effektmått var bland annat andelen patienter som vecka 48 hade minst 15 bokstävers förbättring i BCVA från studiestart, andelen patienter som fick en ETDRS-bokstavspoäng på minst 69 (cirka 20/40 Snellen-ekvivalent) vecka 48 och förändring från studiestart i total poäng enligt NEI VFQ-25 (National Eye Institute Visual Functioning Questionnaire-25) vecka 48.
I PULSAR-studien behandlades totalt 1 009 patienter. Patienterna fördelades i förhållandet 1:1:1 till 1 av 3 parallella behandlingsgrupper:
- Eylea 114,3 mg/ml administrerat var 12:e vecka (8Q12)
- Eylea 114,3 mg/ml administrerat var 16:e vecka (8Q16)
- Eylea 40 mg/ml administrerat var 8:e vecka (2Q8).
Alla patienter fick 3 initiala injektioner av den tilldelade dosen med 4 veckorsintervall.
Enligt studieprotokollet kortades intervallet för 8Q12- och 8Q16-grupperna om både följande kriterier uppfylldes:
- > 5 bokstävers bortfall i BCVA från vecka 12
- > 25 mikromillimeter ökning i CRT från vecka 12 eller ny foveal blödning eller ny foveal neovaskularisering.
Oavsett om patienten bibehöll eller förkortade behandlingsintervallet under år 1 var alla patienter i 8Q12- och 8Q16-grupperna i enlighet med studieprotokollet berättigade att förlänga behandlingsintervallet (i steg om 4 veckor) om följande kriterier uppfylldes:
- < 5 bokstävers bortfall i BCVA från vecka 12
- ingen vätska i den centrala näthinnan på optisk koherenstomografi (OCT)
- ingen ny foveal blödning eller foveal neovaskularisering.
För patienter som inte uppfyllde kriterierna för att förkorta eller förlänga intervallet, bibehölls doseringsintervallet. Den kortaste intervallet mellan injektionerna var 8 veckor i alla grupper.
Patienter med bilateral sjukdom kunde behandlas med Eylea 40 mg/ml eller annan anti-VEGF-läkemedel i det andra ögat.
Patientkarakteristika vid studiestart
Patienter i åldern 50 till 96 år med en genomsnittsålder på 74,5 år.
Cirka 92 % (309/335) och 87 % (295/338) av patienterna som randomiserats till 8Q12- respektive 8Q16-gruppen var 65 år eller äldre och cirka 51 % (172/335) respektive 51 % (171/338) var 75 år eller äldre.
Resultat
Patienter i 8Q12-, 8Q16- och 2Q8-gruppen som slutförde vecka 48 fick i median (genomsnitt) 6,0 (6,1), 5,0 (5,2) respektive 7,0 (6,9) injektioner.
Vecka 48 stod 79,4 % av patienterna i 8Q12-gruppen kvar på Q12-intervall medan 76,6 % av patienterna i 8Q16-gruppen stod kvar på Q16-intervall.
Patienter i 8Q12-, 8Q16- och 2Q8-gruppen som slutförde vecka 60 fick i median (genomsnitt) 7,0 (7,1), 6,0 (6,2) respektive 9,0 (8,8) injektioner.
Vecka 60 förlängdes behandlingsintervallet för 43,1 % av patienterna i 8Q12-gruppen till 16 veckor och till 20 veckor för 38,5 % av patienterna i 8Q16-gruppen.
Patienter i 8Q12-, 8Q16- och 2Q8-gruppen som slutförde vecka 96 fick i median (genomsnitt) 9,0 (9,7), 8,0 (8,2) respektive 13,0 (12,8) injektioner.
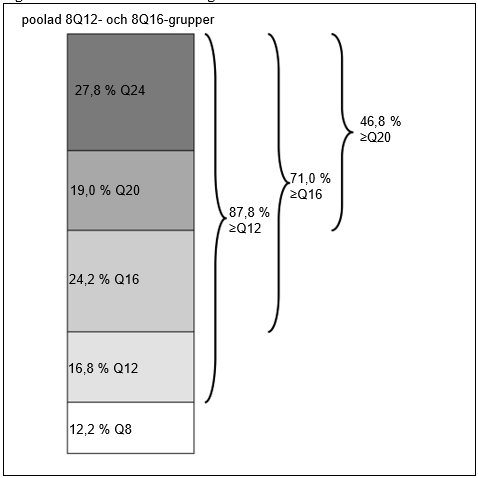
Vecka 96 hade 71 % av patienterna i de poolade 8Q12- och 8Q16-grupperna uppnått behandlingsintervall på ≥16 veckor, 46,8 % av patienterna hade uppnått behandlingsintervall på ≥20 veckor och 27,8 % av patienterna hade uppnått behandlingsintervall på 24 veckor med bibehållen synskärpa och anatomiska resultat.
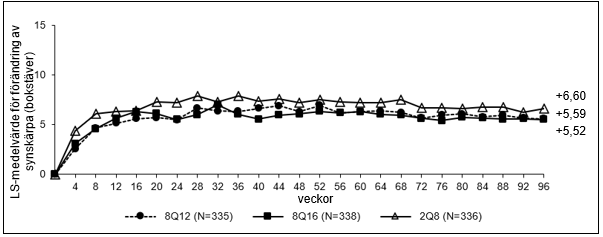
Behandling med 8Q12 och 8Q16 visades vara non-inferior och kliniskt likvärdig behandling med 2Q8 när det gäller det primära effektmåttet ’genomsnittlig förändring i BCVA vecka 48’ och det viktiga sekundära effektmåttet ’genomsnittlig förändring i BCVA vecka 60’. Behandlingseffekten med Eylea 114,3 mg/ml vad gäller genomsnittlig förändring av BCVA bibehölls till och med vecka 96.
Behandling med Eylea (sammanslagna 8Q12- och 8Q16-grupper) visades dessutom vara överlägsen behandling med 2Q8 när det gäller det viktiga sekundära effektmåttet ’andel patienter utan intraretinal vätska (IRL) och utan subretinal vätska (SRF) i centrala näthinnan vecka 16’ (se tabell 5).
Tabell 5: Effektresultat från PULSAR-studien
| Effektresultat | Vecka | Eylea 8Q12 (N = 335) | Eylea 8Q16 (N = 338) | Eylea 2Q8 (N = 336) |
| Förändring i BCVA från studiestart mätt med ETDRS-bokstavspoäng D | ||||
| Aritmetiskt medelvärdet (SD), observerat | 48 | 6,7 (12,6) | 6,2 (11,7) | 7,6 (12,2) |
| LS mean (SE) A | 6,06 (0,77) | 5,89 (0,72) | 7,03 (0,74) | |
Skillnad i LS mean (95 % KI) A,B | -0,97 (‑2,87; 0,92) | -1,14 (‑2,97; 0,69) | ||
| p-värde (ensidigt non-inferiority-test med en marginal på 4 bokstäver) A,B | 0,0009 | 0,0011 | ||
| Aritmetiskt medelvärdet (SD), observerat | 60 | 6,6 (13,6) | 6,6 (11,7) | 7,8 (12,6) |
| LS mean (SE) A | 6,37 (0,74) | 6,31 (0,66) | 7,23 (0,68) | |
Skillnad i LS mean (95 % KI) A,B | -0,86 (‑2,57; 0,84) | -0,92 (‑2,51; 0,66) | ||
| p-värde (ensidigt non-inferiority-test med en marginal på 4 bokstäver) A,B | 0,0002 | <0,0001 | ||
| Aritmetiskt medelvärdet (SD), observerat | 96 | 5,9 (14,2) | 5,6 (13,7) | 7,4 (13,8) |
| LS mean (SE) A | 5,59 (0,77) | 5,52 (0,75) | 6,60 (0,73) | |
Skillnad i LS mean (95 % KI) A,B | -1,01 (-2,82; 0,80) | -1,08 (-2,87; 0,71) | ||
| Patienter utan IRF och ingen SRF i den centrala näthinnan D | ||||
| Andel (LOCF) | 16 | 63,3 % | 51,6 % | |
Justerad skillnad i andel (95 % KI) B, C | 11,7 % (5,3 %; 18,2 %) | |||
| p‑värde (ensidigt superiority-test) B, C | 0,0002 | |||
| Andel (LOCF) | 48 | 71,1 % | 66,8 % | 59,4 % |
Justerad skillnad i andel (95 % KI) B, C | 11,7 % (4,5 %; 18,9 %) | 7,5 % (0,1 %; 14,8 %) | ||
| Andel (LOCF) | 60 | 74,6 % | 72,2 % | 74,6 % |
Justerad skillnad i andel (95 % KI) B, C | 0,0 % (‑6,6 %; 6,7 %) | ‑2,2 % (‑8,9 %; 4,4 %) | ||
| Andel (LOCF) | 96 | 69,6 % | 63,6 % | 66,5 % |
Justerad skillnad i andel (95 % KI) B, C | 3,0 % (-4,1 %; 10,1 %) | -3,0 % (-10,2 %; 4,2 %) | ||
| Patienter som fick en ETDRS-bokstavspoäng på minst 69 (cirka 20/40 Snellen-ekvivalent) D | ||||
| Andel (LOCF) | 48 | 56,9 % | 54,3 % | 57,9 % |
Justerad skillnad i andel (95 % KI) B, C | ‑0,2 % (‑6,6 %; 6,2 %) | ‑2,2 % (‑8,4 %; 4,0 %) | ||
| Andel (LOCF) | 60 | 56,3 % | 54,6 % | 58,2 % |
Justerad skillnad i andel (95 % KI) B, C | ‑1,1 % (‑7,5 %; 5,3 %) | ‑2,3 % (‑8,7 %; 4,1 %) | ||
| Andel (LOCF) | 96 | 53,3 % | 53,1 % | 56,7 % |
Justerad skillnad i andel (95 % KI) B, C | -2,7 % (-9,4 %; 4,0 %) | -2,4 % (-9,1 %; 4,2 %) | ||
| Patienter som hade minst 15 bokstävers förbättring i BCVA från studiestart D | ||||
| Andel (LOCF) | 48 | 20,7 % | 21,7 % | 22,1 % |
Justerad skillnad i andel (95 % KI) B, C | ‑1,7% (‑7,8 %; 4,3 %) | ‑0,9 % (‑7,0 %; 5,1 %) | ||
| Andel (LOCF) | 60 | 23,7 % | 23,1 % | 23,3 % |
Justerad skillnad i andel (95 % KI) B, C | 0,1 % (‑6,2 %; 6,3 %) | ‑0,7 % (‑6,9 %; 5,5 %) | ||
| Andel (LOCF) | 96 | 22,2 % | 22,8 % | 24,2 % |
Justerad skillnad i andel (95 % KI) B, C | -2,4 % (-8,4 %; 3,6 %) | -2,0 % (-8,0 %; 4,1 %) | ||
| Sista avsedda behandlingsintervall | ||||
| Patienter med behandlingsintervall ≥Q12 E | ||||
| Andel (poolade 8Q12- och 8Q16-grupper) | 96 | 87,8 % | n/a | |
| Andel | 86,6 % | 89,0 % | n/a | |
| Patienter med behandlingsintervall ≥Q16 E | ||||
| Andel (poolade 8Q12- och 8Q16-grupper) | 96 | 71,0 % | n/a | |
| Andel | 63,6 % | 78,4 % | n/a | |
| Patienter med behandlingsintervall ≥Q20 E | ||||
| Andel (poolade 8Q12- och 8Q16-grupper) | 96 | 46,8 % | n/a | |
| Andel | 40,5 % | 53,1 % | n/a | |
| Patienter med behandlingsintervall Q24 E | ||||
| Andel (poolade 8Q12- och 8Q16-grupper) | 96 | 27,8 % | n/a | |
| Andel | 24,7 % | 30,8 % | n/a | |
A LS mean (minsta kvadratmedelvärde), KI och p-värde baserade på en MMRM med mätningar av bästa korrigerad synskärpa (BCVA) vid baslinjen som kovariat, behandlingsgrupp som faktor, besök och stratifieringsvariabler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen) som fasta faktorer samt villkor för interaktion mellan baslinje-BCVA och besök och för interaktionen mellan behandling och besök.
B Absoluta skillnader är Eylea 8Q12‑ respektive 8Q16‑grupper minus 2Q8‑grupper.
C Mantel‑Haenszel-viktad behandlingsskillnad med stratifieringsvaraibler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen) och KI beräknad med normal approximation.
D Hela analyssetet
E Säkerhetsanalys-set; patienter som anses ha vara slutförtare behandlingen för respektive tidpunkt.
KI: Konfidensintervall
LOCF: Last Observation Carried Forward
LS: Minsta kvadrat (Least square)
SD: Standardavvikelse
SE: Standardfel
Behandlingsintervallen analyserades på ett fördefinierat explorativt sätt.
Figur 4: LS-medelvärde för förändring av BCVA uppmätt med ETDRS-bokstavspoäng från baslinjen till och med vecka 96 (hela analyssetet) i studien PULSAR
Figur 5: Sista avsedda behandlingsintervall vecka 96
Aflibercept i samtliga doser (8Q12, 8Q16, 2Q8) visade betydande förändringar från studiestart i det fördefinierade sekundära effektmåttet frågeformuläret National Eye Institute Visual Function Questionnaire-25 (NEI VFQ‑25).
Inga kliniskt betydande skillnader sågs mellan 8Q12‑, 8Q16‑ och 2Q8‑grupper i förändringar i totalpoäng enligt NEI VFQ‑25 vid vecka 48 och vecka 96 från studiestart.
Effektresultat i utvärderbara subgrupper för ålder, kön, geografisk region, etnicitet, ras, BCVA vid studiestart och lesionstyp överensstämde med resultaten i den totala populationen.
Effekt bibehölls generellt till och med vecka 96.
Resultat – PULSAR-studiens förlängningsfas
Vid slutet av studiens huvudfas, i vecka 96, kunde patienterna registrera sig i den 60 veckor långa öppna förlängningsfasen. 417 patienter som ursprungligen placerats i 8Q12- och 8Q16‑grupperna fortsatte med Eylea 114,3 mg/ml och bibehöll sina senaste behandlingsintervall. 208 patienter som ursprungligen placerats i 2Q8‑gruppen vid studiens början fick byta till Eylea 114,3 mg/ml och börja med 12‑veckorsintervall. Behandlingsintervallen kunde justeras ytterligare baserat på läkarens bedömning av synskärpa och/eller anatomiska resultat.
Hos de patienter som ursprungligen placerats i 8Q12- och 8Q16‑grupperna bibehölls behandlingseffekten av Eylea 114,3 mg/ml generellt under 3 år (vecka 156). Förändringen i LS-medelvärde från baslinjen i de sammanslagna 8Q12- och 8Q16‑grupperna var +3,41 bokstäver i BCVA och ‑148,05 mikrometer i CRT vid vecka 156.
Hos de patienter som ursprungligen placerats i 2Q8‑gruppen var behandlingseffekten med Eylea 114,3 mg/ml liknande. Förändringen i LS-medelvärde från baslinjen var +4,58 bokstäver i BCVA och ‑145,21 mikrometer i CRT vid vecka 156.
Patienterna i 8Q12- och 8Q16‑grupperna som slutförde vecka 156 fick i median (genomsnitt)
13,0 (13,5) respektive 11,0 (12,2) injektioner.
Patienterna som bytte till Eylea 114,3 mg/ml och slutförde vecka 156 fick en total median (genomsnitt) på 18,0 (17,7) injektioner, varav 5,0 (4,9) injektioner administrerades efter bytet till Eylea 114,3 mg/ml inom de 60 veckorna av studiens förlängningsfas.
Den allmänna säkerhetsprofilen i förlängningsfasen liknade den som observerades i huvudfasen.
Tabell 6: Effektresultat från PULSAR‑studiens förlängningsfas vid vecka 156
| Effektresultat | 8Q12 som fortsatte med Eylea 114,3 mg/ml (N = 185) | 8Q16 som fortsatte med Eylea 114,3 mg/ml (N = 190) | 2Q8 som bytte till Eylea 114,3 mg/ml (N = 208) |
|---|---|---|---|
| Förändring i BCVA från baslinjen (LS mean) | +3,57 bokstäver | +3,23 bokstäver | +4,58 bokstäver |
| Förändring i CRT från baslinjen (LS mean) | ‑148,42 mikrometer | ‑147,54 mikrometer | ‑145,21 mikrometer |
| Sista avsedda behandlingsintervall A | |||
| ≥ 12 veckor | 76,2 % | 78,4 % | 78,5 % |
| ≥ 16 veckor | 53,5 % | 62,1 % | 42,5 % |
| ≥ 20 veckor | 37,8 % | 42,6 % | 16,1 % |
| 24 veckor | 23,8 % | 24,2 % | NA B |
A baserat på patienter som slutförde vecka 156
B Ej tillämpligt för patienter som ursprungligen randomiserades till 2Q8, på grund av studiedesign/studielängd
DME
Studiens syften
Säkerhet och effekt för Eylea 114,3 mg/ml bedömdes i en randomiserad, dubbelmaskerad, aktivt kontrollerad multicenterstudie (PHOTON) på patienter med DME.
Det primära syftet var att bestämma om behandling med Eylea 114,3 mg/ml med 12 (8Q12) eller 16 veckors (8Q16) intervall ger non-inferior BCVA-förändringar jämfört med Eylea 40 mg/ml var 8:e vecka.
Sekundära syften var att bestämma effekten av Eylea 114,3 mg/ml jämfört med Eylea 40 mg/ml på anatomiska och andra visuella responsmått och att utvärdera säkerhet, immunogenitet och farmakokinetik för aflibercept.
Det primära effektmåttet var förändringen av BCVA från studiestart mätt som Early Treatment Diabetic Retinopathy Study (ETDRS)-bokstavspoäng vecka 48.
Ett viktigt sekundärt effektmått var förändring i BCVA från studiestart vecka 60.
Ytterligare sekundära effektmått var bland annat andelen patienter som vecka 48 hade minst 15 bokstävers förbättring i BCVA från studiestart, andelen patienter som fick en ETDRS-bokstavspoäng på minst 69 (cirka 20/40 Snellen-ekvivalent) vecka 48 och förändring från studiestart i total poäng enligt NEI VFQ-25 (National Eye Institute Visual Functioning Questionnaire-25) vecka 48.
I PHOTON-studien behandlades totalt 658 patienter. Patienterna fördelades i förhållandet 2:1:1 till 1 av 3 parallella behandlingsgrupper:
- Eylea 114,3 mg/ml administrerat var 12:e vecka (8Q12)
- Eylea 114,3 mg/ml administrerat var 16:e vecka (8Q16)
- Eylea 40 mg/ml administrerat var 8:e vecka (2Q8).
Patienter som bytte från andra anti-VEGF-läkemedel till Eylea 114,3 mg/ml fick den sista injektionen av den tidigare behandlingen minst 12 veckor innan de påbörjade behandlingen med Eylea 114,3 mg/ml.
Alla patienter i 8Q12- och 8Q16-grupperna fick 3 initiala injektioner och alla patienter i 2Q8-gruppen fick 5 initiala injektioner med 4-veckorsintervall.
Enligt studieprotokollet kortades intervallet för 8Q12- och 8Q16-grupperna om både följande kriterier uppfylldes:
- > 10 bokstävers bortfall i BCVA från vecka 12 i samband med ihållande eller förvärrad DME
- > 50 mikromillimeter ökning i CRT från vecka 12.
Från vecka 52 kunde i enlighet med studieprotokollet patienter i 8Q12- och 8Q16-grupperna som bibehöll eller förkortade sina behandlingsintervall under år 1 förlänga intervallet (i steg om 4 veckor) om följande kriterier uppfylldes.
- < 5 bokstävers bortfall i BCVA vecka 12
- CRT < 300 mikromillimeter i SD‑OCT (eller < 320 mikromillimeter om mätning inkluderade RPE).
För patienter som inte uppfyllde kriterierna för att förkorta eller förlänga intervallet, bibehölls doseringsintervallet. Den kortaste intervallet mellan injektionerna var 8 veckor i alla grupper.
Patienter med bilateral sjukdom kunde behandlas med Eylea 40 mg/ml i det andra ögat.
Patientkarakteristika vid studiestart
Patienter i åldern från 24 till 90 år med en genomsnittsålder på 62,3 år.
Cirka 44 % (143/328) och 44 % (71/163) av patienterna som randomiserats till 8Q12- respektive 8Q16-gruppen var 65 år eller äldre och cirka 11 % (36/328) och 14 % (14/163) var 75 år eller äldre.
Andelen patienter som tidigare behandlats för DME var balanserad mellan behandlingsgrupperna (43,6 % i 8Q12‑, 43,6 % i 8Q16‑ respektive 44,3 % i 2Q8‑gruppen).
Resultat
Patienter i 8Q12-, 8Q16- och 2Q8-grupperna som slutförde vecka 48 fick i median (genomsnitt) 6,0 (6,0); 5,0 (5,0) respektive 8,0 (7,9) injektioner.
Vecka 48 stod 91,0 % av patienterna i 8Q12‑gruppen kvar på Q12-intervall medan 89,1 % av patienterna i 8Q16‑gruppen stod kvar på Q16-intervall.
Patienter i 8Q12-, 8Q16- och 2Q8-grupperna som slutförde vecka 60 fick i median (genomsnitt) 7,0 (7,0); 6,0 (6,0) respektive 10,0 (9,8) injektioner. Vecka 60 förlängdes behandlingsintervallet för 42,6 % av patienterna i 8Q12-gruppen till 16 veckor och till 20 veckor för 34,2 % av patienterna i 8Q16‑gruppen.
Patienter i 8Q12-, 8Q16- och 2Q8-grupperna som slutförde vecka 96 fick i median (genomsnitt) 9,0 (9,5), 8,0 (7,8) respektive 14,0 (13,8) injektioner.
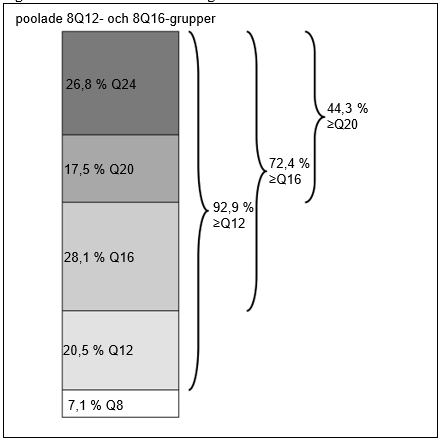
Vecka 96 hade 72,4 % av patienterna i de poolade 8Q12- och 8Q16-grupperna uppnått behandlingsintervall på ≥16 veckor, 44,3 % av patienterna hade uppnått behandlingsintervall på ≥20 veckor och 26,8 % av patienterna hade uppnått behandlingsintervall på 24 veckor med bibehållen synskärpa och anatomiska resultat.
Behandling med Eylea (både 8Q12‑ och 8Q16‑grupperna) visades vara non‑inferior och kliniskt likvärdig behandling med 2Q8 när det gäller det primära effektmåttet ’genomsnittlig förändring i BCVA vecka 48’ och det viktiga sekundära effektmåttet ’genomsnittlig förändring i BCVA vecka 60’. Behandlingseffekten med Eylea 114,3 mg/ml vad gäller genomsnittlig förändring av BCVA bibehölls till och med vecka 96.
Tabell 7: Effektresultat från PHOTON-studien
| Effektresultat | Vecka | Eylea 8Q12 (N = 328) | Eylea 8Q16 (N = 163) | Eylea 2Q8 (N = 167) |
| Förändring i BCVA från studiestart mätt med ETDRS-bokstavspoäng D | ||||
| Aritmetiskt medelvärdet (SD), observerat | 48 | 8,77 (8,95) | 7,86 (8,38) | 9,21 (8,99) |
| LS mean (SE) A | 8,10 (0,61) | 7,23 (0,71) | 8,67 (0,73) | |
Skillnad i LS mean (95 % KI) A,B | ‑0,57 (‑2,26; 1,13) | ‑1,44 (‑3,27; 0,39) | ||
| p-värde (ensidigt non-inferiority-test med en marginal på 4 bokstäver) A,B | <0,0001 | 0,0031 | ||
| Aritmetiskt medelvärdet (SD), observerat | 60 | 9,05 (9,27) | 7,96 (9,14) | 9,62 (9,58) |
| LS mean (SE) A | 8,52 (0,63) | 7,64 (0,75) | 9,40 (0,77) | |
Skillnad i LS mean (95 % KI) A,B | ‑0,88 (‑2,67; 0,91) | ‑1,76 (‑3,71; 0,19) | ||
| p-värde (ensidigt non-inferiority-test med en marginal på 4 bokstäver) A,B | 0,0003 | 0,0122 | ||
| Aritmetiskt medelvärdet (SD), observerat | 96 | 8,82 (9,93) | 7,50 (9,86) | 8,41 (11,10) |
| LS mean (SE) A | 8,15 (0,63) | 6,59 (0,77) | 7,70 (0,89) | |
Skillnad i LS mean (95 % KI) A,B | -0,45 (-1,55; 2,45) | -1,11 (-3,27; 1,05) | ||
| Patienter som fick en ETDRS-bokstavspoäng på minst 69 (cirka 20/40 Snellen-ekvivalent) D | ||||
| Andel (LOCF) | 48 | 65,3 % | 62,6 % | 63,0 % |
Justerad skillnad i andel (95 % KI) B,C | 2,45 % (‑6,47 %, 11,36 %) | ‑0,67 % (‑11,16 %, 9,82 %) | ||
| Andel (LOCF) | 60 | 64,7 % | 62,0 % | 60,6 % |
Justerad skillnad i andel (95 % KI) B, C | 4,34 % (‑4,72 %, 13,40 %) | 1,63 % (‑8,91 %, 12,17 %) | ||
| Andel (LOCF) | 96 | 66,9 % | 61,3 % | 63,0 % |
Justerad skillnad i andel (95 % KI) B, C | 4,01 % (-4,99 %; 13,01 %) | -1,51 % (-11,91 %; 8,89 %) | ||
| Patienter som hade minst 15 bokstävers förbättring i BCVA från studiestart D | ||||
| Andel (LOCF) | 48 | 18,7 % | 16,6 % | 23,0 % |
Justerad skillnad i andel (95 % KI) B, C | ‑4,64 % (‑12,30 %, 3,02 %) | ‑7,14 % (‑15,45 %, 1,17 %) | ||
| Andel (LOCF) | 60 | 21,5 % | 16,0 % | 26,1 % |
Justerad skillnad i andel (95 % KI) B, C | ‑5,01 % (‑13,04 %; 3,02 %) | ‑10,78 % (‑19,27 %; ‑2,29 %) | ||
| Andel (LOCF) | 96 | 24,5 % | 19,6 % | 26,1 % |
Justerad skillnad i andel (95 % KI) B, C | -1,88 % (-10,03 %; 6,28 %) | -7,07 % (-15,94 %; 1,80 %) | ||
| Sista avsedda behandlingsintervall | ||||
| Patienter med behandlingsintervall ≥Q12 E | ||||
| Andel (poolade Q812- och 8Q16-grupper) | 96 | 92,9 % | n/a | |
| Andel | 91,8 % | 95,0 % | n/a | |
| Patienter med behandlingsintervall ≥Q16 E | ||||
| Andel (poolade Q812- och 8Q16-grupper) | 96 | 72,4 % | n/a | |
| Andel | 64,1 % | 87,8 % | n/a | |
| Patienter med behandlingsintervall ≥Q20 E | ||||
| Andel (poolade Q812- och 8Q16-grupper) | 96 | 44,3 % | n/a | |
| Andel | 43,0 % | 46,8 % | n/a | |
| Patienter med behandlingsintervall Q24 E | ||||
| Andel (poolade Q812- och 8Q16-grupper) | 96 | 26,8 % | n/a | |
| Andel | 23,8 % | 32,4 % | n/a | |
A LS mean (minsta kvadratmedelvärde), KI och p-värde baserade på en MMRM med mätningar av bästa korrigerad synskärpa (BCVA) vid baslinjen som kovariat, behandlingsgrupp som faktor, besök och stratifieringsvariabler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen) som fasta faktorer samt villkor för interaktion mellan baslinje-BCVA och besök och för interaktionen mellan behandling och besök.
B Absoluta skillnader är Eylea 8Q12‑ respektive 8Q16‑grupper minus 2Q8‑grupper.
C Mantel‑Haenszel-viktad behandlingsskillnad med stratifieringsvaraibler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen) och KI beräknat med normal approximation.
D Hela analyssetet
E Säkerhetsanalys-set; patienter som anses ha vara slutförtare behandlingen för respektive tidpunkt.
KI: konfidensintervall
LOCF: Last Observation Carried Forward
LS: minsta kvadrat (Least square)
SD: standardavvikelse
SE: standardfel
Behandlingsintervall analyserades på ett fördefinierat explorativt sätt.
Figur 6: LS-medelvärde för förändring av BCVA uppmätt med ETDRS-bokstavspoäng från baslinjen till och med vecka 96 (hela analyssetet) i studien PHOTON
Figur 7: Sista avsedda behandlingsintervall vecka 96
Eylea i samtliga doser (8Q12, 8Q16, 2Q8) visade betydande förändringar från studiestart i det fördefinierade sekundära effektmåttet frågeformuläret NEI VFQ‑25 (National Eye Institute Visual Function Questionnaire-25).
Inga kliniskt betydande skillnader sågs mellan 8Q12‑, 8Q16‑ och 2Q8‑grupper i förändringar i totalpoäng enligt NEI VFQ‑25 vecka 48 och vecka 96 från studiestart.
Effektresultat i utvärderbara subgrupper för ålder, kön, geografisk region, etnicitet, ras, BCVA vid studiestart och CRT vid studiestart och tidigare DME-behandling överensstämde med resultaten i den totala populationen.
Effekt bibehölls generellt till och med vecka 96.
Behandlingseffekterna i subgrupper med tidigare behandlade patienter var likartade de som sågs hos tidigare obehandlade patienter.
Resultat – PHOTON-studiens förlängningsfas
Vid slutet av studiens huvudfas, i vecka 96, kunde patienterna registrera sig i den 60 veckor långa öppna förlängningsfasen. 195 patienter som ursprungligen placerats i 8Q12- och 8Q16‑grupperna fortsatte med Eylea 114,3 mg/ml och bibehöll sina senaste behandlingsintervall. 70 patienter som ursprungligen placerats i 2Q8‑gruppen vid studiens början fick byta till Eylea 114,3 mg/ml och börja med 12‑veckorsintervall. Behandlingsintervallen kunde justeras ytterligare baserat på läkarens bedömning av synskärpa och/eller anatomiska resultat.
Hos de patienter som ursprungligen placerats i 8Q12- och 8Q16‑grupperna bibehölls behandlingseffekten av Eylea 114,3 mg/ml generellt under 3 års tid (vecka 156). Förändringen i LS-medelvärde från baslinjen i de sammanslagna 8Q12- och 8Q16‑grupperna var +7,2 bokstäver i BCVA och ‑192,4 mikrometer i CRT vid vecka 156.
Hos de patienter som ursprungligen placerats i 2Q8‑gruppen var behandlingseffekten med Eylea 114,3 mg/ml liknande. Förändringen i LS-medelvärde från baslinjen var +6,5 bokstäver i BCVA och ‑197,4 mikrometer i CRT vid vecka 156.
Patienterna i 8Q12- och 8Q16‑grupperna som slutförde vecka 156 fick i median (genomsnitt) 13,0 (13,2) respektive 11,0 (11,4) injektioner.
Patienterna som bytte till Eylea 114,3 mg/ml och slutförde vecka 156 fick en total median (genomsnitt) på 19,0 (18,6) injektioner, varav 5,0 (4,8) injektioner administrerades efter bytet till Eylea 114,3 mg/ml inom de 60 veckorna av studiens förlängningsfas.
Den allmänna säkerhetsprofilen i förlängningsfasen liknade den som observerades i huvudfasen.
Tabell 8: Effektresultat från PHOTON‑studiens förlängningsfas vid vecka 156
| Effektresultat | 8Q12 som fortsatte med Eylea 114,3 mg/ml (N = 103) | 8Q16 som fortsatte med Eylea 114,3 mg/ml (N = 49) | 2Q8 som bytte till Eylea 114,3 mg/ml (N = 70) |
|---|---|---|---|
| Förändring i BCVA från baslinjen (LS mean) | +6,8 bokstäver | +8,1 bokstäver | +6,5 bokstäver |
| Förändring i CRT från baslinjen (LS mean) | ‑190,3 mikrometer | ‑198,1 mikrometer | ‑197,4 mikrometer |
| Sista avsedda behandlingsintervall A | |||
| ≥ 12 veckor | 85,4 % | 91,8 % | 82,8 % |
| ≥ 16 veckor | 62,1 % | 81,6 % | 50,0 % |
| ≥ 20 veckor | 40,8 % | 63,3 % | 19,0 % |
| 24 veckor | 20,4 % | 42,9 % | NA B |
A baserat på patienter som slutförde vecka 156
B Ej tillämpligt för patienter som ursprungligen randomiserades till 2Q8, på grund av studiedesign/studielängd
RVO
Studiens syften
Säkerhet och effekt för Eylea 114,3 mg/ml bedömdes i en randomiserad, dubbelmaskerad, aktivt kontrollerad multicenterstudie (QUASAR) på patienter med behandlingsnaivt makulaödem sekundärt till RVO.
Det primära syftet var att bestämma om behandling med Eylea 114,3 mg/ml med 8 veckors intervall (8Q8) ger non-inferior bästa korrigerad synskärpa (BCVA)-förändringar jämfört med Eylea 40 mg/ml var 4:e vecka (2Q4).
De sekundära syftena var att bestämma om behandling med 8Q8 kräver färre injektioner jämfört med 2Q4, att bestämma effekten av Eylea 114,3 mg/ml jämfört med Eylea 40 mg/ml på anatomiska och andra visuella responsmått samt att utvärdera säkerhet och farmakokinetik för aflibercept.
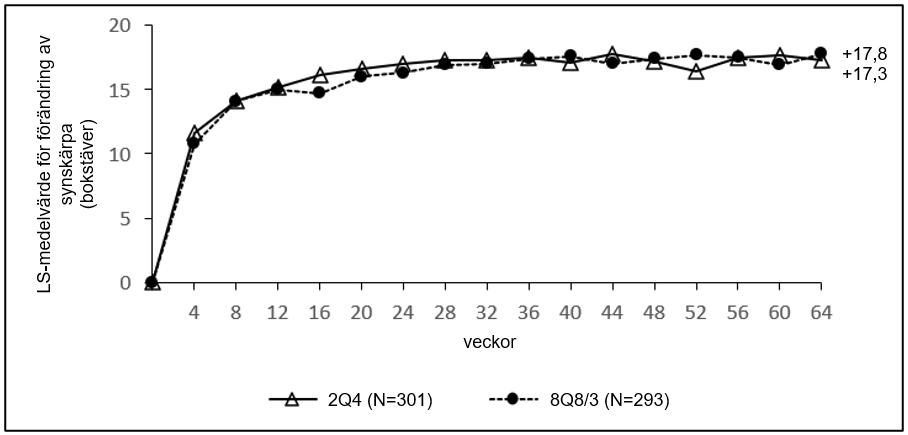
Det primära effektmåttet var förändringen av BCVA från studiestart mätt som Early Treatment Diabetic Retinopathy Study (ETDRS)-bokstavspoäng vecka 36.
Det viktigaste sekundära effektmåttet var antalet aktiva injektioner från studiestart till vecka 64.
Ytterligare sekundära effektmått var bland annat antalet aktiva injektioner från studiestart till vecka 36, andelen patienter som vecka 36 hade minst 15 bokstävers förbättring i BCVA från studiestart, andelen patienter som fick en ETDRS-bokstavspoäng på minst 69 (cirka 20/40 Snellen-ekvivalent) vecka 36 samt förändring från studiestart i total poäng enligt NEI VFQ-25 (National Eye Institute Visual Functioning Questionnaire-25) vecka 36.
I QUASAR-studien behandlades totalt 892 patienter. Patienterna fördelades i förhållandet 1:1:1 till 1 av 3 parallella behandlingsgrupper:
- Eylea 114,3 mg/ml administrerat var 8:e vecka efter 3 initiala injektioner med 4‑veckorsintervall (8Q8/3)
- Eylea 114,3 mg/ml administrerat var 8:e vecka efter 5 initiala injektioner med 4‑veckorsintervall (8Q8/5)
- Eylea 40 mg/ml administrerat var 4:e vecka (2Q4).
Från vecka 16 (8Q8/3), vecka 24 (8Q8/5) och vecka 40 (2Q4, vid tidigare förlängning till Q8), kunde patienterna förkorta intervallet med 4 veckor om båda följande kriterier var uppfyllda vid ett doseringsbesök:
- >5 bokstävers bortfall i BCVA från referensbesöket, och
- >50 mikrometers ökning av CRT från referensbesöket.
Förlängning av intervallet var tillåten från och med vecka 32 (2Q4 och 8Q8/3) eller vecka 40 (8Q8/5) i steg om 4 veckor om båda följande kriterier var uppfyllda vid ett doseringsbesök:
- <5 bokstävers bortfall i BCVA från referensbesöket, och
- CRT <320 mikrometer i SD‑OCT (eller <300 mikrometer om mätning exkluderade RPE).
Referensbesök var vid vecka 12 för 8Q8/3 och vecka 20 för 8Q8/5 och 2Q4.
För patienter som inte uppfyllde kriterierna för att förkorta eller förlänga intervallet, bibehölls doseringsintervallet. Det kortaste intervallet mellan injektionerna var 4 veckor i alla grupper.
Patienter med bilateral sjukdom kunde behandlas med Eylea 40 mg/ml eller annat anti-VEGF-läkemedel i det andra ögat.
Patientkarakteristika vid studiestart
Patienter i åldern från 23 till 95 år med en genomsnittsålder på 65,9 år.
Cirka 57 % (168/293) och 57 % (170/298) av patienterna randomiserade till grupperna 8Q8/3 respektive 8Q8/5 var 65 år eller äldre och cirka 26 % (76/293) respektive 25 % (74/298) var 75 år eller äldre.
425 (48 %) av de rekryterade patienterna hade CRVO/HRVO och 467 (52 %) hade BRVO. Andelen patienter per subtyp var liknande i behandlingsgrupperna.
Resultat
Behandling med Eylea 114,3 mg/ml visades vara non‑inferior och kliniskt likvärdig behandling med 2Q4 när det gäller det primära effektmåttet ”förändring i BCVA från studiestart mätt med ETDRS-bokstavspoäng vecka 36”.
Dessutom visade sig behandling med Eylea 114,3 mg/ml vara överlägsen behandling med 2Q4 vad gäller det viktiga sekundära effektmåttet ”antal aktiva injektioner från studiestart till vecka 64”.
Eylea 8Q8/3‑gruppen behövde 3,2 färre injektioner än 2Q4‑gruppen.
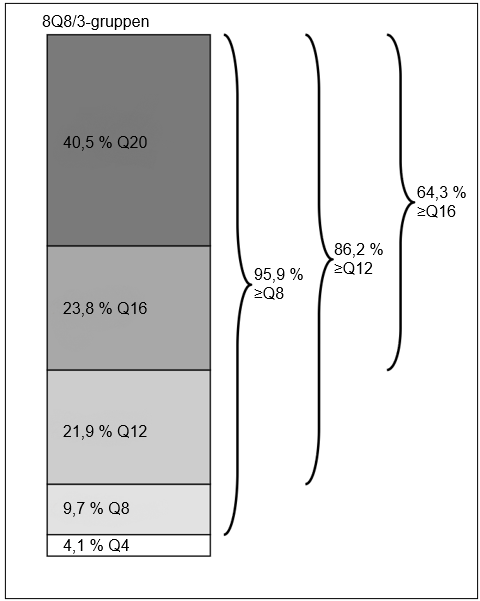
Vecka 36 hade 93,9 % av patienterna i 8Q8/3‑gruppen uppnått behandlingsintervall på ≥8 veckor, med bibehållen synskärpa och anatomiska resultat.
Vecka 64 hade 56,1 % av patienterna i 8Q8/3‑gruppen slutfört behandlingsintervall på 16 veckor med bibehållen synskärpa och anatomiska resultat.
Vecka 64 hade 40,5 % av patienterna i 8Q8/3‑gruppen ett sista avsedda behandlingsintervall på 20 veckor med bibehållen synskärpa och anatomiska resultat.
Tabell 9: Effektresultat från studien QUASAR
| Effektresultat | Vecka | Eylea 8Q8/3 (N = 293) | Eylea 2Q4 (N = 301) |
| Förändring i BCVA från studiestart mätt med ETDRS-bokstavspoäng A | |||
| Aritmetiskt medelvärde (SD), observerat | 36 | 17,0 (11,8) | 17,8 (13,1) |
| LS mean (SE) B | 17,4 (0,7) | 17,5 (0,7) | |
| Skillnad i LS mean (95 % KI) B, C | ‑0,1 (‑2,0; 1,9) | ||
| p-värde (ensidigt non-inferiority-test med en marginal på 4 bokstäver) B, C | <0,0001 | ||
| Aritmetiskt medelvärde (SD), observerat | 64 | 17,3 (12,7) | 17,4 (14,6) |
| LS mean (SE) B | 17,8 (0,7) | 17,3 (0,8) | |
| Skillnad i LS means (95 % KI) B, C | 0,5 (‑1,6; 2,7) | ||
| CRVO/HRVO D | |||
| Aritmetiskt medelvärde (SD), observerat | 36 | 16,5 (12,7) | 16,2 (14,7) |
| LS mean (SE) B | 16,6 (1,1) | 15,9 (1,2) | |
| Skillnad i LS means (95 % KI) B, C | 0,6 (‑2,6; 3,9) | ||
| Aritmetiskt medelvärde (SD), observerat | 64 | 16,5 (13,8) | 14,8 (16,8) |
| LS mean (SE) B | 17,2 (1,2) | 15,2 (1,3) | |
| Skillnad i LS means (95 % KI) B, C | 2,0 (‑1,5; 5,6) | ||
| BRVO D | |||
| Aritmetiskt medelvärde (SD), observerat | 36 | 17,4 (10,9) | 19,4 (11,0) |
| LS mean (SE) B | 18,3 (0,8) | 19,0 (0,8) | |
| Skillnad i LS means (95 % KI) B, C | ‑0,8 (‑2,9; 1,4) | ||
| Aritmetiskt medelvärde (SD), observerat | 64 | 18,1 (11,8) | 20,1 (11,4) |
| LS mean (SE) B | 18,4 (0,9) | 19,6 (0,8) | |
| Skillnad i LS means (95 % KI) B, C | ‑1,1 (‑3,5; 1,2) | ||
| Patienter som fick en ETDRS-bokstavspoäng på minst 69 (cirka 20/40 Snellen-ekvivalent) A | |||
| Andel (OC) | 36 | 72,7 % | 67,8 % |
| 64 | 70,4 % | 70,2 % | |
| Patienter som hade minst 15 bokstävers förbättring i BCVA från studiestart A | |||
| Andel (OC) | 36 | 58,8 % | 59,8 % |
| 64 | 61,7 % | 60,4 % | |
| Patienter som hade minst 15 bokstävers försämring i BCVA från studiestart A | |||
| Andel (OC) | 36 | 1,2 % | 1,5 % |
| 64 | 1,2 % | 2,4 % | |
| Patienter utan IRF och ingen SRF i den centrala näthinnan A | |||
| Andel (OC) | 36 | 81,2 % | 83,7 % |
| 64 | 76,3 % | 66,0 % | |
| Antal aktiva injektioner | |||
| Aritmetiskt medelvärde (SD), observerat E | 36 | 6,1 (0,6) | 8,8 (0,8) |
| LS mean (SE) F | 6,1 (0,0) | 8,8 (0,0) | |
| Skillnad i LS means (95 % KI) F, C | ‑2,7 (‑2,8; ‑2,6) | ||
| Aritmetiskt medelvärde (SD), observerat E | 64 | 8,4 (1,2) | 11,7 (1,6) |
| LS mean (SE) F | 8,5 (0,1) | 11,7 (0,1) | |
| Skillnad i LS means (95 % KI) F, C | ‑3,2 (‑3,5; ‑3,0) | ||
| p‑värde (tvåsidigt superiority-test) G, C | <0,0001 | ||
| Bibehållet behandlingsintervall | |||
| Patienter som bibehöll behandlingsintervall ≥Q8 E | |||
| Andel | 36 | 88,5 % | n/a |
| 64 | 88,1 % | 70,0 % H | |
| Sista slutförda behandlingsintervall | |||
| Patienter med behandlingsintervall Q4 E | |||
| Andel | 64 | 4,8 % | 13,0 % |
| Patienter med behandlingsintervall ≥Q8 E | |||
| Andel | 64 | 95,2 % | 87,0 % |
| Patienter med behandlingsintervall ≥Q12 E | |||
| Andel | 64 | 81,4 % | 67,8 % |
| Patienter med behandlingsintervall Q16 E | |||
| Andel | 64 | 56,1 % | n/a |
| Sista avsedda behandlingsintervall | |||
| Patienter med behandlingsintervall ≥Q8 E | |||
| Andel | 36 | 93,9 % | 75,6 % |
| 64 | 95,9 % | 92,2 % | |
| Patienter med behandlingsintervall Q12 E | |||
| Andel | 36 | 69,1 % | n/a |
| 64 | 21,9 % | 27,8 % | |
| Patienter med behandlingsintervall ≥Q12 E | |||
| Andel | 64 | 86,2 % | 77,8 % |
| Patienter med behandlingsintervall ≥Q16 E | |||
| Andel | 64 | 64,3 % | 50,0 % |
| Patienter med behandlingsintervall Q20 E | |||
| Proportion | 64 | 40,5 % | n/a |
A Hela analyssetet
B LS mean (minsta kvadratmedelvärde), KI och p-värde baserade på en MMRM med mätning av bästa korrigerade synskärpa (BCVA) vid baslinjen som kovariat, behandlingsgrupp som faktor, besök och stratifieringsvariabler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen och RVO-typ) som fasta faktorer samt villkor för interaktion mellan baslinje-BCVA och besök och för interaktionen mellan behandling och besök.
C Absolut skillnad är Eylea 8Q8/3‑gruppen minus 2Q4‑gruppen.
D Antalet patienter med CRVO/HRVO var 134 och 152 i 8Q8/3‑ respektive 2Q4‑behandlingsgruppen. Antalet patienter med BRVO var 159 och 149 i 8Q8/3‑ respektive 2Q4‑behandlingsgruppen.
E Säkerhetsanalys-set; patienter som anses ha slutfört behandlingen för respektive tidpunkt.
F LS mean och KI baserade på en multipel imputationsprocedur som tillämpar en linjär regressionsmodell justerad för baslinje-BCVA, baslinje näthinnans centrala tjocklek (CST), och stratifieringsvariabler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen och RVO-typ) på varje imputerat dataset och kombination av resultat med användning av Rubins regel.
G p‑värde baserat på en multipel imputationsprocedur som tillämpar en icke-parametrisk ranganalys av kovarians justerad för baslinje-BCVA, baslinje-CST och stratifieringsvariabler använda för randomisering (geografisk region, BCVA-kategori vid baslinjen och RVO-typ) på varje imputerat dataset och kombination av resultat med användning av Rubins regel.
H Patienter i 2Q4‑behandlingsgruppen som förlängde vid vecka 32 och bibehöll ≥Q8 till och med vecka 64.
KI: Konfidensintervall
ETDRS: Early Treatment Diabetic Retinopathy Study
OC: observerade fall, data efter förekomst av en interkurrent händelse uteslöts i linje med den primära estimandstrategin
LS: minsta kvadrat (Least square)
MMRM: blandad modell för upprepade mätningar
SD: Standardavvikelse
SE: Standardfel
Figur 8: LS-medelvärde för förändring av BCVA uppmätt med ETDRS-bokstavspoäng från baslinjen till och med vecka 64 (hela analyssetet) i studien QUASAR
Eylea i samtliga doser (8Q8/3, 2Q4) visade betydande förändringar från studiestart i det fördefinierade sekundära effektmåttet frågeformuläret NEI VFQ‑25 (National Eye Institute Visual Function Questionnaire-25). Omfattningen av dessa förändringar var i linje med det som setts i publicerade studier, vilket återspeglas i förbättringar av synrelaterad livskvalitet.
Inga kliniskt betydande skillnader sågs mellan 8Q8/3‑ och 2Q4‑grupperna i förändringar i totalpoäng enligt NEI VFQ‑25 vecka 36 och vecka 64 från studiestart.
Effektresultat i förspecificerade utvärderbara subgrupper för RVO-typ, ålder, kön, geografisk region, etnicitet, ras, BCVA vid studiestart och CRT vid studiestart överensstämde med resultaten i den totala populationen.
Effekt bibehölls generellt till och med vecka 64.
Vad gäller det förspecificerade sekundära effektmåttet ”deltagare doserade endast 8Q8 till och med vecka 36 i Eylea 114,3 mg/ml 8Q8-gruppen” bibehöll 88,5 % av patienterna i 8Q8/3‑gruppen sitt ursprungliga randomiserade behandlingsintervall på 8 veckor, med bibehållen synskärpa och anatomiska resultat.
Figur 9: Sista avsedda behandlingsintervall vecka 64
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för aflibercept för alla grupper av den pediatriska populationen för nAMD, DME och RVO (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption/Distribution
Aflibercept absorberas långsamt från ögat till den systemiska cirkulationen efter intravitreal administrering där den huvudsakligen finns som ett inaktivt, stabilt komplex med VEGF. Det är dock bara ”fritt aflibercept” som kan binda till endogent VEGF.
Efter unilateral intravitreal administrering av 8 mg aflibercept var genomsnittligt (SD) Cmax för fritt aflibercept i plasma 0,25 (0,21) mg/l och mediantiden till maximal koncentration i plasma var 1 dag i den kombinerade nAMD- och DME-populationen. Ackumuleringen av fritt aflibercept i plasma efter tre initiala månatliga doser var minimal. Därefter observerades ingen ytterligare ackumulering. Dessa data stöds också av populationsfarmakokinetiska analyser.
Dessa farmakokinetiska resultat överensstämmer med data erhållna från patienter med RVO.
Eliminering
Aflibercept är ett proteinbaserat läkemedel och inga studier avseende metabolism har genomförts.
Aflibercept förväntas genomgå eliminering genom både målmedierad disposition via bindning till fritt endogent VEGF och metabolism via proteolys. Mediantiden till dess att den sista kvantifierbara koncentrationen av fritt afliberept uppnåddes i plasma för 8 mg administrerat intravitrealt var 3 veckor.
Nedsatt njur- eller leverfunktion
Inga särskilda studier på patienter med nedsatt njur- eller leverfunktion har utförts med Eylea 114,3 mg/ml.
Den systemiska exponeringen för aflibercept hos patienter med lätt till gravt nedsatt njurfunktion var liknande den som sågs hos patienter med normal njurfunktion. Begränsade data för patienter med lätt nedsatt leverfunktion tyder inte på någon påverkan på systemisk exponering för aflibercept jämfört med patienter med normal njurfunktion.
Prekliniska säkerhetsuppgifter
Erosioner och ulcerationer av respiratoriskt epitel i näsmusslor hos apor behandlade med aflibercept intravitrealt observerades vid systemisk exponering som översteg den maximala humana exponeringen. Den systemiska exponeringen för fritt aflibercept var cirka 26 respektive 33 gånger högre baserat på Cmax och AUC jämfört med motsvarande värden hos vuxna patienter efter en intravitreal dos på 8 mg. Vid NOAEL (No Observed Adverse Effect Level) på 0,5 mg/öga hos apor var den systemiska exponeringen 3,2 och 3,8 gånger högre baserat på Cmax och AUC jämfört med motsvarande värden hos vuxna patienter.
Inga studier har utförts på mutagen eller karcinogen potential för aflibercept.
En effekt av aflibercept på intrauterin utveckling sågs i embryofetala utvecklingsstudier hos dräktiga kaniner med intravenös (3 till 60 mg/kg) samt subkutan (0,1 till 1 mg/kg) administrering. Maternell NOAEL var vid dosen 3 mg/kg respektive 1 mg/kg. NOAEL för embryoutveckling identifierades inte. Vid dosen 0,1 mg/kg var den systemiska exponeringen för fritt aflibercept cirka 1,0 och 1,0 gånger högre baserat på Cmax respektive kumulativ AUC vid jämförelse med motsvarande värden hos vuxna patienter efter en intravitreal dos på 8 mg.
Effekter på manlig och kvinnlig fertilitet bedömdes som en del av en 6‑månaders studie på apor med intravenös administrering av aflibercept vid doser mellan 3 och 30 mg/kg. Avsaknad av eller oregelbunden mens förknippades med förändringar av nivåerna av kvinnligt reproduktionshormon och förändringar av spermiernas morfologi och motilitet observerades vid alla dosnivåer. Baserat på Cmax och AUC för fritt aflibercept observerat vid 3 mg/kg intravenös dos var den systemiska exponeringen cirka 377 respektive 104 gånger högre än exponeringen hos människor efter en intravitreal dos på 8 mg. Alla förändringar var reversibla.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Sackaros
Argininhydroklorid
Histidinhydrokloridmonohydrat
Histidin
Polysorbat 20
Vatten för injektionsvätskor
Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
3 år
Särskilda förvaringsanvisningar
Eylea 114,3 mg/ml injektionsvätska, lösning
Förvaras i kylskåp (2 °C-8 °C).
Får ej frysas.
Förvara injektionsflaskan i ytterkartongen. Ljuskänsligt.
Före användning kan den oöppnade injektionsflaskan förvaras utanför kylskåp vid högst 25 °C i upp till 24 timmar.
Eylea 114,3 mg/ml injektionsvätska, lösning i förfylld spruta
Förvaras i kylskåp (2 °C-8 °C).
Får ej frysas.
Förvara den förfyllda sprutan i dess blister i ytterkartongen. Ljuskänsligt.
Före användning kan den oöppnade blistret förvaras utanför kylskåp vid högst 25 °C i upp till 24 timmar.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
EYLEA injektioneste, liuos
114,3 mg/ml (L:ei) 1 kpl (0,263 ml) (748,01 €)
EYLEA injektioneste, liuos, esitäytetty ruisku
114,3 mg/ml (L:ei) 1 kpl (0,184 ml) (748,01 €)
PF-selosteen tieto
Eylea 114,3 mg/ml injektionsvätska, lösning
Injektionsflaska (typ I‑glas) med en grå gummipropp (klorobutyl) förseglad med en aluminiumkapsyl med vitt lock och en 18 G, 5 mikrometers filternål.
Varje injektionsflaska innehåller 0,263 ml lösning.
Förpackningsstorlek: 1 injektionsflaska och 1 filternål.
Eylea 114,3 mg/ml injektionsvätska, lösning i förfylld spruta
Förfylld spruta (typ I-glas) med en grå kolvpropp (elastomergummi), en vit Luer-lock-adapter med ett grått spetslock (elastomergummi) och ett blått OcuClick doseringssystem (PC/ABS-plast).
Varje förfylld spruta innehåller 0,184 ml lösning.
Förpackningsstorlek: 1 förfylld spruta.
Läkemedlets utseende:
Klar till svagt opaliserande, färglös till ljusgul, isoosmotisk lösning, pH 5,8.
Särskilda anvisningar för destruktion och övrig hantering
Eylea 114,3 mg/ml injektionsvätska, lösning
Injektionsflaskan är endast avsedd för engångsbruk i ett öga. Extraktion av flera doser från en injektionsflaska kan öka risken för kontamination och efterföljande infektion.
Använd inte om förpackningen eller dess komponenter har passerat utgångsdatum, är skadade eller har manipulerats.
Kontrollera etiketten på injektionsflaskan för att säkerställa att styrkan på Eylea är den du avser att använda. För 8 mg-dosen måste Eylea 114,3 mg/ml injektionsflaska användas.
18 G, 5 mikrometers filternål:
Uppdragningskanyl med trubbig spets och filter, ej för injektion i huden.
Uppdragningskanyl med trubbig spets och filter ska inte autoklaveras.
Filternålen är pyrogenfri. Använd inte nålen om den enskilda förpackningen är skadad.
Kassera använd uppdragningskanyl med trubbig spets och filter i godkänd behållare för stickande/skärande avfall.
Varning: Återanvändning av filternålen kan leda till infektion eller annan sjukdom/skada.
Den intravitreala injektionen ska utföras med en 30 G × ½ inch injektionsnål (medföljer inte). Användning av en nål med en mindre storlek (högre gauge) än den rekommenderade 30 G × ½ inch injektionsnålen kan leda till högre injektionstryck.
| 1. | Inspektera lösningen visuellt före administrering. Använd inte injektionsflaskan om lösningen innehåller partiklar, är grumlig eller missfärgad. | |
| 2. | Ta bort plastlocket och desinficera den yttre delen av injektionsflaskans gummipropp. | 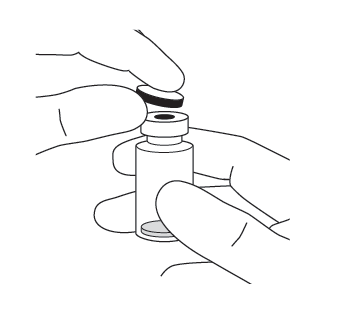 |
| 3. | Använd aseptisk teknik till att utföra steg 3‑10. Anslut filternålen som medföljer kartongen till en 1 ml steril Luer-lock-spruta. | 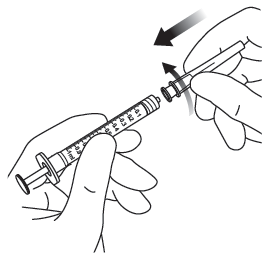 |
| 4. | Tryck in filternålen i mitten på injektionsflaskans propp tills nålen når botten på injektionsflaskan eller kanten av injektionsflaskans botten. | |
| 5. | Dra upp allt innehåll ur injektionsflaskan i sprutan samtidigt som du håller injektionsflaskan upprätt och lutar den något för att underlätta uppdragandet. Se till att avfasningen på filternålen är nedsänkt i lösningen för att förhindra luft från att komma in. Fortsätt med att luta injektionsflaskan under uppdragandet för att hålla avfasningen på filternålen nedsänkt i lösningen. | |
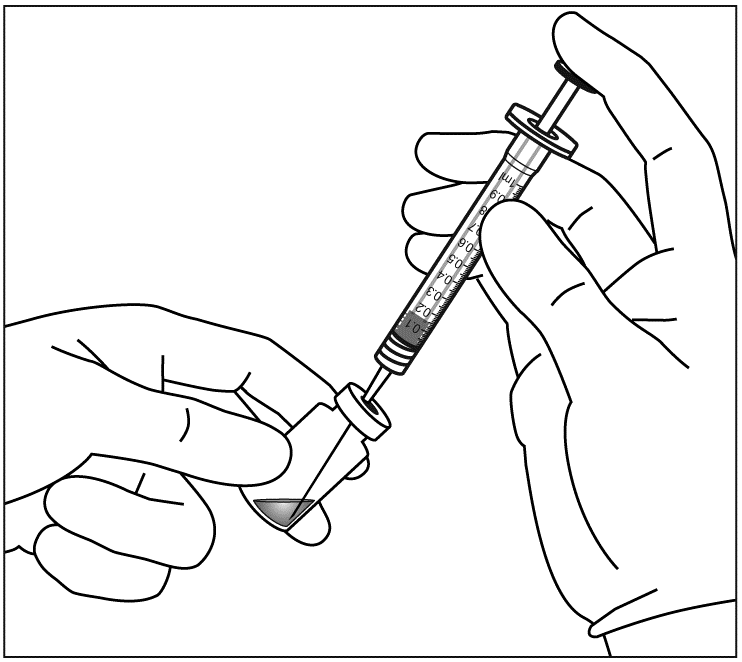 | 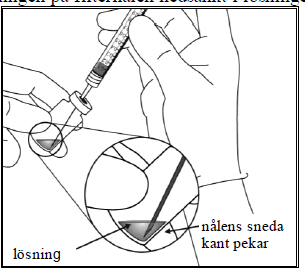 | |
| 6. | Kontrollera att kolvstången är tillräckligt uppdragen när du tömmer injektionsflaskan för att helt tömma filternålen. Efter injektionen måste eventuellt överblivet läkemedel kasseras. | |
| 7. | Ta bort filternålen och kassera den enligt gällande riktlinjer. Obs! Filternålen får inte användas för den intravitreala injektionen. | |
| 8. | Vrid fast 30 G × ½ inch injektionsnålen ordentligt på Luer-lock-sprutans spets. | 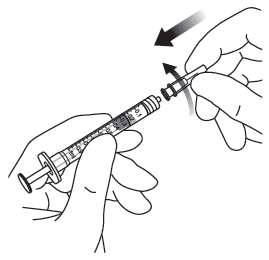 |
| 9. | Håll sprutan med nålen uppåt och kontrollera om det finns bubblor i sprutan. Om det finns bubblor, knacka försiktigt på sprutan med fingret tills bubblorna stiger uppåt. | 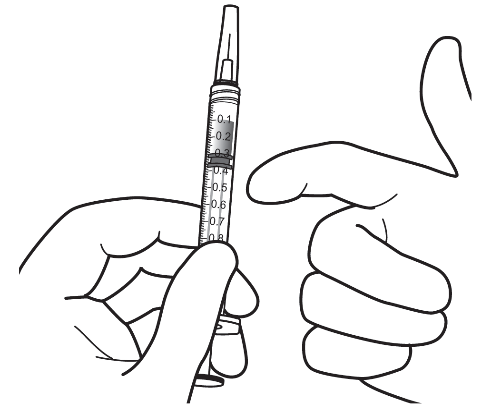 |
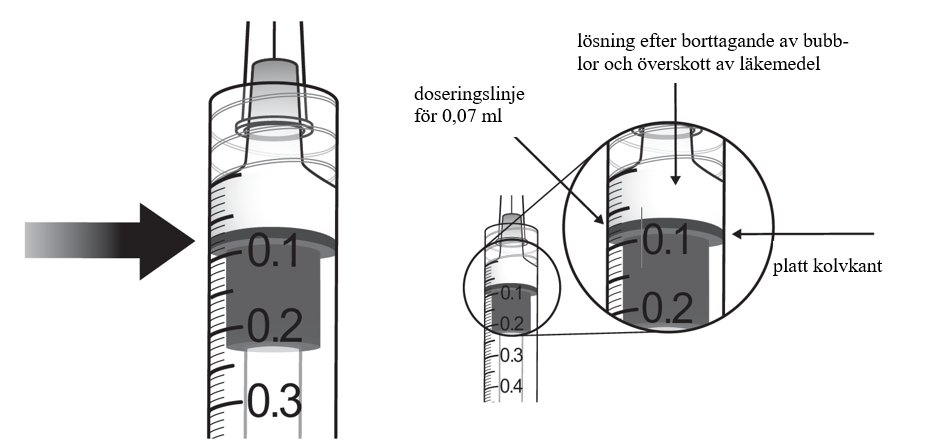
| 10. | Ta bort alla luftbubblor och allt överskott av läkemedel genom att långsamt trycka ned kolven så att den platta kolvkanten ligger i linje med markeringen 0,07 ml på sprutan. | |
| ||
Eylea 114,3 mg/ml injektionsvätska, lösning i förfylld spruta
Den förfyllda sprutan med OcuClick doseringssystem är endast avsedd för engångsbruk i ett öga. Extraktion av flera doser från en förfylld spruta med OcuClick doseringssystem kan öka risken för kontamination och efterföljande infektion.
Använd inte om förpackningen eller dess komponenter har passerat utgångsdatum, är skadade eller har manipulerats.
Kontrollera etiketten på den förfyllda sprutan med OcuClick doseringssystem för att säkerställa att styrkan på Eylea är den du avser att använda. För 8 mg-dosen måste Eylea 114,3 mg/ml förfylld spruta användas.
Den intravitreala injektionen ska utföras med en 30 G × ½ inch injektionsnål (medföljer inte).
Användning av en nål med en mindre storlek (högre gauge) än den rekommenderade 30 G × ½ inch injektionsnålen kan leda till högre injektionstryck.
| Beskrivning av förfylld spruta med integrerat OcuClick doseringssystem | ||
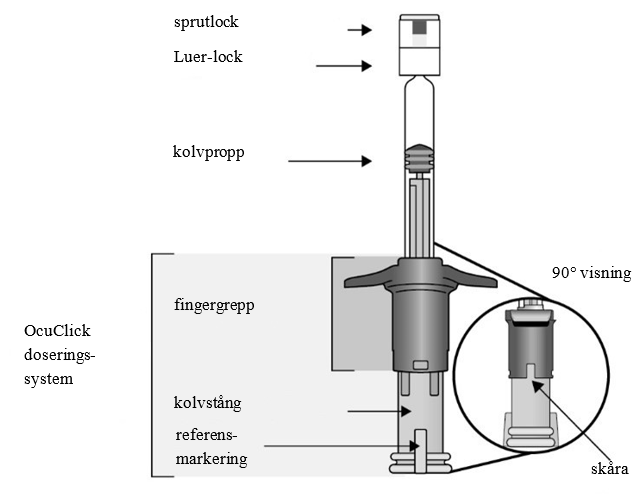 | ||
| 1. | Förbered När du är redo att administrera Eylea 114,3 mg/ml, öppna kartongen och ta ut det sterila blistret. Öppna blistret försiktigt för att garantera att innehållet förblir sterilt. Använd aseptisk teknik för att utföra steg 2-9. | |
| 2. | Ta ut sprutan Ta ut sprutan ur det sterila blistret. | |
| 3. | Inspektera spruta och injektionsvätska, lösning Använd inte den förfyllda sprutan om - partiklar, grumligheter eller missfärgningar är synliga - någon del av den förfyllda sprutan med OcuClick doseringssystem är skadad eller lös - sprutlocket har lossnat från Luer-lock. | |
| 4. | Snäpp av sprutlocket Snäpp av (vrid inte av) sprutlocket genom att hålla sprutan i den ena handen och sprutlocket mellan tummen och pekfingret i den andra handen. Observera: Dra inte tillbaka kolvstången. | 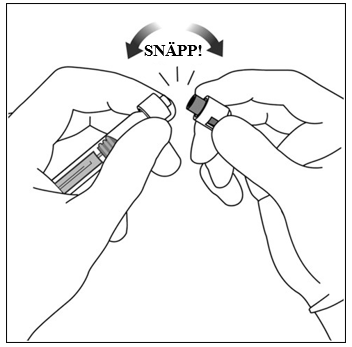 |
| 5. | Montera nålen Vrid fast 30 G × ½ inch injektionsnålen ordentligt på Luer-lock-sprutans spets. | 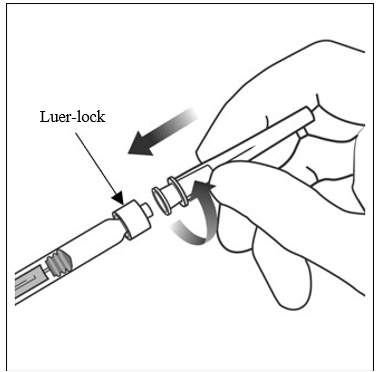 |
| 6. | Ta bort luftbubblor Håll sprutan med nålen pekande uppåt och kontrollera om det finns bubblor i sprutan. Om det finns bubblor, knacka försiktigt på sprutan med fingret tills bubblorna stiger uppåt. | 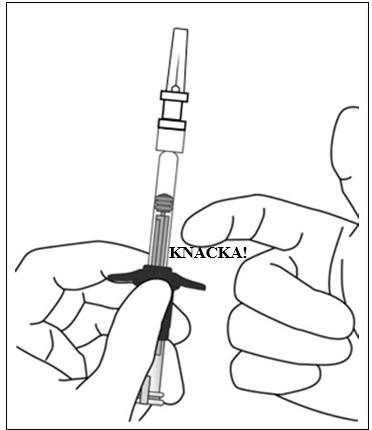 |
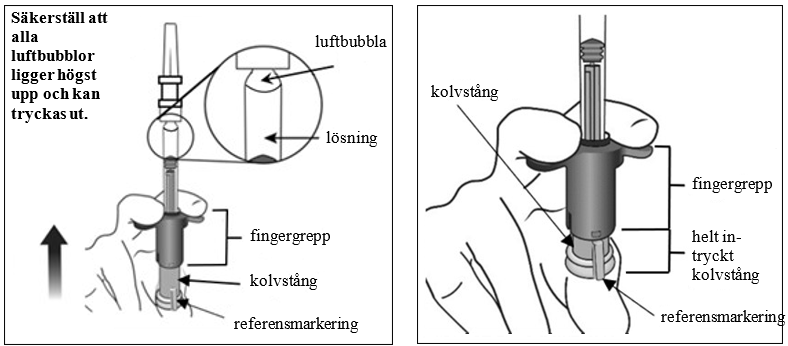
| 7. | Ta bort luft och överskottsvolym (prima) Sprutan har ingen doseringslinje eftersom den är utformad för att mekaniskt ställa in dosen enligt beskrivningen nedan. Priming och inställning av dosen måste ske med följande steg.
| |
| 8. | Ställ in dosen Vrid änden på kolvstången 90 grader medurs eller moturs, tills referensmarkeringen på kolvstången är i linje med skåran. Du kan höra ett ”klick”. | 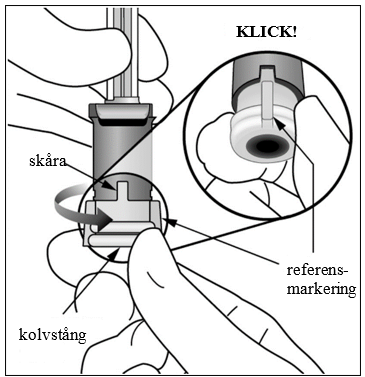 |
| 9. | Administrera injektionen För in nålen i det okulära injektionsstället. Injicera lösningen genom att trycka ned kolvstången tills det tar stopp, dvs. tills referensmarkeringen är helt införd i skåran. Tryck inte mer när referensmarkeringen är införd i skåran. Det är normalt att se en liten mängd restlösning i sprutan. | 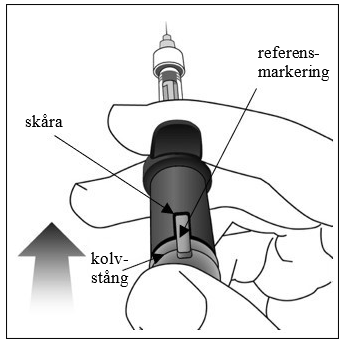 |
| 10. | Den förfyllda sprutan är endast avsedd för engångsadministrering och engångsbruk. Efter injektionen ska den förfyllda sprutan kasseras i en behållare för stickande/skärande avfall. | |
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
EYLEA injektioneste, liuos
114,3 mg/ml 1 kpl
EYLEA injektioneste, liuos, esitäytetty ruisku
114,3 mg/ml 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Aflibersepti ja farisimabi: Aikuisten neovaskulaarisen (kostean) silmänpohjan ikärappeuman (AMD) hoito (3094).
Atc-kod
S01LA05
Datum för översyn av produktresumén
15.01.2026
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi