
COLUMVI infuusiokonsentraatti, liuosta varten 2,5 mg, 10 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Tämä potilaskortti sisältää tärkeitä turvallisuutta koskevia tietoja, joista potilaan on oltava tietoinen ennen Columvi-hoidon aloittamista ja hoidon aikana.
Terveydenhuollon ammattilainen
Opas sisältää tärkeitä turvallisuustietoja terveydenhuollon ammattilaisille tumour flare -reaktion riskin minimoimiseksi ja muistutus potilaskortista.
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Columvi 2,5 mg infuusiokonsentraatti, liuosta varten
Yksi 2,5 ml:n injektiopullo konsentraattia sisältää 2,5 mg glofitamabia pitoisuutena 1 mg/ml.
Columvi 10 mg infuusiokonsentraatti, liuosta varten
Yksi 10 ml:n injektiopullo konsentraattia sisältää 10 mg glofitamabia pitoisuutena 1 mg/ml.
Glofitamabi on kaksoisspesifinen humanisoitu monoklonaalinen CD20-/CD3-vasta-aine, joka tuotetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa (CHO-soluissa).
Apuaineet, joiden vaikutus tunnetaan
Yksi 2,5 ml:n injektiopullo Columvi-valmistetta sisältää 1,25 mg (0,5 mg/ml) polysorbaattia 20.
Yksi 10 ml:n injektiopullo Columvi-valmistetta sisältää 5 mg (0,5 mg/ml) polysorbaattia 20.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Columvi on tarkoitettu aikuisille potilaille yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa uusiutuneen tai hoitoon reagoimattoman tarkemmin määrittämättömän diffuusin suurisoluisen B‑solulymfooman (DLBCL NOS) hoitoon, kun potilas ei sovellu autologiseen kantasolusiirtoon.
Columvi on tarkoitettu aikuisille potilaille monoterapiana uusiutuneen tai hoitoon reagoimattoman diffuusin suurisoluisen B‑solulymfooman hoitoon kahden tai useamman systeemisen hoitolinjan jälkeen.
Ehto
Valmistetta saa antaa vain syöpälääkkeiden käyttöön perehtyneen terveydenhuollon ammattilaisen valvonnassa hoitokeskuksessa, jossa on valmius vaikea-asteisten reaktioiden, kuten sytokiinioireyhtymän hoitoon.
Annostus ja antotapa
Columvi-valmisteen saa antaa vain syöpäpotilaiden diagnosointiin ja hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa, kun saatavilla on asianmukainen lääketieteellinen tuki sytokiinioireyhtymään liittyvien ja immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän (ICANS) vaikea-asteisten reaktioiden hoitoon.
Ennen 1. ja 2. hoitosyklin Columvi-infuusiota saatavilla on oltava vähintään 1 annos tosilitsumabia sytokiinioireyhtymän varalta. On varmistettava, että lisäannos tosilitsumabia on saatavilla 8 tunnin kuluessa edellisestä tosilitsumabiannoksesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Esilääkitys obinututsumabilla
NP30179-tutkimuksessa ja GO41944-tutkimuksessa (STARGLO) kaikki potilaat saivat 1. hoitosyklin 1. päivänä (7 päivää ennen Columvi-hoidon aloittamista) esilääkityksenä kerta-annoksen 1000 mg obinututsumabia kiertävien ja lymfoidisten B-solujen määrän vähentämiseksi (ks. taulukko 2, Annoksen viivästyminen tai saamatta jääminen, ja kohta Farmakodynamiikka).
Obinututsumabi annettiin infuusiona laskimoon nopeudella 50 mg/h. Infuusionopeutta lisättiin 50 mg/h kerrallaan 30 minuutin välein enintään nopeuteen 400 mg/h.
Ks. obinututsumabin valmistetiedoista tarkat tiedot esilääkityksestä, käyttöön valmistamisesta, annostelusta ja obinututsumabin haittavaikutusten hoidosta.
Esilääkitys ja estohoito
Sytokiinioireyhtymän estohoito
Columvi-valmistetta saavan potilaan pitää olla hyvin nesteytetty. Suositeltu esilääkitys sytokiinioireyhtymän varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) kuvataan taulukossa 1.
Taulukko 1. Esilääkitys ennen Columvi-infuusiota
| Hoitosykli (päivä) | Esilääkitystä tarvitsevat potilaat | Esilääkitys | Anto |
1. hoitosykli (8. päivä, 15. päivä); 2. hoitosykli (1. päivä); 3. hoitosykli (1. päivä) | Kaikki potilaat | 20 mg deksametasonia laskimoon1 | Annon päätyttävä viimeistään 1 tunti ennen Columvi-infuusiota |
| Suun kautta otettava kipulääkitys / kuumetta alentava lääkitys2 | Viimeistään 30 minuuttia ennen Columvi-infuusiota | ||
| Antihistamiini3 | |||
| Kaikki myöhemmät infuusiot | Kaikki potilaat | Suun kautta otettava kipulääkitys / kuumetta alentava lääkitys2 | Viimeistään 30 minuuttia ennen Columvi-infuusiota |
| Antihistamiini3 | |||
| Potilaat, joille ilmaantui sytokiinioireyhtymä edellisen annoksen yhteydessä | 20 mg deksametasonia laskimoon1, 4 | Annon päätyttävä viimeistään 1 tunti ennen Columvi-infuusiota |
1 Jos potilas ei siedä deksametasonia tai deksametasonia ei ole saatavilla, annetaan 100 mg prednisonia/prednisolonia tai 80 mg metyyliprednisolonia.
2 Esim. 1000 mg parasetamolia.
3 Esim. 50 mg difenhydramiinia.
4 Annetaan kaikille potilaille tarvittavan esilääkityksen lisäksi.
Infektioiden estohoito
Infektioriskin pienentämiseksi suositellaan estohoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tavanomaista suuremmassa riskissä oleville potilaille on harkittava sytomegaloviruksen (CMV), herpeksen, pneumocystis jiroveciin aiheuttaman keuhkokuumeen ja muiden opportunististen infektioiden estohoitoa (ks. kohta Haittavaikutukset).
Annostus
Columvi-hoito aloitetaan suurentamalla annosta asteittain (step‑up-hoito-ohjelma, jonka tarkoituksena on vähentää sytokiinioireyhtymän riskiä) suositeltuun 30 mg:n annokseen.
Columvi-monoterapia asteittain suurenevina annoksina toteutettavana hoito-ohjelmana (step-up-hoito-ohjelma)
Kun 1. hoitosyklin 1. päivänä annettava obinututsumabiesilääkitys on annettu, Columvi annetaan infuusiona laskimoon step-up-hoito-ohjelman mukaisesti, joka johtaa suositeltuun 30 mg:n annokseen (kuten taulukossa 2 esitetään). Yksi hoitosykli on 21 päivää.
Taulukko 2. Columvi-monoterapia step-up-hoito-ohjelmana uusiutunutta tai hoitoon reagoimatonta diffuusia suurisoluista B‑solulymfoomaa sairastaville potilaille
| Hoitosykli, päivä | Columvi-annos | Infuusion kesto | |
1. hoitosykli (Esilääkitys ja step-up-annos) | 1. päivä | Esilääkityksenä 1000 mg obinututsumabia1 | |
| 8. päivä | 2,5 mg | 4 tuntia2 | |
| 15. päivä | 10 mg | ||
| 2. hoitosykli | 1. päivä | 30 mg | |
| 3.–12. hoitosykli | 1. päivä | 30 mg | 2 tuntia3 |
1 Ks. edellä kohta Esilääkitys obinututsumabilla. 2 Potilailla, joille ilmaantui edellisen Columvi-annoksen yhteydessä sytokiinioireyhtymä, infuusion kestoa voidaan pidentää 8 tuntiin saakka (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). 3 Hoitavan lääkärin harkinnan mukaan, jos potilas sieti edellisen infuusion hyvin. Jos potilaalle ilmaantui jonkin aiemman annoksen yhteydessä sytokiinioireyhtymä, infuusion kestona pitää säilyttää 4 tuntia. | |||
Columvi-step-up-hoito-ohjelma yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa
Kun 1. hoitosyklin 1. päivänä annettava obinututsumabiesilääkitys on annettu, Columvi annetaan infuusiona laskimoon step-up-hoito-ohjelmana, joka johtaa suositeltuun 30 mg:n annokseen (kuten taulukossa 3 esitetään).
Columvi annetaan hoitosykleissä 1–8 yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa ja hoitosykleissä 9–12 monoterapiana. Yksi hoitosykli on 21 päivää.
Taulukko 3. Columvi-step-up-hoito-ohjelma yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa uusiutunutta tai hoitoon reagoimatonta diffuusia suurisoluista B-solulymfoomaa sairastaville potilaille
| Hoitosykli, päivä | Columvi-annos (infuusion kesto) | Gemsitabiini-annos | Oksaliplatiini-annos | |
1. hoitosykli (Esilääkitys ja step-up-annos) | 1. päivä | Esilääkityksenä 1000 mg obinututsumabiaa | ||
| 2. päivä | – | 1000 mg/m2 b | 100 mg/m2 b | |
| 8. päivä | 2,5 mg (4 tuntia)c | – | – | |
| 15. päivä | 10 mg (4 tuntia)c | |||
| 2. hoitosykli | 1. päivä | 30 mg (4 tuntia)c,d | 1000 mg/m2 b, d | 100 mg/m2 b, d |
| 3.–8. hoitosykli | 1. päivä | 30 mg (2 tuntia)d,e | 1000 mg/m2 b, d | 100 mg/m2 b, d |
| 9.–12. hoitosykli | 1. päivä | 30 mg (2 tuntia)e | – | – |
a Ks. edellä kohta Esilääkitys obinututsumabilla.
b 1.–8. hoitosykli: anna gemsitabiini ennen oksaliplatiinia.
c Potilailla, joille ilmaantui edellisen Columvi-annoksen yhteydessä sytokiinioireyhtymä, infuusion kestoa voidaan pidentää 8 tuntiin saakka (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
d 2.–8. hoitosykli: anna Columvi ennen gemsitabiinia ja oksaliplatiinia. Gemsitabiinia ja oksaliplatiinia voidaan antaa 1. tai 2. päivänä.
e Infuusion kesto voidaan lyhentää 2 tuntiin hoitavan lääkärin harkinnan mukaan, jos potilas sieti edellisen infuusion hyvin. Jos potilaalle ilmaantui jonkin aiemman annoksen yhteydessä sytokiinioireyhtymä, infuusion kestona pitää säilyttää 4 tuntia.
Potilaan seuranta
- Kun Columvi annetaan monoterapiana, potilaita on seurattava mahdollisen sytokiinioireyhtymän oireiden ja löydösten havaitsemiseksi kaikkien Columvi-infuusioiden aikana ja vähintään 10 tunnin ajan ensimmäisen Columvi-annoksen (2,5 mg 1. hoitosyklin 8. päivänä) päättymisen jälkeen (ks. kohta Haittavaikutukset).
- Kun Columvi annetaan yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa, potilaita on seurattava mahdollisen sytokiinioireyhtymän oireiden ja löydösten havaitsemiseksi kaikkien Columvi-infuusioiden aikana ja 4 tunnin ajan ensimmäisen Columvi-annoksen (2,5 mg 1. hoitosyklin 8. päivänä) päättymisen jälkeen (ks. kohta Haittavaikutukset).
Potilaita, joille ilmaantui edellisen infuusion yhteydessä ≥ 2. asteen sytokiinioireyhtymä, pitää seurata
infuusion päättymisen jälkeen (ks. taulukko 4 kohdassa Annostus ja antotapa).
Kaikkia potilaita on seurattava sytokiinioireyhtymän ja immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän (ICANS) merkkien ja oireiden varalta Columvi-valmisteen annon jälkeen.
Kaikille potilaille on kerrottava sytokiinioireyhtymän ja immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän riskistä sekä niiden oireista ja löydöksistä, ja heitä on kehotettava ottamaan yhteyttä terveydenhuollon ammattilaiseen heti, jos heille ilmaantuu milloin tahansa sytokiinioireyhtymän ja/tai immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän oireita tai löydöksiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoidon kesto
Columvi-hoitoa monoterapiana suositellaan antamaan enintään 12 hoitosykliä tai kunnes sairaus etenee tai ilmaantuu toksisuutta, joka ei ole hallittavissa, sen mukaan kumpi tapahtuu ensin. Yksi hoitosykli on 21 päivää.
Columvi-hoitoa yhdistelmähoitona gemsitabiinin ja oksaliplatiinin kanssa suositellaan antamaan 8 hoitosyklin ajan. Tämän jälkeen hoitoa jatketaan 4 hoitosyklin ajan Columvi-monoterapiana, eli valmistetta annetaan enintään 12 hoitosykliä tai kunnes sairaus etenee tai ilmaantuu toksisuutta, joka ei ole hallittavissa, sen mukaan kumpi tapahtuu ensin. Yksi hoitosykli on 21 päivää.
Annoksen viivästyminen tai saamatta jääminen
Step-up-annostuksen aikana (viikoittainen annostus):
- Jos Columvi 2,5 mg ‑annos viivästyy obinututsumabiesilääkityksen jälkeen yli 1 viikolla, anna uusi obinututsumabiesilääkitys.
- Jos Columvi-hoidossa on 2–6 viikon tauko 2,5 mg:n tai 10 mg:n Columvi-annoksen jälkeen, anna uudestaan viimeisin siedetty Columvi-annos ja jatka sitten suunniteltua step‑up-annostusta.
- Jos Columvi-hoidossa on yli 6 viikon tauko 2,5 mg:n tai 10 mg:n Columvi-annoksen jälkeen, anna uusi obinututsumabiesilääkitys ja Columvi-step‑up-annos (ks. 1. hoitosykli taulukossa 2 ja taulukossa 3).
2. hoitosyklin jälkeen (30 mg:n annos):
- Jos Columvi-hoidossa on hoitosyklien välissä yli 6 viikon tauko, anna uusi obinututsumabiesilääkitys ja Columvi-step‑up-annos (ks. 1. hoitosykli taulukossa 2 ja taulukossa 3), minkä jälkeen palataan suunniteltuun hoitosykliin (30 mg:n annos).
Annosmuutokset
Columvi-annoksen pienentämistä ei suositella.
Sytokiinioireyhtymän hoito
Sytokiinioireyhtymän tunnistaminen perustuu kliiniseen oireistoon (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Potilailta pitää tutkia kuumeen, hypoksian ja hypotension muut syyt, kuten infektiot tai sepsis. Jos sytokiinioireyhtymää epäillään, se pitää hoitaa taulukossa 4 mainittujen ASTCT:n (American Society for Transplantation and Cellular Therapy) konsensusluokitukseen perustuvien sytokiinioireyhtymän hoitosuositusten mukaisesti.
Taulukko 4. ASTCT:n sytokiinioireyhtymän vaikeusasteluokitus ja sytokiinioireyhtymän hoito-ohjeet
| Vaikeusaste1 | Sytokiinioireyhtymän hoito | Seuraavassa suunnitellussa Columvi-infuusiossa |
1. aste Kuume ≥ 38 °C | Jos sytokiinioireyhtymä ilmaantuu infuusion aikana • keskeytä infuusio ja hoida oireet • kun oireet häviävät, jatka infuusiota hitaammalla antonopeudella • jos oireet uusiutuvat, keskeytä parhaillaan annettava infuusio. Jos sytokiinioireyhtymä ilmaantuu infuusion jälkeen • hoida oireet. Jos sytokiinioireyhtymä kestää yli 48 h oireiden hoidon jälkeen • harkitse kortikosteroideja3 • harkitse tosilitsumabia.4 Taulukossa 5 on tietoa sytokiinioireyhtymästä samanaikaisen ICANS-oireyhtymän kanssa. | • Varmista, että oireet ovat hävinneet viimeistään 72 tuntia ennen seuraavaa infuusiota. • Harkitse hitaampaa infuusionopeutta.2 |
2. aste Kuume ≥ 38 °C ja/tai hypotensio, joka ei vaadi vasopressoreita, ja/tai hypoksia, joka vaatii pienivirtauksista happihoitoa happiviiksillä tai blow‑by-menetelmällä | Jos sytokiinioireyhtymä ilmaantuu infuusion aikana • keskeytä parhaillaan annettava infuusio ja hoida oireet • anna kortikosteroideja3 • harkitse tosilitsumabia.4 Jos sytokiinioireyhtymä ilmaantuu infuusion jälkeen • hoida oireet • anna kortikosteroideja3 • harkitse tosilitsumabia.4 Taulukossa 5 on tietoa sytokiinioireyhtymästä samanaikaisen ICANS-oireyhtymän kanssa. | • Varmista, että oireet ovat hävinneet viimeistään 72 tuntia ennen seuraavaa infuusiota. • Harkitse hitaampaa infuusionopeutta.2 • Seuraa potilasta infuusion jälkeen.5 |
2. aste: tosilitsumabin käyttö Kuuden viikon jakson aikana saa antaa enintään 3 tosilitsumabiannosta. Jos tosilitsumabia ei ole aiemmin annettu tai jos edeltävien 6 viikon aikana on annettu 1 tosilitsumabiannos • anna ensimmäinen tosilitsumabiannos4 • jos 8 tunnin kuluessa ei havaita paranemista, anna toinen tosilitsumabiannos4 • harkitse 2 tosilitsumabiannoksen jälkeen vaihtoehtoista antisytokiinihoitoa ja/tai vaihtoehtoista immunosuppressiivista hoitoa. Jos edeltävien 6 viikon aikana on annettu 2 tosilitsumabiannosta • anna vain yksi tosilitsumabiannos4 • jos 8 tunnin kuluessa ei ole havaittu paranemista, harkitse vaihtoehtoista antisytokiinihoitoa ja/tai vaihtoehtoista immunosuppressiivista hoitoa. | ||
3. aste Kuume ≥ 38 °C ja/tai hypotensio, joka vaatii vasopressoria (ja mahdollisesti vasopressiinia), ja/tai hypoksia, joka vaatii suurivirtauksista happihoitoa happiviiksillä, happimaskilla, varaajapussillisella maskilla tai venturimaskilla. | Jos sytokiinioireyhtymä ilmaantuu infuusion aikana • keskeytä parhaillaan annettava infuusio, ja hoida oireet • anna kortikosteroideja3 • anna tosilitsumabia.4 Jos sytokiinioireyhtymä ilmaantuu infuusion jälkeen • hoida oireet • anna kortikosteroideja3 • anna tosilitsumabia.4 Taulukossa 5 on tietoa sytokiinioireyhtymästä samanaikaisen ICANS-oireyhtymän kanssa. | • Varmista, että oireet ovat hävinneet viimeistään 72 tuntia ennen seuraavaa infuusiota. • Harkitse hitaampaa infuusionopeutta.2 • Seuraa potilasta infuusion jälkeen.5 • Jos ≥ 3. asteen sytokiinioireyhtymä uusiutuu seuraavan infuusion yhteydessä, lopeta infuusio heti ja lopeta Columvi-hoito pysyvästi. |
4. aste Kuume ≥ 38 °C ja/tai hypotensio, joka vaatii useita vasopressoreita (mutta ei vasopressiinia), ja/tai hypoksia, joka vaatii ylipainehengityshoitoa (esim. CPAP, BiPAP, intubaatio ja hengityskonehoito). | Jos sytokiinioireyhtymä ilmaantuu infuusion aikana tai sen jälkeen • lopeta Columvi-hoito pysyvästi, ja hoida oireet • anna kortikosteroideja3 • anna tosilitsumabia.4 Taulukossa 5 on tietoa sytokiinioireyhtymästä samanaikaisen ICANS-oireyhtymän kanssa. | |
3. ja 4. aste: tosilitsumabin käyttö Kuuden viikon jakson aikana saa antaa enintään 3 tosilitsumabiannosta. Jos tosilitsumabia ei ole aiemmin annettu tai jos edeltävien 6 viikon aikana on annettu 1 tosilitsumabiannos • anna ensimmäinen tosilitsumabiannos4 • jos 8 tunnin kuluessa ei havaita paranemista tai sytokiinioireyhtymä etenee nopeasti, anna toinen tosilitsumabiannos4 • harkitse 2 tosilitsumabiannoksen jälkeen vaihtoehtoista antisytokiinihoitoa ja/tai vaihtoehtoista immunosuppressiivista hoitoa. Jos edeltävien 6 viikon aikana on annettu 2 tosilitsumabiannosta • anna vain yksi tosilitsumabiannos4 • jos 8 tunnin kuluessa ei ole havaittu paranemista tai sytokiinioireyhtymä etenee nopeasti, harkitse vaihtoehtoista antisytokiinihoitoa ja/tai vaihtoehtoista immunosuppressiivista hoitoa. | ||
1 Vaikeusastetta koskevat ASTCT:n (American Society for Transplantation and Cellular Therapy) konsensuskriteerit (Lee 2019). 2 Infuusion kestoa voidaan pidentää enintään 8 tuntiin sen mukaan, mikä kyseisessä hoitosyklissä soveltuu (ks. taulukko 2). 3 Kortikosteroidit (esim. 10 mg deksametasonia laskimoon, 100 mg prednisolonia laskimoon, 1–2 mg/kg metyyliprednisolonia laskimoon päivässä tai vastaava hoito). 4 8 mg/kg tosilitsumabia laskimoon (enintään 800 mg), kuten NP30179-tutkimuksessa annettiin. 5 Ks. kohdasta Haittavaikutukset ≥ 2. asteen sytokiinioireyhtymän esiintyvyys ja ilmaantumiseen kulunut aika Columvi-valmisteen 10 mg:n ja 30 mg:n annosten jälkeen. | ||
Immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän (ICANS) hallinta
ICANS-oireyhtymän ensimmäisten merkkien yhteydessä harkitaan sen tyypin ja vaikeusasteen perusteella tukihoitoa, neurologista arviointia ja Columvi-hoidon keskeyttämistä (ks. taulukko 5). Sulje pois muut neurologisten oireiden syyt. Jos on syytä epäillä ICANS-oireyhtymää, sitä hoidetaan taulukon 5 suositusten mukaisesti.
Taulukko 5. ICANS-oireyhtymän vaikeusasteluokitus ja hoito-ohjeet
| Vaikeusaste1 | Oireet2 | ICANS-oireyhtymän hoito | |
| Samanaikainen sytokiinioireyhtymä | Ei samanaikaista sytokiinioireyhtymää | ||
| 1 . aste | ICE-pisteet: 7–93 tai alentunut tajunnan taso4: havahtuu tilasta spontaanisti | • Sytokiinioireyhtymän hoito taulukon 4 mukaisesti. • Seurataan neurologisia oireita ja harkitaan neurologista konsultointia ja arviointia lääkärin harkinnan mukaan. | • Seurataan neurologisia oireita ja harkitaan neurologista konsultointia ja arviointia lääkärin harkinnan mukaan. |
Keskeytetään Columvi-hoito, kunnes ICANS-oireyhtymä on hävinnyt. Harkitaan ei-sedatiivisia kouristuksen estolääkkeitä (esimerkiksi levetirasetaamia) kouristuskohtausten estämiseksi. | |||
| 2. aste | ICE-pisteet: 3–63 tai alentunut tajunnan taso4: havahtuu tilasta reagoimalla ääneen | • Annetaan tosilitsumabia sytokiinioireyhtymän hoitoon taulukossa 4 esitetyn mukaisesti. • Jos tosilitsumabihoidon aloittamisen jälkeen tila ei parane, annetaan deksametasonia5 10 mg laskimoon kuuden tunnin välein, jos muut kortikosteroidit eivät ole jo käytössä. Jatketaan deksametasonin käyttöä, kunnes oireet ovat enintään 1. asteen tasolla, ja pienennetään annosta vähitellen sen jälkeen. | • Annetaan deksametasonia5 10 mg suonensisäisesti kuuden tunnin välein. • Jatketaan deksametasonin käyttöä, kunnes oireet ovat enintään 1. asteen tasolla, ja pienennetään annosta vähitellen sen jälkeen. |
Keskeytetään Columvi-hoito, kunnes ICANS-oireyhtymä on hävinnyt. Harkitaan ei-sedatiivisia kouristuksen estolääkkeitä (esimerkiksi levetirasetaamia) kouristuskohtausten estämiseksi. Harkitaan tarvittaessa neurologin ja muiden asiantuntijoiden konsultointia lisäarviointia varten. | |||
| 3. aste | ICE-pisteet: 0–23 tai alentunut tajunnan taso4: havahtuu tilasta vain reagoimalla kosketusärsykkeeseen tai kouristuskohtaukset4, joko • kliiniset kouristuskohtaukset, paikalliset tai yleistyneet, jotka häviävät nopeasti, tai • sähköenkefalografiassa (EEG) näkyvät kouristamattomat kohtaukset, jotka häviävät interventiolla tai kohonnut kallon sisäinen paine: fokaalinen/paikallinen turvotus neurokuvantamisessa4 | • Annetaan tosilitsumabia sytokiinioireyhtymän hoitoon taulukossa 4 esitetyn mukaisesti. • Lisäksi annetaan deksametasonia5 10 mg suonensisäisesti ensimmäisen tosilitsumabiannoksen kanssa, ja uusi annos annetaan 6 tunnin välein, jos muita kortikosteroideja ei jo käytetä. Jatketaan deksametasonin käyttöä, kunnes oireet ovat enintään 1. asteen tasolla, ja pienennetään annosta vähitellen sen jälkeen. | • Annetaan deksametasonia5 10 mg suonensisäisesti kuuden tunnin välein. • Jatketaan deksametasonin käyttöä, kunnes oireet ovat enintään 1. asteen tasolla, ja pienennetään annosta vähitellen sen jälkeen. |
Keskeytetään Columvi-hoito, kunnes ICANS-oireyhtymä on hävinnyt. Jos 3:nnen vaikeusasteen ICANS-tapahtumat eivät parane seitsemän päivän kuluessa, tulee harkita Columvi-hoidon pysyvää lopettamista. Harkitaan ei-sedatiivisia kouristuksen estolääkkeitä (esimerkiksi levetirasetaamia) kouristuskohtausten estämiseksi. Harkitaan tarvittaessa neurologin ja muiden asiantuntijoiden konsultointia jatkoarviointia varten. | |||
| 4. aste | ICE-pisteet: 03 tai alentunut tajunnan taso4, ja • potilas ei virkoa tai tarvitsee virotakseen voimakkaita tai toistuvia kosketusärsykkeitä, tai • potilas on tajuton tai koomassa tai kouristuskohtaukset4, joko • hengenvaarallisesti pitkittyneet kouristuskohtaukset (> 5 minuuttia) tai • toistuvat kliiniset tai sähköiset kouristuskohtaukset, joiden välillä ei ole palautumista normaalille tasolle tai motoriset löydökset4: • syvä fokaalinen motorinen heikkous, kuten hemipareesi tai parapareesi tai kohonnut kallon sisäinen paine / aivoturvotus4, jonka merkkejä/oireita ovat esimerkiksi • neurokuvantamisessa havaittava diffuusi aivoturvotus tai • deserebraatio- tai dekortikaatiotilan asento, tai • VI aivohermon halvaus, tai • papilledeema tai • Cushingin triadi | • Annetaan tosilitsumabia sytokiinioireyhtymän hoitoon taulukossa 4 esitetyn mukaisesti. • Kuten edellä, tai harkitaan metyyliprednisolonin antamista 1 000 mg vuorokaudessa laskimoon ensimmäisen tosilitsumabiannoksen kanssa, minkä jälkeen jatketaan metyyliprednisolonia 1 000 mg vuorokaudessa laskimoon kahden tai useamman päivän ajan. | • Annetaan deksametasonia5 10 mg suonensisäisesti kuuden tunnin välein. • Jatketaan deksametasonin käyttöä, kunnes oireet ovat enintään 1. asteen tasolla, ja pienennetään annosta vähitellen sen jälkeen. • Vaihtoehtoisesti harkitaan metyyliprednisolonin antamista 1 000 mg vuorokaudessa laskimoon kolmen päivän ajan. Oireiden lievittyessä niitä hoidetaan edellä kuvatulla tavalla. |
Columvi-hoito lopetetaan pysyvästi. Harkitaan ei-sedatiivisia kouristuksen estolääkkeitä (esimerkiksi levetirasetaamia) kouristuskohtausten estämiseksi. Harkitaan tarvittaessa neurologin ja muiden asiantuntijoiden konsultointia lisäarviointia varten. Kohonneessa kallon sisäisessä paineessa tai aivoturvotuksessa noudatetaan paikallisia hoito-ohjeita. | |||
1 ICANS-oireyhtymä ASTCT-asteikolla (Lee 2019).
2 Vakavin tapahtuma (jolla ei ole mitään muuta syytä) määrittelee hoidon.
3 Jos potilas on virkeä ja kykenevä Immune Effector Cell-Associated Encephalopathy (ICE) ‑arviointiin, määritellään
orientaatio (vuosi, kuukausi, kaupunki, sairaala = 4 pistettä)
nimeäminen (3 esineen nimeäminen, joita ovat esim. kello, kynä, painike = 3 pistettä)
ohjeiden noudattaminen (esim. ”nosta kaksi sormea” tai ”sulje silmät ja työnnä kieli ulos” = 1 piste)
kirjoittaminen (kyky kirjoittaa tavanomainen lause = 1 piste
keskittyminen (laskeminen sadasta alaspäin kymmenen numeron välein = 1 piste.
Jos potilas ei ole herätettävissä eikä kykene tekemään ICE-arviota (ICANS-oireyhtymän 4. vaikeusaste) = 0 pistettä.
4 Ilman muuta tiedossa olevaa syytä.
5 Kaikki maininnat deksametasonin antamisesta viittaavat deksametasoniin tai vastaavaan valmisteeseen.
Erityisryhmät
Iäkkäät
65-vuotiaiden ja sitä vanhempien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa (kokonaisbilirubiinipitoisuus > normaalien viitearvojen yläraja [upper limit of normal, ULN] – ≤ 1,5 × ULN tai aspartaattitransaminaasipitoisuus [ASAT] > ULN) sairastavien potilaiden annosta ei tarvitse muuttaa. Columvi-valmistetta ei ole tutkittu keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa (CrCL 30 – < 90 ml/min) sairastavien potilaiden annosta ei tarvitse muuttaa. Columvi-valmistetta ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Columvi-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Columvi on tarkoitettu annettavaksi vain laskimoon.
Terveydenhuollon ammattilaisen on laimennettava Columvi-valmiste aseptista menetelmää noudattaen ennen valmisteen antamista laskimoon. Valmiste on annettava infuusiona laskimoon vain sen antoon tarkoitetun infuusioletkun kautta.
Columvi-valmistetta ei saa antaa laskimoon paineella eikä boluksena.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet Columvi-valmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle, obinututsumabille tai kohdassa Apuaineet mainituille apuaineille.
Ks. obinututsumabin spesifiset vasta-aiheet obinututsumabin valmistetiedoista.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
CD20-negatiivinen tauti
Columvi-hoitoa saavista potilaista, joilla on CD20-negatiivinen diffuusi suurisoluinen B‑solulymfooma, on vähän tietoja saatavilla. On mahdollista, että CD20-negatiivista diffuusia suurisoluista B‑solulymfoomaa sairastavat potilaat saattavat hyötyä hoidosta vähemmän kuin CD20-positiivista diffuusia suurisoluista B‑solulymfoomaa sairastavat potilaat. CD20-negatiivista diffuusia suurisoluista B‑solulymfoomaa sairastavien potilaiden Columvi-hoitoon liittyvät mahdolliset riskit ja hyödyt pitää huomioida.
Sytokiinioireyhtymä
Columvi-valmistetta saaneilla potilailla on raportoitu sytokiinioireyhtymää, mukaan lukien henkeä uhkaavia reaktioita (ks. kohta Haittavaikutukset).
Sytokiinioireyhtymän yleisimpiä ilmenemismuotoja olivat kuume, takykardia, hypotensio, vilunväristykset ja hypoksia. Infuusioon liittyvät reaktiot eivät välttämättä ole kliinisesti erotettavissa sytokiinioireyhtymän ilmenemismuodoista.
Valtaosa sytokiinioireyhtymää koskevista tapahtumista ilmeni ensimmäisen Columvi-annoksen jälkeen. Glofitamabin käytön jälkeen on raportoitu kohonneita maksan toimintakoearvoja (ASAT ja alaniinitransaminaasipitoisuus [ALAT] > 3 × ULN ja/tai kokonaisbilirubiinipitoisuus > 2 × ULN) samanaikaisesti sytokiinioireyhtymän kanssa (ks. kohta Haittavaikutukset).
NP30179- ja GO41944 (STARGLO) -tutkimuksissa potilaat saivat obinututsumabiesilääkityksen verenkierrossa ja imusolmukkeissa olevien B-solujen määrän vähentämiseksi 7 päivää ennen Columvi-hoidon aloittamista. Kaikille potilaille on annettava esilääkityksenä kuumetta alentavaa lääkettä, antihistamiinia ja jotakin glukokortikoidia (ks. taulukko 1).
Ennen 1. ja 2. hoitosyklin Columvi-infuusiota saatavilla on oltava vähintään 1 annos tosilitsumabia sytokiinioireyhtymän varalta. On varmistettava, että lisäannos tosilitsumabia on saatavilla 8 tunnin kuluessa edellisestä tosilitsumabiannoksesta.
Kun Columvi annetaan monoterapiana, potilaita on seurattava kaikkien Columvi-infuusioiden aikana ja vähintään 10 tuntia ensimmäisen infuusion päättymisen jälkeen.
Kun Columvi annetaan yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa, potilaita on seurattava kaikkien Columvi-infuusioiden aikana ja 4 tunnin ajan ensimmäisen infuusion päättymisen jälkeen.
Tarkat tiedot seurannasta, ks. kohta Annostus ja antotapa. Potilaita on kehotettava hakeutumaan heti lääkäriin, jos heille ilmaantuu milloin tahansa sytokiinioireyhtymän oireita tai löydöksiä (ks. jäljempänä Potilaskortti).
Potilailta pitää tutkia kuumeen, hypoksian ja hypotension muut syyt, kuten infektiot tai sepsis. Sytokiinioireyhtymä pitää hoitaa potilaan kliinisen oireiston perusteella sekä taulukossa 4 mainittujen sytokiinioireyhtymän hoito-ohjeiden mukaisesti (kohta Annostus ja antotapa).
Immuuniefektorisoluihin liittyvä neurotoksisuusoireyhtymä
Columvi-hoidon jälkeen on esiintynyt vakavia immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän (ICANS) tapauksia, jotka voivat olla hengenvaarallisia tai johtaa kuolemaan (ks. kohta Haittavaikutukset).
ICANS voi puhjeta samanaikaisesti sytokiinioireyhtymän kanssa, sytokiinioireyhtymän oireiden hävittyä tai ilman sytokiinioireyhtymää. ICANSin kliinisiä merkkejä ja oireita voivat olla mm. sekavuus, tajunnantason aleneminen, tietämättömyys ajasta ja paikasta, kouristuskohtaukset, afasia ja dysgrafia.
Potilaita on seurattava ICANSin merkkien ja oireiden varalta Columvin antamisen jälkeen ja tarvittaessa hoidettava viipymättä. Potilaita on neuvottava hakeutumaan välittömästi hoitoon, jos merkkejä tai oireita ilmaantuu milloin tahansa (ks. potilaskortti jäljempänä).
ICANSin ensimmäisten merkkien tai oireiden ilmaantuessa potilasta hoidetaan taulukossa 5 annettujen ICANS-ohjeiden mukaisesti. Columvi-hoito on keskeytettävä tai lopetettava pysyvästi suosituksen mukaisesti.
Potilaskortti
Lääkkeen määräävän lääkärin on kerrottava potilaalle sytokiinioireyhtymän ja immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän riskistä sekä sytokiinioireyhtymän ja immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän oireista ja löydöksistä. Potilaita on kehotettava hakeutumaan heti lääkäriin, jos heille ilmaantuu sytokiinioireyhtymän ja immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän oireita ja löydöksiä. Potilaille pitää antaa potilaskortti, ja heitä on kehotettava pitämään se aina mukanaan. Kortissa kuvataan sytokiinioireyhtymän ja immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän oireet ja kehotetaan hakeutumaan välittömästi lääkäriin, jos niitä ilmaantuu.
Yhteisvaikutukset CYP450:n substraattien kanssa
Columvi-hoidon aloittamiseen liittyvä sytokiinien alkuvaiheen vapautuminen voi suppressoida CYP450-entsyymejä ja johtaa vaihteluihin samanaikaisesti annettavien lääkkeiden pitoisuudessa. Potilaita, jotka Columvi-hoitoa aloitettaessa saavat hoitoa kapean terapeuttisen indeksin CYP450:n substraateilla, pitää seurata, sillä samanaikaisesti annettavien lääkkeiden pitoisuusvaihteluista voi aiheutua toksisuutta, tehon häviämistä tai haittavaikutuksia (ks. kohta Yhteisvaikutukset).
Vakavat infektiot
Columvi-hoitoa saaneille potilaille on ilmaantunut vakavia infektioita, mukaan lukien opportunistisia infektioita (ks. kohta Haittavaikutukset).
Columvi-valmistetta ei saa antaa potilaille, joilla on jokin aktiivinen infektio. Columvi-valmisteen käyttöä on harkittava tarkoin, jos potilaalla on anamneesissa kroonisia tai toistuvia infektioita, infektioille mahdollisesti altistava perussairaus tai jos potilas on saanut aiemmin merkittävää immunosuppressiivista hoitoa. Estolääkitys mikrobilääkkeillä on annettava tarpeen mukaan. Potilaita on seurattava ennen Columvi-hoitoa ja sen aikana mahdollisten bakteeri- ja sieni-infektioiden sekä uusien tai reaktivoituneiden virusinfektioiden havaitsemiseksi, ja ne on hoidettava asianmukaisesti.
Jos potilaalla on jokin aktiivinen infektio, Columvi-hoito pitää keskeyttää tilapäisesti, kunnes infektio on parantunut. Potilasta pitää neuvoa hakeutumaan lääkäriin, jos hänelle ilmaantuu infektioon viittaavia oireita tai löydöksiä.
Columvi-hoidon aikana on raportoitu kuumeista neutropeniaa. Potilaat, joilla on kuumeista neutropeniaa, pitää tutkia infektion varalta, ja heidät pitää hoitaa viiveettä.
Tumour flare ‑reaktio
Columvi-valmistetta saaneilla potilailla on raportoitu tumour flare ‑reaktioita (ks. kohta Haittavaikutukset). Sen ilmenemismuotoja olivat paikallinen kipu ja turvotus.
Tumour flare ‑reaktio on Columvi-valmisteen vaikutusmekanismin perusteella todennäköinen, sillä T‑solut siirtyvät Columvi-valmisteen annon jälkeen kasvaimen sijaintikohtaan, mikä voi muistuttaa sairauden etenemistä. Tumour flare ‑reaktio ei viittaa siihen, että hoito olisi epäonnistunut, eikä se tarkoita kasvaimen etenemistä.
Tumour flare ‑reaktion spesifisiä riskitekijöitä ei ole tunnistettu, mutta tumour flare ‑reaktiosta aiheutuvan tautimassavaikutuksen vuoksi voinnin heikkenemisen ja sairastuvuuden riski suurenee potilailla, joilla on suuri kasvainmassa lähellä hengitysteitä ja/tai elintärkeää elintä. Columvi-valmistetta saavia potilaita suositellaan seuraamaan ja heiltä tutkimaan tumour flare ‑reaktio kriittisistä anatomisista kohdista sekä hoitamaan se siten kuin on kliinisesti aiheellista. Tumour flare ‑reaktion hoitoon pitää harkita kortikosteroideja ja kipulääkitystä.
Tuumorilyysioireyhtymä
Columvi-valmistetta saaneilla potilailla on raportoitu tuumorilyysioireyhtymää (ks. kohta Haittavaikutukset). Potilailla, joilla on suuri kasvaintaakka, nopeasti kasvavia kasvaimia, munuaisten toimintahäiriö tai elimistön kuivumistila, on tavanomaista suurempi tuumorilyysioireyhtymän riski.
Potilaita, joilla on tällainen riski, pitää seurata tarkoin elektrolyyttistatusta, nesteytystä ja munuaisten toimintaa selvittävillä asianmukaisilla laboratoriokokeilla ja kliinisillä testeillä. Soveltuvia ennalta ehkäiseviä toimenpiteitä virtsahapon muodostumista estävillä lääkkeillä (esim. allopurinoli tai rasburikaasi) ja riittävää nesteytystä pitää harkita ennen obinututsumabiesilääkitystä ja ennen Columvi-infuusiota.
Tuumorilyysioireyhtymän hoito voi käsittää aggressiivisen nesteytyksen, elektrolyyttien poikkeavuuksien korjaamisen, virtsahapon muodostumista estävän hoidon ja tukihoidon.
Immunisaatio
Eläviä taudinaiheuttajia sisältävillä rokotteilla annetun immunisaation turvallisuutta Columvi-hoidon aikana tai sen jälkeen ei ole tutkittu. Immunisaatiota eläviä taudinaiheuttajia sisältävillä rokotteilla ei suositella Columvi-hoidon aikana.
Polysorbaatit
Tämä lääkevalmiste sisältää 1,25 mg polysorbaattia 20 per 2,5 ml:n injektiopullo ja 5 mg polysorbaattia 20 per 10 ml:n injektiopullo, mikä vastaa 0,5 mg:aa/ml.
Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Columvi-valmisteella ei oletettavasti ole sytokromi P450 ‑entsyymien, muiden metaboloivien entsyymien ja kuljettajaproteiinien välityksellä ilmeneviä yhteisvaikutuksia.
Columvi-hoidon aloittamiseen alkuvaiheessa liittyvä sytokiinien vapautuminen voi estää CYP450-entsyymien toimintaa. Lääkkeiden välisten yhteisvaikutusten riski on suurin kahden ensimmäisen Columvi-annoksen (eli 1. hoitosyklin 8. päivänä ja 15. päivänä) jälkeisen viikon aikana niillä potilailla, jotka saavat samanaikaisesti CYP450:n substraatteja, joiden terapeuttinen indeksi on kapea (esim. varfariini, siklosporiini). Jos potilas saa Columvi-hoitoa aloitettaessa hoitoa CYP450:n substraateilla, joiden terapeuttinen indeksi on kapea, potilasta on seurattava.
Gemsitabiinin tai oksaliplatiinin samanaikainen anto ei vaikuta glofitamabin farmakokinetiikkaan.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy
Naispotilaiden, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää Columvi-hoidon aikana ja vähintään 2 kuukautta viimeisen Columvi-annoksen jälkeen.
Raskaus
Ei ole olemassa tietoja Columvi-valmisteen käytöstä raskaana oleville naisille. Lisääntymistoksisuutta koskevia eläinkokeita ei ole tehty (ks. kohta Prekliiniset tiedot turvallisuudesta).
Glofitamabi on immunoglobuliini G (IgG). IgG:n tiedetään läpäisevän istukan. Raskaana olevalle naiselle annettu glofitamabi todennäköisesti aiheuttaa vaikutusmekanisminsa perusteella sikiölle B‑soluvajeen.
Columvi-valmistetta ei suositella raskauden aikana eikä naisille, jotka voivat tulla raskaaksi eivätkä käytä ehkäisyä. Columvi-valmistetta saaville naispotilaille pitää kertoa sikiölle mahdollisesti aiheutuvasta haitasta. Naispotilasta pitää kehottaa ottamaan yhteyttä hoitavaan lääkäriin, jos hän tulee raskaaksi.
Imetys
Ei tiedetä, erittyykö glofitamabi ihmisen rintamaitoon. Glofitamabin vaikutusta maidontuotantoon tai sen erittymistä rintamaitoon ei ole tutkittu. Ihmisen IgG:n tiedetään erittyvän ihmisen rintamaitoon. Glofitamabin mahdollista imeytymistä rintaruokittavalla lapsella ja rintaruokittavalle lapselle aiheutuvia mahdollisia haittavaikutuksia ei tunneta. Naisia pitää kehottaa lopettamaan imetys Columvi-hoidon ajaksi ja 2 kuukaudeksi viimeisen Columvi-annoksen jälkeen.
Hedelmällisyys
Ihmisestä ei ole hedelmällisyyttä koskevia tietoja saatavissa. Glofitamabista hedelmällisyyteen aiheutuvien vaikutusten arvioimiseksi ei ole tehty hedelmällisyyttä koskevia eläinkokeita (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Columvi-valmisteella on huomattava vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Columvia saavilla potilailla on riski sairastua ICANSiin ja siten riski tajunnan tason alenemiseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Potilaita on ohjeistettava välttämään ajamista tai koneiden käyttöä 48 tunnin ajan molempien kahden ensimmäisen annoksen jälkeen annoksen asteittaisen nostamisen aikana, samoin jos immuuniefektorisoluihin liittyvän neurotoksisuusoireyhtymän mitkä tahansa oireet alkavat (sekavuus, tietämättömyys ajasta ja paikasta, alentunut tajunnantaso) ja/tai ilmaantuu sytokiinioireyhtymän oireita (kuumetta, takykardiaa, hypotensiota, vilunväristyksiä, hypoksiaa), kunnes oireet häviävät (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Columvi-monoterapia
Yleisimmät haittavaikutukset (≥ 20 %) olivat sytokiinioireyhtymä, neutropenia, anemia, trombosytopenia ja ihottuma.
Yleisimmät ≥ 2 %:lla potilaista raportoidut vakavat haittavaikutukset olivat sytokiinioireyhtymä (22,1 %), sepsis (4,1 %), koronaviruksen (COVID‑19) aiheuttama infektio (3,4 %), tumour flare ‑reaktio (3,4 %), koronaviruksen (COVID-19) aiheuttama keuhkokuume (2,8 %), kuumeinen neutropenia (2,1 %), neutropenia (2,1 %) ja pleuraeffuusio (2,1 %).
Columvi-hoito lopetettiin pysyvästi haittavaikutuksen vuoksi 5,5 %:lla potilaista. Yleisimmät Columvi-hoidon lopettamiseen johtaneet haittavaikutukset olivat COVID‑19-infektio (1,4 %) ja neutropenia (1,4 %).
Columvi-yhdistelmähoito gemsitabiinin ja oksaliplatiinin kanssa
Yleisimmät haittavaikutukset (≥ 20 %) olivat trombosytopenia, sytokiinioireyhtymä, neutropenia, anemia, pahoinvointi, perifeerinen neuropatia, ripuli, suurentunut aspartaattiaminotransferaasipitoisuus, suurentunut alaniiniaminotransferaasipitoisuus, ihottuma, lymfopenia, kuume ja oksentelu.
Yleisimmät ≥ 2 %:lla potilaista raportoidut vakavat haittavaikutukset olivat sytokiinioireyhtymä (20,3 %), kuume (6,4 %), keuhkokuume (5,8 %), COVID-19-infektio (5,8 %), trombosytopenia (4,7 %), hengitystieinfektio (3,5 %), sepsis (2,3 %), kuumeinen neutropenia (2,3 %) ja ripuli (2,3 %).
Columvi-hoito lopetettiin pysyvästi haittavaikutuksen vuoksi 20,9 %:lla potilaista. Yleisimmät Columvi-hoidon pysyvään lopettamiseen johtaneet haittavaikutukset olivat COVID-19-infektio (11,6 %), sepsis (1,2 %) ja pneumoniitti (1,2 %).
Haittavaikutustaulukko
Columvi-monoterapiaa NP30179-tutkimuksessa saaneilla uusiutunutta tai hoitoon reagoimatonta diffuusia suurisoluista B‑solulymfoomaa sairastavilla potilailla (N = 145) esiintyneet haittavaikutukset luetellaan taulukossa 6. Potilaiden saamien Columvi-hoitosyklien määrän mediaani oli 5 (vaihteluväli 1–13 hoitosykliä).
Haittavaikutukset uusiutunutta tai hoitoon reagoimatonta diffuusia suurisoluista B-solulymfoomaa sairastavilla potilailla, jotka saivat Columvi-valmistetta yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa (n = 172) GO41944-tutkimuksessa (STARGLO), on lueteltu taulukossa 7. Potilaiden saamien Columvi-hoitosyklien määrän mediaani oli 11 (vaihteluväli: 1–13 hoitosykliä).
Haittavaikutukset luetellaan MedDRA-elinjärjestelmien ja yleisyysluokituksen mukaisesti. Seuraavia yleisyysluokkia käytetään: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 6. Columvi-monoterapiaa saaneilla uusiutunutta tai hoitoon reagoimatonta diffuusia suurisoluista B‑solulymfoomaa sairastavilla potilailla raportoidut haittavaikutukset
Elinjärjestelmä | Haittavaikutus | Kaikki vaikeusasteet | 3.−4. aste |
Infektiot | Virusinfektiot1 | Hyvin yleinen | Yleinen* |
Bakteeri-infektiot2 | Yleinen | Yleinen | |
Ylähengitysteiden infektiot3 | Yleinen | Hyvin harvinainen** | |
Sepsis4 | Yleinen | Yleinen* | |
Alahengitysteiden infektiot5 | Yleinen | Hyvin harvinainen** | |
Keuhkokuume | Yleinen | Melko harvinainen | |
Virtsatieinfektio6 | Yleinen | Melko harvinainen | |
Sieni-infektiot7 | Yleinen | Hyvin harvinainen** | |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Tumour flare ‑reaktio | Hyvin yleinen | Yleinen |
Veri ja imukudos | Neutropenia | Hyvin yleinen | Hyvin yleinen |
Anemia | Hyvin yleinen | Yleinen | |
Trombosytopenia | Hyvin yleinen | Yleinen | |
Lymfopenia | Yleinen | Yleinen | |
Kuumeinen neutropenia8 | Yleinen | Yleinen | |
Immuunijärjestelmä | Sytokiinioireyhtymä9 | Hyvin yleinen | Yleinen |
Aineenvaihdunta ja ravitsemus | Hypofosfatemia | Hyvin yleinen | Yleinen |
Hypomagnesemia | Hyvin yleinen | Hyvin harvinainen** | |
Hypokalsemia | Hyvin yleinen | Hyvin harvinainen** | |
Hypokalemia | Hyvin yleinen | Melko harvinainen | |
Hyponatremia | Yleinen | Yleinen | |
Tuumorilyysioireyhtymä | Yleinen | Yleinen | |
Psyykkiset häiriöt | Sekavuustila | Yleinen | Hyvin harvinainen** |
Hermosto | Päänsärky | Hyvin yleinen | Hyvin harvinainen** |
Immuuniefektorisoluihin liittyvä neurotoksisuusoireyhtymä10 | Yleinen | Melko harvinainen* | |
Uneliaisuus | Yleinen | Melko harvinainen | |
Vapina | Yleinen | Hyvin harvinainen** | |
Myeliitti11 | Melko harvinainen | Melko harvinainen | |
Ruoansulatuselimistö | Ummetus | Hyvin yleinen | Hyvin harvinainen** |
Ripuli | Hyvin yleinen | Hyvin harvinainen** | |
Pahoinvointi | Hyvin yleinen | Hyvin harvinainen** | |
Maha-suolikanavan verenvuoto12 | Yleinen | Yleinen | |
Oksentelu | Yleinen | Hyvin harvinainen** | |
Koliitti | Melko harvinainen | Melko harvinainen | |
Iho ja ihonalainen kudos | Ihottuma13 | Hyvin yleinen | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | Kuume | Hyvin yleinen | Hyvin harvinainen** |
Tutkimukset | Suurentunut alaniiniaminotransferaasi-pitoisuus | Yleinen | Yleinen |
Suurentunut aspartaattiaminotransferaasi-pitoisuus | Yleinen | Yleinen | |
Suurentunut alkalisen fosfataasin pitoisuus veressä | Yleinen | Yleinen | |
Suurentunut gammaglutamyylitransferaasi-pitoisuus | Yleinen | Yleinen | |
Suurentunut bilirubiinipitoisuus veressä | Yleinen | Melko harvinainen | |
Suurentunut maksaentsyymipitoisuus | Yleinen | Yleinen |
* 5. asteen reaktioita on raportoitu. Ks. Valikoitujen haittavaikutusten kuvaus.
** 3.–4. asteen tapahtumia ei raportoitu.
1 Sisältää seuraavat: COVID‑19-infektio, COVID‑19-keuhkokuume, vyöruusu, influenssa ja silmänseudun vyöruusu.
2 Sisältää seuraavat: laskimoon asennetun laitteen infektio, bakteeri-infektio, kampylobakteeri-infektio, bakteeriperäinen sappitieinfektio, bakteeriperäinen virtsatieinfektio, Clostridium difficile ‑infektio, Escherichia-infektio ja peritoniitti.
3 Sisältää seuraavat: ylähengitysteiden infektio, sivuontelotulehdus, nasofaryngiitti, krooninen sivuontelotulehdus ja nuha.
4 Sisältää seuraavat: sepsis ja septinen sokki.
5 Sisältää seuraavat: alahengitysteiden infektio ja keuhkoputkitulehdus.
6 Sisältää seuraavat: virtsatieinfektio ja Escherichia-peräinen virtsatieinfektio.
7 Sisältää seuraavat: ruokatorven kandidiaasi ja sammas.
8 Sisältää seuraavat: kuumeinen neutropenia ja neutropeeninen infektio.
9 Perustuu vaikeusastetta koskevaan ASTCT:n konsensusluokitukseen (Lee 2019).
10 ICANS-tutkimuksessa (Lee, 2019) uneliaisuus, kognitiivinen häiriö, sekavuus, delirium ja tietämättömyys ajasta ja paikasta.
11 Myeliitti ilmeni samanaikaisesti sytokiinioireyhtymän kanssa.
12 Sisältää seuraavat: maha-suolikanavan verenvuoto, paksusuolen verenvuoto ja mahalaukun verenvuoto.
13 Sisältää seuraavat: ihottuma, kutiseva ihottuma, makulopapulaarinen ihottuma, dermatiitti, aknetyyppinen dermatiitti, eksfoliatiivinen dermatiitti, eryteema, palmaarieryteema, kutina ja erytematoottinen ihottuma.
Taulukko 7. Haittavaikutukset, joita raportoitiin uusiutunutta tai hoitoon reagoimatonta diffuusia suurisoluista B-solulymfoomaa sairastavilla potilailla, jotka saivat Columvi-valmistetta yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa
Elinjärjestelmä | Haittavaikutus | Kaikki vaikeusasteet | 3.−4. aste |
Infektiot | COVID-19-infektio1 | Hyvin yleinen | Yleinen* |
Hengitystieinfektiot2 | Hyvin yleinen | Yleinen* | |
Keuhkokuume3 | Hyvin yleinen | Yleinen* | |
Sytomegalovirusinfektiot4 | Yleinen | Melko harvinainen | |
Herpesvirusinfektiot5 | Yleinen | Melko harvinainen | |
Virtsatieinfektio6 | Yleinen | Yleinen | |
Sepsis7 | Yleinen | Yleinen* | |
Candida-infektiot8 | Yleinen | Hyvin harvinainen** | |
Pneumocystis jiroveciin aiheuttama keuhkokuume | Melko harvinainen | Melko harvinainen | |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Tumour flare -reaktio9 | Yleinen | Hyvin harvinainen** |
Veri ja imukudos | Trombosytopenia | Hyvin yleinen | Hyvin yleinen |
Neutropenia | Hyvin yleinen | Hyvin yleinen | |
Anemia | Hyvin yleinen | Hyvin yleinen | |
Lymfopenia | Hyvin yleinen | Hyvin yleinen | |
Kuumeinen neutropenia | Yleinen | Yleinen | |
Immuunijärjestelmä | Sytokiinioireyhtymä10 | Hyvin yleinen | Yleinen |
Aineenvaihdunta ja ravitsemus | Hypokalemia | Hyvin yleinen | Yleinen |
Hyponatremia | Hyvin yleinen | Melko harvinainen | |
Hypomagnesemia | Yleinen | Hyvin harvinainen** | |
Hypokalsemia | Yleinen | Melko harvinainen | |
Hypofosfatemia | Yleinen | Yleinen | |
Tuumorilyysioireyhtymä | Yleinen | Yleinen | |
Hermosto | Perifeerinen neuropatia11 | Hyvin yleinen | Yleinen |
Immuuniefektorisoluihin liittyvä neurotoksisuusoireyhtymä12 | Yleinen | Melko harvinainen | |
Päänsärky | Yleinen | Hyvin harvinainen** | |
Vapina | Melko harvinainen | Hyvin harvinainen** | |
Hengityselimet, rintakehä ja välikarsina | Pneumoniitti | Yleinen | Hyvin harvinainen*,** |
Ruoansulatuselimistö | Pahoinvointi | Hyvin yleinen | Melko harvinainen |
Ripuli | Hyvin yleinen | Yleinen | |
Oksentelu | Hyvin yleinen | Melko harvinainen | |
Vatsakipu13 | Hyvin yleinen | Yleinen | |
Ummetus | Hyvin yleinen | Hyvin harvinainen** | |
Koliitti14 | Yleinen | Yleinen | |
Haimatulehdus15 | Yleinen | Yleinen | |
Iho ja ihonalainen kudos | Ihottuma16 | Hyvin yleinen | Melko harvinainen |
Luusto, lihakset ja sidekudos | Muskuloskeletaalinen kipu17 | Hyvin yleinen | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | Kuume | Hyvin yleinen | Melko harvinainen |
Tutkimukset | Suurentunut aspartaattiaminotransferaasipitoisuus | Hyvin yleinen | Yleinen |
Suurentunut alaniiniaminotransferaasipitoisuus | Hyvin yleinen | Yleinen | |
Suurentunut alkalisen fosfataasin pitoisuus veressä | Hyvin yleinen | Melko harvinainen | |
Suurentunut gammaglutamyylitransferaasipitoisuus | Hyvin yleinen | Yleinen | |
Suurentunut laktaattidehydrogenaasipitoisuus veressä | Hyvin yleinen | Hyvin harvinainen** | |
Suurentunut bilirubiinipitoisuus veressä18 | Yleinen | Hyvin harvinainen** | |
Suurentunut maksaentsyymipitoisuus | Melko harvinainen | Hyvin harvinainen** |
* Raportoidut 5. asteen reaktiot. Ks. Valikoitujen haittavaikutusten kuvaus.
** 3.–4. asteen tapahtumia ei raportoitu.
1 Sisältää seuraavat: COVID-19, COVID-19-keuhkokuume ja SARS-CoV-2-testi positiivinen.
2 Sisältää seuraavat: ylähengitysteiden infektio, alahengitysteiden infektio, hengitystieinfektio ja bakteeriperäinen hengitystieinfektio.
3 Sisältää seuraavat: keuhkokuume, bakteerikeuhkokuume ja pneumokokkikeuhkokuume.
4 Uudet oireet tai uudelleen aktivoituminen. Sisältää seuraavat: sytomegalovirusinfektio, sytomegalovirustesti positiivinen, sytomegalovirusinfektion uudelleen aktivoituminen ja sytomegalovirusviremia.
5 Uudet oireet tai uudelleen aktivoituminen. Sisältää seuraavat: vyöruusu ja herpesvirusinfektio.
6 Sisältää seuraavat: virtsatieinfektio ja urosepsis.
7 Sisältää seuraavat: sepsis, streptokokkisepsis, septinen sokki ja enterokokkisepsis.
8 Sisältää seuraavat: sammas ja Candida-infektio.
9 Sisältää seuraavat: tumour flare -reaktio ja kasvaimen kipu.
10 Perustuu ASTCT-konsensusluokitukseen (Lee 2019).
11 Sisältää seuraavat: perifeerinen neuropatia, perifeerinen sensorinen neuropatia, dysestesia, parestesia, hypestesia, perifeerinen motorinen neuropatia ja polyneuropatia.
12 Sisältää seuraavat: sekavuustila, delirium ja immuuniefektorisoluihin liittyvä neurotoksisuusoireyhtymä.
13 Sisältää seuraavat: vatsakipu, epämukava tunne vatsassa, ylävatsakipu, alavatsakipu ja maha-suolikanavan kipu.
14 Sisältää seuraavat: koliitti, iskeeminen koliitti ja enterokoliitti.
15 Sisältää seuraavat: haimatulehdus ja akuutti haimatulehdus.
16 Sisältää seuraavat: ihottuma, kutiseva ihottuma, makulopapulaarinen ihottuma, eryteema, kutina, erytematoottinen ihottuma, nokkosihottuma ja erythema multiforme.
17 Sisältää seuraavat: nivelkipu, muskuloskeletaalinen kipu, selkäkipu, luukipu, lihaskipu, niskakipu, raajakipu, muskuloskeletaalinen kipu rintakehässä ja ei-sydänperäinen rintakipu.
18 Sisältää seuraavat: suurentunut bilirubiinipitoisuus veressä ja hyperbilirubinemia.
Valikoitujen haittavaikutusten kuvaus
Seuraavassa on kuvattu Columvi-monoterapian ja/tai -yhdistelmähoidon yhteydessä havaittuja merkittäviä haittavaikutuksia. Columvi-yhdistelmähoidon yhteydessä havaittujen merkittävien haittavaikutusten tarkemmat tiedot on esitetty erikseen, jos niissä havaittiin kliinisesti merkittäviä eroja Columvi-monoterapiaan verrattuna.
Sytokiinioireyhtymä
Columvi-monoterapia
Minkä tahansa vaikeusasteen sytokiinioireyhtymä (ASTCT-kriteerien mukaan) ilmaantui 67,6 %:lle Columvi-monoterapiaa saaneista potilaista: 1. asteen sytokiinioireyhtymä raportoitiin 50,3 %:lla potilaista, 2. asteen sytokiinioireyhtymä raportoitiin 13,1 %:lla potilaista, 3. asteen sytokiinioireyhtymä raportoitiin 2,8 %:lla potilaista ja 4. asteen sytokiinioireyhtymä raportoitiin 1,4 %:lla potilaista. Sytokiinioireyhtymä ilmaantui useammin kuin kerran 32,4 %:lle (47 potilaalle 145:stä) potilaista; 36 potilaalle 47 potilaasta ilmaantui useita vain 1. asteen sytokiinioireyhtymään liittyviä tapahtumia. Kuolemaan johtaneita sytokiinioireyhtymätapauksia ei ollut. Sytokiinioireyhtymä hävisi yhtä potilasta lukuun ottamatta kaikilta potilailta. Yksi potilas lopetti hoidon sytokiinioireyhtymän vuoksi.
Potilailla, joille sytokiinioireyhtymä ilmaantui, yleisimpiä sytokiinioireyhtymän ilmenemismuotoja olivat kuume (99,0 %), takykardia (25,5 %), hypotensio (23,5 %), vilunväristykset (14,3 %) ja hypoksia (12,2 %). Sytokiinioireyhtymään liittyviä vähintään vaikeusasteen 3 tapahtumia olivat hypotensio (3,1 %), hypoksia (3,1 %), kuume (2,0 %) ja takykardia (2,0 %).
Minkä tahansa vaikeusasteen sytokiinioireyhtymä ilmaantui 1. hoitosyklin 8. päivänä annetun ensimmäisen 2,5 mg:n Columvi-annoksen jälkeen 54,5 %:lle potilaista, jolloin sen ilmaantumiseen kuluneen ajan mediaani (infuusion aloittamisesta) oli 12,6 tuntia (vaihteluväli 5,2–50,8 tuntia) ja keston mediaani oli 31,8 tuntia (vaihteluväli 0,5–316,7 tuntia); 1. hoitosyklin 15. päivänä annetun 10 mg:n annoksen jälkeen 33,3 %:lle potilaista, jolloin sen ilmaantumiseen kuluneen ajan mediaani oli 26,8 tuntia (vaihteluväli 6,7–125,0 tuntia) ja keston mediaani oli 16,5 tuntia (vaihteluväli 0,3–109,2 tuntia), ja 2. hoitosyklissä annetun 30 mg:n annoksen jälkeen 26,8 %:lle potilaista, jolloin sen ilmaantumiseen kuluneen ajan mediaani oli 28,2 tuntia (vaihteluväli 15,0–44,2 tuntia) ja keston mediaani oli 18,9 tuntia (vaihteluväli 1,0–180,5 tuntia). Sytokiinioireyhtymää raportoitiin 0,9 %:lla potilaista 3. hoitosyklissä ja 2 %:lla potilaista 3. hoitosyklin jälkeen.
Ensimmäisen Columvi-annoksen (2,5 mg) jälkeen ≥ 2. asteen sytokiinioireyhtymä ilmaantui 12,4 %:lle potilaista, ja sen ilmaantumiseen kuluneen ajan mediaani oli 9,7 tuntia (vaihteluväli 5,2–19,1 tuntia) ja keston mediaani oli 50,4 tuntia (vaihteluväli 6,5–316,7 tuntia). Ensimmäisen hoitosyklin 15. päivänä annetun 10 mg:n Columvi-annoksen jälkeen ≥ 2. asteen sytokiinioireyhtymän ilmaantuvuus väheni 5,2 %:iin potilaista, jolloin sen ilmaantumiseen kuluneen ajan mediaani oli 26,2 tuntia (vaihteluväli 6,7–144,2 tuntia) ja keston mediaani oli 30,9 tuntia (vaihteluväli 3,7–227,2 tuntia). Toisen hoitosyklin 1. päivänä annetun 30 mg:n Columvi-annoksen jälkeen ≥ 2. asteen sytokiinioireyhtymä ilmeni yhdellä potilaalla (0,8 %); sen ilmaantumiseen kulunut aika oli 15,0 tuntia ja kesto oli 44,8 tuntia. Toisen hoitosyklin jälkeen ei raportoitu ≥ 2. asteen sytokiinioireyhtymää.
145 potilaasta 7 potilaalla (4,8 %) todettiin kohonneita maksantoimintakokeiden arvoja (ASAT ja ALAT > 3 × ULN ja/tai kokonaisbilirubiinipitoisuus > 2 × ULN), joita raportoitiin samanaikaisesti sytokiinioireyhtymän (n = 6) tai sairauden etenemisen kanssa (n = 1).
Niistä 25 potilaasta, joille ilmaantui ≥ 2. asteen sytokiinioireyhtymä Columvi-hoidon jälkeen, 22 potilasta (88,0 %) sai tosilitsumabia, 15 potilasta (60,0 %) sai kortikosteroideja ja 14 potilasta (56,0 %) sai sekä tosilitsumabia että kortikosteroideja. Kymmenen potilasta (40,0 %) sai happihoitoa. Kaikki 6 potilasta (24,0 %), joilla oli 3. tai 4. asteen sytokiinioireyhtymä, sai yhtä vasopressoria.
Sairaalahoitoa Columvi-valmisteen annon jälkeen ilmenneen sytokiinioireyhtymän vuoksi tarvitsi 22,1 % potilaista, ja sairaalahoidon keston raportoitu mediaani oli 4 päivää (vaihteluväli 2–15 päivää).
Columvi yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa
Minkä tahansa vaikeusasteen sytokiinioireyhtymä (ASTCT-kriteerien mukaan) ilmaantui 44,2 %:lle potilaista, jotka saivat Columvi-valmistetta yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa: 1. asteen sytokiinioireyhtymä raportoitiin 31,4 %:lla potilaista, 2. asteen sytokiinioireyhtymä raportoitiin 10,5 %:lla potilaista ja 3. asteen sytokiinioireyhtymä raportoitiin 2,3 %:lla potilaista. Sytokiinioireyhtymä ilmaantui useammin kuin kerran 21,5 %:lle (37 potilaalle 172:sta) potilaista; 30 potilaalle 37 potilaasta ilmaantui useita vain 1. asteen sytokiinioireyhtymään liittyviä tapahtumia. Vaikeusasteen 4 tai kuolemaan johtaneita sytokiinioireyhtymätapauksia ei ollut. Sytokiinioireyhtymä hävisi yhtä potilasta lukuun ottamatta kaikilta potilailta. Yksi potilas lopetti hoidon sytokiinioireyhtymän vuoksi.
Potilailla, joille sytokiinioireyhtymä ilmaantui, yleisimpiä sytokiinioireyhtymän ilmenemismuotoja olivat kuume (98,7 %), hypotensio (22,4 %), vilunväristykset (17,1 %) ja hypoksia (14,5 %). Sytokiinioireyhtymään liittyviä vähintään 3. asteen tapahtumia olivat hypotensio (6,6 %), hypoksia (5,3 %), kuume (3,9 %), vilunväristykset (1,3 %) ja ripuli (1,3 %).
Minkä tahansa vaikeusasteen sytokiinioireyhtymä ilmaantui 1. hoitosyklin 8. päivänä annetun ensimmäisen 2,5 mg:n Columvi-annoksen jälkeen 34,9 %:lle potilaista, jolloin sen ilmaantumiseen kuluneen ajan mediaani (infuusion aloittamisesta) oli 12,6 tuntia (vaihteluväli 4,4–54,7 tuntia) ja keston mediaani oli 19,8 tuntia (vaihteluväli 2,0–168,0 tuntia); 1. hoitosyklin 15. päivänä annetun 10 mg:n annoksen jälkeen 14,4 %:lle potilaista, jolloin sen ilmaantumiseen kuluneen ajan mediaani oli 22,8 tuntia (vaihteluväli 7,4–81,2 tuntia) ja keston mediaani oli 10,6 tuntia (vaihteluväli 1,0–248,5 tuntia) ja 2. hoitosyklissä annetun 30 mg:n annoksen jälkeen 9,3 %:lle potilaista, jolloin sen ilmaantumiseen kuluneen ajan mediaani oli 23,5 tuntia (vaihteluväli 14,7–33,4 tuntia) ja keston mediaani oli 18,4 tuntia (vaihteluväli 8,3–137,0 tuntia). Sytokiinioireyhtymää raportoitiin 6,7 %:lla potilaista 3. hoitosyklissä ja 11,0 %:lla potilaista 3. hoitosyklin jälkeen.
Ensimmäisen Columvi-annoksen (2,5 mg) jälkeen ≥ 2. asteen sytokiinioireyhtymä ilmaantui 10,5 %:lle potilaista, ja sen ilmaantumiseen kuluneen ajan mediaani oli 12,0 tuntia (vaihteluväli 4,4–30,5 tuntia) ja keston mediaani 42,3 tuntia (vaihteluväli 3,5–143,7 tuntia). Suurimmalla osalla (14/18) potilaista, joilla ilmeni ≥ 2. asteen sytokiinioireyhtymä, sytokiinioireyhtymä ilmeni 8 tunnin kuluessa ensimmäisen Columvi-annoksen (2,5 mg) aloittamisesta tai heillä esiintyi kuumetta ≥ 1,5 tuntia ennen muiden ≥ 2. asteen sytokiinioireyhtymän oireiden alkamista. Ensimmäisen hoitosyklin 15. päivänä annetun 10 mg:n Columvi-annoksen jälkeen ≥ 2. asteen sytokiinioireyhtymän ilmaantuvuus väheni 1,8 %:iin potilaista, jolloin sen ilmaantumiseen kuluneen ajan mediaani oli 22,3 tuntia (vaihteluväli 7,4–22,8 tuntia) ja keston mediaani oli 37,0 tuntia (vaihteluväli 34,8–248,5 tuntia). Toisen hoitosyklin 1. päivänä annetun 30 mg:n Columvi-annoksen jälkeen ei ilmennyt ≥ 2. asteen sytokiinioireyhtymän tapahtumia. Kolmella potilaalla (2,0 %) ilmeni ≥ 2. asteen sytokiinioireyhtymä 2. hoitosyklin jälkeen (kaikki 2. asteen tapahtumia).
Näistä 172 potilaasta 2 potilaalla (1,2 %) todettiin kohonneita arvoja maksan toimintakokeissa (ASAT ja ALAT > 3 × ULN), ja ne raportoitiin samanaikaisesti sytokiinioireyhtymän kanssa.
Niistä 76 potilaasta, joille ilmaantui minkä tahansa vaikeusasteen sytokiinioireyhtymä, 28 potilasta (36,8 %) sai hoitona tosilitsumabia, 39 potilasta (51,3 %) sai kortikosteroideja ja 18 potilasta (23,7 %) sai sekä tosilitsumabia että kortikosteroideja.
Niistä 22 potilaasta, joille ilmaantui ≥ 2. asteen sytokiinioireyhtymä Columvi-hoidon jälkeen, 16 potilasta (72,7 %) sai tosilitsumabia, 15 potilasta (68,2 %) sai kortikosteroideja ja 12 potilasta (54,5 %) sai sekä tosilitsumabia että kortikosteroideja. 11 potilasta (50,0 %) sai happihoitoa. Kaikki neljä potilasta (18,2 %), joilla oli 3. asteen sytokiinioireyhtymä, saivat yhtä vasopressoria.
Sairaalahoitoa Columvi-valmisteen annon jälkeen ilmenneen sytokiinioireyhtymän vuoksi tarvitsi 19,8 % potilaista, ja sairaalahoidon raportoidun keston mediaani oli 5 päivää (vaihteluväli 2–85 päivää).
Immuuniefektorisoluihin liittyvä neurotoksisuusoireyhtymä
ICANSsta on ilmoitettu kliinisten tutkimusten yhteydessä ja myyntiluvan myöntämisen jälkeen, mukaan lukien vaikeusaste 3 ja sitä vaikeammat oireet. Yleisimmät ICANS-hoidon kliiniset oireet olivat sekavuus, tajunnan tason aleneminen, tietämättömyys ajasta ja paikasta, kouristuskohtaukset, afasia ja dysgrafia. Saatavilla olevien tietojen perusteella neurologinen toksisuus puhkesi useimmissa tapauksissa samanaikaisesti sytokiinioireyhtymän kanssa.
ICANSin havaittiin alkaneen enimmäkseen 1–7 päivää (mediaani 2 päivää) viimeisimmän annoksen jälkeen. Vain muutaman tapahtuman ilmoitettiin alkaneen yli kuukauden kuluttua Columvin käytön aloittamisesta.
Vakavat infektiot
Vakavia infektioita raportoitiin 15,9 %:lla Columvi-monoterapiaa saaneista potilaista. Yleisimpiä vakavia infektioita, joita raportoitiin ≥ 2 %:lla potilaista, olivat sepsis (4,1 %), COVID‑19-infektio (3,4 %) ja COVID‑19-keuhkokuume (2,8 %). Infektioon liittyvä kuolema raportoitiin 4,8 %:lla potilaista (sepsiksen, COVID‑19-keuhkokuumeen ja COVID‑19-infektion seurauksena). Neljälle potilaalle (2,8 %) ilmaantui vakavia infektioita samanaikaisesti 3. tai 4. asteen neutropenian kanssa.
Vakavia infektioita raportoitiin 22,7 %:lla potilaista, jotka saivat Columvi-valmistetta yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa. Yleisimpiä vakavia infektioita, joita raportoitiin ≥ 2 %:lla potilaista, olivat keuhkokuume (5,8 %), COVID-19-infektio (4,7 %) ja alahengitysteiden infektio (2,9 %). Infektioon liittyvä kuolema raportoitiin 3,5 %:lla potilaista (COVID-19-infektion, keuhkokuumeen, hengitystieinfektion ja septisen sokin seurauksena). Yhdelle potilaalle (0,6 %) ilmaantui vakava infektio (keuhkokuume) samanaikaisesti 3. asteen neutropenian kanssa.
Pneumoniitti
Pneumoniittiin liittyviä tapahtumia (pois lukien infektiosta johtuva keuhkokuume) raportoitiin kahdella potilaalla (1,2 %), jotka saivat Columvi-valmistetta yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa. Kumpikin tapahtuma johti potilaan kuolemaan. Ensimmäisestä Columvi-annoksesta pneumoniitin ilmaantumiseen kuluneen ajan mediaani oli 168 päivää (vaihteluväli 102–255 päivää).
Koliitti
Koliittia (4. aste) raportoitiin yhdellä Columvi-monoterapiaa saaneella potilaalla (0,7 %), ja se ilmaantui 104 päivän kuluttua ensimmäisestä Columvi-annoksesta.
Koliittiin liittyviä tapahtumia (pois lukien infektiosta johtuva etiologia) raportoitiin neljällä potilaalla 172:sta (2,3 %), jotka saivat Columvi-valmistetta yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa. Kahdella potilaalla (1,2 %) oli 3. asteen tapahtumia. Ensimmäisestä Columvi-annoksesta koliitin ilmaantumiseen kuluneen ajan mediaani oli 154 päivää (vaihteluväli 115–187 päivää).
Opportunistiset infektiot
Sytomegaloviruksen aiheuttamia tapahtumia raportoitiin kuudella potilaalla 467 potilaasta (1,3 %), jotka saivat Columvi-monoterapiaa. Yhdellä potilaalla (0,2 %) ilmeni 3. asteen sytomegaloviruksen aiheuttama korioretiniitti. Pneumocystis jiroveciin aiheuttamaa keuhkokuumetta raportoitiin neljällä potilaalla 467 potilaasta (0,9 %). Kolmella heistä (0,6 %) oli 3. asteen tapahtumia.
Sytomegaloviruksen aiheuttamia tapahtumia raportoitiin 11 potilaalla (6,4 %), jotka saivat Columvi-valmistetta yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa, ja yhdellä potilaalla (0,6 %) ilmeni 3. asteen CMV-viremia. Sammasta raportoitiin kolmella potilaalla (1,7 %), ja kaikki niistä olivat 1–2. asteen tapahtumia. Pneumocystis jiroveciin aiheuttama keuhkokuume (3. aste) raportoitiin yhdellä potilaalla (0,6 %), jolla oli myös 3. asteen CMV-viremia. Yhdellä potilaalla (0,6 %) potilaalla raportoitiin borrelioosiaivokalvotulehdus (2. aste).
Neutropenia
Neutropeniaa (mukaan lukien neutrofiilimäärän vähenemistä) raportoitiin 40,0 %:lla potilaista, ja vaikea-asteista neutropeniaa (3. tai 4. aste) raportoitiin 29,0 %:lla Columvi-monoterapiaa saaneista potilaista. Ensimmäisen neutropeniatapahtuman ilmaantumiseen kuluneen ajan mediaani oli 29 päivää (vaihteluväli 1–203 päivää). Pitkittynyttä (yli 30 päivää kestävää) neutropeniaa ilmeni 11,7 %:lle potilaista. Valtaosa neutropeenisista potilaista (79,3 %) sai hoitona granulosyyttiryhmiä stimuloivaa kasvutekijää (G‑CSF). Kuumeista neutropeniaa raportoitiin 3,4 %:lla potilaista.
Tumour flare ‑reaktio
Tumour flare ‑reaktio raportoitiin 11,7 %:lla Columvi-monoterapiaa saaneista potilaista, mukaan lukien 2. asteen tumour flare ‑reaktio 4,8 %:lla potilaista ja 3. asteen tumour flare ‑reaktio 2,8 %:lla potilaista. Tumour flare ‑reaktion raportoitiin liittyneen pään ja kaulan imusolmukkeisiin, mikä ilmeni kipuna, sekä rintakehän imusolmukkeisiin, minkä oire oli hengenahdistus pleuraeffuusion kehittymisen vuoksi. Valtaosa tumour flare ‑tapahtumista (16/17) ilmeni 1. hoitosyklissä eikä 2. hoitosyklin jälkeen raportoitu yhtään tumour flare ‑tapahtumaa. Tumour flare ‑reaktion ilmaantumiseen kuluneen ajan mediaani oli 2 päivää (vaihteluväli 1–16 päivää) ja sen keston mediaani oli 3,5 päivää (vaihteluväli 1–35 päivää).
Niistä 11 potilaasta, joille ilmaantui ≥ 2. asteen tumour flare ‑reaktio, 2 potilasta (18,2 %) sai kipulääkitystä, 6 potilasta (54,5 %) sai kortikosteroideja ja kipulääkitystä, mukaan lukien morfiinin johdannaisia, 1 potilas (9,1 %) sai kortikosteroideja ja pahoinvointilääkkeitä ja 2 potilasta (18,2 %) ei tarvinnut hoitoa. Kaikki tumour flare ‑tapahtumat, paitsi yhden potilaan ≥ 2. asteen tapahtuma, hävisivät. Yksikään potilas ei lopettanut hoitoa tumour flare ‑reaktion vuoksi.
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymää raportoitiin 2:lla Columvi-monoterapiaa saaneella potilaalla (1,4 %), ja kumpikin oli vaikeusasteeltaan 3. asteen tapahtuma. Tuumorilyysioireyhtymän ilmaantumiseen kuluneen ajan mediaani oli 2 päivää ja sen keston mediaani oli 4 päivää (vaihteluväli 3–5 päivää).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisistä tutkimuksista ei ole kokemusta yliannoksesta. Yliannostapauksessa potilasta pitää seurata tarkoin haittavaikutusten oireiden ja löydösten havaitsemiseksi, ja asianmukainen oireiden hoito on aloitettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, muut monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ATC-koodi: L01FX28
Vaikutusmekanismi
Glofitamabi on kaksoisspesifinen monoklonaalinen vasta-aine, joka sitoutuu bivalenttisesti B‑solujen pinnalla ilmentyvään CD20-antigeeniin ja monovalenttisesti T-solujen pinnalla ilmentyvän T‑solureseptorikompleksin CD3-antigeeniin. Sitoutumalla samanaikaisesti B-solun CD20-antigeeniin ja T-solun CD3-antigeeniin glofitamabi toimii välittäjänä immunologisen synapsin muodostumisessa ja sen jälkeisessä T-solun aktivaatiossa ja proliferaatiossa, sytokiinien erittymisessä ja sytolyyttisten proteiinien vapautumisessa, mistä aiheutuu CD20-antigeenia ilmentävien B-solujen lyysi.
Farmakodynamiikka
NP30179-tutkimuksessa 84 %:lla potilaista (84 potilaalla 100 potilaasta) oli B-soluvaje (< 70 solua/mikrol) jo ennen obinututsumabiesilääkitystä. Obinututsumabiesilääkityksen jälkeen ennen Columvi-hoidon aloittamista niiden potilaiden osuus, joilla oli B-soluvajetta, nousi 100 %:iin (94/94), ja B-solujen määrä pysyi alhaisena Columvi-hoidon ajan.
1. hoitosyklissä (step‑up-annostus) havaittiin IL‑6-pitoisuuden suurentuneen plasmassa ohimenevästi 6 tunnin aikapisteessä Columvi-infuusion jälkeen ja se pysyi koholla 20 tunnin aikapisteessä infuusion jälkeen, minkä jälkeen pitoisuus palasi lähtötasolle ennen seuraavaa infuusiota.
GO41944-tutkimuksessa (STARGLO) 63,9 %:lla potilaista (115 potilaalla 180 potilaasta) oli B‑soluvaje (< 70 solua/mikrol) jo ennen obinututsumabiesilääkitystä. Obinututsumabiesilääkityksen jälkeen ennen Columvi-hoidon aloittamista niiden potilaiden osuus, joilla oli B-soluvajetta, nousi 79,4 %:iin (143/180), ja B-solujen määrä pysyi alhaisena Columvi-hoidon ajan.
Sydämen sähköfysiologia
NP30179-tutkimuksessa 16 potilaalla 145:stä Columvi-valmisteelle altistuneesta potilaasta jokin lähtötilanteen jälkeinen QTc-arvo oli > 450 ms. Tutkijan arvion mukaan yksi näistä tapauksista katsottiin kliinisesti merkittäväksi. Yhdenkään potilaan hoitoa ei lopetettu QTc-ajan pitenemisen vuoksi.
GO41944-tutkimuksessa (STARGLO) 16 potilaalla 172:sta Columvi-valmisteelle altistuneesta potilaasta jokin lähtötilanteen jälkeinen QTc-arvo oli > 450 ms. Yhdenkään potilaan hoitoa ei lopetettu QTc-ajan pitenemisen vuoksi.
Kliininen teho ja turvallisuus
Uusiutunut tai hoitoon reagoimaton diffuusi suurisoluinen B-solulymfooma
Columvi-monoterapia
Columvi-valmistetta arvioitiin avoimessa, usean kohortin monikeskustutkimuksessa (NP30179) uusiutunutta tai hoitoon reagoimatonta B-solulähtöistä non-Hodgkinin lymfoomaa sairastavilla potilailla. Diffuusia suurisoluista B-solulymfoomaa koskeneessa yhden haaran monoterapiakohortissa (n = 108) potilaiden, joilla oli uusiutunut tai hoitoon reagoimaton diffuusi suurisoluinen B‑solulymfooma, piti olla saanut vähintään kaksi aiempaa systeemistä hoitolinjaa, mukaan lukien monoklonaalista CD20-vasta-ainetta ja jotakin antrasykliinilääkeainetta. Potilaat, joilla oli follikulaarinen lymfooma (FL3b) tai Richterin transformaatio, eivät soveltuneet tutkimukseen mukaan. Potilaiden odotettiin sairastavan CD20-positiivista diffuusia suurisoluista B‑solulymfoomaa, mutta sisäänotto ei edellyttänyt biomerkkiaineiden sopivuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tutkimukseen ei otettu mukaan potilaita, joilla todettiin seuraavia: ECOG-toimintakykyluokka ≥ 2, merkittävä sydän- ja verisuonitauti (kuten New York Heart Association ‑luokan III tai IV sydänsairaus, sydäninfarkti edeltäneiden 6 kuukauden aikana, epästabiileja sydämen rytmihäiriöitä tai epästabiili angina pectoris), merkittävä aktiivinen keuhkosairaus, heikentynyt munuaisten toiminta (CrCL < 50 ml/min ja seerumin kreatiniinipitoisuus koholla), immunosuppressiivista hoitoa vaativa aktiivinen autoimmuunisairaus, aktiivisia infektioita (eli krooninen aktiivinen Epstein–Barrin virus, akuutti tai krooninen C-hepatiitti, B-hepatiitti, HIV), progressiivinen multifokaalinen leukoenkefalopatia, parhaillaan sairastettava tai anamneesissa oleva keskushermoston lymfooma tai keskushermostosairaus, anamneesissa makrofagiaktivaatio-oireyhtymä / hemofagosyyttinen lymfohistiosytoosi, aiemmin tehty allogeeninen kantasolusiirto, aiemmin tehty elinsiirto tai maksan transaminaasipitoisuus ≥ 3 × ULN.
Kaikki potilaat saivat 1. hoitosyklin 1. päivänä esilääkityksenä obinututsumabia. Potilaat saivat step‑up-hoito-ohjelman mukaisesti 1. hoitosyklin 8. päivänä 2,5 mg Columvi-valmistetta, 1. hoitosyklin 15. päivänä 10 mg Columvi-valmistetta ja 2. hoitosyklin 1. päivänä 30 mg Columvi-valmistetta. Potilaat jatkoivat hoitoa 30 mg:n Columvi-annoksella 3.–12. hoitosyklin 1. päivänä. Kunkin hoitosyklin pituus oli 21 päivää. Potilaiden saamien Columvi-hoitosyklien määrän mediaani oli 5 (vaihteluväli 1–13 hoitosykliä); 34,7 % sai 8 tai useampia hoitosyklejä ja 25,7 % sai Columvi-hoitoa 12 hoitosykliä.
Lähtötilanteen demografiset ja sairauden ominaisuudet olivat seuraavat: iän mediaani 66 vuotta (vaihteluväli 21–90 vuotta), ja 53,7 % oli 65-vuotiaita tai sitä vanhempia ja 15,7 % oli 75-vuotiaita tai sitä vanhempia; 69,4 % oli miehiä; 74,1 % oli valkoihoisia, 5,6 % oli aasialaisia ja 0,9 % oli mustaihoisia tai afroamerikkalaisia; 5,6 % oli latinalaisamerikkalaisia; ja ECOG-toimintakykyluokka oli 0 (46,3 %) tai 1 (52,8 %). Valtaosalla potilaista (71,3 %) oli tarkemmin määrittämätön diffuusi suurisoluinen B-solulymfooma, 7,4 %:lla oli follikulaarisesta lymfoomasta transformoitunut diffuusi suurisoluinen B-solulymfooma, 8,3 %:lla oli follikulaarisesta lymfoomasta transformoitunut korkean maligniteettiasteen B‑solulymfooma tai muu histologia, 7,4 %:lla oli korkean maligniteettiasteen B‑solulymfooma ja 5,6 %:lla oli primaarinen mediastinaalinen suurisoluinen B‑solulymfooma. Aiempien hoitolinjojen lukumäärän mediaani oli 3 (vaihteluväli 2–7); 39,8 % potilaista oli saanut aiemmin 2 hoitolinjaa ja 60,2 % oli saanut aiemmin 3 tai useampia hoitolinjoja. Kaikki potilaat olivat saaneet aiemmin solunsalpaajahoitoa (kaikki potilaat olivat saaneet hoitoa alkyloivilla aineilla ja 98,1 % potilaista oli saanut antrasykliinihoitoa), ja kaikki potilaat olivat saaneet aiemmin hoitoa monoklonaalisella CD20-vasta-aineella; 35,2 % potilaista oli aiemmin saanut CAR-T‑soluhoitoa ja 16,7 % potilaista oli saanut autologisen kantasolusiirron. Valtaosalla potilaista (89,8 %) oli hoitoon reagoimaton tauti, 60,2 %:lla potilaista oli ensilinjan hoitoon reagoimaton tauti ja 83,3 %:lla potilaista tauti ei reagoinut viimeisimpään hoitoon.
Tehon ensisijainen päätetapahtuma oli riippumattoman arviointitoimikunnan Lugano 2014 –kriteereihin perustuva täydellinen vaste. Seurannan kokonaiskeston mediaani oli 15 kuukautta (vaihteluväli 0–21 kuukautta). Toissijaisia tehon päätetapahtumia olivat riippumattoman arviointitoimikunnan arvioima kokonaisvasteluku (ORR), vasteen kesto (DOR), täydellisen vasteen kesto (DOCR) ja ensimmäiseen täydelliseen vasteeseen kulunut aika (TFCR).
Yhteenveto tehon tuloksista esitetään taulukossa 8.
Taulukko 8. Yhteenveto tehosta potilailla, joilla on uusiutunut tai hoitoon reagoimaton diffuusi suurisoluinen B-solulymfooma
| Tehon päätetapahtumat | Columvi N = 108 |
| Täydellinen vaste | |
| Potilaita, joilla täydellinen vaste, n (%) | 38 (35,2) |
| 95 %:n luottamusväli | (26,24–44,96) |
| Kokonaisvasteluku | |
| Potilaita, joilla täydellinen vaste tai osittainen vaste, n (%) | 54 (50,0) |
| 95 %:n luottamusväli | (40,22–59,78) |
| Täydellisen vasteen kesto1 | |
| Täydellisen vasteen keston mediaani, kk (95 %:n luottamusväli) | NE (18,4–NE) |
| Vaihteluväli, kk | 02−202 |
| 12 kuukauden täydellisen vasteen kesto, % (95 %:n luottamusväli)3 | 74,6 (59,19–89,93) |
| Vasteen kesto4 | |
| Keston mediaani, kk (95 %:n luottamusväli) | 14,4 (8,6–NE) |
| Vaihteluväli, kk | 02−202 |
| Ensimmäiseen täydelliseen vasteeseen kulunut aika | |
| Ensimmäiseen täydelliseen vasteeseen kuluneen ajan mediaani, vrk (95 %:n luottamusväli) | 42 (41–47) |
| Vaihteluväli, vrk | 31–308 |
NE = ei arvioitavissa (not estimable).
Hypoteesitestaus tehtiin ensisijaisesta päätetapahtumasta, joka oli riippumattoman arviointikomitean arvio täydellisen vasteen saaneiden määrästä.
1 Täydellisen vasteen kestoksi määriteltiin ensimmäisen täydellisen vasteen päivämäärästä sairauden etenemiseen tai mistä tahansa syystä aiheutuneeseen kuolemaan kulunut aika.
2 Sensuroidut havainnot.
3 Kaplan–Meierin estimaatteihin perustuva aika ilman tapahtumia.
4 Vasteen kestoksi määriteltiin ensimmäisen vasteen (osittainen vaste tai täydellinen vaste) päivämäärästä sairauden etenemiseen tai mistä tahansa syystä aiheutuneeseen kuolemaan kulunut aika.
Vasteen keston seuranta-ajan mediaani oli 12,8 kuukautta (vaihteluväli 0–20 kuukautta).
Columvi yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa
Columvi-valmisteen tehoa yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa (Columvi + GemOx) arvioitiin avoimessa, satunnaistetussa kliinisessä monikeskustutkimuksessa GO41944 (STARGLO), jossa oli mukana 274 potilasta, jotka sairastivat uusiutunutta tai hoitoon reagoimatonta tarkemmin määrittämätöntä diffuusia suurisoluista B-solulymfoomaa (DLBCL NOS).
Tutkimuksessa oli mukana potilaita, joilla oli tarkemmin määrittämätön diffuusi suurisoluinen B‑solulymfooma ja jotka olivat saaneet aiemmin vain yhtä hoitolinjaa, joille ei voitu tehdä autologista kantasolusiirtoa (ASCT) tai jotka olivat saaneet aiemmin ≥ 2:ta hoitoa. Potilailta edellytettiin seuraavat seikat: ECOG-toimintakykyluokka oli ≤ 2, kreatiniinipuhdistuma ≥ 30 ml/min, maksan transaminaasiarvot ≤ 2,5 × ULN, joilla ei ollut merkittävää sydän- ja verisuonitautia (kuten New York Heart Association -luokan III tai IV sydänsairaus, sydäninfarkti edeltäneiden 3 kuukauden aikana, epästabiileja rytmihäiriöitä tai epästabiili angina pectoris), ei parhaillaan tai aiempaa keskushermoston lymfoomaa tai keskushermoston sairautta, ei immunosuppressiivista hoitoa vaativaa aktiivista autoimmuunisairautta, ei aktiivisia infektioita (esim. krooninen aktiivinen Epstein–Barrin virus, aktiivinen B‑hepatiitti, C‑hepatiitti) ja joilla ei ollut aiemmin ollut mitään seuraavista: HIV-infektio, progressiivinen multifokaalinen leukoenkefalopatia, hemofagosyyttinen lymfohistiosytoosi, aiemmin tehty allogeeninen kantasolusiirto tai aiemmin tehty elinsiirto. Tutkimukseen ei otettu mukaan potilaita, joilla oli korkean maligniteettiasteen B‑solulymfooma, primaarinen mediastinaalinen suurisoluinen B‑solulymfooma tai anamneesissa indolentin sairauden muuttuminen diffuusiksi suurisoluiseksi B‑solulymfoomaksi.
Potilaat, jotka olivat saaneet vain yhtä aiempaa hoitolinjaa, katsottiin sellaisiksi, ettei heille voitu tehdä kantasolusiirtoa, jos heillä täyttyi vähintään yksi seuraavista kriteereistä: ikä ≥ 70 vuotta, ECOG-toimintakykyluokka 2, vasemman kammion ejektiofraktio ≤ 40 %, riittämätön vaste salvage-hoitoon, aiemmin tehty autologinen kantasolusiirto, kreatiniinipuhdistuma ≤ 45 ml/min, muut komorbiditeetit tai kriteerit, jotka paikallisten standardien perusteella tai tutkijan mielestä estävät siirteen käytön, tai potilaan kieltäytyminen suuriannoksisesta solunsalpaajahoidosta ja/tai siirteestä.
Potilaat satunnaistettiin suhteessa 2:1 saamaan Columvi + GemOx -yhdistelmähoitoa (N = 183) tai rituksimabia yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa (R-GemOx; N = 91) 8 hoitosyklin ajan, minkä jälkeen Columvi + GemOx -yhdistelmähoitoa saaneet potilaat saivat 4 lisähoitosykliä Columvi-monoterapiaa. Satunnaistaminen ositettiin diffuusiin suurisoluiseen B‑solulymfoomaan annettujen aiempien systeemisten hoitolinjojen lukumäärän perusteella (1 vs. ≥ 2) ja viimeisen systeemisen hoidon hoitotuloksen perusteella (uusiutunut vs. hoitoon reagoimaton).
Columvi + GemOx -ryhmässä potilaat saivat 1. hoitosyklin 1. päivänä esilääkityksenä obinututsumabia ja sen jälkeen step-up-hoito-ohjelman mukaisesti 1. hoitosyklin 8. päivänä 2,5 mg Columvi-valmistetta, 1. hoitosyklin 15. päivänä 10 mg Columvi-valmistetta ja 2. hoitosyklin 1. päivänä 30 mg Columvi-valmistetta. Potilaat jatkoivat hoitoa 30 mg:n Columvi-annoksella 3.–12. hoitosyklin 1. päivänä. Gemsitabiinia (1000 mg/m2) ja oksaliplatiinia (100 mg/m2) annettiin laskimoon 1. hoitosyklin 2. päivänä ja sen jälkeen seuraavien hoitosyklien 1. päivänä 8. hoitosykliin asti. Kummassakin hoitoryhmässä kunkin hoitosyklin pituus oli 21 päivää. Potilaiden saamien Columvi-hoitosyklien lukumäärän mediaani oli 11 (vaihteluväli 1–13 hoitosykliä); 64,5 % sai 8 tai useampia hoitosyklejä ja 44,8 % sai Columvi-hoitoa 12 hoitosykliä.
Lähtötilanteen demografiset ja sairauden ominaisuudet olivat seuraavat: iän mediaani 68 vuotta (vaihteluväli 20–88 vuotta), 62,8 % oli 65-vuotiaita tai sitä vanhempia ja 23,7 % oli 75-vuotiaita tai sitä vanhempia; 57,7 % oli miehiä; 42 % oli valkoihoisia, 50 % oli aasialaisia ja 1,1 % oli mustaihoisia tai afroamerikkalaisia; 5,8 % oli latinalaisamerikkalaisia; ja ECOG-toimintakykyluokka oli 0 (43,3 %), 1 (46,6 %) tai 2 (10,1 %). Suurin osa potilaista (62,8 %) oli saanut yhtä aiempaa systeemistä hoitolinjaa; 37,2 % potilaista oli saanut kahta tai useampaa aiempaa hoitolinjaa. Kaikki potilaat olivat saaneet aiemmin solunsalpaajahoitoa ja useimmat (98,5 %) olivat saaneet aiemmin hoitoa monoklonaalisella CD20-vasta-aineella; 7,7 % potilaista oli aiemmin saanut CAR-T-soluhoitoa ja 4,0 % potilaista oli saanut autologisen kantasolusiirron. Valtaosalla potilaista (66,8 %) oli hoitoon reagoimaton tauti, 55,8 %:lla potilaista oli ensilinjan hoitoon reagoimaton tauti ja 60,6 % potilaista tauti ei reagoinut viimeisimpään hoitoon. Yleisimpiä syitä siihen, miksi kantasolusiirtoa ei katsottu voitavan tehdä potilaalle, olivat ikä (42,3 %), potilaan kieltäytyminen suuriannoksisesta solunsalpaajahoidosta ja/tai siirteestä (34,7 %) ja riittämätön vaste salvage-hoitoon (9,9 %).
Tehoa mittaava ensisijainen tulosmuuttuja oli kokonaiselossaoloaika (OS). Ennalta määritellyn ensisijaisen analyysin ajankohtana havaittiin kokonaiselossaoloajan tilastollisesti merkitsevä piteneminen Columvi + GemOx -ryhmään satunnaistetuilla potilailla verrattuna R-GemOx-ryhmään satunnaistettuihin potilaisiin (riskitiheyksien suhde [HR] 0,59; 95 %:n luottamusväli: 0,40–0,89; p-arvo = 0,011). Kokonaiselossaoloajan mediaani oli R-GemOx-ryhmässä 9,0 kuukautta (95 %:n luottamusväli: 7,3– 14,4) ja Columvi + GemOx -ryhmässä sitä ei saavutettu (95 %:n luottamusväli: 13,8–NE (ei arvioitavissa). Riippumattoman arviointitoimikunnan (IRC) arvion mukaan myös etenemättömyysaika (PFS) piteni ja täydellinen vaste (CR) parani tilastollisesti merkitsevästi enemmän Columvi + GemOx -ryhmässä R-GemOx-ryhmään verrattuna. Etenemättömyysajan mediaani oli Columvi + GemOx -ryhmässä 12,1 kuukautta (95 %:n luottamusväli: 6,8–18,3) ja R‑GemOx-ryhmässä 3,3 kuukautta (95 %:n luottamusväli: 2,5–5,6) (HR 0,37; 95 %:n luottamusväli: 0,25–0,55; p-arvo < 0,001). Täydellisen vasteen saaneiden osuus oli Columvi + GemOx -ryhmässä 50,3 % ja R-GemOx-ryhmässä 22,0 %. Ero oli siis 28,3 % (p-arvo < 0,001).
10,5 kuukauden lisäseurantajakson jälkeen tehdyn päivitetyn analyysin kokonaiselossaoloaikaa, etenemättömyysaikaa ja täydellistä vastetta koskevat tulokset osoittavat edelleen, että hyöty Columvi + GemOx -hoidosta oli suurempi kuin R-GemOx-hoidosta. Keskeiset tulokset on esitetty taulukossa 9. Päivitettyyn analyysiin perustuva kokonaiselossaoloajan Kaplan–Meier-kuvaaja on esitetty kuvassa 1 ja etenemättömyysajan Kaplan–Meier-kuvaaja on kuvassa 2. Päivitetyn analyysin ajankohtana tehty eksploratiivinen alaryhmäanalyysi osoitti Euroopassa tutkimukseen mukaan otettujen potilaiden kokonaiselossaoloajan riskitiheyksien suhteeksi 1,09 (95 %:n luottamusväli: 0,54–2,18) ja etenemättömyysajan riskitiheyksien suhteeksi 0,84 (95 %:n luottamusväli: 0,44–1,59).
Taulukko 9. Teho uusiutunutta tai hoitoon reagoimatonta diffuusia suurisoluista B‑solulymfoomaa sairastavilla potilailla, jotka saivat Columvi-hoitoa yhdistelmänä gemsitabiinin ja oksaliplatiinin kanssa (ITT)
| Tehon päätetapahtumat | Päivitetty analyysi (havainnointiajan mediaani = 20,7 kuukautta) | |
| Columvi + GemOx N=183 | R-GemOx N=91 | |
| Kokonaiselossaoloaika | ||
| Kuolleiden määrä (%) | 80 (43,7) | 52 (57,1) |
| Mediaani (95 %:n luottamusväli), kuukautta | 25,5 (18,3–NE) | 12,9 (7,9–18,5) |
| HR (95 %:n luottamusväli) | 0,62 (0,43–0,88) | |
| Etenemättömyysaika – riippumattoman arviointitoimikunnan arvio | ||
| Potilaita, joilla tapahtumia, lkm (%) | 90 (49,2) | 54 (59,3) |
| Mediaani (95 %:n luottamusväli), kuukautta | 13,8 (8,7–20,5) | 3,6 (2,5–7,1) |
| HR (95 %:n luottamusväli) | 0,40 (0,28–0,57) | |
| Täydellinen vaste – riippumattoman arviointitoimikunnan arvio | ||
| Vasteen saaneita (%) | 107 (58,5) | 23 (25,3) |
| Vasteosuuden ero (%) (95 %:n luottamusväli) | 33,2 (20,9–45,5) | |
| Objektiivinen vasteprosentti – riippumattoman arviointitoimikunnan arvio | ||
| Vasteen saaneita (%) (täydellinen ja osittainen vaste) | 125 (68,3) | 37 (40,7) |
| Vasteosuuden ero (%) (95 %:n luottamusväli) | 27,7 (14,7–40,6) | |
HR = riskitiheyksien suhde (hazard ratio), NE = ei arvioitavissa (not estimable).
Kuva 1. Kaplan–Meierin kuvaaja kokonaiselossaoloajasta GO41944-tutkimuksessa (STARGLO, päivitetty analyysi, ITT)
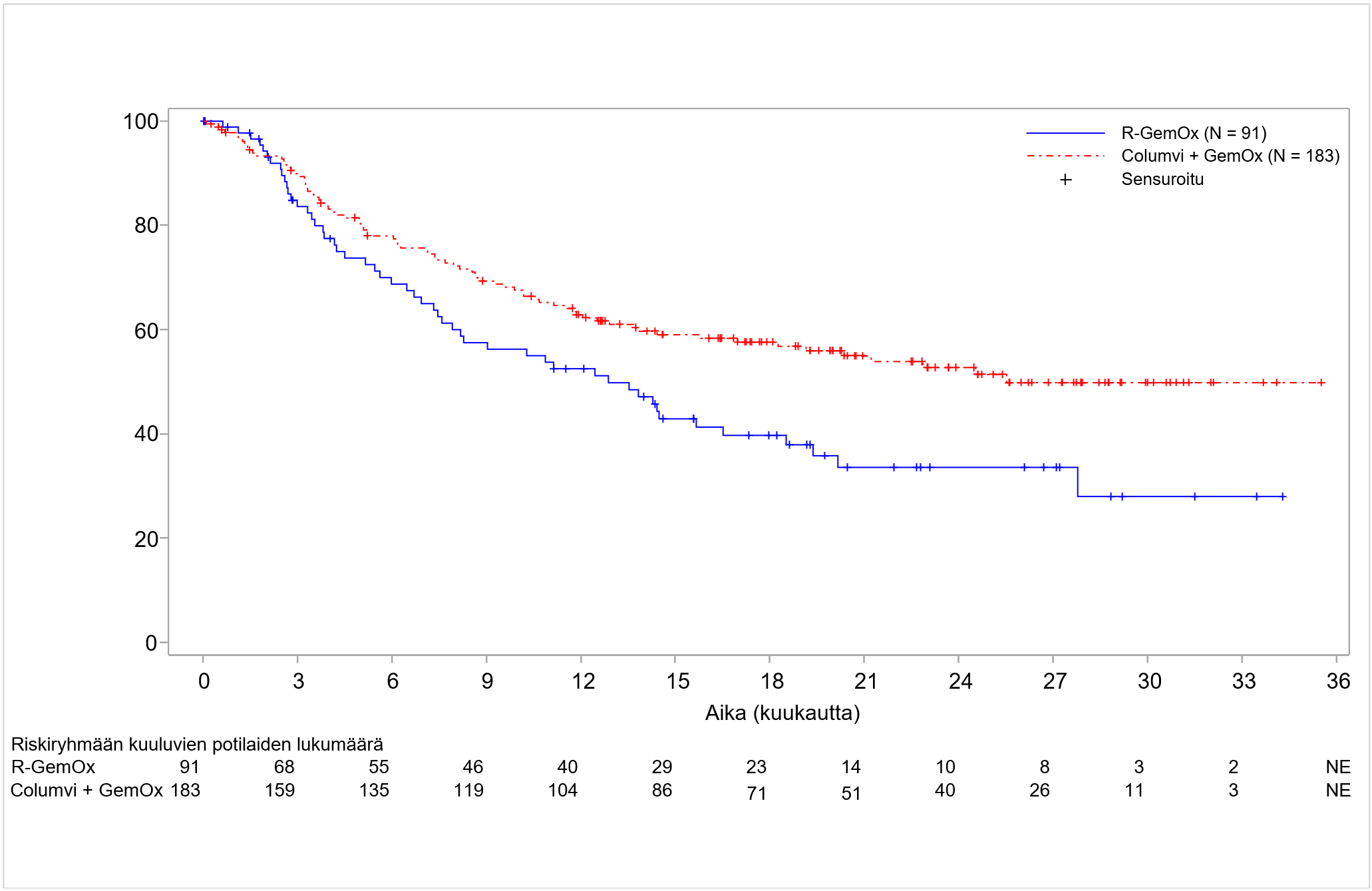
Kuva 2. Kaplan–Meierin kuvaaja riippumattoman arviointitoimikunnan (IRC) arvioimasta etenemättömyysajasta GO41944-tutkimuksessa (STARGLO, päivitetty analyysi; ITT)
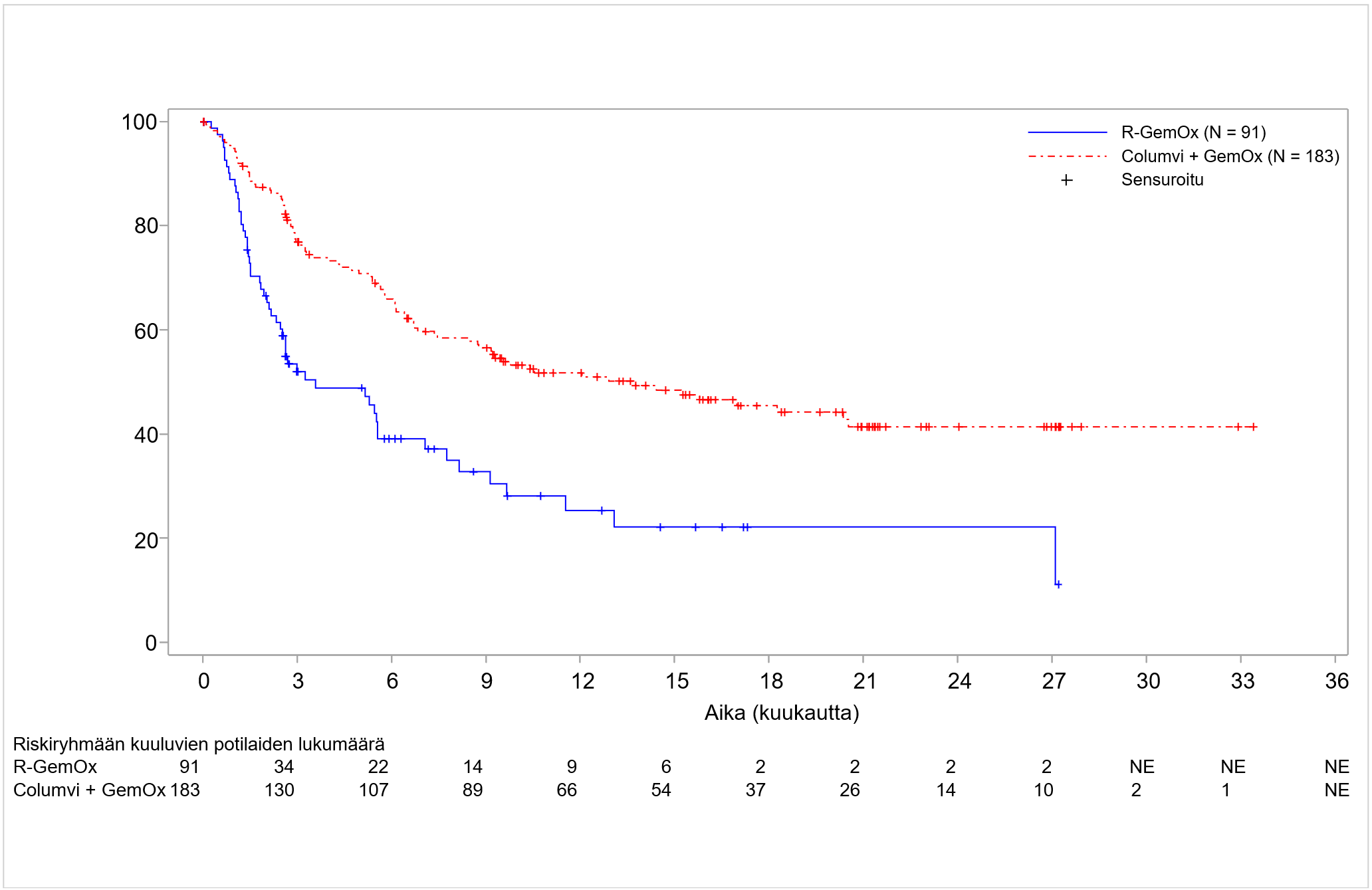
Immunogeenisuus
Tehdyissä tutkimuksissa mukana olleista 608 potilaasta vain 4 potilaalle (0,7 %), joilla ei hoidon alussa ollut glofitamabivasta-aineita, kehittyi glofitamabivasta-aineita hoidon jälkeen. Koska vain rajallisella määrällä potilailla oli glofitamabivasta-aineita, ei voida tehdä päätelmiä immunogeenisuuden mahdollisesta vaikutuksesta tehoon tai turvallisuuteen.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Columvi-valmisteen käytöstä kypsien B‑solukasvainten hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Tilamallittomat analyysit osoittavat, että glofitamabipitoisuus seerumissa saavuttaa huippunsa (Cmax) infuusion lopussa ja pienenee bieksponentiaalisesti. Glofitamabin farmakokinetiikka on lineaarinen ja tutkitulla annosvälillä (0,005–30 mg) suhteessa annokseen sekä riippumaton ajasta.
Imeytyminen
Columvi annetaan infuusiona laskimoon. Glofitamabin huippupitoisuus (Cmax) saavutetaan infuusion lopussa.
Jakautuminen
Laskimoon annon jälkeen jakautuminen keskustilaan oli 3,34 l, mikä on lähellä seerumin kokonaistilavuutta. Perifeerinen jakautumistilavuus oli 2,35 l.
Biotransformaatio
Glofitamabin metaboliaa ei ole tutkittu. Vasta-aineet poistuvat elimistöstä pääasiassa kataboloitumalla.
Eliminaatio
Glofitamabin pitoisuus-aikatietoja seerumissa kuvaa populaatiofarmakokineettinen kaksitilamalli ja sekä aikariippumaton puhdistuma että ajan suhteen muuttuva puhdistuma.
Aikariippumattomaksi puhdistumareitiksi arvioitiin 0,633 l/vrk, ja alkuvaiheen ajan suhteen muuttuvaksi puhdistumareitiksi 0,814 l/vrk, joka vähenee ajan kuluessa eksponentiaalisesti (Kdes ~ 1,5/vrk). Alkuvaiheen kokonaispuhdistuma-arvosta pelkkään aikariippumattomaan puhdistumaan tapahtuvan vähenemän puoliintumisajan estimaatiksi arvioitiin 0,471 vuorokautta.
Lineaarisen vaiheen efektiivinen puoliintumisaika (eli ajan suhteen muuttuvan puhdistuman vähennyttyä merkityksettömäksi) on populaatiofarmakokineettisen analyysin perusteella 7,92 vuorokautta (geometrinen keskiarvo, 95 %:n luottamusväli: 4,69–11,90).
Erityisryhmät
Iäkkäät
Glofitamabialtistuksessa ei populaatiofarmakokineettisen analyysin perusteella havaittu eroja vähintään 65-vuotiaiden ja alle 65-vuotiaiden välillä.
Munuaisten vajaatoiminta
Glofitamabin populaatiofarmakokineettinen analyysi osoitti, että kreatiniinipuhdistuma ei vaikuta glofitamabin farmakokinetiikkaan. Glofitamabin farmakokinetiikka oli lievää tai keskivaikeaa munuaisten vajaatoimintaa (CrCL 30 – < 90 ml/min) sairastavilla potilailla samankaltainen kuin potilailla, joiden munuaisten toiminta oli normaali. Columvi-valmistetta ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla.
Maksan vajaatoiminta
Populaatiofarmakokineettiset analyysit osoittivat, että lievä maksan vajaatoiminta ei vaikuta glofitamabin farmakokinetiikkaan. Glofitamabin farmakokinetiikka lievää maksan vajaatoimintaa (kokonaisbilirubiinipitoisuus > ULN – ≤ 1,5 ULN tai ASAT > ULN) sairastavilla potilailla oli samankaltainen kuin potilailla, joiden maksan toiminta oli normaali. Columvi-valmistetta ei ole tutkittu keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavilla potilailla.
Iän, sukupuolen ja painon vaikutukset
Glofitamabin farmakokinetiikassa ei havaittu kliinisesti merkittäviä ikään (21–90 vuotta), sukupuoleen ja painoon (31–148 kg) perustuvia eroja.
Prekliiniset tiedot turvallisuudesta
Glofitamabin karsinogeenisuuden ja mutageenisuuden selvittämiseksi ei ole tehty tutkimuksia.
Hedelmällisyys
Glofitamabin vaikutusten arvioimiseksi ei ole tehty hedelmällisyyttä koskevia eläinkokeita.
Lisääntymistoksisuus
Glofitamabin vaikutusten arvioimiseksi ei ole tehty lisääntymis- ja kehitystoksisuutta koskevia eläinkokeita. Teratogeenisuusriski on vähäinen, mikä perustuu vasta-aineiden vähäiseen siirtymiseen istukan kautta ensimmäisellä tiineyskolmanneksella, glofitamabin vaikutusmekanismiin (B‑soluvaje, kohderiippuvainen T‑soluaktivaatio ja sytokiinien vapautuminen), saatavissa oleviin glofitamabia koskeviin turvallisuustietoihin ja tietoihin muista CD20-vasta-aineista. Pitkittynyt B‑soluvaje voi johtaa lisääntyneeseen opportunististen infektioiden riskiin, mistä saattaa aiheutua keskenmeno. Myös Columvi-valmisteen antoon liittyvä ohimenevä sytokiinioireyhtymä voi olla sikiölle haitallinen (ks. kohta Raskaus ja imetys).
Systeeminen toksisuus
Jaavanmakakeilla tehdyssä tutkimuksessa eläimillä, joille ilmaantui ilman obinututsumabiesilääkitystä laskimoon annetun glofitamabikerta-annoksen (0,1 mg/kg) jälkeen vaikea-asteinen sytokiinioireyhtymä, oli maha-suolikanavan eroosioita sekä tulehdussoluinfiltraatteja pernassa ja maksan sinusoideissa ja satunnaisesti muissa elimissä. Nämä tulehdussoluinfiltraatit johtuivat todennäköisesti sytokiinien indusoimasta immuunisoluaktivaatiosta. Obinututsumabiesilääkitys vähensi glofitamabin indusoimaa sytokiinien vapautumista ja siihen liittyviä haittavaikutuksia poistamalla B-solut ääreisverestä ja imukudoksesta. Näin jaavanmakakeille voitiin antaa vähintään kymmenkertaisia glofitamabiannoksia (1 mg/kg), jolloin huippupitoisuus (Cmax) oli enimmillään 3,74‑kertainen verrattuna ihmiselle suositellusta 30 mg:n annoksesta aiheutuvaan huippupitoisuuteen (Cmax).
Kaikkien glofitamabiin liittyneiden havaintojen katsottiin olleen farmakologisvälitteisiä vaikutuksia ja korjautuvia. Yli 4 viikkoa kestäviä tutkimuksia ei tehty, sillä glofitamabi oli jaavanmakakeilla erittäin immunogeeninen ja johti altistuksen häviämiseen ja farmakologisen vaikutuksen häviämiseen.
Koska kaikki hoidettavat uusiutunutta tai hoitoon reagoimatonta diffuusia suurisoluista B‑solulymfoomaa sairastavat potilaat ovat altistuneet aiemmin CD20-vasta-ainehoidolle, valtaosalla heistä on ennen obinututsumabihoitoa todennäköisesti vain vähän kiertäviä B‑soluja aiemman CD20-vasta-ainehoidon jäännösvaikutuksena. Siksi eläinmalli ilman aiempaa rituksimabihoitoa (tai muuta CD20-vasta-ainehoitoa) ei välttämättä kuvasta täysin kliinistä tilannetta.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Histidiinihydrokloridimonohydraatti
Metioniini
Sakkaroosi
Polysorbaatti 20 (E432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
30 kuukautta.
Laskimoon annettavaa infuusiota varten laimennettu liuos
Käytönaikaiseksi kemialliseksi ja fysikaaliseksi säilyvyydeksi on osoitettu enintään 72 tuntia 2–8 °C:ssa ja 24 tuntia 30 °C:ssa, ja sen jälkeiseksi infuusion enimmäisajaksi 8 tuntia.
Laimennettu liuos pitää mikrobiologiselta kannalta käyttää välittömästi. Jos liuosta ei käytetä välittömästi, käytönaikaiset säilytysajat ja ‑olosuhteet ennen käyttöä ovat käyttäjän vastuulla eivätkä saisi tavallisesti ylittää 24:ää tuntia 2–8 °C:ssa, ellei valmistetta ole laimennettu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
COLUMVI infuusiokonsentraatti, liuosta varten
2,5 mg (L:ei) 1 kpl (2,5 ml (1 mg/ml)) (993,17 €)
10 mg (L:ei) 1 kpl (10 ml (1 mg/ml)) (3694,56 €)
PF-selosteen tieto
Columvi 2,5 mg infuusiokonsentraatti, liuosta varten
2,5 ml infuusiokonsentraattia, liuosta varten, 6 ml:n injektiopullossa (väritöntä tyypin I lasia), jossa on tulppa (butyylikumia).
Pakkauskoko 1 injektiopullo.
Columvi 10 mg infuusiokonsentraatti, liuosta varten
10 ml infuusiokonsentraattia, liuosta varten, 15 ml:n injektiopullossa (väritöntä tyypin I lasia), jossa on tulppa (butyylikumia).
Pakkauskoko 1 injektiopullo.
Valmisteen kuvaus:
Väritön, kirkas liuos, jonka pH on 5,5 ja osmolaliteetti on 270–350 mosm/kg.
Käyttö- ja käsittelyohjeet
Laimennettu Columvi-liuos voidaan antaa infuusiona laskimoon infuusiopussin (kaikki annokset) tai ruiskun (vain 2,5 mg:n annos) avulla.
Laimentamisohjeet
- Columvi ei sisällä säilytysainetta ja on tarkoitettu vain kertakäyttöön.
- Terveydenhuollon ammattilaisen on laimennettava Columvi-valmiste aseptista menetelmää noudattaen ennen valmisteen antamista laskimoon.
- Tarkista ennen antoa silmämääräisesti, ettei Columvi-liuoksessa ole hiukkasia eikä värimuutoksia. Columvi on väritön, kirkas liuos. Hävitä injektiopullo, jos liuos on sameaa tai jos liuoksessa on havaittavissa värimuutos tai näkyviä hiukkasia.
Infuusiopussin avulla laskimoon annettavan infuusion valmistelu
- Vedä infuusiopussista steriilin neulan ja ruiskun avulla tarvittava tilavuus 9 mg/ml (0,9 %) natriumkloridi-injektionestettä tai 4,5 mg/ml (0,45 %) natriumkloridi-injektionestettä taulukossa 10 kuvatulla tavalla ja hävitä ruiskuun vedetty injektioneste.
- Vedä injektiopullosta steriilin neulan ja ruiskun avulla aiottuun annokseen tarvittava tilavuus Columvi-konsentraattia ja laimenna se infuusiopussiin (ks. taulukko 10). Hävitä injektiopulloon käyttämättä jäävä osa.
- Laimennetun liuoksen lopullisen glofitamabipitoisuuden on oltava 0,1–0,6 mg/ml.
- Sekoita liuos kääntelemällä infuusiopussia ylösalaisin varoen, jotta vältetään liiallinen vaahtoaminen. Ei saa ravistaa.
- Tarkista infuusiopussi hiukkasten varalta. Hävitä se, jos havaitset hiukkasia.
- Infuusiopussin sisällön pitää olla huoneenlämpöistä (25 °C) ennen laskimoon annettavan infuusion aloittamista.
Taulukko 10. Columvi-valmisteen laimentaminen infuusiopussin avulla laskimoon annettavaa infuusiota varten
| Annettava Columvi-annos | Infuusiopussin koko | Vedettävä ja hävitettävä 9 mg/ml (0,9 %) tai 4,5 mg/ml (0,45 %) natriumkloridi-injektionesteen tilavuus | Lisättävä Columvi-konsentraattitilavuus |
| 2,5 mg | 50 ml | 27,5 ml | 2,5 ml |
| 100 ml | 77,5 ml | 2,5 ml | |
| 10 mg | 50 ml | 10 ml | 10 ml |
| 100 ml | 10 ml | 10 ml | |
| 30 mg | 50 ml | 30 ml | 30 ml |
| 100 ml | 30 ml | 30 ml |
Ruiskun avulla laskimoon annettavan infuusion valmistelu (vain 2,5 mg:n annos)
Käytä annoksen valmisteluun liittimen avulla kahden ruiskun menetelmää. Laimennetun liuoksen lopullinen tilavuus on 25 ml.
- Vedä infuusiopussista 22,5 ml 9 mg/ml (0,9 %) natriumkloridi-injektionestettä tai 4,5 mg/ml (0,45 %) natriumkloridi-injektionestettä sopivan kokoiseen (esim. 30 ml) ruiskuun.
- Vedä 2,5 ml Columvi-konsentraattia injektiopullosta steriilin neulan avulla toiseen ruiskuun. Hävitä injektiopulloon käyttämättä jäävä osa.
- Kiinnitä näihin kahteen ruiskuun liitin ja siirrä Columvi-konsentraatti ruiskuun, jossa on 9 mg/ml (0,9 %) natriumkloridi-injektionestettä tai 4,5 mg/ml (0,45 %) natriumkloridi-injektionestettä. Laimennetun liuoksen glofitamabipitoisuuden pitää olla 0,1 mg/ml.
- Irrota ruiskut toisistaan. Vedä laimennetun Columvi-liuoksen sisältävään ruiskuun ilmaa ja sulje se.
- Sekoita liuos kääntelemällä ruiskua ylösalaisin varovasti, jotta vältetään liiallinen vaahtoaminen. Ei saa ravistaa.
- Poista ruiskusta ilmakuplat ennen antoa.
Anto
Annetaan vain infuusiona laskimoon.
Ei saa antaa laskimoon paineella eikä boluksena.
Anna enintään 8 tunnin kestoisena infuusiona laskimoon valmistetta varten tarkoitetun infuusioletkun kautta infuusiopumpun tai ruiskupumpun avulla.
Kun Columvi-infuusiopussi tai ‑ruisku on tyhjentynyt, varmista, että koko Columvi-annos on annettu huuhtelemalla infuusioletku. Käytä huuhteluun infuusiopussia tai ruiskua, joka sisältää 9 mg/ml (0,9 %) natriumkloridi-injektioliuosta tai 4,5 mg/ml (0,45 %) natriumkloridi-injektioliuosta. Jatka infuusiota samalla nopeudella taulukon 2 mukaisesti.
Yhteensopimattomuudet
Columvi-valmisteen laimentamiseen saa käyttää vain 9 mg/ml (0,9 %) tai 4,5 mg/ml (0,45 %) natriumkloridi-injektionestettä, sillä muita liuoksia ei ole tutkittu.
9 mg/ml (0,9 %) natriumkloridi-injektionesteeseen laimennettu Columvi-valmiste on yhteensopiva infuusiopussien kanssa, jotka on valmistettu polyvinyylikloridista (PVC), polyeteenistä (PE), polypropeenista (PP) tai polyolefiinista. 4,5 mg/ml (0,45 %) natriumkloridi-injektionesteeseen laimennettu Columvi-valmiste on yhteensopiva polyvinyylikloridista (PVC) valmistettujen infuusiopussien kanssa.
9 mg/ml (0,9 %) tai 4,5 mg/ml (0,45 %) natriumkloridi-injektionesteeseen laimennettu Columvi-valmiste on yhteensopiva polypropeenista valmistettujen ruiskujen kanssa.
Yhteensopimattomuutta ei ole havaittu sellaisten infuusiovälineiden kanssa, joiden valmisteeseen kosketuksissa oleva pinta on polyuretaania (PUR), polyvinyylikloridia (PVC), polyeteeniä (PE), polybutadieenia (PBD), polyeetteriuretaania (PEUR), polykarbonaattia (PC), silikonia, polytetrafluorieteeniä (PTFE) tai akryylinitriilibutadieenistyreeniä (ABS), eikä letkunsisäisten (in-line) suodatinkalvojen kanssa, jotka on valmistettu polyeetterisulfonista (PES) tai polysulfonista. Letkunsisäisen suodatinkalvon käyttö on valinnaista.
Hävittäminen
Columvi-injektiopullo on tarkoitettu kertakäyttöön.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
COLUMVI infuusiokonsentraatti, liuosta varten
2,5 mg 1 kpl
10 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FX28
Valmisteyhteenvedon muuttamispäivämäärä
24.07.2025
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com