
SOTYKTU tabletti, kalvopäällysteinen 6 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 6 mg deukravasitinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 44 mg laktoosia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti)
Kliiniset tiedot
Käyttöaiheet
Läiskäpsoriaasi
Sotyktu on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon aikuisille, joille harkitaan systeemistä hoitoa.
Nivelpsoriaasi
Sotyktu yksin tai yhdessä metotreksaatin (MTX) kanssa on tarkoitettu aktiivisen nivelpsoriaasin (PsA) hoitoon aikuisille, joiden vaste aiempaan taudin kulkuun vaikuttavaan reumalääkkeeseen (DMARD-lääkkeeseen) on ollut riittämätön tai jotka eivät ole sietäneet tällaista hoitoa (ks. kohta Farmakodynamiikka).
Ehto
Hoito on aloitettava valmisteen käyttöaiheissa mainittujen sairauksien hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito on aloitettava Sotyktu-valmisteen käyttöaiheissa mainittujen sairauksien diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja valvonnassa.
Annostus
Suositeltu annos on 6 mg suun kautta kerran vuorokaudessa.
Jos potilaalla ei todeta näyttöä terapeuttisesta hyödystä 24 viikon jälkeen, on hoidon lopettamista harkittava. Potilaan hoitovaste on arvioitava säännöllisesti.
Erityisryhmät
Iäkkäät
Vähintään 65‑vuotiaiden iäkkäiden potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka). Kliinistä kokemusta vähintään 75-vuotiaista potilaista on vain hyvin vähän, joten deukravasitinibia on käytettävä varoen tälle potilasryhmälle.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on munuaisten vajaatoiminta, mukaan lukien dialyysihoitoinen loppuvaiheen munuaissairaus (ESRD) (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Deukravasitinibia ei suositella vaikeaa maksan vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Deukravasitinibin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Tabletit voi ottaa joko ruoan kanssa tai tyhjään mahaan. Tabletit on nieltävä kokonaisina, eikä niitä saa murskata, halkaista tai pureskella.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Kliinisesti merkittävät aktiiviset infektiot (esim. aktiivinen tuberkuloosi, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Infektiot
Deukravasitinibi voi suurentaa infektioiden riskiä (ks. kohta Haittavaikutukset).
Jos potilaalla on mikä tahansa kliinisesti merkittävä aktiivinen infektio, deukravasitinibihoitoa ei saa aloittaa, ennen kuin infektio on parantunut tai asianmukaisesti hoidettu (ks. kohta Vasta-aiheet). Varovaisuutta on noudatettava, kun deukravasitinibin käyttöä harkitaan potilaalle, jolla on krooninen infektio tai anamneesissa on jokin uusiutuva infektio.
Deukravasitinibihoitoa saavia potilaita on kehotettava kääntymään lääkärin puoleen, jos heillä ilmenee infektioon viittaavia merkkejä tai oireita. Jos potilaalle kehittyy kliinisesti merkittävä infektio tai infektion tavanomainen hoito ei tuota vastetta, potilaan tilannetta on seurattava huolellisesti eikä deukravasitinibia saa antaa, ennen kuin infektio on parantunut.
Hoitoa edeltävä tuberkuloosiseulonta
Ennen deukravasitinibihoidon aloittamista potilas on tutkittava tuberkuloosi-infektion varalta. Deukravasitinibia ei saa antaa, jos potilaalla on aktiivinen tuberkuloosi (ks. kohta Vasta-aiheet). Latentin tuberkuloosin hoito on aloitettava ennen deukravasitinibin antoa. Tuberkuloosilääkityksen käyttöä on harkittava ennen deukravasitinibihoidon aloittamista, jos potilaalla on anamneesissa latentti tai aktiivinen tuberkuloosi eikä riittävän hoitokuurin toteutumista pystytä vahvistamaan. Deukravasitinibia saavan potilaan vointia on seurattava aktiivisen tuberkuloosin merkkien ja oireiden varalta.
Maligniteetit
Maligniteetteja, myös lymfoomia ja ei‑melanoottisia ihosyöpiä, todettiin deukravasitinibilla tehdyissä kliinisissä tutkimuksissa.
Ei tiedetä, voiko tyrosiinikinaasi 2:n (TYK2) estoon liittyä samoja haittavaikutuksia kuin januskinaasin (JAK) estoon. Laajassa satunnaistetussa vaikuttavalla aineella kontrolloidussa tutkimuksessa tutkittiin JAK-estäjää vähintään 50‑vuotiailla nivelreumapotilailla, joilla oli vähintään yksi kardiovaskulaarinen lisäriskitekijä. Tutkimuksessa maligniteettien, etenkin keuhkosyövän, lymfooman ja ei‑melanoottisen ihosyövän, määrät olivat korkeampia JAK-estäjää saaneilla verrattuna tuumorinekroositekijän (TNF) estäjiä saaneisiin.
Deukravasitinibialtistuksen ja maligniteettien kehittymisen välisen mahdollisen yhteyden arvioimiseksi on saatavilla vain vähän kliinisiä tietoja. Pitkäaikaisturvallisuuden arviointeja on käynnissä. Deukravasitinibihoidon riskejä ja hyötyjä on harkittava ennen hoidon aloittamista.
Vakavat sydän- ja verisuoniperäiset haittatapahtumat (MACE), syvä laskimotukos (SLT) ja keuhkoembolia (KE)
Ei tiedetä, voiko TYK2:n estoon liittyä samoja haittavaikutuksia kuin JAK:n estoon. Laajassa satunnaistetussa vaikuttavalla aineella kontrolloidussa tutkimuksessa tutkittiin JAK-estäjää vähintään 50‑vuotiailla nivelreumapotilailla, joilla oli vähintään yksi kardiovaskulaarinen lisäriskitekijä. Tutkimuksessa MACEn (määritelmä: sydän- ja verisuoniperäinen kuolema, ei‑fataali sydäninfarkti ja ei‑fataali aivohalvaus) määrä ja laskimotromboembolian (mukaan lukien SLT ja KE) annosriippuvaiset määrät olivat korkeampia JAK-estäjää saaneilla verrattuna TNF-estäjiä saaneisiin.
MACEn, SLT:n ja KE:n riskin suurenemista ei todettu deukravasitinibilla tehdyissä kliinisissä tutkimuksissa. Deukravasitinibin pitkän aikavälin turvallisuusarvioinnit ovat käynnissä. Deukravasitinibihoidon riskit ja hyödyt on huomioitava ennen hoidon aloittamista.
Rokotukset
Potilaiden kaikkien iänmukaisten rokotusten saattamista ajan tasalle nykyisten rokotussuositusten mukaisesti on harkittava ennen deukravasitinibihoidon aloittamista. Eläviä heikennettyjä taudinaiheuttajia sisältävien rokotteiden käyttöä deukravasitinibihoitoa saavilla potilailla on vältettävä. Vastetta eläviä heikennettyjä taudinaiheuttajia sisältäville tai muille kuin eläviä heikennettyjä taudinaiheuttajia sisältäville rokotteille ei ole arvioitu.
Apuaineet
Laktoosi
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi‑galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Kliinisten tutkimusten mukaan deukravasitinibilla ei ole kliinisesti merkittäviä yhteisvaikutuksia seuraavien muiden samanaikaisesti annettujen lääkevalmisteiden kanssa, joten annosmuutoksia ei tarvita.
Deukravasitinibin vaikutus muihin lääkevalmisteisiin
Deukravasitinibi ei vaikuta merkittävässä määrin rosuvastatiinin (BCRP:n ja OATP:n substraatti), metotreksaatin (BCRP:n ja munuaisten kuljettajaproteiinien substraatti), mykofenolaattimofetiilin (MMF) (CES1:n ja CES2:n substraatti), metformiinin (munuaisten MATE2K‑riippuvaisen ja maksan OCT1‑riippuvaisen lääkeaineiden kuljettajaproteiinin substraatti) tai suun kautta otettavien ehkäisyvalmisteiden (noretindroniasetaatti ja etinyyliestradioli) altistukseen plasmassa.
Muiden lääkevalmisteiden vaikutukset deukravasitinibiin
Lääkevalmisteet, jotka ovat CYP-entsyymien tai kuljettajaproteiinien estäjiä tai indusoreita, kuten siklosporiini (sekä P‑gp:n että rintasyövän resistenssiproteiinin [BCRP] estäjä), fluvoksamiini (voimakas CYP 1A2:n estäjä), ritonaviiri (kohtalainen CYP 1A2:n indusori), diflunisaali (UGT 1A9:n estäjä), pyrimetamiini (OCT1:n estäjä), famotidiini (H2‑reseptorin salpaaja) tai rabepratsoli (protonipumpun estäjä), eivät vaikuta merkittävässä määrin deukravasitinibialtistukseen plasmassa (ks. kohta Farmakokinetiikka).
Raskaus ja imetys
Raskaus
On vain vähän tietoja deukravasitinibin käytöstä raskaana olevilla naisilla. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi deukravasitinibin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Deukravasitinibi ja sen metaboliitti (BMT‑153261) erittyvät äidinmaitoon (ks. kohta Farmakokinetiikka). Arvioitu suhteellinen imeväisen vuorokausiannos vastaa 12 %:a äidin annoksesta.
Deukravasitinibin vaikutuksia imetettävään vauvaan ei tunneta.
On päätettävä, lopetetaanko imetys vai pidättäydytäänkö deukravasitinibihoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Deukravasitinibin vaikutusta ihmisen hedelmällisyyteen ei ole arvioitu. Eläinkokeissa ei ole havaittu suoria tai epäsuoria hedelmällisyyteen kohdistuvia haittavaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Deukravasitinibilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettu haittavaikutus oli ylähengitystieinfektio (18,9 % läiskäpsoriaasitutkimuksissa ja 15,1 % nivelpsoriaasitutkimuksissa). Deukravasitinibilla hoidetuilla nivelpsoriaasipotilailla havaittu turvallisuusprofiili oli yleisesti ottaen yhdenmukainen deukravasitinibilla hoidettujen läiskäpsoriaasipotilaiden turvallisuusprofiilin kanssa. Deukravasitinibin turvallisuusprofiili oli pidemmällä aikavälillä samankaltainen ja aiempien kokemusten mukainen.
Haittavaikutustaulukko
Kliinisiin tutkimuksiin perustuvat deukravasitinibin haittavaikutukset (ks. taulukko 1) on esitetty MedDRA-elinjärjestelmäluokituksen ja esiintyvyyden mukaan seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1: Luettelo haittavaikutuksista
| Elinjärjestelmäluokka | Esiintyvyys | Haittavaikutus |
| Infektiot | Hyvin yleinen | Ylähengitystieinfektiota |
| Yleinen | Keuhkokuume Herpes simplex -infektiotb Keuhkoputkitulehdus | |
| Melko harvinainen | Herpes zoster | |
| Ruoansulatuselimistö | Yleinen | Suun haavaumatc |
| Iho ja ihonalainen kudos | Yleinen | Aknen kaltainen ihottumad Follikuliitti |
| Tutkimukset | Yleinen | Veren kreatiinikinaasipitoisuuden suureneminen |
a Ylähengitystieinfektiot kattavat nämä: nasofaryngiitti, ylähengitystieinfektio, viruksen aiheuttama ylähengitystieinfektio, faryngiitti, sinuiitti, akuutti sinuiitti, riniitti, tonsilliitti, nielupaise, laryngiitti, trakeiitti, nielu- ja nielurisatulehdus ja rinotrakeiitti. b Herpes simplex ‑infektiot kattavat nämä: suun herpes, herpes simplex, sukuelinherpes, silmäherpes ja herpesviruksen aiheuttama infektio. c Suun haavaumat kattavat nämä: aftat, suun haavaumat, kielen haavaumat ja stomatiitti. d Aknen kaltainen ihottuma kattaa nämä: akne, aknetyyppinen dermatiitti, ihottuma, ruusufinni, märkärakkulat, pustulaarinen ihottuma, papulaarinen ihottuma ja näppylät. | ||
Valikoitujen haittavaikutusten kuvaukset
Infektiot
POETYK PSO‑1‑ ja POETYK PSO‑2 ‑tutkimuksissa (ks. kohta Farmakodynamiikka) infektioita esiintyi 29,1 %:lla deukravasitinibiryhmän potilaista (116,0 tapahtumaa 100:aa henkilövuotta kohden) ja 21,5 %:lla lumeryhmän potilaista (83,7 tapahtumaa 100:aa henkilövuotta kohden) ensimmäisten 16 viikon aikana. Suurin osa infektioista ei ollut vakavia vaan vaikeusasteeltaan lieviä tai keskivaikeita eivätkä johtaneet deukravasitinibihoidon lopettamiseen. Vakavien infektioiden ilmaantuvuus oli deukravasitinibiryhmässä 0,6 % (2,0 tapahtumaa 100:aa henkilövuotta kohden) ja lumeryhmässä 0,5 % (1,6 tapahtumaa 100:aa henkilövuotta kohden).
Infektioiden esiintymistiheys deukravasitinibiryhmässä ei suurentunut viikon 52 loppuun mennessä (95,4 tapahtumaa 100:aa henkilövuotta kohden). Vakavien infektioiden esiintymistiheys deukravasitinibiryhmässä ei suurentunut viikon 52 loppuun mennessä (1,7 tapahtumaa 100:aa henkilövuotta kohden).
Infektioiden, mukaan lukien vakavien infektioiden, esiintymistiheyden todettiin olevan yleisesti ottaen deukravasitinibilla hoidetuilla nivelpsoriaasipotilailla yhdenmukainen läiskäpsoriaasipotilailla todetun infektioiden esiintymistiheyden kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen
hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Deukravasitinibia on annettu terveille tutkittaville ilman annosta rajoittavaa toksisuutta enintään 40 mg:n kerta-annoksina (> 6‑kertainen annos verrattuna ihmisille suositeltuun annokseen 6 mg/vrk) ja useampina annoksina, jotka ovat olleet yhteensä enintään 24 mg/vrk (12 mg kahdesti vuorokaudessa) 14 vuorokauden ajan.
Yliannostustapauksessa suositellaan potilaan seurantaa haittavaikutusten merkkien tai oireiden varalta ja asiaankuuluvan oireenmukaisen hoidon aloittamista välittömästi. Dialyysi ei poista deukravasitinibia systeemisestä verenkierrosta merkittävästi (ks. kohta Farmakokinetiikka).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, ATC-koodi: L04AF07
Vaikutusmekanismi
Deukravasitinibi on TYK2‑entsyymin (joka kuuluu JAK-perheeseen) selektiivinen estäjä. Deukravasitinibi sitoutuu TYK2:n säätelyosaan ja vakauttaa entsyymin säätelyosan ja katalyyttisen osan välisen inhibitorisen vuorovaikutuksen. Tätä seuraa TYK2:n reseptorivälitteisen aktivaation allosteerinen inhibitio ja TYK2:n alavirran toimintojen estyminen. TYK2 toimii interleukiini‑23:n (IL‑23), interleukiini‑12:n (IL‑12) ja tyypin I interferonien (IFN) signaalinvälittäjänä. Ne ovat elimistön omia sytokiineja, jotka osallistuvat tulehdus- ja immuunivasteisiin. Deukravasitinibi estää proinflammatoristen sytokiinien ja kemokiinien vapautumisen.
Farmakodynaamiset vaikutukset
Deukravasitinibi vähensi psoriaasiin liittyvää geeniekspressiota psoriaasipotilaiden ihossa. Vähenemistä tapahtui IL‑23‑reitin ja tyypin I IFN-reitin säätelemissä geeneissä. Deukravasitinibi pienensi IL‑17A:n, IL‑19:n ja β‑defensiinin määrää psoriaasi- ja nivelpsoriaasipotilaiden verenkierrossa, kun hoitoa oli annettu kerran vuorokaudessa 16 viikon ajan. Nivelpsoriaasipotilailla todettiin myös valikoitujen lisäbiomarkkereiden vähenemistä verenkierrossa, mukaan lukien C‑reaktiivinen proteiini (CRP), matriksimetalloproteinaasi‑3 (MMP3), matriksimetalloproteinaasi‑1 (MMP1), tyypin 1 kollageenin hajoamistuote (C1M) ja TNF‑alfa.
Kliininen teho ja turvallisuus
Läiskäpsoriaasi
Deukravasitinibin tehoa ja turvallisuutta arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumelääkkeellä ja apremilastilla kontrolloidussa kliinisessä monikeskustutkimuksessa (POETYK PSO‑1 ja POETYK PSO‑2) vähintään 18‑vuotiailla potilailla, joilla oli keskivaikea tai vaikea läiskäpsoriaasi ja jotka soveltuivat systeemiselle hoidolle tai valohoidolle. Potilaiden ihottuma peitti ≥ 10 % kehon pinta-alasta (BSA, Body Surface Area), psoriaasin vaikeusastetta kuvaava PASI (Psoriasis Area and Severity Index) ‑pistemäärä oli ≥ 12, ja lääkärin staattinen kokonaisarvio psoriaasin vaikeusasteesta (static Physician's Global Assessment, sPGA) oli ≥ 3 (keskivaikea tai vaikea) 5‑portaisella asteikolla.
POETYK PSO‑1‑ ja POETYK PSO‑2 ‑tutkimuksissa oli yhteensä 1 686 potilasta, joista 843 satunnaistettiin saamaan deukravasitinibia 6 mg kerran vuorokaudessa, 422 satunnaistettiin saamaan apremilastia 30 mg kahdesti vuorokaudessa ja 421 satunnaistettiin saamaan lumelääkettä.
Molemmissa tutkimuksissa lumelääkettä saaneet potilaat siirtyivät viikolla 16 deukravasitinibihoitoon, jota jatkettiin enintään viikolle 52 asti. Apremilastihoitoon satunnaistetut potilaat, jotka eivät saavuttaneet PASI 50 ‑vastetta (POETYK PSO‑1) tai PASI 75 ‑vastetta (POETYK PSO‑2) viikolla 24, siirtyivät deukravasitinibihoitoon, jota jatkettiin enintään viikolle 52 asti. POETYK PSO‑1 ‑tutkimuksessa deukravasitinibihoitoon satunnaistettujen potilaiden hoitoa jatkettiin enintään viikolle 52 asti. POETYK PSO‑2 ‑tutkimuksessa ne deukravasitinibihoitoa saaneet potilaat, jotka saavuttivat PASI 75 ‑vasteen viikolla 24, satunnaistettiin uudelleen suhteessa 1:1 joko jatkamaan deukravasitinibihoitoa (ylläpito) tai siirtymään lumehoitoon (lopetus).
Taudin ominaisuudet lähtötilanteessa olivat samankaltaisia molempien tutkimusten potilailla: suurin osa potilaista oli miehiä (67 %), potilaiden keskimääräinen ikä oli noin 47 vuotta, ja suurin osa potilaista oli 40–64‑vuotiaita. Potilaista 10 % oli ≥ 65‑vuotiaita. PASI-kokonaispisteiden mediaani oli 18,7, ja BSA:n mediaani oli 20 %. Lähtötilanteen sPGA-pistemäärä oli 3 (keskivaikea) 79,8 %:lla potilaista ja 4 (vaikea) 20,2 %:lla potilaista. Ihosairauden vaikutusta elämänlaatuun arvioivan kyselyn (Dermatology Life Quality Index, DLQI) pistemäärän mediaani oli 11. Tutkimukseen osallistuneista potilaista 18,4 %:lla oli anamneesissa nivelpsoriaasi.
Molemmissa tutkimuksissa 40 % potilaista oli saanut aiemmin valohoitoa, 42,4 % ei ollut saanut aiemmin mitään systeemistä hoitoa (mukaan lukien biologiset ja/tai muut kuin biologiset hoidot), 41 % oli saanut aiemmin jotakin muuta kuin biologista systeemistä hoitoa, ja 34,8 % oli saanut aiemmin jotakin biologista hoitoa (16,1 % oli saanut TNF:n estäjää, 4,9 % oli saanut IL‑12/23:n estäjää, 16,6 % oli saanut IL‑17:n estäjää ja 4,4 % oli saanut IL‑23:n estäjää).
Kahden tutkimuksen yhdistetyt ensisijaiset päätetapahtumat olivat niiden potilaiden osuudet, jotka saavuttivat 1) vähintään 75 %:n paraneman PASI-pistemäärässä (PASI 75) lähtötilanteeseen nähden ja 2) sPGA-tuloksen ”ei ihomuutoksia” tai ”ei juuri lainkaan ihomuutoksia” (0 tai 1) viikolla 16 lumelääkkeeseen verrattuna.
POETYK PSO‑1 -tutkimuksessa PASI 75 -vasteen saavutti 58,4 % deukravasitinibia saaneista potilaista, 35,1 % apremilastia saaneista potilaista ja 12,7 % lumelääkettä saaneista potilaista viikolla 16. Lääkärin staattisen kokonaisarvion (sPGA) tuloksen ”ei ihomuutoksia” tai ”ei juuri lainkaan ihomuutoksia” viikolla 16 saavutti 53,6 % deukravasitinibia saaneista potilaista, 32,1 % apremilastia saaneista potilaista ja 7,2 % lumelääkettä saaneista potilaista. Deukravasitinibin paremmuus lumelääkkeeseen nähden osoitettiin näiden yhdistettyjen ensisijaisten päätetapahtumien osalta. Tulokset olivat yhdenmukaisia POETYK PSO‑2 -tutkimuksessa.
Taulukossa 2 esitetään yhdistettyjen ensisijaisten päätetapahtumien ja muiden päätetapahtumien tärkeimmät tehoa koskevat tulokset.
Taulukko 2:Tärkeimmät tehoa koskevat tulokset läiskäpsoriaasia sairastavilla aikuisilla
| POETYK PSO‑1 | POETYK PSO‑2 | |||||
| Päätetapahtuma | Deukravasitinibi (N = 332) n (%) | Apremilasti (N = 168) n (%) | Lumelääke (N = 166) n (%) | Deukravasitinibi (N = 511) n (%) | Apremilasti (N = 254) n (%) | Lumelääke (N = 255) n (%) |
| sPGA 0/1 | ||||||
| Viikko 16 | 178 (53,6) | 54 (32,1)d | 12 (7,2)a,d | 253 (49,5) | 86 (33,9)d | 22 (8,6)a,d |
| Viikko 24 | 195 (58,7) | 52 (31,0)d | - | 251 (49,8)b | 75 (29,5)d | - |
| sPGA 0 | ||||||
| Viikko 16 | 58 (17,5) | 8 (4,8)d | 1 (0,6)d | 80 (15,7) | 16 (6,3)e | 3 (1,2)d |
| PASI 75 | ||||||
| Viikko 16 | 194 (58,4) | 59 (35,1)d | 21 (12,7)a,d | 271 (53,0) | 101 (39,8)e | 24 (9,4)a,d |
| Viikko 24 | 230 (69,3) | 64 (38,1)d | - | 296 (58,7)b | 96 (37,8)d | - |
| PASI 90 | ||||||
| Viikko 16 | 118 (35,5) | 33 (19,6)e | 7 (4,2)d | 138 (27,0) | 46 (18,1)f | 7 (2,7)d |
| Viikko 24 | 140 (42,2) | 37 (22,0)d | - | 164 (32,5)b | 50 (19,7)d | - |
| PASI 100 | ||||||
| Viikko 16 | 47 (14,2) | 5 (3,0)d | 1 (0,6)d | 52 (10,2) | 11 (4,3)f | 3 (1,2)d |
| Päänahalle spesifinen PGA 0/1c | (N = 209) | (N = 110) | (N = 121) | (N = 305) | (N = 166) | (N = 173) |
| Viikko 16 | 147 (70,3) | 43 (39,1)d | 21 (17,4)d | 182 (59,7) | 61 (36,7)d | 30 (17,3)d |
Hoitoon vastaamattomien imputointia (non-responder imputation, NRI) käytettiin, eli potilaat, jotka keskeyttivät hoidon tai tutkimuksen ennen päätetapahtumaa tai joilta puuttui tietoja, laskettiin hoitoon vastaamattomiksi. a Yhdistetty ensisijainen päätetapahtuma, jossa deukravasitinibia verrattiin lumelääkkeeseen b N = 504, jossa otettiin huomioon COVID‑19-pandemian takia väliin jääneet arvioinnit c Sisältää potilaat, joiden päänahalle spesifinen PGA-pistemäärä oli lähtötilanteessa ≥ 3 d p ≤ 0,0001 deukravasitinibin ja lumelääkkeen tai deukravasitinibin ja apremilastin vertailussa e p < 0,001 deukravasitinibin ja apremilastin vertailussa f p < 0,01 deukravasitinibin ja apremilastin vertailussa | ||||||
Deukravasitinibivasteessa ei todettu eroja alaryhmissä, jotka perustuivat ikään, sukupuoleen, rotuun, painoon, sairauden kestoon, sairauden vaikeusasteeseen lähtötilanteessa ja aiempaan hoitoon biologisilla tai muilla kuin biologisilla lääkkeillä.
Vaste ajan kuluessa
Deukravasitinibin teho ilmeni nopeasti, ja PASI 75 ‑maksimivaste saavutettiin viikkoon 24 mennessä (POETYK PSO‑1 ja PSO‑2) ja se säilyi viikon 52 loppuun asti (POETYK PSO‑1) (ks. kuva 1).
Kuva 1: PASI 75 ‑vaste (NRI) viikon 52 loppuun asti käynneittäin POETYK PSO‑1 ‑tutkimuksessa
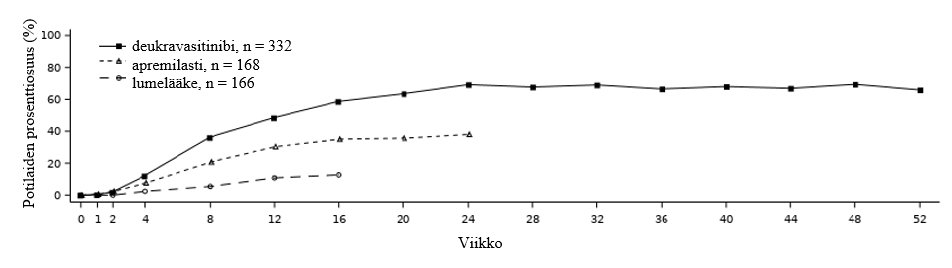
Vasteen säilyminen ja pysyvyys
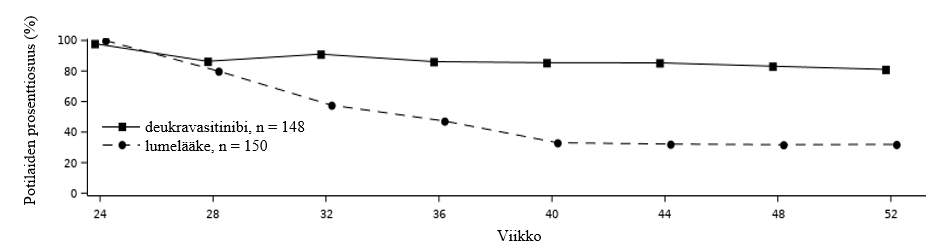
POETYK PSO‑2 ‑tutkimuksessa arvioitiin vasteen säilymistä ja pysyvyyttä siten, että alun perin deukravasitinibihoitoon satunnaistetut ja PASI 75 ‑vasteen viikolla 24 saavuttaneet potilaat satunnaistettiin uudelleen joko jatkamaan deukravasitinibihoitoa tai siirtymään lumehoitoon. Niillä hoitovasteen viikolla 24 saavuttaneilla potilailla, jotka satunnaistettiin uudelleen lumehoitoon, PASI 75 ‑vaste menetettiin noin 12 viikossa (mediaani). Kuvassa 2 on molempien hoitoryhmien PASI 75 ‑vasteet viikoilla 24–52.
Kuva 2: PASI 75 ‑vaste (NRI) uudelleensatunnaistamisen jälkeen viikolla 24 POETYK PSO‑2 ‑tutkimuksessa
Potilaiden raportoimat tulokset
Deukravasitinibihoitoa saaneilla potilailla havaittiin merkitsevästi suurempia parannuksia terveyteen liittyvässä elämänlaadussa, jota mitattiin ihosairauden vaikutusta elämänlaatuun arvioivalla kyselyllä (DLQI) ja potilaiden raportoimina ja psoriaasin oirepäiväkirjan (Psoriasis Symptoms and Signs Diary, PSSD) avulla mitattuina psoriaasin oireina (kutina, kipu, polttelu, kirvely ja ihon kireys) ja merkkeinä (kuiva iho, ihon halkeilu, hilseily, kuoriutuminen tai kesiminen, punoitus ja verenvuoto), kun tuloksia verrattiin lumelääkkeeseen viikolla 16 ja apremilastiin viikolla 16 ja viikolla 24. Näiden vasteiden paranema potilailla, jotka saivat jatkuvaa deukravasitinibihoitoa, säilyi viikon 52 loppuun asti POETYK PSO‑1 ‑tutkimuksessa.
Taulukko 3:Potilaiden raportoimat tulokset POETYK PSO‑1‑ ja POETYK PSO‑2 ‑tutkimuksissa
| POETYK PSO‑1 | POETYK PSO‑2 | |||||
| Deukravasitinibi | Apremilasti | Lumelääke | Deukravasitinibi | Apremilasti | Lumelääke | |
DLQI Potilaat, jotka saavuttivat tuloksen 0 tai 1 (NRI)* | N = 322 | N = 161 | N = 160 | N = 495 | N = 247 | N = 246 |
| Viikko 16, n (%) | 132 (41,0) | 46 (28,6)a | 17 (10,6)b | 186 (37,6) | 57 (23,1)b | 24 (9,8)b |
| Viikko 24, n (%) | 155 (48,1) | 39 (24,2)b | - | 205 (41,4) | 53 (21,5)b | - |
PSSD-oirepisteet Muutos lähtötilanteesta (mBOCF)** | N = 306 | N = 158 | N = 151 | N = 466 | N = 233 | N = 239 |
| Viikko 16, keskiarvo (keskivirhe) | -26,7 (1,8) | -17,8 (2,2)b | -3,6 (2,1)b | -28,3 (1,1) | -21,1 (1,4)b | -4,7 (1,4)b |
| Viikko 24, keskiarvo (keskivirhe) | -31,9 (2,0) | -20,7 (2,4)b | - | -29,1 (1,1) | -21,4 (1,5)b | - |
PSSD-merkkipisteet Muutos lähtötilanteesta (mBOCF)* | N = 306 | N = 158 | N = 151 | N = 466 | N = 233 | N = 239 |
| Viikko 16, keskiarvo (keskivirhe) | -28,9 (1,8) | -20,0 (2,2)b | -5,3 (2,1)a | -31,9 (1) | -23,8 (1,4)b | -7,1 (1,4)b |
| Viikko 24, keskiarvo (keskivirhe) | -33,8 (2,0) | -22,5 (2,4)b | - | -32,4 (1,1) | -24,2 (1,5)b | - |
* Potilaat, joiden lähtötilanteen pistemäärä oli ≥ 2 ** Korjattu keskimääräinen muutos; mBOCF – modified baseline observation carried forward; keskivirhe (SE) a p < 0,01 deukravasitinibin ja lumelääkkeen tai deukravasitinibin ja apremilastin vertailussa b p < 0,0001 deukravasitinibin ja lumelääkkeen tai deukravasitinibin ja apremilastin vertailussa | ||||||
Nivelpsoriaasi
Deukravasitinibin tehoa ja turvallisuutta arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa monikeskustutkimuksessa (POETYK PsA‑1 ja POETYK PsA‑2) vähintään 18‑vuotiailla potilailla, joilla oli aktiivinen nivelpsoriaasi (PsA) (≥ 3 turvonnutta niveltä, ≥ 3 aristavaa niveltä, C‑reaktiivinen proteiini [CRP] ≥ 3 mg/l) ja joko aktiivinen tai anamneesissa oleva läiskäpsoriaasi. Lähtötilanteessa molempiin tutkimuksiin osallistuneet potilaat olivat saaneet nivelpsoriaasidiagnoosin vähintään 3 kuukautta aikaisemmin ja täyttivät seulonnassa CASPAR (Classification criteria for Psoriatic Arthritis) ‑luokittelukriteerit. POETYK PsA‑1 ‑tutkimuksessa potilailla oli myös vähintään yksi käsien ja/tai jalkaterien röntgenkuvassa todettu luueroosio.
Tehoa arvioitiin PsA‑1‑tutkimuksessa (n = 670) ja PsA‑2‑tutkimuksessa (n = 624) yhteensä 1 294 potilaalla enintään viikolle 16 asti. PsA‑1‑tutkimuksessa potilaat satunnaistettiin saamaan joko deukravasitinibia 6 mg kerran vuorokaudessa tai lumelääkettä 16 viikon ajan. Viikolla 16 alun perin lumelääkeryhmään satunnaistetut potilaat siirtyivät saamaan deukravasitinibia 6 mg kerran vuorokaudessa. Potilaita seurattiin enintään viikolle 52 asti. PsA‑2‑tutkimuksessa potilaat satunnaistettiin saamaan joko deukravasitinibia 6 mg kerran vuorokaudessa, lumelääkettä tai apremilastia 30 mg kahdesti vuorokaudessa (turvallisuuden verrokkiryhmä). Tehon lumelääkekontrolloitu arviointivaihe kesti 16 viikkoa. Viikolla 16 PsA‑2‑tutkimuksessa alun perin lumelääkeryhmään satunnaistetut potilaat siirtyivät saamaan deukravasitinibia 6 mg kerran vuorokaudessa ja deukravasitinibi- ja apremilastiryhmiin satunnaistetut potilaat jatkoivat heille määritettyä hoitoa viikkoon 52 asti.
Tutkimusten yhdistettyjen tietojen mukaan sairauden mediaanikesto lähtötilanteessa oli 4 vuotta ja valtaosalla potilaista oli todettu moniniveltulehdus (75,7 %), jota seurasi oligoartriitti (13,5 %). Potilaista 8,4 %:lla sairaus ilmeni pääosin distaalisissa interfalangeaalinivelissä, ja 15,5 %:lla oli sekä perifeerinen että psoriaasiin liittyvä spondyloartriitti. Daktyliittia esiintyi 30,8 %:lla ja entesiittiä 61,8 %:lla potilaista. Potilaista 49,1 %:lla psoriaasiin liittyvä ihottuma peitti ≥ 3 % kehon pinta-alasta (BSA, Body Surface Area) ja lääkärin staattinen kokonaisarvio psoriaasin vaikeusasteesta (static Physician's Global Assessment, sPGA) oli vähintään 2.
Molemmissa tutkimuksissa suurin osa potilaista ei ollut saanut aiemmin biologista hoitoa ja kaikkien vaste tulehduskipulääkkeisiin (NSAID), perinteisiin synteettisiin reumalääkkeisiin (csDMARD) tai apremilastiin oli joko ollut riittämätön tai vaste oli menetetty tai potilas ei ollut sietänyt hoitoa. Lisäksi PsA‑2‑tutkimukseen osallistui potilaita, jotka olivat saaneet TNF‑alfa‑estäjähoitoa. Yhdistettyjen tutkimusanalyysien mukaan 56,3 % potilaista sai samanaikaisesti metotreksaattia (MTX), 10,0 % sai samanaikaisesti muita csDMARD-lääkkeitä ja 33,7 % ei saanut samanaikaisesti csDMARD-lääkkeitä.
Molempien tutkimusten ensisijainen päätetapahtuma oli niiden potilaiden prosenttiosuus, jotka saavuttivat American College of Rheumatology (ACR) 20 ‑vasteen viikolla 16.
Kliininen vaste
ACR20/50/70-vasteena ja taudin vähäisenä aktiivisuutena (Minimal Disease Activity, MDA) mitattuna deukravasitinibin aikaansaama tautiaktiivisuuden paranema oli merkittävä lumelääkkeeseen verrattuna molemmissa tutkimuksissa viikolla 16 (kts.taulukko 4). Deukravasitinibihoitoa saaneiden potilaiden kliiniset vasteet paranivat edelleen viikon 16 jälkeen, ja tulokset säilyivät viikolle 52 asti.
Taulukko 4: Tehoa koskevat tulokset nivelpsoriaasia sairastavilla aikuisilla
| POETYK PsA‑1 | POETYK PsA‑2 | ||||
|---|---|---|---|---|---|---|
Deukravasitinibi
(N = 336) | Lumelääke
(N = 334) | Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | Deukravasitinibi
(N = 312) | Lumelääke
(N = 312) | Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | |
ACR20-vaste | ||||||
Viikko 16 (%) | 54,2 | 34,1 | 20,0 (12,7–27,4)a | 54,2 | 39,4 | 14,8 (7,0–22,5)a |
Viikko 52 (%)* | 76,3 (212/278) |
|
| 71,9 (194/270) |
|
|
ACR50-vaste | ||||||
Viikko 16 (%) | 24,7 | 13,5 | 11,2 (5,3–17,1)c | 28,8 | 16,3 | 12,5 (6,0–19,0)c |
Viikko 52 (%)* | 48,9 (136/278) |
|
| 47,0 (127/270) |
|
|
ACR70-vaste | ||||||
Viikko 16 (%) | 11,6 | 5,4 | 6,2 (2,0–10,4)d | 10,6 | 5,4 | 5,2 (0,9–9,4)d |
Viikko 52 (%)* | 30,2 (84/278) |
|
| 30,0 (81/270) |
|
|
MDA-vaste (taudin vähäinen aktiivisuus)** | ||||||
Viikko 16 (%) | 19,0 | 10,2 | 8,9 (3,6–14,2)b | 25,6 | 14,7 | 10,9 (4,6–17,1)b |
Viikko 52 (%)* | 40,7 (114/280) |
|
| 48,3 (130/269) |
|
|
Hoitoon vastaamattomien imputointia (non‑responder imputation, NRI) käytettiin viikolle 16 asti. Viikon 16 jälkeen tehdyt havainnot esitetään imputoimattomina. N = satunnaistettujen potilaiden lukumäärä. a p ≤ 0,0002 b p ≤ 0,001 c nimellinen p ≤ 0,0002 d nimellinen p ≤ 0,02 * Käytettävissä olevien tutkittavien tiedot näytetään havaintojen mukaan muodossa (%, n/N) ** Taudin vähäinen aktiivisuus (MDA) = 5/7 tulosta: ≤ 1 aristavaa niveltä; ≤ 1 turvonnutta niveltä; psoriaasin aktiivisuutta ja vaikeusastetta kuvaava PASI (Psoriasis Activity and Severity Index) ≤ 1 tai osuus kehon pinta-alasta ≤ 3 %; potilaan kipuarvio (VAS) ≤ 15; potilaan yleisvointiarvio (VAS) ≤ 20; terveydentilan arviointilomakkeen toimintarajoitteisuusindeksi (HAQ‑DI) ≤ 0,5; ≤ 1 aristavaa kohtaa lihasjänteiden ja/tai nivelsiteiden kiinnittymiskohdissa | ||||||
PsA‑1‑ ja PsA‑2‑tutkimuksissa osoitettiin viikolla 16, että kaikki ACR-komponentit olivat parantuneet deukravasitinibilla hoidetuilla potilailla, myös potilaan kipuarvio (kipu-VAS). Nämä tulokset paranivat edelleen viikon 16 jälkeen ja säilyivät viikolle 52 asti.
Potilaiden määrä, joilla oli aksiaalinen affisio / vallitseva spondyliitti, oli liian pieni merkityksellisen arvioinnin tekemiseksi..
POETYK PsA‑1 ‑tutkimuksessa sairauden aktiivisuuspisteet (28 niveltä) CRP:llä mitattuna (DAS28-CRP) paranivat nimellisesti merkittävästi deukravasitinibia saaneilla potilailla verrattuna lumelääkettä saaneisiin viikolla 16 (deukravasitinibiryhmässä −1,33 ja lumelääkeryhmässä −0,83). Samankaltainen paranema havaittiin myös POETYK PsA‑2 ‑tutkimuksessa viikolla 16 (deukravasitinibiryhmässä −1,28 ja lumelääkeryhmässä −0,80).
Sotyktu-valmistetta saaneilla potilailla, joilla oli samanaikainen läiskäpsoriaasi, todettiin viikolla 16 lumelääkkeeseen verrattuna merkitsevää paranemista (PASI 75) -vasteella mitattuna, ja tulokset säilyivät viikkoon 52 asti molemmissa tutkimuksissa.
Vaste fyysisessä toimintakyvyssä
Molemmissa tutkimuksissa deukravasitinibia saaneiden potilaiden fyysinen toimintakyky terveydentilan arviointilomakkeen toimintarajoitteisuusindeksillä (HAQ‑DI) mitattuna parani lähtötilanteesta tilastollisesti merkitsevästi lumelääkkeeseen verrattuna. Korjattu keskimääräinen ero HAQ‑DI-pisteiden muutoksessa lähtötilanteesta (95 %:n luottamusväli) lumelääkkeeseen verrattuna viikolla 16 oli PsA‑1‑tutkimuksessa −0,17 (−0,24...−0,09) ja PsA‑2‑tutkimuksessa −0,11 (−0,18...−0,04). HAQ‑DI-vasteen (paranema lähtötilanteesta ≥ 0,35) saaneita oli viikolla 16 PsA‑1‑tutkimuksen deukravasitinibiryhmässä 51,3 % ja lumelääkeryhmässä 38,8 % ja PsA‑2‑tutkimuksen deukravasitinibiryhmässä 48,9 % ja lumelääkeryhmässä 43,2 %. Molemmissa tutkimuksissa vaste säilyi viikolle 52 asti.
Muut terveydentilaan liittyvät tulokset
SF-36-mittarin fyysistä terveyttä (Physical Component Summary, PCS) kuvastavien pistemäärien havaittiin paranevan viikolla 16 deukravasitinibia saaneilla potilailla verrattuna lumelääkettä saaneisiin potilaisiin. Viikolla 16 havaittiin fyysisen terveyden pistemäärän (PCS) parantuneen merkitsevästi deukravasitinibia saaneilla potilailla verrattuna lumelääkettä saaneisiin potilaisiin (korjattu keskimääräinen ero [95 %:n luottamusväli] lähtötilanteeseen verrattuna: POETYK PsA‑1: 2,3 [1,3–3,4], p < 0,0001. POETYK PsA‑2: 2,0 [1,0–3,1], p = 0,0002). Molemmissa tutkimuksissa parantuneet tulokset säilyivät viikolle 52 asti.
Iäkkäät potilaat
Deukravasitinibia kliinisissä tutkimuksissa saaneista 1 519 läiskäsporiaasia sairastavasta potilaasta, 152 oli vähintään 65‑vuotiaita ja 21 oli vähintään 75‑vuotiaita (ks. kohta Annostus ja antotapa). Kliinisissä tutkimuksissa deukravasitinibia saaneista 1 312 nivelpsoriaasipotilaasta 171 oli vähintään 65‑vuotiaita ja 22 oli vähintään 75‑vuotiaita.
Deukravasitinibia saaneiden vanhempien ja nuorempien potilaiden välillä ei todettu yleisesti ottaen mitään eroja altistuksessa, turvallisuudessa tai tehokkuudessa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset SOTYKTU-valmisteen käytöstä läiskäpsoriaasin ja nivelpsoriaasin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Deukravasitinibi imeytyi suun kautta otettuna lähes täydellisesti, altistus suureni annoksen mukaan, eikä ilmeistä ajasta riippuvaista farmakokinetiikkaa todettu.
Imeytyminen
Tablettien suun kautta annon jälkeen deukravasitinibi imeytyi nopeasti ja lähes täydellisesti. Tmax‑arvon mediaani oli 2–3 tuntia, ja absoluuttinen oraalinen biologinen hyötyosuus oli 99 % terveillä vapaaehtoisilla. Kerran vuorokaudessa tapahtuneen annon jälkeen todettiin lääkeaineen vähäistä kertymistä (< 1,4‑kertaista vakaassa tilassa).
Ruoka
Deukravasitinibia voidaan antaa aterioista ja mahalaukun pH-arvoon vaikuttavista lääkkeistä (H2‑reseptorin salpaajista ja protonipumpun estäjistä) riippumatta. Samanaikainen ruokailu tai mahalaukun pH-arvoon vaikuttavien lääkkeiden käyttö ei vaikuttanut deukravasitinibin kokonaisaltistukseen (AUC[INF]).
Jakautuminen
Jakautumistilavuus vakaassa tilassa (Vss) on 140 l eli suurempi kuin elimistön kokonaisnestemäärä (42 l), mikä viittaa ekstravaskulaariseen jakautumiseen. Deukravasitinibi sitoutuu 81,6‑prosenttisesti ihmisen plasman proteiineihin, pääasiassa ihmisen seerumin albumiiniin.
Deukravasitinibi jakautuu samalla tavalla plasma- ja punasolukomponentteihin, ja sen veri-plasmapitoisuussuhde on 1,26.
Erittyminen äidinmaitoon
Sen jälkeen, kun kahdeksalle terveelle imettävälle naiselle annettiin deukravasitinibiä 9 mg:n kerta-annos suun kautta, deukravasitinibi ja sen päämetaboliitti BMT-153261 olivat mitattavissa äidinmaidosta. Huippupitoisuudet äidinmaidossa saavutettiin nopeasti Tmax-arvojen mediaanin ollessa noin 1 tunti deukravasitinibin osalta ja 6 tuntia BMT‑153261:n osalta. Arvioitu maidon ja plasman pitoisuussuhde oli Cmax-arvoa tarkasteltaessa 1,5 ja AUC-arvoa tarkasteltaessa 3,2 deukravasitinibin osalta ja suurempi BMT‑153261:n osalta (Cmax-suhde 13; AUC-suhde 16). Deukravasitinibista 93 % ja BMT‑153261:stä 80 % oli erittynyt äidinmaitoon 24 tunnin kuluessa annoksen ottamisesta. Imetyksellä oli vähäinen vaikutus tai ei lainkaan vaikutusta deukravasitinibialtistukseen plasmassa.
Biotransformaatio
Ihmisillä deukravasitinibi metaboloituu neljän pääasiallisen biotransformaatioreitin kautta. Reitit ovat triatsoliosassa P450 (CYP) 1A2:n vaikutuksesta tapahtuva N‑demetylaatio, jonka seurauksena muodostuu BMT‑153261-päämetaboliittia; karboksyyliesteraasi 2:n (CES2:n) vaikutuksesta tapahtuva syklopropyyli-karboksamidihydrolyysi, jonka seurauksena muodostuu BMT‑158170-päämetaboliittia; uridiiniglukuronyylitransferaasin (UGT) vaikutuksesta tapahtuva N‑glukuronidaatio, jonka seurauksena muodostuu BMT‑334616-metaboliittia; sekä deuteroidussa metyyliryhmässä CYP 2B6/2D6:n vaikutuksesta tapahtuva mono‑oksidaatio, jonka seurauksena muodostuu M11‑metaboliittia.
Vakaassa tilassa deukravasitinibi on pääasiallinen aine verenkierrossa, sillä sen osuus verenkierrosta mitatuista yhdisteeseen liittyvistä komponenteista on 49 %. Verenkierrosta on tunnistettu kaksi päämetaboliittia, BMT‑153261 ja BMT‑158170. Molempien puoliintumisaika on verrattavissa kanta-aine deukravasitinibin puoliintumisaikaan. BMT‑153261 on voimakkuudeltaan verrattavissa kantayhdisteeseen, ja BMT‑158170 ei ole farmakologisesti aktiivinen. Verenkierrossa BMT‑153261-altistus on paljon pienempi kuin altistus kantayhdisteelle, ja siksi lääkevalmisteen pääasiallisen farmakologisen aktiivisuuden katsotaan johtuvan kantayhdiste deukravasitinibista.
Näiden lisäksi mitään pelkästään ihmisillä esiintyviä metaboliitteja tai verenkierrossa pitkään pysyviä metaboliitteja ei ole tunnistettu.
Eliminaatio
Deukravasitinibi eliminoituu useiden eri reittien kautta, muun muassa vaiheen I ja II metabolian kautta sekä suoraan munuaisten ja ulosteen kautta. Mikään yksittäinen entsyymi ei vastannut kokonaispuhdistumasta yli 26‑prosenttisesti. Deukravasitinibi metaboloituu laajalti, ja 59 % suun kautta annetusta [14C]‑deukravasitinibiannoksesta eliminoitui metaboliitteina virtsaan (37 % annoksesta) ja ulosteeseen (22 % annoksesta). Deukravasitinibiannoksesta 13 % erittyy muuttumattomassa muodossa virtsaan ja 26 % ulosteeseen.
Deukravasitinibin 6 mg:n annoksen efektiivinen puoliintumisaika on terveillä aikuisilla ihmisillä 10 tuntia, ja kokonaispuhdistuma on 15,3 l/h (CV 27 %). Populaatiofarmakokineettisten analyysien perusteella deukravasitinibin efektiivinen puoliintumisaika on psoriaasia ja nivelpsoriaasia sairastavilla samaa luokkaa kuin terveillä aikuisilla: 10–11 tuntia. Deukravasitinibin puhdistuma on psoriaasia sairastavilla samaa luokkaa kuin terveillä aikuisilla. Deukravasitinibin puhdistuma on nivelpsoriaasia sairastavilla jonkin verran (29 %) pienempi kuin terveillä aikuisilla. Deukravasitinibi on effluksi-kuljettajaproteiinien, P‑glykoproteiinin (P‑gp) ja rintasyövän resistenssiproteiinin (BCRP) sekä soluunoton OCT1-kuljettajaproteiinin substraatti. Suuren passiivisen läpäisevyyden, suuren oraalisen hyötyosuuden ja näihin kuljettajaproteiineihin kohdistuvan heikon affiniteetin vuoksi näiden kuljettajaproteiinien vaikutus deukravasitinibin farmakokinetiikkaan on erittäin pieni.
Deukravasitinibi ei ole OATP-, NTCP-, OAT1-, OAT3-, OCT2-, MATE1- tai MATE2K-kuljettajaproteiinien substraatti.
Lineaarisuus/ei-lineaarisuus
Tablettimuotoisen deukravasitinibin kerta-annosten farmakokinetiikka oli lineaarista annosalueella 3–36 mg.
Yhteisvaikutukset
Deukravasitinibin vaikutus muihin lääkevalmisteisiin
In vitro ‑tutkimuksista ei ole saatu mitään näyttöä siitä, että deukravasitinibi ja sen verenkierrossa olevat päämetaboliitit estäisivät kliinisesti merkityksellisellä altistuksella tärkeimpiä CYP-entsyymejä (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4), UGT-entsyymejä (1A1, 1A4, 1A6, 1A9, 2B7), CES2-entsyymiä tai lääkeaineen kuljettajaproteiineja (P‑gp, BCRP, OATP1B1, OATP1B3, BSEP, MRP2, OAT1, OAT3, OCT1, OCT2, MATE1 ja MATE2K). Deukravasitinibi ei myöskään indusoi CYP 1A2‑, 2B6- tai 3A4-entsyymejä (ks. kohta Yhteisvaikutukset).
Erityisryhmät
Iäkkäät potilaat
Populaatiofarmakokineettisen analyysin perusteella deukravasitinibin keskimääräinen vakaan tilan altistus (Cavg,ss) oli 65–74‑vuotiailla potilailla (n = 87 potilasta 1 387:stä [6,3 %] psoriaasiryhmässä ja n = 155 potilasta 1 325:stä [11,7 %] nivelpsoriaasiryhmässä) enintään 31 % suurempi ja 75–84‑vuotiailla potilailla (n = 13 potilasta 1 387:stä [0,94 %] psoriaasiryhmässä ja n = 23 potilasta 1 325:stä [1,7 %] nivelpsoriaasiryhmässä) enintään 53 % suurempi. Altistustietoja ≥ 85‑vuotiaista potilaista on saatavilla vain vähän.
Munuaisten vajaatoimintapotilaat
Munuaisten vajaatoiminnalla ei ole kliinisesti merkittävää vaikutusta deukravasitinibialtistukseen (ks. kohta Annostus ja antotapa) asiaa nimenomaisesti selvittäneen tutkimuksen mukaan. Tässä tutkimuksessa arvioitu glomerulusten suodatusnopeus (eGFR) määritettiin MDRD-kaavan (ruokavalion muuttaminen munuaissairaudessa) perusteella. Munuaistoiminnaltaan normaaliin ryhmään verrattuna deukravasitinibin Cmax muuttui enintään 15 % ja AUC[INF] suureni enintään 48 % munuaisten vajaatoiminnan asteen mukaan (lievä [eGFR: ≥ 60 – < 90 ml/min], keskivaikea [eGFR: ≥ 30 – < 60 ml/min], vaikea [eGFR: < 30 ml/min] ja loppuvaiheen munuaissairaus [eGFR: < 15 ml/min]). Munuaistoiminnaltaan normaaliin ryhmään verrattuna BMT‑153261:n Cmax suureni enintään 34 % ja AUC[INF] suureni enintään 84 % munuaisten vajaatoiminnan asteen mukaan.
Dialyysi ei poista deukravasitinibia systeemisestä verenkierrosta merkittävästi (yksi dialyysikerta poisti 5,4 % annoksesta).
Maksan vajaatoimintapotilaat
Lievällä (Child‑Pugh-luokka A) ja keskivaikealla (Child‑Pugh-luokka B) maksan vajaatoiminnalla ei ole kliinisesti merkittävää vaikutusta deukravasitinibialtistukseen (ks. kohta Annostus ja antotapa). Maksan toiminnaltaan normaaliin ryhmään verrattuna deukravasitinibin Cmax- ja AUC[INF]-kokonaisarvo suureni lievässä maksan vajaatoiminnassa enintään 10 % ja keskivaikeassa maksan vajaatoiminnassa enintään 40 %, kun taas sitoutumattoman deukravasitinibin Cmax-arvo suureni enintään 26 % ja AUC[INF]-arvo enintään 60 %. Vaikeaa (Child‑Pugh-luokka C) maksan vajaatoimintaa sairastavilla aikuisilla deukravasitinibin Cmax-kokonaisarvo oli samaa luokkaa ja AUC-kokonaisarvo oli 43 % suurempi kuin vastaavilla terveillä aikuisilla. Näillä aikuisilla sitoutumattoman deukravasitinibin Cmax‑arvo suureni 62 % ja AUC[INF]‑arvo suureni 131 %. Deukravasitinibia ei suositella vaikeaa maksan vajaatoimintaa sairastaville potilaille (ks. kohta Annostus ja antotapa).
BMT‑153261:n AUC[0‑T] pieneni lievää maksan vajaatoimintaa sairastavilla tutkittavilla 19 %, keskivaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla 53 % ja vaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla 76 % verrattuna tutkittaviin, joiden maksa toimi normaalisti. BMT‑153261:n Cmax pieneni lievää maksan vajaatoimintaa sairastavilla tutkittavilla 25 %, keskivaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla 59 % ja vaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla 79 %.
Sukupuoli
Populaatiofarmakokineettisen mallinnuksen ja simulaation perusteella deukravasitinibin keskimääräisen vakaan tilan altistuksen (Cmax,ss ja Cavg,ss) odotetaan olevan enintään 30 % suurempi naisilla kuin miehillä.
Paino
Populaatiofarmakokineettisen mallinnuksen ja simulaation perusteella deukravasitinibin vakaan tilan altistuksen geometrisen keskiarvon odotetaan olevan korkeampi ollen enintään 37,4 % (Cmax,ss) ja enintään 26,6 % (Cavg,ss) suurempi potilailla, joiden ruumiinpaino on pienempi (< 60 kg). Potilailla, jotka ovat painavampia (> 90 kg), deukravasitinibin vakaan tilan altistuksen geometrisen keskiarvon odotetaan olevan matalampi ollen enintään 24,8 % (Cmax,ss) ja enintään 19,6 % (Cavg,ss) pienempi (verrattuna potilaisiin, joiden paino on 60–90 kg).
Sisäsyntyiset tekijät
Rodulla ja etnisellä taustalla ei ollut kliinisesti merkittävää vaikutusta deukravasitinibialtistukseen.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, karsinogeenisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten ei-kliiniset tulokset eivät viittaa erityiseen vaaraan ihmisille.
Toistuvan altistuksen aiheuttama toksisuus
Rotilla tehdyssä pitkäaikaistoksisuutta selvittäneessä tutkimuksessa todettiin lymfosyyttimäärän pienenemistä, luuytimen solukkuuden vähenemistä ja imukudoksen solukkuuden vähenemistä immuunijärjestelmän kudoksissa pienimmällä havaittavan vaikutuksen (lowest observed effect level, LOEL) aiheuttavalla altistuksella (AUC), joka oli noin 7‑kertainen ihmisille suositeltuun annokseen (RHD) nähden. Näihin vaikutuksiin ei liittynyt immunosuppression kliinisiä merkkejä (esim. infektioita). Verihiutalemäärän ja punasolujen massaparametrien pienentymistä todettiin pienimmällä havaittavan vaikutuksen aiheuttavalla altistuksella (AUC), joka oli noin 33‑kertainen ihmisille suositeltuun annokseen nähden. Apinoilla tehdyssä pitkäaikaistoksisuutta selvittäneessä tutkimuksessa kliinisiä ja mikroskooppisia ihomuutoksia ja punasolujen massaparametrien pienenemistä todettiin pienimmällä havaittavan vaikutuksen aiheuttavalla altistuksella (AUC), joka oli noin 6‑kertainen ihmisille suositeltuun annokseen nähden.
Kehitys- ja lisääntymistoksisuus
Deukravasitinibilla ei ollut vaikutusta uros- ja naarasrottien hedelmällisyyteen eikä varhaisvaiheen alkionkehitykseen altistuksilla (AUC), jotka olivat uroksilla noin 178‑kertaisia ja naarailla noin 136‑kertaisia ihmisille suositeltuun annokseen nähden.
Deukravasitinibi ei ollut embryoletaalinen eikä teratogeeninen emon altistuksilla (AUC), jotka olivat rotilla noin 211‑kertaisia ihmisille suositeltuun annokseen nähden tai 72/16 (kokonaisarvo/vapaa) ‑kertaisia ihmisille suositeltuun annokseen nähden kaniineilla.
Rotilla tehdyssä pre- ja postnataalista kehitystä selvittäneessä tutkimuksessa havaittiin ohimenevää poikasten painon pienenemistä vieroitusta edeltäneessä vaiheessa, kun emon altistus (AUC) oli noin 87‑kertainen ihmisille suositeltuun annokseen nähden. Tämä vaikutus korjaantui täysin vieroituksen jälkeen.
Kun radioaktiivisesti merkittyä deukravasitinibia annettiin imettäville rotille, deukravasitinibia ja/tai sen metaboliitteja todettiin rottien maidossa. Maidon ja plasman pitoisuussuhde oli 2,7–30,9.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Hypromelloosiasetaattisuksinaatti, vedetön laktoosi, mikrokiteinen selluloosa, kroskarmelloosinatrium, kolloidinen hydratoitu piidioksidi, magnesiumstearaatti
Kalvopäällyste
Polyvinyylialkoholi, titaanidioksidi (E171), makrogoli, talkki, punainen rautaoksidi (E172), keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SOTYKTU tabletti, kalvopäällysteinen
6 mg (L:kyllä) 28 fol (854,21 €)
PF-selosteen tieto
Polyvinyylikloridista/polyklorotrifluoroeteenistä (PVC/PCTFE) valmistettu läpinäkyvä läpipainopakkaus, jossa on alumiinista valmistettu läpipainofolio. Yksi läpipainopakkaus sisältää 7 tai 14 kalvopäällysteistä tablettia (kalenteriläpipainopakkaus tai tavallinen läpipainopakkaus).
Pakkauskoot: 7, 14, 28 ja 84 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Vaaleanpunainen, pyöreä, kaksoiskupera, kalvopäällysteinen tabletti, jonka halkaisija on 8 mm. Tabletin toisella puolella ei ole merkintöjä, toiselle puolelle on painettu kahdelle riville ”BMS 895” ja ”6 mg”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SOTYKTU tabletti, kalvopäällysteinen
6 mg 28 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Deukravasitinibi: Aikuisten keskivaikean tai vaikean kroonisen läiskäpsoriaasin hoito erityisin edellytyksin (3088).
ATC-koodi
L04AF07
Valmisteyhteenvedon muuttamispäivämäärä
30.04.2026
Yhteystiedot
09 2512 1244
www.bms.com/fi
medinfo.finland@bms.com