
AFSTYLA injektiokuiva-aine ja liuotin, liuosta varten 250 IU, 500 IU, 1000 IU, 1500 IU, 2000 IU, 2500 IU, 3000 IU
Vaikuttavat aineet ja niiden määrät
AFSTYLA 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 250 IU rekombinanttia, yksiketjuista hyytymistekijää VIII (rVIII‑SingleChain), (INN = lonoktokogi alfa). Kun valmiste on saatettu käyttökuntoon 2,5 ml:lla injektionesteisiin käytettävää vettä, yksi millilitra liuosta sisältää 100 IU yksiketjuista hyytymistekijää VIII.
AFSTYLA 500 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 500 IU rekombinanttia, yksiketjuista hyytymistekijää VIII (rVIII‑SingleChain), (INN = lonoktokogi alfa). Kun valmiste on saatettu käyttökuntoon 2,5 ml:lla injektionesteisiin käytettävää vettä, yksi millilitra liuosta sisältää 200 IU yksiketjuista hyytymistekijää VIII.
AFSTYLA 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 1000 IU rekombinanttia, yksiketjuista hyytymistekijää VIII (rVIII‑SingleChain), (INN = lonoktokogi alfa). Kun valmiste on saatettu käyttökuntoon 2,5 ml:lla injektionesteisiin käytettävää vettä, yksi millilitra liuosta sisältää 400 IU yksiketjuista hyytymistekijää VIII.
AFSTYLA 1500 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 1500 IU rekombinanttia, yksiketjuista hyytymistekijää VIII (rVIII‑SingleChain), (INN = lonoktokogi alfa). Kun valmiste on saatettu käyttökuntoon 5 ml:lla injektionesteisiin käytettävää vettä, yksi millilitra liuosta sisältää 300 IU yksiketjuista hyytymistekijää VIII.
AFSTYLA 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 2000 IU rekombinanttia, yksiketjuista hyytymistekijää VIII (rVIII‑SingleChain), (INN = lonoktokogi alfa). Kun valmiste on saatettu käyttökuntoon 5 ml:lla injektionesteisiin käytettävää vettä, yksi millilitra liuosta sisältää 400 IU yksiketjuista hyytymistekijää VIII.
AFSTYLA 2500 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 2500 IU rekombinanttia, yksiketjuista hyytymistekijää VIII (rVIII‑SingleChain), (INN = lonoktokogi alfa). Kun valmiste on saatettu käyttökuntoon 5 ml:lla injektionesteisiin käytettävää vettä, yksi millilitra liuosta sisältää 500 IU yksiketjuista hyytymistekijää VIII.
AFSTYLA 3000 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 3000 IU rekombinanttia, yksiketjuista hyytymistekijää VIII (rVIII‑SingleChain), (INN = lonoktokogi alfa). Kun valmiste on saatettu käyttökuntoon 5 ml:lla injektionesteisiin käytettävää vettä, yksi millilitra liuosta sisältää 600 IU yksiketjuista hyytymistekijää VIII.
Teho (IU, kansainvälinen yksikkö) on määritetty käyttämällä Euroopan farmakopean kromogeenistä määritystä. AFSTYLA-valmisteen spesifinen aktiivisuus on 7 400–16 000 IU/mg proteiinia.
AFSTYLA on yksiketjuinen rekombinantti ihmisen hyytymistekijä VIII, joka tuotetaan kiinanhamsterin munasarjan (CHO) soluissa. Sen rakenteesta on poistettu suurin osa villityypin täysimittaisesta B‑domeenista sekä 4 aminohappoa viereisestä happamasta a3-domeenista (täysimittaisen hyytymistekijä VIII:n aminohapot 765–1652).
Hyytymistekijä VIII:n vastamuodostunut raskas- ja kevytketjusidos tarjoaa uuden N‑glykosylaatiokohdan. Kun villityypin hyytymistekijän VIII B-domeenin ja a3-domeenin välistä on poistettu furiinia pilkkova kohta, AFSTYLA ilmentyy yksiketjuisena hyytymistekijä VIII ‑molekyylinä.
Apuaine, jonka vaikutus tunnetaan:
AFSTYLA 250, 500 ja 1000 IU (2,5 ml liuotinta)
Yksi injektiopullo sisältää 17,5 mg (0,76 mmol) natriumia.
AFSTYLA 1500, 2000, 2500 ja 3000 IU (5 ml liuotinta)
Yksi injektiopullo sisältää 35 mg (1,52 mmol) natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
A‑hemofiliaa (synnynnäinen hyytymistekijä VIII:n puutos) sairastavien potilaiden verenvuotojen hoito ja ennaltaehkäisy.
AFSTYLA-valmistetta voidaan käyttää kaikissa ikäryhmissä.
Ehto
Hoito pitää toteuttaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito pitää toteuttaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Hoidon seuranta
Hoidon aikana suositellaan tekijä VIII:n asianmukaista määritystä, joka ohjaa käytettävää annosta ja toistuvien injektioiden antotiheyttä. Potilaskohtaiset vasteet tekijä VIII:lle saattavat vaihdella, joten puoliintumisaika ja saanto voivat olla erilaiset. Kehon painoon perustuvaa annosta saatetaan joutua muuttamaan alipainoisille tai ylipainoisille potilaille. Erityisesti suurten kirurgisten toimenpiteiden yhteydessä korvaushoidon tarkka seuranta koagulaatioanalyysin (plasman hyytymistekijä VIII:n aktiivisuus) avulla on välttämätöntä.
Kun potilaiden verinäytteiden tekijä VIII:n aktiivisuusmääritykseen käytetään tromboplastiiniaikaan (aPTT) perustuvaa yksivaiheista in vitro hyytymismääritystä, sekä määrityksessä käytettävä aPTT-reagenssi että viitestandardi voivat vaikuttaa merkittävästi plasman tekijä VIII:n aktiivisuustuloksiin. Myös määritystuloksissa voi olla huomattavia eroja sen mukaan, käytetäänkö tromboplastiiniaikaan (aPTT) perustuvaa yksivaiheista hyytymismääritystä vai Euroopan farmakopean mukaista kromogeenistä määritystä. Tämä on huomioitava erityisesti silloin, kun laboratoriota ja/tai määrityksessä käytettäviä reagensseja vaihdetaan.
Plasman hyytymistekijä VIII:n aktiivisuutta on seurattava AFSTYLA-hoidon aikana joko kromogeenisen määrityksen tai yksivaiheisen hyytymistestin avulla, jotta saadaan opastusta käytettävästä annoksesta ja toistuvien injektioiden antotiheydestä. Kromogeenista määritystä suositellaan, koska sen tulos kuvaa parhaiten AFSTYLA-valmisteen kliinistä hemostaattista potentiaalia. Yksivaiheisen hyytymistestin tulos aliarvioi hyytymistekijän VIII aktiivisuustason noin 45 % verrattuna kromogeenisen määrityksen tulokseen. Jos käytetään yksivaiheista hyytymistestiä, potilaan hyytymistekijän VIII aktiivisuustaso määritetään kertomalla tulos muuntokertoimella 2.
Annostus
Annos ja korvaushoidon kesto riippuvat tekijä VIII:n puutteen vaikeusasteesta, vuotokohdasta ja verenvuodon laajuudesta sekä potilaan kliinisestä tilasta.
Annettavan tekijä VIII:n yksikköjen määrä ilmoitetaan kansainvälisinä yksikköinä (IU), jotka ovat WHO:n voimassa olevan tekijä VIII ‑valmisteiden konsentraattistandardin mukaisia. Tekijä VIII:n aktiivisuus plasmassa ilmaistaan joko prosentteina (suhteessa normaaliin ihmisplasmaan) tai mieluiten kansainvälisinä yksikköinä (suhteessa plasman hyytymistekijä VIII:n kansainväliseen standardiin).
Tekijä VIII:n yksi kansainvälinen yksikkö (IU) vastaa tekijä VIII:n määrää 1 ml:ssa normaalia ihmisen plasmaa.
Teho on määritetty kromogeenisellä substraattitestillä.
Plasman hyytymistekijä VIII:n pitoisuuksia voidaan seurata joko käyttäen kromogeenistä substraattimääritystä tai yksivaiheista hyytymistestiä.
Tarvittaessa toteutettava hoito
Hyytymistekijä VIII:n tarvittavan annoksen laskeminen perustuu empiiriseen havaintoon, jonka mukaan 1 kansainvälinen yksikkö (IU) hyytymistekijää VIII painokiloa kohti lisää plasman hyytymistekijä VIII:n aktiivisuutta keskimäärin 2 IU:lla/dl.
Tarvittava annos määritetään seuraavan kaavan avulla:
Annos (IU) = paino (kg) x haluttu tekijä VIII:n pitoisuuden nousu (IU/dl tai % normaalista) x 0,5 (IU/kg per IU/dl)
Annos ja antotiheys pitää aina määrittää potilaskohtaisesti kliinisen tehon perusteella.
Seuraavissa verenvuototapahtumissa hyytymistekijä VIII:n aktiivisuus ei saa laskea ilmoitetun plasman aktiivisuustason alapuolelle (prosentteina normaalista tai IU/dl) vastaavalla ajanjaksolla. Seuraavaa taulukkoa voidaan käyttää annostelun apuna verenvuodoissa ja leikkauksissa:
Vuodon voimakkuus / Kirurginen toimenpide | Tarvittava hyytymistekijä VIII:n taso (%) (IU/dl) | Antotiheys (tuntia) / Hoidon kesto (vrk) |
Verenvuoto | ||
Alkava nivelvuoto, lihasverenvuoto tai suun limakalvon verenvuoto | 20–40 | Toistetaan 12–24 tunnin välein. Vähintään 1 vrk, kunnes vuotoon liittyvä kipu on ohi tai vuoto on parantunut. |
Laajempi nivelvuoto, lihasverenvuoto tai hematooma | 30–60 | Toistetaan 12–24 tunnin välein 3–4 vrk:n ajan tai kauemmin, kunnes kipu ja akuutti toimintakyvyttömyys poissa. |
Hengenvaaralliset verenvuodot | 60–100 | Toistetaan 8–24 tunnin välein, kunnes uhka on väistynyt. |
Leikkaus | ||
Pieni leikkaus, mukaan lukien hampaanpoisto | 30–60 | 24 tunnin välein, vähintään 1 vrk:n ajan, kunnes haava parantunut. |
Suuri leikkaus | 80–100 (pre- ja postoperatiivinen) | Toistetaan 8–24 tunnin välein, kunnes haava on parantunut riittävästi, sen jälkeen vielä vähintään 7 vrk:n ajan pitäen tekijä VIII:n aktiivisuus 30–60 %:ssa (IU/dl). |
Estohoito
Suositusannos hoitoa aloitettaessa on 20–50 IU/kg AFSTYLA-valmistetta 2–3 kertaa viikossa. Hoito-ohjelmaa voidaan säätää potilaan vasteen mukaan.
Pediatriset potilaat
Lapsille (0 – < 12-vuotiaat) suositusannos hoitoa aloitettaessa on 30–50 IU/kg AFSTYLA-valmistetta 2–3 kertaa viikossa. Lyhemmät annosvälit tai suuremmat annokset voivat olla tarpeen alle 12-vuotiaille lapsille, koska puhdistuma on tässä ikäryhmässä suurempi.
Vähintään 12-vuotiaille nuorille suositellaan samoja annoksia kuin aikuisille (ks. kohta Farmakokinetiikka).
Iäkkäät
AFSTYLA-valmistetta koskevissa kliinisissä tutkimuksissa ei ollut mukana yli 65-vuotiaita.
Antotapa
Laskimoon.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Käyttökuntoon saatettu valmiste on annettava hitaana injektiona potilaalle mukavalla nopeudella, enintään 10 ml/min.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aiempi allerginen reaktio hamsterin proteiineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi annetun lääkevalmisteen nimi ja eränumero pitää kirjata selkeästi potilastietoihin.
Yliherkkyys
Allergistyyppiset yliherkkyysreaktiot ovat mahdollisia AFSTYLA-valmisteen käytön yhteydessä. Valmiste sisältää jäämiä hamsterin proteiineista. Potilasta on neuvottava lopettamaan valmisteen käyttö heti ja ottamaan yhteyttä lääkäriin, jos yliherkkyysoireita ilmaantuu. Potilaalle on kerrottava yliherkkyysreaktioiden varhaisista oireista, kuten nokkosihottuma, yleistynyt urtikaria, puristava tunne rinnassa, hengityksen vinkuminen, hypotensio ja anafylaksia.
Potilaille, joilla on aiemmin esiintynyt yliherkkyysreaktioita, voidaan harkita asianmukaista esilääkitystä.
Sokkitapauksissa potilasta on hoidettava yleisten sokinhoito-ohjeiden mukaisesti.
Inhibiittorit
Tekijä VIII:aa neutraloivien vasta‑aineiden (inhibiittoreiden) muodostuminen on tunnettu komplikaatio hemofilia A ‑potilaiden hoidossa. Inhibiittorit ovat yleensä IgG-immunoglobuliineja, jotka estävät tekijä VIII hyytymistoiminnan aktivoitumisen ja joiden määrä ilmaistaan Bethesda yksikköinä (Bethesda Units, BU) millilitrassa plasmaa käyttämällä muunneltua määritystä. Inhibiittoreiden muodostumisen riski riippuu taudin vaikeusasteesta ja altistumisesta tekijä VIII:lle. Riski on suurin 50 ensimmäisen altistuspäivän aikana, mutta säilyy koko elämän ajan, vaikka onkin melko harvinainen.
Inhibiittorien muodostumisen kliininen merkitys riippuu inhibiittorititteristä, riittämättömän kliinisen vasteen riski on pienempi, jos inhibiittorititteri on alhainen kuin jos se on korkea.
Hyytymistekijä VIII ‑valmisteilla hoidettavien potilaiden inhibiittoreiden esiintyvyyttä on seurattava tarkkaan asianmukaisin kliinisin havainnoin ja laboratoriokokein. Jos odotettuja tekijä VIII:n aktiivisuuden plasmapitoisuuksia ei saavuteta tai jos verenvuotoa ei saada hallintaan asianmukaisella annoksella, on potilaalta testattava tekijä VIII:n inhibiittorin esiintyminen. Jos potilaalla on korkea inhibiittoripitoisuus, tekijä VIII -hoito ei ehkä ole tehokasta ja on harkittava muita terapeuttisia vaihtoehtoja. Näiden potilaiden hoidon on tapahduttava sellaisten lääkäreiden valvonnassa, joilla on kokemusta hemofiliasta ja tekijä VIII:n inhibiittoreista.
Laboratoriotestien seuranta
Jos käytetään yksivaiheista hyytymismääritystä, tulos on kerrottava konversiokertoimella 2 potilaan hyytymistekijä VIII:n aktiivisuustason määrittämiseksi (ks. kohta Annostus ja antotapa).
Sydän- ja verisuonitapahtumat
Potilailla, joilla on sydän- ja verisuonitapahtumien riskitekijöitä, tekijä VIII-korvaushoito voi suurentaa sydän- ja verisuonitapahtumien riskiä.
Katetreihin liittyvät komplikaatiot
Jos toimenpide edellyttää keskuslaskimokatetria, on huomioitava keskuslaskimokatetriin liittyvät komplikaatiot, mukaan lukien paikalliset infektiot, bakteremia ja katetrikohdan tromboosi.
Natriumsisältö
Tämä lääkevalmiste sisältää enintään 35,0 mg natriumia per injektiopullo, joka vastaa 1,8 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Pediatriset potilaat
Luetellut varoitukset ja varotoimet koskevat sekä aikuisia että lapsia.
Yhteisvaikutukset
Ihmisen hyytymistekijä VIII ‑valmisteilla ei ole raportoitu olevan yhteisvaikutuksia muiden lääkevalmisteiden kanssa.
Raskaus ja imetys
Lisääntymistä koskevia eläinkokeita ei ole suoritettu tekijä VIII:lla. Koska A-hemofiliaa esiintyy naisilla vain harvoin, kokemuksia tekijä VIII:n vaikutuksista raskauteen ja imetykseen ei ole. Tämän vuoksi tekijä VIII:aa saa käyttää raskauden ja imetyksen aikana vain, jos se on selvästi tarpeellista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
AFSTYLA-valmisteella ei ole vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yliherkkyysreaktioita tai allergisia reaktioita (joita voivat olla angioedeema, infuusiokohdan polttelu ja kirvely, vilunväreet, punoitus, yleistynyt nokkosihottuma, päänsärky, nokkosihottuma, matala verenpaine, uneliaisuus, pahoinvointi, levottomuus, sydämen tiheälyöntisyys, puristava tunne rinnassa, kihelmöinti, oksentelu, vinkuva hengitys) on havaittu harvoin tekijä VIII:aa sisältävien valmisteiden käytön yhteydessä. Nämä oireet voivat joissain tapauksissa edetä vaikeaksi anafylaksiaksi (mukaan lukien sokki).
Neutraloivia vasta-aineita (inhibiittoreita) voi kehittyä hemofilia A -potilaille, jotka saavat tekijä VIII hoitoa, kuten AFSTYLA-hoito. Mikäli tällaisia inhibiittoreita ilmaantuu, se saattaa näkyä riittämättömänä kliinisenä vasteena hoidolle. Tällaisissa tapauksissa on suositeltavaa ottaa yhteyttä erikoistuneeseen hemofiliakeskukseen.
Haittavaikutustaulukko
Alla oleva taulukko on luokiteltu MedDRAn elinjärjestelmäluokituksen mukaan (elinjärjestelmäluokitus ja suositeltu termi). Taulukossa mainitut esiintyvyydet on havaittu päättyneissä kliinisissä tutkimuksissa aiemmin hoidetuilla vaikeaa A-hemofiliaa sairastavilla potilailla.
Esiintyvyydet on arvioitu potilaskohtaisesti seuraavan luokituksen mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
MedDRA elinjärjestelmäluokka | Haittavaikutus | Esiintymistiheys |
| Veri ja imukudos | Tekijä VIII:n inhibitio | melko harvinainen (PTP)* |
Immuunijärjestelmä | Yliherkkyys | yleinen |
Hermosto | Heitehuimaus | yleinen |
Parestesiat | yleinen | |
Iho ja ihonalainen kudos | Ihottuma | yleinen |
Eryteema | melko harvinainen | |
Kutina | melko harvinainen | |
Yleisoireet ja antopaikassa todettavat haitat | Kuume | yleinen |
Pistoskohdan kipu | melko harvinainen | |
Vilunväreet | melko harvinainen | |
Kuumotus | melko harvinainen |
* Yleisyys perustuu kaikilla tekijä VIII -valmisteilla tehtyihin tutkimuksiin, joihin osallistui vaikeaa hemofilia A:ta sairastavia potilaita. PTP = aiemmin hoidetut potilaat, PUP = potilaat, jotka eivät aiemmin ole saaneet hoitoa.
Pediatriset potilaat
Pediatrisilla potilailla havaitut haittavaikutukset ovat samoja kuin, mitä on havaittu aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Potilaalla, joka jo päättyneessä kliinisessä tutkimuksessa sai yli kaksi kertaa määrättyä suuremman annoksen AFSTYLA-valmistetta, esiintyi huimausta, kuumotusta ja kutinaa. Niiden ei katsottu johtuvan AFSTYLA-valmisteesta, vaan todennäköisemmin analgeetin samanaikaisesta antamisesta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Hemostaatit, veren hyytymistekijä VIII, ATC-koodi: B02BD02
Vaikutusmekanismi
AFSTYLA (INN: lonoktokogi alfa) on rekombinantti ihmisen proteiini, joka korvaa tehokkaaseen hemostaasiin tarvittavan puuttuvan hyytymistekijä VIII:n. AFSTYLA on yksittäinen polypeptidiketju, jonka B-domeeni on lyhennetty, minkä ansiosta hyytymistekijä VIII:n raskas- ja kevytketjut ovat liittyneet toisiinsa kovalenttisidoksella. AFSTYLA-valmisteella on suurempi sitoutumisaffiniteetti Von Willebrandin tekijään kuin täysimittaisella rekombinanttitekijä VIII:lla. Von Willebrandin tekijä stabiloi hyytymistekijä VIII:aa ja suojaa sitä pilkkoutumiselta. Aktivoituneen AFSTYLA-valmisteen aminohappojärjestys on sama kuin endogeenisen aktivoituneen hyytymistekijä VIII:n.
Farmakodynaamiset vaikutukset
Hyytymistekijä VIII / von Willebrandin tekijä -kompleksi koostuu kahdesta molekyylistä (hyytymistekijä VIII ja von Willebrandin tekijä), joilla on erilaiset fysiologiset toiminnot. Kun hyytymistekijä VIII:aa annetaan infuusiona hemofiliapotilaalle, se sitoutuu potilaan verenkierrossa olevaan von Willebrandin tekijään. Aktivoitunut hyytymistekijä VIII toimii aktivoituneen tekijä IX:n kofaktorina ja nopeuttaa tekijä X:n muuttumista aktivoituneeksi tekijä X:ksi. Aktivoitunut tekijä X muuttaa protrombiinin trombiiniksi. Sen jälkeen trombiini muuttaa fibrinogeenin fibriiniksi, jolloin hyytymä pääsee muodostumaan.
A-hemofilia on X-kromosomissa periytyvä veren hyytymishäiriö, joka johtuu normaalia pienemmästä hyytymistekijä VIII:n pitoisuudesta. Se aiheuttaa voimakkaita verenvuotoja niveliin, lihaksiin ja sisäelimiin joko itsestään tai vamman tai kirurgisen toimenpiteen seurauksena. Korvaushoidolla lisätään hyytymistekijä VIII:n pitoisuutta plasmassa, jolloin hyytymistekijä VIII:n puutos ja verenvuotoalttius pystytään väliaikaisesti korjaamaan.
Kliininen teho ja turvallisuus
Aikuiset ja nuoret (12–65-vuotiaat)
Tutkimuksessa 1001 selvitettiin estohoidon tehoa ja turvallisuutta verenvuototapahtumien estossa, hemostaattista tehoa verenvuototapahtumien hallinnassa pysymisessä sekä leikkauksen yhteydessä annettavassa hoidossa. Tutkimukseen otettiin mukaan 175 aiemmin hoitoa saanutta vaikeaa A-hemofiliaa sairastavaa potilasta (ikä 12–65 vuotta) (yksi tutkimushenkilö oli > 60 vuotta). Potilaiden altistukseksi yksiketjuiselle hyytymistekijälle VIII kertyi yhteensä 14 306 altistuspäivää. Yhdelläkään potilaalla ei ilmennyt inhibiittoreiden kehittymistä tai anafylaktisia reaktioita.
Estohoito: Estohoitoa sai 146 tutkittavaa (vuotuisen verenvuototapahtumien määrän mediaani 1,14 [kvartiiliväli: 0,0, 4,2]). Näistä 79 (54 %) ohjattiin hoitoa kolmesti viikossa saavaan ryhmään ja 47 (32 %) ohjattiin hoitoa kahdesti viikossa saavaan ryhmään. Kolmesti viikossa estohoitoa saaneen ryhmän potilaat saivat injektiona mediaaniannoksen 30 IU/kg. Kahdesti viikossa hoitoa saaneen ryhmän potilaat saivat mediaaniannoksen 35 IU/kg. Potilaiden kaikissa estohoitoryhmissä vuoden aikana käyttämä mediaanimäärä oli 4283 IU/kg.
Verenvuodon hoito: Tutkimuksessa 1001 havaituista 848 verenvuototapahtumasta 93,5 % saatiin hallintaan enintään kahdella injektiolla. Mediaaniannos verenvuotoepisodin hoitoon oli 34,7 IU/kg.
Perioperatiivinen hoito (kirurginen profylaksi): 13 tutkittavalle tutkimuksessa 1001 tehtiin ja arvioitiin yhteensä 16 suurta kirurgista toimenpidettä. Yksiketjuisen hyytymistekijän VIII hemostaattinen teho estohoitona kirurgisten toimenpiteiden yhteydessä oli erinomainen tai hyvä kaikissa leikkauksissa. Alle 18 vuoden ikäisiä pediatrisia potilaita ei ollut mukana kirurgisia toimenpiteitä koskevassa populaatiossa.
Pediatriset potilaat < 12-vuotiaat
Tutkimukseen 3002 otettiin mukaan yhteensä 84 aiemmin hoitoa saanutta alle 12-vuotiasta potilasta (35 oli < 6-vuotiaita ja 49 oli 6 − < 12-vuotiaita). Tutkimukseen osallistuneiden altistukseksi yksiketjuiselle hyytymistekijälle VIII kertyi yhteensä 5239 altistuspäivää. Yhdelläkään potilaalla ei ilmennyt inhibiittoreiden kehittymistä tai anafylaktisia reaktioita.
Yksilöity estohoito: Estohoitoa saaneista 81 potilaasta (vuotuisen verenvuototapahtumien määrän mediaani 3,69 [kvartiiliväli: 0,00, 7,20]) 43 (53 %) ohjattiin saamaan hoitoa kahdesti viikossa ja 25 (31 %) kolmesti viikossa.Kolmesti viikossa estohoitoa saaneen ryhmän potilaat saivat injektiona mediaaniannoksen 32 IU/kg. Kahdesti viikossa hoitoa saaneen ryhmän potilaat saivat mediaaniannoksen 35 IU/kg. Potilaiden kaikissa estohoitoryhmissä vuoden aikana käyttämä mediaanimäärä oli 4109 IU/kg.
Verenvuodon hoito: Tutkimuksen 3002 aikana havaituista 347 verenvuototapahtumasta 95,7 % saatiin hallintaan enintään kahdella injektiolla. Verenvuototapahtuman hoitoon käytetty mediaaniannos oli 27,6 IU/kg.
Jatkotutkimukseen 3001 osallistui 222 aiemmin hoitoa saanutta potilasta (67 oli < 12‑vuotiaita). Tässä tutkimuksessa aiemmin hoitoa saaneiden potilaiden keskimääräinen (SD) altistuspäivien lukumäärä oli 341,9 (135,48). Kaikkiaan 212 tutkittavalla (95,5 %) altistuspäivien lukumäärä oli > 100. Jatkotutkimuksessa ei tunnistettu uusia turvallisuussignaaleja eikä huolenaiheita.
Tehoa koskevat tulokset vastasivat aiemmissa tutkimuksissa raportoituja tuloksia.
Aiemmin hoitamattomat potilaat
Tutkimukseen 3001 osallistui yhteensä 24 aiemmin hoitamatonta potilasta, joiden mediaani-ikä oli 1,0 vuotta (vaihteluväli: 0–5 vuotta). Tutkimukseen osallistuneiden altistukseksi yksiketjuiselle hyytymistekijälle VIII kertyi yhteensä 5 909 altistuspäivää (keskimääräinen (SD) altistuspäivien lukumäärä: 245,5 (161,56)).
Yksilöity estohoito: Yhteensä 23 aiemmin hoitamatonta potilasta sai estohoitoa tutkimuksen aikana (11 vaihtoi tarvittaessa käytettävästä hoidosta estohoitoon). Estohoidon aikana vuotuisen verenvuototapahtumien määrän (annualized bleeding rate, ABR) mediaani oli 1,84 (vaihteluväli: 0,0–23,6) ja vuotuisen spontaanien verenvuototapahtumien määrän (annualized spontaneous bleeding rate, AsBR) mediaani oli 0,88 (vaihteluväli: 0,0–19,7).
Verenvuodon hoito: Havaituista 315 verenvuototapahtumasta (joista yksi oli vakava) 88,9 % saatiin hallintaan enintään kahdella injektiolla.
Immunologisen toleranssin induktiosta (ITI) on kerätty tietoa A-hemofiliaa sairastavilta potilailta, joille on kehittynyt hyytymistekijä VIII:n inhibiittoreita.
Huomattakoon, että vuotuinen verenvuototapahtumien määrä (annualized bleeding rate, ABR) ei ole vertailukelpoinen eri hyytymistekijäkonsentraattien ja eri kliinisten tutkimusten välillä.
Farmakokinetiikka
Aikuispotilaat
AFSTYLA-valmisteen farmakokineettisia muuttujia arvioitiin laskimoon annetun injektion (50 IU/kg) jälkeen 81:llä aiemmin hoitoa saaneella 18‒60-vuotiaalla aikuisella, joilla oli diagnosoitu vaikea A-hemofilia sekä FVIII <1 %.
Farmakokineettiset muuttujat perustuivat hyytymistekijän VIII aktiivisuuteen plasmassa, joka mitattiin kromogeenisellä substraattimäärityksellä (ero yksivaiheisella hyytymistestillä tehtyyn hyytymistekijän VIII aktiivisuusmääritykseen, ks. kohta Annostus ja antotapa). 3−6 kuukautta alkuvaiheen farmakokineettisen arvion jälkeen saatu farmakokineettinen profiili oli verrannollinen ensimmäisen annoksen jälkeen saadun farmakokineettisen profiilin kanssa.
Farmakokineettiset muuttujat kerta-annoksena annetun 50 IU/kg AFSTYLA-injektion jälkeen - Kromogeeninen substraattimääritys:
Farmakokineettiset muuttujat | rVIII-SingleChain 50 IU/kg (N=81) Keskiarvo (CV%) Mediaani (Min,Max) |
IR(IU/dl)/(IU/kg) | 2,00 (20,8) 1,99 (0,868, 2,90) |
Cmax (IU/dl) | 106 (18,1) 106 (62,4, 151) |
AUC0-inf (IU*h/dl) | 1960 (33,1) 1910 (932, 4090) |
t1/2 (h) | 14.2 (26,0) 13,7 (7,54; 23,9) |
MRT (h) | 20,4 (25,8) 20,2 (10,8; 35,1) |
CL (ml/h/kg) | 2,90 (34,4) 2,67 (1,26; 5,79) |
Vss (ml/kg) | 55,2 (20,8) 53,2 (32,4; 99,6) |
IR = inkrementaalinen saanto 30 minuuttia injektion jälkeen; Cmax = maksimipitoisuus, AUC0-inf = äärettömyyteen ekstrapoloitu hyytymistekijän VIII aktiivisuus-aikakäyrän alla oleva pinta-ala; t1/2 = puoliintumisaika; MRT = keskimääräinen viipymisaika; CL = painon mukaan suhteutettu puhdistuma (N=80); Vss = painon mukaan suhteutettu vakaan tilan jakautumistilavuus. IR ja Cmax korjattiin suhteessa lähtötilanteeseen, mutta muita parametreja ei korjattu suhteessa lähtötilanteeseen (N=81).
Pediatriset potilaat
AFSTYLA-valmisteen farmakokinetiikkaa tutkittiin 10:llä aiemmin hoitoa saaneella nuorella (12 − < 18-vuotiaalla) ja 39:llä aiemmin hoitoa saaneella lapsella (0 − < 12-vuotiaalla) laskimoon injektiona annetun kerta-annoksen 50 IU/kg jälkeen. Kaikilla potilailla oli todettu vaikea A‑hemofilia sekä < 1 % hyytymistekijää VIII.
Farmakokineettiset muuttujat perustuivat hyytymistekijän VIII aktiivisuuteen plasmassa, joka mitattiin kromogeenisellä substraattimäärityksellä (ero yksivaiheisella hyytymistestillä tehtyyn hyytymistekijän VIII aktiivisuusmääritykseen, ks. kohta Annostus ja antotapa).
Farmakokineettisten muuttujien vertailu ikäluokittain kerta-annoksena annetun 50 IU/kg AFSTYLA-injektion jälkeen - Kromogeeninen määritys:
Farmakokineettiset muuttujat | 0 − < 6 vuotta (N = 20) Keskiarvo (CV%) Mediaani (Min, Max) | 6 − < 12 vuotta (N = 19) Keskiarvo (CV%) Mediaani (Min, Max) | 12 − < 18 vuotta (N = 10) Keskiarvo (CV%) Mediaani (Min, Max) |
IR(IU/dl)/(IU/kg) | 1,60 (21,1) 1,55 (1,18, 2,76) | 1,66 (19,7) 1,69 (0,92, 2,35) | 1,69 (24,8) 1,76 (0,88, 2,44) |
Cmax (IU/dl) | 80,2 (20,6) 78,6 (59,3, 138) | 83,5 (19,5) 84,5 (46,4, 117) | 89,7 (24,8) 92,4 (45,5, 131) |
AUC0-inf (IU*h/dl) | 1080 (31,0) 985 (561, 2010) | 1170 (26,3) 1120 (641, 1810) | 1540 (36,5) 1520 (683, 2380) |
t1/2 (h) | 10,4 (28,7) 10,1 (5,19, 17,8) | 10,2 (19,4) 10,0 (6,92, 14,8) | 14,3 (33,3) 13,5 (6,32; 23,8) |
MRT (h) | 12,4 (25,0) 13,0 (6,05, 17,9) | 12,3 (16,8) 12,8 (8,22, 16,0) | 20,0 (32,2) 18,6 (9,17; 31,7) |
CL(ml/h/kg) | 5,07 (29,6) 5,08 (2,52, 8,92) | 4,63 (29,5) 4,48 (2,79, 7,71) | 3,80 (46,9) 3,31 (2,10; 7,32) |
Vss (ml/kg) | 71,0 (11,8) 70,7 (57,3, 88,3) | 67,1 (22,3) 64,9 (44,3, 111) | 68,5 (29,9) 62,0 (45,9; 121) |
IR = inkrementaalinen saanto 30 minuuttia injektion jälkeen 12 − < 18-vuotiailla ja 60 minuuttia injektion jälkeen 1 − < 12‑vuotiailla; Cmax = maksimipitoisuus, AUC = äärettömyyteen ekstrapoloitu hyytymistekijän VIII aktiivisuus-aikakäyrän alla oleva pinta-ala; t1/2 = puoliintumisaika; MRT = keskimääräinen viipymisaika; CL = painon mukaan suhteutettu puhdistuma; Vss = painon mukaan suhteutettu vakaan tilan jakautumistilavuus. IR ja Cmax korjattiin suhteessa lähtötilanteeseen, mutta muita parametreja ei korjattu suhteessa lähtötilanteeseen.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, kerta-annoksen ja toistuvan altistuksen aiheuttamaa toksisuutta, paikallista siedettävyyttä ja trombogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine
L-histidiini
Polysorbaatti 80
Kalsiumklorididihydraatti
Natriumkloridi
Sakkaroosi
Liuotin
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden tai liuottimien kanssa, lukuun ottamatta niitä, jotka mainitaan kohdissa Annostus ja antotapa ja Pakkaustyyppi ja pakkauskoot.
Kestoaika
3 vuotta.
Valmisteen käytönaikaisen kemiallisen ja fysikaalisen säilyvyyden on osoitettu olevan käyttökuntoon saattamisen jälkeen 48 tuntia huoneenlämmössä (alle 25 °C). Mikrobiologisista syistä valmiste on käytettävä heti. Jos sitä ei käytetä heti, käytönaikainen säilytysaika ja olosuhteet ennen käyttöä ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä. Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
AFSTYLA voidaan säilyttää huoneenlämmössä alle 25 °C yhden kolmen kuukauden mittaisen jakson ajan kotelossa ja injektiopulloissa mainitun kestoajan puitteissa. Kun valmiste on otettu jääkaapista, sitä ei saa laittaa enää takaisin jääkaappiin. Merkitse pakkaukseen päivämäärä, jolloin valmiste on siirretty huoneenlämpöön.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
AFSTYLA injektiokuiva-aine ja liuotin, liuosta varten
250 IU (L:ei) 250 IU (201,96 €)
500 IU (L:ei) 500 IU (375,16 €)
1000 IU (L:ei) 1000 IU (717,79 €)
1500 IU (L:ei) 1500 IU (1054,48 €)
2000 IU (L:ei) 2000 IU (1384,44 €)
2500 IU (L:ei) 2500 IU (1719,46 €)
3000 IU (L:ei) 3000 IU (2041,77 €)
PF-selosteen tieto
AFSTYLA 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (250 IU) 6 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), oranssi levy (muovia) ja vihreäraitainen korkki (alumiinia).
2,5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), levy (muovia) ja korkki (alumiinia).
AFSTYLA 500 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (500 IU) 6 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), sininen levy (muovia) ja vihreäraitainen korkki (alumiinia).
2,5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), levy (muovia) ja korkki (alumiinia).
AFSTYLA 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (1000 IU) 6 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), vihreä levy (muovia) ja vihreäraitainen korkki (alumiinia).
2,5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), levy (muovia) ja korkki (alumiinia).
AFSTYLA 1500 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (1500 IU) 10 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), turkoosi levy (muovia) ja vihreäraitainen korkki (alumiinia).
5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), levy (muovia) ja korkki (alumiinia).
AFSTYLA 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (2000 IU) 10 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), purppuranvärinen levy (muovia) ja vihreäraitainen korkki (alumiinia).
5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), levy (muovia) ja korkki (alumiinia).
AFSTYLA 2500 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (2500 IU) 10 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), vaaleanharmaa levy (muovia) ja vihreäraitainen korkki (alumiinia).
5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), levy (muovia) ja korkki (alumiinia).
AFSTYLA 3000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (3000 IU) 10 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), keltainen levy (muovia) ja vihreäraitainen korkki (alumiinia).
5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (kumia), levy (muovia) ja korkki (alumiinia).
Pakkauskoot
Yksi 250, 500 tai 1000 IU pakkaus sisältää:
1 injektiopullo, joka sisältää kuiva-ainetta
1 injektiopullo, joka sisältää 2,5 ml injektionesteisiin käytettävää vettä
1 suodattimella varustettu siirtolaite 20/20
Sisäpakkaus, joka sisältää:
1 kertakäyttöinen 5 ml:n ruisku
1 laskimopunktiosetti
2 desinfektiopyyhettä
1 ei-steriili laastari
Yksi 1500, 2000, 2500 tai 3000 IU pakkaus sisältää:
1 injektiopullo, joka sisältää kuiva-ainetta
1 injektiopullo, joka sisältää 5 ml injektionesteisiin käytettävää vettä
1 suodattimella varustettu siirtolaite 20/20
Sisäpakkaus, joka sisältää:
1 kertakäyttöinen 10 ml:n ruisku
1 laskimopunktiosetti
2 desinfektiopyyhettä
1 ei-steriili laastari
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai kellertävä injektiokuiva-aine tai mureneva massa ja kirkas, väritön liuotin liuosta varten.
pH: 6,6–7,3
Osmolaliteetti: 500–600 mosm/kg
Käyttö- ja käsittelyohjeet
Yleiset ohjeet
- Käyttövalmiiksi saatetun liuoksen on oltava lähes väritöntä, kirkasta tai hieman opaalinhohtoista. Kun käyttövalmiiksi saatettu valmiste on suodatettu/vedetty ruiskuun (ks. seuraavassa), valmiste on tarkistettava silmämääräisesti ennen antoa, ettei siinä ole havaittavissa hiukkasia eikä värinmuutoksia.
- Älä käytä liuosta, jos se näyttää samealta tai jos siinä on hiutaleita tai hiukkasia.
- Valmisteen käyttökuntoon saattaminen ja vetäminen ruiskuun on tehtävä aseptisissa olosuhteissa.
Käyttökuntoon saattaminen ja anto
Anna liuottimen lämmetä huoneenlämpöiseksi. Varmista, että kuiva-aineen ja liuottimen sisältävien injektiopullojen irti napsautettavat flip-off-sulkimet on poistettu ja tulpat on käsitelty antiseptisella liuoksella. Tulpan on sen jälkeen annettava kuivua ennen Mix2Vial-pakkauksen avaamista.
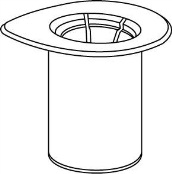
| 1. Avaa Mix2Vial-laitepakkaus vetämällä suojakansi pois. Älä poista Mix2Vial-laitetta läpipainopakkauksesta! |
| 2. Aseta liuottimen sisältävä injektiopullo tasaiselle, puhtaalle alustalle ja ota injektiopullosta tukeva ote. Ota Mix2Vial sekä pakkaus ja paina sinisen sovittimen piikki kohtisuoraan liuotinpullon tulpan läpi. |
| 3. Poista pakkaus varovasti Mix2Vial-laitteesta siten, että pidät pakkauksen reunasta kiinni ja vedät kohtisuoraan ylöspäin. Varmista, että vedät pois vain pakkauksen etkä Mix2Vial-laitetta. |
| 4. Aseta kuiva-aineen sisältävä injektiopullo tasaiselle ja tukevalle alustalle. Kun liuottimen sisältävä injektiopullo on kiinnitettynä Mix2Vial-laitteeseen, käännä ne ylösalaisin ja paina läpinäkyvän sovittimen piikki suoraan kuiva-aineen sisältävän injektiopullon kumitulpan läpi. Liuotin siirtyy automaattisesti kuiva-aineen sisältävään injektiopulloon. |
| 5. Ota toisella kädellä kiinni Mix2Vial-laitteen kuiva-aineen sisältävän injektiopullon puolelta ja toisella kädellä liuottimen sisältävän injektiopullon puolelta ja kierrä vastapäivään laite kahteen osaan varovasti. |
6 | 6. Pyörittele kuiva-aineen sisältävää injektiopulloa ja siihen kiinnitettyä läpinäkyvää sovitinta, kunnes kuiva-aine on liuennut täysin. Älä ravista. |
7 | 7. Vedä tyhjään, steriiliin ruiskuun ilmaa. Kun kuiva-aineen sisältävä injektiopullo on oikeinpäin, kiinnitä ruisku Mix2Vial-sovittimen Luer Lock ‑liittimeen kiertämällä myötäpäivään. Ruiskuta ilma kuiva-aineen sisältävään injektiopulloon. |
 1
1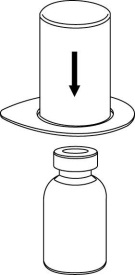 2
2Valmisteen vetäminen ruiskuun ja anto
| 8. Kun ruiskun mäntä on sisään painettuna, käännä laite ja injektiopullo ylösalaisin ja vedä liuos ruiskuun vetämällä mäntää hitaasti ulospäin. |
| 9. Kun liuos on nyt siirretty ruiskuun, ota tukeva ote ruiskun kammiosta (pitäen ruiskun mäntää samalla alaspäin) ja irrota läpinäkyvä Mix2Vial-sovitin ruiskusta kiertämällä vastapäivään. |
AFSTYLA-injektio suositellaan antamaan vain pakkauksen sisältämillä antolaitteilla, koska hoito voi epäonnistua, jos hyytymistekijä VIII adsorptoituu muun injektiolaitteen sisäpintaan.
Varovaisuutta on noudatettava, jotta valmistetta sisältävään ruiskuun ei pääse verta, koska vaarana on, että veri hyytyy ruiskussa ja fibriinihyytymät päätyvät potilaan verenkiertoon.
AFSTYLA-valmistetta ei saa laimentaa.
Käyttökuntoon saatettu liuos on annettava hitaana injektiona laskimoon erillistä injektio-/infuusiolinjaa käyttäen potilaalle mukavalla nopeudella, enintään 10 ml/min.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
AFSTYLA injektiokuiva-aine ja liuotin, liuosta varten
250 IU 250 IU
500 IU 500 IU
1000 IU 1000 IU
1500 IU 1500 IU
2000 IU 2000 IU
2500 IU 2500 IU
3000 IU 3000 IU
- Ylempi erityiskorvaus (100 %). Krooniset hyytymishäiriöt (126).
- Peruskorvaus (40 %).
ATC-koodi
B02BD02
Valmisteyhteenvedon muuttamispäivämäärä
10.06.2022
Yhteystiedot
 CSL BEHRING AB
CSL BEHRING AB Box 712
182 17 Danderyd
Ruotsi
+46 (0) 8 544 966 70
www.cslbehring.fi
info@cslbehring.se