
AKEEGA tabletti, kalvopäällysteinen 50/500 mg, 100/500 mg
Vaikuttavat aineet ja niiden määrät
Akeega 50 mg/500 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää niraparibitosylaattimonohydraattia määrän, joka vastaa 50 mg:aa niraparibia, sekä 500 mg abirateroniasetaattia, joka vastaa 446 mg:aa abirateronia.
Akeega 100 mg/500 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää niraparibitosylaattimonohydraattia määrän, joka vastaa 100 mg:aa niraparibia, sekä 500 mg abirateroniasetaattia, joka vastaa 446 mg:aa abirateronia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 241 mg laktoosia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Akeega yhdessä prednisonin tai prednisolonin kanssa on tarkoitettu käytettäväksi
● yhdistelmänä androgeenideprivaatiohoidon kanssa niiden aikuispotilaiden hoitoon, joilla on metastasoitunut hormonisensitiivinen eturauhassyöpä (mHSPC) ja BRCA1/2-mutaatioita (ituradassa ja/tai somaattisia)
● niiden aikuispotilaiden hoitoon, joilla on metastasoitunut kastraatioresistentti eturauhassyöpä (mCRPC) ja BRCA1/2-mutaatioita (ituradassa ja/tai somaattisia) ja joille solunsalpaajahoito ei ole kliinisesti aiheellinen.
Ehto
Hoito on aloitettava ja toteutettava eturauhassyövän lääkehoitoon perehtyneen erikoislääkärin valvonnassa.
Annostus ja antotapa
Hoidon Akeega-valmisteella yhdistelmänä prednisonin tai prednisolonin kanssa tulee aloittaa erikoislääkäri, joka on perehtynyt eturauhassyövän lääketieteelliseen hoitoon, ja hänen tulee myös valvoa hoitoa.
Ennen Akeega-hoidon aloittamista aikuispotilaille
● joilla on metastasoitunut hormonisensitiivinen eturauhassyöpä (mHSPC), positiivinen BRCA-status on varmistettava validoidulla testimenetelmällä (ks. kohta Farmakodynamiikka)
● joilla on metastasoitunut kastraatioresistentti eturauhassyöpä (mCRPC), positiivinen BRCA-status on varmistettava validoidulla testimenetelmällä (ks. kohta Farmakodynamiikka).
Annostus
Suositeltu Akeega-aloitusannos on 200 mg/1 000 mg (kaksi 100 mg niraparibia ja 500 mg abirateroniasetaattia sisältävää tablettia) kerta-annoksena päivittäin joka päivä suunnilleen samaan aikaan (ks. Antotapa jäljempänä). Annoksen pienentämiseen on saatavana 50 mg/500 mg tablettivahvuus.
Potilailla, joille ei ole tehty kirurgista kastraatiota, pitää jatkaa lääkkeellistä kastraatiota gonadotropiinien vapauttajahormonin (GnRH) analogilla.
Prednisonin tai prednisolonin annostus
Akeega-valmistetta käytetään mHSPC:n hoitoon yhdistelmänä päivittäin otettavan 5 mg:n prednisoni- tai prednisoloniannoksen kanssa. Akeega-valmistetta käytetään mCRPC:n hoitoon yhdistelmänä päivittäin otettavan 10 mg:n prednisoni- tai prednisoloniannoksen kanssa.
Hoidon kesto
Potilaita hoidetaan, kunnes sairaus etenee tai ilmaantuu toksisuutta, joka ei ole hyväksyttävissä.
Ottamatta jäänyt annos
Jos Akeega-, prednisoni- tai prednisoloniannos jää ottamatta, se pitää ottaa mahdollisimman pian samana päivänä, minkä jälkeen seuraavana päivänä palataan tavanomaiseen hoitoaikatauluun. Ottamatta jääneen annoksen korvaamiseksi ei saa ottaa ylimääräisiä tabletteja.
Annosmuutokset haittavaikutusten vuoksi
Ei-hematologiset haittavaikutukset
Potilailla, joille kehittyy ≥ 3. asteen ei-hematologisia haittavaikutuksia, hoito pitää keskeyttää ja aloittaa asianmukainen lääketieteellinen hoito (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Akeega-hoitoa ei pidä aloittaa uudelleen ennen kuin toksisuuden oireet ovat lieventyneet 1. asteeseen tai lähtötasolle.
Hematologiset haittavaikutukset
Jos potilaalle kehittyy ≥ 3. asteen tai sietämätöntä hematologista toksisuutta, Akeega-hoito pitää mieluummin keskeyttää kuin lopettaa ja harkita tukihoitoa. Akeega-hoito pitää lopettaa pysyvästi, jos hematologinen toksisuus ei ole korjautunut hyväksyttävälle tasolle 28 päivän kuluessa hoidon keskeyttämisen jälkeen.
Taulukossa 1 luetellaan annosmuutoksia koskevat suositukset trombosytopenian ja neutropenian yhteydessä.
| Taulukko 1. Annosmuutoksia koskevat suositukset trombosytopenian ja neutropenian yhteydessä | |
| 1. aste | Ei muutosta, harkitse viikoittaista seurantaa. |
| 2. aste | Seuranta vähintään viikoittain, ja harkitse Akeega-hoidon keskeyttämistä, kunnes toksisuus lievenee 1. asteeseen tai lähtötasolle.1 Jatka sitten Akeega-hoitoa. Viikoittaista seurantaa suositellaan 28 päivän ajan hoidon jatkamisen jälkeen. |
| ≥ 3. aste | Keskeytä Akeega-hoito ja seuraa potilaan vointia vähintään viikoittain, kunnes trombosyytti- ja neutrofiilimäärä korjautuu 1. asteeseen tai lähtötasolle.1 Jatka sitten Akeega-hoitoa, tai tarvittaessa käytä kahta pienemmän vahvuuden tablettia (50 mg/500 mg). Verenkuvan viikoittaista seurantaa suositellaan 28 päivän ajan hoidon jatkamisen tai pienemmän annosvahvuuden (kaksi 50 mg/500 mg tablettia) käytön aloittamisen jälkeen. Kun pienemmänannosvahvuuden käyttö aloitetaan, ks. maksan toimintaa koskevat lisätiedot jäljempänä kohdassa Seurantaa koskevat suositukset. |
| ≥ 3. asteen toinen ilmaantumiskerta | Keskeytä Akeega-hoito, ja seuraa potilaan vointia vähintään viikoittain, kunnes trombosyytti- ja/tai neutrofiilimäärä korjautuu 1. asteeseen. Jatkohoito pitää aloittaa kahdella pienemmän vahvuuden tabletilla (50 mg/500 mg). Kun hoitoa jatketaan pienemmällä Akeega-vahvuudella, viikoittaista seurantaa suositellaan 28 päivän ajan. Kun pienemmän annosvahvuuden (kaksi 50 mg/500 mg tablettia) käyttö aloitetaan, ks. maksan toimintaa koskevat lisätiedot jäljempänä kohdassa Seurantaa koskevat suositukset. Jos potilas käytti jo ennestään pienempää Akeega-tablettivahvuutta (50 mg/500 mg), harkitse hoidon lopettamista. |
| ≥ 3. asteen kolmas ilmaantumiskerta | Lopeta hoito pysyvästi. |
| 1 Akeega-hoidon ollessa keskeytettynä lääkäri voi harkita abirateroniasetaattia ja prednisonia tai prednisolonia ja antaa näitä päivittäisen abirateroniasetaattiannoksen ylläpitämiseksi (ks. abirateroniasetaatin valmistetiedot). | |
Akeega-hoitoa voidaan jatkaa vasta, kun trombosytopeniasta ja neutropeniasta aiheutunut toksisuus on lieventynyt 1. asteeseen tai korjautunut lähtötasolle. Hoitoa voidaan jatkaa pienemmällä Akeega-vahvuudella 50 mg/500 mg (2 tablettia). Yleisimmät haittavaikutukset, ks. kohta Haittavaikutukset.
Akeega-hoito pitää keskeyttää ≥ 3. asteen anemian yhteydessä ja potilaalle pitää antaa tukihoitoa, kunnes anemia korjautuu ≤ 2. asteeseen. Annoksen pienentämistä (kahteen 50 mg/500 mg tablettiin) pitää harkita, jos anemia kliinisen arvion mukaan jatkuu. Taulukossa 2 luetellaan annosmuutoksia koskevat suositukset anemian yhteydessä.
| Taulukko 2. Annosmuutoksia koskevat suositukset anemian yhteydessä | |
| 1. aste | Ei muutosta, harkitse viikoittaista seurantaa. |
| 2. aste | Seuranta vähintään viikoittain 28 päivän ajan, jos anemia oli lähtötilanteessa ≤ 1. aste. |
| ≥ 3. aste | Keskeytä Akeega-hoito1 ja anna tukihoitoa sekä seuraa potilaan vointia vähintään viikoittain, kunnes anemia on korjautunut ≤ 2. asteeseen. Annoksen pienentämistä (kahteen pienemmän vahvuuden tablettiin[50 mg/500 mg]) pitää harkita, jos anemia kliinisen arvion mukaan jatkuu. Kun pienemmänannosvahvuuden käyttö aloitetaan, ks. maksan toimintaa koskevat lisätiedot jäljempänä kohdassa Seurantaa koskevat suositukset. |
| ≥ 3. asteen toinen ilmaantumiskerta | Keskeytä Akeega-hoito, anna tukihoitoa ja seuraa potilaan vointia vähintään viikoittain, kunnes anemia on korjautunut ≤ 2. asteeseen. Jatkohoito pitää aloittaa kahdella pienemmän vahvuuden tabletilla (50 mg/500 mg). Kun hoitoa jatketaan pienemmällä Akeega-vahvuudella, viikoittaista seurantaa suositellaan 28 päivän ajan. Kun pienemmänannosvahvuuden käyttö aloitetaan, ks. maksan toimintaa koskevat lisätiedot jäljempänä kohdassa Seurantaa koskevat suositukset. Jos potilas käytti jo ennestään pienempää Akeega-tablettivahvuutta (50 mg/500 mg), harkitse hoidon lopettamista. |
| ≥ 3. asteen kolmas ilmaantumiskerta | Harkitse Akeega-hoidon lopettamista kliinisen arvion perusteella. |
| 1 Akeega-hoidon ollessa keskeytettynä lääkäri voi harkita abirateroniasetaattia ja prednisonia tai prednisolonia ja antaa näitä päivittäisen abirateroniasetaattiannoksen ylläpitämiseksi (ks. abirateroniasetaatin valmistetiedot). | |
Maksatoksisuus
Jos potilaalle kehittyy ≥ 3. asteen maksatoksisuutta (alaniiniaminotransferaasipitoisuus [ALAT] suurenee tai aspartaattiaminotransferaasipitoisuus [ASAT] suurenee yli viisinkertaiseksi normaaliarvojen ylärajaan [upper limit of normal, ULN] nähden), Akeega-hoito pitää keskeyttää ja maksan toimintaa pitää seurata tarkoin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoitoa voidaan jatkaa vasta sen jälkeen, kun maksan toimintakokeet ovat palautuneet potilaan lähtötilanteen tasolle ja pienentämällä annosta käyttäen yhtä tablettia suositellusta Akeega-tablettivahvuudesta (vastaa 100 mg:aa niraparibia/500 mg:aa abirateroniasetaattia). Jos potilaan hoitoa jatketaan, seerumin transaminaasipitoisuuksia pitää seurata kolmen kuukauden ajan vähintään kahden viikon välein ja sen jälkeen kuukausittain. Jos maksatoksisuus uusiutuu käytettäessä pienempää vuorokausiannosta 100 mg/500 mg (1 tabletti), Akeega-hoito pitää lopettaa.
Jos potilaalle kehittyy Akeega-hoidon aikana vaikea-asteista maksatoksisuutta (ALAT tai ASAT 20 kertaa ULN), hoito pitää lopettaa pysyvästi.
Lopeta Akeega-hoito pysyvästi potilailta, joiden ALAT-arvo kohoaa tasolle yli 3 × ULN ja samanaikaisesti kokonaisbilirubiinipitoisuus kohoaa tasolle yli 2 × ULN ilman sappitietukosta tai muuta syytä pitoisuuksien samanaikaiseen kohoamiseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Seurantaa koskevat suositukset
Täydellinen verenkuva on määritettävä ennen hoidon aloittamista, ensimmäisen kuukauden ajan viikoittain, seuraavien kahden kuukauden ajan joka toinen viikko, minkä jälkeen ensimmäisen vuoden ajan kuukausittain ja sitten hoidon loppuajan joka toinen kuukausi, jotta voidaan seurata hematologisissa parametreissa tapahtuvia kliinisesti merkittäviä muutoksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Seerumin aminotransferaasipitoisuudet ja kokonaisbilirubiinipitoisuus pitää mitata ennen hoidon aloittamista, kolmen ensimmäisen hoitokuukauden aikana kahden viikon välein ja sen jälkeen ensimmäisen vuoden ajan kuukausittain, minkä jälkeen hoidon keston ajan joka toinen kuukausi. Kun keskeytettyä hoitoa jatketaan pienemmällä Akeega-annosvahvuudella (kaksi 50 mg/500 mg tablettia), maksan toimintaa pitää seurata suurentuneen abirateronialtistuksen riskin vuoksi kuuden viikon ajan joka toinen viikko (ks. kohta Farmakokinetiikka) ennen kuin palataan normaaliin seurantaan. Seerumin kaliumpitoisuutta pitää seurata ensimmäisen vuoden ajan kuukausittain ja sen jälkeen hoidon keston ajan joka toinen kuukausi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verenpainetta pitää seurata kahden ensimmäisen kuukauden ajan viikoittain, ensimmäisen vuoden ajan kuukausittain ja sen jälkeen hoidon keston ajan joka toinen kuukausi.
Jos potilaalla on ennestään hypokalemia tai hypokalemia kehittyy Akeega-hoidon aikana, harkitse potilaan kaliumpitoisuuden pitämistä tasolla ≥ 4,0 mM.
Erityispotilasryhmät
Iäkkäät
Iäkkäiden potilaiden annosta ei ole tarpeen muuttaa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa (Child–Pugh-luokka A) ennestään sairastavien potilaiden annosta ei ole tarpeen muuttaa. Useiden Akeega-annosten kliinisestä turvallisuudesta ja tehosta ei ole tietoja keskivaikeaa tai vaikeaa maksan vajaatoimintaa (Child–Pugh-luokka B tai C) sairastaville potilaille. Annosmuutoksia ei voida ennakoida. Akeega-valmisteen käyttöä keskivaikeaa maksan vajaatoimintaa sairastaville potilaille pitää arvioida tarkoin, sillä hyödyn pitää olla selvästi mahdollisia riskejä suurempi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Akeega on vasta-aiheista potilaille, joilla on vaikea maksan vajaatoiminta (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei ole tarpeen muuttaa, mutta keskivaikeaa munuaisten vajaatoimintaa sairastavia potilaita pitää seurata tarkoin turvallisuuteen liittyvien tapahtumien varalta, sillä niraparibialtistus voi lisääntyä. Akeega-valmisteen käytöstä vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaissairautta sairastaville hemodialyysihoitoa saaville potilaille ei ole tietoja. Akeega-valmistetta voidaan käyttää vaikeaa munuaisten vajaatoimintaa sairastaville potilaille vain, jos hyödyt ovat mahdollisia riskejä suuremmat; lisäksi potilaan munuaisten toimintaa sekä potilaalle ilmaantuvia haittavaikutuksia pitää seurata tarkoin (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää Akeega-valmistetta pediatrisille potilaille.
Antotapa
Akeega otetaan suun kautta.
Tabletit on otettava kerta-annoksena kerran päivässä. Akeega-tabletit pitää ottaa tyhjään mahaan vähintään 1 tunti ennen ateriaa tai aikaisintaan 2 tuntia aterian jälkeen (ks. kohta Farmakokinetiikka). Akeega-tabletit on optimaalisen imeytymisen vuoksi nieltävä kokonaisina veden kanssa, eikä niitä saa jakaa, murskata eikä pureskella.
Varotoimet ennen valmisteen käsittelyä tai antoa
Naisten, jotka ovat raskaana tai voivat tulla raskaaksi, on käytettävä suojakäsineitä tabletteja käsitellessään (ks. kohta Käyttö- ja käsittelyohjeet).
Vasta-aiheet
Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
Naiset, jotka ovat raskaana tai voivat tulla raskaaksi (ks. kohta Raskaus ja imetys).
Vaikea maksan vajaatoiminta (Child–Pugh-luokka C [ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka]).
Akeega yhdistelmänä prednisonin tai prednisolonin kanssa on vasta-aiheista yhdessä radium-223 (Ra‑223) ‑hoidon kanssa.
Varoitukset ja käyttöön liittyvät varotoimet
Hematologiset haittavaikutukset
Akeega-hoitoa saaneilla potilailla on raportoitu hematologisia haittavaikutuksia (trombosytopenia, anemia ja neutropenia) (ks. kohta Annostus ja antotapa).
Täydellisen verenkuvan määrittämistä suositellaan ensimmäisen kuukauden ajan viikoittain, seuraavien kahden kuukauden ajan joka toinen viikko, minkä jälkeen ensimmäisen vuoden ajan kuukausittain ja sitten hoidon loppuajan joka toinen kuukausi, jotta voidaan seurata hematologisissa parametreissa hoidon aikana tapahtuvia kliinisesti merkittäviä muutoksia (ks. kohta Annostus ja antotapa).
Viikoittainen seuranta toisen kuukauden ajan voi olla aiheellista yksilöllisten laboratorioarvojen perusteella.
Jos potilaalle kehittyy vaikea-asteista pitkittyvää hematologista toksisuutta, mukaan lukien pansytopeniaa, joka ei korjaannu 28 päivän kuluessa hoidon keskeyttämisestä, Akeega-hoito pitää lopettaa.
Muiden trombosyyttimäärää tunnetusti vähentävien lääkkeiden käytössä Akeega-hoitoa saaville potilaille pitää olla varovainen trombosytopenian riskin vuoksi (ks. kohta Haittavaikutukset).
Kun hematologisten haittavaikutusten vuoksi keskeytettyä hoitoa jatketaan pienemmällä Akeega-annosvahvuudella (kaksi 50 mg/500 mg tablettia), maksan toimintaa pitää seurata suurentuneen abirateronialtistuksen riskin vuoksi kuuden viikon ajan joka toinen viikko (ks. kohta Farmakokinetiikka) ennen kuin palataan normaaliin seurantaan (ks. kohta Annostus ja antotapa).
Hypertensio
Akeega voi aiheuttaa hypertensiota, ja potilaalla ennestään olevan hypertension pitää olla riittävässä hoitotasapainossa ennen Akeega-hoidon aloittamista. Verenpainetta pitää seurata kahden kuukauden ajan vähintään viikoittain, minkä jälkeen sitä pitää seurata ensimmäisen Akeega-hoitovuoden ajan kuukausittain ja sen jälkeen joka toinen kuukausi Akeega-hoidon ajan.
Mineralokortikoidiylimäärästä aiheutuva hypokalemia, nesteen kertyminen elimistöön sekä sydämen ja verisuoniston haittavaikutukset
Akeega saattaa aiheuttaa hypokalemiaa ja nesteen kertymistä elimistöön (ks. kohta Haittavaikutukset) CYP17-entsyymin toiminnan estymisestä aiheutuvan mineralokortikoidipitoisuuden suurenemisen seurauksena (ks. kohta Farmakodynamiikka). Kortikosteroidin samanaikainen anto suppressoi adrenokortikotrooppisen hormonin (ACTH:n) erittymistä, jolloin näiden haittavaikutusten ilmaantuvuus ja vaikeusaste vähenevät. Hoidossa on noudatettava varovaisuutta, jos hypokalemia (esim. sydänglykosideja käyttävillä potilailla) tai nesteen kertyminen elimistöön (esim. potilailla, joilla on sydämen vajaatoimintaa, vaikea-asteinen tai epästabiili angina pectoris, äskettäin ollut sydäninfarkti, kammioperäisiä rytmihäiriöitä tai vaikea-asteista munuaisten vajaatoimintaa) saattavat pahentaa potilaan perussairautta. Potilailla, joilla on ollut Akeega-hoidon yhteydessä hypokalemiaa, on havaittu QT-ajan pitenemistä. Hypokalemia ja nesteen kertyminen pitää korjata ja saada hoitotasapainoon.
Ennen kuin hoidetaan potilaita, joilla on kongestiivisen sydämen vajaatoiminnan merkittävä riski (esim. anamneesissa sydämen vajaatoimintaa tai sydäntapahtumia, kuten iskeeminen sydänsairaus), sydämen vajaatoiminta pitää hoitaa ja sydämen toiminta pitää optimoida. Nesteen kertymistä (painon nousu, raajojen turvotus) ja muita kongestiivisen sydämen vajaatoiminnan oireita ja löydöksiä pitää seurata kolmen kuukauden ajan kahden viikon välein ja sen jälkeen kuukausittain, ja poikkeavuudet pitää korjata. Akeega-valmisteen käytössä pitää olla varovainen, jos potilaalla on anamneesissa sydän- ja verisuonitauti.
Akeega-hoitoa saavilla potilailla sydämeen liittyvien riskitekijöiden (mukaan lukien hypertension, dyslipidemian ja diabeteksen) hoito pitää optimoida, ja näitä potilaita pitää seurata sydänsairauden oireiden ja löydösten havaitsemiseksi.
Akeega-valmisteen komponentti abirateroniasetaatti suurentaa mineralokortikoidipitoisuutta, ja siihen liittyy sydän- ja verisuonitapahtumien riski. Mineralokortikoidiylimäärästä voi aiheutua hypertensiota, hypokalemiaa ja nesteen kertymistä. Aiempi altistus androgeenideprivaatiohoidolle sekä korkea ikä ovat sydän- ja verisuonisairastavuuden ja ‑kuolleisuuden lisäriskejä. AMPLITUDE- ja MAGNITUDE-tutkimuksista suljettiin pois potilaat, joilla oli kliinisesti merkittävä sydänsairaus, josta oli osoituksena sydäninfarkti, valtimoiden ja laskimoiden tromboottiset tapahtumat edeltävien 6 kuukauden aikana, vaikea-asteinen tai epästabiili angina pectoris tai NYHA-luokan II–IV sydämen vajaatoiminta tai sydämen ejektiofraktion mittaustulos < 50 %. Jos potilaalla on anamneesissa sydämen vajaatoimintaa, tila pitää optimoida kliinisesti ja oireiden asianmukainen hoito pitää aloittaa. Jos sydämen toiminta heikkenee kliinisesti merkittävästi, Akeega-hoidon lopettamista pitää harkita.
Infektiot
Vaikea-asteisia infektioita, mukaan lukien kuolemaan johtaneita COVID‑19‑infektioita, ilmaantui sekä AMPLITUDE- että MAGNITUDE-tutkimuksessa yleisemmin Akeega-hoitoa saaneille potilaille.
Potilaita pitää seurata infektioiden oireiden ja löydösten havaitsemiseksi. Vaikea-asteisia infektioita voi ilmetä ilman neutropeniaa ja/tai leukopeniaa.
Keuhkoembolia
Keuhkoemboliatapauksia raportoitiin sekä AMPLITUDE- että MAGNITUDE-tutkimuksessa Akeega-hoitoa saaneilla potilailla yleisemmin kuin verrokeilla. Potilailla, joilla on anamneesissa keuhkoembolia tai laskimotromboosi, niiden uudelleen ilmaantumisen riski voi olla suurempi. Potilaita pitää seurata keuhkoembolian kliinisten oireiden ja löydösten havaitsemiseksi. Jos keuhkoembolian kliinisiä piirteitä ilmenee, potilas on tutkittava viipymättä, minkä jälkeen annetaan asianmukaista hoitoa.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES)
Posteriorinen reversiibeli enkefalopatiaoireyhtymä on harvinainen, korjautuva, neurologinen sairaus, joka voi ilmetä nopeasti kehittyvinä oireina, mukaan lukien kouristuskohtauksina, päänsärkynä, mielentilan muutoksina, näköhäiriöinä tai kortikaalisena sokeutena, joihin voi liittyä hypertensiota. Posteriorisen reversiibelin enkefalopatiaoireyhtymän diagnosointi edellyttää varmistusta aivojen kuvantamisella, mieluiten magneettikuvauksella.
Posteriorista reversiibeliä enkefalopatiaoireyhtymää on raportoitu potilailla, jotka ovat saaneet 300 mg niraparibia (Akeega-valmisteen komponentti) monoterapiana munasarjasyövän hoitoon. Posteriorista reversiibeliä enkefalopatiaoireyhtymää ei raportoitu AMPLITUDE- eikä MAGNITUDE-tutkimuksissa 200 mg niraparibia eturauhassyövän hoitoon saaneilla potilailla.
Jos posteriorinen reversiibeli enkefalopatiaoireyhtymä ilmaantuu, Akeega-hoito pitää lopettaa pysyvästi ja asianmukainen lääketieteellinen hoito pitää aloittaa.
Maksatoksisuus ja maksan vajaatoiminta
Maksatoksisuus on tunnistettu Akeega-valmisteen komponentin abirateroniasetaatin tärkeäksi tunnistetuksi riskiksi. Abirateroniasetaattiin liittyvää maksatoksisuutta aiheuttavaa mekanismia ei täysin tunneta. Akeega-yhdistelmävalmistetta koskeneista tutkimuksista suljettiin pois potilaat, joilla oli keskivaikeaa tai vaikeaa maksan vajaatoimintaa (NCI-luokitus), sekä potilaat, joiden Child‑Turcotte-Pugh-luokka oli B tai C.
Kaikissa Akeega-valmisteen kliinisissä tutkimuksissa maksatoksisuuden riskiä vähennettiin sulkemalla pois potilaat, joilla oli lähtötilanteessa hepatiitti tai merkittäviä poikkeavuuksia maksan toimintakokeissa (seerumin kokonaisbilirubiinipitoisuus > 1,5 × ULN tai konjugoituneen bilirubiinin pitoisuus > 1 × ULN ja ASAT tai ALAT > 3 × ULN).
Kliinisissä tutkimuksissa esiintyi huomattavaa maksaentsyymipitoisuuksien suurenemista, mikä johti hoidon keskeyttämiseen tai lopettamiseen; tämä ei kuitenkaan ollut yleistä (ks. kohta Haittavaikutukset). Seerumin aminotransferaasipitoisuudet ja kokonaisbilirubiinipitoisuus pitää mitata ennen hoidon aloittamista, kolmen ensimmäisen hoitokuukauden aikana kahden viikon välein, sen jälkeen ensimmäisen vuoden aikana kuukausittain ja sitten hoidon keston ajan joka toinen kuukausi. Kun keskeytettyä hoitoa jatketaan pienemmällä Akeega-annosvahvuudella (kaksi 50 mg/500 mg tablettia), maksan toimintaa pitää seurata suurentuneen abirateronialtistuksen riskin vuoksi kuuden viikon ajan joka toinen viikko (ks. kohta Farmakokinetiikka) ennen kuin palataan normaaliin seurantaan. Jos potilaalle kehittyy maksatoksisuuteen viittaavia kliinisiä oireita tai löydöksiä, seerumin transaminaasipitoisuudet on mitattava heti. Akeega-hoitoa saavan potilaan aminotransferaasipitoisuuksien kohoaminen pitää hoitaa viipymättä keskeyttämällä hoito. Jos ALAT- tai ASAT-arvo suurenee hoidon missä tahansa vaiheessa yli viisinkertaiseksi normaaliarvojen ylärajaan (ULN) nähden, Akeega-hoito on keskeytettävä ja maksan toimintaa on seurattava tarkoin. Hoitoa voidaan jatkaa vasta, kun maksan toimintakokeiden tulokset ovat palautuneet potilaan hoitoa edeltäneisiin arvoihin, ja annosta on tällöin pienennettävä (ks. kohta Annostus ja antotapa).
Jos potilaan ALAT- tai ASAT-arvo kohoaa tasolle > 20 × ULN, hoito on lopetettava pysyvästi. Jos potilaan ALAT-arvo kohoaa tasolle > 3 × ULN ja samanaikaisesti kokonaisbilirubiinipitoisuus nousee tasolle > 2 × ULN ilman sappitietukosta tai muuta samanaikaista nousua selittävää syytä, hoito on lopetettava pysyvästi.
Jos potilaalle kehittyy vaikea-asteista maksatoksisuutta (ALAT- tai ASAT-arvo 20 × ULN) hoidon missä tahansa vaiheessa, Akeega-hoito on lopetettava pysyvästi.
Aktiivista tai oireista virushepatiittia sairastavat potilaat suljettiin pois kliinisistä tutkimuksista, joten Akeega-valmisteen käytön tueksi tälle potilasryhmälle ei ole tietoja.
Keskivaikean maksan vajaatoiminnan (Child–Pugh-luokka B tai ASAT-arvo mikä tahansa ja kokonaisbilirubiiniarvo > 1,5 x – 3 x ULN) on osoitettu lisäävän systeemistä abirateroni- ja niraparibialtistusta (ks. kohta Farmakokinetiikka). Useiden Akeega-annosten kliinisestä turvallisuudesta ja tehosta ei ole tietoja keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Akeega-valmisteen käyttöä keskivaikeaa maksan vajaatoimintaa sairastaville potilaille pitää arvioida tarkoin, sillä hyödyn pitää olla selvästi mahdollisia riskejä suurempi (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka). Jos potilaalla on vaikea maksan vajaatoiminta, Akeega-hoitoa ei pidä antaa (ks. kohdat Annostus ja antotapa, Vasta-aiheet ja Farmakokinetiikka).
Hypoglykemia
Abirateroniasetaatin (Akeega-valmisteen komponentti) ja prednisonin tai prednisolonin yhdistelmän käytössä on raportoitu hypoglykemiaa potilailla, joilla oli ennestään diabetes ja jotka saivat pioglitatsonia tai repaglinidia (metaboloituvat CYP2C8-entsyymin välityksellä) (ks. kohta Yhteisvaikutukset). Diabeetikoiden verensokeripitoisuutta pitää sen vuoksi seurata.
Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia
Munasarjasyöpätutkimuksissa 300 mg niraparibia (Akeega-valmisteen komponentti) saaneilla potilailla on raportoitu myelodysplastista oireyhtymää / akuuttia myelooista leukemiaa, mukaan lukien kuolemaan johtaneita tapauksia.
Sekä AMPLITUDE- että MAGNITUDE-tutkimuksissa myelodysplastista oireyhtymää / akuuttia myelooista leukemiaa, mukaan lukien kuolemaan johtaneita tapauksia, raportoitiin eturauhassyöpää sairastaneilla potilailla, jotka saivat hoitona 200 mg niraparibia ja 1 000 mg abirateroniasetaattia yhdistelmänä prednisonin tai prednisolonin kanssa.
Jos myelodysplastista oireyhtymää / akuuttia myelooista leukemiaa epäillään tai jos pitkittynyt hematologinen toksisuus ei ole hävinnyt keskeyttämällä hoito tai pienentämällä annosta, potilaasta pitää tehdä lähete hematologille jatkotutkimuksia varten. Jos myelodysplastinen oireyhtymä / akuutti myelooinen leukemia varmistuu, Akeega-hoito pitää lopettaa pysyvästi ja potilasta pitää hoitaa asianmukaisesti.
Kortikosteroidihoidon lopettaminen ja hoito stressitilanteissa
Jos prednisoni- tai prednisolonihoito lopetetaan, se on tehtävä varoen ja potilasta on seurattava lisämunuaiskuoren vajaatoiminnan varalta. Jos Akeega-hoitoa jatketaan kortikosteroidihoidon lopettamisen jälkeen, potilasta on seurattava mineralokortikoidiylimäärän oireiden havaitsemiseksi (ks. tiedot edellä).
Prednisoni- tai prednisolonihoitoa saavien potilaiden kortikosteroidiannostusta saattaa olla aiheellista suurentaa epätavallisen stressin yhteydessä ennen stressiä aiheuttavaa tilannetta, sen aikana ja jälkeen.
Luuntiheys
Miehillä, joilla on pitkälle edennyt metastasoitunut eturauhassyöpä, luuntiheys voi pienentyä. Abirateroniasetaatin (Akeega-valmisteen komponentti) käyttö yhdessä glukokortikoidin kanssa voi lisätä tätä vaikutusta.
Luunmurtumien ja kuolleisuuden lisääntyminen yhdistelmähoidossa radium‑223‑dikloridin (Ra-223) kanssa
Akeega-valmisteen ja prednisonin tai prednisolonin yhdistelmän käyttö yhdessä Ra-223-hoidon kanssa on vasta-aiheista (ks. kohta Vasta-aiheet), koska Akeega-valmisteen komponentilla abirateroniasetaatilla tehdyissä kliinisissä tutkimuksissa havaittiin, että oireettomilla tai lieväoireisilla eturauhassyöpää sairastavilla potilailla on lisääntynyt luunmurtumien riski ja havaittiin kuolleisuuden lisääntymistä.
On suositeltavaa, että Ra-223-hoitoa ei aloiteta vähintään viiteen päivään Akeega-valmisteen ja prednisonin tai prednisolonin yhdistelmän viimeisen antokerran jälkeen.
Hyperglykemia
Glukokortikoidien käyttö voi lisätä hyperglykemiaa, joten diabeetikoiden verensokeripitoisuutta on mitattava tiheästi.
Vaikutukset luustolihaksiin
Akeega-hoitoa saaneilla potilailla ei ole havaittu myopatiaa ja rabdomyolyysia. Abirateroniasetaattimonoterapiaa (abirateroniasetaatti on Akeega-valmisteen komponentti) koskeneissa tutkimuksissa valtaosa tapauksista kehittyi kuuden ensimmäisen hoitokuukauden kuluessa ja korjautui abirateroniasetaatin käytön lopettamisen jälkeen. Samanaikaisessa hoidossa lääkevalmisteilla, joihin tiedetään liittyvän myopatiaa/rabdomyolyysiä, suositellaan noudattamaan varovaisuutta.
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Voimakkaita CYP3A4:n indusoijia on vältettävä hoidon aikana, paitsi jos käytettävissä ei ole muuta hoitovaihtoehtoa, koska niiden käyttöön liittyy riski, että altistus abirateronille pienenee (ks. kohta Yhteisvaikutukset).
Laktoosi ja natrium
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi- intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Farmakokineettiset yhteisvaikutukset
Akeega-valmisteella ei ole tehty lääkkeiden yhteisvaikutuksia selvittäviä kliinisiä tutkimuksia. Yhteisvaikutukset, jotka on tunnistettu Akeega-valmisteen erillisten komponenttien (niraparibin tai abirateroniasetaatin) käytön yhteydessä, määrittävät Akeega-valmisteen käytössä mahdollisesti ilmenevät yhteisvaikutukset.
Muiden lääkevalmisteiden vaikutukset niraparibiin tai abirateroniasetaattiin
CYP3A4:n indusoijat ja estäjät
Abirateroni on CYP3A4:n substraatti. Kliinisessä tutkimuksessa, jossa terveet tutkittavat saivat ensin voimakasta CYP3A4:n indusoijaa rifampisiinia 600 mg/vrk kuuden vuorokauden ajan ja sen jälkeen 1 000 mg abirateroniasetaattia kerta-annoksena, abirateronin AUC∞-arvon keskiarvo plasmassa pieneni 55 %. Voimakkaita CYP3A4:n indusoijia (esim. fenytoiinia, karbamatsepiinia, rifampisiinia, rifabutiinia, rifapentiinia, fenobarbitaalia, mäkikuismaa [Hypericum perforatum]) on vältettävä Akeega-hoidon aikana, paitsi jos muuta hoitovaihtoehtoa ei ole käytettävissä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erillisessä terveillä tutkittavilla tehdyssä kliinisessä tutkimuksessa voimakkaan CYP3A4:n estäjän ketokonatsolin samanaikaisella annolla ei ollut kliinisesti merkityksellistä vaikutusta abirateronin farmakokinetiikkaan.
Niraparibin tai abirateroniasetaatin vaikutukset muihin lääkevalmisteisiin
CYP2D6:n substraatit
Abirateroni on CYP2D6:n estäjä. Abirateroniasetaatin ja prednisonin yhdistelmän (AAP) vaikutuksia CYP2D6:n substraatin dekstrometorfaanin kerta-annokseen selvittäneessä kliinisessä tutkimuksessa systeeminen altistus (AUC) dekstrometorfaanille suureni noin 2,9-kertaiseksi. Dekstrometorfaanin aktiivisen metaboliitin dekstrorfaanin AUC24-arvo suureni noin 33 %. Jos CYP2D6:n välityksellä metaboloituvan lääkevalmisteen terapeuttinen indeksi on kapea, kyseisen lääkevalmisteen annoksen pienentämistä pitää harkita. Esimerkkejä CYP2D6:n välityksellä metaboloituvista lääkevalmisteista ovat mm. metoprololi, propranololi, desipramiini, venlafaksiini, haloperidoli, risperidoni, propafenoni, flekainidi, kodeiini, oksikodoni ja tramadoli.
CYP2C8:n substraatit
Abirateroni on CYP2C8:n estäjä. Terveillä tutkittavilla tehdyssä kliinisessä tutkimuksessa CYP2C8:n substraatin pioglitatsonin AUC suureni 46 % ja pioglitatsonin aktiivisten metaboliittien M-III:n ja M-IV:n AUC-arvot pienenivät 10 %, kun pioglitatsonia annettiin yhdessä 1 000 mg:n abirateroniasetaattikerta-annoksen kanssa. Jos CYP2C8:n substraatin terapeuttinen indeksi on kapea ja tällaista valmistetta käytetään samanaikaisesti Akeega-valmisteen kanssa, potilaita pitää Akeega-valmisteen abirateroniasetaattikomponentin vuoksi seurata CYP2C8:n substraattiin liittyvän toksisuuden oireiden havaitsemiseksi. Esimerkkejä CYP2C8:n välityksellä metaboloituvista lääkevalmisteista ovat mm. pioglitatsoni ja repaglinidi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Farmakodynaamiset yhteisvaikutukset
Akeega-valmistetta ei ole tutkittu rokotteiden tai immunosuppressiivisten lääkeaineiden kanssa.
Tiedot niraparibin käytöstä yhdistelmänä sytotoksisten lääkevalmisteiden kanssa ovat rajalliset. Varovaisuutta on noudatettava, jos Akeega-valmistetta käytetään yhdistelmänä eläviä tai eläviä heikennettyjä taudinaiheuttajia sisältävien rokotteiden, immunosuppressiivisten lääkeaineiden tai muiden sytotoksisten lääkevalmisteiden kanssa.
QT-aikaa tunnetusti pidentävien lääkkeiden samanaikainen käyttö
Androgeenideprivaatiohoito saattaa pidentää QT-aikaa, joten Akeega-valmisteen käytössä kehotetaan varovaisuuteen, kun samanaikaisesti käytetään QT-aikaa tunnetusti pidentäviä lääkevalmisteita tai lääkevalmisteita, jotka voivat aiheuttaa kääntyvien kärkien takykardiaa (torsades de pointes), kuten ryhmän IA (esim. kinidiini, disopyramidi) tai ryhmän III (esim. amiodaroni, sotaloli, dofetilidi, ibutilidi) rytmihäiriölääkkeitä, metadonia, moksifloksasiinia, psykoosilääkkeitä, tms.
Spironolaktonin samanaikainen käyttö
Spironolaktoni sitoutuu androgeenireseptoriin ja saattaa suurentaa prostataspesifisen antigeenin (PSA) pitoisuuksia. Spironolaktonin ja Akeega-valmisteen samanaikaista käyttöä ei suositella (ks. kohta Farmakodynamiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehille ja naisille
Ei tiedetä, esiintyykö Akeega-valmisteen komponentteja tai niiden metaboliitteja siemennesteessä.
Hoidon aikana ja neljän kuukauden ajan viimeisen Akeega-annoksen jälkeen
- on käytettävä kondomia, jos potilas on sukupuoliyhteydessä raskaana olevan naisen kanssa
- on käytettävä kondomia sekä jotakin toista tehokasta ehkäisymenetelmää, jos potilas on sukupuoliyhteydessä naisen kanssa, joka voi tulla raskaaksi.
Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Raskaus
Akeega ei ole tarkoitettu naisille (ks. kohta Vasta-aiheet).
Ei ole olemassa tietoja Akeega-valmisteen käytöstä raskaana oleville naisille. Akeega-valmiste voi kummankin komponentin vaikutusmekanismin sekä abirateroniasetaatilla tehtyjen eläinkokeiden havaintojen perusteella vahingoittaa sikiötä. Niraparibilla ei ole tehty kehitys- ja lisääntymistoksikologisia eläinkokeita (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetys
Akeega ei ole tarkoitettu naisille.
Hedelmällisyys
Akeega-valmisteesta ei ole hedelmällisyyttä koskevia kliinisiä tietoja. Eläinkokeissa niraparibi tai abirateroniasetaatti heikensi urosten hedelmällisyyttä, mutta tällaiset vaikutukset korjautuivat hoidon keskeyttämisen jälkeen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Akeega-valmisteella on kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Akeega-valmistetta käyttävillä potilailla voi esiintyä voimattomuutta, uupumusta, heitehuimausta tai keskittymisvaikeuksia. Potilaiden on oltava varovaisia ajaessaan ajoneuvoa tai käyttäessään koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Akeega-valmisteen kokonaisturvallisuusprofiili perustuu yhdistettyihin tietoihin kahdesta vaiheen 3 kliinisestä tutkimuksesta, joissa oli mukana 559 potilasta (AMPLITUDE [n = 347] ja MAGNITUDE kohortti 1 [n = 212]). Yleisimpiä niraparibin sekä abirateroniasetaatin ja prednisonin yhdistelmää saaneessa ryhmässä > 10 %:lla esiintyneitä haittavaikutuksia olivat anemia (51,9 %), hypertensio (40,1 %), uupumus (39,7 %), tuki- ja liikuntaelimistön kipu (35,8 %), ummetus (34,7 %), pahoinvointi (28,6 %), hypokalemia (22,0 %), trombosytopenia (20,9 %), neutropenia (19,7 %), edeema (18,1 %), hengitystieinfektiot (17,7 %), hengenahdistus (16,6 %), oksentelu (15,7 %), heikentynyt ruokahalu (15,0 %), leukopenia (14,5 %), kuumat aallot (14,1 %), heitehuimaus (14,0 %), painon lasku (14,0 %), vatsakipu (13,4 %), hyperglykemia (13,4 %), unettomuus (13,1 %), ripuli (12,3 %), lymfopenia (12,2 %), päänsärky (12,0 %), suurentunut veren kreatiniinipitoisuus (11,8 %), virtsatieinfektio (11,8 %) ja yskä (10,4 %). Yleisimmin havaittuja 3.–4. asteen haittavaikutuksia olivat anemia (29,7 %), hypertensio (22,7 %), hypokalemia (9,3 %), neutropenia (8,4 %), trombosytopenia (7,5 %) ja lymfopenia (5,2 %).
Haittavaikutustaulukko
Kliinisten tutkimusten aikana havaitut haittavaikutukset luetellaan jäljempänä yleisyysluokittain. Yleisyysluokat määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 3. Kliinisissä lääketutkimuksissa tunnistetut haittavaikutukset | ||
| Elinjärjestelmä | Esiintymistiheys | Haittavaikutus |
| Infektiot | hyvin yleinen | hengitystieinfektiot1, virtsatieinfektio2 |
| yleinen | keuhkokuume, maha-suolikanavan infektio3, sepsis4 | |
| melko harvinainen | ihoinfektio5, konjunktiviitti | |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | melko harvinainen | myelodysplastinen oireyhtymä / akuutti myelooinen leukemia |
| Veri ja imukudos | hyvin yleinen | anemia, trombosytopenia, neutropenia, leukopenia, lymfopenia |
| tuntematon | pansytopenia6 | |
| Immuunijärjestelmä | melko harvinainen | yliherkkyys7 |
| Umpieritys | tuntematon | lisämunuaisten vajaatoiminta8 |
| Aineenvaihdunta ja ravitsemus | hyvin yleinen | heikentynyt ruokahalu, hypokalemia, hyperglykemia |
| yleinen | lipidiaineenvaihdunnan häiriöt9 | |
| Psyykkiset häiriöt | hyvin yleinen | unettomuus |
| yleinen | masennus, ahdistuneisuus, sekavuustila | |
| Hermosto | hyvin yleinen | heitehuimaus, päänsärky |
| yleinen | letargia, kognitiivinen häiriö, makuhäiriö10 | |
| tuntematon | posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES)6 | |
| Sydän | yleinen | sydämen vajaatoiminta11, rytmihäiriö12, angina pectoris13, takykardia14, sydämentykytys |
| melko harvinainen | sydäninfarkti15 | |
| Verisuonisto | hyvin yleinen | hypertensio16, kuumat aallot |
| tuntematon | hypertensiivinen kriisi6 | |
| Hengityselimet, rintakehä ja välikarsina | hyvin yleinen | hengenahdistus17, yskä |
| yleinen | keuhkoembolia, nenäverenvuoto | |
| melko harvinainen | pneumoniitti | |
| tuntematon | allerginen alveoliitti8 | |
| Ruoansulatuselimistö | hyvin yleinen | ummetus, pahoinvointi, oksentelu, vatsakipu18, ripuli |
| yleinen | gastriitti, vatsan pingottuneisuus, dyspepsia, stomatiitti, suun kuivuminen | |
| Maksa ja sappi | yleinen | hyperbilirubinemia |
| melko harvinainen | maksan vajaatoiminta, hepatiitti19 | |
| Iho ja ihonalainen kudos | yleinen | ihottuma20, kutina, valoherkkyysreaktio |
| Luusto, lihakset ja sidekudos | hyvin yleinen | tuki- ja liikuntaelinten kipu21 |
| tuntematon | rabdomyolyysi8, myopatia8 | |
| Munuaiset ja virtsatiet | yleinen | verivirtsaisuus, akuutti munuaisvaurio |
| melko harvinainen | virtsaputken verenvuoto | |
| Yleisoireet ja antopaikassa todettavat haitat | hyvin yleinen | uupumus22, edeema23 |
| yleinen | ei-sydänperäinen rintakipu | |
| melko harvinainen | rintakipu, limakalvotulehdus | |
| Tutkimukset | hyvin yleinen | painon lasku, suurentunut veren kreatiniinipitoisuus |
| yleinen | suurentunut veren alkalisen fosfataasin pitoisuus, suurentunut ASAT-arvo, suurentunut ALAT-arvo | |
| melko harvinainen | sydänsähkökäyrässä pidentynyt QT-aika, suurentunut gammaglutamyylitransferaasipitoisuus | |
| Vammat, myrkytykset ja hoitokomplikaatiot | yleinen | luunmurtumat24 |
| 1 sisältää ylähengitystieinfektion, alahengitystieinfektion, kurkunpäätulehduksen, nuhan, keuhkoputkentulehduksen, nenänielutulehduksen, hengitysteiden virusinfektion, kurkunpää–nielutulehduksen, ylähengitysteiden virusinfektion 2 sisältää virtsarakkotulehduksen 3 sisältää gastroenteriitin, virusgastroenteriitin, sieniesofagiitin, ruokatorven kandidiaasin, suunielun kandidiaasin 4 sisältää urosepsiksen 5 sisältää streptokokki-infektion 6 ei havaittu Akeega-hoidossa; raportoitu niraparibimonoterapiassa 7 yliherkkyyttä ilman anafylaksiaa havaittiin AMPLITUDE-tutkimuksessa; anafylaktisia reaktioita ei raportoitu Akeega-valmisteella tehdyissä kliinisissä tutkimuksissa, mutta on raportoitu niraparibimonoterapiassa valmisteen markkinoille tulon jälkeen 8 ei havaittu Akeega-hoidossa; raportoitu abirateronimonoterapiassa valmisteen markkinoille tulon jälkeen 9 sisältää hyperkolesterolemian, hypertriglyseridemian, dyslipidemian 10 sisältää makuaistin häiriön 11 sisältää akuutin sydämen vajaatoiminnan, kongestiivisen sydämen vajaatoiminnan, keuhkosydänsairauden, vasemman kammion toimintahäiriön 12 sisältää eteisvärinän, lisälyönnit, supraventrikulaariset lisälyönnit, kammiolisälyönnit, sinusarytmian 13 sisältää sepelvaltimotaudin, äkillisen sepelvaltimo-oireyhtymän 14 sisältää sinustakykardian, eteistakykardian 15 sisältää sydänlihasiskemian 16 sisältää systolisen hypertension 17 sisältää rasitushengenahdistuksen 18 sisältää ylävatsakivun, alavatsakivun 19 sisältää poikkeavan maksan toiminnan, akuutin maksatulehduksen, fulminantin maksatulehduksen, maksasolujen hajoamisen, maksatoksisuuden 20 sisältää punoituksen, dermatiitin, makulopapulaarisen ihottuman, kutisevan ihottuman 21 sisältää nivelsäryn, selkäkivun, lihassäryn, tuki- ja liikuntaelinvaivan 22 sisältää astenian 23 sisältää perifeerisen edeeman, turvotuksen, kasvojen edeeman, ääreisosien turvotuksen, kasvojen turvotuksen 24 sisältää osteoporoosin, kylkiluun murtuman, reisiluun kaulan murtuman, reisiluun murtuman, nilkkamurtuman, olkaluun murtuman, pohjeluun murtuman, alaraajan murtuman, rintalastan murtuman, rasitusmurtuman, yläraajamurtuman, lonkkamaljan murtuman, värttinäluun murtuman, rintanikaman murtuman, sääriluun murtuman | ||
Valikoitujen haittavaikutusten kuvaus
Jäljempänä kuvatut valikoidut haittavaikutukset perustuvat yhdistettyihin tietoihin kahdesta vaiheen 3 kliinisestä tutkimuksesta, joissa oli mukana 559 potilasta (AMPLITUDE [n = 347] ja MAGNITUDE kohortti 1 [n = 212]).
Hematologinen toksisuus
Hematologinen toksisuus (anemia, trombosytopenia ja neutropenia), mukaan lukien laboratoriolöydökset, on yleisin niraparibiin (Akeega-valmisteen komponentti) liittyvä haittavaikutus. Tällaista toksisuutta esiintyi yleensä kolmen ensimmäisen hoitokuukauden aikana, ja ilmaantuvuus väheni ajan mittaan.
Seuraavat hematologiset parametrit olivat MAGNITUDE- JA AMPLITUDE-tutkimuksissa sisäänottokriteerejä: absoluuttinen neutrofiilien määrä ≥ 1,5 x 109/l, trombosyytit ≥ 100 x 109/l ja hemoglobiini ≥ 90 g/l. Hematologiset haittavaikutukset hoidettiin laboratorioseurannalla ja annosta muuttamalla (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Anemia
Anemia oli yleisin haittavaikutus (51,9 %) ja yleisimmin havaittu 3.–4. asteen tapahtuma (29,7 %). Anemia ilmaantui hoidon varhaisvaiheessa (ilmenemiseen kuluneen ajan mediaani 64,5 päivää). Hoito keskeytettiin 26,1 %:lla potilaista, ja annosta pienennettiin 13,6 %:lla potilaista. Anemiaa hoidettiin vähintään yhdellä punasolusiirrolla 25,9 %:lla potilaista. Hoito lopetettiin 2,5 %:lla potilaista.
Trombosytopenia
Trombosytopeniaa raportoitiin 20,9 %:lla hoitoa saaneista potilaista, ja 3.–4. asteen trombosytopenia todettiin 7,5 %:lla potilaista. Ensimmäisestä annoksesta trombosytopenian ensimmäiseen ilmaantumiskertaan kuluneen ajan mediaani oli 56 päivää. Trombosytopenia hoidettiin annosmuutoksella (hoidon keskeytys 9,5 %:lla ja annoksen pienentäminen 1,6 %:lla) ja trombosyyttisiirrolla (2,1 %), jos se soveltui potilaalle (ks. kohta Annostus ja antotapa). Hoito lopetettiin 0,5 %:lla potilaista. Trombosytopeniaa ja ei-hengenvaarallisia verenvuototapahtumia ilmeni 2,3 %:lla potilaista.
Neutropenia
Neutropeniaa raportoitiin 19,7 %:lla potilaista, ja 3.–4. asteen neutropeniaa raportoitiin 8,4 %:lla potilaista. Ensimmäisestä annoksesta neutropenian ensimmäiseen raportointikertaan kuluneen ajan mediaani oli 58 päivää. Neutropenia johti hoidon keskeyttämiseen 8,9 %:lla potilaista ja annoksen pienentämiseen 1,3 %:lla potilaista. Hoito lopetettiin 0,4 %:lla potilaista. Neutropenia ja infektio raportoitiin 2,5 %:lla potilaista.
Hypertensio
Hypertensio on Akeega-valmisteen kumpaankin komponenttiin liittyvä haittavaikutus, joten potilaat, joilla oli huonossa hoitotasapainossa oleva hypertensio (pitkäaikaisesti systolinen verenpaine ≥ 160 mmHg tai diastolinen verenpaine ≥ 100 mmHg), suljettiin pois kaikista yhdistelmähoitoa koskeneista tutkimuksista.
Hypertensio raportoitiin 40,6 %:lla potilaista, ja näistä 22,9 %:lla oli ≥ 3. asteen hypertensio. Hypertension alkamiseen kuluneen ajan mediaani oli 70 päivää. Hypertensio hoidettiin tukilääkevalmisteilla.
Potilaiden verenpaineen pitää olla hoitotasapainossa ennen Akeega-hoidon aloittamista, ja verenpainetta pitää seurata hoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sydäntapahtumat
Sydämen häiriöitä koskevien hoidonaikaisten haittatapahtumien (kaikki vaikeusasteet) ilmaantuvuus niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä ja lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä oli samankaltainen. Poikkeuksena oli sydämen rytmihäiriöiden kategoria, jossa haittatapahtumia havaittiin 17,2 %:lla potilaista niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä ja 7,9 %:lla potilaista lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Sydämen rytmihäiriöiden suurempi esiintyvyys johtui pääasiassa lieväasteisista sydämentykytykseen, takykardiaan ja eteisperäisiin rytmihäiriöihin liittyvistä tapahtumista. Sydämen rytmihäiriötapahtumien ilmaantumiseen kuluneen ajan mediaani oli niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä 150,5 päivää ja lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä 233,5 päivää. Sydämen rytmihäiriötapahtumat hävisivät 70,8 %:lla potilaista niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä ja 72,7 %:lla potilaista lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä.
Sydämen vajaatoiminnan (yhdistelmätermi) ilmaantuvuus oli niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä 4,7 % ja lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä 1,8 %. Sydämen vajaatoiminnan ilmaantumiseen kuluneen ajan mediaani oli niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä 359,5 päivää ja lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä 165,5 päivää. Sydämen vajaatoimintatapahtumat hävisivät niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä 42,3 %:lla potilaista ja lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä 40 %:lla potilaista.
Iskeemisen sydänsairauden (yhdistelmätermi) ilmaantuvuus oli niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä 5,4 % ja lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä 5,0 %. Iskeeminen sydänsairaus sisälsi angina pectoriksen, sepelvaltimotaudin tai sepelvaltimon ahtauman, sydäninfarktin, laboratorioarvojen poikkeavuudet (kuten suurentunut troponiiniarvo) ja iskeemiseen sydänsairauteen sopivat muutokset sydänsähkökäyrässä. Iskeemisen sydänsairauden ilmaantumiseen kuluneen ajan mediaani oli niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä 237,5 päivää ja lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä 322,5 päivää. Iskeemiseen sydänsairauteen liittyvät tapahtumat hävisivät niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä 73,3 %:lla potilaista ja lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä 67,9 %:lla potilaista.
Maksatoksisuus
Maksatoksisuuden kokonaisilmaantuvuus oli niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä (13,6 %) samankaltainen kuin lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä (17,5 %) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Valtaosa näistä tapahtumista oli lieväasteisesti kohonneita aminotransferaasipitoisuuksia. Niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä 3. asteen tapahtumia esiintyi 2,0 %:lla potilaista, 4. asteen tapahtuma esiintyi yhdellä potilaalla (0,2 %) ja 5. asteen tapahtuma esiintyi yhdellä potilaalla (0,2 %). Vakavien haittatapahtumien ilmaantuvuus oli 0,7 %. Maksatoksisuuden ilmaantumiseen kuluneen ajan mediaani oli 43 päivää. Maksatoksisuus hoidettiin 2,0 %:lla potilaista keskeyttämällä hoito ja 0,7 %:lla potilaista pienentämällä annosta. Hoidon lopetti 0,5 % potilaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Akeega-valmisteen yliannokseen ei ole spesifistä hoitoa. Yliannoksen yhteydessä lääkärin pitää ryhtyä yleisiin tukitoimenpiteisiin ja hoitaa potilasta oireenmukaisesti, mihin kuuluvat rytmihäiriöiden ja hypokalemian sekä nesteen elimistöön kertymiseen liittyvien oireiden ja löydösten seuranta. Maksan toiminta pitää myös arvioida.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet, ATC-koodi: L01XK52
Vaikutusmekanismi
Akeega on yhdistelmävalmiste. Se sisältää niraparibia, joka on poly-ADP-riboosipolymeraasin (PARP) estäjä, ja abirateroniasetaattia (abirateronin aihiolääke), joka on CYP17:n (17α‑hydroksylaasi/C17,20‑lyaasin) estäjä. Nämä kohdentuvat kahteen onkogeeniseen riippuvuuteen liittyvään tekijään potilailla, joilla on metastasoitunut eturauhassyöpä ja HRR-geenimutaatioita.
Niraparibi
Niraparibi on poly-ADP-riboosipolymeraasientsyymien PARP-1:n ja PARP-2:n estäjä. Nämä entsyymit osallistuvat DNA:n korjaamiseen. In vitro ‑tutkimukset ovat osoittaneet, että niraparibin indusoimalla sytotoksisuudella voi olla osuus PARP:n entsymaattisen aktiivisuuden estymisessä ja PARP-DNA-kompleksien lisääntyneessä muodostumisessa, mistä seuraa DNA:n vaurioituminen, apoptoosi ja solukuolema.
Abirateroniasetaatti
Abirateroniasetaatti muuntuu in vivo abirateroniksi, joka on androgeenin biosynteesin estäjä. Erityisesti abirateroni estää selektiivisesti entsyymiä 17α‑hydroksylaasi/C17,20‑lyaasi (CYP17). Tämä entsyymi ilmentyy kiveksissä, lisämunuaisessa ja eturauhaskasvaimen kudoksessa tapahtuvassa androgeenien biosynteesissä ja sitä tarvitaan tähän biosynteesiin. CYP17 katalysoi pregnenolonin ja progesteronin muuntumista testosteronin esiasteiksi, pregnenolonin DHEA:ksi ja progesteronin androsteenidioniksi, 17α‑hydroksylaation ja C17,20-sidoksen katkeamisen kautta. CYP17:n toiminnan estyminen lisää myös mineralokortikoidituotantoa lisämunuaisissa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Androgeeneille herkkä eturauhassyöpä reagoi androgeenipitoisuutta pienentävään hoitoon. Androgeenideprivaatiohoito, kuten luteinisoivaa hormonia vapauttava hormoni (LHRH) -analogihoito tai kivesten poisto, vähentävät androgeenien muodostumista kiveksissä, mutta eivät vaikuta androgeenien muodostumiseen lisämunuaisissa tai kasvaimessa. Abirateroniasetaattihoito yhdessä LHRH-analogihoidon (tai kivesten poistoleikkauksen) kanssa pienentää seerumin testosteronipitoisuuden (kaupallisilla määritysmekanismeilla saavutettavan) havaitsemisrajan alapuolelle.
Farmakodynaamiset vaikutukset
Abirateroniasetaatti
Abirateroni pienentää seerumin testosteronipitoisuuden ja muiden androgeenien pitoisuudet pienemmiksi kuin pelkällä LHRH-analogihoidolla tai kivesten poistoleikkauksella saavutetaan. Tämä johtuu CYP17-entsyymin selektiivisestä estymisestä, koska CYP17-entsyymiä tarvitaan androgeenien biosynteesiin.
Kliininen teho ja turvallisuus
Akeega-valmisteen teho varmistettiin kahdessa satunnaistetussa lumekontrolloidussa vaiheen 3 kliinisessä monikeskustutkimuksessa potilailla, joilla oli metastasoitunut hormonisensitiivinen eturauhassyöpä (mHSPC, AMPLITUDE-tutkimus 67652000PCR3002), ja potilailla, joilla oli metastasoitunut kastraatioresistentti eturauhassyöpä (mCRPC, MAGNITUDE-tutkimus 64091742PCR3001).
Hoito mHSPC-potilailla, joilla on BRCA1/2-mutaatioita
AMPLITUDE on vaiheen 3 satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikansallinen tutkimus, jossa päivittäin otettua niraparibista (200 mg) sekä abirateroniasetaatin (1 000 mg) ja prednisonin (5 mg) yhdistelmästä koostuvaa yhdistelmähoitoa verrattiin tavanomaiseen hoitoon abirateroniasetaatin ja prednisonin yhdistelmällä (AAP). Tutkimuksessa oli mukana 696 mHSPC-potilasta, joilla oli tiettyjä HRR-geenimuutoksia. Potilaat satunnaistettiin (1:1) saamaan suun kautta päivittäin joko niraparibin ja AAP:n (N = 348) tai lumelääkkeen ja AAP:n yhdistelmää (N = 348). Satunnaistaminen ositettiin HRR-geenimuutoksen (BRCA2 versus CDK12 versus kaikki muut patogeeniset muutokset), mHSPC:n hoitoon aiemmin annetun dosetakselin (kyllä tai ei) ja seulontahetkellä todetun tautivolyymin (korkea versus matala) mukaan. Hoitoa jatkettiin, kunnes tauti eteni, ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai potilas kuoli.
Potilaat, joilla oli mHSPC ja tiettyjä HRR-geenimuutoksia, olivat aiemmin saaneet saada ≤ 6 hoitosykliä dosetakselia, ≤ 45 päivän AAP-hoidon, ≤ 6 kuukauden androgeenideprivaatiohoidon ja enintään 2 viikon ketokonatsolihoidon. Potilaat olivat myös saaneet saada yhden sädehoitojakson ja yhden kirurgisen toimenpiteen eturauhassyövän oireiden hoitoon. Potilaat, joilla oli eturauhasen pienisoluinen tai neuroendokriininen karsinooma, suljettiin pois tutkimuksesta. Kaikkien potilaiden plasma-, veri- ja/tai kasvainkudosnäytteet testattiin validoidulla uuden sukupolven sekvensoinnilla (NGS) ituradan ja/tai somaattisen HRR-geenimutaatiostatuksen määrittämiseksi. Tutkimukseen otettiin mukaan 696 potilasta, joilla oli vähintään yksi seuraavista mutaatioista (348 sai Akeega-valmistetta): BRCA1, BRCA2, BRIP1, CDK12, CHEK2, FANCA, PALB2, RAD51B, RAD54L. 387:llä tutkimukseen mukaan otetuista tutkittavista oli BRCA1/2-mutaatio (191 sai Akeega-valmistetta). Lisäksi 309:llä tutkimukseen mukaan otetuista potilaista oli jokin muu kuin BRCA1/2-mutaatio (BRIP1, CDK12, CHEK2, FANCA, PALB2, RAD51B, RAD54L; 157 sai Akeega-valmistetta). ECOG-suorituskykyluokka (Eastern Cooperative Oncology Group Performance Status) tutkimukseen mukaan tullessa oli 68,0 %:lla potilaista 0, 31,0 %:lla potilaista 1 ja 1,0 %:lla potilaista 2. Valtaosalla potilaista oli GnRH-analogilla aikaansaatu androgeenideprivaatio (95,3 %), ja 6,5 %:sta potilaista raportoitiin, että heille oli tehty kummankin kiveksen poistoleikkaus. Potilaat, joille ei ollut aiemmin tehty kivesten poistoleikkausta, jatkoivat taustalla jollakin GnRH-analogilla toteutettavaa androgeenideprivaatiohoitoa.
Ensisijainen päätetapahtuma, joka oli radiologinen taudin etenemisvapaa elossaoloaika (radiographic progression free survival, rPFS), määriteltiin satunnaistamispäivämäärästä tutkijan arvioimaan ensimmäiseen radiologiseen etenemiseen tai mistä tahansa syystä aiheutuneeseen kuolemaan (kumpi tahansa todettiin ensin) kuluneeksi ajaksi. Radiologiseksi etenemiseksi määriteltiin luustokartoituksessa todettu ensimmäinen etenemiskerta (Prostate Cancer Working Group 3 -kriteerien mukaan) tai pehmytkudosleesioiden toteaminen TT- tai magneettikuvauksessa (RECIST 1.1 -kriteerien mukaan). Ajaksi oireiseen etenemiseen määriteltiin satunnaistamisesta oireiden etenemishetkeen kulunut aika. Oireinen eteneminen käsitti ulkoisen sädehoidon käytön luuston tai lantion oireisiin, syöpään liittyvät sairastuvuustapahtumat, uuden systeemisen syöpähoidon aloittaminen ja muut syöpään liittyvät toimenpiteet. Päätetapahtumat testattiin muodollisesti järjestyksessä rPFS, aika oireiseen etenemiseen ja kokonaiselossaoloaika (OS) ja näitä kontrolloitiin monilukuisuuden suhteen.
AMPLITUDE-tutkimuksen kummankin haaran potilaiden demografiset tiedot ja lähtötilanteen ominaisuudet olivat verrannolliset keskenään. Iän mediaani oli lähtötilanteessa 67 vuotta (vaihteluväli 41–92), ja 20,9 % potilaista oli ≥ 75-vuotiaita. Tutkimuspotilasjoukosta 69,5 % oli valkoihoisia, 23,8 % oli aasialaisia ja 3,6 % oli tummaihoisia tai afroamerikkalaisia. 12 % potilaista raportoi etniseksi taustaksi latinalaisamerikkalainen. Taudin diagnoosihetkellä PSA-arvon mediaani oli 107,00 µg/l (vaihteluväli 0,1–15900,0), ja valtaosalla (82,2 %) potilaista Gleason-pisteet olivat ≥ 8. Tutkimukseen mukaan tullessa 98,2 %:lla oli etäpesäkkeitä luustossa, 40,8 %:lla oli etäpesäkkeitä vain luustossa (tautietäpesäkkeitä ei muualla elimistössä), 16,5 %:lla oli viskeraalietäpesäkkeitä (etäpesäkkeitä elimissä, kuten maksassa, keuhkoissa tai lisämunuaisissa) ja 49,6 %:lla oli etäpesäkkeitä imusolmukkeissa. Valtaosalla potilaista oli korkean tautivolyymin eturauhassyöpä (79,1 %), ja 20,9 %:lla oli matalan tautivolyymin eturauhassyöpä. Potilaista 16,0 % oli saanut aiemmin dosetakselihoitoa. Hoitoa luustoa säästävillä lääkeaineilla (bisfosfonaatit tai denosumabi) sai 24,1 % potilaista niraparibin ja AAP:n yhdistelmää saaneessa ryhmässä ja 23,0 % potilaista lumelääkkeen ja AAP:n yhdistelmää saaneessa ryhmässä.
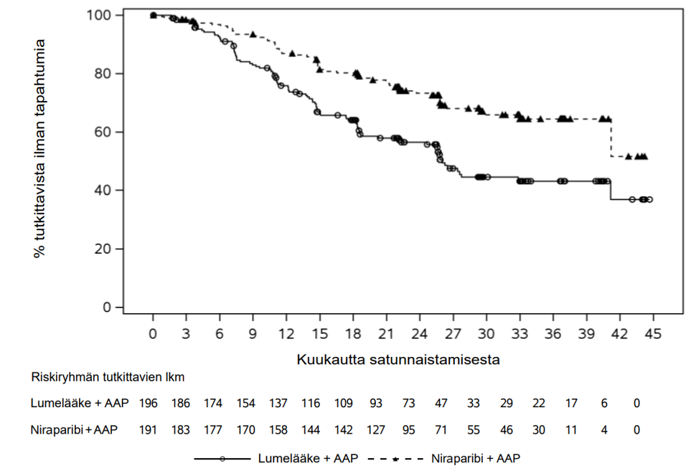
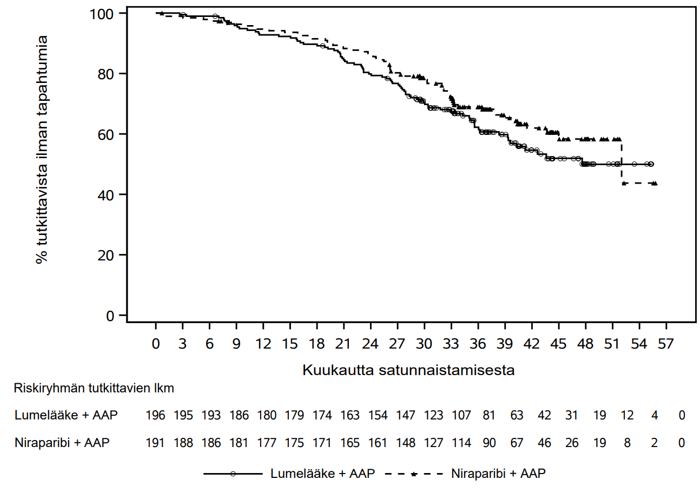
BRCA-potilasjoukossa nähtiin tilastollisesti merkitsevä tutkijan arvioima rPFS-etu niraparibin ja AAP:n yhdistelmää saaneilla tutkittavilla verrattuna lumelääkkeen ja AAP:n yhdistelmää saaneisiin tutkittaviin. Keskeiset tehotulokset BRCA-potilasjoukossa esitetään taulukossa 4. BRCA-potilasjoukon tutkijan arvioima rPFS:n Kaplan–Meier-kuvaaja esitetään kuvassa 1 ja kokonaiselossaoloajan Kaplan–Meier-kuvaaja kuvassa 2.
| Taulukko 4. AMPLITUDE-tutkimuksen BRCA-potilasjoukontehotulokset | ||
| Päätetapahtumat | Akeega + P1 (N = 191) | Lumelääke + AAP1 (N = 196) |
| Radiologinen taudin etenemisvapaa elossaoloaika2 | ||
| Tapahtumat | 57 (29,8 %) | 93 (47,4 %) |
| Tapahtumaan kulunut aika (kuukautta), mediaani (95 %:n luottamusväli) | NE (41,20; NE) | 25,99 (22,11; 41,17) |
| Riskisuhde (95 %:n luottamusväli) | 0,515 (0,370; 0,717) | |
| p-arvo | < 0,0001 | |
| Aika oireiseen etenemiseen3 | ||
| Tapahtumat | 31 (16,2 %) | 66 (33,7 %) |
| Tapahtumaan kulunut aika (kuukautta), mediaani (95 %:n luottamusväli) | NE (NE; NE) | NE (39,72; NE) |
| Riskisuhde (95 %:n luottamusväli) | 0,444 (0,290; 0,681) | |
| p-arvo | 0,0001 | |
| Kokonaiselossaoloaika4 | ||
| Tapahtumat | 65 (34,0 %) | 80 (40,8 %) |
| Tapahtumaan kulunut aika (kuukautta), mediaani (95 %:n luottamusväli) | 52,01 (44,98; NE) | 47,61 (39,36; NE) |
| Riskisuhde (95 %:n luottamusväli) | 0,799 (0,576; 1,109) | |
1 P = prednisoni tai prednisoloni 2 Primaarianalyysi, jossa kliinisten tietojen keruun katkaisupäivämäärä oli 7. tammikuuta 2025 ja seuranta-ajan mediaani oli 30,7 kuukautta 3 Ensimmäinen välianalyysi, jossa kliinisten tietojen keruun katkaisupäivämäärä oli 7. tammikuuta 2025 ja seuranta-ajan mediaani oli 30,7 kuukautta 4 Toinen välianalyysi, jossa kliinisten tietojen keruun katkaisupäivämäärä oli 3. lokakuuta 2025 ja seuranta-ajan mediaani oli 40,4 kuukautta | ||
| NE = ei arvioitavissa | ||
Kuva 1. Kaplan–Meier-kuvaaja tutkijan arvioimasta radiologisesta taudin etenemisvapaasta elossaoloajasta BRCA-potilasjoukossa (AMPLITUDE, primaarianalyysi)
Kuva 2. Kaplan–Meier-kuvaaja kokonaiselossaoloajasta BRCA-potilasjoukossa (AMPLITUDE, toinen välianalyysi)
Ensilinjan hoito metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastaville potilaille, joilla on BRCA1/2‑mutaatioita
MAGNITUDE oli vaiheen 3 satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikeskustutkimus, jossa päivittäin otettua niraparibista (200 mg) sekä abirateroniasetaatin (1 000 mg) ja prednisonin (10 mg) yhdistelmästä koostuvaa yhdistelmähoitoa verrattiin tavanomaiseen hoitoon abirateroniasetaatin ja prednisonin yhdistelmällä (AAP). Tehoa koskevat tiedot perustuvat kohorttiin 1, joka käsitti 423 metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavaa potilasta, joilla oli tiettyjä HRR-geenimutaatioita. Potilaat satunnaistettiin (1:1) saamaan suun kautta päivittäin joko niraparibin sekä abirateroniasetaatin ja prednisonin yhdistelmää (N = 212) tai lumelääkkeen sekä abirateroniasetaatin ja prednisonin yhdistelmää (N = 211). Hoitoa jatkettiin, kunnes sairaus eteni, ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai potilas kuoli.
Tutkimukseen soveltuivat potilaat, joilla oli metastasoitunut kastraatioresistentti eturauhassyöpä ja jotka eivät olleet aiemmin saaneet metastasoituneeseen kastraatioresistenttiin eturauhassyöpään systeemistä hoitoa, lukuun ottamatta aiempaa lyhytkestoista (enintään 4 kuukautta) abirateroniasetaatin ja prednisonin yhdistelmähoitoa ja parhaillaan saamaansa androgeenideprivaatiohoitoa. Kaikkien potilaiden plasma-, veri- ja/tai kasvainkudosnäytteet testattiin validoidulla rinnakkaissekvensoinnilla (NGS) ituradan ja/tai somaattisen HRR-geenimutaatiostatuksen määrittämiseksi. Tutkimukseen otettiin mukaan 225 tutkittavaa, joilla oli BRCA1/2-mutaatioita (113 sai Akeega-valmistetta). Tutkimukseen otettiin mukaan lisäksi 198 potilasta, joilla oli muita kuin BRCA1/2-mutaatioita (ATM, CHEK2, CDK12, PALB2, FANCA, BRIP1, HDAC2) (99 sai Akeega-valmistetta).
Ensisijainen päätetapahtuma oli radiologinen tautivapaa elossaoloaika (radiographic progression free survival, rPFS), joka perustui sokkoutetun, riippumattoman ja keskitetyn arvioijatahon (blinded independent central radiology, BICR) tarkasteluun kiinteitä kasvaimia koskevien RECIST (Response Evaluation Criteria In Solid Tumours) 1.1 ‑kriteerien (pehmeät ja kudosleesiot) ja PCWG-3 (Prostate Cancer Working Group-3) ‑kriteerien (luuston leesiot) perusteella. Toissijaisia tehon päätetapahtumia olivat aika oireiseen etenemiseen, aika sytotoksiseen solunsalpaajahoitoon ja kokonaiselossaoloaika.
Ensisijaiset tehon tulokset kaikki HRR-statukset käsittävässä potilasjoukossa, kun seuranta-ajan mediaani oli 18,6 kuukautta, osoittivat sokkoutetun, riippumattoman ja keskitetyn arvioijatahon arvioimassa radiologisessa tautivapaassa elossaoloajassa tilastollisesti merkitsevää pitenemistä (riskisuhde = 0,729 [95 %:n luottamusväli: 0,556; 0,956; p = 0,0217]).
Taulukossa 5 on yhteenveto MAGNITUDE-tutkimuksen kohorttiin 1 mukaan otettujen BRCA-potilaiden demografisista ja lähtötilanteen ominaisuuksista. PSA-arvon mediaani diagnoosin yhteydessä oli 41,07 µg/l (vaihteluväli 01–12 080). Kaikkien potilaiden ECOG PS (Eastern Cooperative Oncology Group Performance Status) ‑pisteet tutkimukseen mukaan tullessaan olivat 0 tai 1. Kaikki potilaat, joille ei ollut tehty kivesten poistoleikkausta, jatkoivat perushoitona androgeenideprivaatiohoidon ja GnRH-analogin yhdistelmää.
| Taulukko 5. Yhteenveto demografisista ja lähtötilanteen ominaisuuksista MAGNITUDE-tutkimuksen kohortissa 1 (BRCA) | |||
| Akeega + P1 N = 113 n (%) | Lumelääke + AAP1 N = 112 n (%) | Yhteensä N = 225 n (%) | |
| Ikä (vuotta) | |||
| < 65 | 39 (34,5) | 37 (33,0) | 76 (33,8) |
| ≥ 65–74 | 44 (38,9) | 52 (46,4) | 96 (42,7) |
| ≥ 75 | 30 (26,5) | 23 (20,5) | 53 (23,6) |
| Mediaani | 67,0 | 68,0 | 68,0 |
| Vaihteluväli | 45–100 | 43–88 | 43–100 |
| Etninen tausta | |||
| Valkoihoinen | 78 (69,0) | 84 (75,0) | 162 (72,0) |
| Aasialainen | 18 (15,9) | 20 (17,9) | 38 (16,9) |
| Mustaihoinen | 3 (2,7) | 0 | 3 (1,3) |
| Ei tiedossa | 14 (12,4) | 8 (7,1) | 22 (9,8) |
| Ositustekijät | |||
| Aiempi altistus taksaanisolunsalpaajahoidolle | 26 (23,0) | 29 (25,9) | 55 (24,4) |
| Aiempi altistus androgeenireseptoriin kohdentuvalle hoidolle | 6 (5,3) | 5 (4,5) | 11 (4,9) |
| Aiempi abirateroniasetaatin ja prednisonin yhdistelmän käyttö | 30 (26,5) | 29 (25,9) | 59 (26,2) |
| Sairauden ominaisuudet lähtötilanteessa | |||
| Gleason-pisteet ≥ 8 | 83 (74,1) | 72 (64,3) | 155 (69,2) |
| Luuston affisioituminen | 99 (87,6) | 93 (83,0) | 192 (85,3) |
| Viskeraalinen sairaus (maksa, keuhkot, lisämunuaiset, muu) | 26 (23,0) | 22 (19,6) | 48 (21,3) |
| Metastaasiluokitus alkuvaiheen diagnoosin yhteydessä (M1) | 70 (61,9) | 50 (44,6) | 120 (53,3) |
| Alkuvaiheen diagnoosista satunnaistamiseen kuluneen ajan mediaani (vuotta) | 2,00 | 2,31 | 2,26 |
| Metastasoituneen kastraatioresistentin eturauhassyövän toteamisesta ensimmäiseen annokseen kuluneen ajan mediaani (vuotta) | 0,27 | 0,28 | 0,27 |
BPI-SF-kipupisteet lähtötilanteessa (viimeiset pisteet ennen ensimmäistä annosta) 0 |
|
|
|
ECOG-toimintakykypisteet 0 |
69 (61,1) |
80 (71,4) |
149 (66,2) |
| 1 P = prednisoni tai prednisoloni | |||
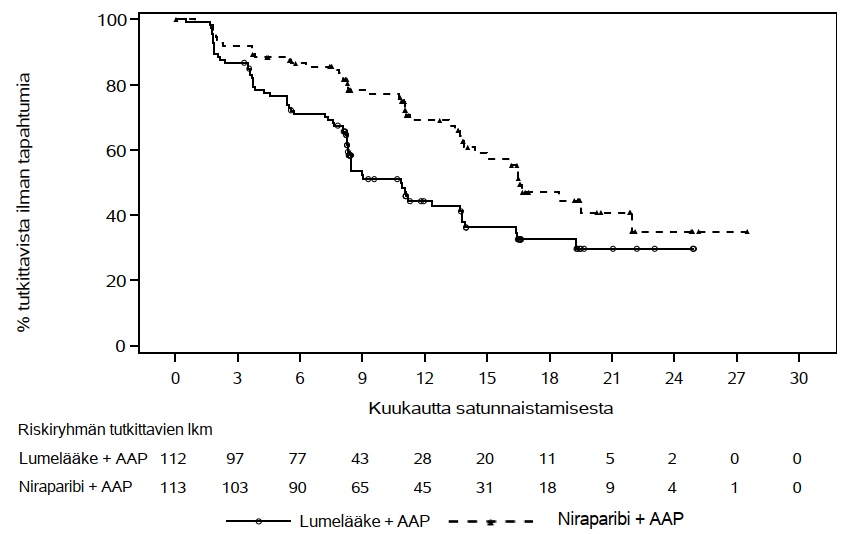
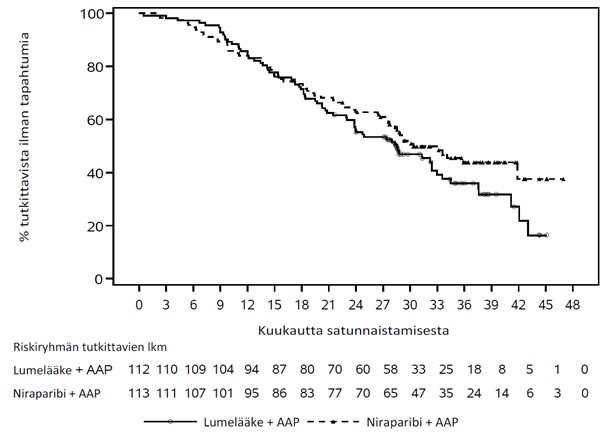
Primaarianalyysissa havaittiin sokkoutetun, riippumattoman ja keskitetyn arvioijatahon arvioiman radiologisen tautivapaan elossaoloajan pidentyneen niraparibin sekä abirateroniasetaatin ja prednisonin yhdistelmää saaneilla BRCA-potilasjoukon tutkittavilla tilastollisesti merkitsevästi verrattuna lumelääkkeen sekä abirateroniasetaatin ja prednisonin yhdistelmää saaneisiin BRCA-potilasjoukon tutkittaviin. BRCA-potilasjoukon keskeiset tehotulokset esitetään taulukossa 6. Sokkoutetun, riippumattoman ja keskitetyn arvioijatahon arvioiman radiologisen tautivapaan elossaoloajan Kaplan–Meierin käyrät BRCA-potilasjoukossa esitetään kuvassa 3.
| Taulukko 6. MAGNITUDE-tutkimuksen BRCA-potilasjoukosta saadut tehoa koskevat tulokset | ||
| Päätetapahtumat | Akeega + P1 (N = 113) | Lumelääke + AAP1 (N = 112) |
| Radiologinen tautivapaa elossaoloaika (rPFS)2 | ||
| Sairauden etenemistä tai kuolemaa koskeva tapahtuma (%) | 45 (39,8 %) | 64 (57,1 %) |
| Mediaani, kuukautta (95 %:n luottamusväli) | 16,6 (13,9; NE) | 10,9 (8,3; 13,8) |
| Riskisuhde (95 %:n luottamusväli) | 0,533 (0,361; 0,789) | |
| p-arvo | 0,0014 | |
| Kokonaiselossaoloaika3 | ||
| Riskisuhde (95 %:n luottamusväli) | 0,788 (0,554; 1,120) | |
| 1 P = prednisoni tai prednisoloni 2 Primaarianalyysi/välianalyysi (tietojen keruun katkaisu: 8. lokakuuta 2021), kun seuranta-ajan mediaani oli 18,6 kuukautta 3 Loppuanalyysi (tietojen keruun katkaisu: 15. toukokuuta 2023), kun seuranta-ajan mediaani oli 35,9 kuukautta NE = ei arvioitavissa | ||
Kuva 3. Kaplan–Meierin kuvaaja sokkoutetun, riippumattoman ja keskitetyn arvioijatahon arvioimasta radiologisesta tautivapaasta elossaoloajasta BRCA-potilasjoukossa (MAGNITUDE, primaarianalyysi)
Kuva 4. Kaplan–Meierin kuvaaja kokonaiselossaoloajasta (MAGNITUDE, kohortti 1, BRCA-potilasjoukko, loppuanalyysi)
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Akeega-valmisteen käytöstä eturauhasen syöpäkasvainten hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Niraparibin ja abirateronin samanaikaisella annolla ei ole vaikutusta kummankaan osakomponentin altistukseen. Niraparibin ja abirateronin AUC- ja Cmax-arvot ovat verrannolliset annettaessa niraparibia ja abirateronia Akeega-kalvopäällysteisten tablettien tavanomaisena vahvuutena (100 mg/500 mg) tai erillisistä valmisteista koostuvana yhdistelmänä verrattuna altistukseen vastaavina monoterapioina.
Imeytyminen
Akeega
Kun Akeega-tabletteja annettiin useina annoksina metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastaville potilaille paastotilassa ja modifioidussa paastotilassa, maksimipitoisuus plasmassa saavutettiin niraparibin osalta 3 tunnissa (mediaani) ja abirateronin osalta 1,5 tunnissa (mediaani).
Vastaavassa hyötyosuutta koskevassa tutkimuksessa kalvopäällysteisten Akeega-tablettien pienemmällä vahvuudella (2 x 50 mg/500 mg) hoitoa saaneilla metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavilla potilailla (n = 67) abirateronin huippupitoisuus (Cmax) oli 33 % ja kokonaisaltistus (AUC0-72h) abirateronille oli 22 % suurempi kuin altistus potilailla (n = 67), jotka ottivat samoja vaikuttavia aineita erillisinä valmisteina (100 mg:n niraparibikapseli ja 4 x 250 mg:n abirateroniasetaattitablettia) (ks. kohta Annostus ja antotapa). Altistuksen vaihtelu (variaatiokerroin) tutkittavien välillä oli huippupitoisuuden osalta 80,4 % ja kokonaisaltistuksen osalta 72,9 %. Niraparibialtistus oli kalvopäällysteisten Akeega-tablettien pienempää vahvuutta ja samoja vaikuttavia aineita erillisinä valmisteina saaneilla verrannollinen.
Niraparibi
Niraparibin absoluuttinen biologinen hyötyosuus on noin 73 %. Niraparibi on P-glykoproteiinin (P‑gp) ja rintasyöpäresistenssiproteiinin (BCRP) substraatti. Sen läpäisevyys ja biologinen hyötyosuus ovat suuret, joten kliinisesti oleellisten yhteisvaikutusten riski näitä kuljettajaproteiineja estävien lääkevalmisteiden kanssa on kuitenkin epätodennäköinen.
Abirateroniasetaatti
Abirateroniasetaatti muuntuu in vivo nopeasti abirateroniksi (ks. kohta Farmakodynamiikka).
Abirateroniasetaatin antaminen ruoan kanssa suurentaa keskimääräistä systeemistä altistusta abirateronille enintään 10‑kertaiseksi (AUC) ja enintään 17‑kertaiseksi (Cmax) paastotilaan verrattuna, aterian rasvasisällöstä riippuen. Kun huomioidaan aterian sisällön ja koostumuksen normaali vaihtelu, abirateroniasetaatin ottaminen aterian yhteydessä saattaa johtaa altistuksen huomattavaan vaihteluun. Abirateroniasetaattia ei siksi saa ottaa ruoan kanssa.
Jakautuminen
Niraparibin laskennallinen jakautumistilavuus oli populaatiofarmakokineettisen analyysin perusteella 1 117 l ja abirateronin osalta se oli 25 774 l, mikä osoittaa laajan ekstravaskulaarisen jakautumisen.
Niraparibi
Niraparibi sitoutui kohtalaisesti (83,0 %) ihmisen plasmassa proteiineihin, pääasiassa seerumin albumiiniin.
Abirateroniasetaatti
14C‑abirateronista ihmisen plasman proteiineihin sitoutuu 99,8 %.
Biotransformaatio
Niraparibi
Niraparibi metaboloituu pääasiassa karboksyyliesteraasien välityksellä pääasialliseksi inaktiiviseksi metaboliitiksi M1:ksi. M1 ja M10 (myöhemmin muodostuneet M1:n glukuronidit) olivat massatasetutkimuksessa pääasialliset kiertävät metaboliitit. CYP3A4:ää suoliston tasolla estävän vaikutuksen mahdollisuutta ei ole selvitetty relevanteilla niraparibipitoisuuksilla. In vitro niraparibi indusoi suurina pitoisuuksina heikosti CYP1A2:ta.
Abirateroniasetaatti
Kun 14C‑abirateroniasetaatti annetaan kapseleina suun kautta, abirateroniasetaatti hydrolysoituu karboksyyliesteraasien välityksellä abirateroniksi, joka metaboloituu tämän jälkeen pääasiassa maksassa sulfaation, hydroksylaation ja oksidaation kautta. Abirateroni on CYP3A4:n ja sulfotransferaasi 2A1:n (SULT2A1) substraatti. Suurin osa verenkierrossa todetusta radioaktiivisuudesta (noin 92 %) on abirateronin metaboliittien muodossa. 15 havaitusta metaboliitista kaksi pääasiallista metaboliittia, abirateronisulfaatti ja N‑oksidiabirateronisulfaatti, vastaavat kumpikin noin 43 %:a kokonaisradioaktiivisuudesta. Abirateroni on maksassa lääkkeitä metaboloivien entsyymien CYP2D6:n ja CYP2C8:n estäjä (ks. kohta Yhteisvaikutukset).
Eliminaatio
Akeega
Annettaessa niraparibia ja abirateronia yhdistelmänä niraparibin keskimääräinen t½ oli noin 62 tuntia ja abirateronilla se oli 20 tuntia, ja niraparibin laskennallinen puhdistuma (CL/F) oli 16,7 l/h ja abirateronilla se oli 1 673 l/h; nämä perustuvat metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavien tutkittavien populaatiofarmakokineettiseen analyysiin.
Niraparibi
Niraparibi eliminoituu pääasiassa maksan ja sapen sekä munuaisten kautta. Suun kautta annetun 300 mg:n [14C]-niraparibikerta-annoksen jälkeen keskimäärin 86,2 % (vaihteluväli 71–91 %) annoksesta havaittiin 21 päivän aikana virtsassa ja ulosteissa. Virtsassa havaittu radioaktiivisuus vastasi 47,5 %:a (vaihteluväli 33,4–60,2 %) ja ulosteessa havaittu radioaktiivisuus vastasi 38,8 %:a (vaihteluväli 28,3–47,0 %) annoksesta. Kuuden päivän aikana kerätyissä yhdistetyissä näytteissä 40,0 % annoksesta havaittiin virtsassa pääasiassa metaboliitteina ja 31,6 % annoksesta havaittiin ulosteissa pääasiassa muuttumattomana niraparibina. M1-metaboliitti on MATE1- (Multidrug And Toxin Extrusion) ja MATE2-kuljettajaproteiinien substraatti.
Abirateroniasetaatti
Kun suun kautta annettiin 1 000 mg 14C‑abirateroniasetaattia, noin 88 % radioaktiivisesta annoksesta todettiin ulosteessa ja noin 5 % virtsassa. Tärkeimmät ulosteessa esiintyvät yhdisteet ovat muuttumaton abirateroniasetaatti ja abirateroni (abirateroniasetaattia noin 55 % ja abirateronia noin 22 % annetusta annoksesta).
Niraparibin tai abirateronin vaikutukset kuljettajaproteiineihin
Niraparibi estää heikosti P-gp:tä IC50-arvolla 161 µM. Niraparibi on BCRP:n, OCT1:n (orgaanisten kationien kuljettajaproteiini 1), MATE-1- ja ‑2-kuljettajaproteiinien estäjä IC50-arvoilla 5,8 µM (BCRP), 34,4 µM (OCT1), 0,18 µM (MATE-1) ja ≤ 0,14 µM (MATE-2). Abirateronin pääasiallisten metaboliittien abirateronisulfaatin ja N-oksidiabirateronisulfaatin osoitettiin estävän lääkeaineita maksasoluihin kuljettavan OATP1B1:n (orgaanisten anionien kuljettajapolypeptidi 1B1) toimintaa, minkä seurauksena altistus OATP1B1:n välityksellä eliminoituville lääkevalmisteille plasmassa saattaa suurentua. Kuljettajaproteiiniin OATP1B1 perustuvien yhteisvaikutusten varmistamiseksi ei ole kliinisiä tietoja saatavilla.
Erityispotilasryhmät
Maksan vajaatoiminta
Eturauhassyöpää sairastavat potilaat saivat kliinisissä tutkimuksissa pelkästään niraparibia tai niraparibin ja abirateroniasetaatin yhdistelmää, ja näistä kliinisistä tutkimuksista saatujen tietojen populaatiofarmakokineettisen analyysin perusteella lievä maksan vajaatoiminta (NCI-ODWG-kriteerit, n = 231) ei vaikuttanut altistukseen niraparibille.
Syöpäpotilailla tehdyssä kliinisessä tutkimuksessa käytettiin NCI-ODWG-kriteerejä maksan vajaatoiminta-asteen luokittelemiseen; 300 mg:n kerta-annoksen jälkeen niraparibin AUCinf-arvo oli keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla (n = 8) 1,56-kertainen (90 %:n luottamusväli: 1,06; 2,30) verrattuna niraparibin AUCinf-arvoon potilailla, joiden maksan toiminta oli normaali (n = 9).
Abirateronin farmakokinetiikkaa tutkittiin tutkittavilla, jotka sairastivat ennestään lievää (Child–Pugh-luokka A, n = 8) tai keskivaikeaa (Child–Pugh-luokka B, n = 8) maksan vajaatoimintaa ja 8:lla terveellä verrokilla. Suun kautta otetun 1 000 mg:n kerta-annoksen jälkeen systeeminen abirateronialtistus suureni lievää maksan vajaatoimintaa ennestään sairastavilla tutkittavilla noin 1,11‑kertaiseksi ja keskivaikeaa maksan vajaatoimintaa ennestään sairastavilla tutkittavilla noin 3,6‑kertaiseksi.
Abirateronin farmakokinetiikkaa selvitettiin toisessa tutkimuksessa tutkittavilla, jotka sairastivat ennestään vaikeaa (n = 8) maksan vajaatoimintaa (Child–Pugh-luokka C) ja 8:lla terveellä verrokilla, joiden maksan toiminta oli normaali. Vaikeaa maksan vajaatoimintaa sairastavilla potilailla abirateronin AUC-arvo suureni noin 7-kertaiseksi ja vapaan lääkeaineen fraktio suureni 1,8-kertaiseksi verrattuna tutkittaviin, joiden maksan toiminta oli normaali. Akeega-valmisteen käytöstä keskivaikeaa ja vaikeaa maksan vajaatoimintaa sairastaville potilaille ei ole kliinistä kokemusta (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Eturauhassyöpää sairastavat potilaat saivat kliinisissä tutkimuksissa pelkästään niraparibia tai niraparibin ja abirateroniasetaatin yhdistelmää. Näistä kliinisistä tutkimuksista saatujen tietojen populaatiofarmakokineettisen analyysin perusteella lievää (kreatiniinipuhdistuma 60–90 ml/min, n = 337) ja keskivaikeaa (kreatiniinipuhdistuma 30–60 ml/min, n = 114) munuaisten vajaatoimintaa sairastavilla potilailla niraparibin puhdistuma oli hieman pienempi kuin henkilöillä, joiden munuaisten toiminta oli normaali (altistus lievää munuaisten vajaatoimintaa sairastavilla enintään 13 % suurempi ja altistus keskivaikeaa munuaisten vajaatoimintaa sairastavilla 13–40 % suurempi).
Abirateronin farmakokinetiikkaa loppuvaiheen munuaissairautta sairastavilla potilailla, jotka saivat säännöllistä hemodialyysihoitoa (n = 8) verrattiin kaltaistettuihin verrokkeihin, joiden munuaisten toiminta oli normaali (n = 8). Suun kautta otetun 1 000 mg:n kerta-annoksen jälkeen systeeminen abirateronialtistus ei suurentunut loppuvaiheen munuaissairautta sairastavilla tutkittavilla, jotka saivat dialyysihoitoa. Akeega-valmisteen käytöstä vaikeaa munuaisten vajaatoimintaa sairastaville potilaille ei ole kliinistä kokemusta (ks. kohta Annostus ja antotapa).
Paino, ikä ja etninen tausta
Populaatiofarmakokineettisen analyysin perusteella eturauhassyöpää sairastavilla potilailla. jotka saivat kliinisissä tutkimuksissa pelkästään niraparibia tai abirateroniasetaattia tai niiden yhdistelmää:
- painolla ei ollut kliinisesti merkittävää vaikutusta niraparibialtistukseen (painon vaihteluväli: 43,3–165 kg) eikä abirateronialtistukseen (painon vaihteluväli: 56,0–135 kg).
- iällä ei ollut merkittävää vaikutusta niraparibin (iän vaihteluväli 45–90 vuotta) ja abirateronin (iän vaihteluväli 19–85 vuotta) farmakokinetiikkaan.
- tiedot ovat riittämättömiä päätelmien tekemiseksi etnisen taustan vaikutuksesta niraparibin ja abirateronin farmakokinetiikkaan.
Pediatriset potilaat
Akeega-valmisteen farmakokinetiikan tutkimiseksi pediatrisilla potilailla ei ole tehty tutkimuksia.
Prekliiniset tiedot turvallisuudesta
Akeega
Akeega-valmisteella ei ole tehty prekliinisiä tutkimuksia. Prekliiniset toksikologiset tiedot perustuvat niraparibilla ja abirateroniasetaatilla erikseen tehtyjen tutkimusten havaintoihin.
Niraparibi
Niraparibi esti in vitro dopamiinin kuljetusproteiinia pitoisuuksina, jotka olivat ihmisen altistusta pienempiä. Niraparibikerta-annokset lisäsivät hiirillä dopamiinin ja metaboliittien solunsisäisiä pitoisuuksia aivokuoressa. Hiirillä tehdyistä kahdesta kerta-annostutkimuksesta toisessa havaittiin vähentynyttä lokomotorista aktiivisuutta. Näiden havaintojen kliinistä merkitystä ei tunneta. Rotilla ja koirilla tehdyissä toistuvan altistuksen toksisuutta koskeneissa tutkimuksissa ei havaittu vaikutuksia käyttäytymisparametreihin ja/tai neurologisiin parametreihin, kun altistuksen arvioitiin olleen keskushermostossa samankaltainen tai pienempi kuin oletettu terapeuttinen altistustaso.
Sekä rotilla että koirilla havaittiin vähentynyttä spermatogeneesia altistuksilla, jotka olivat terapeuttista altistustasoa pienemmät, ja se korjautui pääosin neljän viikon kuluessa annon päättymisestä.
Niraparibi ei ollut mutageeninen bakteerimutageenisuustestissä (Amesin testi), mutta se oli klastogeeninen nisäkkäiden kromosomipoikkeavuusmäärityksessä in vitro ja rotan luuytimen mikrotumamäärityksessä in vivo. Tällainen klastogeenisuus on yhdenmukainen perimän epävakauden kanssa, mikä johtuu niraparibin ensisijaisista farmakologisista ominaisuuksista ja osoittaa mahdollisen genotoksisuuden ihmisillä.
Niraparibilla ei ole tehty lisääntymis- ja kehitystoksisuutta koskevia tutkimuksia.
Niraparibilla ei ole tehty karsinogeenisuustutkimuksia.
Abirateroniasetaatti
Toksisuutta selvittäneissä eläinkokeissa verenkierrossa olevat testosteronipitoisuudet olivat pienentyneet huomattavasti. Tämän seurauksena havaittiin lisääntymiselinten, lisämunuaisten, aivolisäkkeen ja rintarauhasten painon vähenemistä sekä elinten morfologisia ja/tai histopatologisia muutoksia. Kaikkien muutosten osoitettiin olevan täysin tai osittain korjautuvia. Lisääntymiselimissä ja androgeenille herkissä elimissä todetut muutokset ovat yhdenmukaisia abirateronin farmakologisten ominaisuuksien kanssa. Kaikki hoitoon liittyneet hormonaaliset muutokset korjautuivat tai niiden osoitettiin olevan häviämässä 4 viikon palautumisjakson aikana.
Uros- ja naarasrotilla tehdyissä hedelmällisyystutkimuksissa abirateroniasetaatti heikensi hedelmällisyyttä, mutta tämä vaikutus korjautui täysin 4–16 viikon kuluessa abirateroniasetaatin annon lopettamisen jälkeen.
Rotalla tehdyssä kehitystoksisuutta selvittäneessä tutkimuksessa abirateroniasetaatti vaikutti tiineyteen mm. alentamalla sikiöiden painoa ja heikentämällä niiden eloonjäämistä. Ulkoisissa sukuelimissä havaittiin vaikutuksia, vaikka abirateroniasetaatti ei ollut teratogeeninen.
Kaikki vaikutukset näissä rotalla tehdyissä hedelmällisyys- ja kehitystoksisuustutkimuksissa liittyivät abirateronin farmakologiseen vaikutukseen.
Lukuun ottamatta kaikissa toksikologisissa eläinkokeissa lisääntymiselimissä todettuja muutoksia, farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Abirateroniasetaatti ei ollut karsinogeeninen transgeenisellä (Tg.rasH2) hiirellä tehdyssä kuusi kuukautta kestäneessä tutkimuksessa. Rotalla tehdyssä 24 kuukautta kestäneessä karsinogeenisuustutkimuksessa abirateroniasetaatti lisäsi interstitiaalisolukasvainten ilmaantuvuutta kiveksissä. Löydöksen katsotaan liittyvän abirateronin farmakologiseen vaikutukseen ja olevan rotille ominainen. Abirateroniasetaatti ei ollut karsinogeeninen naarasrotille.
Ympäristöön kohdistuvien riskien arviointi
Ympäristöön kohdistuvien riskien arviointia koskevat tutkimukset ovat osoittaneet, että abirateroniasetaatista saattaa aiheutua riski pintavedelle, pohjavedelle ja sedimenteille (ks. kohta Käyttö- ja käsittelyohjeet).
Farmaseuttiset tiedot
Apuaineet
Akeega 50 mg/500 mg kalvopäällysteiset tabletit
Tabletin ydin
Kolloidinen vedetön piidioksidi
Krospovidoni
Hypromelloosi
Laktoosimonohydraatti
Magnesiumstearaatti
Mikrokiteinen selluloosa
Natriumlauryylisulfaatti
Kalvopäällyste
Musta rautaoksidi (E172)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Natriumlauryylisulfaatti
Glyserolimonokaprylokapraatti
Polyvinyylialkoholi
Talkki
Titaanidioksidi (E171)
Akeega 100 mg/500 mg kalvopäällysteiset tabletit
Tabletin ydin
Kolloidinen vedetön piidioksidi
Krospovidoni
Hypromelloosi
Laktoosimonohydraatti
Magnesiumstearaatti
Mikrokiteinen selluloosa
Natriumlauryylisulfaatti
Kalvopäällyste
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Natriumlauryylisulfaatti
Glyserolimonokaprylokapraatti
Polyvinyylialkoholi
Talkki
Titaanidioksidi (E171)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
30 kuukautta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
AKEEGA tabletti, kalvopäällysteinen
50/500 mg (L:kyllä) 56 fol (5364,66 €)
100/500 mg (L:kyllä) 56 fol (5364,66 €)
PF-selosteen tieto
Yksi 28 päivän kartonkikotelo sisältää 56 kalvopäällysteistä tablettia kahdessa kartonkisessa taskupakkauksessa, joista kumpikin sisältää 28 kalvopäällysteistä tablettia PVdC/PE/PVC-läpipainopakkauksessa, jossa on alumiininen läpipainokalvo.
Valmisteen kuvaus:
Akeega 50 mg/500 mg kalvopäällysteiset tabletit
Kellertävän oransseja tai kellertävän ruskeita, soikeita, kalvopäällysteisiä tabletteja (22 mm x 11 mm), joiden toiselle puolelle on kaiverrettu ”N 50 A” ja joiden toisella puolella ei ole merkintöjä.
Akeega 100 mg/500 mg kalvopäällysteiset tabletit
Oransseja, soikeita, kalvopäällysteisiä tabletteja (22 mm x 11 mm), joiden toiselle puolelle on kaiverrettu ”N 100 A” ja joiden toisella puolella ei ole merkintöjä.
Käyttö- ja käsittelyohjeet
Tämä lääkevalmiste saattaa vaikutusmekanisminsa perusteella vahingoittaa sikiötä. Sen vuoksi naisten, jotka ovat raskaana tai voivat tulla raskaaksi, pitää käyttää suojaimia, esim. suojakäsineitä, käsitellessään Akeega-valmistetta (ks. kohta Raskaus ja imetys).
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti. Tämä lääkevalmiste saattaa aiheuttaa ympäristöriskin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Korvattavuus
AKEEGA tabletti, kalvopäällysteinen
50/500 mg 56 fol
100/500 mg 56 fol
- Ylempi erityiskorvaus (100 %). Niraparibin ja abirateronin yhdistelmävalmiste: Etäpesäkkeisen kastraatioresistentin eturauhassyövän hoito yhdistelmänä prednisonin tai prednisolonin kanssa aikuisille erityisin edellytyksin (1548).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Niraparibin ja abirateronin yhdistelmävalmiste: Etäpesäkkeisen kastraatioresistentin eturauhassyövän hoito erityisin edellytyksin (3093).
ATC-koodi
L01XK52
Valmisteyhteenvedon muuttamispäivämäärä
05.03.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com