
AREXVY injektiokuiva-aine ja suspensio, suspensiota varten
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
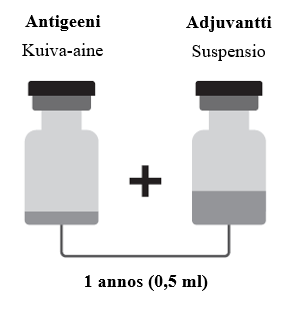
Käyttökuntoon saattamisen jälkeen yksi annos (0,5 ml) sisältää:
RSVPreF31‑antigeeni2,3 120 mikrog
1 RSVPreF3 = RS-viruksen rekombinantti glykoproteiini F, joka on stabiloitu fuusioitumista edeltävään konformaatioon)
2 RSVPreF3 on valmistettu kiinanhamsterin munasarjasoluissa yhdistelmä‑DNA‑tekniikalla
3 Adjuvantti AS01E sisältää seuraavia aineita:
Quillaja saponaria Molina ‑kasviuute, fraktio 21 (QS‑21) 25 mikrog
3‑O‑desasyyli‑4’‑monofosforyylilipidi A (MPL) Salmonella minnesota ‑mikrobista 25 mikrog
Apuaineet, joiden vaikutus tunnetaan
1 annos Arexvy injektiokuiva‑ainetta ja suspensiota suspensiota varten sisältää 0,18 mg polysorbaatti 80:tä (E433) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
1 annos Arexvy injektiokuiva‑ainetta (injektiopullo) ja suspensiota (esitäytetty ruisku) suspensiota varten sisältää 0,20 mg polysorbaatti 80:tä (E433) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva‑aine ja suspensio suspensiota varten.
Kliiniset tiedot
Käyttöaiheet
Arexvy on tarkoitettu aktiiviseen immunisaatioon RS‑viruksen (Respiratory Syncytial Virus, RSV) aiheuttaman alahengitystietaudin ehkäisemiseksi
- vähintään 18‑vuotiaille aikuisille.
Rokotteen käytön on perustuttava virallisiin suosituksiin.
Annostus ja antotapa
Annostus
Arexvy annetaan 0,5 ml:n kerta‑annoksena. Uusintarokotuksen tarvetta toisella annoksella ei ole määritetty (ks. kohta Farmakodynamiikka).
Pediatriset potilaat
Arexvy‑valmisteen turvallisuutta ja tehoa lasten hoidossa ei ole varmistettu.
Tietoja ei ole saatavilla.
Antotapa
Vain injektiona lihakseen, mieluiten hartialihakseen.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Ennen rokottamista
Ripeään, asianmukaiseen lääkärinhoitoon ja tarkkailuun on aina oltava valmius siltä varalta, että rokotteen annon jälkeen kehittyy anafylaktinen tapahtuma.
Rokotusta on lykättävä, jos rokotettavalla on akuutti vaikea kuumetauti. Vähäinen infektio, kuten nuhakuume, ei edellytä rokotuksen siirtämistä.
Kuten kaikkien rokotteiden yhteydessä, kaikille rokotetuille ei välttämättä kehity suojaavaa immuunivastetta.
Itse rokotusprosessin yhteydessä voi esiintyä ahdistuneisuuteen liittyviä reaktioita, kuten vasovagaalisia reaktioita (pyörtymistä), hyperventilaatiota tai stressiin liittyviä reaktioita. On tärkeää ryhtyä varotoimiin pyörtymisestä johtuvien vammojen ehkäisemiseksi.
Käyttöön liittyvät varotoimet
Rokotetta ei saa antaa verisuoneen eikä ihon sisään. Arexvy‑rokotteen annosta ihon alle ei ole tietoja.
Kuten kaikki lihakseen annettavat injektiot, Arexvy on annettava varoen, jos rokotettavalla on trombosytopenia tai jokin hyytymishäiriö. Näillä henkilöillä voi esiintyä verenvuotoa, kun valmiste on annettu lihakseen.
Systeemiset immunosuppressantit ja immuunivajavuus
Jos potilas saa immunosuppressiivista hoitoa tai on immuunipuutteinen, immuunivaste Arexvylle voi olla heikompi (ks. kohta Farmakodynamiikka).
Apuaineet, joiden vaikutus tunnetaan
1 annos Arexvy injektiokuiva‑ainetta ja suspensiota suspensiota varten sisältää 0,18 mg polysorbaatti 80:tä.
1 annos Arexvy injektiokuiva‑ainetta (injektiopullo) ja suspensiota (esitäytetty ruisku) suspensiota varten sisältää 0,20 mg polysorbaatti 80:tä.
Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Tämä lääkevalmiste sisältää kaliumia alle 1 mmol (39 mg) per annos, eli sen voidaan sanoa olevan ”kaliumiton”.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Käyttö muiden rokotteiden kanssa
Arexvy voidaan antaa samanaikaisesti COVID-19 mRNA-rokotteen, pneumokokkikonjugaattirokotteen, vyöruusurokotteen (rekombinantti, adjuvanttia sisältävä), tai inaktivoidun kausi‑influenssarokotteen (adjuvantiton tavanomainen annos, adjuvantiton suuri annos tai adjuvantillinen tavanomainen annos) kanssa.
Jos Arexvy annetaan samanaikaisesti toisen injisoitavan rokotteen kanssa, rokotteet on annettava aina eri kohtiin.
Arexvyn samanaikaista antoa muiden kuin yllä mainittujen rokotteiden kanssa ei ole tutkittu.
Raskaus ja imetys
Raskaus
Arexvyn käytöstä raskaana oleville naisille ei ole olemassa kliinisistä tutkimuksista saatuja tietoja. Yhdessä kliinisessä tutkimuksessa ennenaikaisten synnytysten määrä lisääntyi lumelääkkeeseen verrattuna, kun kokeellista adjuvantitonta RSVPreF3-rokotetta annettiin 3 557:lle raskaana olevalle naiselle. Tällä hetkellä ei voida tehdä johtopäätöksiä syy-yhteydestä adjuvantittoman RSVPreF3-rokotteen antamisen ja ennenaikaisen synnytyksen välillä. Arexvy‑valmistetta tai kokeellista adjuvantitonta RSVPreF3‑rokotetta koskevissa eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria kehitykseen kohdistuvia tai lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Arexvyn käyttöä ei suositella raskauden aikana.
Imetys
Ei ole olemassa tietoja Arexvyn erittymisestä maitoon ihmisillä tai koe‑eläimillä. Arexvyn käyttöä ei suositella imetyksen aikana.
Hedelmällisyys
Arexvyn vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Arexvy‑valmistetta tai kokeellista adjuvantitonta RSVPreF3‑rokotetta koskevissa eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia Arexvy‑valmisteen vaikutuksesta ajokykyyn tai koneidenkäyttökykyyn ei ole tehty.
Arexvy‑valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Joillakin kohdassa Haittavaikutukset mainituilla haittavaikutuksilla (esim. väsymyksellä) voi olla ohimenevä vaikutus ajokykyyn tai koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Vaiheen 3 lumekontrolloidussa tutkimuksessa (RSV OA=ADJ‑006) ≥ 60‑vuotiailla tutkittavat saivat joko yhden annoksen Arexvya (N = 12 469) tai lumerokotteen (N = 12 503). Seuranta-aika oli noin 12 kuukautta. Yleisimmin ilmoitetut haittavaikutukset olivat injektiokohdan kipu (61 %), väsymys (34 %), lihaskipu (29 %), päänsärky (28 %) ja nivelkipu (18 %).
Vaiheen 3 lumekontrolloidussa tutkimuksessa (RSV OA=ADJ‑018) 50–59-vuotiailla tutkittavilla (N = 769) yleisimmin raportoituja haittavaikutuksia olivat injektiokohdan kipu (76 %), väsymys (40 %), lihaskipu (36 %), päänsärky (32 %) ja nivelkipu (23 %).
Vaiheen 3 avoimessa tutkimuksessa (RSV OA=ADJ‑025) 18–49‑vuotiailla tutkittavilla (N = 1 029) yleisimmin raportoituja haittavaikutuksia olivat injektiokohdan kipu (76 %), väsymys (60 %), lihaskipu (60 %), päänsärky (44 %) ja nivelkipu (28 %).
Kaikissa kolmessa tutkimuksessa nämä haittavaikutukset olivat yleensä lieviä tai kohtalaisia ja hävisivät muutaman päivän kuluessa rokotuksesta.
Haittavaikutustaulukko
Taulukossa 1 kuvattu turvallisuusprofiili perustuu vaiheen 3 kliinisten tutkimusten tietoihin (RSV OA=ADJ‑006, ‑018 ja ‑025), jotka on tehty Euroopassa, Pohjois-Amerikassa, Aasiassa ja eteläisellä pallonpuoliskolla aikuisilla ≥18‑, sekä markkinoilletulon jälkeiseen kokemukseen.
Haittavaikutukset luokitellaan jäljempänä MedDRA‑elinjärjestelmäluokan ja yleisyyden mukaan.
Hyvin yleinen (≥ 1/10)
Yleinen (≥ 1/100, < 1/10)
Melko harvinainen (≥ 1/1 000, < 1/100)
Harvinainen (≥ 1/10 000, < 1/1 000)
Hyvin harvinainen (< 1/10 000)
Tuntematon (koska saatavissa oleva tieto ei riitä yleisyyden arviointiin)
Taulukko 1. Haittavaikutukset
Elinjärjestelmäluokka | Yleisyys | Haittavaikutukset |
Veri ja imukudos | Melko harvinainen | lymfadenopatia |
Immuunijärjestelmä | Melko harvinainen | yliherkkyysreaktiot (kuten ihottuma) |
Hermosto | Hyvin yleinen | päänsärky |
Hyvin harvinainen | Guillain–Barrén oireyhtymä | |
Ruoansulatuselimistö | Melko harvinainen | pahoinvointi, vatsakipu, oksentelu |
Luusto, lihakset ja sidekudos | Hyvin yleinen | lihaskipu, nivelkipu |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | injektiokohdan kipu, väsymys |
Yleinen | injektiokohdan punoitus, injektiokohdan turvotus, kuume, vilunväristykset | |
Melko harvinainen | injektiokohdan kutina | |
kipu, huonovointisuus | ||
Tuntematon | injektiokohdan nekroosi1 |
1Spontaanisti raportoitu haittavaikutus.
Valikoitujen haittavaikutusten kuvaus
Yhdysvalloissa ≥ 65‑vuotiailla toteutetussa markkinoilletulon jälkeisessä havainnointitutkimuksessa Guillain–Barrén oireyhtymän riski oli suurentunut 42 vuorokauden seurannassa Arexvy-rokotuksen jälkeen (arvio 7 ylimääräistä tapausta miljoonaa annettua rokoteannosta kohden).
Muut erityisryhmät
Immuunipuutteiset henkilöt
Kun kiinteän elinsiirteen (munuais- tai keuhkosiirteen) saaneille aikuisille annettiin yksi tai kaksi Arexvy-annosta (RSV OA=ADJ‑023; ks. kohta Farmakodynamiikka), haittavaikutukset olivat yhdenmukaisia yhden Arexvy-annoksen saaneilla ei‑immuunipuutteisilla aikuisilla todettujen haittavaikutusten kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa ei ole ilmoitettu yhtään yliannostustapausta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: rokotteet, muut virusrokotteet, ATC‑koodi: J07BX05
Vaikutusmekanismi
Arexvy, joka on RSV‑spesifisen antigeenin (F‑proteiini fuusioitumista edeltävässä konformaatiossa) ja adjuvanttijärjestelmän (AS01E) yhdistelmä, on kehitetty antigeenispesifisen soluvälitteisen immuunivasteen ja neutraloiviin vasta‑aineisiin liittyvän vasteen tehostamiseen henkilöille, joilla on ennestään immuniteetti RS‑virusta vastaan. Adjuvantti AS01E edistää rokoteperäisiä antigeeneja esittelevien solujen rekrytointia ja aktivoitumista imusolmukkeessa, jonne imuneste laskee, mikä taas johtaa RSVPreF3‑spesifisten CD4+‑T‑solujen muodostukseen.
Teho
Tehoa RSV‑alahengitystietautia vastaan arvioitiin ≥ 60‑vuotiailla aikuisilla enintään kolmen RSV‑kauden ajan satunnaistetussa, lumekontrolloidussa, havainnoijasokkoutetussa vaiheen 3 kliinisessä tutkimuksessa, joka toteutettiin 17 maassa pohjoisella ja eteläisellä pallonpuoliskolla (RSV OA=ADJ‑006).
Ensisijainen tehoanalyysipopulaatio (josta käytetään nimitystä muokattu altistunut populaatio) koostui ≥ 60‑vuotiaista aikuisista, jotka saivat yhden annoksen Arexvy‑rokotetta tai lumerokotetta ja jotka eivät ilmoittaneet vahvistetusta akuutista RSV‑hengitystietaudista ennen päivää 15 rokotuksen jälkeen.
Yhteensä 24 960 tutkittavaa satunnaistettiin samassa suhteessa saamaan yksi annos Arexvy‑rokotetta (N = 12 466) tai lumerokotetta (N = 12 494) ensimmäisen RSV-kauden aikana. Ennen toista RSV-kautta potilaat, jotka olivat saaneet Arexvy-rokotetta ensimmäisen kauden aikana, satunnaistettiin uudelleen saamaan joko lumerokotetta (N = 4 991) tai toinen annos Arexvy-rokotetta (N = 4 966). Osallistujat, jotka olivat saaneet lumerokotetta ennen ensimmäistä RSV-kautta, saivat toisen annoksen lumerokotetta ennen toista RSV-kautta. Osallistujia seurattiin kolmannen RSV-kauden loppuun (seuranta-ajan mediaani 30,6 kuukautta).
Tutkittavien iän mediaani oli 69 vuotta (vaihteluväli: 59–102 vuotta). Tutkittavista noin 74 % oli yli 65‑vuotiaita, noin 44 % yli 70‑vuotiaita ja noin 8 % yli 80‑vuotiaita. Noin 52 % oli naisia.
Lähtötilanteessa 39,3 %:lla tutkittavista oli vähintään yksi erityisesti seurattava liitännäissairaus; 19,7 %:lla oli perussairautena kardiorespiratorinen sairaus (keuhkoahtaumatauti, astma, jokin krooninen hengitystie‑/keuhkosairaus tai krooninen sydämen vajaatoiminta) ja 25,8 %:lla endokriinis‑metabolinen tila (diabetes, pitkälle edennyt maksa‑ tai munuaistauti).
Vahvistetut RSV‑tapaukset määritettiin nenänielunäytteestä tehtävällä qRT‑PCR‑testillä (qRT‑PCR: kvantitatiivinen käänteistranskriptaasientsyymiä hyödyntävä polymeraasiketjureaktio).
Alahengitystietauti määriteltiin seuraavien kriteerien perusteella: tutkittavalla oli täytynyt olla vähintään kaksi alahengitystieoiretta/‑löydöstä, mukaan lukien vähintään yksi alahengitystielöydös, vähintään 24 tunnin ajan tai vähintään kolme alahengitystieoiretta vähintään 24 tunnin ajan. Alahengitystieoireita olivat yskösten ilmaantuminen tai lisääntyminen, yskän ilmaantuminen tai lisääntyminen ja hengenahdistuksen ilmaantuminen tai lisääntyminen. Alahengitystielöydöksiä olivat hengityksen vinkumisen ilmaantuminen tai lisääntyminen, ritinä/rahina, hengitystiheys ≥ 20 kertaa/min, pieni tai pienentynyt happisaturaatio (O2‑saturaatio < 95 % tai ≤ 90 %, jos lähtötilanteessa < 95 %) tai lisähapen tarve.
Teho RSV‑alahengitystietautia vastaan ensimmäisen RSV-kauden aikana (varmistusanalyysi)
Ensisijaisena tavoitteena oli osoittaa teho vahvistetun, RSV‑alatyypin A ja/tai B aiheuttaman ensimmäisen alahengitystietautiepisodin ehkäisyssä ensimmäisen RSV‑kauden aikana.
Rokotteen teho koko populaatiossa ja alaryhmittäin on esitetty taulukossa 2.
Teho ensimmäisen RSV‑alahengitystietaudin ehkäisyssä alkaen rokotuksen jälkeisestä päivästä 15 oli 82,6 % (96,95 %:n luottamusväli [lv] 57,9–94,1 %) ≥ 60‑vuotiailla tutkittavilla verrattuna lumerokotteeseen. Rokotteen teho RSV‑alahengitystietautia vastaan säilyi 6,7 kuukauden seurantavaiheen ajan (mediaanikesto). Rokotteen teho RSV‑alatyypin A aiheuttamaa tautia vastaan oli 84,6 % (95 %:n lv 32,1–98,3) ja RSV‑alatyypin B aiheuttamaa tautia vastaan 80,9 % (95 %:n lv 49,4–94,3).
Taulukko 2. Tehoanalyysi ensimmäisen RSV-kauden aikana (varmistusanalyysi): ensimmäinen RSV‑alahengitystietauti koko populaatiossa, iän ja samanaikaisten sairauksien mukaan luokitelluissa alaryhmissä (muokattu altistunut populaatio)
| Alaryhmä | Arexvy | Lume | Teho, % (lv)a | ||||
| N | n | Ilmaantuvuus / 1 000 henkilövuotta | N | n | Ilmaantuvuus / 1 000 henkilövuotta | ||
| Koko populaatio (≥ 60 vuotta)b | 12 466 | 7 | 1,0 | 12 494 | 40 | 5,8 | 82,6 (57,9–94,1) |
| 60–69 vuotta | 6 963 | 4 | 1,0 | 6 979 | 21 | 5,5 | 81,0 (43,6–95,3) |
| 70–79 vuotta | 4 487 | 1 | 0,4 | 4 487 | 16 | 6,5 | 93,8 (60,2–99,9) |
| Tutkittavat, joilla vähintään 1 erityisesti seurattava samanaikainen sairaus | 4 937 | 1 | 0,4 | 4 861 | 18 | 6,6 | 94,6 (65,9–99,9) |
alv = luottamusväli (96,95 % koko populaatiota [≥ 60‑vuotiaita] koskevassa analyysissä ja 95 % kaikissa alaryhmäanalyyseissä). Kaksisuuntainen eksakti luottamusväli rokotteen teholle johdettiin Poissonin mallista, jota korjattiin ikäluokkien ja alueen perusteella.
bVahvistava tavoite, jonka ennalta määritelty saavuttamiskriteeri oli kaksisuuntaisen luottamusvälin alaraja rokotteen teholle yli 20 %
N = Tutkittavien määrä kussakin ryhmässä
n = Niiden tutkittavien määrä, joille vahvistettu RSV‑alahengitystietauti ilmaantui ensimmäistä kertaa aikaisintaan päivänä 15 rokotuksen jälkeen
Rokotteen tehoa ei voida arvioida luotettavasti ≥ 80‑vuotiaiden alaryhmässä (1 016 tutkittavaa Arexvy‑ryhmässä vs. 1 028 tutkittavaa lumeryhmässä) kertyneiden tapausten pienen kokonaismäärän takia (5 tapausta).
RSV‑alahengitystietautitapauksia (määritelmä: vähintään 2 alahengitystielöydöstä tai arkitoimien estyminen) todettiin 18, joista lisähapen tarvetta esiintyi lumeryhmässä 4 vaikeassa tapauksessa ja Arexvy‑ryhmässä ei yhdessäkään tapauksessa.
Teho RSV-alahengitystietautia vastaan kahden RSV-kauden aikana ja kolmen RSV-kauden aikana
≥ 60-vuotiaita osallistujia, jotka saivat yhden annoksen Arexvy-rokotetta tai lumerokotetta (RSV OA=ADJ‑006), seurattiin kolmen RSV-kauden ajan (toisen ja kolmannen kauden loppuun pohjoisella pallonpuoliskolla), mediaaniseuranta-ajan ollessa 17,8 kuukautta kahden RSV-kauden aikana ja 30,6 kuukautta kolmen RSV-kauden aikana. Rokotteen teho RSV-alahengitystietautia vastaan oli kahden RSV-kauden aikana 67,2 % (97,5 %:n luottamusväli [48,2; 80,0]) ja kolmen RSV-kauden aikana 62,9 % (97,5 %:n luottamusväli [46,7; 74,8]).
Kolmen RSV-kauden aikana rokotteen teho RSV‑alatyypin A aiheuttamaa alahengitystietautia vastaan oli 69,8 % (97,5 %:n luottamusväli [42,2; 85,7)] ja RSV‑alatyypin B aiheuttamaa alahengitystietautia vastaan 58,6 % (97,5 %:n luottamusväli [35,9; 74,1]).
Rokotteen teho RSV-alahengitystietautia vastaan oli samanlainen niiden osallistujien alaryhmässä, joilla oli vähintään yksi liitännäissairaus.
Toinen rokoteannos, joka annettiin 12 kuukautta ensimmäisen annoksen jälkeen, ei antanut lisätehoa.
Immunogeenisuus 18–59-vuotiailla aikuisilla
Arexvy-valmisteella aikaansaadun immuunivasteen non-inferioriteettia 18–59-vuotiailla aikuisilla verrattuna ≥ 60 vuotiaisiin, joilla rokotteen teho RS‑viruksen aiheuttaman alahengitystietaudin ehkäisyssä oli osoitettu, arvioitiin kahdessa tutkimuksessa, joista ensimmäinen oli tutkijan suhteen sokkoutettu, satunnaistettu, lumekontrolloitu vaiheen 3 tutkimus (RSV OA=ADJ‑018) ja toinen avoin vaiheen 3 tutkimus (RSV OA=ADJ‑025).
Ensimmäisessä tutkimuksessa (RSV OA=ADJ‑018) kohortti 1 koostui 50–59-vuotiaista tutkittavista, jotka jaettiin kahteen alakohorttiin terveydentilan perusteella (Adults-AIR ja Adults-non-AIR). Adults-AIR-alakohortti (adults at increased risk; niiden aikuisten kohortti, joilla oli suurentunut riski) koostui tutkittavista, joilla oli etukäteen määritelty, vakaa pitkäaikaissairaus, kuten krooninen keuhkosairaus, krooninen sydän- ja verisuonisairaus, diabetes tai krooninen munuais- tai maksasairaus, ja sen takia suurentunut RS-virustaudin riski (Arexvy, N = 386; lume, N = 191). Adults-non-AIR-alakohortti koostui tutkittavista, joilla ei ollut etukäteen määriteltyä, vakaata pitkäaikaissairautta (Arexvy, N = 383; lume, N = 192). Kohortti 2 (OA; older adults) koostui ≥ 60‑vuotiaista tutkittavista (Arexvy, N = 381) (taulukko 3).
Toinen tutkimus (RSV OA=ADJ‑025) sisälsi 18–49‑vuotiaita tutkittavia, joilla oli etukäteen määritelty, vakaa pitkäaikaissairaus, kuten krooninen keuhkosairaus, krooninen sydän- ja verisuonisairaus, diabetes, krooninen munuais- tai maksasairaus tai neurologinen tai neuromuskulaarinen sairaus (N = 1 029, joista 426 kuului immunogeenisuus-osajoukkoon) ja sen takia suurentunut RS‑virustaudin riski, sekä ≥ 60‑vuotiaita tutkittavia (N = 429). Kaikki tutkittavat saivat yhden Arexvy-annoksen (taulukko 4).
Ensisijainen immunogeenisuuteen liittyvä tavoite oli humoraalisen immuunivasteen non-inferioriteetin osoittaminen (RSV‑A- ja RSV‑B-tyyppejä neutraloivien tittereiden osalta) 1 kuukauden kuluttua Arexvy-rokotteen antamisesta 50–59-vuotiailla tutkittavilla, joilla oli tai ei ollut etukäteen määriteltyä, vakaata pitkäaikaissairautta ja sen takia suurentunutta RS-virustaudin riskiä, sekä 18–49‑vuotiailla tutkittavilla, joilla oli etukäteen määritelty, vakaa pitkäaikaissairaus ja sen takia suurentunut RS‑virustaudin riski, verrattuna ≥ 60-vuotiaisiin tutkittaviin.
Immuunivasteiden non-inferioriteettikriteerit saavutettiin RSV‑A- ja RSV‑B-tyyppejä neutraloivien tittereiden kohdalla molemmissa ikäryhmissä. Arexvy-rokotteen teho 18–59‑vuotiailla aikuisilla on pääteltävissä ≥ 60‑vuotiailla aikuisilla osoitetusta tehosta.
Taulukko 3. Korjattujen GMT- ja SRR-arvojen yhteenveto sekä korjatut GMT-suhteet ja SRR-erot RSV‑A- ja RSV‑B-tyyppejä neutraloivien tittereiden (ED60) suhteen ≥ 60-vuotiailla aikuisilla (OA) verrattuna 50–59-vuotiaisiin, joilla oli (Adults-AIR) tai ei ollut (Adults-non-AIR) etukäteen määriteltyä, vakaata pitkäaikaissairauttaa ja sen takia suurentunutta RS-virustaudin riskiä – tutkimussuunnitelman mukaisesti hoidettu populaatio (Per-Protocol Set)
| RSV-A:ta neutraloiva titteri (ED60) | ||||
Korjattu GMT (95 %:n lv) | Korjattu GMT-suhde (95 %:n lv) b | SRR (%) (95 %:n lv) | SRR-ero (95 %:n lv) c | |
OA (≥ 60 vuotiaat) | 7 440,1 (6 768,4–8 178,5) | 0,83 (0,73–0,95) | 80,4 (75,8–84,5) | −6,5 (−12,1– −0,9) |
Adults-AIR (50-59 vuotiaat) | 8 922,7 (8 118,2–9 806,9) | 86,9 (82,8–90,3) | ||
OA (≥ 60 vuotiaat) | 7 492,6 (6 819,1–8 232,7) | 0,95 (0,83–1,09) | 80,4 (75,8–84,5) | −2,4 (−8,3–3,5) |
Adults-non-AIR (50-59 vuotiaat) | 7 893,5 (7 167,5–8 692,9) | 82,8 (78,3–86,8) | ||
| RSV-B:tä neutraloiva titteri (ED60) | ||||
Korjattu GMT (95 %:n lv) | Korjattu GMT-suhdeb | SRR (%) (95 %:n lv) | SRR-eroc | |
OA (≥ 60 vuotiaat) | 8 062,8 (7 395,9–8 789,9) | 0,80 (95 %:n lv [0,71–0,91]) | 74,5 (69,5–79,0) | −7,2 (95 %:n lv [−13,3– −0,9]) |
Adults-AIR (50-59 vuotiaat) | 10 054,7 (9 225,4–10 958,7) | 81,6 (77,1–85,6) | ||
OA (≥ 60 vuotiaat) | 8 058,2 (7 373,1–8 807,0) | 0,89 (97,5 %:n lv [0,77–1,03]) | 74,5 (69,5–79,0) | −3,7 (97,5 %:n lv [−11,1–3,7]) |
Adults-non-AIR (50-59 vuotiaat) | 9 009,5 (8 226,8–9 866,6) | 78,2 (73,3–82,6) | ||
a Etukäteen määritellyt, vakaat pitkäaikaissairaudet, kuten krooninen keuhkosairaus, krooninen sydän- ja verisuonisairaus, diabetes, krooninen munuais- tai maksasairaus.
b,c Etukäteen määritellyt immuunivasteiden non-inferioriteetin kriteerit: korjattujen GMT-suhteiden (OA-ryhmä suhteessa Adults-AIR- tai Adults-non-AIR-ryhmään) kaksisuuntaisen 95 %:n tai 97,5 %:n luottamusvälin ylärajat ≤ 1,5 ja SRR-eron (OA:n prosentista vähennetty Adults-AIR:n tai Adults-non-AIR:n prosentti) kaksisuuntaisen 95 %:n tai 97,5 %:n luottamusvälin yläraja ≤ 10 % ≥ 60‑vuotiailla tutkittavilla (OA) verrattuna 50–59‑vuotiaisiin tutkittaviin, joilla oli (Adults-AIR) tai ei ollut (Adults-non-AIR) etukäteen määriteltyä, vakaata pitkäaikaissairautta ja sen takia suurentunutta RS-virustaudin riskiä.
ED60: arvioitu laimeneminen 60; GMT = geometrinen keskimääräinen titteri; lv = luottamusväli; SRR = serologisten vasteiden prosentti.
Taulukko 4. Korjattujen GMT- ja SRR-arvojen yhteenveto sekä korjatut GMT-suhteet ja SRR-erot RSV‑A- ja RSV‑B-tyyppejä neutraloivien tittereiden (ED60) suhteen ≥ 60-vuotiailla aikuisilla (OA) verrattuna 18–49-vuotiaisiin, joilla oli etukäteen määritelty, vakaa pitkäaikaissairausa ja sen takia suurentunut RS-virustaudin riski (Adults-AIR) – tutkimussuunnitelman mukaisesti hoidettu populaatio (Per-Protocol Set)
| RSV-A:ta neutraloiva titteri (ED60) | ||||
Korjattu GMT (95 %:n lv) | Korjattu GMT-suhde (95 %:n lv) b | SRR (%) (95 %:n lv) | SRR-ero (95 %:n lv) c | |
OA (≥ 60 vuotiaat) | 8 591,5 (7 902,7–9 340,3) | 0,72 (0,64–0,81) | 77,7 (73,4–81,6) | −9,4 (−14,6– −4,1) |
Adults-AIR (18-49 vuotiaat) | 11 914,6 (10 933,2–12 984,2) | 87,1 (83,3–90,2) | ||
| RSV-B:tä neutraloiva titteri (ED60) | ||||
Korjattu GMT (95 %:n lv) | Korjattu GMT-suhde (95 %:n lv) b | SRR (%) (95 %:n lv) | SRR-ero (95 %:n lv) c | |
OA (≥ 60 vuotiaat) | 9 087,6 (8 372,1–9 864,2) | 0,73 (0,65–0,82) | 77,2 (72,9–81,2) | −10,1 (−15,3– −4,8) |
Adults-AIR (18-49 vuotiaat) | 12 503,4 (11 490,5–13 605,4) | 87,3 (83,6–90,4) | ||
a Etukäteen määritellyt, vakaat pitkäaikaissairaudet, kuten krooninen keuhkosairaus, krooninen sydän- ja verisuonisairaus, diabetes, krooninen munuais- tai maksasairaus, neurologinen tai neuromuskulaarinen sairaus.
b,c Etukäteen määritellyt immuunivasteiden non-inferioriteetin kriteerit: korjattujen GMT-suhteiden (OA-ryhmä suhteessa Adults-AIR-ryhmään) kaksisuuntaisen 95 %:n luottamusvälin ylärajat ≤ 1,5 ja SRR-eron (OA:n prosentista vähennetty Adults-AIR:n prosentti) kaksisuuntaisen 95 %:n luottamusvälin yläraja ≤ 10 % ≥ 60‑vuotiailla tutkittavilla (OA) verrattuna 18–49‑vuotiaisiin tutkittaviin, joilla oli etukäteen määritelty, vakaa pitkäaikaissairaus ja sen takia suurentunut RS-virustaudin riski (Adults-AIR).
ED60: arvioitu laimeneminen 60; GMT = geometrinen keskimääräinen titteri; lv = luottamusväli; SRR = serologisten vasteiden prosentti.
Immunogeenisuus erityisryhmillä
Immuunipuutteiset henkilöt
Vaiheen 2b avoimessa, satunnaistetussa, kontrolloidussa tutkimuksessa verrattiin immuunivastetta ≥ 18‑vuotiailla kiinteän elinsiirteen (munuais‑ tai keuhkosiirteen) saaneilla osallistujilla (N = 261), jotka saivat joko 1 annoksen (131 osallistujaa) tai 2 annosta (130 osallistujaa) Arexvy‑rokotetta 30–60 vrk:n välein, ja ≥ 50‑vuotiailla ei‑immuunipuutteisilla osallistujilla (N = 125), jotka saivat 1 annoksen Arexvy‑rokotetta (RSV OA=ADJ‑023). Kaikki elinsiirteen saaneet saivat ylläpitävää immunosuppressiivista hoitoa allograftin hylkimisen estämiseksi.
Elinsiirteen saaneilla 1 Arexvy‑annos nosti RSV‑A‑ ja RSV‑B‑tyyppejä neutraloivia tittereitä, ja ne säilyivät lähtötasoa korkeampina 12 kk:een asti rokotuksen jälkeen. Niillä elinsiirteen saaneilla, jotka saivat immunosuppressiivista hoitoa mykofenolaatti pois lukien (23 % osallistujista), neutraloivat titterit olivat 1 annoksen jälkeen samaa tasoa kuin ei‑immuunipuutteisilla. Niillä elinsiirteen saaneilla, jotka saivat immunosuppressiivista hoitoa, mykofenolaatti mukaan lukien (77 % osallistujista), neutraloivat titterit olivat 1 annoksen jälkeen matalammalla tasolla kuin niillä elinsiirteen saaneilla, jotka eivät saaneet mykofenolaattia. Toinen annos nosti RSV:tä neutraloivia tittereitä tässä ryhmässä, jolloin ne nousivat lähemmäs ei‑immuunipuutteisten osallistujien titteritasoa (ks. taulukko 5).
Taulukko 5. RSV‑A ja RSV-B ‑tyyppejä neutraloivien tittereiden (ED60) geometriset keskiarvot ≥ 18‑vuotiailla kiinteän elinsiirteen (SOT) saaneilla osallistujilla, jotka joko saivat tai eivät saaneet mykofenolaattia (MC) (Per‑Protocol Set)
| RSV-A neutraloivat titterit (ED60) | |||||
| SOT, 1 annoksen ryhmäa | SOT, 2 annoksen ryhmäb | Non-ICc | |||
| Ajankohta | MC kyllä | MC Ei | MC Kyllä | MC Ei | |
| N=95 | N=28 | N=94 | N=29 | N=125 | |
| Lähtötilanne | 785 [663 - 929] | 888 [692 - 1 139] | 813 [694 - 952] | 818 [553 - 1 210] | 889 [782 - 1 011] |
| N=95 | N=28 | N=90 | N=24 | N=117 | |
| 1 kk annoksesta 1 | 3 101 [2 459 - 3 912] | 9 388 [6 329 - 13 926] | 3 602 [2 672 - 4 855] | 7 255 [4 668 - 11 277] | 6 881 [5 976 - 7 924] |
| NA | NA | N=88 | N=23 | NA | |
| 1 kk annoksesta 2 | NA | NA | 4 960 [3 779 - 6 511] | 7 327 [4 811 - 11 159] | NA |
| N=89 | N=27 | N=83 | N=24 | N=114 | |
| 12 kk viimeisestä annoksesta | 1 528 [1 254 - 1 862] | 2 899 [2 044 - 4 110] | 2 564 [2 000 - 3 287] | 2 363 [1 567 - 3 563] | 2 244 [1 925 - 2 615] |
| RSV-B neutraloivat titterit (ED60) | |||||
| SOT, 1 annoksen ryhmäa | SOT, 2 annoksen ryhmäb | Non-ICc | |||
| Ajankohta | MC Kyllä | MC Ei | MC Kyllä | MC Ei | |
| N=95 | N=28 | N=94 | N=29 | N=125 | |
| Lähtötilanne | 859 [703 - 1 049] | 882 [621 - 1 253] | 877 [729 - 1 055] | 946 [625 - 1 433] | 1 027 [890 - 1 186] |
| N=95 | N=28 | N=90 | N=24 | N=117 | |
| 1 kk annoksesta 1 | 3 931 [2 985 - 5 177] | 11 336 [7 042 - 18 249] | 4 041 [3 012 - 5 422] | 9 468 [5 900 - 15 195] | 9 125 [7 782 - 10 700] |
| NA | NA | N=88 | N=23 | NA | |
| 1 kk annoksesta 2 | NA | NA | 5 274 [4 062 - 6 848] | 8 487 [5 736 - 12 559] | NA |
| N=89 | N=27 | N=83 | N=24 | N=114 | |
| 12 kk viimeisestä annoksesta | 2 048 [1 620 - 2 589] | 2 822 [1 968 - 4 047] | 2 898 [2 308 - 3 638] | 2 846 [1 848 - 4 385] | 2 665 [2 311 - 3 074] |
aSOT, 1 annoksen ryhmä = kiinteän elinsiirteen saaneet osallistujat, jotka saivat 1 Arexvy-annoksen
bSOT, 2 annoksen ryhmä = kiinteän elinsiirteen saaneet osallistujat, jotka saivat 2 Arexvy-annosta
Non‑IC = ei‑immuunipuutteiset osallistujat, jotka saivat 1 Arexvy‑annoksen
N = osallistujien määrä kussakin ryhmässä kullakin käynnillä
ED60: arvioitu laimeneminen 60
MC kyllä = kiinteän elinsiirteen saaneet osallistujat, jotka saivat immunosuppressiivista hoitoa, mykofenolaatti mukaan lukien
MC ei = kiinteän elinsiirteen saaneet osallistujat, jotka saivat immunosuppressiivista hoitoa, mykofenolaatti pois lukien
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Arexvy‑valmisteen käytöstä RS‑viruksen aiheuttaman alahengitystietaudin ehkäisyssä yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Ei oleellinen
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Arexvy‑valmistetta tai adjuvantitonta RSVPreF3‑rokotetta koskevien kaniineilla tehtyjen lisääntymis- ja kehitystutkimusten tulosten perusteella ei todettu rokotteeseen liittyviä vaikutuksia naaraan hedelmällisyyteen, tiineyteen, alkio-sikiökehitykseen eikä jälkeläisten kehitykseen.
Farmaseuttiset tiedot
Apuaineet
Kuiva‑aine (RSVPreF3‑antigeeni)
Trehaloosidihydraatti
Polysorbaatti 80 (E433)
Kaliumdivetyfosfaatti (E340)
Dikaliumfosfaatti (E340)
Suspensio (AS01E‑adjuvanttijärjestelmä)
Dioleoyylifosfatidyylikoliini (E322)
Kolesteroli
Natriumkloridi
Dinatriumfosfaatti, vedetön (E339)
Kaliumdivetyfosfaatti (E340)
Injektionesteisiin käytettävä vesi
Tiedot adjuvantista, ks. myös kohta Vaikuttavat aineet ja niiden määrät.
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Käyttökuntoon saattamisen jälkeen
Valmisteen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 4 tuntia 2–8 °C:n lämpötilassa tai huoneenlämmössä (enintään 25 °C).
Mikrobiologiselta kannalta valmiste on käytettävä välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikaiset säilytysajat ja käyttöä edeltävät säilytysolosuhteet ovat käyttäjän vastuulla. Säilytysaika saa olla enintään 4 tuntia.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
AREXVY injektiokuiva-aine ja suspensio, suspensiota varten
1 annos (120 mikrog+0,5 ml) (228,93 €)
PF-selosteen tieto
Arexvy on saatavilla kahdenlaisissa yhdistelmäpakkauksissa, jotka on kuvattu seuraavassa.
Arexvy injektiokuiva‑aine (injektiopullo) ja suspensio (injektiopullo) suspensiota varten:
- Kuiva‑aine (antigeeni) yhtä annosta varten injektiopullossa (tyypin I lasia), jossa on tulppa (butyylikumia) ja kellertävänvihreä irti napsautettava korkki.
- Suspensio (adjuvantti) yhtä annosta varten injektiopullossa (tyypin I lasia), jossa on tulppa (butyylikumia) ja ruskea irti napsautettava korkki.
Arexvy on saatavilla pakkauksessa, jossa on 1 injektiopullo kuiva‑ainetta ja 1 injektiopullo suspensiota, tai pakkauksessa, jossa on 10 injektiopulloa kuiva‑ainetta ja 10 injektiopulloa suspensiota.
Injektiopullon tulppa on synteettistä kumia.
Arexvy injektiokuiva‑aine (injektiopullo) ja suspensio (esitäytetty ruisku) suspensiota varten:
- Kuiva‑aine (antigeeni) yhtä annosta varten injektiopullossa (tyypin I lasia), jossa on tulppa (butyylikumia) ja harmaa irti napsautettava korkki.
- Suspensio (adjuvantti) yhtä annosta varten esitäytetyssä ruiskussa (tyypin I lasia), jossa on männäntiiviste (butyylikumia) ja kuminen kärkikorkki.
Arexvy on saatavilla pakkauksessa, jossa on 1 injektiopullo kuiva‑ainetta ja 1 esitäytetty ruisku suspensiota (ilman neulaa), tai pakkauksessa, jossa on 10 injektiopulloa kuiva‑ainetta ja 10 esitäytettyä ruiskua suspensiota (ilman neulaa).
Esitäytetyn ruiskun kärkikorkki ja männäntiiviste sekä injektiopullon tulppa ovat synteettistä kumia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kuiva‑aine on valkoista.
Suspensio on opalisoivaa, väritöntä tai vaalean rusehtavaa nestettä.
Käyttö- ja käsittelyohjeet
Kuiva‑aine ja suspensio on saatettava käyttökuntoon ennen antoa.
Kuiva‑aine ja suspensio on tarkastettava silmämääräisesti vieraiden hiukkasten ja/tai ulkonäkömuutosten varalta. Jos jompaakumpaa näistä todetaan, rokotetta ei saa saattaa käyttökuntoon.
Arexvyn valmistelu
Injektiopullo/injektiopullo
1. Vedä suspensiota sisältävän injektiopullon koko sisältö ruiskuun sopivalla neulalla (21 G–25 G).
2. Lisää ruiskun koko sisältö injektiopulloon, jossa kuiva‑aine on.
3. Pyöritä injektiopulloa varovasti, kunnes kuiva‑aine on liuennut kokonaan.
4. Vedä 0,5 ml käyttökuntoon saatettua rokotetta ruiskuun ja anna rokote lihakseen käyttäen uutta neulaa.
Injektiopullo/ esitäytetty ruisku
1. Kiinnitä esitäytettyyn ruiskuun sopiva neula (21 G–25 G) jäljempänä olevien kuvien mukaisesti. Lisää sitten esitäytetyn ruiskun koko sisältö injektiopulloon, jossa kuiva aine on.
2. Pyöritä injektiopulloa varovasti, kunnes kuiva‑aine on liuennut kokonaan.
3. Vedä kaikki käyttökuntoon saatettu rokote ruiskuun. Anna rokote lihakseen käyttäen uutta neulaa.
Esitäytetyn ruiskun käyttöohjeet
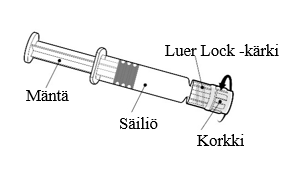 | Pidä kiinni ruiskun säiliöstä, älä männästä. Poista ruiskun korkki kiertämällä vastapäivään. |
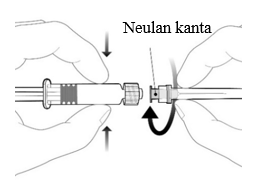 | Kiinnitä neula ruiskuun seuraavasti: aseta neulan kanta Luer Lock ‑kärkeen ja kierrä neulaa neljänneskierros myötäpäivään, kunnes tunnet neulan lukittuvan kiinni ruiskuun. Saata rokote käyttökuntoon edellä esitettyjen ohjeiden mukaisesti. Älä vedä ruiskun mäntää ulos säiliöstä. Jos näin käy, rokotetta ei saa antaa. |
Käyttökuntoon saatettu rokote on opalisoivaa, väritöntä tai vaaleanrusehtavaa nestettä.
Käyttökuntoon saatettu rokote on tarkastettava silmämääräisesti vieraiden hiukkasten ja/tai ulkonäkömuutosten varalta. Jos jompaakumpaa näistä todetaan, rokotetta ei saa antaa.
Valmisteen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 4 tuntia 2–8 °C:n lämpötilassa tai huoneenlämmössä (enintään 25 °C).
Mikrobiologiselta kannalta valmiste on käytettävä välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikaiset säilytysajat ja käyttöä edeltävät säilytysolosuhteet ovat käyttäjän vastuulla. Säilytysaika saa olla enintään 4 tuntia.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
AREXVY injektiokuiva-aine ja suspensio, suspensiota varten
1 annos
- Ei korvausta.
ATC-koodi
J07BX05
Valmisteyhteenvedon muuttamispäivämäärä
16.04.2026
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi