
BERINERT injektiokuiva-aine ja liuotin, liuosta varten 2000 IU, 3000 IU
Vaikuttavat aineet ja niiden määrät
Vaikuttava aine: Ihmisen C1-esteraasi-inhibiittori (ihmisen plasmasta) annettavaksi ihon alle (s.c.)
Yksi Berinert 2000 IU‑injektiopullo sisältää 2000 kansainvälistä yksikköä (IU).
Yksi Berinert 3000 IU‑injektiopullo sisältää 3000 kansainvälistä yksikköä (IU).
Ihmisen C1-esteraasi-inhibiittorien teho ilmaistaan WHO:n C1-esteraasi-inhibiittoreita koskevan voimassa olevan standardin mukaisesti kansainvälisinä yksikköinä (IU).
Berinert 2000 IU sisältää 500 IU/ml ihmisen C1-esteraasi-inhibiittoria, kun se on saatettu käyttövalmiiksi sekoittamalla 4 ml:aan injektionesteisiin käytettävää vettä.
Berinert 3000 IU sisältää 500 IU/ml ihmisen C1-esteraasi-inhibiittoria, kun se on saatettu käyttövalmiiksi sekoittamalla 5,6 ml:aan injektionesteisiin käytettävää vettä.
Käyttövalmiiksi sekoitetun liuoksen kokonaisproteiinisisältö on 65 mg/ml.
Apuaineet, joiden vaikutus tunnetaan:
Natriumia enintään 486 mg (noin 21 mmol) per 100 ml liuosta.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Berinert 2000 IU: Injektiokuiva-aine ja liuotin, liuosta varten.
Berinert 3000 IU: Injektiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Ihon alle annettava Berinert on tarkoitettu perinnöllisen angioedeeman (HAE) toistuvien kohtausten ehkäisyyn nuorilla ja aikuisilla, joilla on C1-esteraasi-inhibiittorin puutos.
Annostus ja antotapa
Berinert on tarkoitettu pistettäväksi itse injektiona ihon alle. Potilasta tai hoitajaa opastetaan pistämään Berinert itse tarvittaessa.
Annostus
Suositeltu Berinert-annos ihon alle on 60 IU painokiloa kohden kahdesti viikossa (3–4 päivän välein).
Pediatriset potilaat
Nuorilla annostus on sama kuin aikuisilla.
Antotapa
Vain injektiona ihon alle.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Berinert annetaan ihonalaisena injektiona mieluiten vatsan alueelle. Kliinisissä tutkimuksissa Berinert injisoitiin yhteen pistoskohtaan.
Käyttövalmis liuos annetaan injektiona ihon alle potilaalle sopivalla antonopeudella.
Vasta-aiheet
Henkilöt, joilla on esiintynyt henkeä uhkaavia yliherkkyysreaktioita, anafylaksia mukaan lukien, C1-inhibiittorivalmisteille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyysreaktiot
Jos potilaalla ilmenee vaikeita allergisia reaktioita, Berinert-valmisteen antaminen on lopetettava heti (esim. injektion antaminen on keskeytettävä) ja asianmukainen hoito on aloitettava.
Akuutissa HAE-kohtauksessa on annettava yksilöllistä hoitoa.
Tromboemboliset tapahtumat
Tromboosin kehittymistä on todettu, kun suuria C1-inhibiittoriannoksia on annettu laskimoon tarkoituksena antaa potilaalle estohoitoa kapillaarivuoto-oireyhtymään tai hoitaa sitä ennen sydänleikkausta, jossa on käytetty kehonulkoista verenkiertoa tai tällaisen leikkauksen aikana tai jälkeen (käyttöaihetta ja annostusta ei ole hyväksytty). Syy-yhteyttä tromboembolisten tapahtumien ja C1-inhibiittorikonsentraatin käytön välillä ei ole osoitettu suositelluilla ihon alle annettavilla annoksilla.
Virusturvallisuus
Vakiintuneita toimenpiteitä ihmisen verestä tai plasmasta valmistetuista lääkevalmisteista aiheutuvien infektioiden estämiseksi ovat verenluovuttajien valinta, erityisten infektiomerkkiaineiden seulominen luovutetusta verestä ja plasmapooleista sekä valmistuksenaikaiset tehokkaat toimenpiteet virusten inaktivoimiseksi/poistamiseksi. Tästä huolimatta taudinaiheuttajien siirtymisen mahdollisuutta ei voida täysin sulkea pois, kun käytetään ihmisen verestä tai plasmasta valmistettuja lääkevalmisteita. Tämä koskee myös tuntemattomia tai uusia viruksia ja muita taudinaiheuttajia.
Käytössä olevien toimenpiteiden katsotaan tehoavan vaipallisiin viruksiin, kuten ihmisen immuunikatovirukseen (HIV), hepatiitti B-virukseen (HBV) ja hepatiitti C-virukseen (HBC) sekä vaipattomiin viruksiin kuten hepatiitti A -virus ja parvovirus B19.
Ihmisen plasmasta valmistettuja valmisteita säännöllisesti/toistuvasti saavien potilaiden on tavallisesti harkittava asianmukaisten rokotusten (hepatiitti A:ta ja B:tä vastaan) ottamista.
Berinert 2000 IU sisältää alle 1 mmol natriumia (23 mg) per injektiopullo eli sen voidaan sanoa olevan ”natriumiton”.
Berinert 3000 IU sisältää 29 mg natriumia per injektiopullo, joka vastaa 1,5 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Raskaus
Käytössä olevat tiedot viittaavat siihen, etteivät riskit ole lisääntyneet, kun naiset ovat saaneet ihmisen C1-esteraasi-inhibiittorivalmisteita raskauden aikana. Ihmisen C1-esteraasi-inhibiittori on ihmisen plasman fysiologinen aineosa. Berinert-valmisteen lisääntymistoksisuutta ja kehitykseen kohdistuvaa toksisuutta ei ole tutkittu eläimillä. Ihmisellä ei odoteta esiintyvän haitallisia vaikutuksia hedelmällisyyteen, prenataaliseen ja postnataaliseen kehitykseen.
Kolmessa tutkimuksessa oli mukana 344 potilasta, ja tietoja saatiin 36 naisesta (50 raskautta). C1-inhibiittorihoitoon ei liittynyt haittatapahtumia raskautta ennen, raskauden aikana tai sen jälkeen, ja naiset saivat terveitä lapsia.
Imetys
Ei tiedetä, erittyykö Berinert ihmisen rintamaitoon tai vaikuttaako se imeväiseen tai maidon tuotantoon. Imetyksen hyötyjä lapsen kehitykselle ja terveydelle on puntaroitava äidin kliiniseen Berinert-hoidon tarpeeseen sekä imeväiselle Berinert-valmisteesta tai äidin sairaudesta aiheutuviin mahdollisiin haittoihin nähden.
Hedelmällisyys
Ihmisen C1-esteraasi-inhibiittori on ihmisen plasman fysiologinen aineosa. Berinert-valmisteen lisääntymistoksisuutta ja kehitykseen kohdistuvaa toksisuutta ei ole tutkittu eläimillä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Berinertillä ei ole haitallista vaikutusta ajokykyyn tai koneiden käyttökykyyn.
Haittavaikutukset
Haittavaikutukset on kerätty 3001-pivotaalitutkimuksesta (vaihe 3) HAE-potilailta (n=86), jotka saivat Berinert-valmistetta ihon alle. Soveltuvat potilaat pystyivät osallistumaan myös avoimeen jatkotutkimukseen (tutkimus 3002), jonka kesto oli enintään 140 viikkoa (n=126). Haittavaikutusten esiintymistiheys perustuu Berinert-valmisteeseen liittyviin tapahtumiin. Ne on arvioitu potilaskohtaisesti ja luokiteltu seuraavasti:
Hyvin yleiset: (≥ 1/10)
Yleiset: (≥ 1/100, < 1/10)
Melko harvinaiset: (≥ 1/1 000, < 1/100)
Harvinaiset: (≥ 1/10 000, < 1/1 000)
Hyvin harvinaiset: (< 1/10 000)
MedDRA elinjärjestelmäluokitus | Haittavaikutus | Esiintymistiheys |
Infektiot | Nasofaryngiitti | Hyvin yleiset |
Immuunijärjestelmä | Yliherkkyys (yliherkkyys, kutina, ihottuma ja nokkosihottuma) | Yleiset |
Hermosto | Heitehuimaus | Yleiset |
Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan reaktiota | Hyvin yleiset |
aInjektiokohdan mustelma, injektiokohdan kylmyys, eritevuoto injektiokohdassa, injektiokohdan punoitus, hematooma injektiokohdassa, verenvuoto injektiokohdassa, kovettuma injektiokohdassa, edeema injektiokohdassa, injektiokohdan kipu, injektiokohdan kutina, ihottuma injektiokohdassa, injektiokohdan reaktio, arpi injektiokohdassa, injektiokohdan turpoaminen, nokkosihottuma injektiokohdassa, lämmöntunne injektiokohdassa. | ||
Pediatriset potilaat
Kummassakin tutkimuksessa (tutkimus 3001 ja tutkimus 3002) Berinert-valmisteen turvallisuusprofiilia arvioitiin alaryhmässä, jossa oli 11 potilasta (ikä 8–< 17 vuotta). Turvallisuusprofiili oli yhdenmukainen kokonaisturvallisuutta koskevien tulosten kanssa.
Muut erityiset potilasryhmät
Iäkkäät
Kummassakin tutkimuksessa (tutkimus 3001 ja tutkimus 3002) Berinert-valmisteen turvallisuusprofiilia arvioitiin alaryhmässä, jossa oli 10 potilasta (ikä 65–72 vuotta). Turvallisuusprofiili oli yhdenmukainen kokonaisturvallisuutta koskevien tulosten kanssa.
Turvallisuus tarttuvien taudinaiheuttajien suhteen, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta (ks. yhteystiedot alla).
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksia ei ole raportoitu. Annoksia, jotka vastaavat enimmillään 117 IU/kg, on annettu ihon alle kahdesti viikossa kiinteillä annoksilla tehdyssä kliinisessä tutkimuksessa ja ne ovat olleet hyvin siedettyjä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut hematologiset valmisteet, Hereditaarisen angioödeeman hoitoon tarkoitetut lääkkeet: proteiini-C1-estäjät
ATC-koodi: B06AC01
C1-esteraasi-inhibiittori on plasman glykoproteiini, jonka molekyylipaino on 105 kD, josta hiilihydraattiosa on 40 %. Sen pitoisuus ihmisen plamassa on noin 240 mg/l. C1-esteraasi-inhibiittoria esiintyy ihmisen plasman lisäksi myös istukassa, maksasoluissa, monosyyteissä ja trombosyyteissä.
C1-esteraasi-inhibiittorit kuuluvat ihmisen plasman seriini-proteaasi-inhibiittori-(serpiini)-järjestelmään samoin kuin muut proteiinit, kuten antitrombiini III, alfa2-antiplasmiini, alfa1-antitrypsiini ja muut proteiinit.
Vaikutusmekanismi
C1-esteraasi-inhibiittori estää fysiologisissa olosuhteissa komplementtijärjestelmän klassista aktivaatiotietä inaktivoimalla entsymaattisesti aktiivisia komponentteja C1s ja C1r. Aktiivinen entsyymi muodostaa kompleksin inhibiittorin kanssa suhteessa 1:1.
C1-esteraasi-inhibiittori on lisäksi yksi tärkeimmistä koagulaation kontaktiaktivaation estäjistä, koska se estää tekijää XIIa ja sen osia. Se on alfa2-makroglobuliinin lisäksi plasman kallikreiinin tärkein estäjä.
Berinert-valmisteen terapeuttinen vaikutus hereditaarisen angioedeeman hoidossa on seurausta puutteellisen C1-esteraasi-inhibiittorin aktiivisuuden substituutiosta.
Kliininen teho ja turvallisuus
Berinert-valmisteen teho ja turvallisuus HAE-kohtausten ehkäisyn rutiiniprofylaksiassa osoitettiin satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa vaihtovuoroisessa monikeskustutkimuksessa (tutkimus 3001). Tutkimuksessa oli mukana 90 tutkimushenkilöä (aikuisia ja nuoria), joilla oli oireinen tyypin I tai II HAE. Tutkimushenkilöiden mediaani-ikä oli 40 vuotta (vaihteluväli 12–72 vuotta); 60 heistä oli naisia ja 30 miehiä. Tutkimushenkilöt satunnaistettiin saamaan Berinert-valmistetta joko 60 IU/kg tai 40 IU/kg 16 viikkoa kestävän hoitojakson ajan ja lumelääkettä toisen 16 viikkoa kestävän hoitojakson ajan. Potilaat annostelivat itse Berinert-valmistetta tai lumelääkettä ihon alle kahdesti viikossa. Tehoa arvioitiin jokaisen hoitojakson viimeisten 14 viikon aikana. Soveltuvat potilaat pystyivät myös osallistumaan avoimeen jatkotutkimukseen, jonka kesto oli enintään 140 viikkoa (tutkimus 3002). Noin puolet jatkotutkimukseen mukaan otetuista tutkimushenkilöistä osallistui myös tutkimukseen 3001 (64/126, 50,8 %), joten tutkimusryhmien välillä oli samankaltaisuuksia.
Tutkimus 3001:
Kahdesti viikossa ihon alle annetut Berinert-annokset 60 IU/kg tai 40 IU/kg saivat aikaan merkittävän eron HAE-kohtausten aikanormalisoidussa määrässä (kohtaustiheys) lumelääkkeeseen verrattuna (Taulukko 1). HAE-kohtausten aikanormalisoitu määrä annosta 60 IU/kg saaneilla tutkimushenkilöillä oli 0,52 kohtausta kuukaudessa ja lumelääkettä saaneilla 4,03 kohtausta kuukaudessa (p <0,001). Aikanormalisoitu HAE-kohtausten määrä annosta 40 IU/kg saaneilla tutkimushenkilöillä oli 1,19 kohtausta kuukaudessa ja lumelääkettä saaneilla 3,61 kohtausta kuukaudessa (p <0,001).
Taulukko 1. HAE-kohtausten aikanormalisoitu määrä (lukumäärä/kk)
60 IU/kg | 40 IU/kg | |||
VALMISTE | Lumelääke | VALMISTE | Lumelääke | |
n | 43 | 42 | 43 | 44 |
Keskiarvo (SD) | 0,5 (0,8) | 4,0 (2,3) | 1,2 (2,3) | 3,6 (2,1) |
Min, Max | 0,0, 3,1 | 0,6, 11,3 | 0,0, 12,5 | 0,0, 8,9 |
Mediaani | 0,3 | 3,8 | 0,3 | 3,8 |
LS-keskiarvo (SE)* | 0,5 (0,3) | 4,0 (0,3) | 1,2 (0,3) | 3,6 (0,3) |
LS-keskiarvon 95 % CI* | (0,0, 1,0) | (3,5, 4,6) | (0,5, 1,9) | (3, 4,3) |
Hoitoero (yksilökohtainen) | 60 IU/kg – lumelääke | 40 IU/kg – lumelääke | ||
LS-keskiarvo* (95 % CI) | ‑3,5 (‑4,2, ‑2,8) | ‑2,4 (‑3,4, ‑1,5) | ||
p‑arvo* | < 0,001 | < 0,001 | ||
CI = luottamusväli; HAE = perinnöllinen angioedeema; N = satunnaistettujen henkilöiden määrä; n = niiden henkilöiden määrä, joista on tutkimustietoa; LS = pienimmän neliösumman menetelmällä laskettu.
* sekamallista.
HAE-kohtausten aikanormalisoidun määrän prosentuaalisen vähenemisen mediaani (25. ja 75. persentiili) suhteessa lumelääkkeeseen oli 95 % (79, 100) Berinert-annoksella 60 IU/kg ja 89 % (70, 100) Berinert-annoksella 40 IU/kg niillä tutkimushenkilöillä, joista saatiin arvioitavaa dataa kummastakin hoitojaksosta.
Vasteen Berinert-hoitoon saavuttaneiden potilaiden prosenttiosuus (95%:n luottamusväli) suhteessa lumelääkkeeseen oli 83 % (73 %, 90 %). Vasteen saavuttaneiksi laskettiin henkilöt, joilla HAE-kohtausten aikanormalisoitu määrä väheni ≥ 50 %. Tutkimushenkilöistä 90 % vastasi hoitoannokseen 60 IU/kg ja 76 % vastasi hoitoannokseen 40 IU/kg.
Annoksella 60 IU/kg hoidetuista tutkimushenkilöistä 71 %:lla ja annoksella 40 IU/kg hoidetuista tutkimushenkilöistä 53 %:lla ilmeni ≥ 1 HAE-kohtaus 4 viikon lumelääkehoitojakson aikana ja < 1 HAE-kohtaus 4 viikon Berinert-hoitojakson aikana.
Kaiken kaikkiaan 40 % annosta 60 IU/kg saavista tutkimushenkilöistä ja 38 % annosta 40 IU/kg saavista tutkimushenkilöistä oli kohtauksettomia, ja HAE-kohtausten mediaani oli 0,3 kohtausta kuukaudessa kummallakin annoksella.
Berinert sai aikaan merkittävän eron kohtauslääkkeiden käytön aikanormalisoidussa määrässä (kohtauslääkkeiden käyttötiheys) lumelääkkeeseen verrattuna. Annoksella 60 IU/kg keskimääräinen kohtauslääkkeiden käyttötiheys oli 0,3 kertaa kuukaudessa ja lumelääkkeellä 3,9 kertaa kuukaudessa. Annoksella 40 IU/kg keskimääräinen kohtauslääkkeen käyttötiheys oli 1,1 kertaa kuukaudessa ja lumelääkkeellä 5,6 kertaa kuukaudessa.
Tutkimus 3002:
Berinert-valmisteen pitkän ajan turvallisuus ja teho HAE-kohtausten ehkäisyn rutiiniprofylaksiassa osoitettiin avoimessa, satunnaistetussa, rinnakkaisryhmillä toteutetussa tutkimuksessa. Tutkimuksessa oli mukana 126 tutkimushenkilöä (aikuisia ja lapsia), joilla oli oireinen tyypin I tai II HAE. Heistä 64 oli jo ollut mukana tutkimuksessa 3001 ja 62 oli uusia potilaita. Tutkimushenkilöiden mediaani-ikä oli 41,0 vuotta (vaihteluväli 8–72 vuotta). Tutkimukseen otettiin potilaita, joilla kohtaustiheys oli ollut 4,3 kohtausta kuukaudessa kolmen kuukauden aikana ennen tutkimuksen alkamista, ja heitä hoidettiin keskimäärin 1,5 vuotta; 44 potilaalla (34,9 %) altistus kesti yli 2 vuotta. Keskimääräinen vakaan tilan C1-inhibiittorin funktionaalinen aktiivisuus kasvoi 52,0 % annoksella 40 IU/kg ja 66,6 % annoksella 60 IU/kg. Haittatapahtumien ilmaantuvuus oli vähäistä ja samankaltaista kummassakin annosryhmässä (11,3 tapahtumaa potilasvuotta kohti annoksella 40 IU/kg ja 8,5 tapahtumaa potilasvuotta kohti annoksella 60 IU/kg). HAE-kohtausten keskimääräinen (SD) aikanormalisoitu määrä oli 0,45 (0,737) kohtausta kuukaudessa annoksella 40 IU ja 0,45 (0,858) kohtausta kuukaudessa annoksella 60 IU.
Vasteen Berinert-hoitoon saavuttaneiden potilaiden prosenttiosuus (95 %:n luottamusväli) oli 93,5 % (84,6 %, 97,5 %) hoitoryhmässä, jossa annos oli 40 IU/kg, ja 91,7 % (81,9 %, 96,4 %) hoitoryhmässä, jossa annos oli 60 IU/kg. Vasteen saavuttaneiksi laskettiin henkilöt, joilla HAE-kohtausten aikanormalisoitu määrä väheni ≥ 50 % suhteessa tutkimuksen 3002 osallistumiskriteerit täyttävään HAE-kohtausten aikanormalisoituun määrään.
Niiden tutkimushenkilöiden prosenttiosuus, joilla HAE-kohtausten aikanormalisoitu esiintymistiheys oli < 1 HAE-kohtaus neljän viikon aikana, oli 79,4 % annoksella 40 IU/kg ja 85,7 % annoksella 60 IU/kg.
Kohtauksettomien tutkimushenkilöiden prosenttiosuus oli 34,9 % annoksella 40 IU/kg ja 44,4 % annoksella 60 IU/kg (koko tutkimuksen keston aikana pisimmän altistuksen ollessa > 2,5 vuotta). Annosta 60 IU/kg yli 2 vuoden ajan saaneista 23 potilasta 19 (83 %) oli kohtauksettomia hoitokuukausien 25–30 aikana.
Keskimääräinen kohtauslääkkeiden aikanormalisoitu käyttökertojen määrä oli 0,26 (0,572) kertaa kuukaudessa annoksella 40 IU/kg ja 0,31 (0,804) kertaa kuukaudessa annoksella 60 IU/kg.
Pediatriset potilaat
Berinert-valmisteen turvallisuutta ja tehoa arvioitiin 11 potilasta käsittävässä alaryhmässä (ikä 8–<17 vuotta) satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa, vaihtovuoroisessa rutiiniprofylaksiatutkimuksessa (tutkimus 3001) sekä satunnaistetussa, avoimessa, aktiivihoitokontrolloidussa tutkimuksessa (tutkimus 3002). Alaryhmien iän mukaisessa analyysissä tulokset olivat yhdenmukaisia tutkimusten kokonaistulosten kanssa.
Farmakokinetiikka
Ihon alle annettavan Berinertin farmakokineettisia ominaisuuksia on kuvattu lähinnä populaatiofarmakokineettisten mallien avulla yhdistämällä kolmesta kliinisestä terveillä tutkimushenkilöillä ja HAE-potilailla tehdystä tutkimuksesta saadut tutkimustulokset.
Imeytyminen
Kahdesti viikossa ihon alle annettaessa Berinert imeytyy hitaasti ja huippupitoisuuden (tmax) saavuttamiseen kuluvan ajan mediaani (95 % CI) on noin 59 tuntia (23, 134 tuntia). Näennäisen puoliintumisajan mediaani (95 % CI) plasmassa on 69 tuntia (24, 250 tuntia), joten C1-inhibiittorin vakaa tila voidaan saavuttaa 3 viikon sisällä annostelusta. Toiminnallisen C1-inhibiittorin vakaan tilan minimipitoisuudeksi voidaan odottaa keskimäärin (95 % CI) 48 % (25,1 102 %), kun Berinert-valmistetta annostellaan 60 IU/kg kahdesti viikossa ihon alle. Keskimääräinen (95 % CI) suhteellinen hyötyosuus (F) annettaessa Berinert ihon alle on arviolta noin 43 % (35,2, 50,2 %).
Jakautuminen ja eliminaatio
Populaatiossa Berinertin keskimääräisen (95 % CI) puhdistuman arvioitiin olevan noin 83 ml/h (72,7, 94,2 ml/h) ja ilmeisen jakautumistilavuuden noin 4,33 litraa (3,51, 5,15 litraa). C1-inhibiittorin puhdistumassa todettiin positiivinen korrelaatio kehonpainon kanssa. Ihon alle annettavan Berinertin vakaan tilan farmakokinetiikka oli riippumaton annoksesta, kun HAE-potilaiden saama annos oli 20–80 IU/kg.
Sellaisia tutkimuksia ei ole tehty, joissa arvioitaisiin C1-inhibiittorin farmakokinetiikkaa sukupuolen, rodun, iän tai maksan tai munuaisten vajaatoiminnan mukaan jaotelluissa erityisryhmissä. Populaatioanalyysissä (8–72-vuotiaat) todettiin, että ikä ei vaikuta C1-inhibiittorin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, kerta-annoksen ja toistuvan altistuksen aiheuttamaa toksisuutta, paikallista siedettävyyttä ja trombogeenisuutta koskevien konventionaalisten tutkimusten tulokset laskimoon ja/tai ihon alle annettuna eivät viittaa erityiseen vaaraan ihmisille.
Karsinogeenisuutta ja lisääntymistoksisuutta koskevia tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine:
Glysiini
Natriumkloridi
Natriumsitraatti
Liuotin:
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden ja liuottimien kanssa.
Kestoaika
36 kuukautta
Valmisteen fysikaalis-kemiallisen säilyvyyden on osoitettu olevan 48 tuntia huoneenlämmössä (enintään 30 °C) käyttövalmiiksi saattamisen jälkeen. Mikrobiologiselta kannalta katsottuna käyttövalmisliuos tulee käyttää heti valmistamisen jälkeen, koska se ei sisällä säilytysainetta. Jos valmistetta ei käytetä heti, sitä saa säilyttää enintään 8 tuntia huoneenlämpötilassa. Käyttövalmiiksi saatettu valmiste on säilytettävä injektiopullossa.
Säilytys
Säilytä alle 30 °C.
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Ei markkinoilla olevia pakkauksia.
PF-selosteen tieto
Berinert 2000 IU:
Kuiva-aine (2000 IU) injektiopullossa (tyypin II lasia), jossa on suljin (bromobutyylikumia), sininen sinetti (alumiinia) ja harmaa irti napsautettava flip-off-suljin (muovia).
4 ml liuotinta injektiopullossa (tyypin I lasia), jossa on suljin (klorobutyyli- tai bromobutyylikumia), sininen sinetti (alumiinia) ja harmaa irti napsautettava flip-off-suljin (muovia).
Berinert 3000 IU:
Kuiva-aine (3000 IU) injektiopullossa (tyypin II lasia), jossa on suljin (bromobutyylikumia), sininen sinetti (alumiinia) ja sitruunankeltainen irti napsautettava flip-off-suljin (muovia).
5,6 ml liuotinta injektiopullossa (tyypin I lasia), jossa on suljin (klorobutyyli- tai bromobutyylikumia), sininen sinetti (alumiinia) ja limetinvihreä irti napsautettava flip-off-suljin (muovia).
Pakkaukset:
Ulkopakkauksen sisältö:
1 injektiopullo, joka sisältää kuiva-ainetta
1 injektiopullo, joka sisältää liuotinta (Berinert 2000 IU: 4 ml, Berinert 3000 IU: 5,6 ml)
1 suodattimella varustettu siirtolaite 20/20
Annostelutarvikkeet (sisälaatikko):
1 kertakäyttöinen ruisku (Berinert 2000 IU: 5 ml, Berinert 3000 IU: 10 ml)
1 hypoderminen neula
1 injektiosetti ihonalaista injektiota varten
2 alkoholipyyhettä
1 laastari.
Pakkaukset: 1 x 2000 IU tai 1 x 3000 IU.
Pakkaus, jossa 5 x 2000 IU tai 20 x 2000 IU.
Pakkaus, jossa 5 x 3000 IU tai 20 x 3000 IU.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen jauhe.
Kirkas, väritön liuotin.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Antotapa
Yleiset ohjeet
- Käyttövalmiin Berinert-liuoksen on oltava väritöntä ja kirkasta tai hieman opaalinhohtoista.
- Kun käyttövalmiiksi sekoitettu valmiste on suodatettu/vedetty ruiskuun (ks. seuraavassa), valmiste on tarkistettava silmämääräisesti ennen antoa, ettei siinä ole havaittavissa hiukkasia eikä värinmuutoksia.
- Älä käytä liuosta, jos se on sameaa tai siinä on sakkaa.
- Valmisteen käyttövalmiiksi sekoittaminen ja vetäminen ruiskuun on tehtävä aseptisissa olosuhteissa. Käytä valmisteen mukana toimitettua ruiskua.
Käyttövalmiiksi saattaminen
Anna liuottimen lämmetä huoneenlämpöiseksi. Varmista, että kuiva-aineen ja liuottimen sisältävien injektiopullojen irti napsautettavat flip-off-sulkimet on poistettu ja tulpat on käsitelty antiseptisella liuoksella. Tulppien on sen jälkeen annettava kuivua ennen Mix2Vial-pakkauksen avaamista.
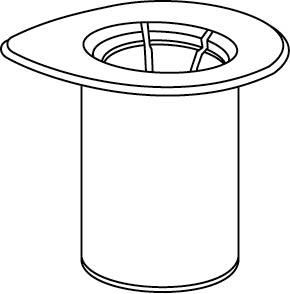
| 1. Avaa Mix2Vial-pakkaus vetämällä suojakansi pois. Älä ota Mix2Vial-laitetta pois pakkauksesta! |
| 2. Aseta liuottimen sisältävä injektiopullo tasaiselle, puhtaalle alustalle ja ota injektiopullosta tukeva ote. Ota Mix2Vial sekä pakkaus ja paina sinisen sovittimen piikki suoraan liuotinpullon tulpan läpi. |
| 3. Poista pakkaus varovasti Mix2Vial-laitteesta siten, että pidät pakkauksen reunasta kiinni ja vedät kohtisuoraan ylöspäin. Varmista, että vedät pois vain pakkauksen etkä Mix2Vial-laitetta. |
| 4. Aseta injektiopullo tasaiselle ja tukevalle alustalle. Käännä liuotinpullo ja siihen kiinnitetty Mix2Vial ylösalaisin, ja paina läpinäkyvän sovittimen piikki suoraan kuiva-aineinjektiopullon tulpan läpi. Liuotin siirtyy automaattisesti kuiva-aineen sisältävään injektiopulloon. |
| 5. Ota toisella kädellä kiinni Mix2Vial-laitteen kuiva-aineen sisältävän injektiopullon puolelta ja toisella kädellä liuottimen sisältävän injektiopullon puolelta ja kierrä laite varovasti vastapäivään kahteen osaan. Hävitä liuotinpullo ja siihen kiinnitetty sininen Mix2Vial-sovitin. |
| 6. Pyörittele kuiva-aineinjektiopulloa ja siihen kiinnitettyä läpinäkyvää sovitinta, kunnes kuiva-aine on liuennut täysin. Ei saa ravistaa. |
| 7. Vedä tyhjään, steriiliin ruiskuun ilmaa. Käytä valmisteen mukana toimitettua ruiskua. Kun kuiva-aineen sisältävä injektiopullo on oikeinpäin, kiinnitä ruisku Mix2Vial-sovittimen Luer Lock ‑liittimeen kiertämällä myötäpäivään. Ruiskuta ilma kuiva-aineen sisältävään injektiopulloon. |
 1
1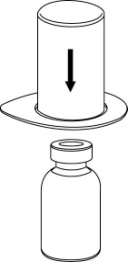 2
2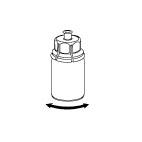 6
6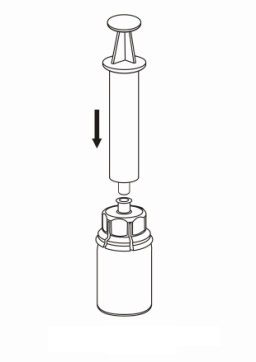 7
7Valmisteen vetäminen ruiskuun ja anto
| 8. Kun ruiskun mäntä on alas painettuna, käännä laite ja injektiopullo ylösalaisin ja vedä liuos ruiskuun vetämällä mäntää hitaasti ulospäin. |
| 9. Kun liuos on nyt siirretty ruiskuun, ota tukeva ote ruiskun kammiosta (pitäen ruiskun mäntää samalla alaspäin) ja irrota ruiskusta läpinäkyvä Mix2Vial-sovitin kiertämällä vastapäivään. |
Anto
Valmiste voidaan antaa käyttämällä hypodermistä neulaa tai infuusiosettiä ihonalaiseen antoon.
ATC-koodi
B06AC01
Valmisteyhteenvedon muuttamispäivämäärä
24.02.2022
Yhteystiedot
 CSL BEHRING AB
CSL BEHRING AB Box 712
182 17 Danderyd
Ruotsi
+46 (0) 8 544 966 70
www.cslbehring.fi
info@cslbehring.se