
TAVNEOS kapsel, hård 10 mg
Observera
▼Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Varje hård kapsel innehåller 10 mg avakopan.
Hjälpämne med känd effekt
Varje hård kapsel innehåller 245 mg makrogolglycerolhydroxistearat.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Hård kapsel
Kliniska uppgifter
Terapeutiska indikationer
Tavneos i kombination med en rituximab- eller cyklofosfamidregim är avsett för behandling av vuxna patienter med svår, aktiv granulomatös polyangit (GPA) eller mikroskopisk polyangit (MPA) (se avsnitt Dosering och administreringssätt).
Villkor
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Dosering och administreringssätt
Behandlingen ska inledas och övervakas av hälso- och sjukvårdspersonal med erfarenhet av diagnostik och behandling av GPA eller MPA (se avsnitt Varningar och försiktighet).
Dosering
Den rekommenderade dosen är 30 mg Tavneos (3 hårda kapslar à 10 mg) som tas peroralt två gånger dagligen, morgon och kväll, tillsammans med mat.
Tavneos ska administreras i kombination med en rituximab- eller cyklofosfamidregim enligt följande:
- intravenösa doser av rituximab en gång per vecka i 4 veckor, eller
- intravenöst eller peroralt cyklofosfamid i 13 eller 14 veckor följt av peroralt azatioprin eller mykofenolatmofetil, samt
- glukokortikoider när klinisk indicerat.
För uppgifter om doser, samtidig användning av glukokortikoider och data om effekt och säkerhet för kombinationerna, se avsnitt Biverkningar och Farmakodynamiska egenskaper
Kliniska studiedata är begränsade till 52 veckors exponering följt av 8 veckors observation.
Missade doser
Om en patient missar en dos ska den missade dosen tas så snart som möjligt, om det inte är mindre än tre timmar till nästa schemalagda dos. Om det är mindre än tre timmar ska den missade dosen inte tas.
Dosjustering
Behandlingen måste utvärderas kliniskt och tillfälligt stoppas om:
- alaninaminotransferas (ALAT) eller aspartataminotransferas (ASAT) är mer än 3 gånger den övre gränsen för normalvärdet (ULN).
Behandlingen måste tillfälligt stoppas om:
- ALAT eller ASAT är > 5 x ULN
- en patient utvecklar leukopeni (antalet vita blodkroppar < 2 × 109/l) eller neutropeni (neutrofiler < 1 × 109/l) eller lymfopeni (lymfocyter < 0,2 × 109/l)
- en patient har en aktiv, allvarlig infektion (dvs. kräver sjukhusvistelse eller förlängd sjukhusvistelse).
Behandlingen kan återupptas:
- när värdena normaliserats och baserat på en individuell nytta/riskbedömning.
Om behandlingen återupptas ska levertransaminaser och totalt bilirubin övervakas noggrant.
Permanent utsättning av behandlingen måste övervägas om:
- ALAT eller ASAT > 8 × ULN
- ALAT eller ASAT > 5 × ULN under mer än 2 veckor
- ALAT eller ASAT > 3 × ULN och totalt bilirubin > 2 × ULN eller internationell normaliserad kvot (INR) > 1,5
- ALAT eller ASAT > 3 × ULN med förekomst av fatigue, illamående, kräkning, smärta eller ömhet i övre högra bukkvadranten, feber, utslag och/eller eosinofili (> 5 %)
- ett samband mellan avakopan och leversvikt har fastställts.
Särskilda populationer
Äldre
Ingen dosjustering krävs för äldre patienter (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Ingen justering krävs för patienter med lätt eller måttligt nedsatt leverfunktion (se avsnitt Farmakokinetiska egenskaper).
Avakopan har inte studerats hos patienter med allvarligt nedsatt leverfunktion (Child-Pugh klass C) och användning rekommenderas därför inte till dessa patienter.
Nedsatt njurfunktion
Ingen dosjustering krävs baserat på njurfunktionen (se avsnitt Farmakokinetiska egenskaper).
Avakopan har inte studerats hos patienter med vaskulit associerad med anti-neutrofila cytoplasmaantikroppar (ANCA-associerad vaskulit) med en uppskattad glomerulär filtrationshastighet (eGFR) under 15 ml/min/1,73 m², som behandlas med dialys eller är i behov av dialys eller plasmabyte.
Allvarlig sjukdom med lungblödning
Avakopan har inte studerats hos patienter med allvarlig sjukdom med lungblödning.
Pediatrisk population
Säkerhet och effekt för avakopan för ungdomar (i åldern 12‑17 år) har ännu inte fastställts. Tillgänglig information finns i avsnitt Biverkningar och Farmakodynamiska egenskaper, men ingen doseringsrekommendation kan fastställas. Avakopans säkerhet och effektivitet för barn under 12 år har inte fastställt ännu. Inga data finns tillgängliga.
Administreringssätt
Detta läkemedel är avsett för oral användning.
De hårda kapslarna ska tas tillsammans med mat och sväljas hela med vatten och får inte krossas, tuggas eller öppnas.
Grapefrukt och grapefruktjuice ska undvikas hos patienter som behandlas med avakopan (se avsnitt Interaktioner).
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Hepatotoxicitet
Allvarliga biverkningar i form av förhöjda levertransaminaser med förhöjt totalt bilirubin har observerats hos patienter som får avakopan i kombination med cyklofosfamid (följt av azatioprin eller mykofenolat) eller rituximab och trimetoprim och sulfametoxazol. Efter marknadsintroduktionen har det rapporterats om läkemedelsinducerad leverskada och VBDS (vanishing bile duct syndrome), inklusive fall med dödlig utgång (se avsnitt Biverkningar).
Levertransaminaser och totalt bilirubin måste bestämmas innan behandling sätts in.
Avakopan får inte ges till patienter med tecken på leversjukdom såsom förhöjt ASAT, ALAT, alkaliskt fosfatas (ALP) eller totalt bilirubin > 3 gånger ULN.
Patienterna måste övervakas avseende levertransaminaser och totalt bilirubin minst var fjärde vecka efter inledning av behandling under de 6 första månaderna av behandling och när kliniskt indicerat därefter (se avsnitt Dosering och administreringssätt).
Blodet och immunsystemet
Antalet vita blodkroppar måste bestämmas innan behandling sätts in och patienterna måste övervakas när kliniskt indicerat och som del av den rutinmässiga uppföljningen av patientens underliggande tillstånd (se avsnitt Dosering och administreringssätt).
Behandling med avakopan får inte sättas in om antalet vita blodkroppar är < 3.5 × 109/l eller antalet neutrofiler är < 1.5 × 109/l eller antalet lymfocyter är < 0.5 × 109/l.
Patienter som får avakopan måste instrueras att omedelbart rapportera alla tecken på infektion, oväntade blåmärken, blödning eller några andra tecken på benmärgssvikt.
Allvarliga infektioner
Allvarliga infektioner har rapporterats hos patienter som får kombinationsmedel för behandling av GPA eller MPA, inklusive avakopan i kombination med rituximab eller cyklofosfamid (se avsnitt Biverkningar).
Patienterna måste bedömas avseende allvarliga infektioner.
Avakopan har inte studerats hos patienter med hepatit B, hepatit C eller infektioner med humant immunbristvirus (hivinfektioner). Före och under behandling måste patienterna tala om för sin läkare om de har fått diagnosen tuberkulos, hepatit B, hepatit C eller hivinfektion.
Iaktta försiktighet vid behandling av patienter med tuberkulos, hepatit B, hepatit C eller hivinfektion i anamnesen.
Avakopan minskar inte bildandet av membranattackkomplexet (C5b‑9) eller terminalkomplementkomplexet (TCC). Inga fall av Neisseria meningitidis har identifierats i det kliniska avakopanprogrammet. Övervaka patienter som behandlas för ANCA-associerad vaskulit enligt gängse praxis avseende kliniska tecken och symtom på Neisseria-infektioner.
Profylax av pneumocystis jirovecii-pneumoni
Profylax av Pneumocystis jirovecii-pneumoni rekommenderas för vuxna patienter med GPA eller MPA under behandling med avakopan, enligt lokala riktlinjer för klinisk praxis.
Immunisering
Säkerheten vid immunisering med virusvaccin efter avakopanbehandling har inte undersökts.
Ge helst vaccinationer innan den insättande behandlingen med avakopan eller under sjukdomens vilande fas.
Angioödem
Angioödem har rapporterats hos patienter som får avakopan (se avsnitt Biverkningar).
Patienterna måste tala om för sin läkare om de får symtom såsom svullnad i ansikte, läppar eller tunga, trånghetskänsla i strupen eller svårighet att andas.
Uppehåll i behandlingen med avakopan måste göras vid fall av angioödem.
Interaktion med starka CYP3A4-inducerare
Samtidig användning av starka CYP3A4-enzyminducerare (t.ex. karbamazepin, enzalutamid, mitotan, fenobarbital, fenytoin, rifampicin och johannesört) med avakopan ska undvikas (se avsnitt Interaktioner).
Patienter som förväntas behöva långvarig administrering av dessa läkemedel ska inte behandlas med avakopan.
Om kortvarig samtidig administrering inte kan undvikas hos en patient som redan använder avakopan måste patienten övervakas noga avseende recidiverande sjukdomsaktivitet.
Hjärtat
Patienter med GPA eller MPA har större risk att drabbas av hjärtsjukdomar såsom hjärtinfarkt, hjärtsvikt och hjärtvaskulit.
Allvarliga biverkningar i form av hjärtsjukdom har rapporterats hos patienter som behandlades med avakopan. En behandlingsregim som baserar sig på en kombination av cyklofosfamid följt av azatioprin kan medföra en ökad risk för hjärtsjukdomar jämfört med en regim baserad på en kombination med rituximab.
Malignitet
Immunmodulerande läkemedel kan öka risken för maligniteter. Kliniska data är för närvarande begränsade (se avsnitt Farmakodynamiska egenskaper).
Innehåll av makrogolglycerolhydroxistearat
Detta läkemedel innehåller makrogolglycerolhydroxistearat, vilket kan ge magbesvär och diarré.
Interaktioner
Avakopan är ett substrat för CYP3A4. Samtidig administrering av inducerare eller hämmare av detta enzym kan påverka avakopans farmakokinetik.
Effekt av starka CYP3A4-inducerare på avakopan
Samtidig administrering av avakopan med rifampicin, en stark CYP3A4-enzyminducerare, resulterade i en minskning av arean under koncentrationskurvan (AUC) och maximal plasmakoncentration (Cmax) av avakopan med cirka 93 % respektive 79 %. Eftersom denna interaktion kan leda till sämre effekt av avakopan ska samtidig användning av starka CYP3A4-enzyminducerare (t.ex. karbamazepin, enzalutamid, mitotan, fenobarbital, fenytoin, rifampicin och johannesört) med avakopan undvikas (se avsnitt Varningar och försiktighet). Patienter som förväntas behöva långvarig administrering av dessa läkemedel ska inte behandlas med avakopan. Om kortvarig samtidig administrering inte kan undvikas hos en patient som redan använder avakopan måste patienten övervakas noga avseende recidiverande sjukdomsaktivitet.
Effekt av måttliga CYP3A4-inducerare på avakopan
Iaktta försiktighet vid användning av måttliga CYP3A4-inducerare (t.ex. bosentan, efavirenz, etravirin och modafinil) som förskrivits som behandling samtidigt med avakopan och utvärdera noga nytta/riskförhållandet för avakopan.
Effekt av starka CYP3A4-hämmare på avakopan
Samtidig administrering av avakopan och itrakonazol, en stark CYP3A4-enzymhämmare, resulterade i en ökning i avakopans AUC och Cmax med cirka 2,2 gånger respektive 1,9 gånger. Därför ska starka CYP3A4-enzymhämmare (t.ex. boceprevir, klaritromycin, konivaptan, indinavir, itrakonazol, ketokonazol, lopinavir/ritonavir, mibefradil, nefazodon, nelfinavir, posakonazol, ritonavir, sakvinavir, telaprevir, telitromycin och vorikonazol) användas med försiktighet hos patienter som behandlas med avakopan. Patienterna måste övervakas avseende potentiell ökning av biverkningar på grund av ökad exponering för avakopan.
Grapefrukt och grapefruktjuice kan öka koncentrationen av avakopan, därför ska grapefrukt och grapefruktjuice undvikas hos patienter som behandlas med avakopan.
Avakopans effekt på andra läkemedel
Avakopan är en måttlig hämmare av CYP3A4 in vivo och kan öka plasmaexponeringen för samtidiga läkemedel som är CYP3A4-substrat (t.ex. alfentanil, ciklosporin, dihydroergotamin, ergotamin, fentanyl, sirolimus och takrolimus). Patienterna måste behandlas i enlighet med produktresumén för samtidiga läkemedel. Dosreducering eller övervakning av biverkningar kan vara nödvändiga.
I en klinisk studie ökade samtidig administrering av avacopan och simvastatin, ett känsligt CYP3A4-substrat, den totala systemiska exponeringen (AUC) av simvastatin med 3,5 gånger och Cmax med 3,2 gånger. Se produktresumén för simvastatin för lämpliga dosjusteringar.
Effekt av makrogolglycerolhydroxistearat på känsliga P-glykoprotein (P-gp)-substrat
En kliniskt relevant effekt av hjälpämnet makrogolglycerolhydroxistearat på känsliga P-gp-substrat med relativt låg biotillgänglighet (t.ex. dabigatranetexilat) kan inte uteslutas. Iaktta försiktighet vid användning av P-gp-substrat med låg biotillgänglighet hos patienter som behandlas med avakopan.
Fertilitet, graviditet och amning
Fertila kvinnor/graviditet
Det finns inga data från användning av avakopan i gravida kvinnor.
Djurstudier har visat reproduktionstoxikologiska effekter (se avsnitt Prekliniska säkerhetsuppgifter).
Avakopan rekommenderas inte under graviditet eller till fertila kvinnor som inte använder preventivmedel.
Amning
Avakopan har inte uppmätts i mjölk från lakterande djur. Dock har avakopan detekterats i plasma i digivande djurs avkomma utan uppenbara effekter på avkomman (se avsnitt Prekliniska säkerhetsuppgifter).
En risk för det nyfödda barnet/spädbarnet kan inte uteslutas. Ett beslut måste fattas om man ska avbryta amningen eller avbryta/avstå från behandling med avakopan efter att man tagit hänsyn till fördelen med amning för barnet och fördelen med behandling för kvinnan.
Fertilitet
Det finns inga data om effekterna av avakopan på mänsklig fertilitet. Djurdata indikerar inte några störningar i manlig eller kvinnlig fertilitet (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
Tavneos har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligaste biverkningarna är illamående (23,5 %), huvudvärk (20,5 %), minskat antal vita blodkroppar (18,7 %), övre luftvägsinfektion (14,5 %), diarré (15,1 %), kräkningar (15,1 %) och nasofaryngit (15,1 %).
De vanligaste allvarliga biverkningarna är avvikande leverfunktionsvärden (5,4 %) och pneumoni (4,8 %).
Tabell över biverkningar
Biverkningarna som observerats i den pivotala fas 3-studien av ANCA-associerad vaskulit, och efter marknadsintroduktionen, hos patienter som behandlas med avakopan listas i tabell 1, indelat efter organsystem och frekvens.
Frekvenserna definieras enligt följande: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10) mindre vanliga (≥ 1/1 000, < 1/100) och ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom varje frekvensgrupp visas biverkningarna i fallande allvarlighetsgrad.
Tabell 1: Biverkningar
| Organsystem | Mycket vanliga (≥ 1/10) | Vanliga (≥ 1/100, < 1/10) | Mindre vanliga (≥ 1/1 000, < 1/100) | Ingen känd frekvens |
| Infektioner och infestationer | Övre luftvägsinfektion Nasofaryngit | Pneumoni Rinit Urinvägsinfektion Sinuit Bronkit Gastroenterit Nedre luftvägsinfektion Cellulit Herpes zoster Influensa Oral kandidos Oral herpes Otitis media | ||
| Blodet och lymfsystemet | Neutropeni1 | |||
| Centrala och perifera nervsystemet | Huvudvärk | |||
| Magtarmkanalen1 | Illamående Diarré Kräkningar | Övre buksmärta | ||
| Lever och gallvägar | Förhöjda leverfunktionsvärden1,2 | Läkemedels-inducerad leverskada1, VBDS (vanishing bile duct syndrome)1 | ||
| Hud och subkutan vävnad | Angioödem | |||
| Undersökningar | Minskat antal vita blodkroppar3 | Förhöjt kreatinfosfokinas i blodet1 |
1 Se avsnitt ”Beskrivning av utvalda biverkningar”.
2 Förhöjt alaninaminotransferas, förhöjt totalt bilirubin i blodet, avvikande leverfunktionsvärden, förhöjt gammaglutamyltransferas, förhöjt leverenzym, förhöjda transaminaser.
3 Inkluderar leukopeni.
Beskrivning av utvalda biverkningar
Hepatotoxicitet
I den pivotala fas 3‑studien där 330 patienter fick läkemedel, fick 13,3 % av patienterna i avakopangruppen och 11,6 % av patienterna i prednisongruppen en biverkning i form av förhöjda leverfunktionsvärden (LFT).
I avakopangruppen i fas 3-studien rapporterades förhöjda LFT som inkluderade hepatit (1,2 %), kolestatisk hepatit (0,6 %) av vilka en patient diagnostiserades med både hepatit och kolestatisk hepatit, samt hepatocellulär skada (0,6 %) hos en patient med asymtomatisk hepatit, cytolys och anikterisk kolestas utan hepatocellulär insufficiens.
I den pivotala fas 3‑studien var biverkningar i form av lever- och gallsjukdomar vanligare hos patienter behandlade med en regim baserad på en kombination med cyklofosfamid följt av azatioprin (10,2 %) än hos dem som behandlades med en regim baserad på en kombination med rituximab (3,7 %).
Studieläkemedlet avbröts tillfälligt eller sattes ut permanent på grund av förhöjda LFT hos 5,4 % av patienterna i avakopangruppen och 3,0 % av patienterna i prednisongruppen. Allvarliga biverkningar i form av LFT rapporterades hos 5,4 % av patienterna i avakopangruppen och 3,7 % av patienterna i prednisongruppen. Samtliga allvarliga leverhändelser gick tillbaka vid utsättning av avakopan och/eller andra potentiella hepatotoxiska läkemedel, inklusive trimetoprim och sulfametoxazol.
Läkemedelsinducerad leverskada och VBDS (vanishing bile duct syndrome) har rapporterats efter marknadsintroduktionen (se avsnitt Varningar och försiktighet).
Neutropeni
I den pivotala fas 3‑studien rapporterades neutropeni hos 4 patienter (2,4 %) i varje behandlingsgrupp.
Ett enda fall av agranulocytos rapporterades i vardera prednisongruppen och avakopangruppen.
Patienten i avakopangruppen konstaterades ha central neutropeni på en benmärgsbiopsi, som gick tillbaka spontant utan ytterligare behandling.
Förhöjt kreatinfosfokinas
I den pivotala fas 3‑studien fick 6 patienter (3,6 %) i avakopangruppen och 1 patient (0,6 %) i prednisongruppen biverkningar i form av ökning av kreatinfosfokinas (CPK).
Överkänslighet inklusive angioödem
I den pivotala fas 3‑studien fick 2 av patienterna (1,2 %) i avakopangruppen en angioödembiverkning. En patient lades in på sjukdom med anledning av biverkningen. Avakopan avbröts tillfälligt och biverkningarna gick tillbaka utan sekvele. Avakopan sattes in på nytt hos en patient och angioödem återkom inte.
Magtarmkanalen
I den pivotala fas 3‑studien observerades biverkningar i magtarmkanalen hos 74,6 % av patienterna som behandlades med avakopan och en regim baserad på en kombination med cyklofosfamid följt av azatioprin, jämfört med dem som behandlades med en regim baserad på en kombination med rituximab (53,3 %).
Särskilda populationer
Pediatrisk population
Totalt deltog tre ungdomar i fas 3-studien, en i prednisongruppen och två i avakopangruppen. Det finns inga data gällande barn under 12 år (se avsnitt Farmakodynamiska egenskaper).
Äldre patienter
Säkerhetsprofilen var likartad hos patienter ≥ 65 år och hos vuxna patienter < 65 år i de kliniska studierna.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Avakopan studerades hos friska försökspersoner med en daglig dos på 200 mg (given som 100 mg två gånger dagligen) i 7 dagar utan några belägg för dosbegränsande toxiciteter. I fall av överdosering rekommenderas att patienten övervakas avseende tecken eller symtom på biverkningar och att lämplig symtomatisk behandling och stödjande vård ges.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: komplementhämmare, ATC-kod: L04AJ05
Verkningsmekanism
Avakopan är en selektiv antagonist på den humana komplement 5a-receptorn (C5aR1 eller CD88) och hämmar kompetitivt interaktionen mellan C5aR1 och anafylatoxin C5a.
Avakopans specifika och selektiva blockering av C5aR1 minskar de proinflammatoriska effekterna av C5a, vilka inkluderar neutrofil aktivering, migration och adhesion till ställen med kapillär inflammation, retraktion av vaskulära endotelceller och permeabilitet.
Farmakodynamisk effekt
Avakopan blockerar den C5a-inducerade uppregleringen av Cd11b (alfa M‑integrin) på neutrofiler som tagits från människor som fått avakopan. CD11b underlättar neutrofiladhesion till vaskulära endotelytor, ett av stegen i sjukdomsprocessen för vaskulit.
Klinisk effekt och säkerhet
Totalt 330 patienter som var 13 år eller äldre med granulomatös polyangit (GPA) (54,8 %) eller mikroskopisk polyangit (MPA) (45,2 %) behandlades i 52 veckor i den randomiserade, dubbelblindade, dubbel-dummy-, multicenter-, pivotala fas 3-studien ADVOCATE med aktiv komparator.
ADVOCATE-studiens upplägg visas i figur 1.
Figur 1: ADVOCATE-studiens upplägg
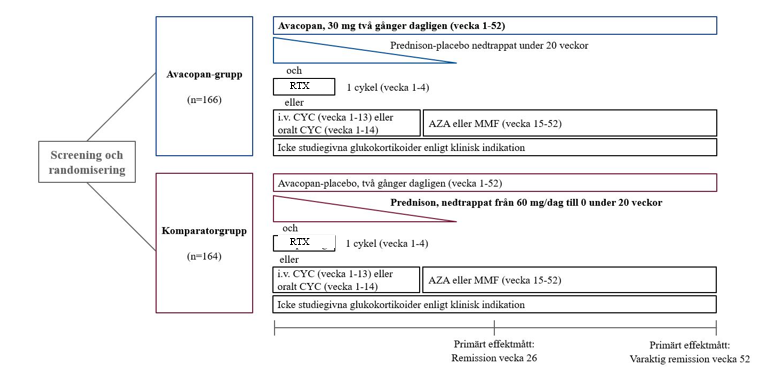
AZA = azatioprin; CYC = cyklofosfamid; i.v. = intravenöst; MMF = mykofenolatmofetil; RTX = rituximab
Patienterna randomiserades i förhållandet 1:1 till en av två grupper:
- Avakopangrupp (N = 166): Patienterna fick 30 mg avakopan två gånger dagligen i 52 veckor plus prednison-matchande placebo som trappades ned under 20 veckor.
- Komparatorgrupp (N = 164): Patienterna fick avakopan-matchande placebo två gånger dagligen i 52 veckor plus prednison (nedtrappning från 60 mg/dag till 0 under 20 veckor).
Alla patienter i båda grupperna fick gängse immunsuppressiva regimer med antingen:
- rituximab i en intravenös dos på 375 mg/m2 per vecka i 4 veckor, eller
- intravenöst cyklofosfamid i 13 veckor (15 mg/kg upp till 1,2 g var 2–3 vecka) och sedan, med start vecka 15, oralt azatioprin 1 mg/kg dagligen med titrering upp till 2 mg/kg dagligen (mykofenolatmofetil 2 g dagligen var tillåtet i stället för azatioprin. Om mykofenolatmofetil inte tolererades eller inte var tillgängligt kunde enteroläkemedel med mykofenolatnatrium ges i en måldos på 1 440 mg/dag), eller
- oralt cyklofosfamid i 14 veckor (2 mg/kg dagligen) följt av oralt azatioprin eller mykofenolatmofetil/-natrium med start vecka 15 (samma doseringsregimen som intravenöst cyklofosfamid).
För den första rituximabinfusionen gavs 100 mg metylprednisolon eller motsvarande före infusionsstart med rituximab. Premedicinering med glukokortikoid var tillåtet för de andra, tredje och fjärde rituximabinfusionerna.
Dosreducering eller -justering av cyklofosfamid, azatioprin och mykofenolat var tillåtet för att maximera säkerheten med dessa läkemedel.
Följande schema användes för nedtrappning av glukokortikoid som tillhandahölls under studien (tabell 2).
Tabell 2: Schema för nedtrappning av glukokortikoid – prednisondos (mg per dag)
| Studiedag | Avakopan | Komparator | |
| Alla | ≥ 55 kg | < 55 kg | |
| 1–7 | 0 | 60 | 45 |
| 8–14 | 0 | 45 | 45 |
| 15–21 | 0 | 30 | 30 |
| 22–42 | 0 | 25 | 25 |
| 43–56 | 0 | 20 | 20 |
| 57–70 | 0 | 15 | 15 |
| 71–98 | 0 | 10 | 10 |
| 99–140 | 0 | 5 | 5 |
| ≥ 141 | 0 | 0 | 0 |
Glukokortikoider som inte tillhandahölls inom ramen för studien måste undvikas så mycket som möjligt under studien, såvida det inte var absolut nödvändigt på grund av ett tillstånd som krävde behandling med glukokortikoider (som binjureinsufficiens). Patienter som upplevde försämring eller recidiv av sin ANCA-associerade vaskulit under studien kunde dock behandlas med en begränsad kur glukokortikoider.
Patienterna stratifierades vid tiden för randomisering för att erhålla balans mellan behandlingsgrupperna baserat på tre faktorer:
- nyligen diagnostiserad eller recidiverad ANCA-associerad vaskulit
- proteinas 3 (PR3)‑positiv eller myeloperoxidas (MPO)-positiv ANCA-associerad vaskulit
- erhåller antingen intravenöst rituximab, intravenöst cyklofosfamid eller peroralt cyklofosfamid.
De två behandlingsgrupperna var väl balanserade gällande patienternas demografiska egenskaper och sjukdomskarakteristika vid baslinjen (tabell 3).
Tabell 3: Ett urval av karakteristika vid baslinjen i den pivotala fas 3-studien ADVOCATE (intent-to-treat-population)
| Demografisk egenskap | Avakopan (N = 166) | Komparator (N = 164) |
| Ålder vid screening | ||
| Genomsnittlig (SD), år | 61 (14,6) | 61 (14,5) |
| Intervall, år | 13–83 | 15–88 |
| ANCA-associerad vaskulitstatus, n (%) | ||
| Nyligen diagnostiserad | 115 (69,3) | 114 (69,5) |
| Recidiverad | 51 (30,7) | 50 (30,5) |
| ANCA-positivitet, n (%) | ||
| PR3 | 72 (43,4) | 70 (42,7) |
| MPO | 94 (56,6) | 94 (57,3) |
| Typ av ANCA-associerad vaskulit, n (%) | ||
| Granulomatös polyangit (GPA) | 91 (54,8) | 90 (54,9) |
| Mikroskopisk polyangit (MPA) | 75 (45,2) | 74 (45,1) |
| BVAS-poäng | ||
| Genomsnittlig (SD) | 16,3 (5,87) | 16,2 (5,69) |
| eGFR | ||
| Genomsnittlig (SD), ml/min/1,73 m2 | 50,7 (30,96) | 52,9 (32,67) |
| Tidigare användning av glukokortikoider (under screening) | ||
| n (%) | 125 (75,3) | 135 (82,3) |
| Genomsnittlig (SD, prednisonekvivalent dos (mg) | 907 (1 145,9) | 978 (1 157,5) |
ANCA = antineutrofil cytoplasmaantikropp; BVAS = Birmingham Vasculitis Activity Score; MPO = myeloperoxidas; PR3 = proteinas‑3, SD = standardavvikelse
Syftet med studien var att fastställa om avakopan kan vara en effektiv behandling för patienter med ANCA-associerad vaskulit, som samtidigt möjliggör minskad användning av glukokortikoider utan att säkerhet eller effekt försämras.
Det primära syftet var att utvärdera effekten av de ovan beskrivna behandlingsregimerna vad gällde att inducera och upprätthålla remission hos patienter med ANCA-associerad vaskulit baserat på följande två primära effektmått:
- Andelen patienter i sjukdomsremission, definierat som att ha uppnått 0 poäng på Birmingham Vasculitis Activity Score (BVAS) och inte ta glukokortikoider för behandling av ANCA-associerad vaskulit inom 4 veckor före vecka 26.
- Andelen patienter i bibehållen remission, definierat som remission vecka 26 utan recidiv till vecka 52 och ha uppnått 0 BVAS-poäng och inte ta glukokortikoider för behandling av ANCA-associerad vaskulit inom 4 veckor före vecka 52.
De två primära effektmåtten testades sekventiellt för non-inferiority (icke-underlägsenhet) och superiority (överlägsenhet) med hjälp av en gatekeepingprocedur för att bevara typ I‑felfrekvensen på 0,05.
Resultat från denna studie visas tabell 4.
Tabell 4: Remission vecka 26 och bibehållen remission vecka 52 i den pivotala fas 3-studien ADVOCATE (intent-to-treat-population)
Avakopan N = 166 n (%) | Komparator N = 164 n (%) | Uppskattning av behandlingsskillnad i %a | |
| Remission vecka 26 | 120 (72,3) | 115 (70,1) | 3,4 |
| 95 % KI | 64,8; 78,9 | 62,5; 77,0 | −6,0; 12,8 |
| Bibehållen remission vecka 52 | 109 (65,7) | 90 (54,9) | 12,5b |
| 95 % KI | 57,9; 72,8 | 46,9; 62,6 | 2,6; 22,3 |
KI = konfidensintervall
a Tvåsidiga 95 % KI beräknas genom att justera för randomiseringsstratifieringsfaktorer.
b p-värde för superiority = 0,013 (tvåsidigt)
Den observerade effekten var överensstämmande i relevanta undergrupper, dvs. de som hade nyligen diagnostiserad och recidiverande sjukdom, PR3 och MPO ANCA positiv, GPA och MPA, och män och kvinnor. Effektresultat per bakgrundsbehandling presenteras i tabell 5.
Tabell 5: Remission vecka 26 och bibehållen remission vecka 52 i den pivotala fas 3-studien ADVOCATE per bakgrundsbehandling (intent-to-treat-population)
Avakopan n/N (%) | Komparator n/N (%) | Skillnad i %, 95 % KIa | |
| Remission vecka 26 | |||
| Patienter som fick intravenöst rituximab | 83/107 (77,6) | 81/107 (75,7) | 1,9 (−9,5; 13,2) |
| Patienter som fick intravenöst eller oralt cyklofosfamid | 37/59 (62,7) | 34/57 (59,6) | 3,1 (−14,7; 20,8) |
| Bibehållen remission vecka 52 | |||
| Patienter som fick intravenöst rituximab | 76/107 (71,0) | 60/107 (56,1) | 15,0 (2,2; 27,7) |
| Patienter som fick intravenöst eller oralt cyklofosfamid | 33/59 (55,9) | 30/57 (52,6) | 3,3 (−14,8; 21,4) |
a Tvåsidiga 95 % konfidensintervall (KI) beräknas för skillnaden i andelar (avakopan minus komparator) med hjälp av Wald-metoden.
Glukokortikoidtoxicitet
I den pivotala fas 3-studien ADVOCATE var den genomsnittliga totala kumulativa prednison-ekvivalenta dosen från dag 1 till slutet på behandlingen cirka 2,3 gånger högre i komparatorgruppen än i avakopangruppen (3 846,9 mg respektive 1 675,5 mg).
Från baslinjen till vecka 26 fick 86,1 % av patienterna som använde avakopan, glukokortikoider utom ramen för studien. I komparatorgruppen berodde största delen av glukokortikoidanvändningen på den prednisonkur som bestämdes av studieprotokollet.
Figur 2: Total genomsnittlig daglig prednisonekvivalent glukokortikoiddos (i mg) per patient per studievecka per behandlingsgrupp i ADVOCATE-studien (Intent-to-Treat-population)
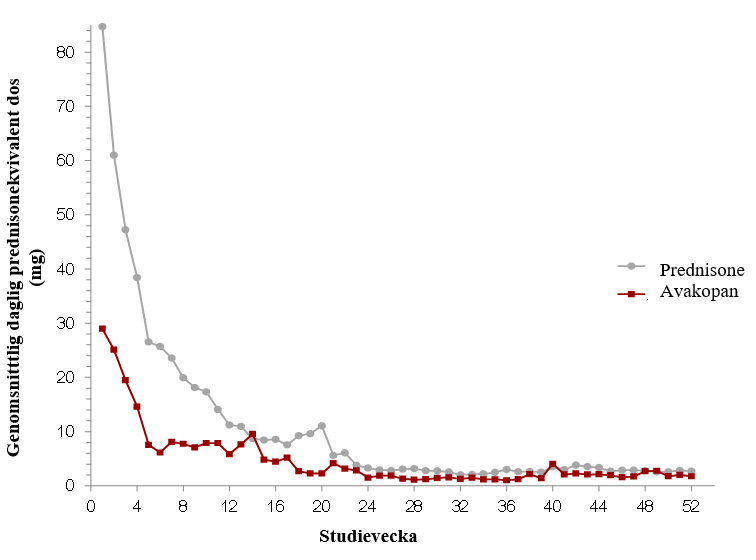
Glukokortikoidtoxicitetsindexet (GTI) bedömer glukokortikoidrelaterad morbiditet inklusive mätningar av kroppsmasseindex, glukostolerans, lipider, steroidmyopati, hudtoxicitet, neuropsykiatrisk toxicitet och infektion. Ett högre GTI indikerar högre glukokortikoidtoxicitet. GTI innehåller den kumulativa försämringspoängen (CWS), som fångar upp kumulativ toxicitet över tid, och den sammanlagda förbättringspoängen (AIS), som fångar upp både förbättring och försämring av toxicitet över tid.
De två GTI-poängen (CWS och AIS) i avakopangruppen jämfört med komparatorgruppen sammanfattas i tabell 6. GTI-värdena var sekundära effektmått i studien och kontrollerades inte för multiplicitet.
Tabell 6: Resultat av glukokortikoidtoxicitetsindex i den pivotala fas 3‑studien ADVOCATE (Intent-to-treat-population)
Avakopan N = 166 | Komparator N = 164 | Skillnad mellan grupperna, 95 % KI | |
| Kumulativ försämringspoäng (CWS) | |||
| Vecka 13 (minsta kvadratmedelvärde) | 25,7 | 36,6 | −11,0 (−19,7; −2,2) |
| Vecka 26 (minsta kvadratmedelvärde) | 39,7 | 56,6 | −16,8 (−25,6; −8,0) |
| Sammanlagd förbättringspoäng (AIS) | |||
| Vecka 13 (minsta kvadratmedelvärde) | 9,9 | 23,2 | −13,3 (−22,2; −4,4) |
| Vecka 26 (minsta kvadratmedelvärde) | 11,2 | 23,4 | −12,1 (−21,1; −3,2) |
Pediatrisk population
Totalt 3 ungdomar deltog i den pivotala fas 3-studien ADVOCATE, två i avakopangruppen och en i komparatorgruppen. En ungdom i avakopangruppen avbröt behandlingen på grund av försämrad njurvaskulit. Den andra unga patienten som fick avakopan fullföljde behandlingen och uppnådde både remission vecka 26 och bibehållen remission vecka 52.
Ungdomen i komparatorgruppen avbröt behandlingen på grund av att instruktionerna om användning av preventivmedel inte efterföljdes.
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för avakopan för en eller flera grupper av den pediatriska populationen för behandling av ANCA-associerad vaskulit (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption
När avakopan administreras utan mat uppnås maximal koncentration i plasma (Cmax) efter en mediantid (tmax) på cirka 2 timmar. Avakopan har uppvisat en ungefärlig dosproportionell ökning i systemisk exponering vid dosintervallet 10–30 mg.
Samtidig administrering av 30 mg-kapslar och en kaloririk måltid med högt fettinnehåll ökar plasmaexponeringen (AUC) av avakopan med cirka 72 % och fördröjer tmax med cirka 3 timmar; Cmax påverkas inte.
Distribution
Avakopans och metaboliten M1:s reversibla plasma-proteinbindning (t.ex. till albumin och surt α1-glykoprotein) är större än 99,9 %. Den skenbara distributionsvolymen är hög (Vz/F 3 000 –11 000 l), vilket indikerar en utbredd vävnadsdistribution av den aktiva substansen.
Metabolism
Avakopan elimineras huvudsakligen genom fas I metabolism. Efter oral administrering av radiomärkt avakopan återfanns största delen av aktiv substans-relaterat material i feces i form av fas I metaboliter. En viktig cirkulerande metabolit (M1), en monohydroxylerad produkt av avakopan fanns i en koncentration på ~12 % av totalt aktiv substans-relaterat material i plasma. Denna metabolit utgör 30 – 50 % av modersubstansens exponering och utövar ungefär samma aktivitet som avakopan på C5aR1. Cytokrom P450 (CYP) 3A4 är det viktigaste enzymet för elimineringav avakopan och för bildning och eliminering av metaboliten M1.
Avakopans kliniska interaktion med det känsliga substratet CYP3A4 beskrivs i avsnitt Interaktioner. Avakopan är en svag hämmare av CYP2C9 såsom indikeras av en blygsam ökning av AUC för den aktiva testsubstansen celecoxib (1,15 gånger).
In vitro är avacopan varken hämmare eller en inducerare av andra CYP-enzymer.
Avakopan visade en försumbar till svag hämning av vanliga transportörer in vitro. Kliniskt relevanta interaktioner är därför osannolika när avakopan administreras tillsamman med substanser som är substrat eller hämmare av dessa transportörer.
Eliminering
Baserat på populationsfarmakokinetisk analys är total skenbar kroppseliminering (CL/F) av avakopan 16,3 l/h (95 % KI: 13,1 –21,1 l/h). Den genomsnittliga terminala elimineringshalveringstiden är 510 timmar (21 dagar) baserat på populationsfarmakokinetisk analys. När avakopan stoppas efter att steady state har uppnåtts beräknas den kvarvarande plasmakoncentrationen av avakopan minska till ∼ 20 %, < 10 % och < 5 % av den högsta steady state-koncentrationen ungefär 4 veckor, 7 veckor respektive 10 veckor efter den sista dosen.
Efter oral administrering av radiomärkt avakopan återfanns cirka 77 % respektive 10 % av radioaktiviteten i feces och i urin, och 7 % respektive < 0,1 % av den radioaktiva dosen återfanns som oförändrat avakopan i feces och urin. Dessa resultat tyder på att den huvudsakliga elimineringsvägen för avakopan är metabolism följt av utsöndring av metaboliter till feces via gallan, och att direkt utsöndring av avakopan till urin eller feces via galla är försumbar.
Särskilda populationer
Äldre
Populationsfarmakokinetisk analys fann ingen signifikant effekt av ålder (bland vuxna) på plasmaexponeringen av avakopan. Farmakokinetiska data om patienter över 75 år är dock begränsade i kliniska studier. Inga dosjusteringar behövs för äldre patienter (se avsnitt Dosering och administreringssätt).
Nedsatt leverfunktion
De farmakokinetiska egenskaperna hos avakopan har undersökts hos 16 patienter med lätt (Child-Pugh klass A) eller måttligt (Child-Pugh klass B) nedsatt leverfunktion. Jämfört med friska kontrollpersoner observerades inga farmakologiskt relevanta skillnader i exponeringen (genomsnittlig kvot för Cmax och AUC ≤ 1,3) av avakopan eller dess viktigaste metabolit M1, varför ingen dosjustering krävs (se avsnitt Dosering och administreringssätt).
Avakopan har inte studerats hos patienter med allvarligt nedsatt leverfunktion (Child-Pugh klass C) (se avsnitt Dosering och administreringssätt).
Nedsatt njurfunktion
Baserat på populationsfarmakokinetisk analys är plasmaexponeringen av avakopan jämförbar hos patienter med nedsatt njurfunktion och friska försökspersoner. Ingen dosjustering krävs därför baserat på njurfunktion (se avsnitt Farmakokinetiska egenskaper).
Avakopan har inte studerats hos patienter med ANCA-associerad vaskulit med en eGFR under 15 ml/min/1,73 m², som behandlas med dialys eller är i behov av dialys eller plasmabyte.
Prekliniska säkerhetsuppgifter
Gängse studier avseende säkerhetsfarmakologi, allmäntoxicitet, gentoxicitet och karcinogenicitet visade inte några särskilda risker för människa.
Fertilitet och tidig embryonal utveckling
Avakopan gav inga effekter på han- eller hondjurens reproduktionsförmåga (fertilitet) eller på tidig utveckling hos hamster vid orala doser motsvarande upp till 6,8 gånger den kliniska AUC.
Embryofetal utveckling
Avakopan var inte teratogent vid oral dosering till hamster och kanin. Hos hamster observerades en ökad förekomst av skelettvariationer (korta torakolumbala extra revben) vid exponeringar motsvarande 5,3 gånger den kliniska AUC. Hos kanin orsakade avakopan toxicitet hos moderdjuret (kliniska tecken på biverkningar samt aborter) men ingen fostertoxicitet vid 0,6 gånger den kliniska AUC.
Pre- och postnatal utveckling
Avakopan resulterade inte i biverkningar hos honavkomman vid administrering till hamster vid exponeringar på upp till 6,3 gånger den kliniska AUC under dräktighet och digivning fram till avvänjning. Hos hanavkomma förekom det en liten fördröjning av preputial separation vid 3,7 gånger den kliniska AUC. Detta isolerade fynd ansågs ha låg toxikologisk betydelse och var inte associerat med någon försämring av reproduktionsförmågan.
Analys av avakopans plasmanivåer hos de digivande moderdjuren och plasmanivåerna hos den diande avkomman visade på förekomst av avakopan, vilket tyder på att avakopan sannolikt utsöndras i mjölken hos lakterande hamstrar.
Karcinogenicitet
Avakopans karcinogena potential utvärderades i en 2‑årig studie på både råtta och hamster.
Hos hanråttor noterades en något ökad förekomst av C-cellsadenom i sköldkörteln som behandlats med avakopan. Denna ökning var inte statistiskt signifikant och förekomsten låg inom det historiska kontrollintervallet. Avakopan var inte karcinogent hos hamster, den farmakologiskt relevanta arten.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Kapselinnehåll
Makrogolglycerolhydroxistearat
Makrogol (4000)
Kapselskal
Gelatin
Röd järnoxid (E172)
Gul järnoxid (E172)
Titandioxid (E171)
Polysorbat 80
Tryckbläck
Svart järnoxid (E172)
Shellack
Kaliumhydroxid
Inkompatibiliteter
Ej relevant.
Hållbarhet
4 år
Särskilda förvaringsanvisningar
Inga särskilda temperaturanvisningar. Förvaras i originalburken. Ljuskänsligt.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
TAVNEOS kapseli, kova
10 mg (L:ei) 180 kpl (6153,35 €)
PF-selosteen tieto
Burk av högdensitetspolyeten (HDPE) med barnskyddande förslutning och induktionsförsegling.
Förpackningsstorlekar: 30 eller 180 hårda kapslar eller multipelförpackning innehållande 540 (3 förpackningar med 180) hårda kapslar.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
Kapslar med gul underdel och ljusorange överdel märkt med ”CCX168” i svart bläck.
En kapsel är 22 mm lång och har en diameter på 8 mm (storlek 0).
Särskilda anvisningar för destruktion och övrig hantering
Inga särskilda anvisningar.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
TAVNEOS kapseli, kova
10 mg 180 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Avakopaani: Aikuisten vaikean aktiivisen granulomatoottisen polyangiitin (GPA) tai mikroskooppisen polyangiitin (MPA) hoito yhdessä rituksimabia tai syklofosfamidia sisältävän hoito-ohjelman kanssa erityisin edellytyksin (3084).
Atc-kod
L04AJ05
Datum för översyn av produktresumén
15.01.2025
Yhteystiedot
Gustav III:s Boulevard 46
SE-169 73 Solna
Sverige
+46 8 558 066 00
www.viforpharma.se
Info.nordic@viforpharma.com