
IDELVION injektiokuiva-aine ja liuotin, liuosta varten 250 IU, 500 IU, 1000 IU, 2000 IU
Vaikuttavat aineet ja niiden määrät
Idelvion 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 250 IU rekombinanttia fuusioproteiinia, joka koostuu ihmisen albumiiniin liitetystä ihmisen hyytymistekijästä IX (rIX‑FP), (albutrepenonakogi alfa). Kun valmiste on saatettu käyttökuntoon 2,5 ml:lla injektionesteisiin käytettävää vettä, liuos sisältää 100 IU/ml albutrepenonakogi alfaa.
Idelvion 500 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 500 IU rekombinanttia fuusioproteiinia, joka koostuu ihmisen albumiiniin liitetystä ihmisen hyytymistekijästä IX (rIX‑FP), (albutrepenonakogi alfa). Kun valmiste on saatettu käyttökuntoon 2,5 ml:lla injektionesteisiin käytettävää vettä, liuos sisältää 200 IU/ml albutrepenonakogi alfaa.
Idelvion 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 1000 IU rekombinanttia fuusioproteiinia, joka koostuu ihmisen albumiiniin liitetystä ihmisen hyytymistekijästä IX (rIX‑FP), (albutrepenonakogi alfa). Kun valmiste on saatettu käyttökuntoon 2,5 ml:lla injektionesteisiin käytettävää vettä, liuos sisältää 400 IU/ml albutrepenonakogi alfaa.
Idelvion 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 2000 IU rekombinanttia fuusioproteiinia, joka koostuu ihmisen albumiiniin liitetystä ihmisen hyytymistekijästä IX (rIX-FP), (albutrepenonakogi alfa). Kun valmiste on saatettu käyttökuntoon 5 ml:lla injektionesteisiin käytettävää vettä, liuos sisältää 400 IU/ml albutrepenonakogi alfaa.
Vahvuus (IU) määritetään käyttämällä Euroopan farmakopean yksivaiheista hyytymistestiä. Idelvion-valmisteen spesifinen aktiivisuus on noin 54–85 IU/mg proteiinia.
Albutrepenonakogi alfa on rekombinantti-DNA-tekniikalla tuotettu puhdistettu proteiini, joka on valmistettu rekombinantin albumiinin ja rekombinantin hyytymistekijä IX:n geneettisen fuusion avulla. Ihmisen albumiinin cDNA:n ja ihmisen hyytymistekijä IX:n cDNA:n geneettisen fuusion tuloksena syntyy yksi rekombinanttiproteiini, jolloin valmiste on homogeeninen ilman kemiallista konjugaatiota. Hyytymistekijä IX:n (rekombinantti) osuus on verrannollinen plasmaperäisen hyytymistekijä IX:n Thr148-alleelisen muodon kanssa. Tekijä IX:n (rekombinantti) ja albumiinimolekyylien välinen sidos on johdettu endogeenisestä ”aktivaatiopeptidistä” luonnollisessa (natiivissa) hyytymistekijä IX:ssä.
Apuaine, jonka vaikutus tunnetaan
Käyttökuntoon saatettu 250 IU, 500 IU tai 1000 IU injektiopullo sisältää 4,3 mg natriumia.
Käyttökuntoon saatettu 2000 IU injektiopullo sisältää 8,6 mg natriumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten
Kliiniset tiedot
Käyttöaiheet
B‑hemofiliaa (synnynnäistä hyytymistekijä IX:n puutosta) sairastavien potilaiden verenvuotojen hoito ja ehkäisy.
Idelvion-valmistetta voidaan käyttää kaikissa ikäryhmissä.
Ehto
Hoito tulee aloittaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito tulee antaa B‑hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Hoidon seuranta
Hoidon aikana tekijä IX:n tasojen asianmukainen määrittäminen on suositeltavaa, jotta saadaan ohjeistusta käytettävästä annoksesta ja toistuvien infuusioiden antotiheydestä. Potilaskohtaiset vasteet tekijä IX:lle saattavat vaihdella, joten puoliintumisaika ja saanto (ns. in vivo recovery) voivat olla erilaiset. Kehon painoon perustuvaa annosta saatetaan joutua muuttamaan alipainoisille tai ylipainoisille potilaille. Erityisesti suurten kirurgisten toimenpiteiden yhteydessä korvaushoidon tarkka seuranta koagulaatioanalyysin (plasman hyytymistekijä IX:n aktiivisuus) avulla on välttämätöntä.
Kun potilaiden verinäytteiden tekijä IX:n aktiivisuuden määritykseen käytetään in vitro tromboplastiiniaikaan (aPTT) perustuvaa yksivaiheista hyytymismääritystä, sekä määrityksessä käytettävä aPTT-reagenssityyppi että vertailustandardi voivat vaikuttaa merkittävästi plasman tekijä IX:n aktiivisuustuloksiin. Jos yksivaiheisessa hyytymistestissä käytetään kaoliinipohjaista aPTT-reagenssia tai Actin FS aPTT-reagenssia, aktiivisuustulokset ovat todennäköisesti aliarvioituja. Tämä on huomioitava erityisesti silloin, kun laboratoriota ja/tai määrityksessä käytettäviä reagensseja vaihdetaan.
Annostus
Annos ja korvaushoidon kesto riippuvat tekijä IX:n puutteen vakavuudesta, vuotokohdasta ja vuodon laajuudesta sekä potilaan kliinisestä tilasta.
Annettavan tekijä IX:n yksikköjen määrä ilmoitetaan kansainvälisinä yksikköinä (IU), jotka ovat WHO:n voimassa olevan tekijä IX -valmisteiden standardin mukaisia. Tekijä IX:n aktiivisuus plasmassa ilmaistaan joko prosentteina (suhteessa normaaliin ihmisplasmaan) tai kansainvälisinä yksikköinä (suhteessa plasman hyytymistekijä IX:n kansainväliseen standardiin).
Tekijä IX:n yksi kansainvälinen yksikkö (IU) vastaa tekijä IX:n määrää 1 ml:ssa normaalia ihmisen plasmaa.
Hoito tarvittaessa
Hyytymistekijä IX:n tarvittavan annoksen laskeminen perustuu empiiriseen havaintoon, jonka mukaan 1 IU hyytymistekijä IX:ää painokiloa kohti lisää plasman hyytymistekijä IX:n aktiivisuutta keskimäärin 1,3 IU:lla/dl (1,3 %:lla normaalista aktiivisuudesta) 12-vuotiailla ja vanhemmilla potilailla ja 1,0 IU:lla/dl (1,0 %:lla normaalista aktiivisuudesta) alle 12-vuotiailla potilailla. Tarvittava annos määritetään seuraavan kaavan avulla:
Tarvittava annos (IU) = paino (kg) x haluttu tekijä IX:n pitoisuuden nousu (% normaalista aktiivisuudesta tai IU/dl) x {havaitun saannon [ns. in vivo recovery] käänteisarvo (IU/kg per IU/dl)}
Odotettu tekijä IX:n pitoisuuden nousu (IU/dl tai % normaalista aktiivisuudesta) = Annos (IU) x Saanto [ns. in vivo recovery] (IU/dl per IU/kg)/paino (kg)
Annos ja antotiheys pitää aina määrittää potilaskohtaisesti kliinisen vaikuttavuuden mukaan.
Alle 12-vuotiaat potilaat
Inkrementaaliselle saannolle (ns. incremental in vivo recovery), jossa 1 IU/dl on 1 IU/kg:aa kohti, annos lasketaan seuraavalla tavalla:
Tarvittava annos (IU) = paino (kg) x haluttu tekijä IX:n pitoisuuden nousu (IU/dl) x 1 dl/kg
Esimerkki
- 20 kg:n painoiselle vaikeaa B-hemofiliaa sairastavalle potilaalle vaadittava huippupitoisuus on 50 % normaalista aktiivisuudesta. Sopiva annos olisi 20 kg x 50 IU/dl x 1 dl/kg = 1 000 IU.
- Jos 1 000 IU:n annos Idelvion-valmistetta annetaan 25 kg:n painoiselle potilaalle, odotettu tekijä IX:n pitoisuuden nousu injektion jälkeen on enintään 1 000 IU/25 kg x 1,0 (IU/dl per IU/kg) = 40 IU/dl (40 % normaalista aktiivisuudesta).
Vähintään 12-vuotiaat potilaat
Inkrementaaliselle saannolle (ns. incremental in vivo recovery), jossa 1,3 IU/dl on 1 IU/kg:aa kohti, annos lasketaan seuraavalla tavalla:
Tarvittava annos (IU) = paino (kg) x haluttu tekijä IX:n pitoisuuden nousu (IU/dl) x 0,77 dl/kg
Esimerkki
- 80 kg:n painoiselle vaikeaa B-hemofiliaa sairastavalle potilaalle vaadittava huippupitoisuus on 50 % normaalista aktiivisuudesta. Sopiva annos olisi 80 kg x 50 IU/dl x 0,77 dl/kg = 3 080 IU.
- Jos 2 000 IU:n annos Idelvion-valmistetta annetaan 80 kg:n painoiselle potilaalle, odotettu tekijä IX:n pitoisuuden nousu injektion jälkeen on enintään 2 000 IU x 1,3 (IU/dl per IU/kg)/80 kg = 32,5 IU/dl (32,5 % normaalista aktiivisuudesta).
Seuraavissa verenvuototapahtumissa hyytymistekijä IX:n aktiivisuus ei saa laskea ilmoitetun plasman aktiivisuustason alapuolelle (prosentteina normaalista tai IU/dl) vastaavalla ajanjaksolla. Seuraavaa taulukkoa voidaan käyttää annostelun apuna verenvuodoissa ja leikkauksissa:
Vuodon voimakkuus / Kirurginen toimenpide | Tarvittava hyytymistekijä IX:n pitoisuus (%) (IU/dl) | Antotiheys (tuntia) / Hoidon kesto (vrk) |
Verenvuoto | 30–60 | Kerta-annos voi olla riittävä suurimmassa osassa tapauksista. Ylläpitoannos 24–72 tunnin kuluttua, jos vuoto jatkuu. |
Laaja verenvuoto | 60–100 | Toista 24–72 tunnin välein ensimmäisen viikon ajan. Tämän jälkeen ylläpitoannos kerran viikossa, kunnes vuoto lakkaa ja vaurio paranee. |
Pieni leikkaus | 50–80 (ennen leikkausta ja sen jälkeen) | Kerta-annos voi olla riittävä suurimmassa osassa pienistä leikkauksista. Tarvittaessa voidaan antaa ylläpitoannos 24–72 tunnin kuluttua, kunnes vuoto lakkaa tai vaurio paranee. |
Suuri leikkaus | 60–100 (ennen leikkausta ja sen jälkeen) | Toista 24–72 tunnin välein ensimmäisen viikon ajan. Tämän jälkeen ylläpitoannos 1–2 kertaa viikossa, kunnes vuoto lakkaa ja haava paranee. |
Estohoito
Vaikeaa B-hemofiliaa sairastavien potilaiden verenvuodon pitkäaikaisessa estohoidossa tavanomainen annostus on 35–50 IU/kg kerran viikossa.
Joillekin potilaille, joiden sairaus pysyy hyvin hallinnassa kerran viikossa annettavalla hoidolla, voi sopia hoidoksi enintään 75 IU/kg ‑annos 10 tai 14 päivän välein. Yli 18-vuotiaille potilaille voidaan harkita vielä pidempää antoväliä (ks. kohta Farmakodynamiikka).
Joissakin tapauksissa, erityisesti nuoria potilaita hoidettaessa, lyhyempi antoväli tai suurempi annos voi olla tarpeen.
Estohoidon aikana esiintyneen verenvuotojakson jälkeen estohoitoa jatketaan mahdollisimman tarkasti samalla annostuksella, mutta potilaalle annetaan kaksi Idelvion-annosta. Antovälin on oltava vähintään 24 tuntia, mutta se voi olla pidempikin, jos tämä on potilaan kannalta parempi.
Pediatriset potilaat
Pitkäaikaisessa estohoidossa suositeltu annostus on 35–50 IU/kg kerran viikossa (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka). Annossuositukset vähintään 12-vuotiaille nuorille ovat samat kuin aikuisille (ks. edellä).
Antotapa
Laskimoon.
Käyttökuntoon saatettu valmiste on annettava hitaana injektiona laskimoon nopeudella, joka takaa potilaan mukavuuden, enintään 5 ml/min.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aiempi allerginen reaktio hamsterin proteiineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annettavan valmisteen nimi ja eränumero kirjattava selkeästi ylös.
Yliherkkyys
Allergistyyppiset yliherkkyysreaktiot ovat mahdollisia Idelvion-valmisteen käytön yhteydessä. Valmiste sisältää jäämiä hamsterin proteiineista. Potilasta on neuvottava lopettamaan valmisteen käyttö heti ja ottamaan yhteyttä lääkäriin, jos yliherkkyysoireita ilmaantuu. Potilaalle on kerrottava yliherkkyysreaktioiden varhaisista oireista, kuten nokkosihottuma, yleistynyt urtikaria, puristuksen tunne rinnassa, hengityksen vinkuminen, hypotensio ja anafylaksia.
Sokkitapauksissa käytetään normaaleja sokin hoitoon tarkoitettuja toimenpiteitä.
Inhibiittorit
Kun potilaita on hoidettu toistuvasti ihmisen hyytymistekijä IX ‑valmisteilla, heitä on tarkkailtava neutraloivien vasta-aineiden (inhibiittorien) varalta, jotka pitää määrittää Bethesda-yksiköinä (BU) käyttämällä soveltuvaa biologista testausta. B‑hemofiliapotilailla on ilmoitettu inhibiittorin muodostumista tekijä IX:lle Idelvion-valmisteella annetun tekijä IX ‑korvaushoidon aikana.
Kirjallisuudessa on raportteja, joissa osoitetaan korrelaatio tekijä IX inhibiittorien ja allergisten reaktioiden esiintymisen välillä. Siksi kaikilta allergisia reaktioita saavilta potilailta pitää tutkia inhibiittorin esiintyminen. On huomattava, että potilailla, joilla esiintyy tekijä IX:n inhibiittoreita, saattaa olla suurentunut anafylaksiariski myöhemmän hyytymistekijä IX:n käytön yhteydessä.
Tekijä IX konsentraatin allergisten reaktioiden vaaran vuoksi tekijä IX:n aloitusannokset pitäisi antaa, riippuen hoitavan lääkärin arviosta, lääketieteellisen valvonnan alaisena paikassa, jossa allergisia reaktioita voidaan hoitaa lääketieteellisesti asianmukaisesti.
Tromboembolia
Tromboottisten komplikaatioiden riskin vuoksi tromboottisen ja konsumptiokoagulopatian varhaisten merkkien kliininen tarkkailu on aloitettava soveltuvalla biologisella testauksella, kun tätä valmistetta annetaan potilaille, joilla on maksasairaus, leikkauksen jälkeen, vastasyntyneille tai potilaille, joilla on tromboottisten tapahtumien tai DIC-oireyhtymän riski. Kaikissa näissä tapauksissa Idelvion-hoidon hyöty on arvioitava suhteessa näiden komplikaatioiden aiheuttamaan riskiin.
Sydän- ja verisuonitapahtumat
Potilailla, joilla on sydän- ja verisuonitapahtumien riskitekijöitä, tekijä IX-korvaushoito voi suurentaa sydän- ja verisuonitapahtumien riskiä.
Katetreihin liittyvät komplikaatiot
Jos toimenpide edellyttää keskuslaskimokatetria, on huomioitava keskuslaskimokatetriin liittyvät komplikaatiot, mukaan lukien paikalliset infektiot, bakteremia ja katetrikohdan tromboosi.
Iäkkäät
Idelvion-valmistetta koskevissa kliinisissä tutkimuksissa ei ollut mukana 65-vuotiaita tai sitä vanhempia henkilöitä. Ei ole tiedossa, onko iäkkäiden potilaiden vaste erilainen kuin nuoremmilla potilailla.
Siedätyshoito
Idelvion-valmisteen turvallisuutta ja tehoa siedätyshoidossa ei ole varmistettu.
Natriumsisältö
Tämä lääkevalmiste sisältää enintään 8,6 mg natriumia per injektiopullo, joka vastaa 0,4 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Pediatriset potilaat
Luetellut varoitukset ja varotoimet koskevat sekä aikuisia että lapsia.
Yhteisvaikutukset
Ihmisen hyytymistekijä IX (rDNA) ‑valmisteilla ei ole raportoitu olevan yhteisvaikutuksia muiden lääkevalmisteiden kanssa.
Raskaus ja imetys
Lisääntymistä koskevia eläinkokeita ei ole suoritettu tekijä IX:llä. Koska B-hemofiliaa esiintyy naisilla vain harvoin, kokemuksia tekijä IX:n vaikutuksista raskauteen ja imetykseen ei ole.
Tämän vuoksi tekijä IX:ää saa käyttää raskauden ja imetyksen aikana vain, jos se on selvästi tarpeellista.
Tietoa tekijä IX:n vaikutuksesta hedelmällisyyteen ei ole.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Idelvion-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yliherkkyysreaktioita tai allergisia reaktioita (joita voivat olla angioedeema, infuusiokohdan polttelu ja kirvely, vilunväreet, punoitus, yleistynyt nokkosihottuma, päänsärky, nokkosihottuma, matala verenpaine, uneliaisuus, pahoinvointi, levottomuus, sydämen tiheälyöntisyys, puristus rinnassa, kihelmöinti, oksentelu, vinkuva hengitys) on havaittu harvoin. Nämä oireet voivat joissain tapauksissa edetä vaikeaksi anafylaksiaksi (mukaan lukien sokki). Joissakin tapauksissa nämä reaktiot ovat edenneet vaikeaksi anafylaksiaksi, ja ne ovat ilmenneet läheisessä aikayhteydessä hyytymistekijä IX:n inhibiittoreiden kehittymisen kanssa (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet). Nefroottista oireyhtymää on raportoitu, kun valmisteella on annettu siedätyshoitoa sellaisille B-hemofiliaa sairastaville potilaille, joilla on tekijä IX-inhibiittoreita ja joilla on ollut allergisia reaktioita.
Hyvin harvoin on havaittu vasta-aineiden kehittymistä hamsterin proteiineja vastaan, mihin on liittynyt myös yliherkkyysreaktioita.
B-hemofiliaa sairastaville potilaille voi kehittyä neutraloivia vasta-aineita (inhibiittoreita) tekijä IX:lle. Jos tällaisia inhibiittoreita ilmenee, tila ilmenee riittämättömänä kliinisenä vasteena. Tällaisissa tapauksissa suositellaan ottamaan yhteyttä hemofiliaan erikoistuneeseen hoitokeskukseen. Aiemmin hoitamattomilla potilailla tehdyssä kliinisessä tutkimuksessa ilmoitettiin yhdessä tapauksessa korkea inhibiittorititteri. Inhibiittorien kehittymistä on havaittu Idelvion-valmisteen markkinoilletulon jälkeen potilailla, jotka ovat saaneet aiempaa hoitoa.
Tekijä IX -valmisteiden annon jälkeen on olemassa mahdollinen tromboembolisten tapahtumien riski, ja riski on suurempi, jos valmisteen puhtaus on alhainen. Alhaisen puhtauden hyytymistekijä IX ‑valmisteiden käyttö on liitetty sydäninfarktitapauksiin, DIC-oireyhtymään, laskimotukokseen ja keuhkoemboliaan. Korkean puhtauden hyytymistekijä IX on liitetty harvoin tällaisiin haittavaikutuksiin.
Haittavaikutustaulukko
Alla oleva taulukko on luokiteltu MedDRAn elinjärjestelmäluokituksen mukaan (elinjärjestelmäluokitus ja suositeltu termi). Taulukossa mainitaan kliinisissä tutkimuksissa ja/tai markkinoille tulon jälkeisessä käytössä raportoidut haittavaikutukset.
Esiintyvyydet on arvioitu seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
MedDRA-elinjärjestelmäluokka | Haittavaikutus | Esiintymistiheys potilasta kohden |
Veri ja imukudos | Tekijä IX:n esto / inhibiittorien kehittyminen | Tuntematon |
Immuunijärjestelmä | Yliherkkyys | Yleinen |
Hermosto | Päänsärky | Yleinen |
Heitehuimaus | Yleinen | |
Iho ja ihonalainen kudos | Ihottuma | Yleinen |
Ekseema | Melko harvinainen | |
Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan reaktiot | Yleinen |
Valikoitujen haittavaikutusten kuvaus
Aiemmin hoitamattomilla potilailla tehdyssä kliinisessä tutkimuksessa ilmoitettiin yhdessä tapauksessa korkea inhibiittorititteri (ks. kohta Farmakodynamiikka). Tietojen vähäisyyden vuoksi inhibiittorin ilmaantuvuutta ei ole ilmoitettu.
Pediatriset potilaat
Haittavaikutusten esiintyvyyden, tyypin ja vaikeusasteen odotetaan olevan lapsilla samanlainen kuin aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Idelvion-valmisteella ei ole raportoitu yliannostusoireita.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Hemostaatit, veren hyytymistekijä IX, ATC-koodi: B02BD04.
Vaikutusmekanismi
Hyytymistekijä IX on yksiketjuinen glykoproteiini, jonka molekyylimassa on noin 68 000 daltonia. Se on K-vitamiiniriippuvainen hyytymistekijä ja syntetisoituu maksassa. Hyytymistekijä IX:n aktivoi tekijä XIa sisäisessä hyytymisreitissä ja tekijä VII/kudoksen hyytymistekijäyhdistelmä ulkoisessa reitissä. Aktivoitunut hyytymistekijä IX yhdessä aktivoituneen tekijä VIII:n kanssa aktivoi tekijä X:n. Aktivoitunut tekijä X muuntaa protrombiinia trombiiniksi. Trombiini muuntaa sitten fibrinogeenin fibriiniksi, jolloin hyytymä muodostuu. B‑hemofilia on sukupuoleen sidoksissa oleva perinnöllinen verenhyytymishäiriö, joka johtuu tekijä IX:n alentuneesta tasosta ja josta seuraa runsasta nivelten, lihasten tai sisäelinten verenvuotoa. Vuoto voi olla spontaania tai onnettomuuden tai kirurgisen trauman aiheuttamaa. Korvaushoidolla lisätään hyytymistekijä IX:n pitoisuuksia plasmassa, jolloin tekijän puutosta ja vuototaipumusta korjataan tilapäisesti.
On huomattava, ettei vuotuinen verenvuotojen esiintyvyys ole vertailukelpoinen eri hyytymistekijäkonsentraattien eikä eri kliinisten tutkimusten välillä.
Albutrepenonakogi alfa on rekombinantti hyytymistekijä IX. Yhdistäminen rekombinanttiin albumiiniin mahdollistaa albutrepenonakogi alfan pidentyneen puoliintumisajan ja tehostuneen systeemisen altistuksen (ks. kohta Farmakokinetiikka). Albumiini on luonnollinen inertti plasman kuljettajaproteiini, jonka puoliintumisaika on noin 20 vuorokautta.
Albutrepenonakogi alfa säilyy koskemattomana verenkierrossa, kunnes tekijä IX aktivoituu, jolloin albumiini pilkkoutuu vapauttaen aktivoitua tekijä IX:ää (FIXa), kun sitä tarvitaan hyytymiseen.
Yleistä tietoa kliinisestä tehosta ja turvallisuudesta
Vaiheen 1/2 tutkimuksessa arvioitiin hoidon tehoa ja verenvuotojaksojen ehkäisyä rIX-FP-valmisteella 17 tutkimushenkilöllä (iältään 13–46 vuotta). Estohoitoa saavassa tutkimusryhmässä 13 tutkimushenkilöä sai Idelvion-valmistetta verenvuotojen ehkäisyyn kerran viikossa noin 11 kuukauden ajan. Tarvittaessa hoitoa saavien tutkimusryhmässä 4 tutkimushenkilöä sai Idelvion-valmistetta verenvuotojen ilmaantuessa. Kaikki 85 vuotojaksoa saatiin onnistuneesti hoidettua antamalla yksi tai kaksi Idelvion-annosta.
Idelvion-valmisteen tehoa on arvioitu vaiheen 2/3 tutkimuksen avoimessa, kontrolloimattomassa osassa, jossa yhteensä 63 aiemmin hoitoa saaneelle miehelle (ikä 12–61 vuotta) annettiin Idelvion-valmistetta joko verenvuotojen ehkäisyyn kerran viikossa 7, 10 ja/tai 14 päivän välein ja/tai verenvuodon hoitoon tarvittaessa. Kaikilla tutkimushenkilöillä oli vaikea (FIX-aktiivisuus < 1 %) tai keskivaikea (FIX-aktiivisuus ≤ 2 %) B‑hemofilia. Näistä miehistä 40 sai Idelvion-valmistetta verenvuotojen ehkäisyyn.
Estohoitoa saaneet tutkimushenkilöt saivat aluksi 35–50 IU/kg kerran viikossa. Osa potilaista siirtyi pidennettyyn hoitoväliin (annos 10 tai 14 päivän välein) suositusannoksella 75 IU/kg, ja hoitoa muutettiin yksilöllisesti. Pidennetyn 14‑päiväisen antovälin mukaista estohoitoa jatkettiin 21:lle aiemmin hoitoa saaneelle potilaalle vielä 98–575 (mediaani: 386) päivän ajan. Näistä potilaista 8:lla (38 %:lla) ilmeni vähintään yksi verenvuoto 14 päivän välein annetussa estohoidossa mutta ei yhtään vuototapahtumaa kerran viikossa annetussa estohoidossa. Vuotuinen verenvuotojen esiintyvyys (annualised bleeding rate, ABR) oli 7 päivän välein annetussa Idelvion-estohoidossa kaikenlaisten verenvuotojen osalta 0,0 (mediaani; vaihteluväli 0–6) ja 14 päivän välein annetussa estohoidossa 1,08 (mediaani; vaihteluväli 0–9,1).
Rutiininomaisen estohoidon pitkäaikaisteho ja turvallisuus enintään 5 vuoden käytössä vahvistettiin avoimessa jatkotutkimuksessa, jossa yhteensä 59 vähintään 12-vuotiasta aiemmin hoitoa saanutta potilasta (54 aikuista ja 5 nuorta) sai Idelvion-valmistetta estohoitona ja/tai tarvittaessa verenvuotojaksojen hoitoon. Estohoitoa saaneet potilaat jatkoivat tai aloittivat annoksella 35–50 IU/kg kerran viikossa. Alaryhmä potilaita vaihtoi ohjelmaan, jossa antotiheyttä oli harvennettu (10, 14 tai 21 päivän välein) ja suositusannos oli 75 IU/kg (10 tai 14 päivää) tai 100 IU/kg (21 päivää). Tutkimuksen lopussa 14 aiemmin hoitoa saanutta potilasta (24 %) sai estohoitoa 7 päivän välein. Yhteensä 11 aiemmin hoitoa saanutta potilasta (19 %) pysyi 10 päivän välein annettavassa, 25 aiemmin hoitoa saanutta potilasta (42 %) 14 päivän välein annettavassa ja 9 aiemmin hoitoa saanutta potilasta (15 %) 21 päivän välein annettavassa harvennetussa anto-ohjelmassa. Tutkimuksen aikana 2 aiemmin hoitoa saanutta potilasta (18 %), jotka olivat olleet 21 päivän anto-ohjelmassa, vaihtoi tiheämpään annostukseen lisääntyneiden vuotokomplikaatioiden vuoksi. Arvioitu vuotuisen verenvuotojen esiintyvyyden (ABR) mediaani oli 7 päivän Idelvion-estohoidossa 1,3 (vaihteluväli 0–8), 14 päivän hoidossa 0,9 (vaihteluväli 0–13) ja 21 päivän hoidossa 0,3 (vaihteluväli 0–5).
Tällä hetkellä saatavilla olevat tiedot tukevat joidenkin potilaiden hoitovälin pidentämistä, vaikka tähän saattaa liittyä verenvuotoriskin suureneminen verrattuna kerran viikossa annettavaan hoitoon.
Verenvuotojen estohoito ja hallinta aiemmin hoitoa saaneilla alle 12‑vuotiailla potilailla
Idelvion-valmisteen tehoa on arvioitu vaiheen 3 tutkimuksessa, jossa oli mukana yhteensä 27 aiemmin hoitoa saanutta iältään 1–10‑vuotiasta poikaa (iän mediaani 6,0 vuotta), joista 12 potilasta oli < 6 vuotta. Tutkimushenkilöille annettiin Idelvion-valmistetta verenvuotojaksojen estohoitoon tai hallintaan. Kaikki 27 tutkimushenkilöä saivat Idelvion-valmistetta viikoittain estohoitona keskimäärin 13,1 kuukauden ajan (9; 18 kk).
106 vuotojaksossa suurimmalle osalle (94; 88,7 %) riitti hoidoksi yksi kertainjektio, 103 (97,2 %) sai 1–2 injektiota. Hemostaattinen teho vuodon tyrehdyttyä arvioitiin erinomaiseksi tai hyväksi 96 %:ssa kaikista hoidetuista verenvuotojaksoista.
Rutiininomaisen estohoidon pitkäaikaisteho ja turvallisuus enintään 5 vuoden käytössä vahvistettiin avoimessa jatkotutkimuksessa, jossa yhteensä 24 aiemmin hoitoa saanutta alle 12-vuotiasta potilasta sai Idelvion-valmistetta ehkäisyhoitona ja/tai tarvittaessa vuotojaksojen hoitoon. Ehkäisyhoitoa saaneet potilaat jatkoivat annoksella 35–50 IU/kg kerran viikossa. Alaryhmä potilaita vaihtoi ohjelmaan, jossa antotiheyttä oli harvennettu (10 tai 14 päivän välein) ja suositusannos oli 75 IU/kg. Tutkimuksen lopussa 17 aiemmin hoitoa saanutta potilasta (71 %) sai estohoitoa 7 päivän välein. Yhteensä 3 aiemmin hoitoa saanutta potilasta (12 %) pysyi 10 päivän välein annettavassa ja 4 aiemmin hoitoa saanutta potilasta (17 %) 14 päivän välein annettavassa harvennetussa anto-ohjelmassa. Tutkimuksen aikana 4 aiemmin hoitoa saanutta potilasta (50 %), jotka olivat olleet 14 päivän anto-ohjelmassa, vaihtoi tiheämpään annostukseen lisääntyneiden vuotokomplikaatioiden vuoksi. Arvioitu vuotuisen verenvuotojen esiintyvyyden (ABR) mediaani oli 7 päivän Idelvion-estohoidossa 2,0 (vaihteluväli 0–14) ja 14 päivän hoidossa 5,6 (vaihteluväli 0–8).
Perioperatiivinen hoito
Valmisteen turvallisuutta ja tehoa perioperatiivisessa käytössä arvioitiin kahdessa vaiheen 3 pivotaalitutkimuksessa ja pitkäaikaisessa jatkotutkimuksessa. Tutkimussuunnitelman mukainen tehoanalyysi käsitti 30 leikkausta 21:lle iältään 5–58-vuotiaalle potilaalle, joille tehtiin suuri tai pieni kirurginen tai hammaslääketieteellinen toimenpide tai muu invasiivinen kirurginen toimenpide. Annostus yksilöllistettiin tutkimushenkilön farmakokineettisen ja kliinisen hoitovasteen perusteella. Leikkausta edeltävää 14–163 IU/kg boluskerta-annosta käytettiin 96,7 %:ssa (n = 29) leikkauksista. Hemostaattinen teho arvioitiin erinomaiseksi tai hyväksi kaikissa arvioiduissa toimenpiteissä. Potilaat saivat leikkauksen jälkeisen 14 vuorokauden jakson aikana 0–11 infuusiota, ja kokonaisannos oli 0–444 IU/kg.
Potilaat, jotka eivät ole aiemmin saaneet hoitoa (previously untreated patients, PUP)
IDELVION-valmisteen tehoa ja turvallisuutta arvioitiin avoimessa kliinisessä monikeskustutkimuksessa 12:lla B-hemofiliaa sairastavalla potilaalla, jotka eivät olleet aiemmin saaneet hoitoa (endogeenisen tekijä IX:n aktiivisuus ≤ 2 %) ja joista 11 oli iältään 0–1-vuotiaita. Altistuspäivien lukumäärän mediaani oli 50 (vaihteluväli 22–146 altistuspäivää), ja 8 potilasta, jotka eivät olleet aiemmin saaneet hoitoa, saavutti ≥ 50 altistuspäivää tarvittaessa annettavan hoidon, estohoidon, kirurgisten ja farmakokineettisten jaksojen aikana.
Kaikki 12 potilasta, jotka eivät olleet aiemmin saaneet hoitoa, saivat rutiininomaista estohoitoa, ja heistä 11 sai hoitoa 7 päivän välein. Estohoidon kokonaisajan mediaani oli 11,5 kuukautta (vaihteluväli: 3,1–32,3 kuukautta). Yhdeksän potilasta, jotka eivät olleet aiemmin saaneet hoitoa, sai estohoitoa 7 päivän välein > 6 kuukauden ajan, ja heillä vuotuisen verenvuotojen esiintyvyyden (ABR) mediaani oli 1,16 (vaihteluväli 0–3,1). Viidellä näistä yhdeksästä potilaasta ABR oli 0. Seitsemän päivän välein annettavassa estohoidossa (N = 9) kuukausittaisen annoksen mediaani oli 195,9 IU/kg (vaihteluväli 171,8–215,6 IU/kg).
Viisi tutkittavaa sai tarvittaessa annettavaa hoitoa eripituisten jaksojen ajan ennen estohoitoa, ja altistuspäivien lukumäärä oli 1–4.
Kymmenellä potilaalla, jotka eivät olleet aiemmin saaneet hoitoa, tutkimusjaksojen aikana havaituista 37 verenvuototapahtumasta 94 % saatiin onnistuneesti hallintaan yhdellä tai kahdella infuusiolla.
Farmakokinetiikka
Aikuispotilaat
Idelvion-valmisteen farmakokinetiikkaa arvioitiin 25, 50 ja 75 IU/kg kerta-annoksena laskimoon annetun injektion jälkeen. Farmakokineettiset muuttujat Idelvion 50 IU/kg kertainjektion jälkeen (ks. taulukko alla) perustuvat yksivaiheisella hyytymistestillä määritettyyn plasman tekijä IX:n aktiivisuuteen. Keskimääräinen tekijä IX:n aktiivisuus päivänä 7 oli 13,76 % ja päivänä 14 6,10 % Idelvion 50 IU/kg kerta-annoksen jälkeen. Toistetuissa farmakokineettisten parametrien arvioinnissa aina 30 viikkoon asti todettiin, että farmakokineettinen profiili oli vakaa ja inkrementaalinen saanto (ns. incremental in vivo recovery), tasainen ajan kuluessa.
Kliinisissä tutkimuksissa verenvuoto on saatu pidettyä estohoidossa hallinnassa tavoittelemalla pienintä pitoisuutta 5–10 %. Farmakokineettiset simulaatiot viittaavat siihen, että 5 %:n FIX‑aktiivisuuden saavuttaminen plasmassa kestää aikuisilla Idelvion 50 IU/kg kertainjektion jälkeen 12,5 päivää.
Vaikeaa hemofiliaa sairastavien farmakokineettiset parametrit (Mediaani [min, max]) aikuisilla Idelvion-kertainjektion jälkeen
Parametri | 50 IU/kg |
Inkrementaalinen saanto (ns. incremental in vivo recovery) a, | 1,18 (0,86; 1,86) |
Cmax a (IU/dl) | 62,7 (40,5; 87,0) |
AUC0-inf (h*IU/dl) | 6638 (2810; 9921) |
Eliminaation puoliintumisaika, t1/2 (h) | 95,3 (51,5; 135,7) |
CL (ml/h/kg) | 0,875 (0,748; 1,294) |
a = korjattu lähtötason suhteen
AUC = tekijä IX:n aktiivisuuden käyrän alle jäävä pinta-ala; CL = painoon suhteutettu puhdistuma
Pediatriset potilaat
Idelvion-valmisteen farmakokineettisiä muuttujia arvioitiin nuorilla (12 – < 18‑vuotiailla) sekä pikkulapsilla ja lapsilla (1 – < 12‑vuotiailla) 50 IU/kg kerta-annoksena laskimoon annetun injektion jälkeen. Farmakokineettiset muuttujat (ks. alla) perustuvat yksivaiheisella hyytymistestillä määritettyyn plasman tekijä IX:n aktiivisuuteen ajan suhteen.
Idelvion-valmisteen farmakokineettisten parametrien vertailu lapsilla (Mediaani [min, max]) Idelvion 50 IU/kg kertainjektion jälkeen
Parametri | 1 – < 6 vuotta | 6 – < 12 vuotta | 12 – < 18 vuotta |
Inkrementaalinen saanto (ns. incremental in vivo recovery) a (IU/dl)/(IU/kg) | 0,968 (0,660; 1,280) | 1,07 (0,70; 1,47) | 1,11 (0,84; 1,61) |
Cmaxa (IU/dl) | 48,2 (33,0; 64,0) | 50,5 (34,9; 73,6) | 55,3 (40,5; 80,3) |
AUC0-inf (h*IU/dl) | 4301 (2900; 8263) | 4718 (3212; 7720) | 4804 (2810; 9595) |
Eliminaation puoliintumisaika (h) | 86,2 (72,6; 105,8) | 89,3 (62,1; 123,0) | 88,8 (51,5; 130,0) |
CL (ml/h/kg) | 1,16 (0,61; 1,72) | 1,06 (0,65; 1,56) | 1,04 (0,52; 1,67) |
a = korjattu lähtötason suhteen
AUC = tekijä IX:n aktiivisuuden käyrän alle jäävä pinta-ala; CL = painoon suhteutettu puhdistuma
Kliinisissä tutkimuksissa verenvuoto on saatu pidettyä estohoidossa hallinnassa tavoittelemalla pienintä pitoisuutta 5–10 %. Farmakokineettiset simulaatiot viittaavat siihen, että 5 %:n FIX‑aktiivisuuden saavuttaminen plasmassa kestää Idelvion 50 IU/kg kertainjektion jälkeen 7 päivää 1 – < 6‑vuotiailla, 9 päivää 6 – < 12‑vuotiailla ja 11 päivää 12 – < 18‑vuotiailla.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, kerta-annoksen ja toistuvan altistuksen aiheuttamaa toksisuutta, geenitoksisuutta, trombogeenisuutta ja paikallista siedettävyyttä koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Karsinogeenisuutta ja lisääntymistoksisuutta koskevia tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine:
Natriumsitraatti
Polysorbaatti 80
Mannitoli
Sakkaroosi
Kloorivetyhappo (pH:n säätämiseen)
Liuotin:
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Injektion saa antaa vain pakkauksen sisältämillä antolaitteilla, koska hoito voi epäonnistua, jos hyytymistekijä IX adsorboituu muun injektiolaitteen sisäpintaan.
Kestoaika
3 vuotta
Valmisteen käytönaikaisen kemiallisen ja fysikaalisen säilyvyyden on osoitettu olevan käyttökuntoon saattamisen jälkeen 8 tuntia 2–25 °C:n lämpötilassa. Mikrobiologisista syistä valmiste on käytettävä heti. Jos sitä ei käytetä heti, käytönaikainen säilytysaika ja olosuhteet ennen käyttöä ovat käyttäjän vastuulla.
Säilytys
Säilytä alle 25 °C.
Ei saa jäätyä. Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Ei markkinoilla olevia pakkauksia.
PF-selosteen tieto
Idelvion 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (250 IU) 6 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (bromobutyylikumia), levy (muovia) ja korkki (alumiinia).
2,5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (bromo- tai klorobutyylikumia), levy (muovia) ja korkki (alumiinia).
Idelvion 500 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (500 IU) 6 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (bromobutyylikumia), levy (muovia) ja korkki (alumiinia).
2,5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (bromo- tai klorobutyylikumia), levy (muovia) ja korkki (alumiinia).
Idelvion 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (1000 IU) 6 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (bromobutyylikumia), levy (muovia) ja korkki (alumiinia).
2,5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (bromo- tai klorobutyylikumia), levy (muovia) ja korkki (alumiinia).
Idelvion 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kuiva-aine (2000 IU) 10 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (bromobutyylikumia), levy (muovia) ja korkki (alumiinia).
5 ml liuotinta injektiopullossa (tyypin I lasia), jossa on tulppa (bromo- tai klorobutyylikumia), levy (muovia) ja korkki (alumiinia).
Pakkauskoot
Yksi pakkaus sisältää:
Idelvion 250 IU injektiokuiva-aine ja liuotin, liuosta varten
1 injektiopullo kuiva-ainetta
1 injektiopullo, joka sisältää 2,5 ml injektionesteisiin käytettävää vettä
1 suodattimella varustettu siirtolaite 20/20
Sisäpakkaus, joka sisältää:
1 kertakäyttöinen 5 ml:n ruisku
1 laskimopunktiosetti
2 desinfektiopyyhettä
1 ei-steriili laastari
Idelvion 500 IU injektiokuiva-aine ja liuotin, liuosta varten
1 injektiopullo kuiva-ainetta
1 injektiopullo, joka sisältää 2,5 ml injektionesteisiin käytettävää vettä
1 suodattimella varustettu siirtolaite 20/20
Sisäpakkaus, joka sisältää:
1 kertakäyttöinen 5 ml:n ruisku
1 laskimopunktiosetti
2 desinfektiopyyhettä
1 ei-steriili laastari
Idelvion 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
1 injektiopullo kuiva-ainetta
1 injektiopullo, joka sisältää 2,5 ml injektionesteisiin käytettävää vettä
1 suodattimella varustettu siirtolaite 20/20
Sisäpakkaus, joka sisältää:
1 kertakäyttöinen 5 ml:n ruisku
1 laskimopunktiosetti
2 desinfektiopyyhettä
1 ei-steriili laastari
Idelvion 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
1 injektiopullo kuiva-ainetta
1 injektiopullo, joka sisältää 5 ml injektionesteisiin käytettävää vettä
1 suodattimella varustettu siirtolaite 20/20
Sisäpakkaus, joka sisältää:
1 kertakäyttöinen 10 ml:n ruisku
1 laskimopunktiosetti
2 desinfektiopyyhettä
1 ei-steriili laastari
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Vaaleankeltainen tai valkoinen injektio-kuiva-aine ja kirkas, väritön liuotin
pH: 6,6–7,2
Osmolaliteetti:
Idelvion 250 IU injektiokuiva-aine ja liuotin, liuosta varten
175–215 mosm/kg.
Idelvion 500 IU injektiokuiva-aine ja liuotin, liuosta varten
260–300 mosm/kg.
Idelvion 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
260–300 mosm/kg.
Idelvion 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
260–300 mosm/kg.
Käyttö- ja käsittelyohjeet
Yleiset ohjeet
- Käyttövalmiiksi saatetun liuoksen on oltava kirkasta tai hieman opaalinhohtoista, keltaista tai väritöntä. Kun käyttövalmiiksi saatettu valmiste on suodatettu/vedetty ruiskuun (ks. seuraavassa), valmiste on tarkistettava silmämääräisesti ennen antoa, ettei siinä ole havaittavissa hiukkasia eikä värinmuutoksia.
- Älä käytä liuosta, jos se on sameaa tai siinä on hiukkasia.
- Valmisteen käyttökuntoon saattaminen ja vetäminen ruiskuun on tehtävä aseptisissa olosuhteissa.
Käyttökuntoon saattaminen
Anna liuottimen lämmetä huoneenlämpöiseksi (alle 25 °C). Varmista, että IDELVION-valmisteen ja liuottimen sisältävien injektiopullojen irti napsautettavat flip-off-sulkimet on poistettu ja tulpat on käsitelty antiseptisella liuoksella. Tulpan on sen jälkeen annettava kuivua ennen Mix2Vial-pakkauksen avaamista.
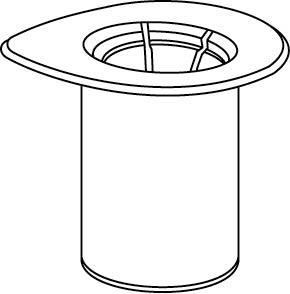
| 1. Avaa Mix2Vial vetämällä suojakansi pois. Älä poista Mix2Vial-laitetta läpipainopakkauksesta! |
| 2. Aseta liuottimen sisältävä injektiopullo tasaiselle, puhtaalle alustalle ja ota injektiopullosta tukeva ote. Ota Mix2Vial sekä pakkaus ja paina sinisen sovittimen piikki kohtisuoraan liuotinpullon tulpan läpi. |
| 3. Poista pakkaus varovasti Mix2Vial-laitteesta siten, että pidät pakkauksen reunasta kiinni ja vedät kohtisuoraan ylöspäin. Varmista, että vedät pois vain pakkauksen etkä Mix2Vial-laitetta. |
| 4. Aseta Idelvion-valmisteen sisältävä injektiopullo tasaiselle ja tukevalle alustalle. Kun liuottimen sisältävä injektiopullo on kiinnitettynä Mix2Vial-laitteeseen, käännä ne ylösalaisin ja paina läpinäkyvän sovittimen piikki suoraan Idelvion-valmisteen sisältävän injektiopullon kumitulpan läpi. Liuotin siirtyy automaattisesti IDELVION-valmisteen sisältävään injektiopulloon. |
| 5. Ota toisella kädellä kiinni Mix2Vial-laitteen Idelvion-valmisteen sisältävän injektiopullon puolelta ja toisella kädellä liuottimen sisältävän injektiopullon puolelta ja kierrä vastapäivään laite kahteen osaan varovasti. Hävitä liuotinpullo ja siihen kiinnitetty sininen Mix2Vial-sovitin. |
| 6. Pyörittele Idelvion-valmisteen sisältävää injektiopulloa ja siihen kiinnitettyä läpinäkyvää sovitinta, kunnes kuiva-aine on liuennut täysin. Älä ravista. |
| 7. Vedä tyhjään, steriiliin ruiskuun ilmaa. Kun Idelvion-valmisteen sisältävä injektiopullo on oikeinpäin, kiinnitä ruisku Mix2Vial-sovittimen Luer Lock ‑liittimeen kiertämällä myötäpäivään. Ruiskuta ilma Idelvion-valmisteen sisältävään injektiopulloon. |
 1
1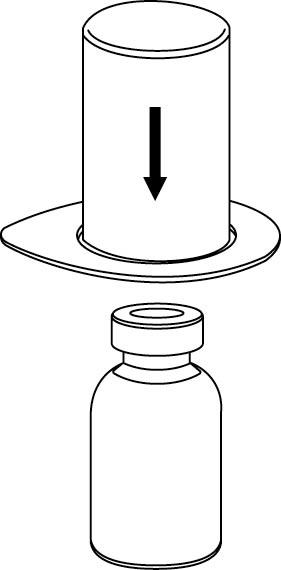 2
2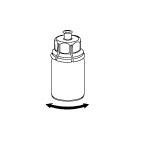 6
6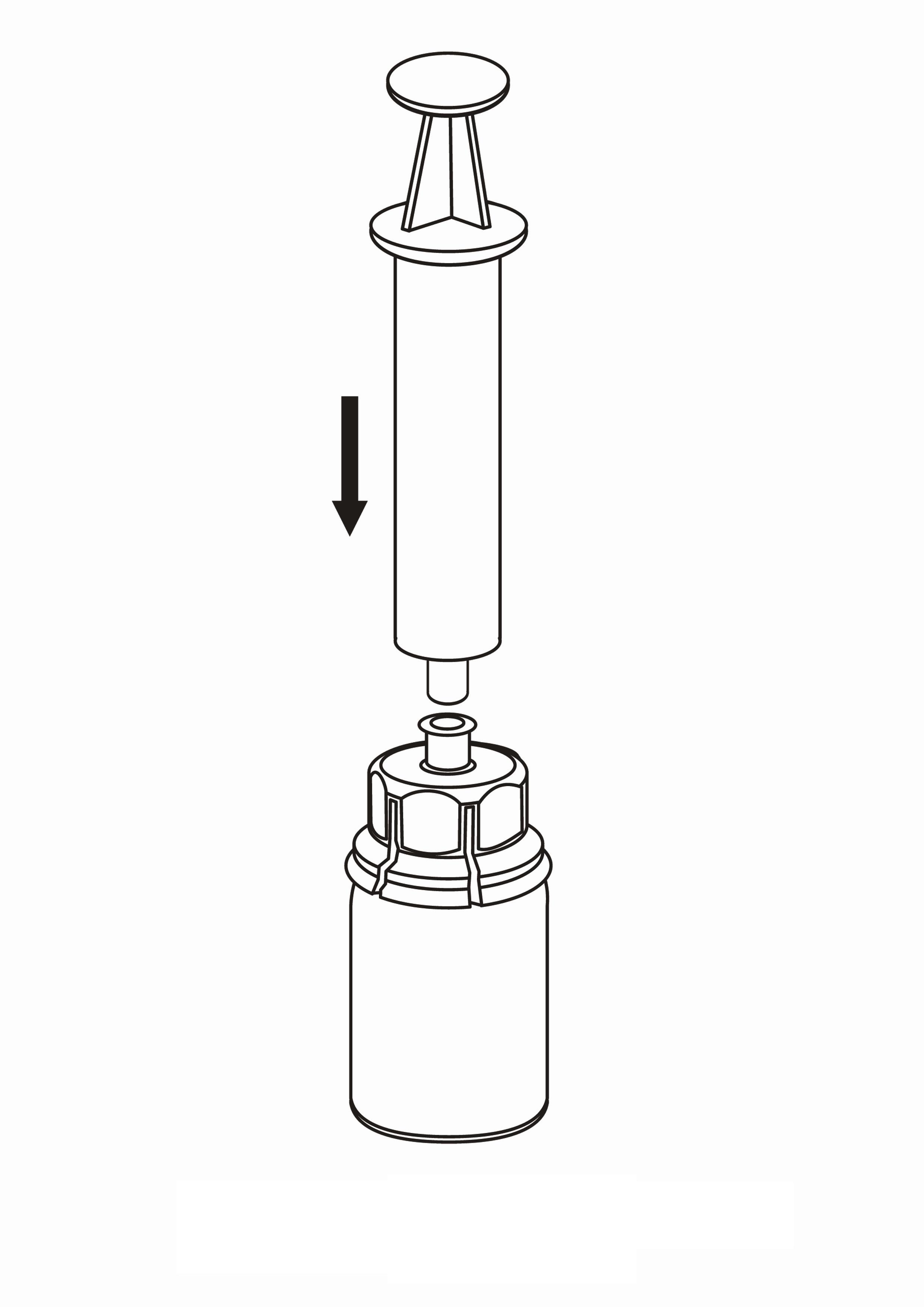 7
7Valmisteen vetäminen ruiskuun ja anto
| 8. Kun ruiskun mäntä on sisään painettuna, käännä laite ja injektiopullo ylösalaisin ja vedä liuos ruiskuun vetämällä mäntää hitaasti ulospäin. |
| 9. Kun liuos on nyt siirretty ruiskuun, ota tukeva ote ruiskun kammiosta (pitäen ruiskun mäntää samalla alaspäin) ja irrota läpinäkyvä Mix2Vial-sovitin ruiskusta kiertämällä vastapäivään. |
Varovaisuutta on noudatettava, jotta valmistetta sisältävään ruiskuun ei pääse verta, koska vaarana on, että veri hyytyy ruiskussa ja fibriinihyytymät päätyvät potilaan verenkiertoon.
Käyttövalmista Idelvion-valmistetta ei saa laimentaa.
Käyttövalmis liuos annetaan hitaana injektiona laskimoon. Antonopeus määritetään potilaan hyvinvoinnin mukaan; enimmäisnopeus on 5 ml/min.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
IDELVION injektiokuiva-aine ja liuotin, liuosta varten
250 IU 1 pakkaus
500 IU 1 pakkaus
1000 IU 1 pakkaus
2000 IU 1 pakkaus
- Ei korvausta.
ATC-koodi
B02BD04
Valmisteyhteenvedon muuttamispäivämäärä
12.09.2023
Yhteystiedot
 CSL BEHRING AB
CSL BEHRING AB Box 712
182 17 Danderyd
Ruotsi
+46 (0) 8 544 966 70
www.cslbehring.fi
info@cslbehring.se