
KIMMTRAK infuusiokonsentraatti, liuosta varten 100 mikrog/0,5 ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
KIMMTRAK Potilaan opas v.1
Terveydenhuollon ammattilainen
KIMMTRAK Hoito-opas terveydenhuollon ammattilaisille
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi 0,5 millilitran injektiopullo sisältää 100 mikrogrammaa tebentafuspia, mikä vastaa ennen laimennusta pitoisuutta 200 mikrog/ml.
Tebentafuspi on fuusioproteiini, jota tuotetaan yhdistelmä-DNA-tekniikalla Escherichia coli -soluissa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
KIMMTRAK on tarkoitettu annettavaksi monoterapiana ihmisen leukosyyttiantigeeni (HLA)-A*02:01 -positiivisille aikuispotilaille, joilla on leikkaukseen soveltumaton tai metastaattinen uvean melanooma.
Ehto
Valmistetta saavat määrätä ja antaa vain sellaiset lääkärit ja terveydenhuollon ammattilaiset, jotka ovat perehtyneet antineoplastisten valmisteiden käyttöön. Valmistetta saa antaa vain sairaalassa, jossa on asianmukaiset elvytyslaitteet.
Annostus ja antotapa
KIMMTRAK on annettava sellaisen lääkärin ohjauksessa ja valvonnassa, jolla on kokemusta syöpälääkkeiden käytöstä ja joka on varautunut sytokiinioireyhtymän hoitamiseen ympäristössä, jossa on välittömästi saatavilla täydet elvytysvälineet. Sairaalahoitoa suositellaan ainakin kolmen ensimmäisen KIMMTRAK-infuusion ajaksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
KIMMTRAK-hoitoa saavilla potilailla on oltava HLA-A*02:01-genotyyppi, joka on todettu validoidulla HLA-genotyyppimäärityksellä.
Annostus
Suositeltu KIMMTRAK-annos on 20 mikrogrammaa päivänä 1, 30 mikrogrammaa päivänä 8, 68 mikrogrammaa päivänä 15 ja tämän jälkeen 68 mikrogrammaa kerran viikossa (ks. kohta Käyttö- ja käsittelyohjeet). KIMMTRAK-hoitoa tulee jatkaa niin kauan, kuin potilas saa siitä kliinistä hyötyä, eikä esiinny toksisuutta, joka ei ole hyväksyttävissä (ks. kohta Farmakodynamiikka).
Esilääkitys
Sytokiinioireyhtymään liittyvän hypotension vaaran minimoimiseksi ennen KIMMTRAK-infuusion antamista potilaalle on annettava nesteitä laskimoon kliinisen arvion ja potilaan nestetilan mukaan.
Jos potilaalla on aiemmin esiintynyt lisämunuaisen vajaatoimintaa systeemisten kortikosteroidien yhteydessä, on harkittava kortikosteroidiannoksen muuttamista hypotensioriskin hallitsemiseksi.
Annoksen muuttaminen
KIMMTRAK-annosmuutoksia ei suositella. KIMMTRAK-hoito on keskeytettävä tai lopetettava taulukossa 1 ja taulukossa 2 kuvattujen haittavaikutusten hoitamista varten.
Mikäli sytokiinioireyhtymää epäillään, oireet on tunnistettava ja hoidettava viipymättä taulukon 1 suositusten mukaisesti. Ks. taulukosta 2 akuuttien ihoreaktioiden hoito-ohjeet.
Taulukko 1: Sytokiinioireyhtymän luokittelu ja hoito-ohjeet
| Sytokiinioireyhtymän luokka* | Hoito |
Luokka 1 Lämpötila ≥ 38 °C Ei hypotensiota tai hypoksiaa |
|
Luokka 2 Lämpötila ≥ 38 °C Nesteisiin reagoiva hypotensio, joka ei edellytä vasopressorihoitoa. Happivaatimus sisältää matalan virtauksen nenäkanyylin (hapen anto ≤ 6 l/min) tai ohipuhalluksen. |
|
Luokka 3 Lämpötila ≥ 38 °C Edellyttää vasopressoria joko vasopressiinin kanssa tai ilman sitä. Edellyttää korkean virtauksen nenäkanyyliä (hapen anto > 6 l/min), kasvomaskia tai varaajapussillista maskia tai venturimaskia. |
|
Luokka 4 Lämpötila ≥ 38 °C Edellyttää useita vasopressoreita (pois lukien vasopressiini) Edellyttää ylipainetta (esim. CPAP, BiPAP, intubaatio ja mekaaninen ventilaatio). |
|
* Perustuu American Society for Transplantation and Cellular Therapyn (ASTCT) yhdistettyyn luokitukseen sytokiinioireyhtymän kriteereistä (Lee et.al 2019).
Taulukko 2: Akuuttien ihoreaktioiden suositeltu hoito ja annosmuutokset
| Haittavaikutukset | Vaikeusa | Hoito |
| Akuutit ihoreaktiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Luokka 2 |
|
| Luokka 3 |
| |
| Luokka 4 |
|
a Perustuu National Cancer Instituten haittavaikutusten yleisiin terminologiakriteereihin (CTCAE), versioon 4.03 (NCI CTCAEv4.03).
Erityisryhmät
Pediatriset potilaat
KIMMTRAK-valmisteen turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Iäkkäät
Iäkkäiden potilaiden (≥ 65-vuotiaat) annoksen muuttaminen ei ole tarpeen.
Munuaisten vajaatoiminta
Turvallisuutta ja tehoa koskevien analyysien perusteella lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei ole tarpeen muuttaa. Annossuosituksia ei voida antaa potilaille, joilla on vaikea munuaisten vajaatoiminta, koska farmakokineettisiä tietoja ei ole; siksi vaikeaa munuaisten vajaatoimintaa sairastavia potilaita on hoidettava varoen ja seurattava huolellisesti (ks. kohta Farmakokinetiikka).
Potilaat, joilla on sydänsairaus
KIMMTRAK-valmistetta ei ole tutkittu potilailla, joilla on ollut merkittävä sydänsairaus. Potilaita, joilla on sydänsairaus, pidentynyt QT-aika tai sydämen vajaatoiminnan riskitekijöitä, on seurattava huolellisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Antotapa
KIMMTRAK annetaan laskimoon. Suositeltu infuusion kesto on 15–20 minuuttia.
KIMMTRAK on laimennettava 9 mg/ml (0,9 %) natriumkloridi-injektionesteellä, joka sisältää ihmisen albumiinia laskimoinfuusiota varten. Jokainen KIMMTRAK-injektiopullo on tarkoitettu käytettäväksi vain yhtenä annoksena. KIMMTRAK-injektiopulloa ei saa ravistaa.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ja antamisesta.
Ensimmäiset kolme hoitoannosta
Ensimmäiset kolme KIMMTRAK-annosta on annettava sairaalassa, jossa pystytään seuraamaan potilasta sytokiinioireyhtymän merkkien ja oireiden varalta yön yli ja vähintään 16 tunnin ajan. Elintoimintoja on seurattava ennen annoksen antamista ja vähintään 4 tunnin välein, kunnes oireet häviävät. Seurantaa on tehtävä useammin tai sairaalahoitoa pidennettävä, jos se on kliinisesti tarpeen.
Mikäli potilaalla ilmenee luokan 3 tai 4 hypotensiota jonkin ensimmäisen kolmen KIMMTRAK-infuusion aikana, potilaita on seurattava tunnin välein vähintään 4 tunnin ajan avohoidossa seuraavien kolmen infuusion yhteydessä.
Seuraavat hoitoannokset
Kun potilas sietää 68 mikrogramman annostason (ts. lääketieteellistä hoitoa edellyttävää luokan ≥ 2 hypotensiota ei ilmene), seuraavat annokset voidaan antaa asianmukaisella poliklinikalla. Potilaita on seurattava vähintään 60 minuutin ajan kunkin infuusion jälkeen. Kun potilas on saanut poliklinikalla KIMMTRAK-infuusioita vähintään kolme kuukautta eikä hänellä ole ollut yli kaksi viikkoa kestäneitä hoidon keskeytyksiä, poliklinikkaseuranta infuusion jälkeen voidaan lyhentää seuraavien annosten yhteydessä vähimmillään 30 minuuttiin.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Sytokiinioireyhtymä
Useimmilla potilailla ilmeni sytokiinioireyhtymä tebentafuspi-infuusion jälkeen. Sytokiinioireyhtymän diagnoosi perustui useimmiten kuumeeseen, jota seurasi hypotensio ja harvemmin hypoksia. Muita sytokiinioireyhtymän yhteydessä yleisesti havaittuja oireita olivat vilunväristykset, pahoinvointi, oksentelu, väsymys ja päänsärky. Sytokiinioireyhtymään on liittynyt elinten toimintahäiriöitä, kuten maksan, munuaisten, haiman, sydämen ja keuhkojen toimintahäiriöitä.
Suurimmassa osassa tapauksista sytokiinioireyhtymä alkoi infuusion antopäivänä, ja paranemisajan mediaani oli 2 vuorokautta. Kuumetta havaittiin lähes kaikissa sytokiinioireyhtymätapauksissa ja näillä potilailla ruumiinlämmön nousua esiintyi yleensä ensimmäisten 8 tunnin aikana tebentafuspi-infuusion jälkeen. Sytokiinioireyhtymä johti harvoin (1,2 %) hoidon lopettamiseen.
Potilaita on seurattava sytokiinioireyhtymän merkkien tai oireiden varalta vähintään 16 tuntia ensimmäisten kolmen tebentafuspi-infuusion jälkeen sairaalaympäristössä, jossa on välittömästi saatavilla lääkevalmisteita ja elvytyslaitteita sytokiinioireyhtymän hoitoon. Mikäli sytokiinioireyhtymä todetaan, on aloitettava viipymättä tukihoito esimerkiksi kuumelääkkeillä, laskimoon annettavilla nesteillä, tosilitsumabilla tai kortikosteroideilla, jotta vältetään sen paheneminen vaikeaksi tai hengenvaaralliseksi. Seurantaa on jatkettava paranemiseen asti.
Seuraavien annosten yhteydessä potilaita on seurattava tarkasti hoidon jälkeen, jotta sytokiinioireyhtymän merkit ja oireet voidaan tunnistaa varhaisessa vaiheessa (ks. kohta Annostus ja antotapa, Antotapa). Potilailla, joilla on muita samanaikaisia sairauksia, esim. kardiovaskulaarisia sairauksia, voi olla suurentunut riski saada sytokiinioireyhtymään liittyviä jälkitauteja.
Tebentafuspihoitoa ei ole tutkittu potilailla, joilla on kliinisesti merkittävä sydänsairaus (ks. kohta Farmakodynamiikka). Sytokiinioireyhtymän jatkumisesta ja vaikeudesta riippuen tebentafuspihoito on keskeytettävä tai lopetettava (ks. kohta Annostus ja antotapa, taulukko 1).
Akuutit ihoreaktiot
Tebentafuspi-infuusion yhteydessä on raportoitu akuutteja ihoreaktioita, jotka saattavat perustua valmisteen vaikutustapaan ja gp100:n ekspressioon ihon normaaleissa melanosyyteissä. Akuutit ihoreaktiot sisälsivät pääasiassa ihottumaa, kutinaa, punoitusta ja ihon turvotusta (ks. kohta Haittavaikutukset).
Akuutteja ihoreaktioita ilmeni tyypillisesti kunkin ensimmäisen kolmen tebentafuspi-infuusion jälkeen, ja niiden vaikeus ja esiintymistiheys alenivat ajan myötä. Suurin osa oireista parani ilman systeemistä kortikosteroidia tai pitkäkestoisia jälkitauteja.
Akuutteja ihoreaktioita voidaan hoitaa antihistamiinilla ja topikaalisilla kortikosteroideilla. Jos oireet ovat jatkuvia tai vaikeita, systeemisiä steroideja on harkittava. Ihoreaktioiden merkkien ja oireiden hoito voi edellyttää seuraavien tebentafuspihoitojen väliaikaista lykkäämistä (ks. kohta Annostus ja antotapa, taulukko 2).
Sydänsairaudet
Sydäntapahtumia, kuten sinustakykardiaa ja rytmihäiriöitä, on havaittu potilailla, jotka ovat saaneet tebentafuspihoitoa (ks. kohta Haittavaikutukset). Potilailla, joilla on aiemmin todettu sydän- ja verisuonisairaus, voi olla suurentunut sytokiinioireyhtymään liittyvien jälkitautien vaara ja heitä on seurattava huolellisesti. Kaikki potilaat, joiden merkit ja oireet viittaavat sydäntapahtumiin, on arvioitava ja hoidettava viipymättä. Lisäksi asianmukaista hoitoa on annettava taustalla olevaan oireita pahentavaan sytokiinioireyhtymään.
QT-ajan pitenemistä on raportoitu tebentafuspihoidon jälkeen (ks. kohta Haittavaikutukset). Tebentafuspihoitoa on annettava varoen potilaille, joilla on ollut tai jotka ovat alttiita QT-ajan pitenemiselle, ja potilaille, jotka käyttävät tunnetusti QT-aikaa pidentäviä lääkevalmisteita.
EKG on otettava kaikista potilaista ennen tebentafuspihoitoa ja ensimmäisten kolmen hoitoviikon aikana ja tämän jälkeen kliinisen tarpeen mukaan. Jos QTcF on yli 500 ms tai suurenee ≥ 60 ms lähtötasoarvosta, tebentafuspihoito on keskeytettävä ja taustalla olevia pahentavia tekijöitä, kuten elektrolyyttipoikkeamia, on hoidettava. Tebentafuspihoitoa on jatkettava, kun QTcF-aika palautuu alle 500 ms:iin tai on < 60 ms lähtötasoarvosta. Sydäntapahtuman ja siihen liittyvän sytokiinioireyhtymän jatkumisesta ja vaikeudesta riippuen tebentafuspihoito on keskeytettävä tai lopetettava (ks. kohta Annostus ja antotapa, taulukko 1).
Ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja 1 viikon ajan tebentafuspihoidon viimeisen annoksen jälkeen (ks. kohta Raskaus ja imetys).
Apuaineet
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per ml eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Virallisia lääkkeiden yhteisvaikutustutkimuksia ei ole tehty tebentafuspilla.
Tebentafuspihoidon aloitus aiheuttaa ohimenevää sytokiinien vapautumista, mikä voi suppressoida CYP450-entsyymejä. Suurin lääkkeiden välisen yhteisvaikutuksen vaara on ensimmäisten 24 tunnin aikana ensimmäisten kolmen tebentafuspiannoksen jälkeen potilailla, jotka saavat samanaikaisesti CYP450:n substraatteja, ja näistä erityisesti sellaisia, joiden terapeuttinen leveys on kapea. Näitä potilaita on seurattava toksisuuden (esim. varfariini) tai lääkepitoisuuksien (esim. siklosporiini) varalta. Samanaikaisen lääkityksen annosta on muutettava tarpeen mukaan.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä tebentafuspihoidon aikana ja 1 viikon ajan viimeisen tebentafuspiannoksen jälkeen.
Raskaus
Tebentafuspin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Tebentafuspilla ei ole tehty lisääntymistä koskevia eläinkokeita (ks. kohta Prekliiniset tiedot turvallisuudesta).
KIMMTRAK-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisy. Naiset, jotka voivat tulla raskaaksi, on tarkastettava raskauden varalta ennen tebentafuspihoidon aloittamista.
Imetys
Tebentafuspin/metaboliittien erittymisestä ihmisen rintamaitoon ei ole riittävästi tietoa. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Rintaruokinta on lopetettava tebentafuspihoidon ajaksi.
Hedelmällisyys
Tebentafuspia koskevia hedelmällisyystutkimuksia ei ole tehty (ks. kohta Prekliiniset tiedot turvallisuudesta). Tebentafuspin vaikutusta miesten ja naisten hedelmällisyyteen ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tebentafuspilla ei ole haitallista vaikutusta tai on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Yleisimmät KIMMTRAK-hoitoon liittyvät haittavaikutukset olivat sytokiinioireyhtymä (88 %), ihottuma (85 %), kuume (79 %), kutina (72 %), väsymys (66 %), pahoinvointi (56 %), vilunväristykset (55 %), vatsakipu (49 %), turvotus (49 %), hypo-/hyperpigmentaatio (48 %), hypotensio (43 %), ihon kuivuminen (35 %), päänsärky (32 %) ja oksentelu (34 %).
Haittavaikutukset johtivat pysyvään KIMMTRAK-hoidon lopettamiseen 4 %:lla potilaista. Yleisin KIMMTRAK-hoidon lopettamiseen johtanut haittavaikutus oli sytokiinioireyhtymä.
Haittavaikutuksia, jotka johtivat vähintään yhteen annon keskeyttämiseen, ilmeni 26 %:lla KIMMTRAK-hoitoa (viikoittain annettuna) saaneista potilaista, ja väliin jääneiden annosten mediaani oli yksi. Haittavaikutuksia, jotka johtivat annon keskeyttämiseen ≥ 2 %:lla potilaista, olivat väsymys (3 %, luokka 1–3), kuume (2,7 %, luokka 1–3), alaniiniaminotransferaasiarvon nousu (2,4 %, luokka 1–4), aspartaattiaminotransferaasiarvon nousu (2,4 %, luokka 1–3), vatsakipu (2,1 %, luokka 1–3) ja lipaasiarvon nousu (2,1 %, luokka 1–3).
Haittavaikutuksia, jotka johtivat vähintään yhteen annoksen muokkaukseen, ilmeni 4,2 %:lla KIMMTRAK-hoitoa saaneista potilaista. Haittavaikutukset, jotka edellyttivät annoksen muuttamista ≥ 1 %:lla potilaista, olivat sytokiinioireyhtymä (1,9 %, luokka 1–3) ja hypotensio (1,1 %, luokka 2–4).
Haittavaikutustaulukko
Taulukossa 3 on yhteenveto haittavaikutuksista, joita ilmeni 378 metastaattista uvean melanoomaa sairastaneella potilaalla kahdessa kliinisessä tutkimuksessa (IMCgp100-102 ja IMCgp100-202). Potilaat saivat suositellun KIMMTRAK-annoksen 20 mikrogrammaa päivänä 1, 30 mikrogrammaa päivänä 8, 68 mikrogrammaa päivänä 15 ja sen jälkeen 68 mikrogrammaa kerran viikossa.
Haittavaikutuksen esiintymistiheys on ilmoitettu MedDRA-elinjärjestelmäluokan mukaisesti. Haittavaikutusten esiintymistiheydet määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen mukaan alenevassa järjestyksessä
Taulukko 3: KIMMTRAK-monoterapiaa saaneiden potilaiden haittavaikutukset
| Haittavaikutukset | |
| Infektiot | |
| Yleinen | Nasofaryngiitti |
| Immuunijärjestelmä | |
| Hyvin yleinen | Sytokiinioireyhtymä1 |
| Aineenvaihdunta ja ravitsemus | |
| Hyvin yleinen | Ruokahaluttomuus, hypomagnesemia, hyponatremia, hypokalsemia, hypokalemia |
| Melko harvinainen | Tuumorilyysioireyhtymä |
| Psyykkiset häiriöt | |
| Hyvin yleinen | Unettomuus |
| Yleinen | Ahdistus |
| Hermosto | |
| Hyvin yleinen | Päänsärky2, heitehuimaus, parestesia |
| Yleinen | Makuhäiriö |
| Sydän | |
| Hyvin yleinen | Takykardia2 |
| Yleinen | Rytmihäiriö2, eteisvärinä2 |
| Melko harvinainen | Angina pectoris2, sydämen vajaatoiminta2 |
| Verisuonisto | |
| Hyvin yleinen | Hypotensio2, punoitus, hypertensio |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | Yskä, hengenahdistus |
| Yleinen | Orofaryngeaalinen kipu, hypoksia2 |
| Ruoansulatuselimistö | |
| Hyvin yleinen | Pahoinvointi2, oksentelu2, ripuli, vatsakipu, ummetus, dyspepsia |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | Ihottuma, kutina, ihon kuivuminen, hypo-/hyperpigmentaatio4, punoitus |
| Yleinen | Hiustenlähtö, yöhikoilu |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleinen | Nivelkipu, selkäkipu, lihaskipu, raajakipu |
| Yleinen | Lihasspasmi |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | Kuume2, väsymys3, vilunväristykset2, ödeema5, influenssan kaltainen sairaus |
| Tutkimukset | |
| Hyvin yleinen | Aspartaattiaminotransferaasiarvon nousu, alaniiniaminotransferaasiarvon nousu, veren bilirubiiniarvon nousu, lipaasiarvon nousu, anemia, lymfosyyttimäärän lasku, veren fosfaattipitoisuuden lasku, veren kreatiniiniarvon nousu |
| Yleinen | Amylaasiarvon nousu, gammaglutamyylitransferaasiarvon nousu, valkosolumäärän nousu, veren alkalisen fosfataasipitoisuuden suureneminen, veren glukoosiarvon nousu |
| Melko harvinainen | QT-ajan piteneminen EKG:ssä |
1 Sytokiinioireyhtymän toteaminen perustui ASTCT:n yhdistettyyn luokitukseen sytokiinioireyhtymän kriteereistä (Lee et.al 2019). Todettu sytokiinioireyhtymä tutkijan raportoiman sytokiinioireyhtymän sijaan.
2 Osa tapahtumista saattaa liittyä sytokiinioireyhtymään, tai ne voivat olla erillisiä raportoituja tapahtumia.
3 Sisältää väsymyksen ja astenian.
4 Sisältää hankinnaisen hiusten harmaantumisen, pisamat, silmäripsien värimuutokset, silmäripsien hypopigmentaation, hiusten värin muutokset, pigmenttiläiskät, pigmentaatio-oireyhtymän, verkkokalvon depigmentaation, ihon depigmentaation, ihon värimuutokset, ihon hyperpigmentaation, ihon hypopigmentaation, vanhuuden pisamat, vitiligon.
5 Sisältää silmäedeeman, silmän turpoamisen, silmäluomien ödeeman, periorbitaalisen turvotuksen, periorbitaalisen ödeeman, silmäluomien turpoamisen, nieluturvotuksen, huuliedeeman, huulen turpoamisen, kasvojen ödeeman, yleistyneen ödeeman, paikallisen ödeeman, ödeeman, perifeerisen ödeeman, perifeerisen turvotuksen, turvotuksen, karvojen turpoamisen.
Kuvaus valikoiduista haittavaikutuksista
Sytokiinioireyhtymä
Kliinisessä tutkimuksessa IMCgp100-202 sytokiinioireyhtymää (todettuna ASTCT:n yhdistetyn luokituksen 2019 mukaan) ilmeni 89 %:lla KIMMTRAK-hoitoa saaneista potilaista. Sytokiinioireyhtymän kokonaisesiintymistiheys sisälsi 12 % luokan 1, 76 % luokan 2 ja 0,8 % luokan 3 tapauksia. Yleisimpiä sytokiinioireyhtymän yhteydessä havaittuja oireita olivat vilunväristykset, pahoinvointi, oksentelu, väsymys, hypotensio ja päänsärky. Sytokiinioireyhtymän yhteydessä mahdollisesti havaittavia luokan 3 tapahtumia ovat takykardia, hypoksia, angina pectoris, eteislepatus ja vasemman kammion toimintahäiriö.
Suurin osa (84 %) sytokiinioireyhtymäepisodeista alkoi infuusion antopäivänä. Mediaaniaika sytokiinioireyhtymän parantumiseen oli 2 vuorokautta.
Sytokiinioireyhtymä johti harvoin (1,2 %) hoidon lopettamiseen. Kaikki sytokiinioireyhtymän oireet olivat ohimeneviä ja niitä hoidettiin pääasiassa antamalla nesteitä laskimoon, kuumelääkkeillä tai yksittäisillä kortikosteroidiannoksilla. Kaksi potilasta (0,8 %) sai tosilitsumabia.
Lisätietoa sytokiinioireyhtymän kliinisestä hoidosta, ks. kohta Annostus ja antotapa, taulukko 1.
Akuutit ihoreaktiot
Tutkimuksessa IMCgp100-202 akuutteja ihoreaktioita esiintyi 91 %:lla KIMMTRAK-hoitoa saaneista potilaista. Näitä olivat minkä tahansa vaikeusasteen ihottuma (83 %), kutina (69 %), eryteema (25 %) ja ihon ödeema (27 %). Useimpien ihoreaktioiden luokka oli 1 (28 %) tai 2 (44 %), ja jotkin KIMMTRAK-hoitoa saaneista potilaista saivat luokan 3 (21 %) reaktioita. Ihottumapotilailla ilmeni yleisesti ihottumaa (55 %), makulopapulaarista ihottumaa (31 %) ja ihon kuoriutumista (21 %). Haittavaikutuksena luokan 3 ihottumaa raportoitiin 5 %:lla potilaista, ja niitä olivat ihottuma (2,4 %) ja makulopapulaarinen ihottuma (1,6 %).
Akuutteja ihoreaktioita ilmeni tyypillisesti ensimmäisten kolmen KIMMTRAK-infuusion jälkeen ja luokan ≥ 3 reaktioiden esiintymistiheys laski annoksen mukaan (annos 1: 17 %, annos 2: 10 %, annos 3: 8 %, annos 4: 3 %). Mediaaniaika akuuttien ihoreaktioiden alkamiseen oli 1 vuorokausi KIMMTRAK-hoitoa saaneilla potilailla ja mediaaniaika paranemiseen luokkaan ≤ 1 oli 6 vuorokautta.
Lisätietoa akuuttien ihoreaktioiden kliinisestä hoidosta, ks. kohta Annostus ja antotapa, taulukko 2.
Kohonneet maksaentsyymiarvot
Tutkimuksessa IMCgp100-202, jossa 95 %:lla potilaista oli aiempi maksametastaasi, ja 65 %:lla KIMMTRAK-hoitoa saaneista potilaista ALAT-/ASAT-arvo nousi luokkaan ≥ 1. Bilirubiiniarvojen nousua on raportoitu 27 %:lla potilaista, ja näihin liittyi ensisijaisesti maksametastaasin koon kasvu. Suurin osa luokan 3 tai 4 ALAT-/ASAT-arvojen nousuista ilmeni yleensä ensimmäisten kolmen KIMMTRAK-infuusion aikana. Suurin osa luokan 3 tai 4 ALAT-/ASAT-arvojen nousuista parani luokkaan ≤1 seitsemän vuorokauden sisällä.
Immunogeenisuus
Hoidon aikana ilmeneviä tebentafuspin lääkevasta-aineita havaittiin 33 %:lla tutkimukseen IMCgp100-102 ja 29 %:lla tutkimukseen IMCgp100-202 osallistuneista potilaista, jotka saivat mitä tahansa tebentafuspiannosta. Lääkevasta-aineiden muodostumisen mediaani alkamisaika oli 6–9 viikkoa tebentafuspihoidon aloittamisen jälkeen.
Näyttöä lääkevasta-aineiden vaikutuksesta tebentafuspin turvallisuuteen tai tehoon ei ollut, vaikkakin niitä potilaita, joille kehittyi suurempi tiitteri lääkevasta-aineita oli vain vähän, mikä estää tiukat johtopäätökset lääkevasta-aineiden kliinisestä vaikutuksesta.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tebentafuspin yliannostuksesta ei ole olemassa tietoa. Yliannostustapauksissa potilaita on seurattava tarkasti haittavaikutusten merkkien ja oireiden varalta, ja oireenmukainen hoito on aloitettava viipymättä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet, ATC- koodi: ei vielä määritetty
Vaikutusmekanismi
Tebentafuspi on bispesifinen fuusioproteiini, joka koostuu T-solureseptorista (TCR; kohdistuva domeeni) yhdistettynä CD3:een kohdistuvaan vasta-ainefragmenttiin (differentiaatioklusteri 3; efektoridomeeni). TCR-pää sitoutuu suurella affiniteetilla gp100-peptidiin, jota on ihmisen leukosyyttiantigeenin A*02:01 (HLA-A*02:01) uvean melanooman kasvainsolujen pinnalla, ja efektoridomeeni sitoutuu polyklonaalisen T-solun CD3-reseptoriin.
Immunologinen synapsi muodostuu, kun tebentafuspin kohdistuva TCR-domeeni sitoutuu uvean melanooman soluihin ja CD3-efektoridomeeni sitoutuu polyklonaalisiin T-soluihin. Tämä immunologinen synapsi aiheuttaa polyklonaalisten T-solujen uudelleensuuntautumisen ja aktivoitumisen niiden alkuperäisestä TCR-spesifisyydestä riippumatta. Tebentafuspiaktivoidut polyklonaaliset T-solut vapauttavat tulehdussytokiineja ja sytolyyttisiä proteiineja, mikä johtaa uvean melanooman kasvainsolujen suoraan lyysiin.
Farmakodynaamiset vaikutukset
Tebentafuspihoidon jälkeen havaittiin ohimenevää ja kliinisesti merkityksetöntä lymfosyyttimäärän laskua veressä. Lymfosyytit vähenivät ensimmäisten kolmen annoksen jälkeisinä päivinä ja palasivat lähtötasolle ennen seuraavia annoksia.
Tebentafuspihoidon jälkeen ohimenevää proinflammatoristen sytokiinien ja kemokiinien seerumitason kohoamista havaittiin näytteissä, jotka otettiin ensimmäisten kolmen annoksen jälkeen. Suurimmat pitoisuudet havaittiin 8–24 tuntia tebentafuspihoidon jälkeen, ja pitoisuudet palasivat lähtötasolle ennen seuraavia annoksia.
Kliininen teho ja turvallisuus
Tutkimus IMCgp100-202: Aiemmin hoitamaton metastaattinen uvean melanooma
Tutkimus IMCgp100-202 oli satunnaistettu, avoin monikeskustutkimus, johon otettiin HLA-A*02:01- positiivisia metastaattista uvean melanoomaa sairastavia potilaita, jotka eivät olleet saaneet systeemistä hoitoa. Potilaat eivät olleet voineet saada aiempaa systeemistä hoitoa tai paikallista (maksaan suunnattua) hoitoa metastaattiseen uvean melanoomaan, lukuun ottamatta aiempaa oligometastaattisen sairauden kirurgista resektiota. Potilaat suljettiin pois tutkimuksesta oireisen tai hoitamattoman aivometastaasin, oireisen kongestiivisen sydämen vajaatoiminnan, Friderician kaavalla korjatun QT-ajan (QTcF) > 470 ms tai synnynnäisen pitkän QT-ajan oireyhtymän, akuutin sydänlihasinfarktin tai alle 6 kuukautta aiemmin ilmenneen epästabiilin angina pectoriksen vuoksi.
Potilaat satunnaistettiin (2:1) saamaan tebentafuspia viikoittain laskimoinfuusiona suositellun annostusohjelman mukaan (ks. kohta Annostus ja antotapa) tai tutkijan päättämää hoitoa (pembrolitsumabi, ipilimumabi tai dakarbatsiini) kyseisten valmisteiden hyväksyttyjä annoksia sairauden etenemiseen tai ei- hyväksyttävissä olevan toksisuuden ilmaantumiseen asti.
Potilaat voivat saada tebentafuspia, pembrolitsumabia tai ipilimumabia sairauden etenemisen jälkeen, jos potilaat olivat kliinisesti vakaita, saivat kliinistä hyötyä hoidosta ja jos heillä ei ilmennyt merkkejä ei-hyväksyttävissä olevasta toksisuudesta tutkijan arvion perusteella. Hoidon keskeytys enintään kahdeksi peräkkäiseksi viikoksi oli sallittua. Satunnaistus ositettiin laktaattidehydrogenaasin (LDH) tason perusteella, mikä on tunnettu leikkaukseen soveltumattoman tai metastaattisen uvean melanooman ennustekijä.
Ensisijainen tehon vastemuuttuja oli elossaoloaika (OS) kaikilla potilailla, jotka satunnaistettiin tutkimukseen. Kasvaimia arvioitiin 12 viikon välein. Muita toissijaisia tehon vastemuuttujia olivat tutkijan arvioima etenemisvapaa elossaoloaika (PFS). Yhteensä 378 potilasta satunnaistettiin; 252 tebentafuspihoitoa saavaan ryhmään ja 126 tutkijan valitsemaan hoitoryhmään (pembrolitsumabi: 82 %; ipilimumabi: 12 %; tai dakarbatsiini: 6 %). Mediaani-ikä oli 64 vuotta (vaihteluväli 23–92 vuotta); 49,5 % potilaista oli ≥ 65-vuotiaita, 87 % oli valkoihoisia, 50 % oli naisia. Lähtötilanteen ECOG-toimintakykyluokka oli 0 (72 %) tai 1 (20,4 %) tai 2 (0,3 %), 36 %:lla oli kohonnut LDH-taso ja 95 %:lla oli maksametastaasi.
Tässä IMCgp100-202-tutkimuksessa 43 % potilaista sai tebentafuspihoitoa hoidon aikana todetun sairauden etenemisen jälkeen, eikä uusia turvallisuussignaaleja tunnistettu. Tebentafuspihoidon mediaanikesto sairauden etenemisen jälkeen oli 8 viikkoa. Kaikista tutkimuksen aikana annetuista tebentafuspi-infuusioista 21,5 % annettiin sairauden etenemisen jälkeen.
Ensisijaisen tehoa koskevan analyysin valmistumisen jälkeen potilaat tutkijan valitseman hoidon haarasta saivat siirtyä saamaan tebentafuspihoitoa. Seurannan mediaanikesto oli 22,4 kuukautta, jolloin päivitetty elossaoloaika (OS) suosi edelleen tebentafuspihaaraa (HR = 0,58; 95 %:n CI: 0,44; 0,77). Analyysihetkellä 16 potilasta oli siirtynyt saamaan tebentafuspihoitoa.
Tehoa koskevien tulosten yhteenveto on esitetty taulukossa 4 ja kuvassa 1. Kuva 1 kuvaa 3 vuoden seurannan analyysin tuloksia. Analyysin ajankohtana 16 potilasta oli siirtynyt kontrolliryhmästä saamaan tebentafuspihoitoa.
Taulukko 4: Tehoa koskevat tulokset tutkimuksessa IMCgp100-202
| Ensisijainen ja toissijaiset päätetapahtumat | KIMMTRAK (N = 252) | Tutkijan valitsema hoito (N = 126) |
| Elossaoloaika (OS) 1 | ||
| Kuolemien määrä | 87 (34,5 %) | 63 (50 %) |
| Mediaani (kuukausina; 95 %:n CI) | 21,7 (18,6; 28,6) | 16,0 (9,7; 18,4) |
| HR (95 %:n CI)2,4 | 0,51 (0,37; 0,71) | |
| Ositettu log-rank p-arvo2 | p = < 0,0001 | |
| Etenemisvapaa elossaoloaika (PFS)3,4 | ||
| Potilasmäärä (%), jolla tapahtuma | 198 (78,6 %) | 97 (77 %) |
| Mediaani (kuukausina; 95 %:n CI) | 3,3 (3,0; 5,0) | 2,9 (2,8; 3,0) |
| HR (95 %:n CI)4 | 0,73 (0,58; 0,94) | |
| Ositettu log-rank p-arvo2 | p = 0,0139 | |
| Objektiivinen vasteosuus (ORR)6 | ||
| n (%) | 26 (10,3) | 6 (4,8) |
| 95 %:n CI | 6,9; 14,8 | 1,8; 10,1 |
| Täydellinen vaste (CR) | 1 (0,4) | 0 |
| Osittainen vaste (PR) | 25 (9,9) | 6 (4,8) |
| Vakaa sairaus (SD)5 | 52 (20,6) | 16 (12,7) |
| Vasteen mediaanikesto | ||
| Kuukausia (95 %:n CI) | 9,9 (5,6; 22,1) | 9,7 (2,7; --) |
CI = luottamusväli, HR = riskisuhde
1 Ennalta määritetyssä välianalyysissä havaittiin 150 elossaoloaikaan liittyvää tapahtumaa ja tehon julistamisen p-arvoraja (0,006) määritettiin Lan-Demetsin alfakorjausfunktiolla O’Brien Fleming -tyyppisellä rajalla.
2 Kaksipuolinen p-arvo perustui LDH:lla ositettuun log rank -testiin.
3 Tutkijan arvioimana RECIST v1.1 -kriteerien mukaisesti.
4 Riskisuhde on suhteellisesta riskimallista, joka on ositettu LDH-tason perusteella.
5 Perustuu ≥ 24 viikkoon.
6 Päivitetty sen perusteella, että kaikilla potilailla oli mahdollisuus ainakin kolmeen radiologiseen arvioon
Kuva 1: Kaplan-Meier-käyrät elossaoloajasta tutkimuksessa IMCgp100-202 (3-vuotisen seurannan analyysi) – ITT-populaatio
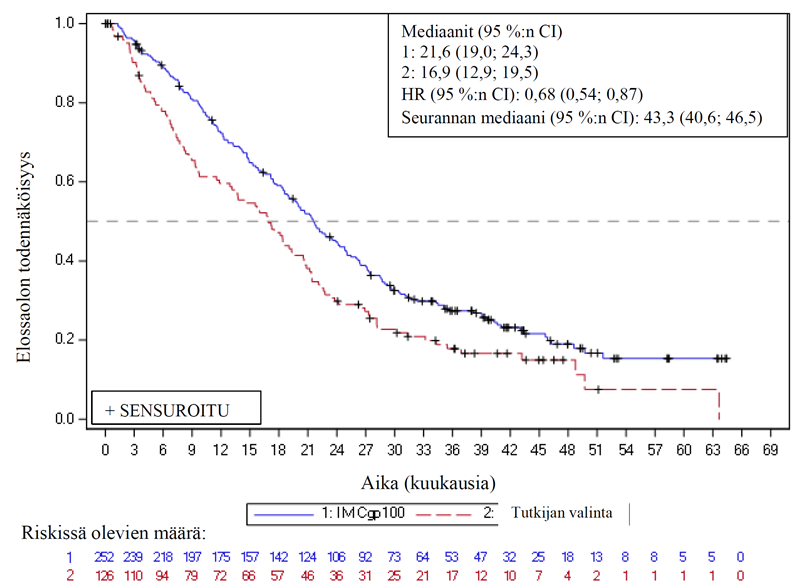
CI = luottamusväli, HR = riskisuhde, IMCgp100 = tebentafuspi, ITT = hoitoaie
3 vuoden seurannan jälkeen tebentafuspihoidon hyöty elossaoloaikana mitattuna on edelleen huomattava verrattuna tutkijan valintaan.
Tutkimus IMCgp100-102: Aiemmin hoidettu metastaattinen uvean melanooma
Tutkimus IMCgp100-102 oli avoin faasin 2 monikeskustutkimus, johon osallistui 127 potilasta. Potilaita hoidettiin kohdassa Annostus ja antotapa suositellulla annostusohjelmalla. Potilaiden täytyi olla HLA-A*02:01- positiivisia. Potilaat olivat soveltuvia tutkimukseen, jos heidän sairautensa oli edennyt ainakin yhden tai useamman joko maksaan suunnatun tai systeemisen hoitolinjan , kuten metastaattisessa sairaudessa immuuniaktivaation vapauttajahoidon, jälkeen. Potilaat suljettiin pois tutkimuksesta kliinisesti merkittävän sydänsairauden, tai oireellisen tai hoitamattoman aivometastaasin vuoksi.
Merkittäviä tehoa koskevia vastemuuttujia olivat vahvistettu ORR erillisen keskitetyn arviointilautakunnan arvioimana Response Evaluation Criteria in Solid Tumours (RECIST) v1.1 -kriteerien mukaan. Toissijaisia tehon koskevia vastemuuttujia olivat PFS, DCR, DOR ja OS.
Mediaani-ikä oli 61 vuotta, 50 % oli naisia, 99 % oli valkoihoisia, ECOG-toimintakykyluokka oli 0 (70 %) tai 1 (30 %) ja 96 %:lla potilaista oli maksametastaasi. Aiemmat hoidot sisälsivät immunoterapiaa (73 % potilaista), kuten immuuniaktivaation vapauttajaa (PD-1/PD-L1; 65 %; CTLA- 4; 31 %) ja maksaan kohdistuvaa hoitoa 45%. Yhteenveto tutkimuksen IMCgp100-102 tehoa koskevista tuloksista on esitetty taulukossa 5.
Taulukko 5: Tehoa koskevat tulokset tutkimuksessa IMCgp100-102
| Ensisijainen ja toissijaiset päätetapahtumat | KIMMTRAK (N = 127) |
| Vahvistettu objektiivinen vasteosuus | 6 (4,7 %) |
| (95 %:n CI) | (1,8 %; 10 %) |
| Täydellinen vaste (CR) | 0 |
| Osittainen vaste (PR) | 6 (4,7 %) |
| Vakaa sairaus (SD)2 | 23 (18,1 %) |
| Vasteen mediaanikesto | |
| Kuukausia (95 %:n CI) | 8,7 (5,6; 24,5) |
1 Erillisen keskustarkastelulautakunnan arvioimana RECIST v1.1 -kriteerien mukaisesti.
2 Perustuu ≥ 24 viikkoon.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset KIMMTRAK-valmisteen käytöstä okulaarisen melanooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Tebentafuspin farmakokinetiikka vaikuttaa lineaariselta ja annosvasteiselta annosvälillä 20–68 mikrog. Metastaattista uvean melanoomaa sairastavien potilaiden viikoittaisen laskimoinfuusion jälkeen plasman huippupitoisuus (Cmax), 4,2–13,7 ng/ml, saavutettiin välittömästi infuusion lopussa (T = 0,5 tuntia). Kertymistä ei havaittu viikoittaisessa annostuksessa tavoitelluilla terapeuttisilla annoksilla.
Jakautuminen
Tebentafuspi ei jakautunut merkittävästi, ja sen jakautumistilavuus oli verrattavissa veren määrään (5,25 l).
Biotransformaatio
Tebentafuspin metaboliareittiä ei ole kuvattu. Kuten muut proteiinivalmisteet, tebentafuspin odotetaan hajoavan pieniksi peptideiksi ja aminohapoiksi katabolisia reittejä.
Eliminaatio
Tebentafuspin erittymistä ei ole täysin kuvattu. Sen glomerulusten suodatuskoon kynnysarvon lähellä olevan molekyylikoon perusteella pieniä tebentafuspimääriä voi erittyä virtsaan.
Metastaattista uvean melanoomaa sairastavien potilaiden tebentafuspi-infuusion jälkeen arvioitu systeeminen puhdistuma oli 4,29 l/vrk, ja terminaalinen puoliintumisaika 6–8 tuntia.
Erityisryhmät
Populaatiofarmakokineettinen analyysi osoitti, että painolla (43–163 kg), sukupuolella, rodulla tai iällä (23–91 vuotta) ei ollut merkittävää vaikutusta tebentafuspin puhdistumaan.
Munuaisten vajaatoiminta
Tebentafuspilla ei ole tehty virallisia farmakokineettisiä tutkimuksia potilailla, joilla on munuaisten vajaatoiminta.
Vaikutusta turvallisuus- tai tehoparametreihin ei havaittu potilailla, joilla oli lievä (kreatiniinipuhdistuma [CrCL] 60–89 ml/min) tai keskivaikea (CrCL 30–59 ml/min) munuaisten vajaatoiminta, ja annoksen muuttamista ei suositella. Keskivaikeaa munuaisten vajaatoimintaa sairastaneista potilaista (< 5 %) on niukasti tietoa ja vaikeaa munuaisten vajaatoimintaa (CrCL alle 30 ml/min) sairastavista potilaista tietoja ei ole.
Maksan vajaatoiminta
Tebentafuspilla ei ole tehty virallisia farmakokineettisiä tutkimuksia potilailla, joilla on maksan vajaatoiminta. Populaatiofarmakokineettisten analyysien mukaan lähtötilanteessa todetut tai hoidon aikaiset ALAT-/ASAT-arvojen nousut eivät vaikuttaneet tebentafuspin farmakokinetiikkaan. Annoksen muuttamista ALAT-/ASAT-tasojen perusteella ei suositella.
Prekliiniset tiedot turvallisuudesta
Tebentafuspi on ihmiselle spesifi proteiini, eikä ole olemassa relevanttia eläinlajia, jolla tebentafuspin ei-kliinistä toksikologiaa voitaisiin testata.
Tebentafuspilla ei ole tehty karsinogeenisuus-, genotoksisuus- tai kehitys- ja lisääntymistoksisuustutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Sitruunahappomonohydraatti (E330)
Dinatriumvetyfosfaatti (E339)
Mannitoli (E421)
Trehaloosi
Polysorbaatti 20 (E432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Avaamisen jälkeen
Mikrobiologiselta kannalta avattu lääkevalmiste on laimennettava ja annettava infuusiona välittömästi.
Infuusionesteen, liuoksen valmistamisen jälkeen
Kemiallinen ja fysikaalinen käytönaikainen säilyvyys on osoitettu olevan 24 tuntia 2–8 °C:ssa.
Mikrobiologiselta kannalta valmiste on käytettävä välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikaiset säilytysajat ja -olosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä ja kuljeta jäähdytettynä (2–8 °C).
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KIMMTRAK infuusiokonsentraatti, liuosta varten
100 mikrog/0,5 ml (L:ei) 0,5 ml (15235,24 €)
PF-selosteen tieto
Tyypin I lasinen injektiopullo, jossa on bromobutyylikumitulppa ja alumiininen/muovinen repäisykorkki, sisältää 0,5 ml konsentraattia.
Pakkauskoko 1 injektiopullo.
Valmisteen kuvaus:
Kirkas, väritön tai hieman kellertävä liuos yhden annoksen injektiopullossa.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Yleiset varotoimet
Terveydenhuollon ammattilaisen on valmisteltava infuusioneste, käyttämällä asianmukaista aseptista tekniikkaa koko lääkevalmisteen käsittelyn ajan.
Aseptista tekniikkaa on käytettävä annostusliuosten laimennuksen ja valmistelun aikana.
Suljetun järjestelmän siirtolaitteita ei saa käyttää KIMMTRAK-infuusionesteen annoksen valmisteluun.
Parenteraaliset lääkevalmisteet ja infuusiopussit on tarkastettava silmämääräisesti hiukkasten ja värimuutosten varalta ennen antamista, mikäli liuos ja säiliö sen mahdollistavat.
Käyttökuntoon saattaminen
KIMMTRAK on laimennettava ennen sen antamista laskimoon.
Tarkista, että seuraavat välineet ovat saatavilla ennen KIMMTRAK-valmisteen valmistelemista antamista varten:
- 1 ml:n steriilit ruiskut, jossa on kahden desimaalin tarkkuudella oleva asteikko.
- Steriilit neulat.
- Ihmisen albumiini; käytä pitoisuutta paikallisen saatavuuden mukaan. Paikallisia pitoisuuksia ovat mm. 4 % (40 g/l), 5 % (50 g/l), 20 % (200 g/l), 25 % (250 g/l).
-
100 ml:n infuusiopussi, jossa on 9 mg/ml (0,9 %) natriumkloridi-injektioneste, liuosta:
- Infuusiopussin on oltava valmistettu polyolefiineista (PO) (kuten polyeteenistä [PE] ja polypropyleenistä [PP]) tai polyvinyylikloridista (PVC).
- Steriili, pyrogeeniton vähän proteiinia sitova 0,2 mikronin linjasuodattimella varustettu infuusiosarja lopullisen infuusiopussin sisällön antamiseen.
Laimentaminen ja antaminen
Lopullisen KIMMTRAK-annoksen valmistelu edellyttää kaksivaiheista prosessia:
Vaihe 1: Valmistele infuusiopussi
Valmistele infuusiopussi aseptista tekniikkaa käyttämällä seuraavasti:
a. Käytä 1 ml:n ruiskua ja steriiliä neulaa ja ota laskettu määrä ihmisen albumiinia ruiskuun (ks. taulukko 6 alla), ja lisää se 100 ml:n infuusiopussiin, joka sisältää 9 mg/ml (0,9 %) natriumkloridi-injektioneste liuosta, jotta saat ihmisen albumiinin lopulliseksi pitoisuudeksi 225– 275 mikrog/ml.
Taulukko 6: Esimerkkejä ihmisen albumiinipitoisuudesta ja hyväksyttävät ottomäärät
Ihmisen albumiinin pitoisuus | Hyväksyttävä määrä lisättäväksi 100 ml:n infuusiopussiin, jotta ihmisen albumiinin pitoisuus on välillä 225–275 mikrog/ ml |
4 % (40 g/l) | 0,63 ml (0,57–0,69 ml) |
5 % (50 g/l) | 0,50 ml (0,45–0,55 ml) |
20 % (200 g/l) | 0,13 ml (0,12–0,14 ml) |
25 % (250 g/l) | 0,10 ml (0,09–0,11 ml) |
b. Sekoita laimennettu liuos varovasti seuraavien ohjeiden mukaan:
- Kääntele infuusiopussia niin, että aukko on pussin yläosassa, ja napauta syöttöletkun sivua sen varmistamiseksi, että letkuun jäänyt liuos vapautuu pussissa olevaan liuokseen.
- Sekoita varovasti pyörittämällä pussia pitkittäissuunnassa 360 astetta käännetystä asennosta ainakin viisi kertaa. ÄLÄ ravista infuusiopussia.
- Toista vaiheet (i) ja (ii) vielä kolme kertaa.
Vaihe 2: KIMMTRAK-infuusioneste, liuoksen käyttökuntoon saattaminen
c. Ota 1 ml:n ruiskulla ja steriilillä neulalla tarvittavaan annokseen perustuva määrä (esitetty jäljempänä taulukossa 7) KIMMTRAK 100 mikrog / 0,5 ml -valmistetta, ja lisää se valmisteltuun 100 ml:n infuusiopussiin, jossa on natriumkloridia 9 mg/ml (0,9 %) injektioneste, liuosta, sekä ihmisen albumiinia.
d. ÄLÄ huuhtelee neulaa ja ruiskua siirron yhteydessä. Hävitä injektiopullossa oleva käyttämätön KIMMTRAK-valmiste paikallisten vaatimusten mukaisesti. Älä valmista enempää kuin yksi annos injektiopullosta.
Taulukko 7: Infuusiopussiin lisättävät KIMMTRAK-määrät
Hoitopäivä | KIMMTRAK- annos (mikrog) | KIMMTRAK- määrä (ml) |
Päivä 1 | 20 | 0,10 |
Päivä 8 | 30 | 0,15 |
Päivä 15 ja viikoittain sen jälkeen | 68 | 0,34 |
e. Sekoita infuusiopussi noudattamalla vaiheessa 1b esitettyä ohjetta.
Annnostelu
- Annostele KIMMTRAK vain infuusiona laskimoon.
- Anna infuusio välittömästi 15–20 minuutin kuluessa omalla laskimoyhteydellä. Käytä steriiliä, pyrogeenitonta vähän proteiinia sitovaa 0,2 mikronin linjasuodattimella varustettua infuusiosarjaa. Anna KIMMTRAK-infuusiopussin koko sisältö potilaalle.
- Kun KIMMTRAK-infuusio päättyy, huuhtele infuusioletku riittävällä määrällä steriiliä natriumkloridi-injektionestettä 9 mg/ml (0,9 %), sen varmistamiseksi, että koko infuusiopussin sisältö on annettu. Älä anna KIMMTRAK-valmistetta laskimoon nopeana infuusiona tai boluksena. Älä sekoita KIMMTRAK-valmistetta muiden lääkkeiden kanssa tai anna muita lääkkeitä saman laskimolinjan kautta.
Valmistetun infuusiopussin säilytys
- KIMMTRAK ei sisällä säilöntäainetta. Käyttökuntoon saattamisen jälkeen valmistetun infuusiopussin sisältö on annettava 4 tunnin kuluessa , mikä sisältää myös infuusion keston. Tämän 4 tunnin ajanjakson aikana KIMMTRAK-infuusiopussin tulee pysyä alle 30 °C:n lämpötilassa.
- Jos KIMMTRAK-infuusiopussia ei käytetä välittömästi, säilytä sitä käyttökuntoon saattamisen jälkeen jääkaapissa 2–8 °C:n lämpötilassa enintään 24 tunnin ajan, mikä sisältää myös infuusiopussin huoneenlämpöön tasautumisen ajan sekä infuusion keston.
- Kun KIMMTRAK-infuusiopussi on poistettu jääkaapista, sitä ei saa laittaa takaisin jääkaappiin. Hävitä käyttämätön KIMMTRAK-liuos suositellun säilytysajan jälkeen.
Korvattavuus
KIMMTRAK infuusiokonsentraatti, liuosta varten
100 mikrog/0,5 ml 0,5 ml
- Ei korvausta.
ATC-koodi
L01XX75
Valmisteyhteenvedon muuttamispäivämäärä
14.11.2024
Yhteystiedot
Unit 1, Sky Business Centres, Port Tunnel Business and Technology Park
D17 FY82 Dublin
Ireland
+44 (0)1235 438600
info@immunocore.com